Theranostics
13.3
Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2018; 8(16):4552-4562. doi:10.7150/thno.24723 This issue Cite
Research Paper
Liver-heart crosstalk controls IL-22 activity in cardiac protection after myocardial infarction
Ting-Ting Tang1,2, Yuan-Yuan Li1,2, Jing-Jing Li1,2, Ke Wang1,2, Yue Han1,2, Wen-Yong Dong1,2, Zheng-Feng Zhu1,2, Ni Xia1,2, Shao-Fang Nie1,2, Min Zhang1,2, Zhi-Peng Zeng1,2, Bing-Jie Lv1,2, Jiao Jiao1,2, Heng Liu3, Zong-Shu Xian3, Xiang-Ping Yang4, Yu Hu5,6, Yu-Hua Liao1,2, Qing Wang7, Xin Tu7, Ziad Mallat8, Yu Huang9, Guo-Ping Shi10, Xiang Cheng1,2 ![]()
1. Department of Cardiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China
2. Key Laboratory of Biological Targeted Therapy of the Ministry of Education, Wuhan 430022, China
3. Generon Corporation, Building 9, 720 Cai Lun Road, Zhang Jiang Hi-Tech Park, Shanghai 201203, China
4. Department of Immunology, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China
5. Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science & Technology, Wuhan 430022, China
6. Targeted Biotherapy Key Laboratory of Ministry of Education, Wuhan 430022, China
7. Key Laboratory of Molecular Biophysics of the Ministry of Education, Cardio-X Institute, College of Life Science and Technology and Center of Human Genome Research, Huazhong University of Science and Technology, Wuhan 430074, China
8. Division of Cardiovascular Medicine, Department of Medicine, University of Cambridge, Cambridge, CB20 SZ, UK
9. Institute of Vascular Medicine and Li Ka Shing Institute of Health Sciences, Chinese University of Hong Kong, Hong Kong, China
10. Department of Medicine, Brigham and Women's Hospital and Harvard Medical School, Boston, MA 02115, USA
T.T.Tang, Y.Y. Li, J.J. Li and K. Wang contributed equally to this study.
Received 2018-1-3; Accepted 2018-7-28; Published 2018-8-10
Citation:
Tang TT, Li YY, Li JJ, Wang K, Han Y, Dong WY, Zhu ZF, Xia N, Nie SF, Zhang M, Zeng ZP, Lv BJ, Jiao J, Liu H, Xian ZS, Yang XP, Hu Y, Liao YH, Wang Q, Tu X, Mallat Z, Huang Y, Shi GP, Cheng X. Liver-heart crosstalk controls IL-22 activity in cardiac protection after myocardial infarction. Theranostics 2018; 8(16):4552-4562. doi:10.7150/thno.24723. https://www.thno.org/v08p4552.htm
Other stylesAbstract

Interleukin (IL)-22 regulates tissue inflammation and repair. Here we report participation of the liver in IL-22-mediated cardiac repair after acute myocardial infarction (MI).
Methods: We induced experimental MI in mice by ligation of the left ascending artery and evaluated the effect of IL-22 on post-MI cardiac function and ventricular remodeling.
Results: Daily subcutaneous injection of 100 µg/kg mouse recombinant IL-22 for seven days attenuated adverse ventricular remodeling and improved cardiac function in mice at 28 days after left anterior descending coronary artery ligation-induced MI. Pharmacological inhibition of signal transducer and activator of transcription (STAT3) muted these IL-22 activities. While cardiomyocyte-selective depletion of STAT3 did not affect IL-22 activities in protecting post-MI cardiac injury, hepatocyte-specific depletion of STAT3 fully muted these IL-22 cardioprotective activities. Hepatocyte-derived fibroblast growth factor (FGF21) was markedly increased in a STAT3-dependent manner following IL-22 administration and accounted for the cardioprotective benefit of IL-22. Microarray analyses revealed that FGF21 controlled the expression of cardiomyocyte genes that are involved in cholesterol homeostasis, DNA repair, peroxisome, oxidative phosphorylation, glycolysis, apoptosis, and steroid responses, all of which are responsible for cardiomyocyte survival.
Conclusions: Supplementation of IL-22 in the first week after acute MI effectively prevented left ventricular dysfunction and heart failure. This activity of IL-22 involved crosstalk between the liver and heart after demonstrating a role of the hepatic STAT3-FGF21 axis in IL-22-induced post-MI cardiac protection.
Keywords: IL-22, myocardial infarction, liver-heart crosstalk, FGF21
Introduction
In the past decades, advances in early reperfusion therapies have reduced mortality in patients with myocardial infarction (MI) [1]. However, post-MI heart failure remains a significant clinical challenge for patients who survive MI. The pathogenesis of post-MI heart failure is associated with a chronic maladaptive process referred to as ventricular remodeling [2]. Despite the availability of multiple treatments to target ventricular remodeling, e.g., β-blockers and angiotensin-converting enzyme inhibitors, the incidence of post-MI heart failure has increased and led to a high mortality rate and poor life quality [3]. Therefore, the search for alternative management strategies targeting ventricular remodeling remains of major clinical importance.
Interleukin (IL)-22, a member of the IL-10 cytokine family, is an important regulator of inflammation and tissue repair and is produced mainly by innate lymphoid cells, CD4+ T cells, and natural killer cells [4]. IL-22 exerts its function by binding to IL-22 receptor 1 (IL-22R1) and IL-10R2 [5]. Of the two receptors, IL-10R2 shows ubiquitous expression, whereas IL-22R1 has selective expression and determines the IL-22 target cells. Accumulating evidence suggests that IL-22 protects tissue from damage and enhances tissue repair during the inflammatory process by activating signal transducer and activator of transcription (STAT3) [6-10]. At present, a phase 2, open-label study (NCT02655510) is being conducted to assess the efficacy and safety of a recombinant protein containing human IL-22 and immunoglobulin G2-Fc (F-652) in alcoholic hepatitis patients. Administration of IL-22 is associated with tissue protection and repair in murine models of liver [11] and kidney [12] ischemic injuries. The heart also expresses IL-22 R1 [13], yet the contribution of IL-22 to post-MI cardiac inflammation and repair remains unknown.
Fibroblast growth factor (FGF21) belongs to the FGF superfamily and is expressed predominantly in the liver [14]. Early studies identified FGF21 as a hepatokine that is induced in response to stresses, such as cold exposure, starvation, and mitochondrial stress, and that regulates lipid, glucose, and energy metabolism [15-17]. More recent studies have indicated a role for FGF21 in cardiovascular stress and diseases. FGF21 knockout (KO) mice showed worse cardiac function with a hypertrophic stimulus, which could be ameliorated by treatment with FGF21 [18]. FGF21 administration significantly preserved cardiac function post-MI [19]. Here we report a protective role of IL-22 in post-MI cardiac injury via liver-derived FGF21 following hepatic STAT3 activation.
