Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Identification of neoantigens to...
Research progress regarding...
Challenges of exploiting...
Future perspectives of...
Conclusion
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(15):4238-4246. doi:10.7150/thno.24387 This issue Cite
Review
Personalized cancer neoantigen vaccines come of age
Yanhong Chu#, Qin Liu#, Jia Wei, Baorui Liu ![]()
The Comprehensive Cancer Centre of Drum Tower Hospital, Medical School of Nanjing University & Clinical Cancer Institute of Nanjing University, Nanjing, China
#These authors contributed equally to this work.
Received 2017-12-26; Accepted 2018-6-25; Published 2018-7-30
Abstract

Cancer vaccines have encountered their ideal personalized partner along with evidence for great breakthroughs in the identification and synthesis of neoantigens. Individual cancer neoantigen vaccines are capable of eliciting robust T-cell responses and have been demonstrated to achieve striking clinical efficacy due to their high immunogenicity and central thymic tolerance escape of neoantigens. Two recent phase I clinical trials have provided support for the hypothesis and have heralded a nascent era of personalized vaccines in the field of immunotherapy. This review aims to address the identification of neoepitopes and describes advances made in personalized vaccines. In addition, this review discusses the challenges related to the exploitation of vaccine therapy, and provides potential thoughts for the improvement of vaccine design and applications.
Keywords: Neoantigen, vaccine, immunotherapy, T-cell response
Introduction
Over the past few decades, numerous studies have focused on cancer vaccines [1]. However, there remain a few questions that must be addressed, including the selection of an optimal antigen and adjuvant component, a suitable delivery mode, and an efficient approach to overcome immune invasion. Recently, two successful phase I clinical trials were published in Nature that have attracted a great deal of attention to personalized neoantigen vaccines [2, 3]. These studies showed that a total of 66.7% (4/6) and 61.5% (8/13) resected melanoma patients remained recurrence-free during the entire follow-up period (20-32 months and 12-23months, separately) following vaccination. Thus, exploitation of neoantigens represents a critical point to reaching impressive therapeutic efficacy.
Neoantigens are a series of immunogenic peptides derived from tumour-specific mutations or viral open reading frames, instead of from the normal human genome [4-7]. Neoantigens have been demonstrated to be highly immunogenic and can escape from central thymic tolerance. Because the proportion of virus-associated malignancies is relatively low, mutation-related neoantigens represent ideal immunotherapy targets.
While neoantigens represent the optimal choice for an anti-tumour immunotherapy, the unique neoantigen landscape of individualised tumours hinders its application. Discovery and verification of neoantigens, including screening tumour cDNA library pools [8], has been laborious, and time-consuming studies have been carried out over the past few years. With improvements in sequencing technology and bioinformatics, neoantigen identification has excitingly become both increasingly feasible and economical.
Neoantigen-specific CD4+ and CD8+ T-cell responses induced by ACT (adoptive cell transfer) [9-14] or immune checkpoint blockade [15-19], have been frequently observed, particularly in the case of melanoma [20]. Various findings described by Rosenberg's team demonstrated that neoantigen-specific T cells play a major role in rapid tumour eradication and long-term patient survival [9-14]. Schreiber et al. demonstrated that tumour mutant epitopes represent the targets of immune checkpoint blockade therapy [21]. Due to the ability of neoantigens to fire up the body's natural immune responses directly to the tumour, cancer vaccines exhibit great potential as a therapeutic. Cancer-specific vaccinations with neoantigens have been shown to be equally efficient as checkpoint blockades [22]. With early success demonstrated in clinical stage trials, the personalized mutanome vaccine is likely to selectively target heterogeneous tumours and elicit a strong T-cell response, generating a new age of personalized immunotherapy.
Identification of neoantigens to synthesize personalized vaccines
With the development of NGS (next-generation sequencing) and bioinformatics, frequently used screening methods for neoantigen identification can be categorized into three different approaches: A, B and C (Figure 1).
Three common approaches to neoantigen identification. Approach A: tumour-specific mutations are identified by WES, confirmed by RNA sequencing, and then ranked by predicted high-affinity binding to autologous HLA types; finally, neopeptides are synthesized based on prioritized mutated alleles, which is followed by T-cell reactivity analysis ex vivo to confirm their immunogenicity. Approach B: based on tumour-specific mutations identified by WES, TMG constructs are synthesized as templates for the generation of IVT RNA, which is followed by T-cell reactivity analysis ex vivo to confirm their immunogenicity. Approach C: mutation identification is based on research from databases and the literature. The next steps can be the same as those for either of the two methods described above. APCs: antigen-presenting cells; HLA: human leukocyte antigen; PBMCs: peripheral blood mononuclear cells; TMG: tandem minigene; WES: whole-exome sequencing.
