Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(17):4620-4632. doi:10.7150/thno.26975 This issue Cite
Research Paper
Senescent fibroblasts drive ageing pigmentation: A potential therapeutic target for senile lentigo
Jung Eun Yoon1,4, Yeongeun Kim2,4, Soohyun Kwon2, Misun Kim2, Young Hwa Kim1, Jang-Hee Kim3, Tae Jun Park1,4,5, ![]() , Hee Young Kang2,4,
, Hee Young Kang2,4, ![]()
1. Department of Biochemistry and Molecular Biology, Ajou University, School of Medicine, Suwon, 443-721, Korea
2. Department of Dermatology, Ajou University School of Medicine, Suwon, 443-721, Korea
3. Department of Pathology, Ajou University School of Medicine, Suwon, 443-721, Korea
4. Department of Biomedical Sciences, Ajou University Graduate School of Medicine, Suwon,443-721, Korea
5. Institute on Ageing, Ajou University Medical Center, Suwon, 443-721, Korea
Received 2018-4-30; Accepted 2018-8-16; Published 2018-9-9
Abstract

Cutaneous ageing is an important extrinsic process that modifies the pigmentary system. Because cellular senescence is a fundamental ageing mechanism, we examined the role of senescent cells in ageing pigmentation.
Methods: Biopsies obtained from senile lentigo and perilesional normal skin were assayed for a marker of cellular senescence, p16INK4A. To determine the secretory phenotypes of senescent fibroblasts, we performed microarray, RNA sequencing and methylation array analyses in senile lentigo and senescent fibroblasts. To further investigate the impact of senescent cells on ageing-related pigmentation, an intervention that targeted senescent cells using radiofrequency was performed.
Results: In vivo, senescent fibroblasts accumulated at the sites of age-related pigmentation. Phenotype switching of the cells resulted in the repression of stromal-derived factor 1 (SDF1) by promoter methylation. SDF1 induced melanocyte differentiation via stromal-epithelial interactions, ultimately driving skin pigmentation. Furthermore, the elimination of senescent fibroblasts from pigmented skin using radiofrequency was accompanied by skin lightening, rendering it a potential target for treatment.
Conclusion: Aged pigmented skin contains an increasing proportion of senescent fibroblasts. Cells with phenotype switching exhibited a loss of SDF1, which stimulates the melanogenic process and thereby contributes to aging pigmentation. These data may promote the development of new therapeutic paradigms, such as a stroma-targeting therapy for pigmentary disorders.
Keywords: Skin pigmentation, senile lentigo, SDF1, senescent fibroblasts
Introduction
Pigmentation is an outcome of the interplay between melanocytes and neighbouring cells, such as keratinocytes and fibroblasts [1]. UV radiation is a well-known stimulus that regulates this intricate process. Several paracrine factors secreted from these cell types upon UV exposure regulate melanogenesis, causing skin tanning and hyperpigmentation [2]. Cutaneous ageing is another important extrinsic process that modifies the pigmentary system. Senile lentigo, also known as age spots, is one of the major changes associated with laxity and wrinkling during the ageing of skin. It is characterized by the presence of hyperpigmented spots in the elderly.
Cellular senescence is a fundamental ageing mechanism. Senescent cells and those with the related senescence-associated secretory phenotype (SASP) are known to be the main drivers of the age-related phenotype [3-5]. During intrinsic and extrinsic skin ageing, the skin can contain senescent cells in epidermal and dermal compartments [6-9]. Cellular senescence has been studied in dermal fibroblasts, which secrete factors that contribute to skin wrinkling [10]. For example, the chronic secretion of matrix metalloproteinases by senescent cells is an important contributor to the degradation of collagen and other extracellular matrix components in dermal tissue [11]. A decrease in the expression of transforming growth factor type II receptor appeared to be a critical event in age-related skin thinning [12]. However, despite the important role exerted by neighbouring cells on the regulation of melanocyte biology, few studies have examined how senescent cells are involved in skin pigmentation [13, 14], and it remains unclear whether senescent cells affect nearby epidermal melanocytes and influence ageing pigmentation.
Here, we show that in vivo, senescent fibroblasts accumulate at the sites of age-related pigmentation and that senescent fibroblasts alter melanocyte differentiation via stromal-epithelial interactions during ageing. We present an integrated study aimed at increasing our understanding of the key roles of senescent fibroblasts and their secretory phenotype in driving ageing pigmentation. These data might also promote the development of new therapeutic paradigms, such as a stroma-targeting therapy for pigmentary disorders.
Methods
Cell culture
Normal human melanocytes, keratinocytes and fibroblasts were isolated from foreskin. In these experiments, melanocytes at passages 2-7 were maintained in F12 medium containing 10% heat-inactivated foetal bovine serum (FBS, Invitrogen, Carlsbad, CA), 24 μg/mL 3-isobutyl-1-methylxanthine, 80 nM 12-O-tetradecanoylphorbor 13-acetate, 1.2 ng/mL basic fibroblast growth factor and 0.1 μg/mL cholera toxin (All from Sigma-Aldrich, St. Louis, MO). Keratinocytes at passages 2-3 were grown in Epilife medium supplemented with human keratinocyte growth supplement (HKGS; Gibco-BRL, Bethesda, MD). Fibroblasts at passages 3-7 were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco-BRL) supplemented with 10% FBS. SDF1-overexpressing or knockdown fibroblasts (3.5×104) were seeded in the inserts of Transwell chambers (Corning, Tewksbury, MA), and melanocytes (1×105) were seeded at the bottom of 6-well plates. After 24 h, the insert chambers were moved into the melanocyte-seeded 6-well plates, and the cultures were maintained in melanocyte culture medium for 5 days. The inserts with fibroblasts were changed to fresh ones at 3 days.
Enzyme-linked immunosorbent assay (ELISA)
Cells (1×105) were seeded in 6-well plates and incubated for 48 h, after which the media were harvested. SDF1 secretion into the cell culture media was measured using SDF1 ELISA kits (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions. To measure cAMP levels (R&D Systems), 1×105 melanocytes were treated with recombinant human SDF1α (rhSDF1, 100 ng/mL, R&D System) for 24 h or infected with an SDF1-containing lentivirus for 2 days and then analysed to determine cAMP levels in the cell lysates. For the forskolin treatments, melanocytes were treated with rhSDF1 (0 to 200 ng/mL) 10 min prior to treatment with forskolin (700 ng/mL) and then cultured for 30 min. They were then analysed to determine the cAMP level. These experiments were performed according to the manufacturer's instructions.