Methods
Animals
Male C57BL/6J mice aged 8-10 weeks were purchased from Peking University (Beijing, China). Cardiac-specific STAT3 KO mice (αMHC-MerCreMer/STAT3flox/flox) were generated by breeding STAT3flox/flox mice (from Jackson Laboratory, USA) with αMHC-MerCreMer mice (from Dr. Hongliang Li, Renmin Hospital of Wuhan University, China). To induce Cre-mediated recombination, αMHC-MerCreMer mice or αMHC-MerCreMer/STAT3flox/flox mice were intraperitoneally injected with 20 mg/kg tamoxifen (Sigma, USA) daily for 5 consecutive days. Liver-specific STAT3 KO mice (ALB-Cre/STAT3flox/flox) were provided by Dr. Zhexiong Lian, University of Science and Technology of China, China. Liver-specific FGF21 KO (ALB-Cre/FGF21flox/flox) mice were obtained from Dr. Aimin Xu, University of Hong Kong, China. All genetically engineered mice were on a C57BL/6J background. All animal studies were conducted according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institute of Health (NIH publication 86-23 revised 1985) and approved by the Animal Care and Utilization Committee of Huazhong University of Science and Technology, China.
MI and study design
MI was produced by ligating the left anterior descending coronary artery as described previously [20]. In brief, after anesthesia with ketamine (75 mg/kg) and medetomidine (1 mg/kg), mice were intubated and ventilated on a rodent respirator. Parasternal thoracotomy was performed and the left anterior descending coronary artery was visualized and ligated at a level approximately 3 mm from its origin using an 8-0 silk suture. Mice in the sham group underwent the same procedure except for the coronary artery ligation. All the operations in this study were performed by an experienced and skilled surgeon who was blinded to the experimental groups. Recombinant mouse IL-22 (derived from Escherichia coli, purity> 95% by SDS-PAGE and endotoxin level <1.09 EU/mg), provided by Generon (Shanghai, China) Corporation Ltd. [21], was dissolved in 1% mouse serum in phosphate-buffered saline (PBS) or an equal volume of 1% serum in PBS and injected subcutaneously daily for 7 days at a dose of 100 μg/kg immediately after the operation. In the STAT3 inhibitor experiment, the STAT3 inhibitor S3I-201 (Merck, Germany) was administered intraperitoneally at a dose of 5 mg/kg every 2 days from 1day prior to the coronary artery ligation until 6 days after the ligation [22].
Echocardiographic and hemodynamic analysis
Transthoracic echocardiography was performed using a Vevo 2100 high-resolution microimaging system (VisualSonics, Canada) on lightly sedated mice (with 2% isoflurane) before the operation and at day 28 post-MI. Two-dimensional echocardiographic views of the mid-ventricular short axis and parasternal long axis were obtained for guided M-mode measurements of the left ventricular end-diastolic diameter (LVEDD), left ventricular end-systolic diameter (LVESD), fractional shortening (FS) and ejection fraction (EF). Echocardiographic acquisition and analysis were performed by a technician who was blinded to the treatment groups. For hemodynamic measurements, a 1.4 French micromanometer-tipped catheter (SPR-671, Millar Instruments, USA) was inserted into the right carotid artery and then advanced into the LV. Left ventricular end-diastolic pressure (LVEDP) was measured, and the maximal (LV +dp/dtmax.) and minimal (LV -dp/dtmin.) first derivative of the LV pressure rise and fall were calculated.
Statistics
Data are expressed as the mean ± SEM. Differences were evaluated using unpaired Student's t-test or chi-square test between two groups and one-way ANOVA followed by a post hoc Student-Newman-Keuls test for multiple comparisons. All analyses were performed using SPSS 13.0, and p<0.05 was considered statistically significant.
Additional methods
Detailed methods are provided in Supplementary Material.
Results
Recombinant IL-22 improves post-MI survival and cardiac function
We first examined the expression of the IL-22 receptors IL-22R1 and IL-10R2 in different tissues. RT-PCR and immunoblot analysis showed low levels of both receptors in mouse hearts and aortas, but high levels of IL-22R1 and IL-10R2 in mouse livers (Figure S1A-C). Further evaluation of heart resident cells showed expression of IL-22R1 and IL-10R2 in hepatocytes but negligible expression in cardiomyocytes, cardiac fibroblasts, endothelial cells, and lymphocytes (Figure S1D-G). To directly evaluate the role of IL-22 in ventricular remodeling, we gave a subcutaneous injection of recombinant IL-22 (100 μg‧kg-1‧d-1) or saline to wild type (WT) mice immediately after MI production for 7 consecutive days. Cardiac function was assessed by echocardiography and cardiac catheterization at 28 days post-MI. Injection of IL-22 significantly increased the plasma IL-22 levels, reaching a peak concentration of 6545.84±1277.90 pg/mL at 1 h and returning to the baseline at 12 h after administration (Figure S2). Seven days of injection of IL-22 resulted in a significant improvement in post-MI survival (Figure 1A). And, recombinant IL-22 improved post-MI cardiac function, as evidenced by decreased LVEDD, LVESD, and LVEDP and increased FS, EF, +dp/dtmax and -dp/dtmin in IL-22-treated mice compared with those in the saline-treated control mice (Figure 1B-I and Table S1).
Figure 1
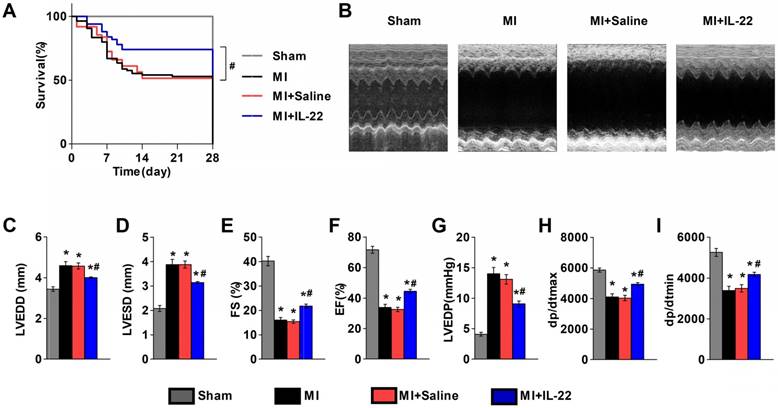
IL-22 protects mice from post-MI cardiac injury. (A) Survival curves of sham mice (n=12), MI mice (n=85), saline-treated MI mice (n=62), and IL-22-treated MI mice (n=50) groups at 28 days after operation. (B) Representative M-mode echocardiographic images at 28 days after operation. (C-I) Quantification of LVEDD, LVESD, FS and EF by echocardiography, and LVEDP, dp/dtmax, and dp/dtmin by hemodynamic analysis at 28 days after operation. n=10-15 per group. #p<0.05 by log-rank (Mantel-Cox) test in (A) and*p<0.05 vs. Sham group and #p<0.05 vs. MI+Saline group by one-way ANOVA in (C-I).