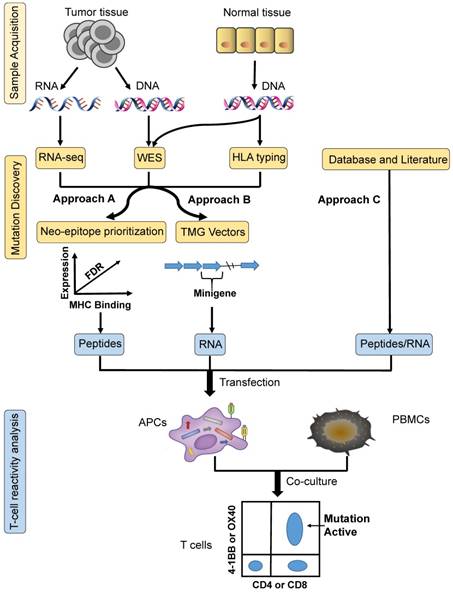
As demonstrated in Approach A, the first step to designing a mature neopeptide-based vaccine therapy is identifying tumour-specific mutations. Mutations related to immune recognition primarily include nsSNVs (non-synonymous single nucleotide variants) with exons, indels, and fusion genes [23]. WES (whole-exome sequencing) [24] of matched tumour- and normal-cell DNA represents the most common method for identifying somatic mutations. Expression levels of identified mutated alleles are then orthogonally validated and analyzed via RNA sequencing [25]. Mutations are then ranked according to their predicted high-affinity binding to autologous HLA class I and II. The IEDB (immune epitope database and analysis resource) is an online comprehensive database comprised of T-cell epitopes and tools that can be used to predict MHC binding. The prediction tools available from IEDB include ANN (artificial neural networks)/NetMHC [26, 27], NetMHCpan [28], SMM [29], SMMPMBEC [30], ARB, Comblib_Sidney2008, Pickpocket, and Consensus. Synthesized neopeptide immunogenicity must be finally validated using T-cell reactivity analysis, by generating antigen-loaded autologous APCs (antigen-presenting cells) to stimulate T cells. Activation markers of both CD4+ and CD8+ T cells must then be detected, including OX-40, 4-1BB, CD170a, and IFN-γ, ex vivo. In early 2014, vaccination of mice confirmed the approach described above [31]. In 2017, Wu's team [2] utilized this strategy to manufacture personal vaccines that consisted of four pools of synthetic long peptides. The RNA-based poly-epitopes developed in this study induced strong multi-functional CD4+ and CD8+ T-cell responses in high-risk melanoma patients.
Simplified from Approach A, following the mining of nsSNVs, multiple minigenes encoding mutations are synthesized in tandem in order to generate TMG (tandem minigene) constructs in Approach B. The TMG construct consists of a variable number of minigenes that are genetically fused together, with each minigene encoding for a mutation flanked by 12 AA (amino acids) from the endogenous protein sequence. Plasmids encoding the TMG constructs are utilized as templates to generate IVT (in vitro-transcribed) RNA. In 2014, Rosenberg's team [10] identified a ERBB2IP (erbb2 interacting protein) mutation from a metastatic cholangiocarcinoma patient using this approach. Following adoptive transfer of TILs (tumour-infiltrating lymphocytes) that contained approximately 25% mutation-specific T cells, the patient experienced tumour regression. In consecutive studies, these authors went on to demonstrate that neoepitopes from 9 out of 10 metastatic gastrointestinal cancer patients could be recognized by autologous TILs [9].
Approach C identifies neoepitopes based on databases and literature without sample acquisition. The key to this strategy is the presence of high-frequency mutational sites found in solid tumours. Based on this pattern, Schumacher et al. identified the most frequent mutation, IDH1(R132H), in diffuse grade II and III glioma patients [32]. These authors then synthesized a peptide vaccine to target mutant IDH1, which functioned to induce anti-tumour responses in mice. Similarly, Platten et al. discovered that K27M-mutant histone-3 acts as an optimal target for the generation of a glioma vaccine, demonstrating that a peptide vaccine targeted against K27M-mutant histone-3 elicited a mutation-specific immune response in an MHC-humanized mouse model [33]. In theory, other high-frequency mutations, including BRAF, RGFR, and KRAS, could also function as ideal cancer vaccine targets.
Research progress regarding neoantigen vaccines
As different methods and tools have been developed for the identification of neoantigens and vaccine synthesis, the current issue now revolves around how to successfully apply such vaccines for clinical treatments. Both completed and ongoing clinical trials are depicted in Table 1. The start times of all of these studies demonstrate that the neoantigen vaccine is a brand new, but encouraging area in immunotherapy.