Biopsy collection
For immunohistochemical staining, a retrospective study was performed in 17 patients with facial senile lentigo (SL). All patients were women with Fitzpatrick skin type III or IV, and their average age was 54 years old. Each diagnosis was based on a physical examination and confirmed by histopathological findings. All patients underwent a skin biopsy, and samples (diameter, 2 mm) were obtained from lesional and perilesional normal areas (usually within 20 mm of the lesion margin). For RNA arrays, skin biopsy samples (3 mm) were obtained from the lesional and perilesional normal skin of volunteers. Informed written consent was obtained from each subject. RNA array data were deposited in the Gene Expression Omnibux (GEO, GSE109778).
Institutional review board statement
This study was approved by the institutional review board of Ajou University Hospital (IRB numbers: AJIRB-DEV-DE3-15-491 and AJIRB-BMR-SMP-14-387). All participants were informed of the study goals and procedures, and signed written informed consent forms were obtained prior to their participation.
RNA sequencing
Total RNA was extracted from young, replicative senescent, sham irradiated and UVA-induced senescent fibroblasts using Macherey-Nagel RNA kits (Macherey-Nagel GmbH & Co. KG, Düren, Germany). Briefly, the sample quality was checked using a Bioanalyzer RNA chip (Agilent Technologies) and RNA sequencing was carried out with a Nextseq 500 device (Illumina, San Diego, CA). RNA sequencing data was deposited in SRA (SRP131607 and SRP131659).
Senescence Associated β-Galactosidase (SA-β-Gal) Staining
The cells or frozen tissue slides were fixed with 10% formalin for 1 min and then incubated with SA-β-Gal solution (X-gal, 1 mg/mL; citric acid/sodium phosphate, pH 5.8, 40 mM; potassium ferrocyanide, 5 mM; potassium ferricyanide, 5 mM; NaCl, 150 mM; MgCl2, 2 mM) for 12 h at 37 °C. After PBS washing, SA-β-Gal-positive cells were analyzed under light microscopy.
Sequencing analysis of bisulfite-treated DNA
The human SDF1 promoter region in genomic DNA isolated from young, replicative-senescent, sham-irradiated, UVA-induced senescent fibroblasts, perilesional normal skin and SL skin were sequenced after bisulfite treatment. The genomic DNA (1 μg) was treated with bisulfite, and PCR was then carried out. The PCR conditions were as follows: one cycle at 94 °C for 3 min; 35 cycles at 94 °C for 30 s, 52 °C for 40 s, and 72 °C for 30 s; and one cycle at 72 °C for 3 min. Primers were designed for the SDF1 CpG island promoter (-741 to -477). The forward 5′-GTTTGTGATTAGTTTATTTTATTA-3′ and reverse 5′-CTAAATAAAAACCAATAAAAAAC-3′ sequences were used. The PCR product was ligated into blunt TOPO vectors (Mgmed, Seoul, Korea), and 10 (cells) or 5 (tissues) colonies were extracted and analysed by sequencing for CpG island methylation.
In vitro model of senescent fibroblasts
A primary culture of human dermal fibroblasts (HDFs) was prepared and maintained in our laboratory in DMEM (high-glucose) supplemented with 10% FBS. The number of population doublings (PDs) of HDFs was calculated based on the equation PDs log(A/BC)/log2, where A, B, and C indicate the number of collected cells, the number of plated cells and the attachment efficiency, respectively. The doubling time of HDF was measured with PDs, and the young cells used in this study represent cells with doubling times of approximately 24 h. The doubling time of replicative senescent fibroblasts was 14 days. For the UVA-induced senescent fibroblasts, HDF samples were washed once with PBS and placed in fresh PBS. The cells were treated with 8-methoxy-psoralen (8-MOP; Sigma-Aldrich) at 25 ng/mL for 16 h and irradiated with UVA (wavelength 320-400 nm, maximum peak 350 nm) using a LZC-1 photoreactor system (Luzchem Research Inc. Ontario, Canada). Several doses of UVA were tested according to methods described in previous reports [13, 14], and a dose of 5 J/cm2 was chosen for the experiment because it maintained the fibroblasts in a fully senescent state in which more than 75% show SA-β-Gal staining without cell death. Sham-irradiated HDF was rinsed and placed into an irradiator box without UV irradiation. After irradiation, the cells were maintained in DMEM for 7 days.
Clinical trial of a radiofrequency (RF) treatment
In total, 10 women with facial senile lentigo of Fitzpatrick skin type III or IV were included in the study. The age range was 55-62 years old. The exclusion criteria were prior aesthetic medical procedures or the use of topical depigmenting agents in the three months prior to the study. Lentigo lesions were treated using a pulsed-type RF device in bipolar mode at a frequency of 2 MHz and with a disposable tip consisting of 25 non-insulated, penetrating microneedles in a uniform 5 × 5 array (SYLFIRMTM, Viol, Gyounggi, Korea). In each case, the face was anaesthetized using a topical 4% lidocaine cream (LMX4, Ferndale Laboratories, Inc., Ferndale, MI) approximately 30 min before the procedure. Parameters were set at level 6. The treatment was delivered in a single, non-overlapping pass over the indicated area. Patients underwent 1 treatment session weekly for 6 weeks. Outcome assessments included standardized photography, colorimetric measurements, and histologic analysis. The clinical skin lightening effect was assessed according to a 5-point grading scale to determine the degree of pigmentation (5, very severe; 4, severe; 3, moderate; 2, mild; and 1, very mild) using patient photographs. Skin pigmentation levels were measured using a chromameter (CR-300, Minolta, Japan). The values for lightness are indicated by L*. All measurements were taken three times, and the mean value was used. The lesional and perilesional normal skin samples of participants were analysed at baseline and after 6 weeks in an immunohistochemical study.