Figure 2
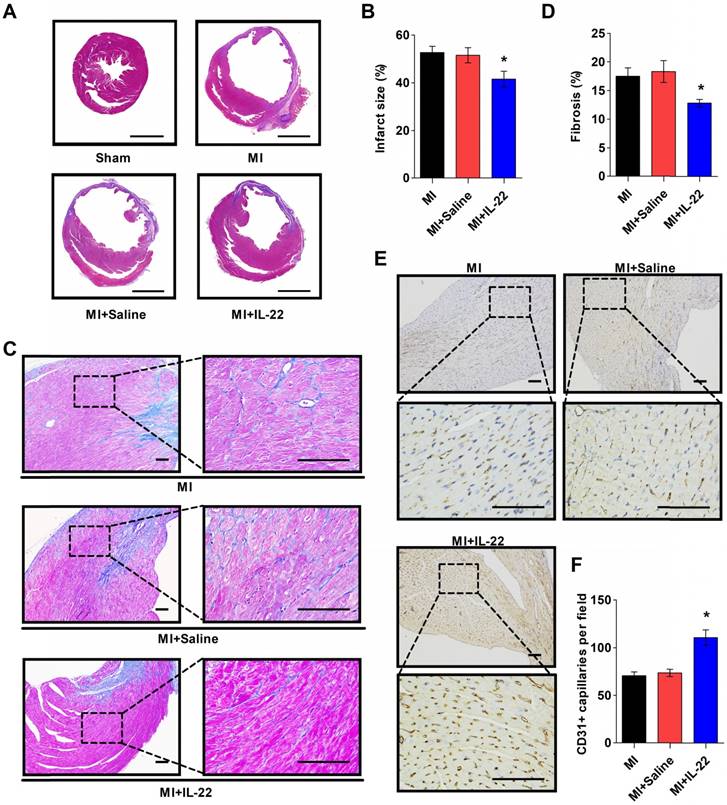
IL-22 decreases post-MI infarct size and fibrosis and promotes post-MI neovascularization. (A) Representative images of Masson's trichrome-stained heart sections (scale: 2000 μm) and (B) quantification of infarct size at 28 days post-MI. n=8-10 per group. (C) Representative images of Masson's trichrome-stained heart sections (scale: 100 μm) at 28 days post-MI and (D) quantification of interstitial fibrosis by collagen volume fraction per high-magnification view. n=8-10 per group. (E) Representative immunostaining of CD31+ cells in the peri-infarct zone (scale:100 μm) on heart sections at 3 days post-MI and (F) the calculated CD31+ cell number per high-magnification view. n=8 per group. *p<0.05 vs. MI+Saline group by one-way ANOVA.
After demonstrating the cardiac protection of IL-22 against MI, we assessed IL-22 activities in cardiac necrosis, fibrosis, neovascularization, and inflammatory cell profiles in ventricular remodeling. Heart infarct size and interstitial fibrosis were determined at 28 days post-MI by Masson's trichrome staining. Interstitial fibrosis was analyzed as the collagen volume fraction in the border zone. Border zone neovascularization was evaluated by CD31 staining at 3 days post-MI. Infarct size (Figure 2A-B) and collagen volume fraction (Figure 2C-D) decreased significantly in IL-22-treated mice compared with saline-treated control mice. We detected a marked increase in the number of CD31-positive capillaries in IL-22-treated mice (Figure 2E-F). Flow cytometry was used to analyze the impact of IL-22 administration on the infiltration of leukocytes in the heart at 3 and 7 days post-MI. Figure 3A shows the identification of leukocytes as CD45-positive cells and the definition of different leukocyte populations by their specific surface markers. IL-22 inhibited the infiltration of total leukocytes, neutrophils, monocytes/macrophages, natural killer cells, γδ T cells, and B cells while promoting the infiltration of αβ T cells (Figure 3B-H). Previous studies have reported that decreased accumulation of monocytes/macrophages [23], neutrophils [24], γδT cells [25] and B cells [26] in the infarcted heart was associated with improved cardiac dysfunction post-MI, while a deficiency of CD4+ T cells [27], which accounted for the majority of αβT cells in the infarcted heart, was associated with a worse outcome post-MI. Therefore, the altered accumulation of leukocytes after IL-22 treatment may be associated with improved post-MI cardiac function in IL-22-treated mice.
Figure 3
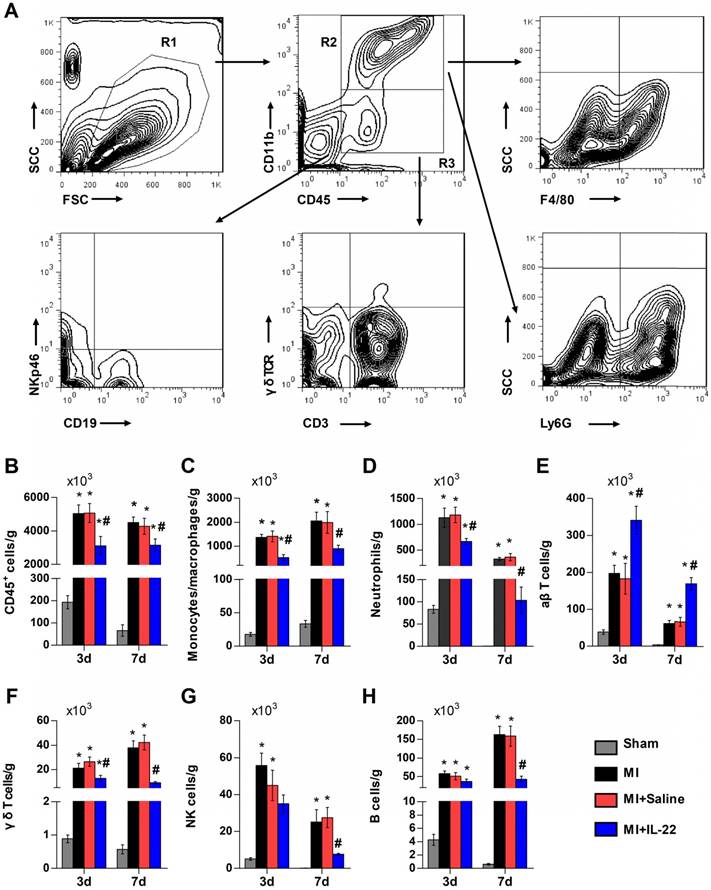
IL-22 inhibits post-MI cardiac inflammation. (A) Gating strategy for infiltrated CD45+ total leukocytes, CD45+CD11b+F4/80+ macrophages, CD45+CD11b+Ly6G+ neutrophils, CD45+CD11b-CD3+γδTCR- αβT cells, CD45+CD11b-CD3+ γδ TCR+ γδT cells, CD45+CD11b-NKp46+ natural killer (NK) cells, and CD45+CD11b-CD19+ B cells in the heart. (B-H) Quantification of the absolute numbers of different infiltrated inflammatory cells per gram of heart. n=5 per group. *p<0.05 vs Sham group and #p<0.05 vs. MI+Saline group by one-way ANOVA.
