Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results and discussion
Conclusions
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(14):3949-3963. doi:10.7150/thno.26161 This issue Cite
Research Paper
Polylactide-tethered prodrugs in polymeric nanoparticles as reliable nanomedicines for the efficient eradication of patient-derived hepatocellular carcinoma
Hangxiang Wang1,2, ![]() , Liqian Zhou1, Ke Xie1, Jiaping Wu1, Penghong Song1, Haiyang Xie1, Lin Zhou1, Jialin Liu2, Xiao Xu1, Youqing Shen3, Shusen Zheng1,
, Liqian Zhou1, Ke Xie1, Jiaping Wu1, Penghong Song1, Haiyang Xie1, Lin Zhou1, Jialin Liu2, Xiao Xu1, Youqing Shen3, Shusen Zheng1, ![]()
1. The First Affiliated Hospital; Key Laboratory of Combined Multi-Organ Transplantation, Ministry of Public Health; Key Laboratory of Organ Transplantation of Zhejiang Province, School of Medicine; Zhejiang University, Hangzhou 310003, P. R. China.
2. Shenzhen Key Laboratory of Hepatobiliary Disease, Shenzhen Third People's Hospital, Shenzhen 518112, P. R. China
3. Center for Bionanoengineering and State Key Laboratory of Chemical Engineering, Department of Chemical and Biological Engineering, Zhejiang University, Hangzhou, 310027, P. R. China.
Received 2018-3-19; Accepted 2018-6-2; Published 2018-6-24
Abstract

Nanomedicines have been extensively explored for cancer treatment, and their efficacies have arguably been proven in various cancer cell-derived xenograft (CDX) mouse models. However, they generally fail to show such therapeutic advantages in patients because of the huge pathological differences between human tumors and CDX models.
Methods: In this study, we fabricated colloidal ultrastable nanomedicines from polymeric prodrugs and compared the therapeutic efficacies in hepatocellular carcinoma (HCC) CDX and clinically relevant patient-derived xenograft (PDX) mouse models, which closely mimic human tumor pathological properties. Working towards this goal, we esterified a highly potent SN38 (7-ethyl-10-hydroxycamptothecin) agent using oligo- or polylactide (oLA or PLA) segments with varying molecular weights.
Results: The resulting SN38 conjugates assembled with polyethylene glycol-block-polylactic acid to form systemically injectable nanomedicines. With increasing PLA chain length, the SN38 conjugates showed extended retention in the nanoparticles and superior antitumor activity, completely eradicating xenografted tumors in both mouse models. Our data implicate that these small-sized and ultrastable nanomedicines might also efficaciously treat cancer in patients. More interestingly, the systemically delivered nanomedicines notably alleviated the incidence of bloody diarrhea.
Conclusion: Our studies demonstrate that the appropriate molecular editing of anticancer drugs enables the generation of better tolerated cytotoxic nanotherapy for cancer, which represents a potentially useful scaffold for further clinical translation.
Keywords: cancer nanomedicine, polylactide, SN38 prodrug, self-assembly, patient-derived xenograft model
Introduction
Developing nanoparticle (NP)-mediated delivery systems offers great promise for efficacious cancer therapy, and these approaches can improve the efficacy and mitigate the side effects of encapsulated therapeutics [1-6]. To date, various nanostructured carriers have been leveraged to effectively deliver versatile therapeutics to tumor lesions [7-14]. Nanomedicines that achieve long-term systemic circulation and appropriate particle sizes (typically smaller than 200 nm) are expected to passively accumulate in the target solid tumors because of the leaky and disorganized tumor vasculature, coupled with impaired tumor lymphatic drainage [15]. Such structural and functional abnormalities can produce enhanced permeability and retention (EPR), thus potentiating the therapeutic efficacy of nanomedicines relative to that of free drugs in numerous preclinical animal models [16, 17]. Physically entrapping therapeutic molecules into polymer matrices via a simple self-assembly process has been actively explored for the construction of nanoscopic objects for precise drug delivery [18-23]. In line with this, a collection of nanomedicines have been approved for clinical use and many are undergoing clinical and preclinical development [24]. Extensive efforts from the past decade have arguably validated the potential of nanomedicines in a variety of established cancer cell-derived xenograft (CDX) mouse models. However, clinical trials show that these nanomedicines fail to yield better survival in cancer patients than solvent-based free drug formulations [25, 26]. This great discrepancy obtained from CDX models and clinical data implies that human cancers are more complex and heterogeneous than we anticipated [26]. In addition, heterogeneous EPR effects in patients may result in poor NP delivery. Consequently, one could consider that CDX models, which are routinely used in the cancer field, might be oversimplified and inadequate to predict the therapeutic efficacy of nanomedicines.
When passaged in mice, patient-derived tumor xenograft (PDX) models retain their biological stability and share histologic and genetic properties similar to their donor tumor [27, 28]. These attributes confer PDX models with overwhelming improvements over conventional CDX models and reflect the greater fidelity and real therapeutic clinical outcomes of drugs that are under preclinical evaluation [29, 30]. For these reasons, PDX models therefore represent useful scaffolds for evaluating the effects of therapeutic drugs as well as for assessing EPR-mediated nanoparticle accumulation in tumors. However, despite these advantages, few reports have been published on the use of PDX models for the evaluation of anticancer drugs, especially with regard to nanomedicines [31-33].
SN38 (7-ethyl-10-hydroxycamptothecin) is a potent inhibitor of DNA topoisomerase I [34-36]. However, this agent exhibits dose-limiting toxicity and extremely low solubility in water, thus hampering its clinical use. Accordingly, CPT-11 (also called irinotecan) was developed by linking with the bispiperidine moiety to generate a water-soluble SN38 derivative, which is now in clinical use [37]. In the body, active SN38 is regenerated with the help of carboxylesterases, but the enzymatic conversion is inefficient (usually less than 8% of dosed CPT-11). However, SN38 exhibits 100- to 1000-fold higher potency in in vitro cytotoxic activity tests than CPT-11 against a broad spectrum of cancer cell lines. Therefore, we can reasonably envision that this SN38 molecule can be directly harnessed for nanoparticle formulation, bypassing the inefficient activation of the CPT-11 prodrug and improving the therapeutic index [38]. Unfortunately, nanoformulation of SN38 into conventional polymeric NPs poses a formidable challenge because of the intrinsically planar structure and moderate polarity of this molecule [35, 39-41]. Previous studies demonstrated that rational esterification of the phenolic hydroxyl group of this molecule can be used to generate new chemical entities, enabling their self-assembly or coassembly with other matrices [34, 35, 42-47]. As one of the approaches for chemical derivatization of drug, biocompatible oligo- or polylactides have been exploited to couple with various therapeutic drugs to augment the compatibility of drug compounds with polymer matrices [48-51]. Enhancing drug retention within nanoplatforms could remarkably hinder the burst and premature release of therapeutics during systemic circulation, thereby improving the EPR effect in tumors [52].
Inspired by these studies, as well as to fully explore the utility of the SN38 agent and to evaluate the potential of nanomedicines in PDX models, a series of oligo- or polylactide (oLA or PLA)-tethered derivatives (termed oLAn-SN38 or PLAn-SN38) was synthesized using the carboxyl-terminated polymer backbone as a building block (Figure 1). Subsequent nanoformulation of these conjugates with the polyethylene glycol-block-polylactic acid (PEG-PLA) matrix resulted in small-sized and ultrastable nanomedicines suited for preclinical studies. We further used two hepatocellular carcinoma (HCC) models, including a human HCC CDX model and a PDX model, to evaluate their in vivo therapeutic potential. The efficient eradication of patient-derived HCC tumors was demonstrated using SN38-formulated nanomedicines. Finally, we performed animal studies to examine side effects induced by CPT-11 such as bloody diarrhea, as well as the effects of these nanomedicines in healthy mice.
(A) Schematic illustration of the assembly of oligo- or polylactide-tethered SN38 prodrugs with clinically approved amphiphilic copolymers (e.g., PEG-PLA) to form stable nanoparticles. (B) Chemical structure and synthetic scheme of oLAn-SN38 conjugate 1 (n = 8) or PLAn-SN38 conjugates 2-4 (n = 17, 36, and 71, respectively). SN38 was tethered to the carboxyl-terminated oLA or PLA through a hydrolytic ester bond linkage.
