Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(14):3856-3869. doi:10.7150/thno.25149 This issue Cite
Research Paper
Expansion of allogeneic NK cells with efficient antibody-dependent cell cytotoxicity against multiple tumors
Diego Sanchez-Martinez1,#, Nerea Allende-Vega1,2,#, Stefania Orecchioni3, Giovanna Talarico3, Amelie Cornillon1, Dang-Nghiem Vo1, Celine Rene1, Zhao-Yang Lu1, Ewelina Krzywinska1, Alberto Anel4, Eva M. Galvez5, Julian Pardo4, Bruno Robert6, Pierre Martineau6, Yosr Hicheri7, Francesco Bertolini3, Guillaume Cartron7, Martin Villalba1,2, ![]()
1. IRMB, Univ Montpellier, INSERM, CHU Montpellier, Montpellier, France.
2. IRMB, CHU Montpellier, France.
3. Laboratory of Hematology-Oncology, European Institute of Oncology, Milan, Italy.
4. University of Zaragoza/Institute of Health Research of Aragón (IIS-Aragón), Spain.
5. Instituto de Carboquimica, Consejo Superior de Investigaciones Científicas (CSIC).
6. IRCM, INSERM U1194, Univ Montpellier, France.
7. Département d'Hématologie Clinique, CHU Montpellier, Univ Montpellier, France.
# These two authors have equally contributed to this manuscript
Received 2018-1-25; Accepted 2018-4-26; Published 2018-6-14
Citation:
Sanchez-Martinez D, Allende-Vega N, Orecchioni S, Talarico G, Cornillon A, Vo DN, Rene C, Lu ZY, Krzywinska E, Anel A, Galvez EM, Pardo J, Robert B, Martineau P, Hicheri Y, Bertolini F, Cartron G, Villalba M. Expansion of allogeneic NK cells with efficient antibody-dependent cell cytotoxicity against multiple tumors. Theranostics 2018; 8(14):3856-3869. doi:10.7150/thno.25149. https://www.thno.org/v08p3856.htm
Other stylesAbstract

Monoclonal antibodies (mAbs) have significantly improved the treatment of certain cancers. However, in general mAbs alone have limited therapeutic activity. One of their main mechanisms of action is to induce antibody-dependent cell-mediated cytotoxicity (ADCC), which is mediated by natural killer (NK) cells. Unfortunately, most cancer patients have severe immune dysfunctions affecting NK activity. This can be circumvented by the injection of allogeneic, expanded NK cells, which is safe. Nevertheless, despite their strong cytolytic potential against different tumors, clinical results have been poor.
Methods: We combined allogeneic NK cells and mAbs to improve cancer treatment. We generated expanded NK cells (e-NK) with strong in vitro and in vivo ADCC responses against different tumors and using different therapeutic mAbs, namely rituximab, obinutuzumab, daratumumab, cetuximab and trastuzumab.
Results: Remarkably, e-NK cells can be stored frozen and, after thawing, armed with mAbs. They mediate ADCC through degranulation-dependent and -independent mechanisms. Furthermore, they overcome certain anti-apoptotic mechanisms found in leukemic cells.
Conclusion: We have established a new protocol for activation/expansion of NK cells with high ADCC activity. The use of mAbs in combination with e-NK cells could potentially improve cancer treatment.
Keywords: NK cells, monoclonal antibodies (mAbs), antibody-dependent cell cytotoxicity (ADCC), cancer
Introduction
Recent progress in cancer treatment is primarily related to the development of novel targeted therapies [1]. These require the identification of suitable targets that are mainly expressed by the tumor cell population and/or playing a critical role in neoplastic cell growth. Therapeutic monoclonal antibodies (mAbs) particularly illustrate this concept. Indeed, rituximab (RTX), an IgG1 mAb directed against CD20 antigen, has now become the treatment of choice for most B-cell chronic lymphocytic leukemias (B-CLL) and B-cell non-Hodgkin's lymphomas (B-NHL). The combination of RTX with conventional chemotherapy has shown better efficacy in randomized clinical trials. Similar success has been found with other cytotoxic mAbs, such as trastuzumab in breast cancer or cetuximab in colorectal carcinoma and squamous cell carcinoma of the head and neck [2, 3]. Nevertheless, mAbs alone generally have modest clinical activity. For example, the anti-CD20 mAbs RTX and obinutuzumab (OBZ; previously GA101, Roche, Genentech), when used as monotherapy in patients with relapsed follicular lymphoma (FL), have only led to short progression-free survival (PFS) [4]. Thus, there is a need to optimize their use in combination therapy.
RTX success is related to its capacity to induce Fc-antibody-dependent cell-mediated cytotoxicity (ADCC). One receptor for human IgG1 is FcγRIIIa (CD16a), which is expressed on natural killer (NK) cells and macrophages. The link between FcγRIIIa-158VF polymorphism and RTX clinical responses strongly suggests that ADCC is critical [5]. This polymorphism is located on the extra-cellular domain of FcγRIIIa, and amino-acid 158 is involved in the interaction with CH2 of human IgG1 [4]. Human IgG1 has a higher affinity for VV-NK cells compared to FF-NK cells [5]. Based on these observations, there has been an attempt to produce new anti-CD20 mAbs by either Fc mutations or by glycoengeenering that exhibit higher affinity for FcγRIIIa [4, 6]. Lowering the fucose content of the N-glycan is currently under clinical investigation in B-cell malignancies with the mAb OBZ, which shows stronger ADCC in vitro and in a lymphoma xenograft mouse model relative to RTX. It also demonstrated improved clinical activity for treating B-CLL and other B-cell malignancies [4]. OBZ is approved for first-line B-CLL in association with chlorambucil, and in combination with bendamustine for the treatment of patients with FL who relapse or are refractory to a RTX-containing regimen [4]. Initial results show that lenalidomide, which stimulates NK cell activity [7], activates NK cells in OBZ-treated patients[8].
NK cells mediate ADCC but also possess natural cytotoxicity, which is mediated by engagement of their natural cytotoxicity receptors (NCRs). These play a central role in triggering NK activation. In humans, NKp30, NKp46, and NKp80 are constitutively expressed on resting and activated NK cells [9]. The NK cell-activating receptor CD16 mediates ADCC. Hematological cancer patients possess antitumor NK cells that are unable to control disease [10, 11]. Notably, blood-borne tumor cells use different mechanisms for immune escape [12, 13], e.g., by inducing NK cell dysfunction [7, 14]. This mechanism has also been observed in a variety of patients of solid tumors [3]. In addition, NK cell differentiation may be inhibited by the presence of tumor cells, e.g., acute myeloid leukemia (AML) cells infiltrating bone marrow [15, 16]. Therefore, the failure of mAbs in monotherapy could be related to impaired NK cell function. Hence, there is a clinical interest to reactivate or replace patient NK cells [17]. Clinical-grade production of allogeneic NK cells is efficient and NK cell-mediated therapy after hematopoietic stem cell transplantation (HSCT) seems safe [16, 18, 19]. Despite the strong cytolytic potential of expanded NK cells against different tumors, clinical results have been very limited [16, 18, 19].
The combination of allogeneic NK cells with mAb could improve cancer treatment by replacing the defective effector immune cells. In addition, mAbs would effectively guide these effectors to their tumor targets. Several groups have tried this combination with varying results that could be due to deficient CD16 expression or lack of proper activation of expanded NK [20-23]. In addition, these studies did not include a systematic evaluation of the effect of these cells in combination with several mAbs on different tumors, nor did they include primary tumor cells.
The aim of this work was to generate allogeneic NK cells with strong ADCC response against different tumors and mediated by different therapeutic mAbs. In addition, NK cell production should be easily scaled up and developed with good manufacturing practices (GMP). We have produced umbilical cord blood (UCB)-derived NK cells because UCB are rapidly available, present low risk of viral transmission and have less strict requirements for HLA matching and lower risk of graft-versus-host disease (GvHD) [18]. For NK cell expansion we used Epstein-Barr virus (EBV)-transformed lymphoblastoid B cell lines as accessory cells, which induce a unique genetic reprogramming of NK cells [24]. This generates effectors that overcome the anti-apoptotic mechanism of leukemic cells [25] and that are able to eliminate tumor cells from patients with poor prognosis [26]. We show that NK cells obtained with our protocol are able to perform ADCC in vitro and in vivo. The ADCC response was induced by using different therapeutic antibodies and against multiple target cells.