Figure 4
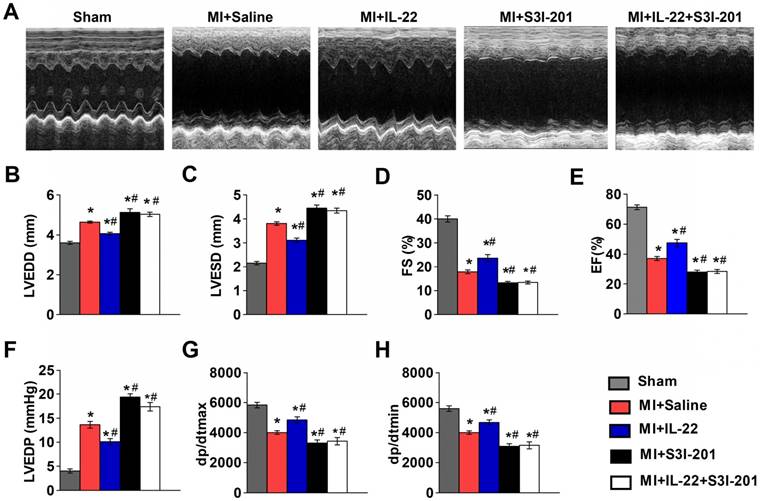
IL-22 protects mice post-MI cardiac injury via STAT3 activation. (A) Representative M-mode echocardiographic images at 28 days after operation. (B-E) Analysis of LVEDD, LVESD, FS and EF by echocardiography at 28 days after operation. n=8-10 per group. (F-H) Quantification of LVEDP, dp/dtmax, and dp/dtmin by hemodynamic analysis at 28 days after operation. n=8 per group. *p<0.05 vs Sham group and #p<0.05 vs. MI+Saline group by one-way ANOVA.
IL-22 protects the heart against MI via hepatic but not cardiac STAT3 activation
The functional link between IL-22 and STAT3 activation is known in liver injury and intestinal inflammation [6-10]. It is possible that IL-22-mediated post-MI cardiac injury protection also involved STAT3 activation. We tested this hypothesis by giving MI mice a STAT3 inhibitor S3I-201 [28]. We first confirmed that S3I-201 effectively inhibited phosphorylation of STAT3 in vivo (Figure S3). S3I-201 led to cardiac function deterioration and diminished the IL-22 post-MI cardioprotective activities (Figure 4A-H and Table S2). However, when attempting to explore how IL-22 modulated post-MI cardiac STAT3 activation and to identify its target cells in the heart, we could not detect cardiac STAT3 activation in vivo in IL-22-treated mice (Figure 5A). Similarly, we were unable to observe STAT3 activation in either cardiomyocytes or cardiac fibroblasts after they were subjected to in vitro IL-22 treatment (Figure 5B). To identify the possible intrinsic links between cardiac STAT3 activation and IL-22-mediated post-MI cardioprotection, we generated cardiac-specific STAT3 KO mice by breeding α-MHC-MerCreMer mice with STAT3flox/flox mice. Consistent with prior studies [29], cardiac-specific STAT3 ablation exacerbated post-MI cardiac dysfunction. However, IL-22 administration retained its activity in improving post-MI cardiac function in cardiac-specific STAT3 KO mice (Figure 5C, Figure S4 and Table S3), as in WT control mice (Figure 1B-I), suggesting that cardiac STAT3 activation was not required for IL-22-mediated post-MI cardioprotection.
IL-22 is known to induce prominent hepatic STAT3 activation [30]. We assessed changes in hepatic STAT3 activation in IL-22-treated mice after MI. IL-22 induced rapid and transient hepatic STAT3 activation in mice with MI in vivo, reaching maximum activation within 30 min after IL-22 administration and returning to the baseline in 3 h (Figure 5D). Furthermore, IL-22 treatment induced STAT3 activation in primary cultured hepatocytes (Figure 5E). To examine the role of hepatic STAT3 activation in IL-22-mediated cardiac protection, we generated liver-specific STAT3 KO mice by breeding ALB-Cre mice with STAT3flox/flox mice. Unexpectedly, liver-specific STAT3 deletion exacerbated cardiac dysfunction and abolished the protective effects of IL-22 on cardiac function post-MI (Figure 5F, Figure S5 and Table S4). These in vitro and in vivo observations suggest that hepatic but not cardiac STAT3 activation is essential for the beneficial effect of IL-22 on post-MI cardiac function.
Figure 5
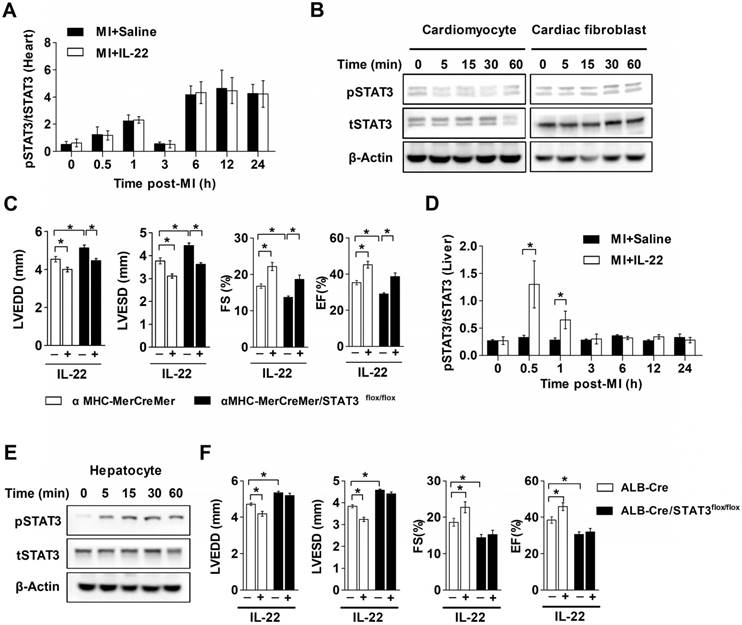
The cardioprotective effect of IL-22 depends on hepatic STAT3 activation. (A) Quantification of STAT3 phosphorylation in the heart at different time points post-MI by ELISA. n=5 per group. (B) Representative western blot images of STAT3 phosphorylation in cardiomyocytes and cardiac fibroblasts treated with IL-22 at different time points. (C) Analysis of LVEDD, LVESD, FS and EF by echocardiography at 28 days post-MI. n=8 per group. (D) Quantification of STAT3 phosphorylation in the liver at different time points post-MI by ELISA. n=5 per group. (E) Representative western blot images of STAT3 phosphorylation in IL-22- stimulated primary hepatocytes at different time points. (F) Analysis of LVEDD, LVESD, FS and EF by echocardiography at 28 days post-MI. n=8 per group. *p<0.05 by unpaired t test in (D) and by one-way ANOVA in (C, F).