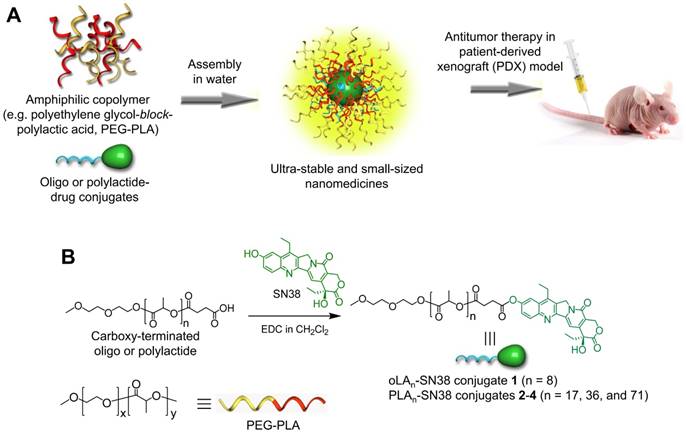
Methods
Materials, cell lines, and synthetic procedures
SN38 was purchased from Knowshine Pharmachemicals Inc. (Shanghai, China). Poly(ethylene glycol)-block-poly(D, L-lactic acid) copolymers and carboxyl-terminated oligo- or polylactides were customized by Advanced Polymer Materials Inc. (Montreal, Canada). All other compounds and solvents were purchased from J&K Chemical (Shanghai, China). Human hepatocellular carcinoma (HCC) cell line Hep 3B was maintained in MEM, HCC-LM3 was maintained in DMEM, and BEL-7402 was maintained in PRMI 1640 medium. All of the media were supplemented with 10% fetal bovine serum (FBS), penicillin (100 units/mL) and streptomycin (100 μg/mL), and the cells were maintained in a humid atmosphere at 37 °C with 5% CO2. The detailed procedures for synthesis of oLA- or PLA-tethered prodrugs 1-4 are provided in the supplementary materials.
Preparation of SN38 prodrug-loaded nanoparticles
SN38 prodrug-loaded polymeric nanoparticles were prepared using a nanoprecipitation method. In all cases, the matrix to drug (at an SN38 equivalence) weight ratios were fixed at 19:1. Briefly, predetermined amounts of block copolymer PEG5k-PLA8k and SN38 prodrugs (1 mg, at an SN38 equivalence) were dissolved in 2 mL acetonitrile and added dropwise into 18 mL of water while stirring, which yielded a final drug concentration of 0.05 mg/mL. After stirring for 10 min, the remaining organic solvent was removed in a rotary evaporator at reduced pressure. The solution containing the nanoparticles was concentrated with centrifugal filter devices (Amicon Ultra4, 10k MWCO, Millipore Corp.) and washed with deionized (DI) water.
Determination of drug loading and encapsulation efficiency
The drug loading content and encapsulation efficiency of the nanomedicines were determined by analytical reverse-phase high-performance liquid chromatography (RP-HPLC). Briefly, lyophilized SN38-loaded NPs (10 mg) were dissolved in acetonitrile (200 μL) and, subsequently, the NP solutions were added to NaOH (0.5 M, 200 μL) and stirred for 30 min at 37 °C to release the SN38 molecules. The suspension was centrifuged to collect the supernatant, and the SN38 content was quantitatively determined by HPLC. RP-HPLC was performed using a Hitachi Chromaster 5000 system with a YMC-Pack ODS-A column (5 μm, 250 × 4.6 mm) at a flow rate of 1.0 mL/min. UV detection for SN38 was performed at 378 nm. All of the runs used linear gradients of acetonitrile (solvent A) and water (solvent B) containing 0.1% trifluoroacetic acid (TFA). The encapsulation efficiency (EE) and percentages of drug loading (DL) of SN38 in NPs were calculated according to Equations (1) and (2):
EE (%) = WSN38inNP / Wfeed × 100% (1)
DL (%) = WSN38inNP / Wtotal × 100% (2)
where WSN38inNP, Wfeed, and Wtotal represent the amount of SN38 encapsulated in PEG5k-PLA8k NPs, the SN38-equivalent amount fed for NP fabrication and the total amount of nanomedicines, respectively.
Characterization of the particle size using dynamic light scattering (DLS)
The hydrodynamic diameters of the prodrug-loaded nanoparticles were measured on a Malvern Nano-ZS90 instrument (Malvern Instruments, Malvern, UK) at 25 °C.
Morphology study using transmission electron microscopy (TEM) analysis
Transmission electron microscopy (TEM) images were obtained using TECNAL 10 (Philips) at an acceleration voltage of 80 kV. The sample solution of SN38 prodrug-loaded NPs at a concentration of 0.5 mg/mL (SN38 equivalent) was placed onto a 300-mesh copper grid coated with carbon. Approximately 2 min after deposition, the surface water was removed with filter paper and then air dried. Positive staining was performed using a 2 wt% aqueous uranyl acetate solution.
In vitro SN38 release kinetics
To quantify the release kinetics of encapsulated SN38, NP solutions (10 mL) containing 0.1 mg/mL of an SN38 equivalent concentration were dialyzed against phosphate-buffered saline (PBS, 50 mL, pH 7.4, 0.2% Tween 80). The dialysis tubes (Spectrum, molecular weight cutoff of 14 kD) were continuously stirred in an orbital shaking water bath at 37 °C. At predetermined time intervals, the release media were collected, and fresh media were supplemented. The amounts of released SN38 were measured via a UV-Vis spectrometer (Shimadzu, UV-2700) at 378 nm. The drug release profiles were also determined by dialyzing against PBS (pH 7.4) in the presence of 20% (v/v) FBS. To determine whether the released components were active SN38 agents, analytical HPLC was used. At predetermined time intervals, the release media were subjected to HPLC analysis using pure SN38 dissolved in DMSO as a standard.
In vitro cytotoxicity using an MTT assay
The in vitro cytotoxicity of the nanomedicines was measured by an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. The cells were plated in flat-bottomed 96-well plates (4000-5000 cells per well) and incubated at 37 °C for 24 h. The cells were added via serial dilution of CPT-11, free SN38 and SN38-formulated NPs and then incubated at 37 °C for 72 h. At the end of the exposure, 30 μL MTT solution (5 mg/mL in PBS) was added to each well. The MTT solution was removed from the wells after 4 h, and the purple MTT-formazan crystals were then dissolved by the addition of DMSO (100 μL). The absorbance in each well was measured at 490 nm using a microplate reader (Multiskan FC, Thermo Scientific).
Cell proliferation as determined by EdU incorporation
The Hep 3B cells were plated in flat-bottomed 48-well plates (20000 cells per well) and incubated at 37 °C for 24 h. CPT-11 and SN38-formulated NPs (2 μM, at SN38 equivalent concentration) were added to the cells and then incubated for 24 h at 37 °C. To quantify synthesized DNA, we used a Click-iT® EdU Alexa Fluor® 488 Assay Kit (Invitrogen) according to the manufacturer's protocol. At the end of the drug exposure, EdU (5-ethynyl-2'-deoxyuridine) was added to each well, and the cells were further incubated for 2 h at 37 °C. The cells were washed with PBS and fixed for 15 min at room temperature by the addition of 3.7% formaldehyde. Upon incubating with 0.5% Triton X-100 for 10 min, azide-labeled Alexa Fluor® 488 was added and incubated for 30 min in the dark. The nuclei were stained with Hoechst 33342 (Invitrogen) for 15 min, and the cells were imaged by fluorescence microscopy (Olympus, IX71). Finally, n ≥ 5 regions with 1500-2000 total cells were counted to assess the presence of cell proliferation.
Pharmacokinetics study with SN38-loaded nanomedicines
The drug concentrations of SN38-loaded NPs (at an SN38 equivalent dose of 8 mg/kg) and CPT-11 (12 mg/kg) in the blood were compared in Sprague Dawley (SD) rats (~250 g, n = 5 in each group). After a single i.v. injection of various drugs, blood samples were collected at 5 min and 1, 6, 24, 48, and 72 h and then immediately subjected to centrifugation for 10 min at 3000×g, with the resulting plasma samples collected in microtubes. The plasma samples were then stored at -80 °C until analysis.
To analyze the free SN38 (unconjugated) and CPT-11 in the blood, acetonitrile (300 μL) was added to 50 μL of the plasma and extracted by ultrasonication. Coumarin was added as an internal standard, and the suspension was then centrifuged for 10 min at 14000×g and analyzed by high-performance liquid chromatography (HPLC).
To analyze the oligo- or polylactide-conjugated SN38 in the blood, 60 μL plasma was added to methanol (60 μL) and NaOH (0.5 M, 60 μL) and incubated for 30 min at 25 °C to release the conjugated SN38. After incubation, an HCl solution (60 μL, 0.5 M), the internal standard and acetonitrile (300 μL) were added to the samples. The samples were then extracted by ultrasonication and centrifuged for 10 min at 14000×g. The supernatants were analyzed by HPLC.
RP-HPLC was performed using a Hitachi Chromaster 5000 system with a YMC-Pack ODS-A column (5 μm, 250 × 4.6 mm) at a flow rate of 1.0 mL/min. UV detection for SN38 was performed at 378 nm. All of the runs used linear gradients of acetonitrile (solvent A) and water (solvent B) containing 0.1% TFA.
In vivo antitumor activity in the HCC Hep 3B cell-derived xenograft mouse model using SN38-loaded nanomedicines
Balb/c nude mice (4- to 5-week-old) were purchased from the Shanghai Experimental Animal Center, Chinese Academy of Science, for the establishment of the mouse model bearing HCC Hep 3B xenografts. The mice were housed under aseptic conditions and were provided autoclaved rodent diet and sterile water. All of the mouse studies were conducted in accordance with the National Institute Guide for the Care and Use of Laboratory Animals. The experimental protocol was approved by the local hospital ethics committee.