Methods
Ethics statement
Experimental procedures were conducted according to the European guidelines for animal welfare (2010/63/EU). Protocols were approved by the Animal Care and Use Committee “Languedoc-Roussillon” (approval number: CEEA-LR-12163). The use of human specimens for scientific purposes was approved by the French National Ethics Committee. All methods were carried out in accordance with the approved guidelines and regulations of this committee. Written informed consent was obtained from each patient prior to surgery.
Chemicals
The D1D2 peptide has been previously described [27]. IL-2 and IL-15 were obtained from Miltenyi Biotec. To produce deglycosylated cetuximab, a commercial cetuximab solution was treated overnight with PNGAseF (Promega) at 37 °C in 50 mM sodium bicarbonate buffer (pH 7.8) at 125 U/mg of cetuximab. Deglycosilated cetuximab was purified by gel filtration using a Sephadex 75 column in PBS, and sterilized by filtration. The AF 647 Goat F(AB')2 anti human IgG (H+L) min x (BOV, HRS, MS) was from Interchim.
B-CLL patients
Data and samples from patients were collected at the Clinical Hematology Department of the CHU Montpellier, France, after patients' written consent and following French regulations. Patients were enrolled in two independent clinical programs approved by the “Comités de Protection des Personnes Sud Méditerranée I”: ref 1324 and HEMODIAG_2020 (ID-RCB: 2011-A00924-37). Samples were collected at diagnosis and kept by the CHU Montpellier [11, 28]. For analysis, peripheral blood mononuclear cells (PBMCs) were obtained by ficoll gradient and stored frozen in liquid nitrogen until use. The percentage of CD19+CD5+ cells was always higher than 90%. Other samples from hematological cancer patients were obtained from the same collections.
Cell lines
The (EBV)-transformed lymphoblastoid B cell line PLH, the hematopoietic cell lines HL60 and MV4-11 (acute myeloid leukemia), Daudi and Raji (Burkitt's lymphoma derived, CD20+), K562 (erythroleukemia), MM.1S and U266 (multiple myeloma), MEC1 (B-chronic lymphocytic leukemia) and its variants MEC-1-BCL-XL and MEC-1-MCL-1 were cultured in 10% FBS RPMI medium. The adherent cell lines Calu-1 (lung cancer), A549 (lung adenocarcinoma), SK-OV-3 (ovarian carcinoma) and SKBR3 (breast adenocarcinoma) were cultured in 10% FBS DMEM medium.
PBMC and UCBMC purification
UCBs were obtained from healthy donors from the CHU Montpellier. Prof. John de Vos is the responsible of the “Collection du Centre de Resources Biologiques du CHU de Montpellier” - http://www.chu-montpellier.fr/fr/plateformes (Identifiant BIOBANQUES - BB-0033-00031). PBMC and UCB mononuclear cells (UCBMC) were respectively collected from peripheral blood samples and UCB units using Histopaque -1077 (Sigma). Briefly, 13 mL Histopaque was added to 50 mL centrifugation tubes and 30 mL of 1/2 diluted blood in RPMI, (Invitrogen) was slowly added at the top. Tubes were centrifuged at 400 rcf for 30 min at 20 °C without brake. Mononuclear cells were collected from the interlayer white ring, washed in RPMI and suspended in RPMI medium supplemented with 10% FBS (Invitrogen).
Isolation and activation of human NK cells
Frozen UCBMCs were depleted of T cells by using EasySepTM CD3 Positive Selection Kit (STEMCELL technologies). Cells were cultured for 10 to 20 days with γ-irradiated PLH cells at 1:1 NK cell:accessory cell ratio in the presence of IL-2 (100 U/mL) and IL-15 (5 ng/mL), or with IL-2 alone (1000 U/mL). PLH cells were added every four days and fresh cytokines every two days. At the end of the process, NK cell purity (CD56+/CD3-) was always higher than 90%.
FACS analysis
For phenotype analysis, cells were stained with 7AAD (Beckman) to identify viable cells and antibodies against the surface markers CD25-FITC, CD45RO-FITC, CD69-PE, CD62L-PE, CD19-PE, CD3-PE, CD19-ECD, CD56-PECy7, CD56-APC, CD3-APC, CD45-APCAlexaFluor750, CD45RA-APCAlexaFluor750, CD16-PacificBlue, CD57-PacificBlue, CD45-KromeOrange, CD16-KromeOrange (Beckman), CD158b-FITC, CD158a-PE, CD107a-HV500 (BD Biosciences), CD158e-Vioblue (Miltenyi). 1×105-3×105 cells were incubated for 20-30 min at 4 °C with different antibodies in PBS containing 2.5% FBS. Cells were then washed and suspended in 200-250 µL of the same media. Staining was analyzed on a Gallios flow cytometer (Beckman) using the Kaluza software. Alive lymphocytes were gated using FSC/SSC and 7AAD staining. B lymphocytes (CD19+), T lymphocytes (CD3+CD56-) and NK cells (CD56+CD3-) were distinguished using, respectively, CD19, CD3 and CD56 antibodies.
NK cell-mediated cytotoxicity
Fresh or frozen (stored in liquid nitrogen) NK cells were labeled with 3 µM of CellTracker™ Violet BMQC Dye (Life Technologies) and incubated overnight with target cells at different E:T ratios. Subsequently, phosphatidylserine (PS) translocation and membrane damage were analyzed in the violet fluorescence-negative target cell population by flow cytometry using Annexin V-FITC (Immunostep) and 7AAD (BD Biosciencies) or propidium iodide (PI) as previously described [25, 29]. We consider all cells positive for annexin-V and/or PI (or 7-ADD) as dead (or dying).
In ADCC experiments, we incubated target cells with the relevant antibodies (RTX and OBZ at 10 µg/mL; daratumumab, cetixumab and trastuzumab at 5 µg/mL) for 30 min at 37 °C. To arm NK cells, we incubated them at the same concentration of antibodies before washing and incubation with target cells. EGTA was used at 1 mM to block the granular exocytosis pathway and MgCl2 at 1.5 mM to maintain the osmotic pressure.
We used the tetrazolium dye MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazomium bromide) to determinate cellular viability. We added 10 μL of MTT (5 mg/mL) to the adherent cells (100 μL of medium after 2 washes with PBS) and incubated for 1 h at 37 °C, then added 100 μL of 0.05 M HCl in isopropanol to dissolve the crystals and quantified absorption at 550 nm using a spectrophotometer.
In all experiments, we calculated the basal cell death in the absence and presence of the different mAbs. These values were subtracted from those obtained after NK cell or NK cell+mAb treatments to generate the specific natural cytotoxicity or specific ADCC, respectively. All mAbs gave very low levels (<3%) of cytotoxicity in presence of heat-inactivated serum media.
Evaluation of RTX-armed e-NK
e-NK cells (2×105) were incubated for 1 h with 10 µg/mL RTX, washed and incubated with 1:800 solution of a goat F(ab')2 anti-human IgG (H+L) for 30 min at 4 °C. After incubation, NK cells were washed with PBS and RTX binding was analyzed by FACs. As a control, cells were only stained with the goat F(ab')2 anti-human IgG.
NK degranulation assay
Briefly, 50×103 target cells per well were placed in RPMI, 10% FBS, IL-2 100 U/mL with monensin (BD Biosciences) in a 96-well V-bottom plate. NK and target cells were incubated overnight at 37°C in 5% CO2 and living cells were counted using a Muse cytometer (Millipore) with the count and viability kit (Millipore). As a control, NK cells were incubated without targets. CD107a+ NK cells were analyzed on a Gallios flow cytometer (Beckman Coulter) using 7AAD, CD45RO-FITC, CD19-PE, CD56-PECy7, CD3-APC, CD45RA-APCAlexaFluor750, CD16-KromeOrange and CD107a-HV500 (BD Biosciences). Results were analyzed using Kaluza software.