IL-22 protects post-MI cardiac dysfunction via liver-derived FGF21
The role of IL-22 in liver-specific STAT3 activation (Figure 5E-F) inspired us to identify the molecules from hepatic STAT3 activation that mediate the cardioprotective effects of IL-22. We compared the liver expression levels between IL-22-treated and control mice of ten common genes that are regulated by IL-22 [31] or are cardioprotective [32], namely, alpha-1-acid glycoprotein-2 (AGP2), AGP3, C-X-C ligand-13 (CXCL13), FGF21, trefoil factor-3 (TFF3), lipopolysaccharide binding protein (LBP), neuregulin-4 (NRG4), proteoglycan-4 (PRG4), BMP binding endothelial regulator (BMPER), and serum amyloid A-2 (SAA2). Of the ten genes, only the mRNA levels of FGF21 and AGP2 were elevated significantly at 6-12 h after IL-22 treatment (Figure S6). Immunoblot analysis examined the protein levels of FGF21 and AGP2 in livers from mice at 12 h post-MI. MI increased liver FGF21 protein levels, and IL-22 further increased liver FGF21 expression. These increases were more robust than those of AGP2 (Figure 6A). Plasma FGF21 levels were also markedly elevated in mice at 12 h post-MI. IL-22 administration further increased plasma FGF21 levels at 12 and 24 h post-MI (Figure 6B). FGF21 is also inducible in many organs, such as white and brown adipose tissue, pancreas, skeletal muscle, heart and kidney [33]. However, RT-PCR did not detect an effect of IL-22 on FGF21 expression in the heart, pancreas, kidney, brown adipose tissue, white adipose tissue, or skeletal muscle (Figure S7).
The absences of IL-22 cardioprotection in liver-selective STAT3 KO mice (Figure 5F), IL-22-induced STAT3 activation in the liver and hepatocytes (Figure 5D-E), and IL-22-induced liver FGF21 expression (Figure S6 and Figure 6A) all suggest a role of IL-22 in inducing liver FGF21 expression via STAT3 activation. We tested this hypothesis by showing that IL-22 activity in inducing plasma FGF21 levels was blunted in mice with liver-specific STAT3 ablation (Figure 6C). In cultured mouse hepatocytes, immunoblot analysis demonstrated that IL-22 activated STAT1, STAT3, and ERK1/2 (Figure S8A). However, FGF21 secretion was blocked by only the STAT3 inhibitor S3I-201, not the STAT1 inhibitor fludarabine or the ERK inhibitor PD98059 (Figure S8B).
Figure 6
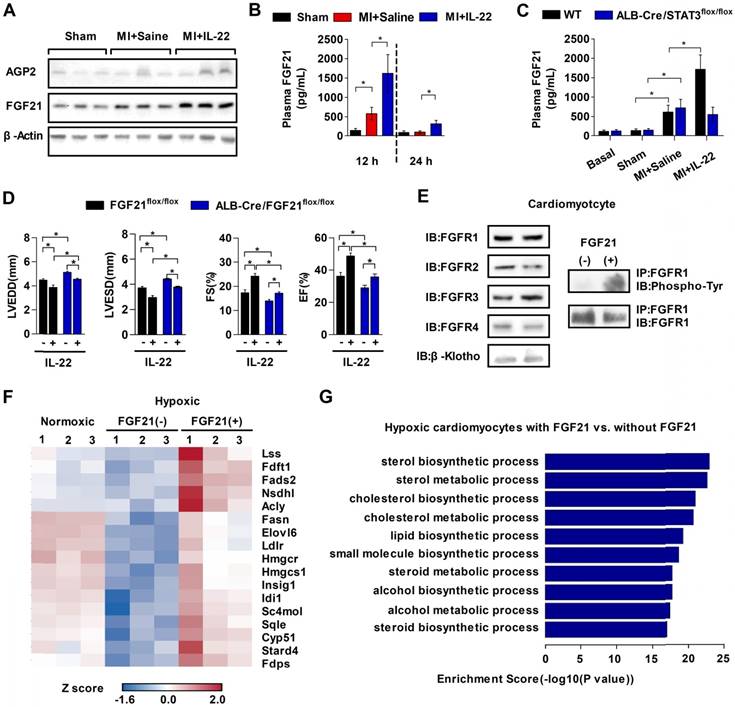
IL-22 promotes liver FGF21 production to protect mice from post-MI cardiac injury. (A) Representative western blot images of FGF21 and AGP2 protein in the liver at 12 hours post-MI. (B) ELISA-determined plasma FGF21 levels in sham, saline-treated, and IL-22-treated mice at 12 h and 24 h post-MI. (C) Comparison of plasma FGF21 levels by ELISA between WT and ALB-Cre/STAT3flox/flox mice in basal condition or at 12 h post-MI treated with saline or IL-22. n=5-10 per group. (D) Analysis of LVEDD, LVESD, FS and EF by echocardiography at 28 days post-MI. n=8-12 per group. (E) Representative western blot images of FGFR1, FGFR2, FGFR3, FGFR4 and β-klotho in cardiomyocytes (left panels) and representative coimmunoprecipitation images of FGFR1 and tyrosine phosphorylation in cardiomyocytes stimulated with FGF21 for 20 min (right panels). (F) Heat map of genes involved in lipid biosynthesis and differentially regulated by FGF21 in hypoxic cardiomyocytes. Red represents upregulation and blue represents downregulation. (G) GO analysis of biological processes that are enriched in FGF21-treated cardiomyocytes. *p<0.05 by unpaired t-test in (B-C) and by one-way ANOVA in (D).