Promoter analysis
A reporter construct containing LEF/TCF luciferase was generated in our laboratory. A total of 6 copies of a response element were inserted into the lentivirus pGF1 vector (pGF1-LEF/TCF, System Biosciences, Mountain View, CA) to generate lentivirus particles in HEK 293TN cells. Melanocytes were infected with LEF/TCF or with a control luciferase lentivirus. The cells were treated with 100 ng/mL of human recombinant SDF1α for 12 h, and luciferase activity was then analysed. The human SDF1 promoter (2 kb) was cloned in our laboratory and inserted into a pGF1 vector (pGF1-SDF1, System Bioscience) to generate lentivirus particles in HEK 293TN cells. Young, replicative-senescent, sham-irradiated and UVA-irradiated fibroblasts were infected with the SDF1 promoter or a control luciferase lentivirus for 3 days in the presence of puromycin (3.5 μM), and luciferase activity was then analysed using a Synergy 2 luminometer (BioTek, Winooski, VT) according to the luciferase reporter assay system instructions (Promega, Madison, WI). All luciferase assays were carried out in triplicate.
Ex vivo skin organ culture and pigmentation assays in cultured skin
Normal human skin samples obtained during minor skin surgery were placed on a sterilized stainless-steel grid in a culture dish containing DMEM supplemented with 5% FBS. The senescent fibroblasts were seeded in the bottom of the culture dish. After 3 days of culture in an incubator at 37 °C with 5% CO2, the specimens were fixed in 10% formalin and embedded in paraffin before they were cut into sections. Melanin pigments were detected with Fontana-Masson staining. An image analysis was performed using Image Pro Plus Version 4.5 (Media Cybernetics Co., Rockville, MD), and the pigmented area per epidermal area (PA/EA) was measured.
Melanin content and tyrosinase activity assay
The cells were lysed with a 0.1 M phosphate buffer (pH 6.8) containing 1% Triton X-100 with a protease inhibitor cocktail (Roche, Basel, Switzerland). The supernatants were measured to determine the protein concentration using the Lowry assay system. Pellets were solubilized in 100 μL of 1 N NaOH for 3 h at 60 °C and the absorbance was measured at 490 nm to determine the melanin content relative to a standard curve using synthetic melanin (Sigma-Aldrich). For the assay of the tyrosinase activity, each sample was incubated with 2 mM L-DOPA (Sigma-Aldrich) in a 0.1 M phosphate buffer (pH 6.8) for 90 min at 37 °C. After incubation, the tyrosinase activity was measured at 490 nm.
Lentivirus and adenovirus production
The human SDF1 cDNA was cloned from normal fibroblasts in our laboratory. cDNA was inserted into the pCDH-CMV-MCS-EF1-Puro lentivirus vector (System Biosciences). For the knockdown of SDF1 expression, shRNA was prepared in a pLKO lentiviral vector (Sigma-Aldrich). Fibroblasts were plated and grown in 6 cm culture dishes. After culturing overnight, they were infected with the lentivirus and the cells were then selected with 3.5 μM puromycin for one week. The shRNA sequences were as follows: sh-SDF1 #1: 5′-CGCCAACGTCAAGCATCTCAAA-3′; sh-SDF1 #2: 5′-ACATCTCAAAATTCTCAACACA-3′. To generate lentiviral particles, HEK-293TN cells were transfected with plasmid DNA (pGag-pol, pVSV-G, and pCDH-SDF1 or shSDF1) using Lipofectamine (Invitrogen). Viral supernatant was collected after 48 h and transduced into normal human melanocytes and fibroblasts. cDNAs of wild-type p53 were inserted into replication-defective E1- and E3-adenoviral vectors containing a cytomegalovirus enhancer and a chicken β-actin promoter. Adenoviruses of p53 were then amplified in 293 human kidney epithelial cells, and the virus particles were purified by filtration (0.45 μm). Similarly, an adenovirus of bacterial β-galactosidase (Ad-LacZ) was also prepared as a control.
Real-time PCR analysis
First-strand cDNA was synthesized by a reverse-transcription reaction using oligo-dT primers from 1 μg of total cellular RNA (Thermo Fisher Scientific, Waltham, MA). Real-time PCR was carried out with the Power SYBR Green PCR Master Mix (Bio-Rad, Hercules, CA) using the following conditions: initial activation at 95 °C for 5 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The primers used for real-time PCR as follows: MITF: 5'-AGAACAGCAACGCGCAAAAGAAC-3', 5'-TGATGATCCGATTCACCAAATCTG-3', Tyrosinase: 5'-CACCACTTGGGCCTCAATTTC-3', 5'-AAAGCCAAACTTGCAGTTTCCAC-3', SDF1: 5'-TGCCAGAGCCAACGTCAAG-3', 5'-CAGCCGGGCTACAATCTGAA-3', CXCR4: 5'-GCCTTATCCTGCCTGGTATTGTC-3', 5'-GCGAAGAAAGCCAGGATGAGGAT-3', human 18S: 5'-CGGCTACCACATCCAAGGAA-3', 5'-GCTGGAATTACCGCGGCT-3', β-actin: 5'-CCCTGGCACCCAGCAC-3', 5'-GCCGATCCACACGGAGTAC-3', Apol6: 5'-CAGATTTGCTGCCACAGAG-3', 5'-GTGACATAGTCTGCCTTCTC-3', Col5a3: 5'-TTCAGCTCTTCTCGAGCGGGATTT-3', 5'-TCAAAGCCTCAGCACCAAATGCAC-3', LIF1: 5'-TATCACCATCTGTGCCTTTGCTGC-3', 5'-TCTGCCAGATTGTTCCTATGCCCA-3', p16INK4A: 5'-CCCAACGCACCGAATAGTTA-3', 5'-ACCAGCGTGTCCAGGAAG-3'.
Western blot analysis
Cells were lysed in RIPA buffer (1% NP-40, 150 mM NaCl, 10 mM Tris-HCl at pH 8.0, 1 mM EDTA) with a complete protease inhibitor (Sigma-Aldrich). The proteins were separated by SDS-polyacrylamide gel and transferred to PVDF membranes (Millipore, Billerica, MA). The antibody against MITF was purchased from Abcam (ab12039, Cambridge, UK), those for pCREB (9191s) and CREB (9197s) were purchased from Cell Signalling Technology (Danvers, MA), and the antibodies for tyrosinase (sc-7833) and p53 (sc-126) were purchased from Santa Cruz Biotechnology (Dallas, TX).