Human HCC Hep 3B cells were grown to 80% confluence in 90 mm tissue culture dishes. After harvesting, the cells were resuspended in PBS at 4 °C to a final concentration of 2.5 × 107 cells/mL. The right flank of 5-week-old Balb/c nude mice was subcutaneously injected with 200 μL of a cell suspension containing 5×106 cells in a disposable syringe. After the tumor reached ~50 mm3 in volume at day 14 after implantation, the animals were randomized into six groups (n = 7 in each group). The mice were intravenously injected with SN38 prodrug-loaded nanomedicines (15 mg/kg, at an SN38 equivalent dose) three successive times at day 0, 3, and 6. Saline and CPT-11 (22.5 mg/kg) were intravenously injected as the controls. The tumor growth and body weight were monitored and recorded every three days. The length (L) and width (W) of the tumors were measured with calipers, and the tumor volume was calculated using the following formula: V = L × W2 / 2, with W smaller than L. The mice were sacrificed by CO2 inhalation when their tumors reached the 2000 mm3 endpoint value.
Establishment of a human HCC PDX tumor model in nude mice and in vivo therapeutic studies
Balb/c nude mice (4- to 5-week-old) were purchased from the Shanghai Experimental Animal Center, Chinese Academy of Science, for the establishment of the mouse PDX model. The mice were housed under aseptic conditions and were provided autoclaved rodent diet and sterile water. All of the mouse studies were conducted in accordance with the National Institute Guide for the Care and Use of Laboratory Animals. Prior written informed consent was obtained from the patients, and the experimental protocol was approved by the local hospital ethics committee.
A schematic illustration of the protocol for the establishment of the HCC PDX tumor model in Balb/c nude mice is depicted in Figure 6A. Briefly, surgically resected human HCC tissue was obtained from HCC patient 1-150605. The primary HCC tumor tissue was collected in an ice bath in RPMI 1640 supplemented with antibiotics. The tumor tissue was diced into ~1 mm3 pieces and subcutaneously implanted into the right flanks of the nude mice within 2 h as the engraftment phase (Figure 1). For the serial transplantation, the mice bearing PDX tumors were sacrificed and ~1 mm3 tumor fragments were further implanted into the 5- to 6-week-old nude mice.
SN38 prodrug-formulated NPs exhibited potent antitumor efficacy and survival benefits in a mouse model bearing human HCC PDX tumors. (A) Schematic illustration of the protocol for the establishment of a human HCC PDX tumor model in Balb/c nude mice. The primary HCC tissue from patient 1-150605 obtained by surgical resection was finely minced and implanted subcutaneously into nude mice at the engraftment phase (F1). Drug treatment was performed from the third generation. (B) Tumor volume (mm3) as a function of time after drug treatment (n = 7 in each group). When the tumor volumes reached ~50 mm3, NP solutions containing SN38 prodrugs (15 mg/kg, at the SN38 equivalent dose) were injected three successive times. Saline and CPT-11 (22.5 mg/kg) were also intravenously injected as controls. Arrows represent the i.v. injections; the tumor volumes are expressed as the means ± s.d.; *p < 0.05, **p < 0.01. (C) Kaplan-Meier survival plots demonstrating the survival benefit in animals with a 100% survival after treatment with PLA71-SN38-NPs.
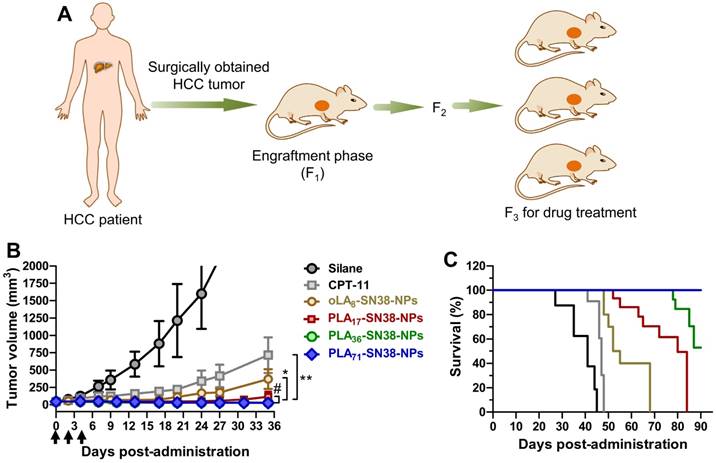
Drug treatment was performed starting in the third generation. When the PDX tumor volumes reached ~50 mm3, the mice (n = 7 in each group) were intravenously injected with SN38 prodrug-loaded nanomedicines (15 mg/kg, at an SN38 equivalent dose) three successive times at day 0, 2, and 4. Saline and CPT-11 (22.5 mg/kg) were intravenously injected as controls. Both the volumes of the tumors and body weight were monitored. The mice were sacrificed by CO2 inhalation when their tumors reached the 2000 mm3 endpoint value.
Immunohistochemical analysis
For histological analysis, the HCC Hep 3B tumor tissues were excised 2 days after the single treatment of various drugs. The tissues were fixed in 4% formaldehyde, embedded in paraffin, and sectioned into 5-μm slices. The formalin-fixed sections were stained with hematoxylin and eosin (H&E, Sigma). For the TUNEL apoptosis assay, the fixed tumor tissues were stained by the In Situ Cell Death Detection Kit (Fluorescein, Roche Applied Science) according to the manufacturer's protocol. 4',6-diamino-2-phenylindole (DAPI) was used for nuclear counterstaining. The stained tumor slices were imaged by fluorescence microscopy (Olympus, IX71).
Study of bloody diarrhea induced by CPT-11 and nanomedicines
Healthy Balb/c mice (6-week-old) were randomized into four groups (n = 10 in each group) and housed in conventional metabolic cages (N = 1/cage). The mice were intravenously administered with SN38-loaded NPs (at an SN38 equivalent dose of 15 mg/kg) three times every two days. In addition, saline and CPT-11 (22.5 mg/kg) were intravenously injected on the same schedule as the controls. The signs of bloody diarrhea were monitored every day using BASO fecal OB-II (BASO diagnostic Inc., Zhuhai, China) according to the manufacturer's protocol. Briefly, 1 drop of reagent A (pyramidon and acetic acid) and 1 drop of reagent B (hydrogen peroxide) were added onto the fecal smear paper, with a blue coloration obtained under acidic conditions. The time required for the color to appear was used to define the grade of bloody diarrhea. The rapid appearance of purple-blue was defined as grade 4, the gradual emergence of purple-blue within 10 seconds was defined as grade 3, the gradual emergence of purple within 1 min was defined as grade 2, and the gradual emergence of purple within 2 min was defined as grade 1. The mice treated with saline were used as negative control. At day 9 postadministration, the mice were sacrificed, and their large intestines were formalin-fixed and paraffin embedded for routine H&E histopathologic examination.
Statistical analysis
All of the quantitative data are presented as the means ± s.d. The statistical significance between the measurements was assessed using Student's t test. A p-value less than 0.05 was considered statistically significant, whereas a p-value less than 0.01 was considered highly significant. Survival studies were analyzed using Kaplan-Meier plots.
Results and discussion
Rational design of oLA- or PLA-conjugated SN38 prodrugs
In this study, a phenyl ester bond that is susceptible to hydrolysis and widely used as a linker in prodrug design was employed [53]. To keep the backbone close to the FDA-approved skeleton, we chose oLA or PLA segments, which vary in molecular weight and are only minimally modified (i.e., bearing a terminal carboxylate group) to condense the SN38 agent (Figure 1). We hypothesized that incorporating this potent SN38 agent into the termini of polymer segments through covalent conjugation could augment drug retention in the PEG-PLA NPs. The use of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) as a carboxyl activator and dichloromethane (DCM) as a solvent yielded the production of desirable SN38 derivatives. Consequently, oLAn-SN38 conjugate 1 (n = 8) or PLAn-SN38 conjugates 2-4 (n = 17, 36, and 71, respectively) were successfully synthesized and purified with flash silica gel chromatography at relatively high conjugation efficiencies (48-57%, see Supporting Information for synthesis procedures). The structures of purified SN38 prodrugs were confirmed by 1H NMR. The 1H NMR spectrum of SN38 molecule was provided in Figure S1 as a reference. The peak ratios of the protons of the oLA or PLA backbone (-CH(CH3)-: δ 5.16 and 1.58 ppm, red box in Figure 2A) to the SN38 aromatic protons (green box in Figure 2A) were 8, 17, 36, and 71, respectively, for products 1-4. Thus, the results from the 1H NMR spectra suggested that nearly one SN38 molecule was successfully conjugated to each polymer chain irrespective of the chain length. The synthesis feasibility of oligomer or polymer backbone-based prodrugs demonstrates that versatile structures can be used for SN38 prodrug reconstitutions.
