Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(15):4181-4198. doi:10.7150/thno.25707 This issue Cite
Research Paper
Flufenamic acid inhibits secondary hemorrhage and BSCB disruption after spinal cord injury
Yingtao Yao1,2, Jianyi Xu1,2, Tingting Yu1,2, Zhilong Chen1,2, Zhiyong Xiao3, Jiedong Wang3, Yiqiang Hu3, Yongchao Wu3 ![]() , Dan Zhu1,2
, Dan Zhu1,2 ![]()
1. Britton Chance Center for Biomedical Photonics, Wuhan National Laboratory for Optoelectronics-Huazhong University of Science and Technology, Wuhan, Hubei 430074, China
2. MoE Key Laboratory for Biomedical Photonics, Collaborative Innovation Center for Biomedical Engineering, School of Engineering Sciences, Huazhong University of Science and Technology, Wuhan, Hubei 430074, China
3. Department of Orthopedics, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China
Received 2018-2-24; Accepted 2018-6-19; Published 2018-7-30
Citation:
Yao Y, Xu J, Yu T, Chen Z, Xiao Z, Wang J, Hu Y, Wu Y, Zhu D. Flufenamic acid inhibits secondary hemorrhage and BSCB disruption after spinal cord injury. Theranostics 2018; 8(15):4181-4198. doi:10.7150/thno.25707. https://www.thno.org/v08p4181.htm
Other stylesAbstract

Acute spinal cord injury (SCI) induces secondary hemorrhage and initial blood-spinal cord barrier (BSCB) disruption. The transient receptor potential melastatin 4 (Trpm4) together with sulfonylurea receptor 1 (Sur1) forms the Sur1-Trpm4 channel complex. The up-regulation of Sur1-Trpm4 after injury plays a crucial role in secondary hemorrhage, which is the most destructive mechanism in secondary injuries of the central nervous system (CNS). The matrix metalloprotease (MMP)-mediated disruption of the BSCB leads to an inflammatory response, neurotoxin production and neuronal cell apoptosis. Thus, preventing secondary hemorrhage and BSCB disruption should be an important goal of therapeutic interventions in SCI.
Methods: Using a moderate contusion injury model at T10 of the spinal cord, flufenamic acid (FFA) was injected intraperitoneally 1 h after SCI and then continuously once per day for one week.
Results: Trpm4 expression is highly up-regulated in capillaries 1 d after SCI. Treatment with flufenamic acid (FFA) inhibited Trpm4 expression, secondary hemorrhage, and capillary fragmentation and promoted angiogenesis. In addition, FFA significantly inhibited the expression of MMP-2 and MMP-9 at 1 d after SCI and significantly attenuated BSCB disruption at 1 d and 3 d after injury. Furthermore, we found that FFA decreased the hemorrhage- and BSCB disruption-induced activation of microglia/macrophages and was associated with smaller lesions, decreased cavity formation, better myelin preservation and less reactive gliosis. Finally, FFA protected motor neurons and improved locomotor functions after SCI.
Conclusion: This study indicates that FFA improves functional recovery, in part, due to the following reasons: (1) it inhibits the expression of Trpm4 to reduce the secondary hemorrhage; and (2) it inhibits the expression of MMP-2 and MMP-9 to block BSCB disruption. Thus, the results of our study suggest that FFA may represent a potential therapeutic agent for promoting functional recovery.
Keywords: spinal cord injury, secondary hemorrhage, blood-spinal cord barrier, Trpm4, matrix metalloproteases
Introduction
Spinal cord injury (SCI) disrupts neurons and axons, leading to sensory and motor impairment and even paralysis. Functional deficits are caused by the initial mechanical injury and subsequent secondary injury over a period of weeks or even months [1]. The secondary injury increases cell death and the sizes of the damaged areas based on the primary injury. In addition, the axons and neurons of adult mammals regenerate poorly. Therefore, therapeutic strategies have mainly focused on early therapeutic interventions aimed at minimizing secondary injury and improving functional recovery. All types of secondary injury, which can include ischemia/hypoxia, oxidative stress, free radical injury, lipid peroxidation, immune inflammatory reactions, and excitotoxicity, are considered responsible for progressive secondary hemorrhage [2-5], which is the most destructive type of secondary injury in the central nervous system (CNS) [6].
Recently, the use of glibenclamide has been widely reported to inhibit bleeding in SCI. Glibenclamide has since been confirmed as an effective therapeutic agent in traumatic injuries [5, 7]. In fact, progressive hemorrhagic necrosis is always accompanied by capillary fragmentation, and the blood-brain barrier (BBB), including the blood-spinal cord barrier (BSCB), is disrupted after CNS injury. After SCI, BSCB permeability is increased, allowing infiltration by immune cells and neurotoxic products in addition to tissue edema, all of which contribute to the subsequent death of neurons and glia, resulting in permanent neurological disability [8, 9]. Thus, preventing BSCB disruption is another potential approach for therapeutic interventions for SCI. Excessive proteolytic activity by matrix metalloproteases (MMPs), especially the activation of MMP-9 and MMP-2, plays a critical role in BBB/BSCB disruption in a variety of pathological conditions, including SCI [10, 11].
The mammalian transient receptor potential (Trp) superfamily includes 28 nonselective cation channels. While most Trp channels are permeable to both monovalent and divalent ions, Trpm4 is one of two unique TrPs that can conduct monovalent cations and is activated by internal Ca2+ [6, 12-14]. Sulfonylurea receptor 1 (Sur1) is an important member of the adenosine triphosphate (ATP)-binding cassette (ABC) protein superfamily and is also a nonselective cation channel. A recent study proved that Sur1 and Trpm4 co-assembled to form a unique Sur1-Trpm4 protein channel [15]. Previous studies have indicated that glibenclamide is highly effective in reducing secondary hemorrhage and secondary injury by blocking the Sur1 channel of the Sur1-Trpm4 complex [5, 7]. Recently, studies have also confirmed that the up-regulation and activation of the Trpm4 part of the Sur1-Trpm4 protein complex are key molecular events responsible for secondary hemorrhage [6]. Flufenamic acid (FFA) has been known to exert anti-inflammatory and analgesic effects for more than 50 years [16, 17]. Recently, many in vitro cell culture experiments have confirmed that the Sur1-Trpm4 channel is very sensitive and specific to FFA and that its expression is highly reduced by FFA [14, 18]. In addition, Gerzanich V et al. demonstrated that FFA was associated with a strong blockade of capillary fragmentation and secondary hemorrhage in SCI [6]. Based on previous research, we hypothesized that FFA might be a potential therapeutic agent in SCI.
In this study, we first applied FFA to treat SCI. We used a weight-drop contusion model to induce injury at T10 of the spinal cord, and we then analyzed the extent of demyelination, the sizes of the cavities that formed, the size of the lesion area, the amount of necrosis in axons and neurons, and functional recovery to assess the effect of FFA as a therapy for SCI. Then, we examined the extent of secondary hemorrhage and Trpm4 expression, the permeability of the BSCB, the activation of MMP-9 and MMP-2, and the inflammatory immune response triggered by secondary hemorrhage and BSCB disruption to determine the mechanism underlying the role of FFA as a treatment for SCI.
Methods
Experimental animals
We used 10-week-old adult female C57BL/6 mice and 10-week-old adult female Thy1-GFP-M mice (Jackson Laboratory, USA). All the animals were divided randomly into the following three groups (n=12 animals per group): 1) animals that received injections of a vehicle solution (4% PEG-400, 2% DMSO and 94% saline) beginning 1 h after injury and continuing on a daily basis for 7 d (Vehicle control group), 2) animals that received T10 laminectomy without SCI and injections of a vehicle solution (4% PEG-400, 2% DMSO and 94% saline) beginning 1 h after injury and continuing on a daily basis for 7 d (Sham-operated control group), and 3) animals that received injections of FFA dissolved into the vehicle solution as previously described (Treatment group). All animal experiments were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996), and all efforts were made to minimize suffering. All animal procedures were approved by the Hubei Provincial Animal Care and Use Committee and complied with the experimental guidelines of the Animal Experimentation Ethics Committee of Huazhong University of Science and Technology in China.
Spinal cord surgical procedures and care
Mice were kept anesthetized using 2% chloral hydrate (0.18 mL/20 g). Body temperature was maintained by keeping the mice on a heating pad (37 °C) throughout the entire procedure. The skin over the upper thoracic area was shaved and cleaned. The skin was incised, and the muscle tissue was then bluntly dissected to expose laminae T9 to T11. A T10 laminectomy was completed, taking care not to damage the spinal cord while the dorsal lamina was removed. Contusive thoracic (T10) SCI was induced using a blunt force impactor (modified NYU device (0.5-mm impactor head on a 5 g weight dropped 12.5 mm)) [19]. After injury, the muscle layers and skin were sutured with 5-0 silk. The mice were placed on soft bedding on a warming blanket and maintained at 37 °C for approximately 16 h until fully awake.
After they recovered from the anesthetic, the animals were housed four per cage on Alpha-Dri bedding. For 3 d after surgery, the animals received lactated Ringer's solution (0.2 mL/20 g) for hydration, the analgesic drug (4% (w/v) Tolfedine, 0.1 mL/20 g), and penicillin (32000 U/20 g) a prophylactic treatment against urinary tract infections. Manual bladder emptying was performed 2 times daily until reflex bladder emptying was established.
Drug administration
FFA was dissolved in sterile vehicle solution made up of 4% PEG-400, 2% DMSO and 94% saline. The mice in the treatment group were given FFA (12.5 mg/kg) via intraperitoneal (i.p.) injection 1 h after SCI. Drugs were injected continuously once per day for 1 week. In the vehicle and sham groups, the same dose of vehicle solution (4% PEG-400, 2% DMSO and 94% saline) was given continuously for 7 d.
Evaluation of blood-spinal cord barrier permeability
The permeability of the BSCB was investigated with Evans blue dye extravasation according to previous reports [20], with some modifications. To clearly show the fluorescence intensity of the Evan's Blue dye extravasation, Thy1-GFP-M mice were used in this study.
At 1 and 3 d after SCI, 0.15 mL of 2% Evans blue dye (Sigma, St. Louis, MO) in saline was administered in the Thy1-GFP-M mice via intravenous injection. One hour later, the mice were anesthetized and killed by intra-cardiac perfusion with PBS and 4% paraformaldehyde (PFA). A 5-mm length of T10 spinal cord segment containing the lesion site was dissected out and stored in 4% PFA. To qualitatively examine Evan's Blue extravasation, the PFA-fixed spinal cords were cut into transverse sections at a thickness of 20 µm on a cryostat. The fluorescence of Evan's Blue was analyzed with confocal microscopy (LSM 710, Carl Zeiss) in transverse sections beginning 1 mm caudal to the lesion epicenter. The relative fluorescence intensity was quantitatively analyzed by Image J software.