Immunocytochemistry and immunohistochemical analysis
For immunocytochemistry, cells grown in Lab-Tek chambers (Nalge Nunc International, Rochester, NY) were fixed in 4% paraformaldehyde for 15 min at room temperature and permeated with 0.2% Triton X-100. Non-specific antibody binding was blocked by 1% BSA for 1 h, and the cells were then incubated with adequate primary antibodies overnight at 4 °C. All fluorescence photographs were taken using a fluorescence microscope (Carl Zeiss, Oberkochen, Germany). Immunohistochemical staining was performed with primary antibodies on 4 μm-thick representative tissue sections obtained from formalin-fixed paraffin-embedded tissue samples in a Benchmark XT automated immunohistochemistry stainer (Ventana Medical Systems Inc., Tucson, AZ). The primary antibodies used were as follows: p16INK4A, predilution (725-4713, Ventana Medical Systems, Inc.); anti-human Ki67 antigen, clone MIB-1, 1:100 (Dako Denmark A/S, Glostrup, Denmark); CXCR4, 1:100 (MAB172, R&D System); SDF1, 1:100 (MAB350, R&D System); p53, 1:1000 (sc-126, Santa Cruz); cleaved caspase 3, 1:1000 (9661s, Cell Signaling); LIF1, 1:1000 (AF-250, R&D System); vimentin, 1:100 (ab92547, Abcam); FSP-1, 1:100 (ab27957, Abcam); and procollagen type 1, 1:50 (M-38, DSHB, Iowa city, IA). Detection was performed using a Ventana Optiview DAB Kit (Ventana Medical Systems). Immunohistochemical staining was scored by an experienced pathologist (JHK). For the SDF1 or procollagen expression analysis, SDF1- or procollagen-positive fibroblasts in two of the most highly labelled areas (hot fields) under 400x magnification were counted and graded as none (no SDF1- or procollagen-positive cells), weak (1-10 positive cells), moderate (11-30 positive cells), and strong (more than 30 positive cells).
Microscope image acquisition
Image acquisition during the histology and immunohistochemical staining processes was performed using a ScanScope® CS system (Aperio Technologies, Inc., Vista, CA) at room temperature. The cell images were acquired using an Olympus microscope mounted onto an Olympus DP70 digital camera with DP-Manager software (Olympus Microscope Corp., Tokyo, Japan) at room temperature. Immunofluorescence images were collected on a Zeiss LSM 510 microscope and analyzed with Zeiss Axio Imager software (Carl Zeiss) at room temperature.
TUNEL assay
We prepared 4 μm-thick representative tissue sections of formalin-fixed paraffin-embedded tissue sections. After they were deparaffinised, the slides were fixed with 4% buffered formaldehyde. For TUNEL labelling, a DeadEndTM Fluorometric TUNEL system (Promega) was used according to the manufacturer's instructions.
BrdU corporation assay
Young, senescent, SDF1-overexpressing or shSDF1-infected fibroblasts (1×104) were seeded in Transwell chambers (Corning), and melanocytes (3.5×104) were seeded at the bottom of 24-well plates. The proliferation of melanocytes was analysed with a CytoSelect™ bromodeoxyuridine (BrdU) Cell Proliferation Elisa Kit (Cell Biolabs Inc., San Diego, CA). BrdU incorporation was measured according to the manufacturer's instructions.
Statistical analysis
Data are presented as the mean ± SD of independent determinations and were analysed using Wilcoxon or paired Student's t-tests with a p value < 0.05 considered significant. The statistical analysis of SDF1 staining in SL was performed using the Chi-square test (χ2 test). IBM SPSS ver. 23 (IBM Corp., Armonk, NY) was used for all statistical analyses.
Results
Cellular senescence in ageing pigmented skin occurs in dermal fibroblasts
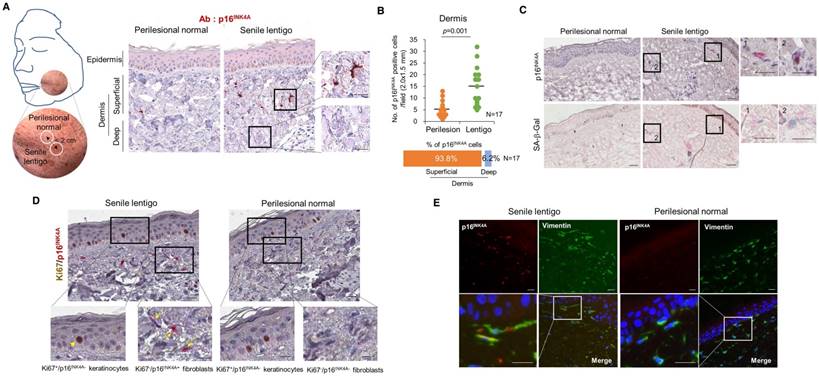
To characterize cellular senescence in ageing pigmented skin, biopsies obtained from the senile lentigo (SL) and perilesional normal skin of 17 volunteers were assayed for a marker of cellular senescence, p16INK4A [15]. A significantly higher number of p16INK4A-positive cells were observed in SL dermal skin than in perilesional normal skin (Figure 1A-B and Figure S1). The senescent cells were located near the dermo-epidermal junction, including the superficial dermis, but were rarely detected in the deep dermis (93.8 vs. 6.2%) (Figure 1A). Perilesional normal skin contained fewer p16INK4A-positive cells than were observed in lesional skin but showed a similar distribution, with 92.3% of the positive cells in the superficial dermis. An in vivo marker of senescence, senescence-associated galactosidase (SA- β-Gal), was also observed in the corresponding p16INK4A-positive cells (Figure 1C). Because cellular senescence is characterized by stable cell cycle arrest, Ki67 immunostaining was performed and demonstrated an absence of proliferation in the p16INK4A-positive cells (Figure 1D). Ki67-positive cells were observed in the epidermis in both SL and perilesional normal skin samples. Double-immunostaining for p16INK4A/vimentin or p16INK4A/fibroblast-specific protein 1 (FSP1) revealed that the senescent cells were fibroblasts (Figure 1E and Figure S2). These results indicate that ageing pigmented skin is characterized by the accumulation of senescent fibroblasts.