Clinical trials of neoantigen cancer vaccines
| Conditions | Phase | Status | Interventions | Start | Completion | NCT number |
|---|---|---|---|---|---|---|
| Melanoma | Phase 1 | Completed | DC vaccine | 2008.08 | 2016.06 | NCT00683670 |
| Melanoma | Phase 1 | Completed | RNA vaccine | 2013.12 | 2017.04 | NCT02035956 |
| Melanoma | Phase 1 | Completed | Peptide vaccine | 2014.01 | 2018.12 | NCT01970358 |
| Glioma | Phase 1 | Active, not recruiting | Neoantigen vaccine | 2014.11 | 2018.08 | NCT02287428 |
| Glioma | Phase 1 | Recruiting | IDH1 Peptide vaccine | 2015.06 | 2018.08 | NCT02454634 |
| Glioma | Phase 1 | Terminated | Peptide vaccine | 2015.12 | 2017.02 | NCT02510950 |
| Kidney Cancer | Phase 1 | Not yet recruiting | Neoantigen vaccine | 2016.10 | 2022.09 | NCT02950766 |
| Urinary Bladder Cancer, etc | Phase 1 | Recruiting | NEO-PV-01 | 2016.10 | 2020.12 | NCT02897765 |
| Advanced Cancer | Phase 1 | Recruiting | AutoSynVax™ vaccine | 2017.01 | 2019.03 | NCT03219450 |
| Pancreatic Cancer | Phase 1 | Recruiting | Peptide vaccine | 2016.05 | 2022.05 | NCT02600949 |
| Pancreatic Cancer | Phase 1 | Recruiting | DNA vaccine | 2018.01 | 2022.03 | NCT03122106 |
| Triple Negative Breast Cancer | Phase 1 | Recruiting | DNA vaccine | 2015.06 | 2019.06 | NCT02348320 |
| Triple Negative Breast Cancer | Phase 1 | Recruiting | Peptide vaccine | 2015.09 | 2019.08 | NCT02427581 |
| Triple Negative Breast Cancer | Phase 1 | Recruiting | RNA vaccine | 2016.09 | 2019.03 | NCT02316457 |
| Triple Negative Breast Cancer | Phase 1 | Not yet recruiting | DNA vaccine | 2017.09 | 2020.09 | NCT03199040 |
| Paediatric Brain Tumour | Phase 1 | Not yet recruiting | Peptide vaccine | 2018.05 | 2022.10 | NCT03068832 |
DC: dendritic cells.
The first clinical trial using a neoantigen-based vaccine began in 2008 by Carreno and colleagues [34]. In their study, three patients with advanced melanoma who had been previously treated with ipilimumab were vaccinated intravenously with DCs pulsed with class I-restricted 8-10-mer neoantigen peptides. Their results from these studies demonstrated that cancer vaccine neoepitopes functioned to increase the breadth and diversity of tumour-specific T-cell responses.
In the absence of the ex vivo DC-stimulation process, both peptide-based vaccines and nucleic acid-based vaccines are considered to be fairly easy approaches that can be mass produced. As mentioned for Approach C, vaccination with IDH1 vaccines [32], was able to induce an effective MHC class II-restricted mutation-specific anti-tumour immune response, while CD4+ TH1 cells and antibodies spontaneously occurred. Because of the high level of uniformity and penetrance of IDH1(R132H), comments [35, 36] regarding this research study were found to be in approval of its positive prospects for the treatment of patients suffering from low-grade and anaplastic gliomas. In addition, a clinical trial (NCT02454634) based on this concept has been underway since 2015. However, the breakthrough regarding neo-peptide-based vaccines also occurred first in melanoma. In 2017, Ott et al. demonstrated that a vaccine that targeted up to 20 predicted personal tumour neoantigens functioned to induce poly-functional CD4+ and CD8+ T cells in melanoma patients [2]. In these studies, DNA and RNA sequencing, HLA typing, and computational prediction of HLA-binding peptides for the synthesis of clinical-grade long neo-peptides (15-30-mer) were carried out. A total of six high-risk-of-relapse melanoma patients enrolled completed the full series of five priming and two booster vaccinations without related, serious adverse events. Only two patients with previous lung metastases underwent disease recurrence, and later, with the subsequent treatment with the anti-PD-1 (programmed cell death-1) antibody, both patients were found to achieve complete responses.
In the case of nucleic acid-based vaccines, there exist two delivery platforms for encoding antigenic peptides, including DNA-based and mRNA-based vaccines. The anti-tumour activity of DNA cancer vaccines has so far proven modest, with few of these having progressed past phase I clinical trial evaluation [37]. However, mRNA vaccines have been proven to exhibit an excellent safety profile, flexibility, and adjuvant ability [38]. Sahin's team has focused their efforts on personalized RNA vaccines for numerous years. First, in 2015, their studies demonstrated that vaccination with a synthesized RNA vaccine resulted in the induction of potent tumour control in mice [39]. Later, in 2016, they went on to develop a lipid carrier system to deliver neoantigen vaccines to lymphoid organs, which functioned as the ideal microenvironment for the efficient priming and amplification of T-cell responses [40]. Finally, in 2017, their first-in-human application of personalized RNA vaccines was demonstrated to live up to expectations in melanoma studies [3]. Different from typical subcutaneous injections, this neoantigen vaccine was injected percutaneously into inguinal lymph nodes under ultrasound control. As proven in mouse models, the uptake and translation of RNA-encoded antigens by lymph-node-resident DCs (dendritic cells) were found to be more efficient [41].
Various neoepitope vaccine formulations each possess inherent advantages and limitations. Without randomized, controlled, larger studies, there can be no clear consensus as to whether certain formulations are superior to others in regard to immunogenicity. However, in a sense, the application of an RNA-based vaccine is limited by the fact that it cannot be as flexibly combined with different immune adjuvants as a peptide vaccine can.
An unexpected discovery made by many researchers was the category of CD4+ T cells that recognize the majority of neoantigens and functions to elicit the strongest anti-tumour response in the treatment period of neoantigen vaccines. Previous reviews have discussed the relative immunologic mechanisms relevant to this finding [42].
Challenges of exploiting neoantigen vaccines
Further research into the use of neoantigen vaccines could function to reveal more issues that will need to be overcome.