In vivo experiments
In vivo experiments were carried out using 6-8-week-old male NOD scid gamma (NSG) mice. Mice were bred and housed in pathogen-free conditions in the animal facility of the European Institute of Oncology-Italian Foundation for Cancer Research (FIRC), Institute of Molecular Oncology (Milan, Italy). For engraftment of human cells, mice were subcutaneously engrafted with 5×106 BCL-P2 or 10×106 LNH1 primary tumor cells derived from a B-cell lymphoma (BCL) patient (BCL P2) or a diffuse large B-cell lymphoma (DLBCL) patient (LNH1). At day 4, we engrafted 15 (BCL-P2) or 10 (LNH1) million e-NK cells and at day 6, mice were treated i.p. with RTX (in saline medium) 3 mg/kg once a week for 3 weeks; or with a combination of both treatments e-NK and RTX. Tumor growth was monitored at least once a week using a digital caliper, and tumor volume was calculated according to the formula: L × W2/2 (mm3), where W represents the width and L the length of the tumor mass.
Statistical analysis
Experimental figures and statistical analysis were performed using GraphPad Prism (v6.0). All statistical values are presented as * p<0.05; ** p<0.01; *** p<0.001 and **** p<0.0001. Mean values are expressed as mean plus or minus the standard error of the mean (SEM).
Results
Costimulation with the EBV lymphoblastoid PLH cell line more efficiently expands UCB NK cells for clinical use than IL-2 stimulation
Cytokines and encounter with target cells induce dissimilar gene expression on NK cells [24]. We used umbilical cord blood (UCB) cells and compared two NK cell activation/expansion protocols: one using a high dose of the cytokine IL-2 (1000 U/mL) and the other using cell costimulation. The costimulation protocol was performed with the EBV cell line PLH together with low concentrations of IL-2 (100 U/mL) and IL-15 (5 ng/mL) [9]. NK cell expansion is jeopardized by T cells, therefore we depleted them from UCB before expansion. NK cell cultures underwent massive cell death at day 12 in IL-2-driven expansion (data not shown). So, we compared different parameters that reflect NK activity at day 10:
Proliferation. Costimulation-driven expansion was considerably more efficient (Figure 1A).
Cytotoxicity. IL-2-driven expansion led to NK cells with superior natural cytotoxicity against K562 (Figure 1B), Daudi (Figure S1A) and primary CD20+ B cell lymphoma cells (BCL P2; Figure 1C). Moreover, these NK cells also showed higher ADCC activity with RTX (Figure 1C and Figure S1A). However, natural cytotoxicity gradually increased in the costimulation protocol when cells were activated for longer periods of time (Figure 1D). This correlated with a notable large-scale expansion of cells (median ± SD, IL-2 10 d (16.8 ± 22.2), costimulation 10 d (140.5 ± 235.8) and costimulation 20 d (260.9 ± 141.2), n=10).
Activation markers. Both protocols increased the expression of the activation marker CD69 (Figure S2B) and decreased CD45RA expression to that of CD45RAdim cells (Figure 2B). This was associated with an increase in the activation marker CD45RO, as previously published [10]. Costimulation maintained higher CD16 expression (Figure S2B).
Exhaustion markers. We investigated the expression of two markers that could suppress NK cell-mediated cytotoxicity: TIM-3 [30] and PD-1 [31]. While both protocols did not affect their expression, the mean fluorescence intensity (MFI) of positive populations tended to increase (Figure S2B). This probably reflects that, after long activation, some NK cells become exhausted.
Maturation and homing markers. UCB NK cells showed a low percentage of CD62L+ cells (18.1% ± 6.7%, n=3) that increased 10 d after IL-2 treatment (68.2% ± 13.5%) and costimulation (56.7% ± 11.9%). However, at day 20 post-costimulation, CD62L expression was lost (1.6% ± 0.4). IL-2-stimulated cells did not survive this long; so, we could not measure CD62L levels. CD62L is a "homing receptor" facilitating naive lymphocytes to enter secondary lymphoid tissues. Mature NK cells express low CD62L, which favors their peripheral trafficking [32].
In agreement with others [33], few naïve UCB NK cells expressed CD57 (1.2% ± 1.3%, n=3). IL-2 stimulation barely increased expression (7.3% ± 3.9%) and costimulation did not change it (0.6% ± 0.6). Longer costimulation, i.e., 20 days, also had no effect (1.6% ± 0.5%). The lack of CD57 expression did not impair NK cell cytolytic activity (see below).
In summary, costimulation led to higher numbers of activated and functional NK cells with higher CD16 expression. This prompted us to only use costimulation for the next experiments. On the other hand, IL-2 induced higher and faster cytotoxicity and could be the best option for autologous NK cell grafts.
Frozen/thawed NK cells keep their cytolytic activity
For clinical purposes, it would be advantageous to have a bank of cryopreserved expanded NK cells ready to use [34]. Compared with fresh expanded NK cells, frozen/thawed NK cells lost roughly 35% of CD16 expression and 50% of their cytolytic activity (Figure 1E). As shown in Figure 1D, 20 day-activation showed higher cytolytic activity than shorter expansions. For the next experiments, we used 20-21 days costimulation-induced expansion of UCB-derived NK cells containing more than 90% of NK cells that were kept frozen until use. Hereafter, we call them e-NK.
e-NK cells mediate ADCC against target cells with diverse CD20 levels
We observed that e-NK performed ADCC similarly on Raji and Daudi cells, which express high CD20 levels (Figure S2A), as on primary tumor cells, which express low levels (Figure S2B). Even though P2 cells probably express more CD20 than P148, they were slightly less sensitive to RTX-mediated ADCC. In fact, e-NK performed ADCC even if their natural cytotoxicity against some patient cells was low or absent (Figure 2A and Figure S2B-E). Hence, e-NK show strong ADCC with RTX independently of their natural cytotoxicity and with lower variability (Figure 2A-B). The glycoengineered mAb OBZ [4, 35] induced higher ADCC than RTX (Figure 2C and Figure S2B-C).
e-NK cells can be “armed” with mAbs to facilitate treatment
We next used e-NK cells coated with anti-CD20 mAb (“armed” e-NK cells) as an alternative to antibody-coated target cells. “CD20-armed” e-NK showed similar results to opsonizing tumor cells with anti-CD20 (Figure S3A). The presence of RTX after e-NK “arming” was visualized by using a fluorescent anti-IgG (Figure S3B). “OBZ-armed” e-NK also efficiently generated ADCC (Figure S4).
Statistical analysis of several e-NK productions on cells from 5 CLL patients did not show any differences between opsonizing targets or “arming” e-NK (Figure 3A). Moreover, the analysis of these 4 e-NK expansions on the CLL targets showed that all productions could be armed (Figure 3B). Combining all these results statistically showed that significant ADCC was mediated by e-NK (Figure 3C).
Cytotoxicity requires degranulation and cell interaction by ICAM
Primary human NK cell cytotoxicity is largely independent of degranulation [36] and resides in death receptor binding on tumor cells by ligands expressed by NK cells. In agreement with this, e-NK natural cytotoxicity was only partially diminished by the degranulation inhibitor EGTA (Figure 4A), which in contrast, largely blocked ADCC.