(A) 1H NMR spectra of SN38 prodrug conjugates 1-4. The protons in the green box can be assigned to the aromatic groups of SN38, whereas the protons in the red box can be assigned to the oLA or PLA segments. SN38 prodrugs with high purities as evidenced by 1H NMR spectra were obtained through a direct one-step esterification. (B) Transmission electron microscopy (TEM) images of polymeric nanoparticles stained with 2% uranyl acetate. SN38 prodrugs 1-4 were assembled into PEG5k-PLA8k-based amphiphilic copolymers (designated as oLAn-SN38-NPs, here n = 8 and PLAn-SN38-NPs, here n = 17, 36, and 71). Scale bars, 100 nm. Insets: Distribution of the hydrodynamic diameters (DH) of the NPs measured by dynamic light scattering (DLS). (C) In vitro drug release profiles of the total SN38 including free SN38 or oLA- and PLA-tethered SN38 from NPs consisting of PEG5k-PLA8k copolymers after dialyzing over a 120-h period against PBS (pH 7.4) at 37 °C. The data are presented as the means ± s.d. (n = 3).
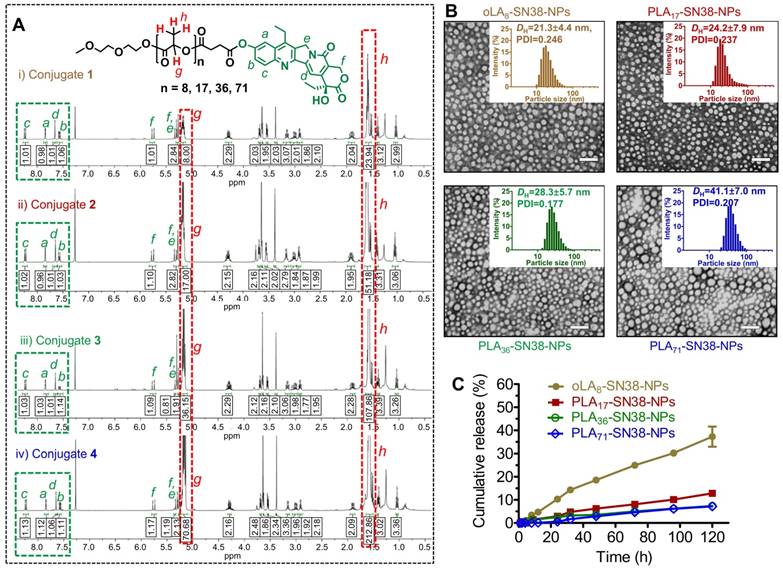
Prodrug encapsulation in nanoparticle and characterization
After obtaining this pool of SN38 prodrugs, we assessed their ability to assemble with PEG-PLA copolymers using a single-step nanoprecipitation method [54, 55]. In this process, drug payloads are incorporated into the hydrophobic micellar core of the NPs, and the PEG chain constitutes the “stealth” shell that reduces the blood clearance of nanoparticles and endows them with in vivo longevity [56]. When we used the matrices/SN38 feed weight ratio of 19:1 (i.e., this weight ratio is referred to total materials except for the SN38 to SN38 agent), prodrugs 1-4 could readily self-assemble with PEG5k-PLA8k copolymers to form transparent solutions (Figure S2, designated oLAn-SN38-NPs for n = 8 and PLAn-SN38-NPs for n = 17, 36, and 71). At this feed ratio, PEG5k-PLA8k copolymers encapsulated prodrugs 1-4 at greater than 92% encapsulation efficiencies and loaded the drugs at approximately 4.5% (Table S1). However, attempts to encapsulate the parent SN38 agent within the PEG5k-PLA8k matrix resulted in large precipitates (Figure S2), and the encapsulation efficiency was substantially lower (<5%, n = 3). Thus, we employed PEG5k-PLA8k as a matrix to formulate SN38 prodrug constructs in subsequent in vitro and in vivo studies.
Upon blending PEG5k-PLA8k with prodrugs 1-4, transmission electron microcopy (TEM)-based morphology studies were conducted. We found that all of these SN38-formulated particles were monodisperse and spherical (Figure 2B) and had diameters less than 50 nm. Dynamic light scattering (DLS) also revealed that the hydrodynamic diameters (DH) ranged from approximately 21 nm to 41 nm, implying that densely packed core-shell structures were formed for this set of particles (Figure 2B). Furthermore, prodrug-containing solutions (i.e., SN38 equivalent concentration of 1 mg/mL) stored at 4 °C were monitored for days, and no visible precipitation was formed. DLS analysis also indicated that these NPs were stable in different media including DI water, PBS and PBS containing 10% FBS (Figure S3). Nevertheless, particle sizes in the sub-50 nm range are favorable for achieving superior in vivo anticancer activity for deep tumor penetration and passive tumor targeting via the EPR effect [57].
To provide sufficient intratumoral accumulation of drugs of interest via the EPR effect, a central challenge has been the optimal interplay between drug payloads and delivery matrices because this parameter confers distinct drug retention profiles within the nanocarriers [45, 51, 52]. Therefore, exploration of structurally stable nanomedicines that exhibit a minimal premature release of drug payloads in the systemic circulation is essential for achieving optimal nanomedicine delivery into solid tumors [58]. We hypothesized that the incorporation of SN38 into the polymer backbones through esterification and sequential assembly with structurally similar PEG-PLA copolymers could strongly constrain drug payloads within the generated nanomedicines, thereby delaying the liberation of active drugs. Therefore, we quantified the release kinetics of SN38 from the assembled NPs by dialyzing the NP solutions against PBS (pH 7.4). Compared with our previous versions of SN38 prodrugs [45], strikingly sustained drug release behaviors were observed under neutral pH conditions, and SN38 was released at quite low rates (Figure 2C). Among the four types of NPs, the PLA36-SN38-NPs and PLA71-SN38-NPs were the most stable and released only 7% of the total drug after 120 h at 37 °C, whereas oLA8-SN38-NPs released approximately 37% under identical conditions. We further assessed the SN38 release profiles against PBS in the presence of mouse serum. Exposing SN38-loaded NPs to dialysis buffer containing 20% v/v of mouse serum promoted a slightly accelerated drug release from the NPs, with approximately 39% of the total SN38 released within 72 h in the case of PLA71-SN38-NPs (Figure S1). The ester bond between SN38 and the polymer could be readily hydrolyzed by esterases; thus, the serum esterase activity could accelerate the drug release when the NPs were incubated with mouse serum [53, 59, 60]. Moreover, the presence of serum could destabilize the PEG-PLA-assembled NPs, which also promotes the release of active SN38 agents [61]. Further HPLC analysis indicated that intact SN38 molecules were released from the NPs as the major species using PLA71-SN38-NPs as a model system (Figure S2). This result differed from a recently proposed mechanism for oligo(lactic acid)-tethered paclitaxel (PTX) ester prodrugs that underwent backbiting conversion to release active components [50]. The observed difference could be attributable to the fact that the ester bond between SN38 and PLA was more susceptible to hydrolysis than that in PTX prodrugs. We can expect that the conversion of PLAn-SN38 conjugates to the active SN38 agent proceeds rapidly in aqueous solutions but slowly in the PEG-PLA micellar core in water. Once exposed to aqueous solutions, PLAn-SN38 conjugates could be hydrolyzed, leading to the release of intact SN38. Collectively, these findings suggest that polymer-SN38 conjugates within NPs possess favorable stability and sustained controllable release of active drugs, and these characteristics were rationally achieved by tailoring the chemical structures of the modifiers.
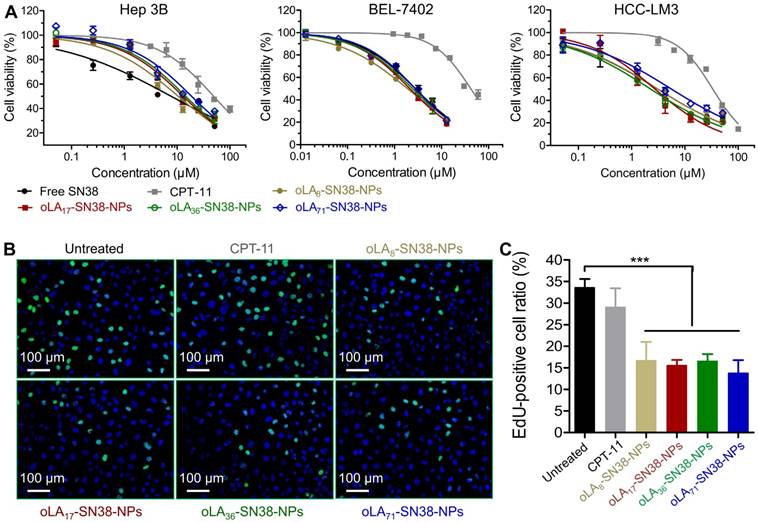
In vitro cytotoxicity against HCC cell lines
We then studied the cytotoxicity of these nanomedicines against HCC-derived cell lines using the conventional MTT assay by employing CPT-11 and free SN38 as the controls. Following the exposure of these NPs and free drugs for 72 h, the cell viabilities of three distinct types of HCC cells were quantified. As expected, HCC exhibited relatively high drug resistance to the SN38 agent, although all of the SN38-loaded NPs demonstrated comparable IC50 values (half-maximal inhibitory concentration) to that of free SN38 administered in DMSO (Figure 3A). In particular, the IC50 values ranged from 10.50 to 18.82 μM in Hep 3B cells, which were larger than those of Huh-7 and HCC-LM3 cells (Table 1). This might be most likely correlated with the high population (~90%) of cancer stem cells, a subtype of cancer cells that induce drug resistance, in Hep 3B cells [62]. Moreover, compared with CPT-11, the IC50 values of the nanomedicines occurred at a concentration of 1 to 2 orders lower. To further verify whether cell proliferation can be inhibited by the release of SN38, EdU incorporation was utilized to label the cells undergoing DNA synthesis. We observed a high level of inhibited proliferation after 24 h in all of the SN38-loaded NP treated Hep 3B cells (Figure 3B-C), whereas the CPT-11 treatment only produced a negligible effect on the proliferation of Hep 3B cells. Obviously, drug conjugation and sequential formulation in the NPs did not reduce their activity, in contrast to previous reports in which lower cytotoxicity was observed due to the sustained in vitro release kinetics. Thus, our data indicate that these nanomedicines can undergo efficient cellular uptake and release therapeutically active SN38 in the intracellular environments.