Low neoantigen burden
Sequencing studies have demonstrated substantial variability between the mutation rates of different tumour types [43]. Increases in mutation load and neoantigen burden have been shown to result in a potential increase in carcinoma antigenicity [15, 44]. Melanoma, with the highest mutation load [45], has been the first cancer type to benefit from neoantigen vaccines [2, 3]. Therefore, cancer types with a low neoantigen burden may not be included in indications for this vaccine therapy. While Rosenberg's team recently demonstrated that the predicted neoantigen load was not significantly associated with DCB (durable clinical benefit) [46], this finding may be limited to the sample size of the study.
Rapid epitope loss
Malignant cells have been demonstrated to lose epitopes via various mechanisms under the immune pressure imposed by vaccination. Among these mechanisms, deficiency in antigen processing machinery represents one of the key evasion mechanisms [47]. Downregulation of MHC class I on tumour cells has been shown to occur first. MHC class I molecule expression in cancer is highly heterogenous, and MHC class I defects are often found in cancers [48], even in early stages of some cancer types [49]. Numerous facts could account for the reduced levels of MHC class I expression, including defective MHC genes, epigenetic regulation, or the Warburg effect of glycolysis. Next, these lost epitopes could be derived from passenger mutations. According to their role in the development of cellular transformation, tumour cell mutations can be subdivided into two different categories: driver mutations and passenger mutations [50]. In this setting, passenger mutations exert no effects on tumour growth or survival, but possess dominant numbers in all types of tumour mutations [51]. T cells do not only recognize neoantigens, but also mediate neoantigen immunoediting. In order to avoid the immune escape of tumours, the induction of broad neoantigen-specific T-cell responses is essential.
Tumour immune-suppressive microenvironment
The complicated tumor microenvironment possesses numerous immunosuppressive mechanisms that result in immune escape. Some studies have suggested that the therapeutic resistance mechanisms implicate tumour stromal cell types [52]. Tumors infiltrated with suppressive cells, including Treg cells (regulatory T cells), macrophages, and MDSCs (myeloid-derived suppressor cells), are associated with a poor clinical prognosis [53]. For example, CD4+FOXP3+ intratumoral Treg cells were shown to suppress cetuximab-mediated ADCC (antibody-dependent cellular cytotoxicity) and were correlated with poor clinical outcome in two prospective clinical trial cohorts [54]. Some studies have revealed negative T-cell regulators within the tumor microenvironment, including Ppp2r2d [55], which encodes a regulatory subunit of PP2A (protein phosphatase 2A). Silencing of Ppp2r2d in CD4+ and CD8+ T cells has been shown to enhance ACT efficacy in mice with established tumours, leading to the accumulation of effector T cells in tumours and a prolonged survival [56]. Immune-suppressive cytokine secretion has also been demonstrated to be a very important mechanism. For example, TGF-β (transforming growth factor-β), IL-10 (interleukin‑10), and VEGF (vascular endothelial growth factor) in the tumour microenvironment have been shown to inhibit the maturation of DCs and impair their presenting function [57].
Difficulty in the induction of tumour-specific T cell responses
T cell activation has two signal categories: antigenic signals delivered through T-cell receptors and other signals delivered through co‑stimulatory receptors. In addition, APCs used to deliver these signals are required. Lower expression levels of co-stimulatory molecules and higher expression levels of co-inhibitory receptors and PD1 ligands have been shown to cause T-cell anergy [58, 59]. Hopefully, these problems can be overcome by numerous means, including agonistic antibodies [60] and blocking antibodies [16, 17]. For example, OX40 is a co-stimulatory receptor that is primarily expressed on activated T cells, and OX40 agonists have been shown to enhance antitumour immunity through the promotion of T cell proliferation and survival [61]. In situ vaccination with a TLR9 (toll-like receptor) ligand and an OX40 agonist generated positive results in mouse models [62]. This combination therapy not only triggered a T-cell immune response locally, but also resulted in an attack on cancer cells throughout the body.
Future perspectives of neoantigen vaccines
As far as we are concerned, it remains a difficult task to make personalized neoantigen vaccines a mature and successful therapy for the treatment of solid tumours. Therefore, the following suggestions may be helpful.
Multi-epitope vaccination
The generation of a multi-epitope vaccine that, in general, contains MHC class I-restricted peptides and MHC class II-restricted peptides, to increase both the breadth and diversity of neoantigen-specific T cells [34], represents a good solution for overcoming epitope loss. The majority of neoantigen vaccines used in clinical trials, including multi-peptide vaccines (NCT00683670, NCT01970358 and NCT02427581), poly-epitope-encoding RNA-based or DNA-based vaccines (NCT02316457, NCT02348320 and NCT03122106), contain as many mutation messages as possible that can function to elicit robust anti-tumour effects and that are less likely to trigger immune escape.
Li et al. designed a saline-based multi-epitope peptide vaccine and demonstrated rapid tumour shrinkage of multiple lung tumour nodules in a lung cancer patient following administration [63]. However, eight weeks following the initiation of vaccination, the patient died from complications due to the progression of liver metastases. The metastases were refractory to vaccine therapy, potentially due to tumour heterogeneity and to the fact that the samples acquired for the identification of neoantigens contained only the primary sites.
Recently, a novel idea of changing the peptide configuration has raised great interest within the field. Simanovich and colleagues synthesized one epitope, EMMPRIN, as an octa-branched multiple antigenic peptide. Vaccination with this peptide vaccine was demonstrated to inhibit tumour growth and metastasis in mice [64].