Western blot analysis
At 1 d after injury, the mice were anesthetized with 2% chloral hydrate (10 mg/mL). A 5-mm length of the spinal cord containing the lesion site was quickly dissected and frozen. The dissected cord sample was washed with cold PBS 3 times and then cut into small pieces. The tissue homogenates were incubated at 4 °C for 30 min and then centrifuged at 12000 g for 15 min. The protein concentration was determined using a BCA protein assay kit (Pierce). Equal amounts of total protein were separated with 10% SDS-PAGE gels. Separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). The membranes were blocked in 5% skim milk in 0.5% Tris-buffered saline solution with Tween (TBST) for 1 h at room temperature. After the proteins were transferred and the membranes blocked, the blots were incubated overnight at 4 °C with the following primary antibodies: rabbit anti-Trpm4 (1:200; Omnilabs), rabbit anti-MMP-2 (1:500; Abcam), and rabbit anti-MMP-9 (1:1000; Bioss). For an internal control, the blots were probed with rabbit anti-β-actin antibodies (1:1000; Servicebio). The primary antibodies were detected by rabbit secondary antibodies conjugated to horseradish peroxidase (HRP) (1:3000; Sigma Aldrich). All experiments were repeated three times. The densities of specific bands on the western blots were determined and analyzed with AlphaImager software (Alpha Innotech).
Reverse-transcription polymerase chain reaction (RT-PCR)
Total RNA was extracted using TRIzol Reagent (Invitrogen Life Technologies) according to the manufacturer's protocol. Total RNA (3 μg) was reverse-transcribed to cDNA using a RevertAid First Strand cDNA Synthesis Kit (Thermo). PCR amplification was performed as follows: a denaturation step at 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s, 60 °C for 60 s, and 72 °C for 30 s. β-actin was used as the endogenous control. The following primers (Invitrogen Biotechnology Co., Ltd.) were used for PCR: for TRPM4: CGCCTTCGGTTTGAGTCCTA (forward) and GGCATCCTCTATCCG CTTTAAC (reverse), and for β-actin: GTGACGTTGACATCCGTAAAGA (forward) and GTAACAGTCCGCCTAGAAGCAC (reverse). The amount of PCR product was quantified with a gel document system (ABI).
Gelatin zymography
At 1 d after injury, the mice were anesthetized with 2% chloral hydrate (10 mg/mL). A 5-mm length of the spinal cord containing the lesion site was quickly dissected and frozen. Then, the dissected segments were rapidly homogenized in 1% NP40 lysis (pH 7.5). A total of 300-500 μg of protein per lane was loaded into the wells of precast gels (8% polyacrylamide gels containing 0.1% gelatin). After electrophoresis at 80 V in ice water for 2.5 h, each gel was incubated with 50 ml of zymogram developing buffer for 40 h (37.5 ºC) in a shaking bath. Then, the gels were stained with Coomassie Brilliant Blue (1%, with 10% acetic acid and 10% isopropyl alcohol, diluted with ddH2O).
Preparation for histology and immunohistochemistry
At specific time points after SCI, the mice were anesthetized with 2% chloral hydrate and transcardially perfused first with 0.01 M PBS and then with 4% PFA in 0.01 M PBS. A 5-mm section of the spinal cord centered at the lesion site was dissected out and post-fixed by immersion in the same fixative (4% PFA) overnight. The tissue was dehydrated through a gradient of ethyl alcohol solutions (75% alcohol for 4 h, 85% alcohol for 2 h, 95% alcohol for 1 h and 100% alcohol for 1 h) and then xylene (6 min twice). After they were dehydrated, the spinal cords were embedded in paraffin and cut into serial longitudinal sections with a thickness of 5 μm. After the slices were dried, all the sections were immersed in antigen repair buffer (pH=8) and filled with EDTA for antigen retrieval. Then, the spinal cord sections were blocked in 3% bovine serum albumin (BSA; Sigma) in PBS for 30 min at room temperature. The sections were then incubated overnight at 4 °C with rabbit anti-GFAP (1:1000; Abcam), mouse anti-GFAP (1:100; Abcam), mouse anti-NF-200 (1:100; Abcam), rabbit anti-Trpm4 (1:100; Omnimabs), rabbit anti-Iba-1 (1:100; Abcam), rabbit anti-NeuN (1:100; Servicebio), rabbit anti-ki67 (1:200; Servicebio), and mouse anti-vWF (1:100; Santa Cruz Biotechnology) antibodies. For double-labeling, the sections were incubated with secondary antibodies, including cy3-conjugated goat anti-rabbit (1:300), Alexa Flour 488-conjugated goat anti-mouse (1:400), cy3-conjugated goat anti-mouse (1:300), Alexa Flour 488-conjugated goat anti-rabbit (1:400) and cy3-conjugated goat anti-mouse (1:300) antibodies, at room temperature for 50 min. Counterstaining was performed with 4',6-diamidino-2-phenylindole (DAPI), and images were obtained by confocal microscopy (LSM 710, Carl Zeiss). For the immunofluorescence labeling of capillaries, Wang & Kubes demonstrated good results in CD31-labeled capillaries [21]; therefore, according to their protocol, 10 μL of CD31 conjugated with Alexa Flour-647 (dissolved in 200 μL saline) was injected intravenously. At 1 h after CD31 injection, the animals were sacrificed and perfused with 4% PFA.
At 1 d and 3 d after SCI, the immunohistochemical labeling of capillaries was performed with vimentin and CD105 antibodies in 5 μm-thick longitudinal sections. The sections were incubated overnight at 4 °C with rabbit anti-vimentin (1:1000; Servicebio) and rabbit anti-CD-105 (1:100; Abcam). The primary antibodies were detected at room temperature with goat anti-rabbit secondary antibodies conjugated to horseradish peroxidase (HRP) (1:200; Servicebo).
At 1, 3, 7 and 35 d after SCI, a 5-mm section of the spinal cord that included the lesion site was dissected out, embedded in paraffin, and cut into 5 μm-thick longitudinal or transverse sections. The selected sections were separated by 30-μm intervals. These sections of the spinal cord were mounted on gelatinized slides and stained with hematoxylin and eosin (H&E) for histopathological and morphological analysis.
We used Luxol fast blue (LFB) staining which labels myelin, to analyze the demyelination of the spinal cord. In this experiment, a 4-mm length of spinal cord that included the lesion site was cut into 3 transverse sections (at 1.0 mm rostral to the epicenter, a section centered on the lesion site, and at 1.0 mm caudal to the epicenter), all of which were 5 μm in thickness. The spinal cord sections were incubated with 1% LFB solution at 60 °C for 3-4 h. Then, we successively rinsed the sections with 95% alcohol and distilled water. The sections were then dipped in 0.5% lithium carbonate solution for 5 s and washed with 70% alcohol and distilled water. These steps were repeated until there was a sharp contrast between the white matter and the gray matter. Dehydration was performed using 95% and 100% alcohol. The sections were cleared in xylene for 1 min and then coverslipped.
Cholera toxin beta-subunit (CTB) labeling of motor neurons and 3D imaging
Cholera toxin beta-subunit (CTB) was used to label the motor neurons in the lumbar cord in tissues obtained at 30 d after contusion injury (n=4 per group). After the animals were anesthetized, CTB conjugated with Alexa Flour-647 (1%, Invitrogen) was bilaterally injected into the sciatic nerves. For each injection, 3.5 µL of CTB conjugate was slowly pressure-injected using a 5-μL Hamilton syringe over approximately 5 min. After the injections were completed, the skin and muscle overlying the sciatic nerve was sutured with 5-0 silk. At 5 d after CTB injection, the animals were sacrificed and perfused with 4% PFA. A 5 mm-long length of lumbar cord containing the L4-L6 segment was dissected out and post-fixed by immersion in 4% PFA overnight.
It is noted that various optical clearing methods provide a way to quantify the number of motor neurons [22, 23]. In this work, we used a modified optical clearing protocol (FDISCO) we developed (unpublished) based on 3DISCO [24] to clear the entire L1-L6 segment of the lumbar cord. The optically cleared cords were then imaged with confocal microscopy (LSM710, Carl Zeiss) to obtain a 3D image.
Morphological quantification
To analyze bleeding areas and cavities., continuous 5 μm longitudinal sections were cut, and sections were selected at 30 μm intervals. The selected sections were stained with H&E, and a total of 8 sections per mouse were analyzed (n=3 per group). The bleeding areas and cavity areas in each section were analyzed with Image J software. The values obtained for the bleeding areas and cavity areas in all the 8 sections were averaged to obtain an average bleeding area and an average cavity area for each mouse.
To analyze the viable neurons, 5-μm longitudinal sections through the middle of the ventral horn were immunofluorescently labeled with NeuN. To quantify the surviving neurons, we analyzed 5 regions of interest (ROIs, 320 μm × 320 μm), including 1.5 mm rostral to the epicenter, 1.0 mm rostral to the epicenter, the center of the lesion site, 1.0 mm caudal to the epicenter, and 1.5 mm caudal to the epicenter in the ventral horns. The number of viable neurons from the 5 ROIs was estimated as the product of the mean number of surviving neurons per square millimeter.
To accurately assess the extent of broken fragments of capillaries between the vehicle control group and the FFA-treated group, the counts of different capillary lengths were quantified. For each mouse, 5-μm longitudinal sections were immunolabeled for vimentin as described above, and 4 ROIs (340 μm × 170 μm) in the penumbra adjacent to the necrotic void were identified. The capillary lengths were calculated according to a previously described method [25]. Briefly, the identified capillaries should meet certain requirement that the maximum length of each object must be longer than 5 μm in size. The capillary length was evaluated using the following formula: L = (P + (P2 - 16 × A)0.5)/4, where P and A are the perimeter and the area of the object, respectively. Histograms of capillaries' lengths (bin width 5 μm) were constructed by combining data from three mice in the vehicle control and the FFA -treated groups.
To quantify the amount of angiogenesis, 3 ROIs (300 μm × 300 μm) were selected from the penumbra caudal adjacent to the necrotic void, and the vWF/ki-67 co-labeled vessels or the CD105-positve vessels in the ROIs were identified and analyzed.
To quantify infiltration by macrophages/microglia and the following astrocytic activation, 10 square ROIs (160 μm × 160 μm) were selected from the same section for analysis. All the ROIs were distributed at equal intervals along the rostral, lateral, and caudal borders of the spinal cord. The number of activated microglia (macrophages) and reactive astrocytes present in the entire section was estimated as the product of the mean number of macrophages/microglia per square millimeter.
To accurately assess the demyelination of spinal cord, 3 continuous transverse sections (beginning 1 mm caudal to the epicenter with each section separated by 30 μm) were obtained per mouse, stained with LFB, and analyzed (n=4 per group). The LFB-positive area (blue) of the spared white matter was also quantified in Image J software, and the ratio of the area of LFB-stained white matter to that of normal white matter was calculated.
To quantify the number of motor neurons, the spinal cord was analyzed with 3D imaging according to an optical clearing protocol. Then, the bilateral CTB-labeled motor neurons were analyzed with Imaris 7.6.0 software.
Behavioral assessments
All animals were acclimated to the open field used for the locomotor test once daily before surgery. Motor function was assessed using the Basso mouse scale (BMS) open-field test [26], which ranges from 0 (complete paralysis) to 9 (normal locomotion). All animals were allowed to move freely in the open field for 5 min, and two independent investigators performed blinded assessments of their performance. The animals in each group (n=12) were tested at 1, 3, 7, 14, 21, 28 and 35 d following injury.
Statistical analyses
Behavioral scores were determined by two independent observers who were blinded to the surgery and treatment associated with each animal. Comparisons between the vehicle- and FFA-treated groups were performed with the unpaired Student's t test. Multiple comparisons among groups (n≥3) were performed using one-way ANOVA followed by Tukey's post hoc analysis. All data are presented as the mean ± SD. *p<0.05, **p<0.01, ***p<0.001. All statistical analyses were performed using the statistical software package SPSS 13.0.