Figure 1
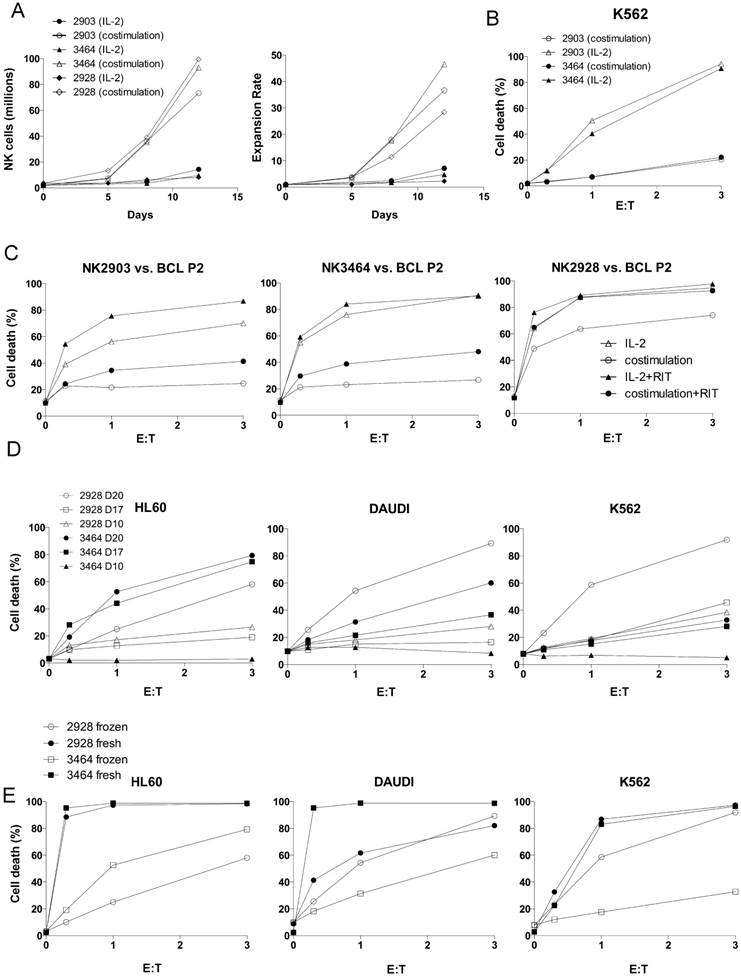
Optimization of NK cell expansion protocol. (A) Comparison of costimulation (PLH accessory cells + IL-2 100 U/mL + IL-15 5 ng/mL) and IL-2 (1000 U/mL) expansion protocols using three UCB donors (2903, 3464, 2928). (B) K562 cells were incubated overnight with costimulation- (circles) or IL-2-activated (triangles) NK cells from two different donors at different effector: target (E:T) ratios. Cell death was analyzed by 7-AAD staining. (C) BCL Patient 2 cells were incubated overnight with costimulation- or IL-2-activated NK cells from three different donors, in the presence (black symbols) or absence (white symbols) of RTX (10 µg/mL). (D) NK cells from 2 donors were expanded by costimulation for different days. At these days, NK cells were frozen. They were thawed at the same time and tested for cytotoxicity against the cell lines. (E) NK cells from 2 donors were expanded for 20 days and frozen or kept fresh before testing their cytotoxicity.
Figure 2
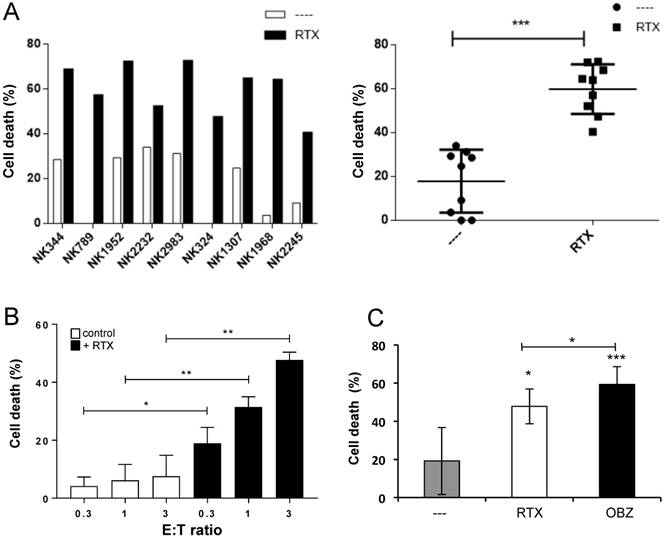
e-NK mediate ADCC with anti-CD20 mAbs. (A) PBMCs from CLL patient 148 were incubated for 1 h with 10 µg/mL of RTX and overnight with e-NK cells obtained from nine different donors at a 1:1 E:T ratio. Cell death was analyzed by 7AAD staining. The right panel shows the statistical analysis. (B) 7 e-NKs were tested against 13 CD20+ target samples at different E:T ratios as described in (A). (C) 5 e-NKs were tested against 6 CD20+ target samples at different 1:1 E:T ratios as described in (A). Graphics represent mean ± SEM. Significance was determined by paired t-test; * p ≤ 0.05, ** p ≤ 0.01 and *** p ≤ 0.001.
The interaction of NK cell-expressed LFA-1 with its target cell ligand ICAM modulates NK cell cytotoxicity [37]. Blocking this interaction with the D1D2 peptide [27] partly reduced natural cytotoxicity and almost completely abolished ADCC (Figure 4B). Therefore, our e-NK use similar mechanisms for eliminating target cells as primary human cells.
e-NK mediate ADCC with daratumumab
Next we tested if e-NK produced ADCC with the anti-CD38 daratumumab [38]. We used 3 target cells that express CD38 (MM.1S, MV4-11 and BCL-P2; Figure S5A) and observed that three different e-NK preparations showed ADCC with daratumumab (Figure S5B-D). In contrast, daratumumab failed to induce ADCC against U266 that are negative for CD38 (Figure S5A, E). Several e-NK productions efficiently performed ADCC with daratumumab on MM.1S and P2 that was statistically significant (Figure 4C).
e-NK mediate ADCC with cetuximab
Next, we analyzed if e-NK cells mediate ADCC with other mAbs used to treat solid tumors. We used the cell lines Calu-1 and A549. Calu-1 cells express more epidermal growth factor receptor (EGFR) than A549 (data not shown). Both lines are targets of the anti-EGFR cetuximab, which has been proposed for use with adoptively-transferred expanded allogeneic NK cells in clinical trials for cervical cancer [39]. In fact, in vitro and clinical data suggest that cetuximab mediates ADCC through NK cells [23, 34]. We observed a relatively large variation in the natural cytotoxicity of the different e-NK donors versus these target cells. The decrease in cell viability, as measured by MTT formation, was low (Figure S6A). However, the increase in cell death, measured by annexin-V / 7-ADD staining, was higher. This showed that e-NK had induced the initial steps of apoptosis (annexin-V staining), but longer times were required to evaluate cell viability with MTT. Cetuximab increased early apoptosis and accelerated the process of cell death, decreasing viability. Several e-NK productions efficiently performed ADCC with cetuximab on CALU-1 and A549 that was statistically significant (Figure 5A).
EGTA diminished ADCC but insignificantly (Figure S6B-C). This suggested that the mechanism of action only partly involved degranulation, indicating a possible participation of death ligand-induced apoptosis.
Figure 3
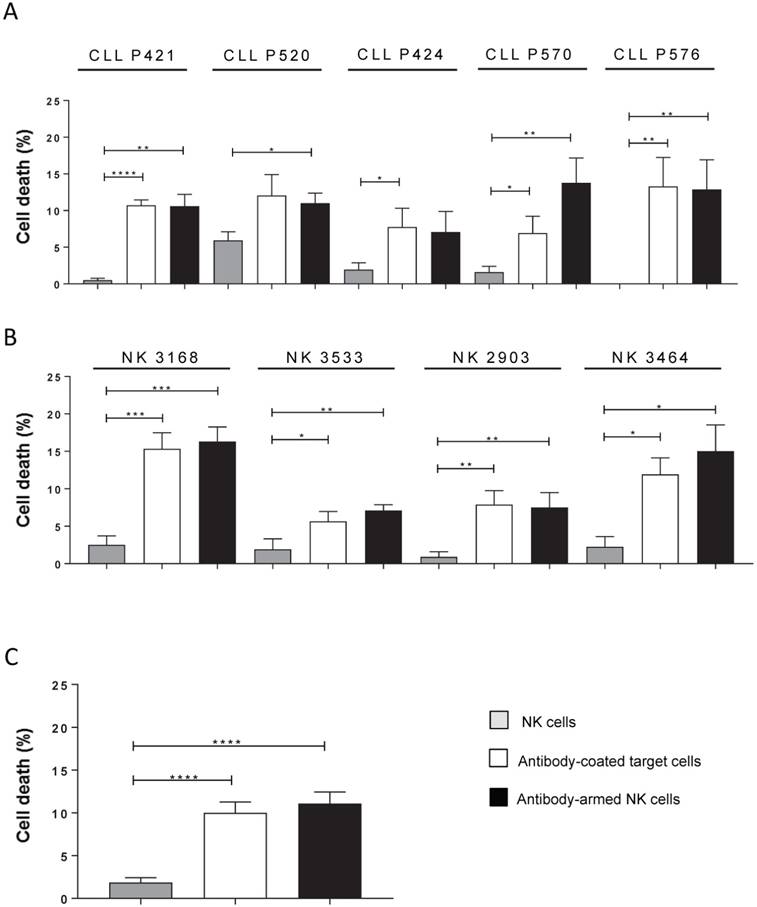
Anti-CD20-armed e-NK show ADCC activity. PBMCs from CLL samples were incubated for 1 h with 10 µg/mL of RTX and overnight with donor e-NK cells at a 3:1 E:T ratio (antibody-coated target cells condition). Alternatively, e-NK cells were incubated for 1 h with 10 µg/mL of RTX before incubating them overnight with target cells (antibody-armed NK cell condition). (A) The bars represent cell death of each individual CLL sample to 4/5 e-NK preparations. (B) The bars represent cell death of each individual e-NK preparation to 4/5 CLL samples. B-CLL cell death was analyzed by 7-AAD. Graphics represent mean ± SEM. Significance was determined by paired t-test; * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001 and **** p ≤ 0.0001.