Cellular senescence in ageing pigmented skin. (A) Biopsies obtained from the facial senile lentigo (SL) and perilesional normal skin of 17 volunteers were immunostained for p16INK4A. The proportion of p16INK4A-positive cells is shown according to the depth of the skin. The bar graph indicates the percentage of p16INK4A-positive cells in the dermis. The superficial dermis is defined as cells within 500 μm of the epidermal-dermal junction. (B) The number of p16INK4A-positive cells is presented in a dotted graph. Lines in the graph indicate the mean values. (p value, Wilcoxon test). (C) Frozen tissues were stained with SA-β-Gal/nuclear fast red (NFR) or for p16INK4A (n=4). (D) Ki67 and p16INK4A co-immunostaining. Arrowheads and arrows indicate Ki67+/p16INK4A- keratinocytes and Ki67-/p16INK4A+ fibroblasts, respectively. (E) Immunostaining for vimentin and p16INK4A (n=6) (scale bars, 50 μm).
Senescent fibroblasts promote skin pigmentation
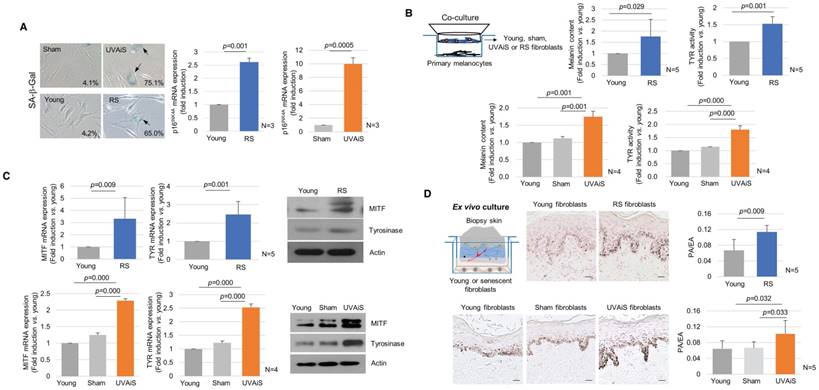
To investigate the regulatory role played by senescent fibroblasts in skin pigmentation, in vitro models of senescent fibroblasts, i.e., replicative-senescent (RS) and UVA-induced senescent (UVAiS) fibroblasts, were established (Figure 2A) [13, 14, 16]. The senescent fibroblasts were then co-cultured with normal human melanocytes using Transwell plates. Melanin levels and tyrosinase activity were significantly higher in the senescent fibroblasts than in the controls (young or sham-irradiated fibroblasts). In addition, the mRNA and protein expression levels of melanogenesis-associated proteins, microphthalmia-associated transcription factor (MITF), and tyrosinase were significantly upregulated (Figure 2B-C). Melanocyte proliferation was unaffected by the senescent fibroblasts (Figure S3). The effects of the senescent fibroblasts on pigmentation were further evaluated using ex vivo human skin samples. Fontana-Masson staining that epidermal pigmentation was significantly increased by the presence of senescent fibroblasts (Figure 2D). An increase in the pigmented area/epidermal area (PA/EA) ratio was observed in an image analysis. Taken together, these data indicate that senescent fibroblasts promote skin pigmentation.
Senescent fibroblasts promote skin pigmentation. (A) Replicative senescent (RS) and UVA-induced senescent (UVAiS) fibroblasts. Primary human fibroblasts were sub-cultured for 6 months or irradiated with 5 J/cm2 UVA or sham for 7 days. Two-hundred cells were counted in each experiment, and the number of SA-β-Gal positive fibroblasts (arrow) is presented as a percentage (left, n=5). Real-time PCR analysis of p16INK4A mRNA levels (right). (B) Melanocytes were co-cultured with senescent fibroblasts (RS or UVAiS) or control cells (young or sham). Melanin content and tyrosinase (TYR) activity were measured. (C) mRNA and protein expression levels of MITF and tyrosinase. (D) Pigmentation of ex vivo human skin visualized with Fontana-Masson staining (Left). The pigmented area/epidermal area (PA/EA) ratio was measured by image analysis (Right). Data are presented as the mean ± SD (p value, t-test) (scale bars, 50 μm).
Senescent fibroblasts exhibit SDF1 deficiency resulting from promoter methylation
To identify the secretory phenotype of the senescent fibroblasts that promoted pigmentation, a gene expression profiling analysis was performed using RNA sequencing. Among the 43 genes that were differentially expressed between the senescent fibroblasts and controls, 4 encoded secreted proteins: senescent fibroblasts expressed higher levels of LIF1 and lower levels of COL5A3, APOL6 and SDF1 than were observed in normal fibroblasts (Table 1). The corresponding mRNA expression levels were detected by real-time PCR (Figure S4). The full RNA sequencing data are available at the Sequencing Read Archive (SRA; SRP131607 and SRP131659). Of these, we selected stromal derived factor-1 (SDF1) as an intriguing candidate gene. SDF1, also known as C-X-C motif chemokine ligand 12 (CXCL12), is a widely expressed constitutive chemokine that regulates tissue homeostasis and inflammatory responses and is the only known ligand for C-X-C chemokine receptor type 4 (CXCR4) [17-19]. While no reports have directly linked SDF1 to pigmentation, SDF1 and its receptor CXCR4 are known to inhibit the cAMP signalling pathway, and cAMP signalling is also known to be involved in the production of melanin [20]. Thus, we investigated whether SDF1 may be one of the proteins responsible for age-related pigmentation.