Results
Flufenamic acid (FFA) reduces secondary hemorrhage and protects the structural integrity of capillaries
At 1 and 3 d after SCI, the cords of vehicle control mice showed prominent surface bleeding. Bleeding areas were notably lower on both the dorsal and ventral sides of FFA-treated spinal cords than on the same sides in the vehicle-treated controls (Figure 1A-B). Except for hemorrhaging at the surface of the injured cords, internal bleeding was mainly located at the impact site. In our study, H&E was used to evaluate and quantify internal bleeding. Briefly, a 5 mm-long longitudinal section of the spinal cord that included the lesion site was cut into 5 μm-thick sections, which were selected at 30 μm intervals to produce a total of 8 sections for each spinal cord (n=3/group). After injury, H&E staining revealed several prominent spotty hemorrhagic areas in the gray matter (Figure 1C-D). As shown in Figure 1E, the hemorrhagic area was smaller in the FFA-treated group than in the vehicle control group at both 1 d (n=3/group; Veh, 0.386±0.047 versus FFA, 0.131±0.036; p<0.001) and 3 d (n=3/group; Veh, 0.034±0.007 versus FFA, 0.012±0.0032; p<0.01). Additionally, H&E staining revealed that at 3 d after injury, numerous round swollen neurons (abnormal neurons) were observed in the vehicle controls whereas a number of large polygonal neurons (neurons with normal morphology) were observed in the treatment group (Figure S2). To quantify the number of viable neurons at 1 d and 3 d after injury, we immunolabeled the neurons with NeuN, and the surviving neurons were quantified as the number of NeuN-positive and DAPI-labeled cells (Figure 1F). As shown in Figure 1G-H, there were more surviving neurons in the sham controls than in the vehicle control and FFA-treated groups at both 1 d and 3 d after SCI (n=3/group). In addition, there were significantly more surviving neurons in the FFA-treated group than in the vehicle control group at both 1 d and 3 d after injury.
Figure 1
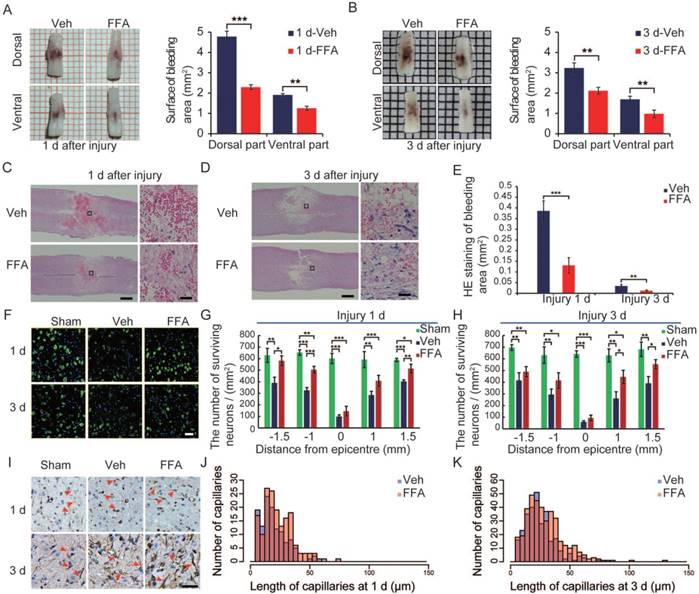
FFA reduces secondary hemorrhage and protects the structural integrity of capillaries. (A-B) Spinal cords at 1 d and 3 d after injury. Cords were obtained from vehicle control mice and FFA-treated mice, as indicated. There was a notable reduction in bleeding area at both the dorsal and ventral sides in the FFA-treated mice than in the vehicle controls. Intravascular blood was removed via PBS perfusion. The bleeding area at the surface of the injured cords obtained from FFA-treated and vehicle control mice was quantified, as shown. (C-D) Longitudinal sections of cords obtained at 1 d and 3 d, as indicated, after SCI. The cords were stained with hematoxylin and eosin (H&E). Evidence of hemorrhage and edema was observed in the sections. Scale bar = 500 μm (left images show the whole spinal cords) and 50 μm (right images, as marked in a black box in the left spinal cords). (E) Quantification of the internal bleeding area in injured cords at 1 d and 3 d after injury. (F) Representative immunofluorescence labeling images for NeuN (green) obtained from longitudinal sections 1 mm caudal to the lesion site at 1 d and 3 d after injury. The blue staining indicates the DAPI-stained nuclei. Scale bar = 50 μm. (G-H) Quantitative analysis of NeuN-positive neurons at 1 d and 3 d after injury. (I) Immunohistochemical labeling of capillaries was performed with vimentin in sections including the injury penumbra at 1 d and 3 d after injury. The results showed fragmentation in the capillaries (red arrow) of sham-operated, vehicle control and FFA-treated mice, as indicated. Scale bar = 50 μm. (J-K) Quantitative analysis of the lengths of penumbral microvessels in vehicle control group and FFA-treated group at 1 d and 3 d after injury. Error bars represent the SD; *p<0.05, **p<0.01, and ***p<0.001 by one-way ANOVA followed by Tukey's post hoc analysis.
The catastrophic failure of capillary integrity induces petechial hemorrhaging. We examined the structural integrity of capillaries in the region of the lesion by applying immunolabeling for vimentin (n=3/group), which is up-regulated in the endothelium following injury. After 1 d, the number of vimentin-positive capillaries was smaller and more fragmenting was observed in the vehicle controls and FFA-treated cords than in the sham group (Figure 1F). There were many more such capillaries in the FFA-treated group than in the vehicle control group, and the capillaries appeared much longer in the FFA-treated group than in the vehicle control group. In the vehicle control mice, broken fragments of capillaries and foreshortened vessels were prominent around the lesion site. In contrast, in the mice treated with FFA, the capillaries were elongated and had a more normal appearance around the lesion site, and this may have accounted for the significant reduction in extravasated blood (Figure 1J-K).
FFA promotes angiogenesis in SCI
At 3 d after injury, an increase in vimentin-positive capillaries was observed in all 3 groups (sham control, vehicle and FFA-treated), which meant that newly born vessels might form after SCI. To identify and quantify the newly formed vessels, vWF (von Willebrand factor) and ki-67 co-labeled vessels in the tissue were identified and analyzed. As shown in Figure 2A-B, an increase in angiogenesis was observed at 3 d after injury in all 3 groups compared to that at 1 d after injury. In addition, the density of angiogenesis was significantly higher (n=3/group; Veh, 34.01±5.67 vWF-ki67 co-labeled microvessels/mm2 versus FFA, 73.70±11.34 vWF-ki67 co-labeled microvessels/mm2; p<0.01) at 3 d after injury in the FFA-treated mice than in the vehicle controls (Figure 2C). Furthermore, we also used another angiogenesis marker, endoglin (CD-105), to identify and quantify the newly formed vessels at 1 d and 3 d after injury (Figure 2D). Our results showed that the density of CD105-positve microvessels was significantly higher (n=3/group; Veh, 96.33±69.2 CD105-positve microvessels/mm2 versus FFA, 211.11±29.44 CD105-positve microvessels /mm2; p<0.01) in the FFA-treated mice at 3 d after injury than in the vehicle controls (Figure 2E).
Up-regulation of Trpm4 in SCI
Contusion-type thoracic SCI is one of the most common forms of SCI observed in patients. To ensure clinical consistency, a thoracic 10 contusion injury model was chosen for this study. We studied Trpm4 expression in spinal cords obtained from uninjured mice (sham-operated group) and mice submitted to severe SCI (a 5 g weight dropped from a height of 12.5 mm; n=3/group).
In the uninjured controls, Trpm4 expression levels were very low. However, at 1 d after SCI, Trpm4 was prominently up-regulated in the tissues surrounding the injury and its expression domain extended to tissues distant from the actual impact site (Figure S3). An examination of tissues obtained earlier (at 1 h after injury) showed that almost no change in Trpm4 expression had occurred at that time point (data not shown).
In the core of the lesion, Trpm4 was up-regulated in various cells and structures, especially in capillary-like elongated structures (Figure 3A). In the penumbra (the tissue adjacent to the lesion core), Trpm4 was prominently up-regulated in elongated structures that were co-labeled with CD31, suggesting that these structures were capillaries (Figure 3A).
The up-regulation of Trpm4 was also confirmed by immunoblotting. Trpm4 levels were very low in uninjured cords, whereas a prominent single band was observed at approximately 193 kDa was observed at 1 d post-SCI (Figure 3D). Western blot analysis revealed the TRPM4 expression was 15-fold higher in the penumbra region (Figure 3E) than in uninjured cords.
FFA blocks Trpm4 expression in SCI
At 1 h after injury, the mice were injected with FFA (i.p.). Immunohistochemical localization of Trpm4 showed that its expression was much lower in the FFA-treated group than in the vehicle control group (Figure S2A).
We used reverse transcriptase PCR to examine Trpm4 mRNA expression levels after injury (n=3/group). As shown in Figure 3B-C, the level of Trpm4 mRNA was higher at 1 d after injury than in the sham controls. In addition, at 24 h after injury, Trpm4 levels were significantly lower in the FFA-treated mice than in the vehicle controls.
Immunoblotting (n=3/group) of tissues obtained 1 d after SCI confirmed that FFA was highly effective in reducing Trpm4 protein expression following SCI (Figure 3D-E).
FFA inhibits BSCB disruption and blocks the expression and activation of MMP-2 and MMP-9 after SCI
Traumatic SCI results in changes in the microvasculature and the disruption of the BSCB [27, 28]. Because our data confirm that FFA is highly effective in protecting the structural integrity of capillaries, we anticipated that FFA might also attenuate BSCB disruption. Thus, we examined the effect of FFA on BSCB permeability at 1 d and 3 d after injury in Evan's Blue assays (n=3/group). As shown in Figure 4A, the amount of Evan's Blue dye extravasation was substantially higher in the SCI-treated mice than in the uninjured sham controls, implying that BSCB leakage had occurred.