Figure 4
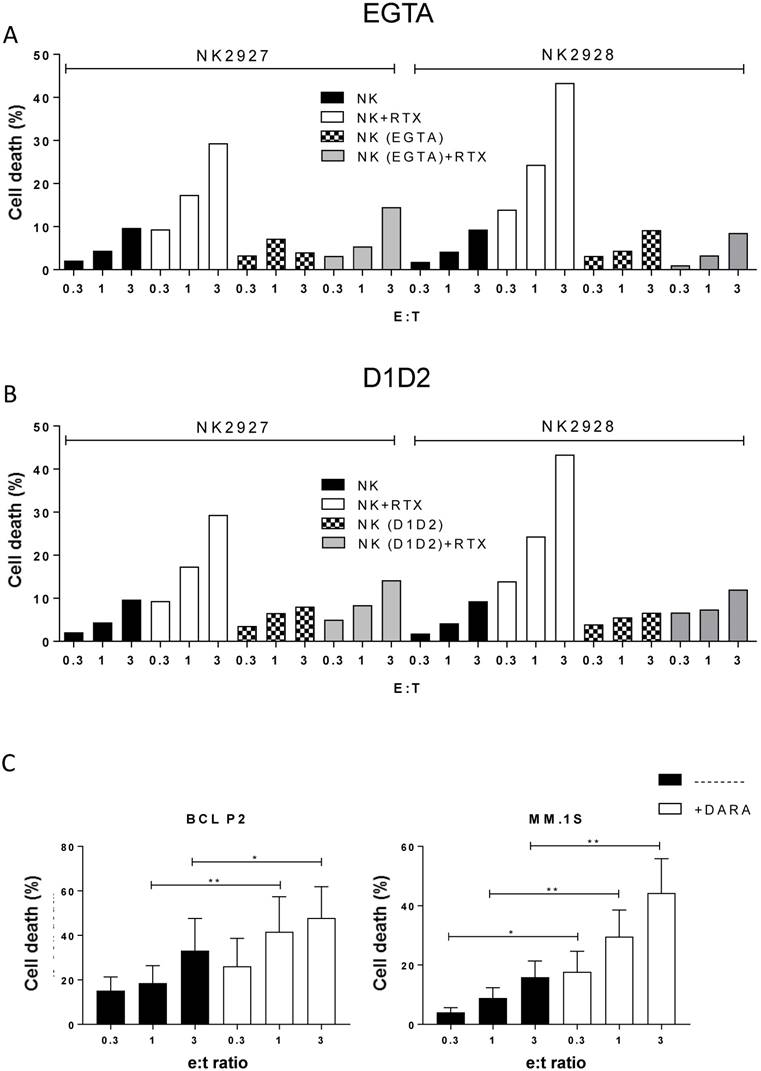
ADCC requires degranulation and LFA-1/ICAM interaction. (A-B) Daudi cells were incubated overnight with e-NK cells from two different donors and/or RTX (10 µg/mL) as described in Figure 2. Cytotoxic assays were performed also in the presence of 1 mM EGTA (A) or 15 µg/mL D1D2 protein (B). Cell death was analyzed by 7-AAD staining. (C) e-NK produced ADCC with daratumumab. Three different e-NK cell productions were tested against the CD38+ cell line MM.1S and BCL-P2 cells that express CD38. Target cells were pre-incubated for 1 h with 5 µg/mL daratumumab before overnight incubation at different E:T ratios with e-NK. Cell death was analyzed by 7-AAD staining. Graphics represent mean ± SEM. Significance was determined by paired t-test; * p ≤ 0.05, ** p ≤ 0.01 and *** p ≤ 0.001.
Figure 5
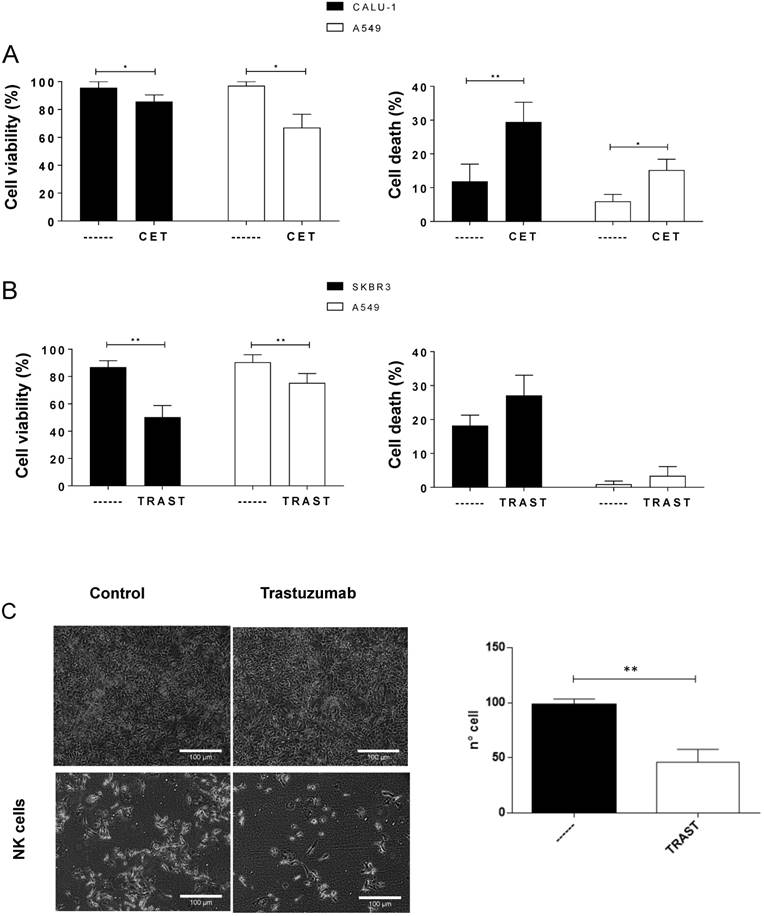
e- NK perform ADCC with cetuximab and trastuzumab against EGFR- and HER2-positive cell lines, respectively. (A-B) Tumor cells were incubated with 5 µg/mL cetuximab (A) or trastuzumab (B) for 1 h and overnight with 4 e-NK preparations at 3:1 E:T ratio. Subsequently, we measured cell viability (MTT) and cell death (annexin-V/PI). (C) 1×104 SK-OV-3 cells were plated and 24 h later treated with trastuzumab and cultured with 5×105 NK cells. After 6 days, medium was removed, and cells were fixed and stained. The right graph shows the statistical analysis comparing cells incubated with e-NK alone or with trastuzumab. Two areas of 2 different experiments were counted and the mean of cells/area is depicted in the graphic. Graphics represent mean ± SEM. Significance was determined by paired t-test; * p ≤ 0.05, ** p ≤ 0.01 and *** p ≤ 0.001.
e-NK mediate ADCC with trastuzumab
We next tested the anti-HER2 mAb trastuzumab on SK-BR-3 cells, which express high HER2, and A549 cells, which express low levels. Under this condition, e-NK performed ADCC in both cell lines by decreasing viability or increasing apoptosis (Figure S7A). Natural cytotoxicity, as well as ADCC, heavily depended on degranulation because EGTA largely decreased both (Figure S7A-B). Statistical analysis of several e-NK productions on SK-BR-3 and A549 showed that cell viability was significantly reduced and apoptosis tended to increase, although this was not statistically significant. This suggested that e-NK efficiently performed ADCC with trastuzumab (Figure 5B). However, the increase in apoptosis was not statistically significant. Finally, we extended this study to the ovarian cell line SK-OV-3 that was resistant to both natural cytotoxicity and ADCC during short treatment (data not shown). A 6-day treatment revealed that NK cells, mainly with trastuzumab, efficiently killed these cells (Figure 5C).