RNA sequencing analysis of senescent fibroblasts (RS or UVAiS) and controls (young or sham irradiated). Values indicate the averages of 3 independent cases of each cell and are presented as the Log2 ratio compared to the controls (p<0.05).
| Genes | Log2 Ratio (Young vs. RS) | Log2 Ratio (Sham vs. UVAiS) |
|---|---|---|
| APLP1 | 1.65 | 3.10 |
| BTG2 | 2.01 | 3.05 |
| C12orf5 | 1.47 | 1.97 |
| C1QTNF1 | 1.80 | 2.17 |
| CYFIP2 | 2.83 | 4.22 |
| DCBLD2 | 1.58 | 1.86 |
| FUCA1 | 3.11 | 3.11 |
| GDF15 | 2.39 | 5.47 |
| GM2A | 2.16 | 2.20 |
| HIST1H2AC | 2.67 | 3.24 |
| HIST1H2BK | 2.11 | 2.63 |
| HIST2H2BE | 2.51 | 2.74 |
| LIF1 | 2.94 | 3.26 |
| MDM2 | 1.41 | 2.31 |
| MTRNR2L8 | 1.54 | 1.67 |
| PARD6G | 2.85 | 3.07 |
| PSG2 | 2.43 | 3.21 |
| SLC14A1 | 4.16 | 3.59 |
| TNFRSF10C | 2.45 | 5.98 |
| TNFRSF10D | 1.93 | 2.04 |
| WDR63 | 2.49 | 3.61 |
| ANLN | -1.21 | -5.54 |
| APOL6 | -1.17 | -3.28 |
| ASPM | -1.79 | -5.50 |
| BIRC5 | -1.99 | -5.40 |
| C1R | -2.28 | -3.19 |
| CDC20 | -2.52 | -5.72 |
| CDKN2C | -1.46 | -3.52 |
| CENPF | -1.59 | -5.30 |
| COL5A3 | -1.90 | -3.39 |
| SDF1 | -3.55 | -2.47 |
| HR | -3.43 | -2.98 |
| KIF18B | -1.62 | -4.76 |
| KIF2C | -2.01 | -5.27 |
| KIFC1 | -2.12 | -5.05 |
| NUSAP1 | -1.44 | -5.82 |
| OLFML2B | -1.57 | -3.37 |
| PHGDH | -1.80 | -2.39 |
| TENM3 | -3.84 | -2.17 |
| TK1 | -2.36 | -4.90 |
| TMPO | -1.29 | -3.34 |
| TOP2A | -1.62 | -5.67 |
| TPX2 | -1.58 | -3.60 |
| WNT2 | -6.64 | -3.11 |
The expression of the SDF1 mRNA was suppressed in senescent fibroblasts but present at high levels in normal fibroblasts (Figure 3A). ELISA data showed that normal fibroblasts but not senescent fibroblasts secreted a considerable level of SDF1 (Figure 3B). To further clarify SDF1 downregulation in vivo, biopsies obtained from SL and perilesional normal skin were assayed for SDF1 expression. Immunohistochemical staining detected very low levels of the SDF1 protein in a few fibroblasts obtained from the SL, whereas fibroblasts obtained from perilesional normal skin contained high levels of SDF1 expression (Figure 3C and Figure S5-6). Consistent with these results, the expression of the SDF1 mRNA was significantly lower in SL than in perilesional normal skin (Log2 ratio, -0.527) (Table 2). These data indicate that in ageing pigmented skin, senescent fibroblasts are deficient in SDF1. We also characterized the expression of 3 other secreted proteins identified by RNA sequencing (LIF1, COL5A3, and APOL6) and genes known to control melanogenesis, such as melanocyte stimulating hormone (α-MSH), stem cell factor (SCF), and TGFβ, but none of these genes were differentially expressed. It is likely that relative weak changes in the LIF1, APOL6 and COL5A3 transcript expressions in SL skin samples compared to senescent fibroblasts are most likely due to their expressions in keratinocytes (Figure S4). However, collagen-related genes (COLA1, COLA2 and COL3A1) were down-regulated, as expected. The full microarray data are available at GEO (GSE109778).
RNA array analysis of senile lentigo (SL) (n=2). Value indicates the fold induction compared to perilesional normal skin.
| Anti-melanogenic Genes | Fold induction (Lentigo vs. normal) | Melanogenic Genes | Fold induction (Lentigo vs. normal) |
| TGFβ1 | 1.103 | SCF | 0.904 |
| IL-6 | 1.036 | Edn1 | 0.990 |
| IL-1β | 0.988 | FGF1 | 0.979 |
| IL-4 | 1.033 | BMP6 | 0.940 |
| DKK1 | 1.017 | HGF | 1.027 |
| PTN | 0.969 | NRG-1 | 1.055 |
| TNF-α | 0.981 | CSF2 | 0.917 |
| IL-10 | 1.050 | LIF | 1.111 |
| Genes | Fold induction (Lentigo vs. normal) | Collagen genes | Fold induction (Lentigo vs. normal) |
| SDF1 | 0.694 | Col1a1 | 0.701 |
| Col5a3 | 1.090 | Col1a2 | 0.627 |
| APOL6 | 1.180 | Col3a1 | 0.672 |
Senescent fibroblasts exhibit SDF1 deficiency caused by altered DNA methylation. (A) Real-time PCR and (B) ELISA analyses of SDF1. N.D.: not detected. (C) SDF1 immunostaining of SL (scale bars: upper, 100 μm; lower, 50 μm). The expression levels were graded as none, weak, moderate, or strong (p value, χ2 test). (D) SDF1 promoter methylation analysis in senescent fibroblasts (RS and UVAiS). The CpG islands (-741 to -477) in the human SDF1 promoter were analysed in ten clones from each cell. Blue circles indicate an unmethylated status, and red circles denote methylation. The methylation % of each CpG island is presented, with red numbers indicating a more than a 1.5-fold increase in CpG island methylation. (E) Real-time PCR analysis of SDF1 mRNA levels in the presence of 5-aza-deoxycytidine (5-Aza). (F) DNA methylation status of the SDF1 promoter in SL patients (n=5). Pt # indicates the patient number.