Figure 2
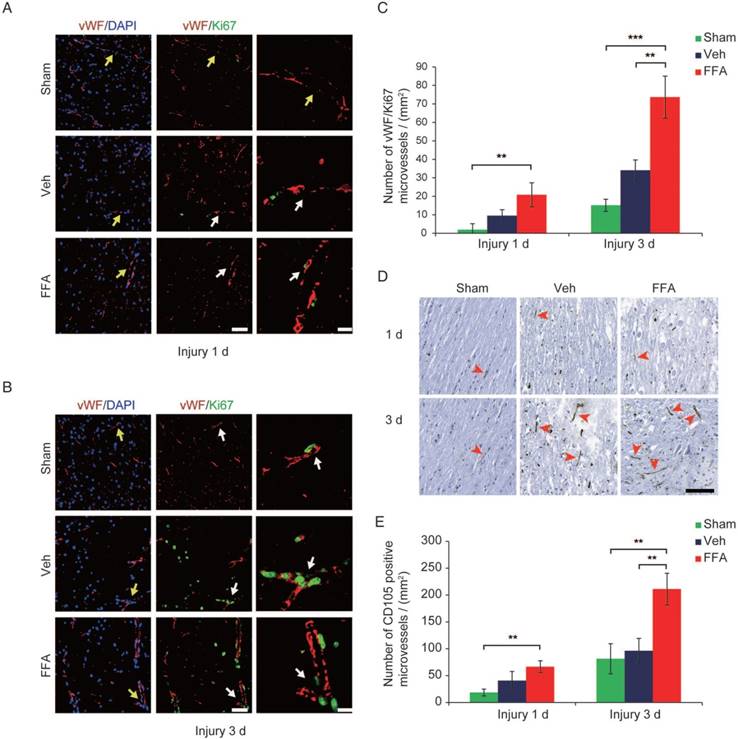
FFA promotes angiogenesis in SCI. (A-B) Representative immunofluorescence labeling images obtained from longitudinal sections adjacent to the necrotic void. The images show labeling for the blood vessel marker vWF (red), the proliferation marker ki-67 (green), and the nuclei marker (DAPI, blue) in tissues obtained at 1 d and 3 d after injury. The vWF-positive vessel-like structures with nuclei indicate the microvessels (yellow arrows). The vWF/ki-67 co-labeled vessels indicate representative newly formed microvessels (white arrows). Scale bar = 50 μm (left images) and 15 μm (right, higher magnification images, as marked with the white arrows). (C) Quantitative analysis of vWF/ki-67 microvessels at 1 d and 3 d after injury. (D) Representative immunohistochemical staining images for CD105 obtained from longitudinal sections adjacent to the necrotic void. The red arrows indicate the CD105-positive microvessels. Scale bar = 100 μm. (E) Quantitative analysis of the CD105-positive microvessels at 1 d and 3 d after injury. The error bars represent the SD. *p<0.05, **p<0.01, and ***p<0.001 by one-way ANOVA followed by Tukey's post hoc analysis.
As shown in Figure 4B, the red fluorescent signal representing the Evan's Blue dye extravasation and the green fluorescent signal representing the background showed the shape of the transverse section. To clearly show the fluorescence GFP signal, 20x objective high magnification images are also shown (Figure S4). The results in Figure 4C showed that FFA treatment resulted in a significantly lower amount of Evan's Blue dye extravasation at both at 1 d and 3 d after injury than was observed in the vehicle control. A qualitative analysis also showed that the fluorescence intensity of Evan's Blue was higher in the injured spinal cord (at both 1 d and 3 d) than in the sham controls and that FFA significantly reduced its fluorescence intensity at 1 d (n=3/group; Veh, 146.80±5.24 versus FFA, 98.71±4.42; p<0.001) and 3 d (n=3/group; Veh, 56.73±2.44 versus FFA, 13.35±2.88; p<0.001) after injury (Figure 4C). These results indicated that FFA inhibited BSCB permeability after injury.
Figure 3
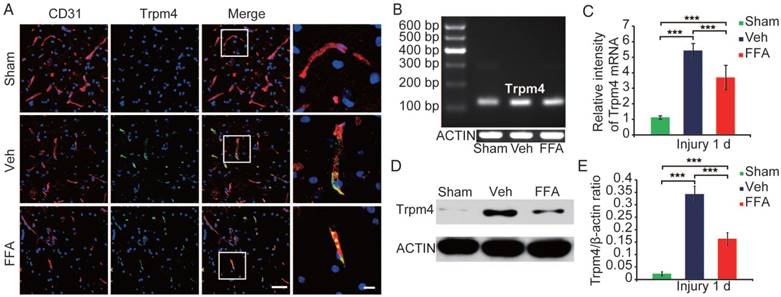
Trpm4 is up-regulated after SCI, and FFA blocks Trpm4 expression in SCI. (A) Double immunofluorescence labeling of capillaries for Trpm4 (green) and CD31 (red) in sections through the penumbra at 1 d after injury in tissues obtained from sham-operated, vehicle control and FFA-treated mice. There was almost no obvious Trpm4 expression in the capillaries (labeled with CD31) of the sham-operated mice. As shown in the vehicle control and FFA-treated mice, Trpm4 was prominently up-regulated in the capillaries. Scale bar = 50 μm (left images) and 15 μm (right, higher magnification images, as marked in a white box in the left images). (B) RT-PCR for Trpm4 mRNA at 1 d after injury. β-actin was used as the control. (C) Bar graph showing the quantitative analysis of the RT-PCR results. (D) Western blots performed for the Trpm4 protein in tissues obtained at 1 d after SCI. β-actin was used as the control. (E) Bar graph showing a densitometry analysis of the results of the Western blotting experiments. The error bars represent the SD. *p<0.05, **p<0.01, and ***p<0.001 by one-way ANOVA followed by Tukey's post hoc analysis.
Figure 4
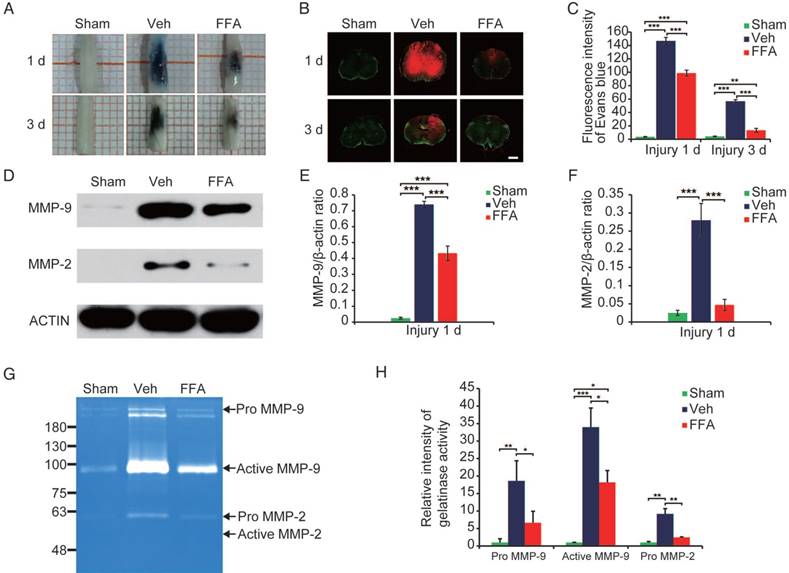
FFA inhibits blood-spinal cord barrier (BSCB) disruption and the expression of MMP-2/MMP-9 after SCI. To evaluate BSCB permeability, 0.15 mL of 2% Evans blue dye was intravenously injected via the tail at 1 d and 3 d after SCI. (A) Representative whole spinal cords showing Evan's Blue dye extravasation into the spinal cord at 1 d and 3 d. (B) Representative confocal images showing Evan's Blue extravasation at a position 1 mm caudal to the lesion epicenter at 1 d and 3 d after SCI. The green color represents the neurons or fibers labeled with GFP, and the red color represents the Evan's Blue dye extravasation. Scale bar = 500 μm. (C) Quantitative analyses of the fluorescence intensity of Evan's Blue. (D) Western blots showing MMP-2 and MMP-9 protein levels in tissues obtained at 1 d after SCI. β-actin was used as the control. (E) Densitometric analysis of MMP-2 protein expression. (F) Densitometric analysis of MMP-9 protein expression. (G) Gelatin zymography showing the activity of MMP-2 and MMP-9 at 1 d after SCI. (H) Densitometric analysis of the gelatin zymography. The error bars represent the SD. *p<0.05, **p<0.01, and ***p<0.001 by one-way ANOVA followed by Tukey's post hoc analysis.
Excessive proteolytic activity by MMPs, such as MMP-2 and MMP-9, results in BSCB disruption after SCI [20, 29, 30]. We then examined the expression of MMP-2 and MMP-9 after injury (n=3/group). As shown in Figure 4D, the protein levels of MMP-2 and MMP-9 were higher at 1 d after injury than in the sham controls. In addition, MMP2 and MMP9 protein expression levels were significantly lower at 1 d after injury in the FFA-treated mice than in the vehicle controls (Figure 4E-F).
In addition, gelatin zymography was performed to determine the activity of MMP-2 and MMP-9. As shown in Figure 4G, prominent bands of the MMP9 active form and inactive zymogen (pro-MMP9) were observed and analyzed, indicating that MMP9 activity was significantly increased at 1 day after injury. Additionally, the pro-MMP2 level was also higher at 1 d after injury compared than in the sham controls. Furthermore, the levels of pro-MMP9, active MMP9 and pro-MMP2 were significantly lower (n=3/group; pro-MMP9, Veh, 18.63±5.74 versus FFA, 6.65±3.29; active MMP9, Veh, 33.97±5.47 versus FFA, 18.20 ± 3.37; pro-MMP2, Veh, 9.18±1.53 versus FFA, 2.47±0.12; p<0.05) in the FFA-treated group at 1 d after injury than in the vehicle group (Figure 4H).
FFA inhibits microglial activation following SCI
During pathological processes, BSCB disruption and hemorrhage occur following blood cell infiltration after SCI triggers a secondary degenerative cascade that includes inflammatory immune responses [20, 27]. The microglia/macrophages located within or proximal to the lesion produce neurotoxic factors, such as tumor necrosis factor alpha (TNF-α), suggesting that the microglia/macrophages located within the epicenter of the wound during the early period following an injury are neurotoxic [31, 32]. As shown in Figure 5A-C, the microglia immunolabeled with Iba-1 appeared to be 'resting' (i.e., having 2 or more processes and a ramified morphology) before injury. However, the Iba-1-positive microglia observed in the vehicle control and FFA-treated cords had become amoeboid, suggesting microglial activation (indicated by microglia with less than 2 processes and an amoeboid morphology). Iba-1 staining demonstrated that fewer microglia were activated and the number of macrophage/microglia was significantly lower at 3 d (n=3/group; Veh, 478.3±34.2 activated microglia/mm2 versus FFA, 356.8±38.8 activated microglia/mm2; p<0.001) and 7 d (n=3/group; Veh, 602.3±52.5 activated microglia/mm2 versus FFA, 467.7±57.6 activated microglia/mm2; p<0.001) after injury in the FFA-treated mice than in the vehicle controls (Figure 5D). Astrocytic activation always followed and was promoted by the microglial activation [33, 34]. We then sought to determine if astrogliosis was reduced following inhibition of microglial activation with FFA. In this study, immunofluorescence for the astrocyte marker glial fibrillary acidic protein (GFAP) was performed on longitudinal sections, and the results shown that the GFAP-expressing reactive astrocytes was significantly lower (n=3/group; Veh, 1744.9±59.8 reactive astrocytes/mm2 versus FFA, 1250±39.1 reactive astrocytes/mm2; p<0.001 ) in the FFA-treated group at 7 d after injury than in the vehicle group (Figure 5E).
FFA inhibits secondary injury
The primary injury induced the initial stages of BSCB disruption and secondary hemorrhage. BSCB disruption and hemorrhage led to infiltration by immune cells and a variety of toxic molecules and was followed by secondary tissue damage, such as demyelination, neuronal loss, lesion expansion and cavity formation [8, 35, 36]. Demyelination was assessed with LFB staining, as shown in cross-sections in Figure 6A at 7 d after injury. In the FFA-treated and vehicle control groups, LFB staining was associated with more prominent dissociation between the gray matter and white matter than was observed in the sham controls. However, the extent of dissociation between gray matter and white matter was much lower in the FFA-treated mice than in the vehicle controls. At 35 d after injury, there was less white matter, indicating an increase in the loss of myelin, in the FFA-treated and vehicle control groups than in the sham controls (Figure 6B). As shown in Figure 6C, in tissues located 1 mm caudal to the epicenter at 35 d after SCI, treatment with FFA resulted in less myelin loss, and the ratio of spared LFB-positive myelin sheaths was significantly higher in the FFA-treated group (n=4/group, 37.6±1.76%) than in the vehicle control group (n=4/group, 31.5±2.13%; p<0.01).