Figure 6
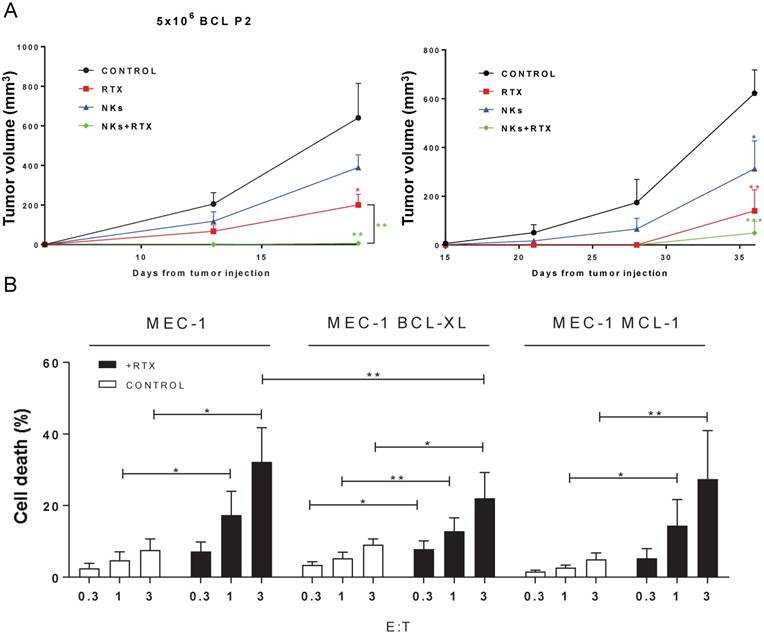
e-NK show ADCC in vivo and overcome mechanisms of drug resistance. (A) 5 NSG mice/group were subcutaneously engrafted with 5×106 BCLP2 (left) or 10×106 LNH1 (right) cells and treated with e-NK and/or RTX. (B) e-NK cell-induced ADCC overcome anti-apoptotic mechanisms of drug resistance. CD20+ MEC-1 cells overexpressing BCL-XL and MCL1 were incubated with RTX (10 µg/mL). After 1 h, e-NK cells from 3 different donors were added overnight. Cell death was analyzed by 7AAD/annexin-V labeling. Graphics represent mean ± SEM. Significance was determined by paired t-test; * p ≤ 0.05, ** p ≤ 0.01 and *** p ≤ 0.001.
e-NK mediated ADCC in vivo
We next evaluated e-NK activity in vivo by engrafting primary tumor cells from a B-cell lymphoma (BCL) patient (P2) or from a diffuse large B-cell lymphoma (DLBCL) patient (LNH1) into NSG mice. NSG mice have a complete null mutation by knockout of the γ chain of the interleukin 2 receptor (Il2rγ), which is a common component of the cell surface receptors for several cytokines, including IL-2 and IL-15. Therefore, the signaling pathways for these cytokines are blocked in Il2rγ knockout mice. They should lack functional NK cells, which require IL15 signaling to develop. Four days later, mice were engrafted with e-NK and, 2 days later, treated with RTX; the latter decreased tumor growth (Figure 6A), showing that RTX possesses direct, non-effector functions independently of NK cell-mediated effects. While e-NK also decreased tumor growth (Figure 6A), co-treatment was more effective, protecting all mice from BCL cells and 4 out of 5 mice from DLBCL cells.
e-NK cells showed ADCC against chemoresistant cells
EBV-activated NK cells overcome anti-apoptotic mechanisms active in chemoresistant cells [25, 26]. Overexpression of BCL-XL and MCL1 are common features of several hematological cancers [40]. Jurkat cells over-expressing BCL-XL are resistant to doxorubicin and to soluble TRAIL [41]. MCL1 over-expression protects them from ibrutinib cytotoxicity [42]. e-NK killed the CD20+ B-CLL cell line MEC-1 overexpressing BCL-XL or MCL1, neither of which conferred protection against ADCC compared to wild type cells (Figure S8). Several e-NK productions kill chemoresistant cell lines to statistically significant levels (Figure 6B). However, BCL-XL overexpression significantly decreases ADCC at high E:T ratios. This suggests that chemoresistance could partially protect tumor cells from e-NK-mediated ADCC, but not from e-NK natural cytotoxicity.
Discussion
Clinical mAbs fail to improve prognosis in a large number of patients. This could be due to the impairment of NK cells observed in most cancer patients [3, 16, 18]. Therefore, restoring this immune function should improve mAb clinical benefits. We focused on developing a protocol to obtain NK cells in sufficient number and with high ADCC activity together with different therapeutic mAbs. From UCB-derived NK, we produced e-NK that only partially lost ADCC function after cryopreservation (Figure 1) and preserved ADCC in vivo (Figure 6). e-NK do not require relatively high Ag levels to perform ADCC, since they were effective with different cell lines expressing variable Ag levels.
The coupling of mAbs and e-NK should synergize in several clinical contexts. First, e-NKs should bypass NK dysfunction by increasing mAb-induced ADCC in patients with immune defects. Second, the clinical activity of NK cells is uncertain in solid cancers [16, 18]. Probably these effectors scarcely recognize solid tumor targets in vivo and/or fail to infiltrate these tumors—e.g., NK have been detected in the tumor stroma but not within the tumor lesion in some cases [43-45]. Moreover, the adoptive transfer of autologous NK cells in patients as single therapy maintained high levels of circulating NK cells but did not mediate tumor regression [3, 18, 46]. mAbs should recruit e-NK to the selected targets and can also facilitate target elimination by favoring the recognition of opsonized cells. In fact, haploidentical NK cells combined with anti-GD2 mAb therapy has shown promising antitumor activity in pediatric recurrent/refractory neuroblastoma [47, 48]. Third, e-NKs overcome anti-apoptotic mechanisms active in leukemic cells (Figure 6B and [25]), allowing elimination of tumor cells from patients with poor prognosis [26].
e-NK could also have anti-tumor activity per se. First, high numbers of tumor-infiltrating NK correlates with a better prognosis in some tumors [16]. Second, NK are the first lymphocytes to recover after HSCT including after umbilical cord blood transplantation (UCBT). The speed of recovery correlates with the prognosis [18]. In spite of these findings, NK cell adoptive immunotherapy has provided clinical benefit. Perhaps current expansion protocols fail to produce enough NK cells to support clinical success or generate cells with impaired activity [16]. An inconvenience of engraftment of allogeneic expanded NK cell is their low survival in vivo [49]. The persistence of ex vivo haploidentical IL-2-activated and -expanded NK-DLIs reaches a maximum of 7 days in lymphoma patients [50]. This leaves grafted NK cells little time to eliminate their targets. The advantage is that NK will be less likely to generate the clinical problems found with CAR-T cells, which produce some chronic effects related to their long-term persistence ([51]; http://www.medscape.com/viewarticle/876591). One of the main concerns in using allogeneic immune cells is the incidence of GVHD. Allogeneic NK cells infusion is well tolerated in cancer patients [3, 18] and the severity of aGVHD correlates with impaired reconstitution of the NK cell compartment [52]. To our knowledge, engraftment of NK cells has been linked to GVHD only when combined to HLA-matched, T-cell-depleted nonmyeloablative peripheral blood stem cell transplantation [53]. NK cells likely contributed to GVHD in this setting by augmenting underlying T-cell alloreactivity [53].
An interesting alternative to allogeneic NK is KIR/KIRL blocking antibodies that activate endogenous NK cells [54]. This approach has the inconvenience that cancer patient NK cells are hyporeactive [3, 16, 18], suggesting that they are too inefficient to totally eliminate tumors. Moreover, recent clinical data suggest that such antibodies modify the endogenous NK repertoire. This would make KIR-expressing NK cells, which are those with higher cytolytic activity, hyporeactive [55]. This raises concerns about the clinical use of these antibodies. There are other ways to activate endogenous NK cells, such as the use of lenalidomide (LEN) [7, 8]. Preliminary results from the Phase Ib/II clinical trial GALEN suggest that LEN could facilitate OBZ-mediated NK cell activation [8], as was observed with RTX [56].
In collaboration with the University Hospital of Montpellier, we wish to test the clinical efficiency of e-NK in lymphoma patients resistant to standard treatments, including RTX.