(A) In vitro cytotoxicity in HCC cells after a 72-h treatment with CPT-11, free SN38, and NPs encapsulating SN38 prodrugs. The cell viabilities were determined by an MTT assay. (B-C) Proliferation of HCC Hep 3B cells treated with various drugs (2 μM with SN38 equivalent concentrations) over 24 h as determined by EdU incorporation. The data are presented as the means ± s.d.; n≥5 regions with a total of analyzed 1500-2000 cells; **p<0.01.
In vitro cytotoxicity of SN38-loaded nanomedicines against HCC cells after 72-h incubation (expressed as IC50 ± SD in µM).a
| Cell line | Hep 3B | BEL-7402 | HCC-LM3 |
|---|---|---|---|
| CPT-11 | 49.26±3.30 | 41.95±2.06 | 33.36±1.69 |
| Free SN38 | 7.74±1.18 | 2.76±0.16 | 3.08±0.29 |
| oLA8-SN38-NPs | 10.50±1.01 | 1.89±0.10 | 3.11±0.39 |
| PLA17-SN38-NPs | 13.02±1.00 | 2.12±0.09 | 2.74±0.33 |
| PLA36-SN38-NPs | 14.53±0.90 | 2.42±0.10 | 2.27±0.13 |
| PLA71-SN38-NPs | 18.82±2.21 | 2.77±0.11 | 5.58±0.73 |
a Determined by an MTT assay.
Prolonged circulation of these ultrastable nanomedicines
Because of the characteristic ultrastability of these nanomedicines, we expected that they might have prolonged circulation in blood, thus facilitating the delivery of a sufficient level of drug payload to solid tumors via the EPR effect. To verify this hypothesis, four types of SN38-formulated NPs were systemically injected into Sprague Dawley (SD) rats via the tail vein (n = 5 in each group), and the plasma drug concentration was then determined by HPLC. A significantly prolonged retention of SN38 drugs in the bloodstream was observed for PLA36-SN38-NPs and PLA71-SN38-NPs, whereas relatively rapid clearance was observed for oLA8-SN38-NPs (Figure 4). The data were fitted to a two-compartmental pharmacokinetic model, which produced plasma area-under-curve (AUC) values of 68.9 and 180.7 μg·h/mL for CPT-11 and oLA8-SN38-NPs, respectively. Surprisingly, compared with oLA8-SN38-NPs, a greater than 11- and 12-fold enhancement in the AUC of PLA36-SN38-NPs (2001.1 μg·h/mL) and PLA71-SN38-NPs (2195.6 μg·h/mL), respectively, was confirmed (Table 2). We separately analyzed the PLA-conjugated and the released SN38 in the plasma and found that free SN38 was minimal, i.e., less than 10% relative to the intact PLAn-SN38 prodrugs in the NPs (data not shown). These results strongly indicate that sustained release kinetics are a critical factor that dominates prolonged drug retention in animals and could ultimately lead to preferential drug accumulation in solid tumors.
In vivo plasma concentration-time profiles of the drugs upon intravenous (i.v.) injection of SN38 prodrug-formulated NPs (at an SN38 equivalent dose of 8 mg/kg) in Sprague Dawley rats. CPT-11 (12 mg/kg) was used for comparison. The total amount of SN38 molecules, including oLA- and PLA-tethered or free SN38, in the plasma was extracted and determined by HPLC. The data are presented as the means ± s.d. (n = 5).
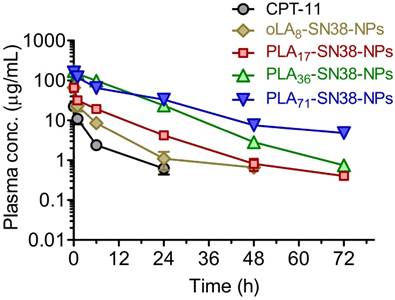
Pharmacokinetic parameters of CPT-11, oLA8-SN38-NPs, PLA17-SN38-NPs, PLA36-SN38-NPs, and PLA71-SN38-NPs (8 mg/kg, SN38 equivalent).
| PK parameter | Drug formulations | ||||
|---|---|---|---|---|---|
| CPT-11 | oLA8-SN38-NPs | PLA17-SN38-NPs | PLA36-SN38-NPs | PLA71-SN38-NPs | |
| t1/2α (h) | 0.70 | 0.35 | 0.25 | 0.23 | 2.57 |
| t1/2β (h) | 9.06 | 6.32 | 8.31 | 8.70 | 16.73 |
| AUC(0-t) (μg·h/mL) | 67.6 | 179.9 | 395.0 | 1994.6 | 2098.8 |
| AUC(0-inf) (μg·h/mL) | 68.9 | 180.7 | 395.9 | 2001.1 | 2195.6 |
| Cmax | 23.8 | 74.7 | 74.0 | 178.7 | 155.3 |
AUC: area under the concentration vs time curve; t1/2α: half-life of the distribution phase; t1/2β: half-life of the elimination phase
In vivo antitumor efficacy in an HCC CDX model
Encouraged by the improved pharmacokinetic property afforded by our approach, we established the immunodeficient Balb/c nude mouse model subcutaneously bearing an HCC Hep 3B cancer cell-derived xenograft to further validate the potential translational implications of these nanomedicines. Comparative efficacy studies were conducted using three intravenous (i.v.) injections of the NP solution at an equivalent SN38 dose of 15 mg/kg, which is a relatively low dose of SN38 compared with that of previous studies [63]. Encouragingly, the systemic administration of SN38-loaded NPs resulted in significant tumor shrinkage relative to that of the saline- and CPT-11-treated groups after the initial dose (Figure 5A, C). In particular, the PLA36-SN38-NPs and PLA71-SN38-NPs treatments presented striking improvements, with four out of seven mice in each treatment group showing an absence of detectable tumors at day 10 postadministration. At the end of the study, the average tumor volumes were 1656 ± 468 and 590 ± 223 mm3, respectively, in saline- and CPT-11-treated groups, whereas the tumors were completely eradicated in the groups receiving PLA36-SN38-NPs and PLA71-SN38-NPs. The SN38 agent targets nuclear DNA topoisomerase I, which is ubiquitous and essential in mammalian cells, including HCCs [64]. Unfortunately, in the aforementioned cell-based cytotoxicity assay, chemoresistance in Hep 3B cells was observed with IC50 values ranging from 7.74 to 18.82 μM. These values are substantially larger than those extrapolated from our previous dose-response curves for other types of cancer cells (i.e., 0.09 μM and 0.08 μM in human colon cancer HT-29 and HCT-116 cells, respectively, and 0.03 μM in human non-small-cell lung cancer A549 cells) [47]. Despite this reduced sensitivity in Hep 3B cells, upon systemic administration of nanomedicines, a strikingly potent tumor shrinkage or even complete tumor reduction was achieved in this HCC xenograft model. Therefore, the enhanced antitumor effects for these high molecular weight PLAn-SN38 prodrug-formulated NPs (i.e., PLA36-SN38-NPs and PLA71-SN38-NPs) might be explained by several cooperative factors, including improved drug-carrier compatibility, extended and long-term circulation in the blood and, thereby, possible preferential drug accumulation in the tumors endowed by the NP delivery.