H&E-stained (n=3) the hemorrhagic area was significantly smaller in longitudinal sections obtained after 7 d than in those obtained at 1 d and 3 d after injury and much lower in the FFA-treated group than in the vehicle control group (Figure 6D). In addition, as shown in Figure 6D, treatment with FFA was associated with smaller lesions and less neuronal loss at 7 d after SCI than was observed in the vehicle controls. At 35 d after injury, the loss of tissue at the lesion site was severe and showed areas of cavitation (Figure 6E). The average cavity area was significantly smaller in the FFA-treated group (n=3/group, 0.0297±0.0071 mm2) than in the vehicle control group (n=3/group, 0.356±0.0855 mm2; p<0.01) (Figure 6E-F).
Figure 5
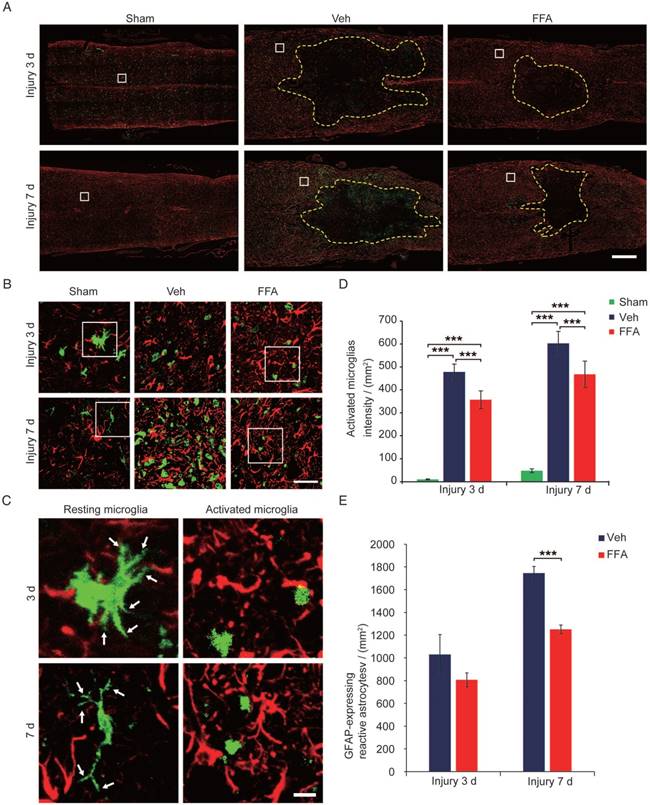
FFA inhibits microglial activation following SCI. (A) Representative images of longitudinal sections obtained from spinal cords removed at 3 d and 7 d after injury showing double immunofluorescence for GFAP (red) and Iba-1 (green). Immunofluorescence labeling for GFAP was mainly used to reveal the lesion border (yellow dotted line). The white-boxed area represents a similar position 500 μm caudal to the necrotic void border. Scale bar = 500 μm. (B) High magnification images of GFAP/Iba-1 in the area marked by the white box in (A). Scale bar = 50 μm. (C) High-resolution images of representative 'resting' microglia (2 or more processes and a ramified morphology) and active microglia (less than 2 processes and an amoeboid morphology) in the area marked by the white box in (B). The white arrow indicates the ramified part of the microglia. Scale bar=15 μm. (D) Quantification of the Iba-1 labeling intensities obtained from 10 ROIs at the rostral, lateral, and caudal borders of the injury. (E) Quantification of the GFAP-expressing reactive astrocytes at 3 d and 7 d after injury. The error bars represent the SD. *p<0.05, **p<0.01, and ***p<0.001 by one-way ANOVA followed by Tukey's post hoc analysis.
The formation of scar tissue is mediated in part by reactive astrocytes, and the resulting tissue has long been thought to represent a major barrier to axon regeneration after injury [37, 38]. Astrocytic activation follows and is promoted by microglial responses [33, 39]. Hence, we next sought to determine whether astrogliosis is reduced by the FFA-mediated inhibition of microglial activation. Immunofluorescence for the astrocyte marker glial fibrillary acidic protein (GFAP) was performed on longitudinal sections, and the results revealed that at both 7 and 35 d after injury, there were fewer GFAP-expressing reactive astrocytes in the sham control group than in the FFA-treated and vehicle control groups. In addition, there were fewer GFAP-expressing reactive astrocytes in the FFA-treated mice than in the vehicle controls (areas marked by the yellow and green boxes in Figure 6G).
Figure 6
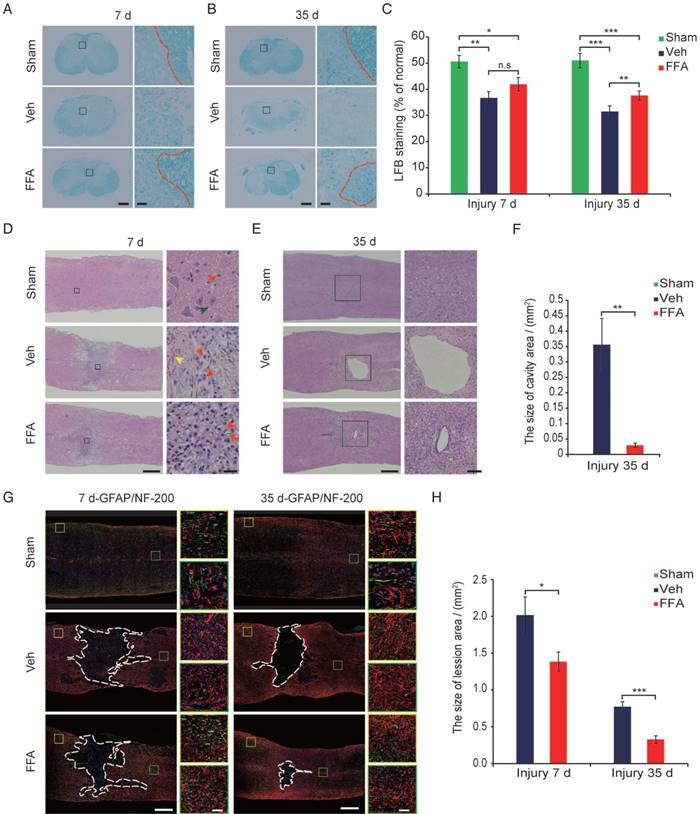
FFA inhibits secondary injury. (A-B) Luxol fast blue (LFB) staining of transverse sections at 1 mm caudal to the epicenter at 7 d and 35 d after injury. The dark blue areas indicate white matter. The areas surrounded by the red dotted line indicate the myelin in the gray boxed area. There was less white matter in the vehicle control and FFA-treated cords than in the sham-operated cords, indicating a more substantial loss of myelin. Scale bar = 300 μm (left images show whole spinal cords) and 20 μm (right high-power images are marked in a gray box in the left images of spinal cords). (C) Quantitative analyses of the extent of demyelination at 7 d and 35 d after injury. (D) Representative images of longitudinal sections obtained at 7 d after SCI and stained with hematoxylin and eosin (H&E). The sections from the vehicle control and FFA-treated groups showed significantly smaller bleeding areas than were observed at 1 d and 3 d after injury. Scale bar = 500 μm (left images show whole spinal cords) and 50 μm (right high-power images show areas marked by gray boxes in the left images of spinal cords). The red arrows point to the neutrophils, the green arrows point to the neurons, and the yellow arrows point to the bleeding area. (E) Representative images of longitudinal sections stained with H&E at 35 d after SCI. The sections obtained from vehicle control and FFA-treated mice showed a more severe loss of tissue at the lesion site and areas of cavitation. Scale bar = 500 μm (left images show whole spinal cords) and 150 μm (right high-power images show area marked in a gray box in the left images of spinal cords). (F) Quantitative analysis of cavity area at 35 d after SCI. (G) Immunofluorescence labeling of longitudinal sections through the thoracic cord. Shown are labeling for NF200 (green), GFAP (red), and nuclei (DAPI, blue) in tissues obtained at 7d and 35 d after injury. The lesion (area surrounded by a dotted line) was defined as the area devoid of staining that was bordered by GFAP+ immunoreactivity. The yellow and green boxed areas represent similar positions caudal and rostral to the epicenter, respectively. The dashed line indicates the lesion border. Scale bar = 500 μm (left images show whole spinal cords) and 50 μm (right high-power images show areas marked by green and yellow boxes in the left images of spinal cords). (H) Quantitative analysis of the lesion area at 7d and 35 d after SCI. The error bars represent the SD. *p<0.05, **p<0.01, and ***p<0.001 by one-way ANOVA followed by Tukey's post hoc analysis.
To analyze lesion size, GFAP immunofluorescence was used to delineate the lesion borders, and the average sizes of the lesion areas were calculated. The lesion area (area surrounded by a dotted line) was significantly smaller in the FFA-treated group than in the vehicle control group at 7d (n=3/group, 31.3% decrease; p<0.05) and 35 d (n=3/group, 57.7% decrease; p<0.001) after injury (Figure 6H).
FFA protects motor neurons and improves functional recovery after SCI
We next asked whether FFA promotes axonal growth by diminishing glial scarring. We evaluated neurofilament 200 (NF200) expression to assess axonal preservation at 7 d and 35 d after injury. The high-power images show areas marked by yellow and green boxes in Figure 6G, and these data indicate that the number of NF200-positive axons beyond the glial scars was much higher in the FFA-treated mice than in the vehicle control mice at 7 d and 35 d after injury. In addition, the average length of the NF200-positive axons was much higher in the FFA-treated group than in the vehicle control group at both 7 d and 35 d after injury (Figure 6G).
To precisely assess whether FFA alters the number of motor neurons, 3D imaging was used to bilaterally evaluate CTB-labeled motor neurons in the lumbar motoneuron pools that innervate the hindlimb to mediate movement (Figure 7A-B and Video S1). As shown in Figure 5C, after 35 d, the number of motor neurons was significantly lower after injury than in the sham control group (n=4/group, 27% decrease; p<0.001). In addition, FFA reduced cell death in motor neurons, and the number of motor neurons was significantly higher in the FFA-treated group (n=4/group, 3800±121%) than in the vehicle control group (n=4/group, 3338±56.9; p<0.01) (Figure 7C).
Neurobehavioral function was assessed using the BMS, which ranges from 0 (complete paralysis) to 9 (normal locomotion). Animals were monitored weekly to assess hindlimb locomotor activity on the BMS in an open-field test. The results showed that hindlimb locomotor function was significantly better in the FFA-treated group from 7 to 35 d after injury than was observed in the vehicle control group (n=12/group, 35 d, BMS score; FFA, 4.04±0.5 versus Veh, 2.21±0.62; p<0.0001) (Figure 7D and Video S2). The open-field test also showed that FFA significantly improved functional recovery after SCI compared with that in the controls (Figure S5A-B).