Abbreviations
ADCC: antibody-dependent cell-mediated cytotoxicity; AML: acute myeloid leukemia; B-CLL: B-cell chronic lymphocytic leukemia; B-NHL: B-cell non-Hodgkin's lymphoma; BCL: B-cell lymphoma; DLBCL: Diffuse large B-cell lymphoma; EBV: Epstein-Barr virus; EGFR: epidermal growth factor receptor; FL: follicular lymphoma; GMP: good manufacturing practices; GvHD: graft-versus-host disease; HSCT: hematopoietic stem cell transplantation; LEN: lenalidomide; NCRs: natural cytotoxicity receptors; NK cells: natural killer cells; OBZ: obinutuzumab; PFS: progression-free survival; RTX: rituximab; UCB: umbilical cord blood; UCBT: umbilical cord blood transplantation; e-NK: expanded NK cells; mAbs: monoclonal antibodies.
Supplementary Material
Supplementary figures.
Acknowledgements
The authors are in debt to Drs. O. Gonzalo and I. Marzo, Doctoral Thesis, University of Zaragoza, 2017 for giving us the BCL-XL and MCL1-overexpressing Jurkat cells. We acknowledge the imaging facility MRI, member of the national infrastructure France-BioImaging supported by the French National Research Agency (ANR-10-INBS-04, «Investments for the future»). The Région Languedoc Roussillon supports the clinical data and samples (HEMODIAG_2020). We gratefully thank Dr. Robert A. Hipskind for his critical revision of this manuscript.
Funding
This work was supported by an AOI from the CHU Montpellier (N°221826;GC/MV), La Ligue Regionale contre le Cancer (GC), Fondation de France (0057921;MV), the PRT-K program 2018 (MV/GC/BR), the “Investissements d'avenir” Grant LabEx MAbImprove: ANR-10-LABX-53 (GC/PM/BR) and the NK 001 projet financed by the "Fonds Europeen de Developpement Regional (FEDER-FSE-IEJ 2014/2020) et par la region Occitanie Pyrénées-Méditerranée, SAF2014- 54763-C2-1-R (JP), SAF2017-83120-C2-1-R (JP) and SAF2014-54763-C2-2-R (EMG) from Spanish Ministry of Economy and Competitiveness.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Neves H, Kwok HF. Recent advances in the field of anti-cancer immunotherapy. BBA clinical. 2015;3:280-8
2. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12:278-87
3. Hu Z. Overcome the Impairment of NK Cells for Icon and Antibody Immunotherapy of Cancer. Journal of Immune Based Therapies, Vaccines and Antimicrobials. 2013;2:1-8
4. Cartron G, Watier H. Obinutuzumab: what is there to learn from clinical trials? Blood. 2017;130:581-9
5. Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P. et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754-8
6. Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov. 2009;8:226-34
7. Giuliani M, Janji B, Berchem G. Activation of NK cells and disruption of PD-L1/PD-1 axis: two different ways for lenalidomide to block myeloma progression. Oncotarget. 2017;8:24031-44
8. Vo DN, Alexia C, Allende-Vega N, Morschhauser F, Houot R, Menard C. et al. NK cell activation and recovery of NK cell subsets in lymphoma patients after obinutuzumab and lenalidomide treatment. Oncoimmunology. 2018;7:e1409322
9. Anel A, Aguilo JI, Catalan E, Garaude J, Rathore MG, Pardo J. et al. Protein Kinase C-theta (PKC-theta) in Natural Killer Cell Function and Anti-Tumor Immunity. Frontiers in immunology. 2012;3:187
10. Krzywinska E, Allende-Vega N, Cornillon A, Vo D, Cayrefourcq L, Panabieres C. et al. Identification of anti tumor cells carrying natural killer (NK) cell antigens in patients with hematological cancers. EBioMedicine. 2015;2:1364-76
11. Krzywinska E, Cornillon A, Allende-Vega N, Vo DN, Rene C, Lu ZY. et al. CD45 Isoform Profile Identifies Natural Killer (NK) Subsets with Differential Activity. PLoS One. 2016;11:e0150434
12. Villalba M, Rathore MG, Lopez-Royuela N, Krzywinska E, Garaude J, Allende-Vega N. From tumor cell metabolism to tumor immune escape. Int J Biochem Cell Biol. 2013;45:106-13
13. Villalba M, Lopez-Royuela N, Krzywinska E, Rathore MG, Hipskind RA, Haouas H. et al. Chemical metabolic inhibitors for the treatment of blood-borne cancers. Anti-cancer agents in medicinal chemistry. 2014;14:223-32
14. Baier C, Fino A, Sanchez C, Farnault L, Rihet P, Kahn-Perles B. et al. Natural Killer Cells Modulation in Hematological Malignancies. Frontiers in immunology. 2013;4:459
15. Ambrosini P, Loiacono F, Conte R, Moretta L, Vitale C, Mingari MC. IL-1beta inhibits ILC3 while favoring NK-cell maturation of umbilical cord blood CD34(+) precursors. Eur J Immunol. 2015;45:2061-71
16. Moretta L, Pietra G, Vacca P, Pende D, Moretta F, Bertaina A. et al. Human NK cells: From surface receptors to clinical applications. Immunology letters. 2016;178:15-9
17. Muntasell A, Ochoa MC, Cordeiro L, Berraondo P, Lopez-Diaz de Cerio A, Cabo M. et al. Targeting NK-cell checkpoints for cancer immunotherapy. Curr Opin Immunol. 2017;45:73-81
18. Sarvaria A, Jawdat D, Madrigal JA, Saudemont A. Umbilical Cord Blood Natural Killer Cells, Their Characteristics, and Potential Clinical Applications. Frontiers in immunology. 2017;8:329
19. Kottaridis PD, North J, Tsirogianni M, Marden C, Samuel ER, Jide-Banwo S. et al. Two-Stage Priming of Allogeneic Natural Killer Cells for the Treatment of Patients with Acute Myeloid Leukemia: A Phase I Trial. PLoS One. 2015;10:e0123416
20. Besser MJ, Shoham T, Harari-Steinberg O, Zabari N, Ortenberg R, Yakirevitch A. et al. Development of allogeneic NK cell adoptive transfer therapy in metastatic melanoma patients: in vitro preclinical optimization studies. PLoS One. 2013;8:e57922
21. Deng X, Terunuma H, Terunuma A, Takane T, Nieda M. Ex vivo-expanded natural killer cells kill cancer cells more effectively than ex vivo-expanded gammadelta T cells or alphabeta T cells. International immunopharmacology. 2014;22:486-91
22. Voskens CJ, Watanabe R, Rollins S, Campana D, Hasumi K, Mann DL. Ex-vivo expanded human NK cells express activating receptors that mediate cytotoxicity of allogeneic and autologous cancer cell lines by direct recognition and antibody directed cellular cytotoxicity. Journal of experimental & clinical cancer research: CR. 2010;29:134
23. Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Frontiers in immunology. 2015;6:368
24. Sanchez-Martinez D, Krzywinska E, Rathore MG, Saumet A, Cornillon A, Lopez-Royuela N. et al. All-trans retinoic acid (ATRA) induces miR-23a expression, decreases CTSC expression and granzyme B activity leading to impaired NK cell cytotoxicity. Int J Biochem Cell Biol. 2014;49:42-52
25. Sanchez-Martinez D, Azaceta G, Muntasell A, Aguilo N, Nunez D, Galvez EM. et al. Human NK cells activated by EBV lymphoblastoid cells overcome anti-apoptotic mechanisms of drug resistance in haematological cancer cells. Oncoimmunology. 2015;4:e991613
26. Sanchez-Martinez D, Lanuza PM, Gomez N, Muntasell A, Cisneros E, Moraru M. et al. Activated Allogeneic NK Cells Preferentially Kill Poor Prognosis B-Cell Chronic Lymphocytic Leukemia Cells. Frontiers in immunology. 2016;7:454
27. Nunez D, Domingo MP, Sánchez-Martínez D, Cebolla V, Chiou A, Velázquez-Campoy A. et al. Recombinant production of human ICAM-1 chimeras by single step on column refolding and purification. Process in Biochemistry. 2013;48:708-15