(A) Comparative efficacy study in an HCC Hep 3B cancer cell-derived xenograft nude mouse model using SN38-formulated NPs. NP solutions containing SN38 prodrugs (15 mg/kg, the SN38 equivalent dose) were injected three successive times. Saline and CPT-11 (22.5 mg/kg) were also intravenously injected as controls. Arrows represent the i.v. injections. The tumor sizes were monitored using a caliper. The data are expressed as the means ± s.d. (n = 7); *p < 0.05, ***p < 0.001. (B) Body weight changes of the mice after drug administration. (C) Representative images of the excised tumors for the groups treated with saline, CPT-11, and oLA8-SN38-NPs. However, the largest tumors in the other groups at the end point are shown in (C). (D) H&E and TUNEL analyses of the excised tumors from the treated groups at day 2 post-single injection of the drugs (15 mg/kg, at SN38 equivalent dose). The tumor sections were stained with H&E (left), DAPI for nuclei (blue, middle), and fluorescein-dUTP for apoptosis (green, right). Potent induction of apoptosis was observed in NP-treated mice, which is consistent with the tumor inhibition results. Scale bars, 100 μm.
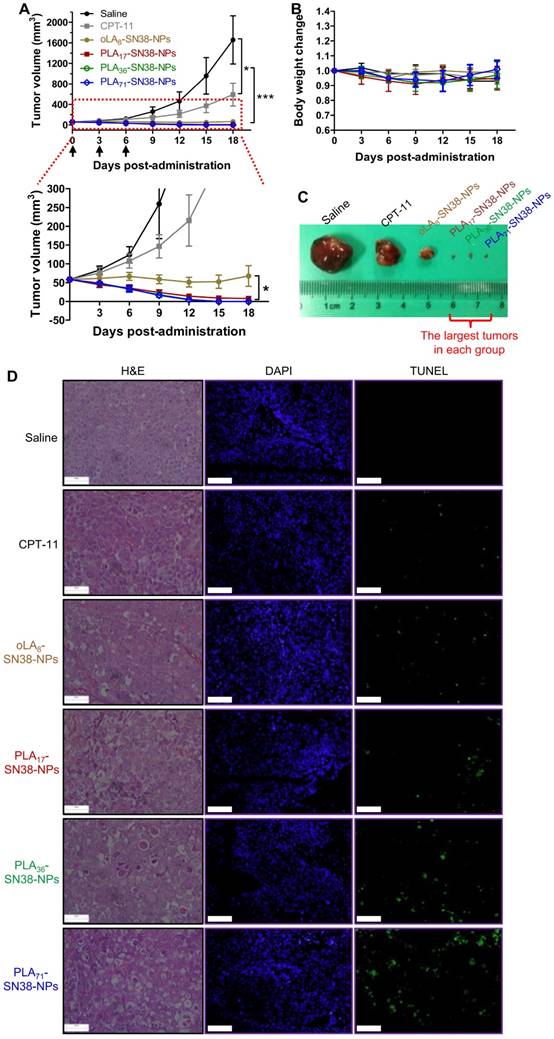
The body weights of the mice receiving successive treatments were stable, suggesting a low systemic toxicity associated with present nanomedicines (Figure 5B). Moreover, we performed histological examinations using hematoxylin and eosin (H&E) stains and the terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end-labeling (TUNEL) assays. The results indicated that relative to the CPT-11 groups, more efficient and extensive intratumoral apoptosis was induced by the SN38 agent after a single i.v. injection of these nanomedicines (Figure 5D), consistent with the in vivo therapeutic efficacy.
Antitumor efficacy in an HCC patient-derived xenograft tumor model
To further examine the potential of these nanomedicines in PDX models, we xenografted human HCC tissues into Balb/c nude mice. Fresh primary HCC tissue was obtained by surgical resection, finely sectioned into ~1 mm3 pieces and implanted subcutaneously into the flanks of the Balb/c nude mice. The rapidly grown xenografts allowed us to take advantage of a third passage of HCC tumors, which is generally suitable for drug treatments as depicted in Figure 6A. Compared with the moderate efficacy of CPT-11 in this PDX model, systemic administration of SN38-loaded NPs consistently resulted in significant tumor shrinkage in this PDX model (Figure 6B). The PLA36-SN38-NPs and PLA71-SN38-NPs showed the most dramatic efficacy and average tumor volumes of 29 ± 7 and 25 ± 4 mm3, respectively, at day 35, and the tumor volumes were significantly smaller compared with those of the groups treated with saline (>2500 mm3) and CPT-11 (714 ± 257 mm3). In particular, all of the mice in the group receiving PLA71-SN38-NPs survived the 90-day study duration, and five out of the seven mice presented complete eradication of the tumors. As a result, these PLA71-SN38-NP-treated mice exhibited a significantly better survival rate compared with those in the other treatment groups (Figure 6C). In general, HCC patients show low response rates (less than 10%) to systemic therapy with cytotoxic drugs [65]. The current standard-of-care therapy is to use sorafenib, an oral multikinase inhibitor, although this therapy only extends the median overall survival of patients with advanced HCC by a paltry 2.8 months compared with a placebo [66]. Thus, there is a desperate need to develop novel effective therapeutic strategies to manage this devastating disease. Effectively suppressing the tumors derived from human patient tissues might render this class of PLA-SN38 nanoplatforms promising for future clinical translation. On the other hand, clinical trials using nanosized drug delivery systems have proven to be disappointing [67]. Thus, people may think that the EPR effect is not as reliable as previously thought in human patients.
Though additional PDX models are necessitated to validate therapeutic efficacy as well as targeting ability, efficacy gains via the EPR effect in the current PDX model could be expected. To our knowledge, the study described here represents the first example of a PDX model derived from an HCC patient that is used to validate the efficacy of nanomedicines.
Alleviating SN38 toxicity by exploiting nanoparticle-mediated drug delivery
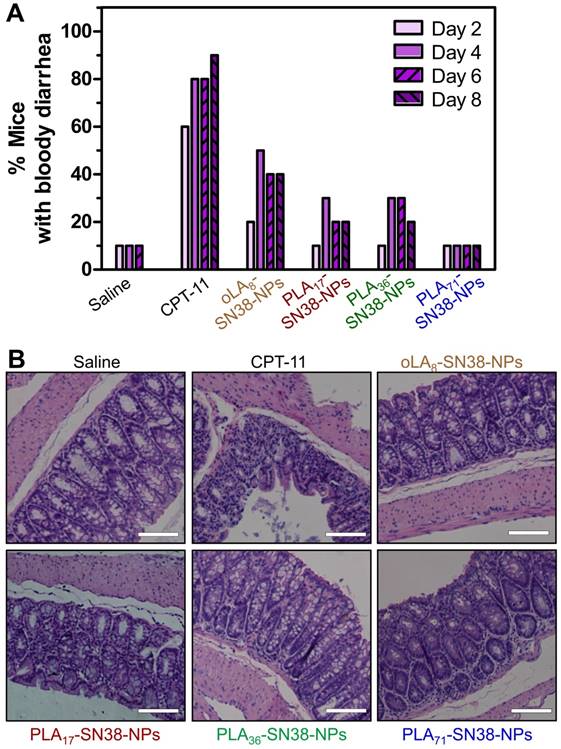
The clinically administered SN38 prodrug, CPT-11, causes diarrhea in up to 70% of treated patients, and incidence of grade 3 or 4 diarrhea has been observed in 31% of the patients [68]. Several strategies, including the removal of GI bacteria [69] and the use of selective inhibitors of β-glucuronidase [70], have been employed to alleviate CPT-11-induced toxicity. We hypothesized that our approach might have the potential to eliminate bloody diarrhea and intestinal damage induced by CPT-11. To verify this assumption, healthy Balb/c mice were used to carefully assess whether our nanomedicines could prevent the onset of bloody diarrhea. Mice (n = 10 in each group) were intravenously injected with three types of SN38-loaded NPs (SN38 equivalent dose of 15 mg/kg) three successive times. In this experimental setting, two groups received saline and CPT-11 (22.5 mg/kg) as controls. GI symptoms, such as bloody diarrhea, were monitored daily. The mice receiving CPT-11 experienced bloody diarrhea from day 2 (Figure 7A). In particular, an increased incidence of grade 2 diarrhea was apparent, with half of the total animals experiencing grade 2 diarrhea in the CPT-11-treated group. However, the mice receiving SN38-loaded NPs suffered less bloody diarrhea during the study. Impressively, only one of ten animals in PLA71-SN38-NPs group exhibited bloody diarrhea, whereas none exhibited grade 2 diarrhea, which was comparable to the saline group (Figure 7A). All of the mice were euthanized at day 9, and their large intestines were further examined by H&E staining. It was clear that the animals treated with saline or SN38-loaded NPs displayed healthy glandular structures and intact epithelial layers, whereas the successive administration of CPT-11 led to the destruction of these tissues by eliminating the glands and causing diffuse mucosal damage (Figure 7B). Our results indicated that the administered nanomedicines have the ability to mitigate the onset of bloody diarrhea and intestinal damage caused by CPT-11, thereby indicating their clinical potential as candidates for dose escalation.
Alleviation of SN38 toxicity in healthy Balb/c mice by using nanoparticle-mediated drug delivery. (A) CPT-11 caused bloody diarrhea starting from day 2 after i.v. administration (22.5 mg/kg), whereas SN38 prodrugs formulated in the NPs (15 mg/kg, at an SN38 equivalent dose) significantly reduced the incidence of this dose-limiting side effect. (B) Hematoxylin and eosin (H&E) stains of the colons of the mice exhibited highly disrupted tissues in the CPT-11-treated mice, whereas healthy glandular structures were observed for the saline, SN38-loaded NP-treated mice. Scale bars, 100 μm.