To test the role of liver-derived FGF21 in IL-22-mediated post-MI cardioprotection, we generated liver-specific FGF21 KO mice by breeding ALB-Cre mice with FGF21flox/flox mice. Plasma FGF21 levels decreased in liver-specific FGF21 KO mice (Figure S9), and these mice showed poorer cardiac function than control mice (Figure 6D). Administration of IL-22 improved cardiac function in these liver-specific FGF21 KO mice, but these improvements were much weaker than those in IL-22-treated FGF21flox/flox control mice (Figure 6D, Figure S10 and Table S5). These observations suggest the following: liver FGF21 played a cardioprotective role, as previously found [32], because ALB-Cre/FGF21flox/flox mice showed impaired cardiac function; IL-22 protected post-MI cardiac dysfunction via liver-specific FGF21 because the cardiac function in IL-22-treated ALB-Cre/FGF21flox/flox mice did not reach the same level as that in IL-22-treated FGF21flox/flox control mice; and, FGF21 was not the only contributor to the cardioprotective effects of IL-22, because IL-22 administration did not completely lose its cardioprotective activity in liver-specific FGF21 KO mice. Therefore, IL-22 must also have a liver FGF21-independent cardioprotective function, which is a hypothesis that merits further investigation. To determine whether FGF21 acted directly on the heart, we performed immunoblot analysis and found that cardiomyocytes expressed all FGF21 receptors, including FGFR1, FGFR2, FGFR3, FGFR4, and its coreceptor β-klotho. Coimmunoprecipitation revealed FGFR1 activation (tyrosine phosphorylation) in FGF21-treated cardiomyocytes (Figure 6E), suggesting that FGFR1 is a functional receptor of FGF21 in cardiomyocytes.
FGF21 changes the expression of genes responsible for cardiomyocyte survival
To further explore the cardioprotective role of FGF21, we treated mouse cardiomyocytes with and without FGF21 under normoxic or hypoxic conditions to mimic those of MI injury. The majority of genes regulated by FGF21 in hypoxic cardiomyocytes were those involved in lipid metabolism, which were inhibited under hypoxic conditions (Figure 6F). In addition, FGF21 changed the expression of genes related to several other aspects of hypoxic cardiomyocytes, including ion homeostasis and cell proliferation and apoptosis (Table S6). Gene ontology (GO) analysis demonstrated that the top 10 ranked FGF21-controlled biological processes were mostly related to lipid biosynthesis and metabolism (Figure 6G). To gain more insights into the pathobiological significance of our findings, we performed gene set enrichment analysis (GSEA) to evaluate the data at the level of gene sets [34]. We found that hypoxic cardiomyocytes treated with FGF21 are enriched in gene sets pertaining to cholesterol homeostasis, DNA repair, peroxisome, oxidative phosphorylation, glycolysis, apoptosis, and steroid responses (Table S7). Microarray data were verified by RT-PCR on select genes with known functions that are listed in Table S6. RT-PCR validated the upregulation of genes involved in cholesterol synthesis (Acly, Lss, and Sqle) and cholesterol transport (Stard4 and Ldlr). In addition, the upregulation of Dhcr24, which has been shown to catalyze the reduction of the delta-24 double bond of sterol intermediates during cholesterol biosynthesis and to be antiapoptotic via interaction with p53 [35], was found. The down-regulation of Kcne3, which is linked to potassium channel activity, was also observed. Intrigued by our finding that FGF21-regulated genes are related to several biological processes (Figure S11A), we asked whether FGF21 affects cardiomyocyte survival under hypoxic conditions. We found that FGF21 reduced caspase-3 activity in hypoxic cardiomyocytes (Figure S11B), demonstrating that FGF21 can inhibit apoptosis.
Discussion
This study established a post-MI cardioprotective role of IL-22 via hepatic STAT3 activation and FGF21 secretion independent of cardiac STAT3 activation. We used cardiac-specific STAT3 KO, liver-specific STAT3 KO, and liver-specific FGF21 KO mice to confirm these unexpected observations. Mechanistic studies disproved a direct role of IL-22 in cardiac resident cells, likely due to their low expression of IL-22 receptors, but established an indirect role of IL-22 in cardiomyocytes via hepatic FGF21 on the expression of cholesterol homeostasis, DNA repair, peroxisome, oxidative phosphorylation, glycolysis, apoptosis, and steroid responses genes that are essential to cardiomyocyte survival.
Clinical studies have reported an association between liver dysfunction and heart failure in humans, but the underlying mechanisms have never been understood. Patients with cirrhosis, a condition with impaired liver function, are at high risk for heart failure characterized by impaired systolic/diastolic function and electrophysiological abnormalities, termed “cirrhotic cardiomyopathy” [36]. A post hoc analysis of the EVEREST (Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study with Tolvaptan) trial indicates that markers of liver dysfunction, including lower albumin and higher bilirubin in heart failure patients, are associated with poor prognosis [37]. Recent studies suggested that the liver plays a cardioprotective role in the experimental model of MI by secreting undefined protective proteins, indicating a new mechanism of protection by liver-derived hepatokines in heart failure [38]. The roles of the STAT3 signaling pathway in liver pathophysiology have been extensively investigated [39]. However, this study reported a novel role of hepatic STAT3 as a protective signaling pathway for the heart, a remote organ, in an endocrine manner post-MI. Of note, hepatic STAT3 ablation lead to aggravated cardiac dysfunction post-MI, but hepatic STAT3 was not activated in the post-MI setting (Figure 5D), suggesting that STAT3 is required for driving hepatic expression of proteins with cardioprotective activities in basal condition. Therefore, future secretome analysis of hepatocytes from liver-specific STAT3 KO mice may help identify additional targets from the STAT3 pathway for sustained cardiac protection.
Prior studies demonstrated increased levels of FGF21 in the circulation, liver, adipose tissue, and heart after MI [19, 32, 38 and 40]. We also detected higher plasma FGF21 in MI mice than in sham mice. However, systemic FGF21 deficiency [38] or liver-specific FGF21 deficiency from our study lead to the deterioration of post-MI cardiac function. Overall, we could speculate that endogenous FGF21 serves as an important cardio-protective mechanism in the MI setting. Furthermore, data from our study suggest that IL-22, as a therapeutic cytokine, further strengthens hepatic FGF21 production to protect the heart against MI. The regulation of hepatic FGF21 has been reported to be associated with several signaling pathways, including peroxisome proliferator-activated receptor (PPAR) α, PPARγ, activating transcription factor 4, cAMP-responsive element-binding protein H, farnesoid X receptor and liver X receptor under various conditions [41]. Our data suggest that hepatic STAT3 is indispensable for the induction of FGF21 following IL-22 administration.