Adjuvants and delivery systems
When DCs in the steady state capture and process antigens in the absence of inflammatory and/or microbial stimuli, they may function to induce immune tolerance rather than immunity [65]. Therefore, efficient adjuvants and optimal methods for vaccine delivery must be developed.
Classical adjuvants include TLR agonists and monoclonal antibodies that target antigens to DCs. The TLR3 agonist, poly-ICLC (polyinosinic-polycytidylic acid with polylysine and carboxymethylcellulose) could function to initiate inflammatory responses by mimicking microbial stimulation and markedly increased peptide vaccine immunogenicity in ovarian cancer patients [66]. This TLR3 agonist represents the most widely used adjuvant in cancer vaccine trials. The TLR4 agonist MPL (monophos-phoryl lipid) A, the TLR7 agonist imiquimod, the TLR7 and TLR8 agonist resiquimod, and the TLR9 agonist CpG ODN (CpG oligodeoxynucleotide) have also been tested in clinical trials. Monoclonal antibodies targeting antigens to DCs, including anti-DEC205 [67] and CD40 agonists [68], may function to direct neoantigens to the strongest APCs. This would be done with the expectation of increasing antigen presentation efficiency and improving vaccination antitumour activity.
Currently, the most desirable vaccine delivery vehicle is nanoparticles. Nanoparticles mimic PAMPs (pathogen-associated molecular patterns), which are viewed as danger signals and are recognized by TLRs on APCs, functioning to enhance nanoparticle-based vaccine uptake [69]. Wang's team [70] worked to develop numerous AC-NPs (antigen-capturing nanoparticles), which were generated using PLGA (poly(lactic-co-glycolic acid)) to bind to TDPAs (tumour-derived protein antigens) by coating the nanoparticles with amine polyethylene glycol, 1,2-dioleoyloxy-3-(trimethylammonium) propane or maleimide polyethylene glycol. These AC-NPs were demonstrated to be capable of efficiently capturing and delivering tumour-specific antigens to APCs, and were demonstrated to significantly improve anti-PD-1 therapy efficacy. Of the various nanomaterials available, nucleic acids are quite appealing for vaccine therapy due to their structural programmability and intrinsic immunomodulatory functionalities. Recently, Chen's team developed self-assembled iDR-NCs (intertwining DNA-RNA nanocapsules), representing the generation of the first hybrid DNA-RNA nanostructures [71]. Consisting of CpG and Stat3-silencing shRNA, this vehicle was shown to be able to synergistically leverage TLR9 and STAT3 signaling pathways in order to co-stimulate APCs. By loading neoantigens, the nanovaccine was shown to elicit 8-fold more frequent neoantigen-specific CD8+ T cells compared to CpG, and was shown to significantly inhibit tumor progression in mice. In addition, liposomes also represent a particularly attractive approach due to the fact that they are synthetic phospholipid vehicles that can preferentially deliver antigens to DLNs (draining lymph nodes) through endogenous albumin hitchhiking [72]. Nanoparticles that are simply assembled by cholesterol-conjugated neopeptides and CpG adjuvants were proven to be safe and quite efficient in animal models [73]. The combination of this vaccine therapy with a tumor-antigen-targeting antibody, specifically a recombinant interleukin-2 and anti-PD-1 antibody, could function to eradicate large established tumors in mice [74]. A nanodisc-based platform [75], comprised of sHDL (synthetic high-density lipoprotein), neoantigens, and Cho-CpG (cholesterol-modified immunostimulatory molecules) were shown to be able to efficiently deliver neoantigens to lymphoid organs, induce DC maturation, and strikingly elicit up to 47-fold greater frequencies of specific CTLs (cytotoxic lymphocytes) in comparison to soluble vaccines. Overall, the advantages of nanoparticles are small size, precise lymph node targeting, and pinpoint loading of theranostic agents.
Combination with other kinds of immunotherapies
As the PD-1 signaling pathway exerts strong immunosuppressive effects on CTL antitumour responses, the coupling of the neo-antigen vaccine with immune checkpoint inhibitors is supposed to be capable of generating a broader-spectrum T-cell antitumor response. Wu's team demonstrated persistence of vaccine-induced immune responses and a broadening of the repertoire of neoantigen-specific T cells following PD-1 blockade therapy in two patients refractory to vaccine therapy [2]. Sahin's team also reported on a patient who developed a complete response to neoantigen vaccination in combination with PD-1 blockade therapy [3]. These studies demonstrated that neo-epitope-specific T-cell subsets were PD-1+ and of a memory phenotype, and post-vaccine lesions were demonstrated to result in the upregulation of PD-L1 (programmed death-ligand 1). There are currently other clinical trials on-going with an aim of assess the efficacy of neoantigen vaccines in combination with ipilimumab (NCT02950766), nivolumab (NCT02897765), or atezolizumab (NCT03289962). These studies are being carried out in an effort to provide further insight into the mechanisms.
Adoptively transferred T cells could also be utilized to potentiate the immune activity of neoantigen vaccination. Programmed T cells that target neoantigens were found to accumulate in vitro and directly elicit immune responses when infused into individuals. Combined with T cells derived from a neoantigen vaccine later in vivo, this therapy can function to induce a sustained and efficient antitumour response.