Figure 7
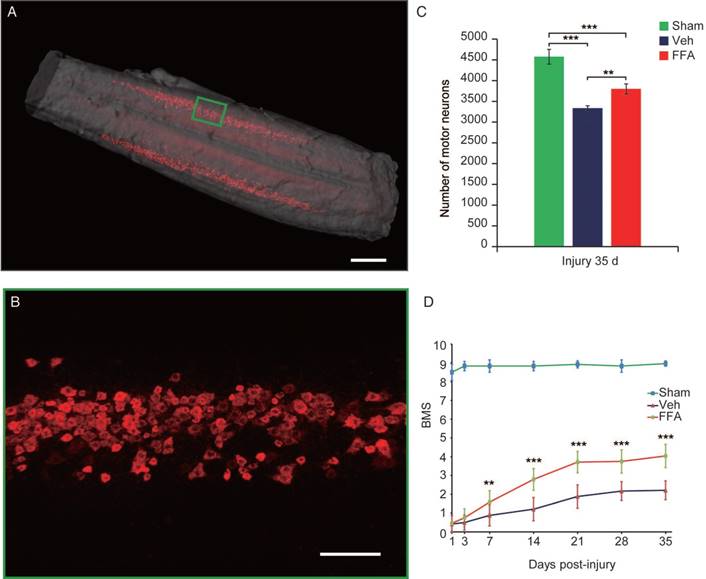
FFA protects motor neurons and improves functional recovery after SCI. (A) Three-dimensional imaging of the L1-L6 lumbar cord, including the motor neuron pool. The motor neuron pool was specifically labeled with cholera toxin β (CTβ)-Alexa fluor 647 via retrograde tracing from the sciatic nerve. Scale bar = 500 μm. (B) High-magnification images of motor neurons in the area marked by the green box in (A). Scale bar = 150 μm. (C) Quantitative analysis of the number of motor neurons. (D) Functional recovery was assessed according to the BMS open-field test. The error bars represent the SD. *p<0.05, **p<0.01, and ***p<0.001 by one-way ANOVA followed by Tukey's post hoc analysis.
Discussion
The results of this study suggest that Trpm4 is prominently up-regulated in capillaries, and we further confirm that FFA is highly effective in reducing Trpm4 expression. Because Trpm4 expression is blocked around the lesion site, our data show that FFA reduces secondary hemorrhage, protects the structural integrity of capillaries, and promotes angiogenesis. In addition, we show that FFA inhibits BSCB disruption and the expression of both MMP-2 and MMP-9, which affect the disruption of the BSCB. Furthermore, FFA decreases the secondary hemorrhage- and BSCB disruption-induced activation of microglia/macrophages. Finally, we show that FFA treatment is associated with smaller lesions, decreased cavity formation, better myelin preservation and less reactive gliosis and it promotes axonal preservation and improves functional recovery after SCI. Taken together, our results indicate that the neuroprotective effect of FFA is mediated in part by the following two mechanisms: (1) the inhibition of the expression of Trpm4 reduces secondary hemorrhage, and (2) the inhibition of the expression of MMP-2 and MMP-9 blocks BSCB disruption, thereby reducing the amount of blood cell infiltration that usually follows SCI.
Individual petechial hemorrhages form around the lesion site during the early stage (10-15 min) after injury. At 3-24 h later, petechial hemorrhages (small spots of bleeding from capillaries) continue to enlarge and coalesce in more distant tissues to form a characteristic region of hemorrhage that eventually “caps” the advancing front of the lesion [33, 40]. Over time, an increasing number of petechial hemorrhages eventually coalesces into a hemorrhagic lesion that encompasses a volume far greater than the site of primary injury [4, 41]. Recent reports have suggested that hemorrhaging results from the progressive catastrophic failure of the structural integrity of capillaries [5, 42]. In this study, FFA decreased both external and internal bleeding in injured cords. Furthermore, our results show that significantly less fragmentation of capillaries was observed in the FFA-treated group than in the vehicle control group. Thus, these results suggest that FFA is highly effective in reducing secondary hemorrhage after SCI. The mammalian Trp superfamily includes 28 nonselective cation channels. Most TRPs are permeable to Ca2+ as well as monovalent cations [43-45]. TRPM4 and TRPM5 are unique among TRPs because they are the only two calcium-activated nonselective cation channels that conduct monovalent but not divalent ions [12-14, 46]. Normally, in the uninjured brain and spinal cord, Trpm4 expression is maintained at very low levels. However, Trpm4 is prominently up-regulated after injury [5, 6, 47]. Trpm4 up-regulation occurs in large, ballooned neuron-like cells and capillary-like elongated structures and is especially prominent in capillaries [5, 6]. Sulfonylurea receptor 1 (Sur1) is a member of adenosine triphosphate (ATP)-binding cassette (ABC) protein family, and is also prominently up-regulated after injury associated with the up-regulation of nonselective cation (NC) channels. Woo SK et al. firstly confirmed that Trpm4, together with Sur1, forms a novel Sur1-Trpm4 channel complex and that the Sur1-Trpm4 complex exhibits both the pharmacological properties of Sur1 and the biophysical properties of Trpm4 [15]. Recent studies have shown that the blockade of the Sur1-Trpm4 channel reduces edema and improves functional outcomes after ischemic injury [48, 49]. Several reports have also demonstrated that blocking the Sur1-Trpm4 channel after injury contributes to protecting the structural integrity of capillaries and promotes angiogenesis [6, 50]. In addition, some reports have confirmed that the molecular mechanisms underlying progressive hemorrhagic necrosis involve Sur1-Trpm4 expression in the capillary endothelium [5, 42, 51]. The Trpm4 subunit regulates the opening of the Sur1-Trpm4 channel and is blocked by the Trpm4 antagonist FFA. In this study, the Trpm4 subunit was up-regulated in capillaries at 1 d after injury, and FFA was highly effective in reducing the expression of Trpm4. Taken together, all of these results indicate that FFA effectively inhibits secondary hemorrhage, in part by blocking the Sur1-Trpm4 channel after SCI.
Ischemia and injury also induce angiogenesis, which can supply oxygen and nutrition to the ischemia or lesion site, thus improving tissue repair and remodeling [50, 52, 53]. vWF is a blood vessel marker, and ki-67 is a proliferation marker; therefore, we identified and quantified the newly formed vessels via vWF and ki-67 co-labeling. Endoglin (CD105) is a prominent feature of newly formed vessels and has been widely used as an angiogenesis marker [54, 55]. To ensure the accuracy of the quantitative analyses, we also quantified the number of CD105-positive microvessels. In this study, the density of the vWF/ki-67 co-labeled vessels was significantly lower than that of the CD105-postive vessels. This difference may be because the vWF/ki-67 co-labeled vessels reflect the vessels containing proliferating endothelial cells, whereas the CD105-positve vessels contain both proliferating and activated endothelial cells. Furthermore, CD 105 also minimally expressed in preexisting vessels.
The BBB includes the BSCB is a highly specialized brain endothelial structure. The BBB represents a tight barrier between the circulating blood and CNS and is formed by dense tight junction proteins. In addition, the BBB is a natural barrier that protects the CNS by limiting the entry of plasma components and blood cells into the brain and spinal cord [11, 56]. After traumatic SCI, the disruption of the BSCB and the accompanying increase in microvascular permeability triggers several secondary degenerative cascades involving inflammatory responses, neurotoxic products and subsequent tissue edema [8, 35, 57]. MMPs are a family of zinc endopeptidases that degrade and remodel the extracellular matrix and other extracellular proteins [58, 59]. Many recent papers have shown that MMPs play a critical role in BBB/BSCB disruption in pathological conditions, including SCI [10, 11, 60, 61]. MMP-9 and MMP-2 are two important members of the MMP superfamily [10]. MMP9 induces the proteolytic degradation of the BBB, and blocking MMP9 activity inhibits vascular permeability [11]. MMP-9 activity is increased by 12 to 24 h post-injury, which corresponds to the maximal period of BSCB disruption [47]. Several reports have demonstrated that up-regulating MMP2 causes the initial opening of the BBB/BSCB [62, 63]. Some reports have demonstrated that MMP-2 activity is increased at a delayed time point (i.e., 5-7 d) after SCI [64, 65], while another report showed that MMP-2 was up-regulated at just 1 h after injury [29]. The differences in these time points of MMP-2 up-regulation are probably related to the degree of SCI. In our study, MMP-9 and MMP-2 were both up-regulated at 1 d after injury, and FFA significantly reduced the expression of both MMP-9 and MMP-2. Furthermore, FFA inhibited BSCB disruption at both 1 d and 3 d after injury. Based on these results, we propose that FFA effectively prevents BSCB disruption and that its mechanism relies, in part, on the inhibition of the expression of MMP-9 and MMP-2 after SCI.
BSCB disruption and hemorrhage are followed by blood cell infiltration after SCI, triggering inflammatory immune responses [20, 27, 66]. After injury, many activated macrophages/microglia migrate into the lesion site. The microglia then secrete neurotoxic factors that damage the tissue and destroy the surrounding environment [67, 68]. Moreover, they also induce the expression of neurotrophic factors that protect the tissue. However, shortly after the initial insult, the microglial neurotoxic effect can outweigh the neuroprotective effect [31, 32]. Therefore, it is crucial to inhibit microglial activation and the expression of these neurotoxic factors. Astrocytic activation follows and is promoted by the microglial response after injury [39]. After a CNS injury, reactive astrocytes form a glial scar to minimize secondary tissue damage and restore post-injury tissue homeostasis [37, 69]. However, the resulting glial scarring is widely regarded as a major barrier to axon regeneration [38]. Reactive astrocytes also produce growth inhibitory extracellular matrix molecules, such as chondroitin sulfate proteoglycans (CSPGs) [70], which act as major inhibitors of axon regeneration in glial scars [71]. In our study, immunoreactivity for markers of both microglia and astrocytes was increased around the lesion border at 3 d and 7 d after injury, and these increases were attenuated by treatment with FFA. One reason for this decrease in astrogliosis may be that microglia secrete many cytokines and growth factors that can, in turn, trigger and modulate astrogliosis [34]. Some reports have shown that the expression of astrocytes reaches a peak at 7 d after injury before gradually decreasing and trending toward a steady state [69, 72]. The reactive astrocytes eventually migrate centripetally to the lesion epicenter, where they gradually compact at the lesion area until 14 d after SCI [69]. The results of our study suggest that the cord located in the lesion area is much smaller at 35 d after injury than at 7 d after injury and that the lesion area was significantly smaller at both time points in the FFA-treated group than in the vehicle control group.
BSCB disruption and hemorrhaging lead to the infiltration of immune cells and a variety of toxic molecules. This is followed by secondary tissue damage, such as neuronal loss, lesion expansion, cavity formation and demyelination [8, 35, 36]. The results of this study demonstrate that FFA reduces the lesion area that does not express GFAP signal and reduces average cavity size. Some recent reports have calculated the area of cavitation in transverse sections of the spinal cord [73, 74]. In this study, the area of cavitation was calculated in a longitudinal section of the cords. The longitudinal sections contained the whole lesion site and cavitation, whereas the transverse sections contained only part of the lesion site, thus the area of cavitation in this study was larger than that in the recent reports. Our results also show that the demyelination observed in cross-sections is reduced by FFA. Hindlimb movement is mainly mediated by motor neurons in the lumbar motoneuron pools. After sustaining a secondary injury at the lesion epicenter, anterograde neurons, including the ventral horn motor neurons caudal to the impact site, might decrease in number. Previous studies have estimated the total number of anterograde neurons, including motor neurons, at the lumbar cord after mid-thoracic injury. However, there were no significant differences in the numbers of labeled neurons or motor neurons at 5 weeks or more after SCI between sham control and injured animals [75, 76]. Contrary to the results of previous reports, our results show that the total number of labeled motor neurons in the lumbar cord was significantly lower at 5 weeks after injury than in the sham control group. Moreover, the total number of ventral horn motor neurons was significantly higher in the FFA-treated group than in the vehicle control group. To obtain these results, researchers cut the spinal cord into sections and then estimated the number of neurons in each slice. However, traditional histological sectioning yields incomplete spatial information that can result in an inaccurate assessment of the original date. In our effort to acquire complete spatial information for motor neurons, we successfully obtained a full 3D image of the bilateral motor neurons in the lumbar cord and then precisely estimated the total number of motor neurons.