Previous studies in vitro have reported beneficial effects of FGF21 in cardiomyocytes by promoting energy supply and inhibiting oxidative stress and inflammation [42], all of which are crucial for cell survival. Our data demonstrated a beneficial effect of FGF21 on cardiomyocyte survival by inhibiting apoptosis under hypoxic conditions. Of note, our gene microarray and RT-PCR validation data suggest that FGF21 mainly upregulates several genes coding the key enzymes in cholesterol biosynthesis, including Acly (which catalyzes the formation of acetyl-CoA), Sqle (which catalyzes the conversion of farnesyl-PP to squalene), Lss (which catalyzes the conversion of (S)-2, 3-oxidosqualene to lanosterol), and Dhcr24 (which catalyzes the reduction of the delta-24 double bond of sterol intermediates). It is widely accepted that hypoxic cardiomyocytes switch substrate utilization from fatty acids to glucose and downregulate lipid biosynthesis under hypoxic conditions to produce more energy with less oxygen consumption, and suppress energy consumption [43]. In line with this notion, we observed that the expression of genes involved in lipid metabolism decreased, while the expression of genes involved in glucose metabolism increased in hypoxic cardiomyocytes (Figure S12). On the other hand, we know that cholesterol itself and several intermediate metabolites in the biosynthesis of cholesterol are crucial for cell survival. Reducing intracellular cholesterol made cardiomyocytes susceptible to anoxia-induced damage [44]. As intermediate metabolites in the biosynthesis of cholesterol, squalene has been found to protect against oxidative DNA damage and apoptosis [45, 46]; elevated cellular lanosterol has been linked to decreased cell apoptosis under intermittent hypoxia conditions [47]. In summary, although the adaptive shift from lipid metabolism to glucose metabolism helps overcome the energy crisis, disturbed lipid metabolism can be deleterious to the ischemic heart. The disturbed cholesterol synthesis in hypoxic cardiomyocytes, which was partially restored by FGF21 treatment, may be related to the beneficial effect of FGF21 during myocardial ischemia.
In conclusion, our findings presented a concept of inter-organ communication between the heart and the liver as an adaptive response to cardiac ischemic stress post-MI, and suggested a new perspective on the therapeutic potential of IL-22 and the hepatic STAT3-FGF21 axis in the setting of acute post-MI.
Supplementary Material
Supplementary materials and methods, figures and tables.
Acknowledgements
We thank Generon Corporation Ltd. (Shanghai, China) for providing recombinant mouse IL-22, Dr. Hongliang Li from Renmin Hospital of Wuhan University, China for providing αMHC-MerCreMer mice, Dr. Zhexiong Lian from University of Science and Technology of China for providing ALB-Cre/STAT3flox/flox mice, and Dr. Aimin Xu from University of Hong Kong, China for providing ALB-Cre/FGF21flox/flox mice. We also thank Ms. Chelsea Swallom from Brigham and Women's Hospital, USA for editorial assistance.
Funding
This work was supported by grants from the National Natural Science Foundation of China [No. 81525003, 91639301, 81720108005 to X.C.; No. 81200177, 81670361 to T.T.T.; No. 81600287 to Z.F.Z..; No. 81400364 to N.X.; No. 81500186 to S.F.N.], and National Institute of Health [HL123568, HL60942 to G.P.S.].
Competing Interests
Recombinant mouse IL-22 was provided by Generon (Shanghai, China) Corporation Ltd in the present study.
References
1. Fox KA, Steg PG, Eagle KA. et al. Decline in rates of death and heart failure in acute coronary syndromes, 1999-2006. JAMA. 2007;297:1892-1900
2. Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101:2981-2988
3. Velagaleti RS, Pencina MJ, Murabito JM. et al. Long-term trends in the incidence of heart failure after myocardial infarction. Circulation. 2008;118:2057-2062
4. Wolk K, Witte E, Witte K, Warszawska K, Sabat R. Biology of interleukin-22. Semin Immunopathol. 2010;32:17-31
5. Bleicher L, de Moura PR, Watanabe L. et al. Crystal structure of the IL-22/IL-22R1 complex and its implications for the IL-22 signaling mechanism. FEBS Lett. 2008;582:2985-2992
6. Dumoutier L, de Meester C, Tavernier J, Renauld JC. New activation modus of STAT3: a tyrosine-less region of the interleukin-22 receptor recruits STAT3 by interacting with its coiled-coil domain. J Biol Chem. 2009;284:26377-26384
7. Guo X, Qiu J, Tu T. et al. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity. 2014;40:25-39
8. Pickert G, Neufert C, Leppkes M. et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465-1472
9. Ki SH, Park O, Zheng M. et al. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology. 2010;52:1291-1300
10. Kong X, Feng D, Wang H. et al. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology. 2012;56:1150-1159
11. Eggenhofer E, Sabet-Rashedi M, Lantow M. et al. RORgammat(+) IL-22-producing NKp46(+) cells protect from hepatic ischemia reperfusion injury in mice. J Hepatol. 2016;64:128-134
12. Xu MJ, Feng D, Wang H, Guan Y, Yan X, Gao B. IL-22 ameliorates renal ischemia-reperfusion injury by targeting proximal tubule epithelium. J Am Soc Nephrol. 2014;25:967-977
13. Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21:241-254
14. Nishimura T, Nakatake Y, Konishi M, Itoh N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim Biophys Acta. 2000;1492:203-206
15. Berglund ED, Li CY, Bina HA. et al. Fibroblast growth factor 21 controls glycemia via regulation of hepatic glucose flux and insulin sensitivity. Endocrinology. 2009;150:4084-4093
16. Coskun T, Bina HA, Schneider MA. et al. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149:6018-6027
17. Kharitonenkov A, Wroblewski VJ, Koester A. et al. The metabolic state of diabetic monkeys is regulated by fibroblast growth factor-21. Endocrinology. 2007;148:774-781
18. Planavila A, Redondo I, Hondares E. et al. Fibroblast growth factor 21 protects against cardiac hypertrophy in mice. Nat Commun. 2013;4:2019
19. Patel V, Adya R, Chen J. et al. Novel insights into the cardio-protective effects of FGF21 in lean and obese rat hearts. PLoS One. 2014;9:e87102
20. Tao L, Wang Y, Gao E. et al. Adiponectin: an indispensable molecule in rosiglitazone cardioprotection following myocardial infarction. Circ Res. 2010;106:409-417
21. Yang L, Zhang Y, Wang L. et al. Amelioration of high fat diet induced liver lipogenesis and hepatic steatosis by interleukin-22. J Hepatol. 2010;53:339-347