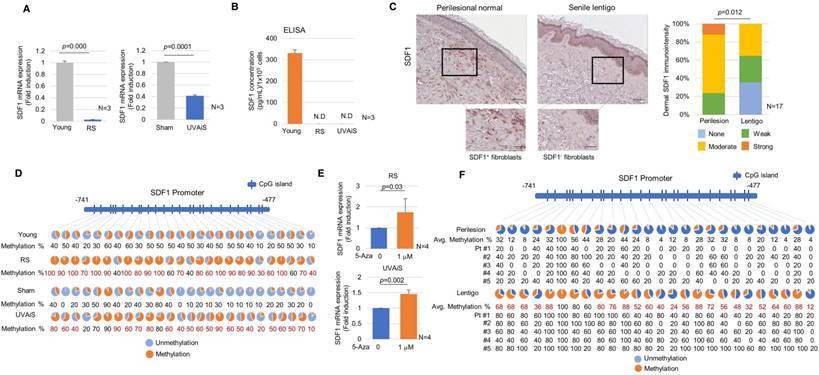
SDF1 promoter activity was lower in senescent fibroblasts than in control fibroblasts, indicating that SDF1 expression is transcriptionally regulated (Figure S7). It was previously reported that ageing causes distinct epigenetic changes in human skin and that age-related changes in DNA methylation contribute to the phenotypic changes associated with skin ageing [21-24]. DNA methylation is also recognized as an important mechanism involved in the regulation of SDF1 expression [25]. We therefore evaluated whether the suppression of the SDF1 gene observed in senescent fibroblasts might be due to DNA methylation. An analysis of the DNA methylation profiles of senescent fibroblasts revealed several points of CpG island methylation on the SDF1 promoter site (Figure S8). A subsequent methylation-specific PCR analysis performed after bisulfite treatment showed that the CpG island methylation of the SDF1 promoter was markedly reduced in senescent fibroblasts (Figure 3D). The reduced SDF1 expression observed in the senescent fibroblasts was restored in the presence of a demethylating agent, 5-aza 2-deoxycytidine (5-Aza) (Figure 3E). DNA methylation of the SDF1 promoter was also observed in ageing pigmented skin. A methylation-specific PCR analysis showed that the level of methylation of the SDF1 promoter was significantly increased in SL (Figure 3F). SDF1 expression is regulated by several types of transcription factors, such as p53, NF-κB, SP1 and AP1, and p53 and NF-κB, which are commonly upregulated in senescent cells. p53 was found to suppress SDF1 production in cultured stromal fibroblasts, while NF-κB is an inducer of SDF expression [26, 27]. Therefore, we also evaluated the ability of p53 and NF-κB to regulate SDF1 transcription in senescent fibroblasts (Figure S9). However, these data were excluded because the dermal fibroblasts in SL skin samples rarely expressed p53 and NF-κB was activated in senescent fibroblasts. Taken together, these findings indicate that in ageing-related pigmentation, senescent fibroblasts exhibit SDF1 deficiency as a result of changes in DNA promoter methylation.
SDF1 deficiency in senescent fibroblasts drives skin pigmentation
The biological role of SDF1 in controlling skin pigmentation was then investigated. The endogenous expression of SDF1 and its receptor CXCR4 was examined in skin cells in vitro and in vivo (Figure 4A-B). Among the tested cells, the mRNA and protein expression of SDF1 was strongest in fibroblasts (Figure 4A). Melanocytes scarcely expressed SDF1 and instead expressed CXCR4 (Figure S10), indicating the possibility of paracrine crosstalk between fibroblasts and melanocytes via SDF1. SDF1/vimentin double-positive fibroblasts and melanocytes expressing SDF1 and CXCR4 were consistently observed in normal human skin in vivo (Figure 4B). Keratinocytes expressed only low levels of SDF1 or CXCR4 in vitro or in vivo.
Effects of SDF1 on skin pigmentation. (A) SDF1 and CXCR4 expression in cultured melanocytes (MC), keratinocytes (KC) and fibroblasts (FB). Real-time PCR (left upper), ELISA (left lower), and immunocytochemical (right, scale bar, 10 μm) analyses. N.D., not detected. (B) Immunohistochemical staining performed in normal human skin. SDF1 (red)/vimentin (green) double-immunostained fibroblasts and CXCR4 (green)-/MITF (red)-stained melanocytes were visualized (scale bars, 50 μm). (C) Pigmentation in melanocytes co-cultured with fibroblasts infected with shSDF1 lentivirus or controls. (D) Pigmentation in melanocytes co-cultured with SDF1-overexpressing fibroblasts. (E) Effect of treatment with recombinant human SDF1α (rhSDF1, 100 ng/mL) in melanocytes. (F) Real-time PCR analysis of MITF and tyrosinase mRNA expression in cells incubated in the presence of a CXCR4 inhibitor (AMD3100). Melanocytes were cultured with conditioned media (CM) obtained from young or RS fibroblasts incubated with/without 1 μM AMD3100 for 2 days. The CM-treated cells were then analysed for MITF and tyrosinase mRNA expression. (G) Pigmentation of ex vivo human skin treated with rhSDF1 (scale bar, 100 μm). (H) SDF1 overexpression in senescent fibroblasts reversed the stimulatory effects of the cells on melanogenesis. (I) Expression of phospho-CREB (upper) and cAMP formation (lower) in melanocytes treated with an rhSDF1- or SDF1-expressing lentivirus. Data are shown as the mean ± SD (p value, t-test). N.S. indicates no significance.
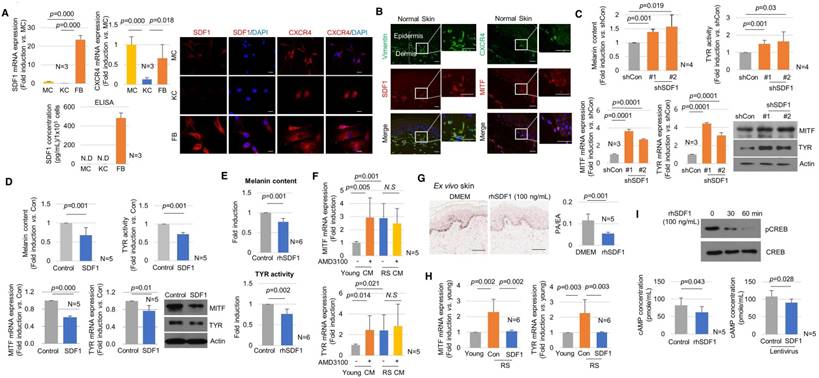
To investigate the paracrine effect of fibroblast-derived SDF1 on pigmentation, melanocytes were co-cultured with fibroblasts infected with the sh-SDF1 or SDF1 lentivirus using Transwell plates (Figure S11). Melanin levels and tyrosinase activity were higher in the SDF1-knockdown fibroblasts than in the controls. The mRNA and protein expression levels of MITF and tyrosinase were significantly upregulated. Consistent with these results, SDF1-overexpressing fibroblasts exhibited decreased melanogenesis (Figure 4C-D). Inducing SDF1 overexpression or knockdown in fibroblasts did not affect melanocyte proliferation (Figure S12). To further investigate the effects of exogenous SDF1 on melanocytes, melanocytes were treated with recombinant human SDF1α (rhSDF1, 100 ng/mL). Exogenous SDF1 reduced pigmentation (Figure 4E), and this effect was reversed by treatment with a selective CXCR4 antagonist, AMD 3100 (Figure S13). We further confirmed that SDF1 acts as a paracrine factor in melanocytes. Conditioned media (CM) was obtained from young or RS fibroblasts incubated with melanocytes in the presence or absence of AMD3100. The mRNA expression levels of MITF and tyrosinase were higher in melanocytes treated with AMD3100 and CM obtained from young fibroblasts. However, the senescent fibroblast-derived CM exerted no effect (Figure 4F). Finally, the inhibitory effect of SDF1 on cutaneous pigmentation was explored in ex vivo human skin samples. In samples grown in the presence of rhSDF1, the amount of epidermal pigmentation was significantly reduced, as shown by Fontana-Masson staining (Figure 4G). Furthermore, in senescent fibroblasts, SDF1 overexpression reversed their stimulatory effects on melanogenesis (Figure 4H). To determine the molecular mechanism by which SDF1 inhibited melanogenesis, we next examined the SDF1/cAMP signalling pathway [18]. Treatment with rhSDF1 was associated with decreased levels of CREB phosphorylation (Figure 4I, upper panel). Consistent with these results, treatment with rhSDF1 or SDF1 overexpression decreased cAMP formation (Figure 4I, lower panel). Furthermore, rhSDF1 decreased forskolin-induced cAMP levels (Figure S14). These data suggest that SDF1 exerts an anti-melanogenic function by down-regulating cAMP/pCREB/MITF/tyrosinase signalling in melanocytes.