Many drugs have been used as therapeutic interventions in SCI, but few have been shown to decrease hemorrhage and BSCB disruption. Methylprednisolone (MP), the only drug approved for clinical treatment, is most widely used in SCI. MP reduces MMP-9 expression but has no significant effect on MMP-2 expression [77, 78]. It is highly effective in decreasing inflammation and edema but exerts almost no significant hemorrhage-reducing effect [77, 79]. In addition, many side effects are also correlated with MP, and its use in SCI is therefore controversial. Glibenclamide is traditional drug used to treat type 2 diabetes. Recently, researchers confirmed that glibenclamide was also highly effective in reducing secondary hemorrhage. Unlike FFA, glibenclamide primarily blocks the Sur1 channel of the Sur1-Trpm4 complex and sometimes inhibits microglial Sur1-Kir6.2 (K ATP) channels to reduce hemorrhage [5, 80]. Furthermore, glibenclamide does not alter MMP-2 and MMP-9 expression [5], but its inhibition of the Sur1 subunit was confirmed to result in significant anti-inflammatory effects in subarachnoid hemorrhage [81]. In our study, we found that FFA blocked Trpm4 expression in capillaries to decrease capillary fragmentation and inhibited the expression of MMP-2/9 to reduce capillary permeability. These results imply a certain correlation between Trpm4 and MMP-2/9. However, the mechanism by which opening Trpm4 channels might trigger the up-regulation of MMP-2/9 was not examined in this study. Thus, future studies are needed to identify this mechanism. Because it inhibits both secondary hemorrhage and BSCB disruption, FFA (including glibenclamide) appears to have a potential advantage as a treatment for SCI. Although early therapeutic intervention with FFA may substantially decrease secondary injury, the axons and neurons impaired after the initial injury regenerate poorly, limiting the eventual functional recovery of the patient. There is currently no effective treatment that both prevents the death of surviving axons and promotes the regeneration of impaired axons. Thus, there is still a long way to before SCI can be effectively treated. Fortunately, the development of a combination treatment that includes early therapeutic intervention and a subsequent approach that promotes the regeneration of axons would be a potentially effective method for treating SCI.
In summary, the results of this study show that the neuroprotective effect of FFA is mediated in part by blocking the Sur1-Trpm4 channel to inhibit capillary fragmentation and subsequent secondary hemorrhage. Furthermore, its inhibition of MMP-9 and MMP-2 expression reduces BSCB disruption and the subsequent inflammatory immune responses, which may also reduce secondary damage. FFA is currently used as an anti-inflammatory and analgesic drug with no undesirable side effects. To our knowledge, this is the first time that FFA has been used as a therapeutic strategy in SCI. The safety of FFA and the identification of its unique mechanism of activity, which acts to reduce hemorrhage and BSCB disruption, indicate that this drug may be potentially useful as a therapeutic clinical agent for traumatic injury.
Abbreviations
3D imaging: three-dimensional imaging; BBB: blood-brain barrier; BMS: Basso mouse scale; BSCB: blood-spinal cord barrier; CNS: central nervous system; CSPGs : chondroitin sulfate proteoglycans; CTB: Cholera toxin beta-subunit; FFA: flufenamic acid; GFAP: glial fibrillary acidic protein; H&E: hematoxylin and eosin staining; LFB: Luxol fast blue; MMP-2: matrix metalloprotease-2; MMP-9: matrix metalloprotease-9; MMPs: matrix metalloproteases; MP: Methylprednisolone; NF200 : neurofilament 200; SCI: spinal cord injury; SUR 1: sulfonylurea receptor 1; Trp: transient receptor potential; Trpm4: transient receptor potential melastatin 4; Sur1: Sulfonylurea receptor 1; GFP: green fluorescent protein; ROI: regions of interest.
Supplementary Material
Supplementary figures and video legends.
Supplementary video 1.
Supplementary video 2.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants Nos. 31571002, 81701354, 81471271, 81572204), Science Fund for Creative Research Group of China (Grant No. 61721092), China Postdoctoral Science Foundation (Grant No. 2017M612463, 2018T110772), the Fundamental Research Funds for the Central Universities, HUST (No. 2018KFYXKJC026), Director Fund of Wuhan National Laboratory for Optoelectronics (WNLO), and Graduates Innovation Fund, HUST (No.5003530002). We thank Chaoyu Wang for the gelatin zymography experiment. And also, we thank Yueyue Zhu for special support and help. We also thank the Optical Bioimaging Core Facility of WNLO for support in data acquisition.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tator CH, Fehlings MG. Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J Neurosurg. 1991;75:15-26
2. Nelson E, Gertz SD, Rennels ML, Ducker TB, Blaumanis OR. Spinal cord injury: The role of vascular damage in the pathogenesis of central hemorrhagic necrosis. Arch Neurol. 1977;34:332-3
3. Fitch MT, Doller C, Combs CK, Landreth GE, Silver J. Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitroanalysis of inflammation-induced secondary injury after CNS trauma. J Neurosci. 1999;19:8182-98
4. Tator CH, Koyanagi I. Vascular mechanisms in the pathophysiology of human spinal cord injury. J Neurosurg. 1997;86:483-92
5. Marc Simard J, Tsymbalyuk O, Ivanov A, Ivanova S, Bhatta S, Zhihua G. et al. Endothelial sulfonylurea receptor 1-regulated NCCa-ATP channels mediate progressive hemorrhagic necrosis following spinal cord injury. J Clin Invest. 2007;117:2105-13
6. Gerzanich V, Woo SK, Vennekens R, Tsymbalyuk O, Ivanova S, Ivanov A. et al. De novo expression of Trpm4 initiates secondary hemorrhage in spinal cord injury. Nat Med. 2009;15:185-91
7. Khalili H, Derakhshan N, Niakan A, Ghaffarpasand F, Salehi M, Eshraghian H. et al. Effects of oral glibenclamide on brain contusion volume and functional outcome of patients with moderate and severe traumatic brain injuries: a randomized double-blind placebo-controlled clinical trial. World Neurosurg. 2017;101:130-6
8. Beck KD, Nguyen HX, Galvan MD, Salazar DL, Woodruff TM, Anderson AJ. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain. 2010;133:433-47
9. Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41-53
10. Asahi M, Wang X, Mori T, Sumii T, Jung J-C, Moskowitz MA. et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724-32
11. Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci. 2002;22:7526-35
12. Launay P, Fleig A, Perraud A-L, Scharenberg AM, Penner R, Kinet J-P. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell. 2002;109:397-407
13. Launay P, Cheng H, Srivatsan S, Penner R, Fleig A, Kinet J-P. TRPM4 regulates calcium oscillations after T cell activation. Science. 2004;306:1374-7
14. Guinamard R, Sallé L, Simard C. The non-selective monovalent cationic channels TRPM4 and TRPM5. Adv Exp Med Biol. 2011;704:147-71
15. Woo SK, Kwon MS, Ivanov A, Gerzanich V, Simard JM. The sulfonylurea receptor 1 (Sur1)-transient receptor potential melastatin 4 (Trpm4) channel. J Biol Chem. 2013;288:3655-67
16. Winder CV, Wax J, Serrano B, Jones EM, McPhee ML. Anti-inflammatory and antipyretic properties of N-(α, α, α-Trifluoro-m-tolyl) anthranilic acid (CI-440; flufenamic acid). Arthritis Rheum. 1963;6:36-47
17. Flower R, Gryglewski R, Herbaczńska-Cedro K, Vane J. Effects of anti-inflammatory drugs on prostaglandin biosynthesis. Nat New Biol. 1972;238:104-106
18. Guinamard R, Simard C, Del Negro C. Flufenamic acid as an ion channel modulator. Pharmacol Ther. 2013;138:272-84
19. Gruner JA. A monitored contusion model of spinal cord injury in the rat. J Neurotrauma. 1992;9:123-8
20. Lee JY, Kim HS, Choi HY, Oh TH, Ju BG, Yune TY. Valproic acid attenuates blood-spinal cord barrier disruption by inhibiting matrix metalloprotease-9 activity and improves functional recovery after spinal cord injury. J Neurochem. 2012;121:818-29
21. Wang J, Kubes P. A reservoir of mature cavity macrophages that can rapidly invade visceral organs to affect tissue repair. Cell. 2016;165:668-78
22. Zhu D, Larin KV, Luo Q, Tuchin VV. Recent progress in tissue optical clearing. Laser Photon Rev. 2013;7:732-57
23. Yu T, Qi Y, Gong H, Luo Q, Zhu D. Optical clearing for multiscale biological tissues. J Biophotonics. 2018;11:e201700187
24. Ertürk A, Becker K, Jährling N, Mauch CP, Hojer CD, Egen JG. et al. Three-dimensional imaging of solvent-cleared organs using 3DISCO. Nat Protoc. 2012;7:1983-95
25. Simard JM, Tsymbalyuk O, Keledjian K, Ivanov A, Ivanova S, Gerzanich V. Comparative effects of glibenclamide and riluzole in a rat model of severe cervical spinal cord injury. Exp Neurol. 2012;233:566-74
26. Basso DM, Fisher LC, Anderson AJ, Jakeman LB, Mctigue DM, Popovich PG. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635-59
27. Higashida T, Kreipke CW, Rafols JA, Peng C, Schafer S, Schafer P. et al. The role of hypoxia-inducible factor-1α, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J Neurosurg. 2011;114:92-101
28. Hosokawa T, Nakajima H, Doi Y, Sugino M, Kimura F, Hanafusa T. et al. Increased serum matrix metalloproteinase-9 in neuromyelitis optica: Implication of disruption of blood-brain barrier. J Neuroimmunol. 2011;236:81-6
29. Lee JY, Kim HS, Choi HY, Oh TH, Yune TY. Fluoxetine inhibits matrix metalloprotease activation and prevents disruption of blood-spinal cord barrier after spinal cord injury. Brain. 2012;135:2375-89
30. Noble L, Wrathall JR. Distribution and time course of protein extravasation in the rat spinal cord after contusive injury. Brain Res. 1989;482:57-66
31. Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435-44
32. Guerrero AR, Uchida K, Nakajima H, Watanabe S, Nakamura M, Johnson WE. et al. Blockade of interleukin-6 signaling inhibits the classic pathway and promotes an alternative pathway of macrophage activation after spinal cord injury in mice. J Neuroinflammation. 2012;9:1-16
33. Popovich PG, Wei P, Stokes BT. Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J Comp Neurol. 1997;377:443-64