Combination with conventional therapies
Molecularly targeted agents can function to elicit remarkable responses in the majority of patients that possess the targeted mutation. However, the responses are often of limited duration [76]. Genomically targeted therapies with a high objective response rate have been shown to induce tumour cell death, resulting in the release of tumour-associated antigens and neoantigens. From a mechanistic perspective, targeted therapy possesses synergistic effects with neoantigen vaccine therapy.
In early 2005, chemotherapy was acknowledged to be a good immunotherapy partner in that it could function to potentially enhance immunotherapy efficacy through increased antigen production, improved antigen presentation, augmented T-cell responses, and trafficking [77]. Multiple successful clinical immunotherapy-chemotherapy combinations [78] resulted in the further consideration of chemotherapy and neoantigen vaccine therapy combinations.
Recently, increasing attention has been paid to effects surrounding radiotherapy on the activation of anti-tumour immune responses. A radiosensitive tumour could generate numerous neo-antigens and pro-inflammatory cytokines when exposed to radiation, thereby acting as an in situ vaccine [79]. It has been widely recognized that the combination of local irradiation with immune therapy is capable of enhancing an abscopal effect. Recently, a phase II trial (NCT01006044) demonstrated that the combination of tumour lysate-pulsed autologous DC vaccination with radio-chemotherapy was a feasible and safe method [80].
Conclusion
The ultimate goal of immunotherapy is to stimulate immune responses against tumours. In addition, neoantigens have been demonstrated to be critical to the destruction of tumor cells. Recent innovations in neoantigen identification have generated a new era of personalized vaccines. Inevitably, the process of vaccine development is accompanied with challenges. These include immune escape by low neoantigen burden, rapid epitope loss, a tumour immune-suppressive microenvironment, and difficulty in induction of tumour-specific T cell responses. Even so, there exist numerous approaches that can be adopted to overcome these issues in part. Specifically, the combination of neoantigen vaccine therapy with other immune therapies or conventional therapies may result in surprisingly synergistic effects on the eradication of large solid tumours.
In summary, we anticipate that the cooperation between neoantigens and anti-tumour vaccines will be a promising approach used in the future.
Abbreviations
AC-NPs: antigen-capturing nanoparticles; ACT: adoptive cell transfer; ADCC: antibody-dependent cellular cytotoxicity; ANN: artificial neural networks; APCs: antigen-presenting cells; Cho-CpG: cholesterol-modified immunostimulatory molecules; CpG ODN: CpG oligodeoxynucleotide; CTLs: cytotoxic T lymphocytes; DCB: durable clinical benefit; DCs: dendritic cells; DLNs: draining lymph nodes; ERBB2IP: erbb2 interacting protein; iDR-NCs: intertwining DNA-RNA nanocapsules; IEDB: immune epitope database and analysis resource; IL-10: interleukin‑10; IVT: in vitro transcribed; MDSCs: myeloid-derived suppressor cells; MPL: monophos-phoryl lipid; NGS: next generation sequencing; nsSNVs: non-synonymous single nucleotide variants; PAMPs: pathogen-associated molecular patterns; PD-1: programmed cell death-1; PD-L1: programmed death-ligand 1; PLGA: poly(lactic-co-glycolic acid); poly-ICLC: polyinosinic-polycytidylic acid with polylysine and carboxymethylcellulose; PP2A: protein phosphatase 2A; sHDL: synthetic high-density lipoprotein; TDPAs: tumour-derived protein antigens; TGF-β: transforming growth factor-β; TILs: tumour-infiltrating lymphocytes; TLR: toll-like receptor; TMG: tandem minigene; Treg cells: regulatory T cells; VEGF: vascular endothelial growth factor; WES: whole-exome sequencing.
Acknowledgements
Baorui Liu receives research support from the National Key Research and Development Program of China (No. 2017YFC1308900) and the Key Research and Development Program of Jiangsu Province (No. BE2017607). Qin Liu is supported by Jiangsu Provincial Medical Youth Talent (No. QNRC2016045). The funding sources had no role in the study design, data collection, data analysis, data interpretation, or writing of the report.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jaroslawski S, Toumi M. Sipuleucel-t (provenge((r)))-autopsy of an innovative paradigm change in cancer treatment: Why a single-product biotech company failed to capitalize on its breakthrough invention. Biodrugs. 2015;29:301-7
2. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217-21
3. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M. et al. Personalized rna mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222-6
4. Liu XS, Mardis ER. Applications of immunogenomics to cancer. Cell. 2017;168:600-12
5. Tran E, Robbins PF, Rosenberg SA. 'Final common pathway' of human cancer immunotherapy: Targeting random somatic mutations. Nat Immunol. 2017;18:255-62
6. Vormehr M, Diken M, Boegel S, Kreiter S, Tureci O, Sahin U. Mutanome directed cancer immunotherapy. Curr Opin Immunol. 2016;39:14-22
7. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69-74
8. van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B. et al. A gene encoding an antigen recognized by cytolytic t lymphocytes on a human melanoma. Science. 1991;254:1643-7
9. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF. et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350:1387-90
10. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME. et al. Cancer immunotherapy based on mutation-specific cd4+ t cells in a patient with epithelial cancer. Science. 2014;344:641-5