Conclusions
In summary, we report that through stabilizing a given chemotherapeutic SN38 agent within polymeric nanoplatforms, it is possible to generate a highly efficacious anticancer nanotherapy against human HCC tumors. Of note, we showed for the first time the use of these small-sized and ultrastable SN38 NPs to efficiently eradicate tumors in a high-fidelity, clinically relevant HCC patient-derived tumor model. Structure-based molecular editing of the SN38 agent enables the production of new prodrug entities with well-defined structures and superior in vivo performance compared with the parent drug. Hence, the strategy described herein also has implications for the reformulation of other potent therapeutic candidates that possess inadequate pharmacokinetics or poor compatibility with delivery matrices. Finally, with the advances in establishing preclinical PDX models that recapitulate tumors in human patients [71, 72], we envisage that the potential of many nanomedicines will be further validated.
Abbreviations
AUC: area-under-curve; CDX: cancer cell-derived xenograft; CPT-11: irinotecan; DL: drug loading; DLS: dynamic light scattering; DAPI: 4',6-diamino-2-phenylindole; DCM: dichloromethane; EPR: enhanced permeability and retention; EE: encapsulation efficiency; EDC: 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide; FBS: fetal bovine serum; HCC: hepatocellular carcinoma; H&E: hematoxylin and eosin; IC50: half-maximal inhibitory concentration; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NP: nanoparticle; oLA: oligolactide; PDX: patient-derived xenograft; PLA: polylactide; PEG-PLA: polyethylene glycol-block-polylactic acid; PBS: phosphate-buffered saline; PTX: paclitaxel; RP-HPLC: reverse-phase high-performance liquid chromatography; SN38: 7-ethyl-10-hydroxycamptothecin; SD: Sprague Dawley; TEM: transmission electron microscopy; TFA: trifluoroacetic acid; TUNEL: TdT-mediated dUTP nick end labeling.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 81773193, 81571799, 21202147, 31671019, 91542205, and 81421062), the National Science and Technology Major Project (2017ZX10203205), and the San Ming Project of Shenzhen City (SZSM201412006).
Supplementary Material
Supplementary methods, figures and table.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mitragotri S, Burke PA, Langer R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat Rev Drug Discovery. 2014;13:655-72
2. Bogart LK, Pourroy G, Murphy CJ, Puntes V, Pellegrino T, Rosenblum D. et al. Nanoparticles for imaging, sensing, and therapeutic intervention. ACS Nano. 2014;8:3107-22
3. Wang HX, Lu ZJ, Wang LJ, Guo TT, Wu JP, Wan JQ. et al. New generation nanomedicines constructed from self-assembling small-molecule prodrugs alleviate cancer drug toxicity. Cancer Res. 2017;77:6963-74
4. An FF, Zhang XH. Strategies for preparing albumin-based nanoparticles for multifunctional bioimaging and drug delivery. Theranostics. 2017;7:3667-89
5. Sun Y, Wang Q, Chen J, Liu L, Ding L, Shen M. et al. Temperature-sensitive gold nanoparticle-coated pluronic-PLL nanoparticles for drug delivery and chemo-photothermal therapy. Theranostics. 2017;7:4424
6. Shen L, Huang Y, Chen D, Qiu F, Ma C, Jin X. et al. pH-responsive aerobic nanoparticles for effective photodynamic therapy. Theranostics. 2017;7:4537
7. Miyata K, Nishiyama N, Kataoka K. Rational design of smart supramolecular assemblies for gene delivery: chemical challenges in the creation of artificial viruses. Chem Soc Rev. 2012;41:2562-74
8. Wu J, Kamaly N, Shi J, Zhao L, Xiao Z, Hollett G. et al. Development of multinuclear polymeric nanoparticles as robust protein nanocarriers. Angew Chem Int Ed. 2014;53:8975-79
9. Vermonden T, Censi R, Hennink WE. Hydrogels for protein delivery. Chem Rev. 2012;112:2853-88
10. Duan X, Liu D, Chan C, Lin W. Polymeric micelle-mediated delivery of DNA-targeting organometallic complexes for resistant ovarian cancer treatment. Small. 2015;11:3962-72
11. Wang X, Bodman A, Shi C, Guo D, Wang L, Luo J. et al. Tunable lipidoid-telodendrimer hybrid nanoparticles for intracellular protein delivery in brain tumor treatment. Small. 2016;12:4185-92
12. Wang J, Liu Y, Ma Y, Sun C, Tao W, Wang Y. et al. NIR-activated supersensitive drug release using nanoparticles with a flow core. Adv Funct Mater. 2016;26:7516-25
13. Hangxiang Wang JW, Li Xu, Ke Xie, Chao Chen, Yuehan Dong. Albumin nanoparticle encapsulation of potent cytotoxic therapeutics shows sustained drug release and alleviates cancer drug toxicity. Chem Commun. 2017;53:2618-21
14. Wang H, Chen W, Xie H, Wei X, Yin S, Zhou L. et al. Biocompatible, chimeric peptide-condensed supramolecular nanoparticles for tumor cell-specific siRNA delivery and gene silencing. Chem Commun. 2014;50:7806-9
15. Sun Q, Sun X, Ma X, Zhou Z, Jin E, Zhang B. et al. Integration of nanoassembly functions for an effective delivery cascade for cancer drugs. Adv Mater. 2014;26:7615-21
16. Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC. et al. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013;73:2412-17
17. Wang H, Chen J, Xu C, Shi L, Tayier M, Zhou J. et al. Cancer nanomedicines stabilized by π-π stacking between heterodimeric prodrugs enable exceptionally high drug loading capacity and safer delivery of drug combinations. Theranostics. 2017;7:3638-52
18. Hrkach J, Von Hoff D, Mukkaram Ali M, Andrianova E, Auer J, Campbell T. et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4:128ra39
19. Wang J, Wang H, Li J, Liu Z, Xie H, Wei X. et al. iRGD-decorated polymeric nanoparticles for the efficient delivery of vandetanib to hepatocellular carcinoma: preparation and in vitro and in vivo evaluation. ACS Appl Mater Interfaces. 2016;8:19228-37
20. Yuan Y, Xu L, Dai S, Wang M, Wang H. A facile supramolecular approach to fabricate multifunctional upconversion nanoparticles as a versatile platform for drug loading, in vivo delivery and tumor imaging. J Mater Chem B. 2017;5:2425-35
21. Marrache S, Dhar S. Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proc Natl Acad Sci U S A. 2012;109:16288-93
22. Han SY, Liu YX, Nie X, Xu Q, Jiao F, Li W. et al. Efficient delivery of antitumor drug to the nuclei of tumor cells by amphiphilic biodegradable poly(L-aspartic acid-co-lactic acid)/DPPE co-polymer nanoparticles. Small. 2012;8:1596-606
23. Son S, Kim N, You DG, Yoon HY, Yhee JY, Kim K. et al. Antitumor therapeutic application of self-assembled RNAi-AuNP nanoconstructs: combination of VEGF-RNAi and photothermal ablation. Theranostics. 2017;7:9-22
24. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2016;17:20-37
25. Sun Q, Zhou Z, Qiu N, Shen Y. Rational design of cancer nanomedicine: nanoproperty integration and synchronization. Adv Mater. 2017;29:1606628
26. Chen H, Zhang W, Zhu G, Xie J, Chen X. Rethinking cancer nanotheranostics. Nat Rev Mater. 2017;2:17024
27. Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM. et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9:338-50
28. Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C. et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discovery. 2014;4:998-1013
29. Choi SY, Lin D, Gout PW, Collins CC, Xu Y, Wang Y. Lessons from patient-derived xenografts for better in vitro modeling of human cancer. Adv Drug Delivery Rev. 2014;79-80:222-37
30. Malaney P, Nicosia SV, Davé V. One mouse, one patient paradigm: new avatars of personalized cancer therapy. Cancer Lett. 2014;344:1-12
31. Siolas D, Hannon GJ. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 2013;73:5315-19
32. Cong Y, Xiao H, Xiong H, Wang Z, Ding J, Li C. et al. Dual drug backboned shattering polymeric theranostic nanomedicine for synergistic eradication of patient-derived lung cancer. Adv Mater. 2018. 1706 220
33. Pi F, Binzel DW, Lee TJ, Li Z, Sun M, Rychahou P. et al. Nanoparticle orientation to control RNA loading and ligand display on extracellular vesicles for cancer regression. Nat Nanotechnol. 2018;13:82-89
34. Shen YQ, Jin EL, Zhang B, Murphy CJ, Sui MH, Zhao J. et al. Prodrugs forming high drug loading multifunctional nanocapsules for intracellular cancer drug delivery. J Am Chem Soc. 2010;132:4259-65