34. Emmetsberger J, Tsirka SE. Microglial inhibitory factor (MIF/TKP) mitigates secondary damage following spinal cord injury. Neurobiol Dis. 2012;47:295-309
35. Bartanusz V, Jezova D, Alajajian B, Digicaylioglu M. The blood-spinal cord barrier: morphology and clinical implications. Ann Neurol. 2011;70:194-206
36. Kwon BK, Tetzlaff W, Grauer JN, Beiner J, Vaccaro AR. Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine J. 2004;4:451-64
37. Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617-27
38. Hellal F, Hurtado A, Ruschel J, Flynn KC, Laskowski CJ, Umlauf M. et al. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science. 2011;331:928-31
39. Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312-8
40. Kawata K, Morimoto T, Ohashi T, Tsujimoto S, Hoshida T, Tsunoda S. et al. Experimental study of acute spinal cord injury: a histopathological study. No Shinkei Geka. 1993;21:45-51
41. Hayes KC, Kakulas BA. Neuropathology of human spinal cord injury sustained in sports-related activities. J Neurotrauma. 1997;14:235-48
42. Griffiths I, Burns N, Crawford A. Early vascular changes in the spinal grey matter following impact injury. Acta Neuropathol. 1978;41:33-9
43. Gees M, Colsoul B, Nilius B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb Perspect Biol. 2010;2:1-31
44. Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev. 2007;87:165-217
45. Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem. 2007;76:387-417
46. Vennekens R, Olausson J, Meissner M, Bloch W, Mathar I, Philipp SE. et al. Increased IgE-dependent mast cell activation and anaphylactic responses in mice lacking the calcium-activated nonselective cation channel TRPM4. Nat Immunol. 2007;8:312-320
47. Lee JY, Choi HY, Na WH, Ju BG, Yune TY. Ghrelin inhibits BSCB disruption/hemorrhage by attenuating MMP-9 and SUR1/TrpM4 expression and activation after spinal cord injury. Biochim Biophys Acta. 2014;1842:2403-12
48. Chen M, Simard JM. Cell swelling and a nonselective cation channel regulated by internal Ca2+ and ATP in native reactive astrocytes from adult rat brain. J Neurosci. 2001;21:6512-21
49. Khanna A, Walcott BP, Kahle KT, Simard JM. Effect of glibenclamide on the prevention of secondary brain injury following ischemic stroke in humans. Neurosurg Focus. 2014;36:1-6
50. Loh KP, Ng G, Yu CY, Fhu CK, Yu D, Vennekens R. et al. TRPM4 inhibition promotes angiogenesis after ischemic stroke. Pflugers Arch. 2014;466:563-76
51. Simard JM, Woo SK, Norenberg MD, Tosun C, Chen Z, Ivanova S. et al. Brief suppression of Abcc8 prevents autodestruction of spinal cord after trauma. Sci Transl Med. 2010;2:28ra9-ra9
52. Zhu W, Fan Y, Frenzel T, Gasmi M, Bartus RT, Young WL. et al. Insulin growth factor-1 gene transfer enhances neurovascular remodeling and improves long-term stroke outcome in mice. Stroke. 2008;39:1254-61
53. Huang X-T, Zhang Y-Q, Li S-J, Li S-H, Tang Q, Wang Z-T. et al. Intracerebroventricular transplantation of ex vivo expanded endothelial colony-forming cells restores blood-brain barrier integrity and promotes angiogenesis of mice with traumatic brain injury. J Neurotrauma. 2013;30:2080-8
54. Muramatsu R, Takahashi C, Miyake S, Fujimura H, Mochizuki H, Yamashita T. Angiogenesis induced by CNS inflammation promotes neuronal remodeling through vessel-derived prostacyclin. Nat Med. 2012;18:1658-64
55. Nassiri F, Cusimano MD, Scheithauer BW, Rotondo F, Fazio A, Yousef GM. et al. Endoglin (CD105): a review of its role in angiogenesis and tumor diagnosis, progression and therapy. Anticancer Res. 2011;31:2283-90
56. Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697-709
57. Beattie MS, Farooqui AA, BRESNAHAN JC. Review of current evidence for apoptosis after spinal cord injury. J Neurotrauma. 2000;17:915-25
58. Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier J-P, Gray JW. et al. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 1999;98:137-46
59. Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463-516
60. Jin Y-J, Park I, Hong I-K, Byun H-J, Choi J, Kim Y-M. et al. Fibronectin and vitronectin induce AP-1-mediated matrix metalloproteinase-9 expression through integrin α5β1/αvβ3-dependent Akt, ERK and JNK signaling pathways in human umbilical vein endothelial cells. Cell Signal. 2011;23:125-34
61. Xu J, Kim G-M, Ahmed SH, Xu J, Yan P, Xu XM. et al. Glucocorticoid receptor-mediated suppression of activator protein-1 activation and matrix metalloproteinase expression after spinal cord injury. J Neurosci. 2001;21:92-7
62. Heo JH, Lucero J, Abumiya T, Koziol JA, Copeland BR, Del Zoppo GJ. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:624-33
63. Chang D-I, Hosomi N, Lucero J, Heo J-H, Abumiya T, Mazar AP. et al. Activation systems for latent matrix metalloproteinase-2 are upregulated immediately after focal cerebral ischemia. J Cereb Blood Flow Metab. 2003;23:1408-19
64. De Castro Jr R, Burns CL, McAdoo DJ, Romanic AM. Metalloproteinase increases in the injured rat spinal cord. Neuroreport. 2000;11:3551-4
65. Hsu J-YC, McKeon R, Goussev S, Werb Z, Lee J-U, Trivedi A. et al. Matrix metalloproteinase-2 facilitates wound healing events that promote functional recovery after spinal cord injury. J Neurosci. 2006;26:9841-50
66. Ousman SS, Kubes P. Immune surveillance in the central nervous system. Nat Neurosci. 2012;15:1096-101
67. Wolpe SD, Davatelis G, Sherry B, Beutler B, Hesse DG, Nguyen HT. et al. Macrophages secrete a novel heparin-binding protein with inflammatory and neutrophil chemokinetic properties. J Exp Med. 1988;167:570-81
68. Xu LL, Warren MK, Rose WL, Gong W, Wang JM. Human recombinant monocyte chemotactic protein and other C-C chemokines bind and induce directional migration of dendritic cells in vitro. J Leukoc Biol. 1996;60:365-71
69. Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K. et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829-34
70. McKeon RJ, Jurynec MJ, Buck CR. The chondroitin sulfate proteoglycans neurocan and phosphacan are expressed by reactive astrocytes in the chronic CNS glial scar. J Neurosci. 1999;19:10778-88
71. Jeong SR, Kwon MJ, Lee HG, Joe EH, Lee JH, Kim SS. et al. Hepatocyte growth factor reduces astrocytic scar formation and promotes axonal growth beyond glial scars after spinal cord injury. Exp Neurol. 2012;233:312-22
72. Hu R, Zhou J, Luo C, Lin J, Wang X, Li X. et al. Glial scar and neuroregeneration: histological, functional, and magnetic resonance imaging analysis in chronic spinal cord injury. J Neurosurg Spine. 2010;13:169-80
73. Byrnes KR, Washington PM, Knoblach SM, Hoffman E, Faden AI. Delayed inflammatory mRNA and protein expression after spinal cord injury. J Neuroinflammation. 2011;8:130-43
74. Gaviria M, Haton H, Sandillon F, Privat A. A mouse model of acute ischemic spinal cord injury. J Neurotrauma. 2002;19:205-21
75. Bjugn R, Nyengaard JR, Rosland JH. Spinal cord transection—no loss of distal ventral horn neurons. Exp Neurol. 1997;148:179-86
76. McBride RL, Feringa ER. Ventral horn motoneurons 10, 20 and 52 weeks after T-9 spinal cord transection. Brain Res Bull. 1992;28:57-60
77. Yu P, Huang L, Zou J, Yu Z, Wang Y, Wang X. et al. Immunization with recombinant Nogo-66 receptor (NgR) promotes axonal regeneration and recovery of function after spinal cord injury in rats. Neurobiol Dis. 2008;32:535-42
78. Yin Y, Sun W, Li Z, Zhang B, Cui H, Deng L. et al. Effects of combining methylprednisolone with rolipram on functional recovery in adult rats following spinal cord injury. Neurochem Int. 2013;62:903-12
79. Merola A, O'brien MF, Castro BA, Smith DA, Eule JM, Lowe TG. et al. Histologic characterization of acute spinal cord injury treated with intravenous methylprednisolone. J Orthop Trauma. 2002;16:155-61
80. Caffes N, Kurland DB, Gerzanich V, Simard JM. Glibenclamide for the treatment of ischemic and hemorrhagic stroke. Int J Mol Sci. 2015;16:4973-84
81. Tosun C, Kurland DB, Mehta R, Castellani RJ, Kwon MS, Woo SK. et al. Inhibition of the Sur1-Trpm4 channel reduces neuroinflammation and cognitive impairment in subarachnoid hemorrhage. Stroke. 2013;44:3522-8
Author contact
![]() Corresponding authors: Dan Zhu, Ph. D Professor, Britton Chance Center for Biomedical Photonics, Huazhong University of Science and Technology.1037 Luoyu Road, Wuhan 430074, P.R. China. Tel: +86 27 8779 2033; Fax: +86 27 8779 2034; dawnzhhust.edu.cn and Yongchao Wu. Department of Orthopedics, Union Hospital, Huazhong University of Science and Technology. 1277 Jiefang Dadao, Wuhan, 430022 Hubei Province, China. Tel: +86 27 8535 1626; fax: +86 27 8577 6343; email: wuyongchaocom
Corresponding authors: Dan Zhu, Ph. D Professor, Britton Chance Center for Biomedical Photonics, Huazhong University of Science and Technology.1037 Luoyu Road, Wuhan 430074, P.R. China. Tel: +86 27 8779 2033; Fax: +86 27 8779 2034; dawnzhhust.edu.cn and Yongchao Wu. Department of Orthopedics, Union Hospital, Huazhong University of Science and Technology. 1277 Jiefang Dadao, Wuhan, 430022 Hubei Province, China. Tel: +86 27 8535 1626; fax: +86 27 8577 6343; email: wuyongchaocom
Citation styles
APA
Yao, Y., Xu, J., Yu, T., Chen, Z., Xiao, Z., Wang, J., Hu, Y., Wu, Y., Zhu, D. (2018). Flufenamic acid inhibits secondary hemorrhage and BSCB disruption after spinal cord injury. Theranostics, 8(15), 4181-4198. https://doi.org/10.7150/thno.25707.
ACS
Yao, Y.; Xu, J.; Yu, T.; Chen, Z.; Xiao, Z.; Wang, J.; Hu, Y.; Wu, Y.; Zhu, D. Flufenamic acid inhibits secondary hemorrhage and BSCB disruption after spinal cord injury. Theranostics 2018, 8 (15), 4181-4198. DOI: 10.7150/thno.25707.
NLM
Yao Y, Xu J, Yu T, Chen Z, Xiao Z, Wang J, Hu Y, Wu Y, Zhu D. Flufenamic acid inhibits secondary hemorrhage and BSCB disruption after spinal cord injury. Theranostics 2018; 8(15):4181-4198. doi:10.7150/thno.25707. https://www.thno.org/v08p4181.htm
CSE
Yao Y, Xu J, Yu T, Chen Z, Xiao Z, Wang J, Hu Y, Wu Y, Zhu D. 2018. Flufenamic acid inhibits secondary hemorrhage and BSCB disruption after spinal cord injury. Theranostics. 8(15):4181-4198.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.