11. Lu YC, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C. et al. Efficient identification of mutated cancer antigens recognized by t cells associated with durable tumor regressions. Clin Cancer Res. 2014;20:3401-10
12. Lu YC, Yao X, Li YF, El-Gamil M, Dudley ME, Yang JC. et al. Mutated ppp1r3b is recognized by t cells used to treat a melanoma patient who experienced a durable complete tumor regression. J Immunol. 2013;190:6034-42
13. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J. et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive t cells. Nat Med. 2013;19:747-52
14. Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV. et al. Isolation of neoantigen-specific t cells from tumor and peripheral lymphocytes. J Clin Invest. 2015;125:3981-91
15. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ. et al. Cancer immunology. Mutational landscape determines sensitivity to pd-1 blockade in non-small cell lung cancer. Science. 2015;348:124-8
16. Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H. et al. Prolonged survival in stage iii melanoma with ipilimumab adjuvant therapy. N Engl J Med. 2016;375:1845-55
17. Davids MS, Kim HT, Bachireddy P, Costello C, Liguori R, Savell A. et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med. 2016;375:143-53
18. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L. et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521-32
19. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD. et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34
20. Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ. et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by cd4+ t cells in human melanoma. Nat Med. 2015;21:81-5
21. Gubin MM, Schreiber RD. Cancer. The odds of immunotherapy success. Science. 2015;350:158-9
22. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T. et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577-81
23. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV. et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415-21
24. Song Y, Li L, Ou Y, Gao Z, Li E, Li X. et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509:91-5
25. Wang Z, Gerstein M, Snyder M. Rna-seq: A revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57-63
26. Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S. et al. Reliable prediction of t-cell epitopes using neural networks with novel sequence representations. Protein science: a publication of the Protein Society. 2003;12:1007-17
27. Buus S, Lauemoller SL, Worning P, Kesmir C, Frimurer T, Corbet S. et al. Sensitive quantitative predictions of peptide-mhc binding by a 'query by committee' artificial neural network approach. Tissue antigens. 2003;62:378-84
28. Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S. et al. Netmhcpan, a method for quantitative predictions of peptide binding to any hla-a and -b locus protein of known sequence. PloS one. 2007;2:e796
29. Peters B, Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC bioinformatics. 2005;6:132
30. Kim Y, Sidney J, Pinilla C, Sette A, Peters B. Derivation of an amino acid similarity matrix for peptide: Mhc binding and its application as a bayesian prior. BMC bioinformatics. 2009;10:394
31. Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S. et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515:572-6
32. Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J. et al. A vaccine targeting mutant idh1 induces antitumour immunity. Nature. 2014;512:324-7
33. Ochs K, Ott M, Bunse T, Sahm F, Bunse L, Deumelandt K. et al. K27m-mutant histone-3 as a novel target for glioma immunotherapy. Oncoimmunology. 2017;6:e1328340
34. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA. et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific t cells. Science. 2015;348:803-8
35. Schumacher T, Bunse L, Wick W, Platten M. Mutant idh1: An immunotherapeutic target in tumors. Oncoimmunology. 2014;3:e974392
36. Melief CJ. Mutation-specific t cells for immunotherapy of gliomas. N Engl J Med. 2015;372:1956-8
37. Zahm CD, Colluru VT, McNeel DG. DNA vaccines for prostate cancer. Pharmacol Ther. 2017;174:27-42
38. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S. et al. Species-specific recognition of single-stranded rna via toll-like receptor 7 and 8. Science. 2004;303:1526-9
39. Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J. et al. Mutant mhc class ii epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692-6
40. Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC. et al. Systemic rna delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534:396-401
41. Kreiter S, Selmi A, Diken M, Koslowski M, Britten CM, Huber C. et al. Intranodal vaccination with naked antigen-encoding rna elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 2010;70:9031-40
42. Sun Z, Chen F, Meng F, Wei J, Liu B. Mhc class ii restricted neoantigen: A promising target in tumor immunotherapy. Cancer Lett. 2017;392:17-25
43. Stratton MR. Exploring the genomes of cancer cells: Progress and promise. Science. 2011;331:1553-8
44. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK. et al. Clonal neoantigens elicit t cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463-9
45. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S. et al. Genomic and transcriptomic features of response to anti-pd-1 therapy in metastatic melanoma. Cell. 2016;165:35-44
46. Snyder A, Nathanson T, Funt SA, Ahuja A, Buros Novik J, Hellmann MD. et al. Contribution of systemic and somatic factors to clinical response and resistance to pd-l1 blockade in urothelial cancer: An exploratory multi-omic analysis. PLoS Med. 2017;14:e1002309
47. Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS. et al. Neoantigen landscape dynamics during human melanoma-t cell interactions. Nature. 2016;536:91-5
48. Garrido F, Cabrera T, Aptsiauri N. "Hard" and "soft" lesions underlying the hla class i alterations in cancer cells: Implications for immunotherapy. Int J Cancer. 2010;127:249-56
49. van Esch EM, Tummers B, Baartmans V, Osse EM, Ter Haar N, Trietsch MD. et al. Alterations in classical and nonclassical hla expression in recurrent and progressive hpv-induced usual vulvar intraepithelial neoplasia and implications for immunotherapy. Int J Cancer. 2014;135:830-42
50. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719-24
51. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ. et al. Cancer exome analysis reveals a t-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400-4
52. Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346-54
53. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605-12
54. Jie HB, Schuler PJ, Lee SC, Srivastava RM, Argiris A, Ferrone S. et al. Ctla-4(+) regulatory t cells increased in cetuximab-treated head and neck cancer patients suppress nk cell cytotoxicity and correlate with poor prognosis. Cancer Res. 2015;75:2200-10
55. Zhou P, Shaffer DR, Alvarez Arias DA, Nakazaki Y, Pos W, Torres AJ. et al. In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature. 2014;506:52-7
56. An in vivo screen implicates ppp2r2d as an inhibitor of t-cell function. Cancer Discov. 2014;4:Of13
57. Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013;39:61-73
58. Driessens G, Kline J, Gajewski TF. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol Rev. 2009;229:126-44
59. Schwartz RH. T cell anergy. Annu Rev Immunol. 2003;21:305-34
60. Melero I, Berman DM, Aznar MA, Korman AJ, Perez Gracia JL, Haanen J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer. 2015;15:457-72
61. Weinberg AD, Morris NP, Kovacsovics-Bankowski M, Urba WJ, Curti BD. Science gone translational: The ox40 agonist story. Immunol Rev. 2011;244:218-31
62. Sagiv-Barfi I, Czerwinski DK, Levy S, Alam IS, Mayer AT, Gambhir SS. et al. Eradication of spontaneous malignancy by local immunotherapy. Sci Transl Med. 2018:10
63. Li F, Chen C, Ju T, Gao J, Yan J, Wang P. et al. Rapid tumor regression in an asian lung cancer patient following personalized neo-epitope peptide vaccination. Oncoimmunology. 2016;5:e1238539
64. Simanovich E, Brod V, Rahat MM, Drazdov E, Walter M, Shakya J. et al. Inhibition of tumor growth and metastasis by emmprin multiple antigenic peptide (map) vaccination is mediated by immune modulation. Oncoimmunology. 2017;6:e1261778
65. Mellman I, Steinman RM. Dendritic cells: Specialized and regulated antigen processing machines. Cell. 2001;106:255-8
66. Sabbatini P, Tsuji T, Ferran L, Ritter E, Sedrak C, Tuballes K. et al. Phase i trial of overlapping long peptides from a tumor self-antigen and poly-iclc shows rapid induction of integrated immune response in ovarian cancer patients. Clin Cancer Res. 2012;18:6497-508
67. Dhodapkar MV, Sznol M, Zhao B, Wang D, Carvajal RD, Keohan ML. et al. Induction of antigen-specific immunity with a vaccine targeting ny-eso-1 to the dendritic cell receptor dec-205. Sci Transl Med. 2014;6:232ra51
68. Vonderheide RH, Glennie MJ. Agonistic cd40 antibodies and cancer therapy. Clin Cancer Res. 2013;19:1035-43
69. Irvine DJ, Swartz MA, Szeto GL. Engineering synthetic vaccines using cues from natural immunity. Nat Mater. 2013;12:978-90
70. Min Y, Roche KC, Tian S, Eblan MJ, McKinnon KP, Caster JM. et al. Antigen-capturing nanoparticles improve the abscopal effect and cancer immunotherapy. Nat Nanotechnol. 2017;12:877-82
71. Zhu G, Mei L, Vishwasrao HD, Jacobson O, Wang Z, Liu Y. et al. Intertwining DNA-rna nanocapsules loaded with tumor neoantigens as synergistic nanovaccines for cancer immunotherapy. Nat Commun. 2017;8:1482
72. Neelapu SS, Baskar S, Gause BL, Kobrin CB, Watson TM, Frye AR. et al. Human autologous tumor-specific t-cell responses induced by liposomal delivery of a lymphoma antigen. Clin Cancer Res. 2004;10:8309-17
73. Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B. et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature. 2014;507:519-22
74. Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM. et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med. 2016;22:1402-10
75. Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater. 2017;16:489-96
76. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell. 2015;161:205-14
77. Lake RA, Robinson BW. Immunotherapy and chemotherapy-a practical partnership. Nat Rev Cancer. 2005;5:397-405
78. Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C. et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517-26
79. Bockel S, Antoni D, Deutsch E, Mornex F. [immunotherapy and radiotherapy]. Cancer Radiother. 2017;21:244-55
80. Inoges S, Tejada S, de Cerio AL, Gallego Perez-Larraya J, Espinos J, Idoate MA. et al. A phase ii trial of autologous dendritic cell vaccination and radiochemotherapy following fluorescence-guided surgery in newly diagnosed glioblastoma patients. J Transl Med. 2017;15:104
Author contact
![]() Corresponding author: Baorui Liu, MD, Ph.D, The Comprehensive Cancer Centre of Drum Tower Hospital, Medical School of Nanjing University, Clinical Cancer Institute of Nanjing University, 321 Zhongshan Road, Nanjing 210008, China. Tel: +86-25-83107081; Fax: +86-25-83317016; E-mail: baoruiliuedu.cn
Corresponding author: Baorui Liu, MD, Ph.D, The Comprehensive Cancer Centre of Drum Tower Hospital, Medical School of Nanjing University, Clinical Cancer Institute of Nanjing University, 321 Zhongshan Road, Nanjing 210008, China. Tel: +86-25-83107081; Fax: +86-25-83317016; E-mail: baoruiliuedu.cn