35. Wang JR, Sun XR, Mao WW, Sun WL, Tang JB, Sui MH. et al. Tumor redox heterogeneity-responsive prodrug nanocapsules for cancer chemotherapy. Adv Mater. 2013;25:3670-76
36. Zhou Z, Ma X, Murphy CJ, Jin E, Sun Q, Shen Y. et al. Molecularly precise dendrimer-drug conjugates with tunable drug release for cancer therapy. Angew Chem Int Ed. 2014;53:10949-55
37. Venditto VJ, Simanek EE. Cancer therapies utilizing the camptothecins: a review of the in vivo literature. Mol Pharmaceut. 2010;7:307-49
38. Bala V, Rao S, Boyd BJ, Prestidge CA. Prodrug and nanomedicine approaches for the delivery of the camptothecin analogue SN38. J Control Release. 2013;172:48-61
39. Kasai H, Murakami T, Ikuta Y, Koseki Y, Baba K, Oikawa H. et al. Creation of pure nanodrugs and their anticancer properties. Angew Chem Int Ed. 2012;51:10315-18
40. Wang JQ, Mao WW, Lock LL, Tang JB, Sui MH, Sun WL. et al. The role of micelle size in tumor accumulation, penetration, and treatment. ACS Nano. 2015;9:7195-206
41. Hu S, Lee E, Wang C, Wang J, Zhou Z, Li Y. et al. Amphiphilic drugs as surfactants to fabricate excipient-free stable nanodispersions of hydrophobic drugs for cancer chemotherapy. J Control Release. 2015;220:175-79
42. Wang HX, Xie HY, Wang JG, Wu JP, Ma XJ, Li LL. et al. Self-assembling prodrugs by precise programming of molecular structures that contribute distinct stability, pharmacokinetics, and antitumor Efficacy. Adv Funct Mater. 2015;25:4956-65
43. Fang T, Dong YH, Zhang XM, Xie K, Lin L, Wang HX. Integrating a novel SN38 prodrug into the PEGylated liposomal system as a robust platform for efficient cancer therapy in solid tumors. Int J Pharm. 2016;512:39-48
44. Alferiev IS, Iyer R, Croucher JL, Adamo RF, Zhang K, Mangino JL. et al. Nanoparticle-mediated delivery of a rapidly activatable prodrug of SN-38 for neuroblastoma therapy. Biomaterials. 2015;51:22-9
45. Wang HX, Xie HY, Wu JP, Wei XY, Zhou L, Xu X. et al. Structure-based rational design of prodrugs to enable their combination with polymeric nanoparticle delivery platforms for enhanced antitumor efficacy. Angew Chem Int Ed. 2014;53:11532-7
46. Wang H, Wu J, Xie K, Fang T, Chen C, Xie H. et al. Precise engineering of prodrug cocktails into single polymeric nanoparticles for combination cancer therapy: extended and sequentially controllable drug release. ACS Appl Mater Interfaces. 2017;9:10567-76
47. Zhang F, Zhu G, Jacobson O, Liu Y, Chen K, Yu G. et al. Transformative nanomedicine of an amphiphilic camptothecin prodrug for long circulation and high tumor uptake in cancer therapy. ACS Nano. 2017;11:8838-48
48. Tong R, Cheng J. Paclitaxel-initiated, controlled polymerization of lactide for the formulation of polymeric nanoparticulate delivery vehicles. Angew Chem Int Ed. 2008;47:4830-4
49. Tong R, Cheng JJ. Ring-opening polymerization-mediated controlled formulation of polylactide-drug nanoparticles. J Am Chem Soc. 2009;131:4744-54
50. Tam YT, Gao J, Kwon GS. Oligo(lactic acid)n-paclitaxel prodrugs for poly(ethylene glycol)-block-poly(lactic acid) micelles: loading, release, and backbiting conversion for anticancer activity. J Am Chem Soc. 2016;138:8674-7
51. Zhao Y, Fay F, Hak S, Manuel Perez-Aguilar J, Sanchez-Gaytan BL, Goode B. et al. Augmenting drug-carrier compatibility improves tumour nanotherapy efficacy. Nat Commun. 2016;7:11221
52. Shi Y, van der Meel R, Theek B, Blenke EO, Pieters EHE, Fens MHAM. et al. Complete regression of xenograft tumors upon targeted delivery of paclitaxel via pi-pi stacking stabilized polymeric micelles. ACS Nano. 2015;9:3740-52
53. Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Jarvinen T. et al. Prodrugs: design and clinical applications. Nat Rev Drug Discovery. 2008;7:255-70
54. Pridgen EM, Alexis F, Kuo TT, Levy-Nissenbaum E, Karnik R, Blumberg RS. et al. Transepithelial transport of Fc-targeted nanoparticles by the neonatal fc receptor for oral delivery. Science Transl Med. 2013;5:213ra167
55. Zhang M, Akbulut M. Adsorption, desorption, and removal of polymeric nanomedicine on and from cellulose surfaces: effect of size. Langmuir. 2011;27:12550-9
56. Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem Int Ed. 2010;49:6288-308
57. Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M. et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815-23
58. Zhou Z, Ma X, Jin E, Tang J, Sui M, Shen Y. et al. Linear-dendritic drug conjugates forming long-circulating nanorods for cancer-drug delivery. Biomaterials. 2013;34:5722-35
59. Ma X, Huang X, Moore Z, Huang G, Kilgore J, Wang Y. et al. Esterase-activatable β-lapachone prodrug micelles for NQO1-targeted lung cancer therapy. J Control Release. 2015;200:201-11
60. Xu HY, Fan MM, Elhissi AMA, Zhang ZR, Wan KW, Ahmed W. et al. PEGylated graphene oxide for tumor-targeted delivery of paclitaxel. Nanomedicine-UK. 2015;10:1247-62
61. Sun X, Wang G, Zhang H, Hu S, Liu X, Tang J, Shen Y. The blood clearance kinetics and pathway of polymeric micelles in cancer drug delivery. ACS Nano. DOI: 10.1021/acsnano.8b02830
62. Ma S, Chan KW, Hu L, Lee TK, Wo JY, Ng IO. et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132:2542-56
63. Koizumi F, Kitagawa M, Negishi T, Onda T, Matsumoto S, Hamaguchi T. et al. Novel SN-38-incorporating polymeric micelles, NK012, eradicate vascular endothelial growth factor-secreting bulky tumors. Cancer Res. 2006;66:10048-56
64. Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789-802
65. Verslype C, Rosmorduc O, Rougier P, Group EGW. Hepatocellular carcinoma: ESMO-ESDO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(Suppl 7):vii41-8
66. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF. et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-90
67. Lammers T, Kiessling F, Ashford M, Hennink W, Crommelin D, Storm G. Cancer nanomedicine: is targeting our target?. Nat Rev Mater. 2016:1 16069
68. Saltz LB, Cox JV, Blanke C, Rosen LS, Fehrenbacher L, Moore MJ. et al. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan study group. N Engl J Med. 2000;343:905-14
69. Flieger D, Klassert C, Hainke S, Keller R, Kleinschmidt R, Fischbach W. Phase II clinical trial for prevention of delayed diarrhea with cholestyramine/levofloxacin in the second-line treatment with irinotecan biweekly in patients with metastatic colorectal carcinoma. Oncology. 2007;72:10-6
70. Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS. et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330:831-5
71. Gengenbacher N, Singhal M, Augustin HG. Preclinical mouse solid tumour models: status quo, challenges and perspectives. Nat Rev Cancer. 2017;17:751-65
72. Cassidy JW, Caldas C, Bruna A. Maintaining tumor heterogeneity in patient-derived tumor xenografts. Cancer Res. 2015;75:2963-8
Author contact
![]() Corresponding authors: Hangxiang Wang, Key Lab of Combined Multi-Organ Transplantation, Ministry of Public Health, Key Lab of Organ Transplantation, The First Affiliated Hospital, Zhejiang University School of Medicine, 79 Qingchun Road, Hangzhou, Zhejiang 310003, P.R. China. Phone: 86-571-88208173; E-mail: wanghxedu.cn; and Shusen Zheng, Key Lab of Combined Multi-Organ Transplantation, Ministry of Public Health. Key Lab of Organ Transplantation, The First Affiliated Hospital, Zhejiang University School of Medicine, 79 Qing chun Road, Hangzhou, Zhejiang 310003, P.R. China. Phone: 86-571-87236570; Fax: 86-571-87236884; E-mail: shusenzhengedu.cn
Corresponding authors: Hangxiang Wang, Key Lab of Combined Multi-Organ Transplantation, Ministry of Public Health, Key Lab of Organ Transplantation, The First Affiliated Hospital, Zhejiang University School of Medicine, 79 Qingchun Road, Hangzhou, Zhejiang 310003, P.R. China. Phone: 86-571-88208173; E-mail: wanghxedu.cn; and Shusen Zheng, Key Lab of Combined Multi-Organ Transplantation, Ministry of Public Health. Key Lab of Organ Transplantation, The First Affiliated Hospital, Zhejiang University School of Medicine, 79 Qing chun Road, Hangzhou, Zhejiang 310003, P.R. China. Phone: 86-571-87236570; Fax: 86-571-87236884; E-mail: shusenzhengedu.cn