22. Lin L, Amin R, Gallicano GI. et al. The STAT3 inhibitor NSC 74859 is effective in hepatocellular cancers with disrupted TGF-beta signaling. Oncogene. 2009;28:961-972
23. Kaikita K, Hayasaki T, Okuma T, Kuziel WA, Ogawa H, Takeya M. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am J Pathol. 2004;165:439-447
24. Rainer PP, Hao S, Vanhoutte D. et al. Cardiomyocyte-specific transforming growth factor beta suppression blocks neutrophil infiltration, augments multiple cytoprotective cascades, and reduces early mortality after myocardial infarction. Circ Res. 2014;114:1246-1257
25. Yan X, Shichita T, Katsumata Y. et al. Deleterious effect of the IL-23/IL-17A axis and gammadeltaT cells on left ventricular remodeling after myocardial infarction. J Am Heart Assoc. 2012;1:e004408
26. Zouggari Y, Ait-Oufella H, Bonnin P. et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med. 2013;19:1273-1280
27. Hofmann U, Beyersdorf N, Weirather J. et al. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation. 2012;125:1652-1663
28. Siddiquee K, Zhang S, Guida WC. et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A. 2007;104:7391-7396
29. Enomoto D, Obana M, Miyawaki A, Maeda M, Nakayama H, Fujio Y. Cardiac-specific ablation of the STAT3 gene in the subacute phase of myocardial infarction exacerbated cardiac remodeling. Am J Physiol Heart Circ Physiol. 2015;309:H471-480
30. Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology. 2004;39:1332-1342
31. Wolk K, Witte E, Wallace E. et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. 2006;36:1309-1323
32. Liu SQ, Tefft BJ, Roberts DT. et al. Cardioprotective proteins upregulated in the liver in response to experimental myocardial ischemia. Am J Physiol Heart Circ Physiol. 2012;303:H1446-1458
33. Kim KH, Lee MS. FGF21 as a Stress Hormone: The Roles of FGF21 in Stress Adaptation and the Treatment of Metabolic Diseases. Diabetes Metab J. 2014;38:245-251
34. Subramanian A, Tamayo P, Mootha VK. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545-15550
35. Kuehnle K, Crameri A, Kalin RE. et al. Prosurvival effect of DHCR24/Seladin-1 in acute and chronic responses to oxidative stress. Mol Cell Biol. 2008;28:539-550
36. Gassanov N, Caglayan E, Semmo N, Massenkeil G, Er F. Cirrhotic cardiomyopathy: a cardiologist's perspective. World J Gastroenterol. 2014;20:15492-15498
37. Ambrosy AP, Vaduganathan M, Huffman MD. et al. Clinical course and predictive value of liver function tests in patients hospitalized for worsening heart failure with reduced ejection fraction: an analysis of the EVEREST trial. Eur J Heart Fail. 2012;14:302-311
38. Liu SQ, Roberts D, Kharitonenkov A. et al. Endocrine protection of ischemic myocardium by FGF21 from the liver and adipose tissue. Sci Rep. 2013;3:2767
39. Wang H, Lafdil F, Kong X, Gao B. Signal transducer and activator of transcription 3 in liver diseases: a novel therapeutic target. Int J Biol Sci. 2011;7:536-550
40. Zhang W, Chu S, Ding W, Wang F. Serum Level of Fibroblast Growth Factor 21 Is Independently Associated with Acute Myocardial Infarction. PLoS One. 2015;10:e0129791
41. Bae KH, Kim JG, Park KG. Transcriptional regulation of fibroblast growth factor 21 expression. Endocrinol Metab (Seoul). 2014;29:105-111
42. Tanajak P, Chattipakorn SC, Chattipakorn N. Effects of fibroblast growth factor 21 on the heart. J Endocrinol. 2015;227:R13-30
43. Ford DA. Alterations in myocardial lipid metabolism during myocardial ischemia and reperfusion. Prog Lipid Res. 2002;41:6-26
44. Bastiaanse EM, van der Valk-Kokshoorn LJ, Egas-Kenniphaas JM, Atsma DE, van der Laarse A. The effect of sarcolemmal cholesterol content on the tolerance to anoxia in cardiomyocyte cultures. J Mol Cell Cardiol. 1994;26:639-648
45. Warleta F, Campos M, Allouche Y. et al. Squalene protects against oxidative DNA damage in MCF10A human mammary epithelial cells but not in MCF7 and MDA-MB-231 human breast cancer cells. Food Chem Toxicol. 2010;48:1092-1100
46. Saito K, Dubreuil V, Arai Y. et al. Ablation of cholesterol biosynthesis in neural stem cells increases their VEGF expression and angiogenesis but causes neuron apoptosis. Proc Natl Acad Sci U S A. 2009;106:8350-8355
47. Zhen YQ, Wu YM, Sang YH. et al. 2,3-Oxidosqualene cyclase protects liver cells from the injury of intermittent hypoxia by regulating lipid metabolism. Sleep Breath. 2015;19:1475-1481
Author contact
![]() Corresponding author: Xiang Cheng MD, PhD, Department of Cardiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Key Laboratory of Biological Targeted Therapy of the Ministry of Education, Wuhan 430022, China. Phone: +86-27-85726011. Fax: +86-27-85727340. E-mail: nathancxedu.cn.
Corresponding author: Xiang Cheng MD, PhD, Department of Cardiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Key Laboratory of Biological Targeted Therapy of the Ministry of Education, Wuhan 430022, China. Phone: +86-27-85726011. Fax: +86-27-85727340. E-mail: nathancxedu.cn.
Citation styles
APA
Tang, T.T., Li, Y.Y., Li, J.J., Wang, K., Han, Y., Dong, W.Y., Zhu, Z.F., Xia, N., Nie, S.F., Zhang, M., Zeng, Z.P., Lv, B.J., Jiao, J., Liu, H., Xian, Z.S., Yang, X.P., Hu, Y., Liao, Y.H., Wang, Q., Tu, X., Mallat, Z., Huang, Y., Shi, G.P., Cheng, X. (2018). Liver-heart crosstalk controls IL-22 activity in cardiac protection after myocardial infarction. Theranostics, 8(16), 4552-4562. https://doi.org/10.7150/thno.24723.
ACS
Tang, T.T.; Li, Y.Y.; Li, J.J.; Wang, K.; Han, Y.; Dong, W.Y.; Zhu, Z.F.; Xia, N.; Nie, S.F.; Zhang, M.; Zeng, Z.P.; Lv, B.J.; Jiao, J.; Liu, H.; Xian, Z.S.; Yang, X.P.; Hu, Y.; Liao, Y.H.; Wang, Q.; Tu, X.; Mallat, Z.; Huang, Y.; Shi, G.P.; Cheng, X. Liver-heart crosstalk controls IL-22 activity in cardiac protection after myocardial infarction. Theranostics 2018, 8 (16), 4552-4562. DOI: 10.7150/thno.24723.
NLM
Tang TT, Li YY, Li JJ, Wang K, Han Y, Dong WY, Zhu ZF, Xia N, Nie SF, Zhang M, Zeng ZP, Lv BJ, Jiao J, Liu H, Xian ZS, Yang XP, Hu Y, Liao YH, Wang Q, Tu X, Mallat Z, Huang Y, Shi GP, Cheng X. Liver-heart crosstalk controls IL-22 activity in cardiac protection after myocardial infarction. Theranostics 2018; 8(16):4552-4562. doi:10.7150/thno.24723. https://www.thno.org/v08p4552.htm
CSE
Tang TT, Li YY, Li JJ, Wang K, Han Y, Dong WY, Zhu ZF, Xia N, Nie SF, Zhang M, Zeng ZP, Lv BJ, Jiao J, Liu H, Xian ZS, Yang XP, Hu Y, Liao YH, Wang Q, Tu X, Mallat Z, Huang Y, Shi GP, Cheng X. 2018. Liver-heart crosstalk controls IL-22 activity in cardiac protection after myocardial infarction. Theranostics. 8(16):4552-4562.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.