28. Allende-Vega N, Krzywinska E, Orecchioni S, Lopez-Royuela N, Reggiani F, Talarico G. et al. The presence of wild type p53 in hematological cancers improves the efficacy of combinational therapy targeting metabolism. Oncotarget. 2015;6:19228-45
29. Aguilo JI, Garaude J, Pardo J, Villalba M, Anel A. Protein kinase C-theta is required for NK cell activation and in vivo control of tumor progression. J Immunol. 2009;182:1972-81
30. Ndhlovu LC, Lopez-Verges S, Barbour JD, Jones RB, Jha AR, Long BR. et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood. 2012;119:3734-43
31. Benson DM Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B. et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. 2010;116:2286-94
32. Luetke-Eversloh M, Killig M, Romagnani C. Signatures of human NK cell development and terminal differentiation. Frontiers in immunology. 2013;4:499
33. Nielsen CM, White MJ, Goodier MR, Riley EM. Functional Significance of CD57 Expression on Human NK Cells and Relevance to Disease. Frontiers in immunology. 2013;4:422
34. Trivedi S, Srivastava RM, Concha-Benavente F, Ferrone S, Garcia-Bates TM, Li J. et al. Anti-EGFR Targeted Monoclonal Antibody Isotype Influences Antitumor Cellular Immunity in Head and Neck Cancer Patients. Clin Cancer Res. 2016;22:5229-37
35. Evans SS, Clemmons AB. Obinutuzumab: A Novel Anti-CD20 Monoclonal Antibody for Chronic Lymphocytic Leukemia. Journal of the advanced practitioner in oncology. 2015;6:370-4
36. Zhu Y, Huang B, Shi J. Fas ligand and lytic granule differentially control cytotoxic dynamics of Natural Killer cell against cancer target. Oncotarget. 2016
37. Barber DF, Faure M, Long EO. LFA-1 contributes an early signal for NK cell cytotoxicity. J Immunol. 2004;173:3653-9
38. Shallis RM, Terry CM, Lim SH. The multi-faceted potential of CD38 antibody targeting in multiple myeloma. Cancer Immunol Immunother. 2017
39. Veluchamy JP, Heeren AM, Spanholtz J, van Eendenburg JD, Heideman DA, Kenter GG. et al. High-efficiency lysis of cervical cancer by allogeneic NK cells derived from umbilical cord progenitors is independent of HLA status. Cancer Immunol Immunother. 2017;66:51-61
40. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J. et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899-905
41. De Miguel D, Basanez G, Sanchez D, Malo PG, Marzo I, Larrad L. et al. Liposomes decorated with Apo2L/TRAIL overcome chemoresistance of human hematologic tumor cells. Molecular pharmaceutics. 2013;10:893-904
42. Bojarczuk K, Sasi BK, Gobessi S, Innocenti I, Pozzato G, Laurenti L. et al. BCR signaling inhibitors differ in their ability to overcome Mcl-1-mediated resistance of CLL B cells to ABT-199. Blood. 2016;127:3192-201
43. Balsamo M, Vermi W, Parodi M, Pietra G, Manzini C, Queirolo P. et al. Melanoma cells become resistant to NK-cell-mediated killing when exposed to NK-cell numbers compatible with NK-cell infiltration in the tumor. Eur J Immunol. 2012;42:1833-42
44. Carrega P, Morandi B, Costa R, Frumento G, Forte G, Altavilla G. et al. Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in CD56 bright CD16(-) cells and display an impaired capability to kill tumor cells. Cancer. 2008;112:863-75
45. Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N. et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin Cancer Res. 2011;17:678-89
46. Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res. 2011;17:6287-97
47. Federico SM, McCarville MB, Shulkin BL, Sondel PM, Hank JA, Hutson P. et al. A Pilot Trial of Humanized Anti-GD2 Monoclonal Antibody (hu14.18K322A) with Chemotherapy and Natural Killer Cells in Children with Recurrent/Refractory Neuroblastoma. Clin Cancer Res. 2017;23:6441-9
48. Talleur AC, Triplett BM, Federico S, Mamcarz E, Janssen W, Wu J. et al. Consolidation Therapy for Newly Diagnosed Pediatric Patients with High-Risk Neuroblastoma Using Busulfan/Melphalan, Autologous Hematopoietic Cell Transplantation, Anti-GD2 Antibody, Granulocyte-Macrophage Colony-Stimulating Factor, Interleukin-2, and Haploidentical Natural Killer Cells. Biol Blood Marrow Transplant. 2017;23:1910-7
49. Shevtsov M, Multhoff G. Immunological and Translational Aspects of NK Cell-Based Antitumor Immunotherapies. Frontiers in immunology. 2016;7:492
50. Bachanova V, Burns LJ, McKenna DH, Curtsinger J, Panoskaltsis-Mortari A, Lindgren BR. et al. Allogeneic natural killer cells for refractory lymphoma. Cancer Immunol Immunother. 2010;59:1739-44
51. Ramos CA, Savoldo B, Dotti G. CD19-CAR trials. Cancer journal. 2014;20:112-8
52. Ullrich E, Salzmann-Manrique E, Bakhtiar S, Bremm M, Gerstner S, Herrmann E. et al. Relation between Acute GVHD and NK Cell Subset Reconstitution Following Allogeneic Stem Cell Transplantation. Frontiers in immunology. 2016;7:595
53. Shah NN, Baird K, Delbrook CP, Fleisher TA, Kohler ME, Rampertaap S. et al. Acute GVHD in patients receiving IL-15/4-1BBL activated NK cells following T-cell-depleted stem cell transplantation. Blood. 2015;125:784-92
54. Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A. et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood. 2012;120:4317-23
55. Carlsten M, Korde N, Kotecha R, Reger R, Bor S, Kazandjian D. et al. Checkpoint Inhibition of KIR2D with the Monoclonal Antibody IPH2101 Induces Contraction and Hyporesponsiveness of NK Cells in Patients with Myeloma. Clin Cancer Res. 2016;22:5211-22
56. Lagrue K, Carisey A, Morgan DJ, Chopra R, Davis DM. Lenalidomide augments actin remodeling and lowers NK-cell activation thresholds. Blood. 2015;126:50-60
Author contact
![]() Corresponding author: martin.villalbafr (MV)
Corresponding author: martin.villalbafr (MV)
Citation styles
APA
Sanchez-Martinez, D., Allende-Vega, N., Orecchioni, S., Talarico, G., Cornillon, A., Vo, D.N., Rene, C., Lu, Z.Y., Krzywinska, E., Anel, A., Galvez, E.M., Pardo, J., Robert, B., Martineau, P., Hicheri, Y., Bertolini, F., Cartron, G., Villalba, M. (2018). Expansion of allogeneic NK cells with efficient antibody-dependent cell cytotoxicity against multiple tumors. Theranostics, 8(14), 3856-3869. https://doi.org/10.7150/thno.25149.
ACS
Sanchez-Martinez, D.; Allende-Vega, N.; Orecchioni, S.; Talarico, G.; Cornillon, A.; Vo, D.N.; Rene, C.; Lu, Z.Y.; Krzywinska, E.; Anel, A.; Galvez, E.M.; Pardo, J.; Robert, B.; Martineau, P.; Hicheri, Y.; Bertolini, F.; Cartron, G.; Villalba, M. Expansion of allogeneic NK cells with efficient antibody-dependent cell cytotoxicity against multiple tumors. Theranostics 2018, 8 (14), 3856-3869. DOI: 10.7150/thno.25149.
NLM
Sanchez-Martinez D, Allende-Vega N, Orecchioni S, Talarico G, Cornillon A, Vo DN, Rene C, Lu ZY, Krzywinska E, Anel A, Galvez EM, Pardo J, Robert B, Martineau P, Hicheri Y, Bertolini F, Cartron G, Villalba M. Expansion of allogeneic NK cells with efficient antibody-dependent cell cytotoxicity against multiple tumors. Theranostics 2018; 8(14):3856-3869. doi:10.7150/thno.25149. https://www.thno.org/v08p3856.htm
CSE
Sanchez-Martinez D, Allende-Vega N, Orecchioni S, Talarico G, Cornillon A, Vo DN, Rene C, Lu ZY, Krzywinska E, Anel A, Galvez EM, Pardo J, Robert B, Martineau P, Hicheri Y, Bertolini F, Cartron G, Villalba M. 2018. Expansion of allogeneic NK cells with efficient antibody-dependent cell cytotoxicity against multiple tumors. Theranostics. 8(14):3856-3869.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.