The elimination of senescent fibroblasts induces skin lightening
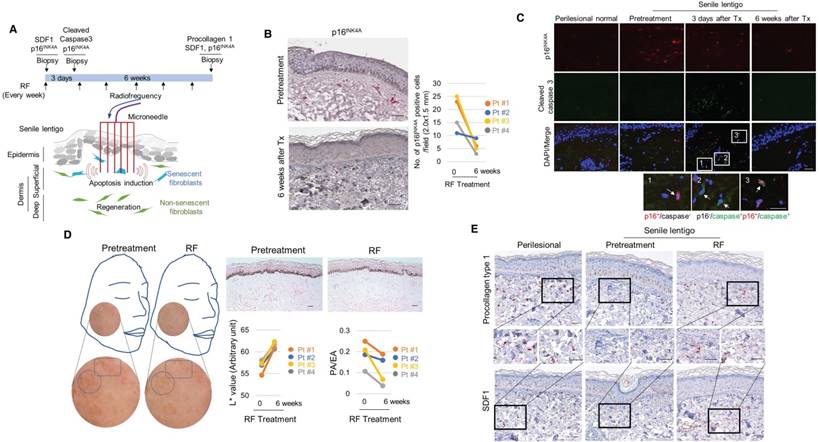
To further investigate the impact of senescent cells on ageing-related pigmentation, we performed an intervention that targeted senescent cells. Microneedle fractional radiofrequency (RF) is a cosmetic therapy that induces skin rejuvenation via electromagnetic thermal injury [28, 29]. The microneedle RF device was chosen to manipulate only dermal cells, in which the microneedles generate thermal coagulation columns in the dermis, not in the epidermis [30]. It was previously demonstrated that fractional laser treatment decreases the occurrence of senescent fibroblasts in aged dermis [31]. Ten volunteers with SL were treated with RF, and skin samples were collected from 4 participants who agreed to undergo a skin biopsy before and at 6 weeks after treatment (Figure 5A). Following RF treatment, the number of senescent fibroblasts was significantly reduced (Figure 5B). The elimination of these cells was thought to be caused by RF-induced cell death. On day 3, cleaved caspase 3- and TUNEL-positive cells were observed (Figure 5C and Figure S15). The elimination of senescent fibroblasts from SL was accompanied by skin lightening. The L* (lightness) value measured by a chromameter were higher than those obtained at baseline, and histological evaluations consistently revealed a marked decrease in the epidermal pigmentation compared to baseline levels (Figure 5D). RF treatment increased the synthesis of collagen and restored SDF1 expression to levels comparable to those observed in perilesional normal skin (Figure 5E and Figure S16-17). These findings indicate that senescent fibroblasts play a crucial role in ageing-related pigmentation. They provide further support for the therapeutic potential of eliminating senescent cells and restoring SDF1 to correct uneven pigmentation.
The elimination of senescent fibroblasts induces skin lightening. (A) Scheme of the intervention performed to target senescent fibroblasts. Ten volunteers with senile lentigo (SL) underwent radiofrequency (RF) treatment for 6 weeks. Matched biopsies obtained from 4 individual subjects in whom a pair (SL and perilesional normal) of samples were obtained from untreated facial tissues and a second pair of samples were obtained from facial tissue treated with RF were assayed to determine the expression profiles of p16INK4A (B) and cleaved caspase 3 (C) by immunohistochemistry. (D) Skin lightness was assessed using L* (lightness) values measured by a chromameter and using histological evaluation. Circles and quadrangles indicate SL lesions (left panel). The pigmented area/epidermal area (PA/EA) ratio was measured by image analysis (right lower). (E) SL and perilesional normal tissue obtained from facial tissues that were untreated and treated with RF were assayed to determine the expression levels of SDF1 and procollagen type 1 by immunohistochemistry (scale bars: 50 μm).
Discussion
Melanogenesis, the formation of melanin pigment, is regulated by cytokines and growth factors in a highly orchestrated manner [1]. In this study, we reveal what we believe is a novel mechanism whereby aged fibroblasts contribute to the local regulation of melanogenesis. We show that as an individual ages, pigmented skin contains an increasing proportion of senescent fibroblasts. Phenotype switching in these cells results in the loss of SDF1, and SDF1 deficiency appears to be a potent stimulus for the melanogenic processes that contribute to uneven pigmentation. These changes might be epigenetic. For example, the level of hypermethylation of the SDF1 promoter was remarkably different between hyperpigmented and perilesional skin. Although the mechanism that drives the initiation of this epigenetic change is not well understood, natural and photo-ageing-related epigenetic changes are known to contribute to the phenotypic changes associated with skin ageing [21-24].
The human skin, unlike other organs, undergoes photo-ageing in addition to natural ageing processes, and photo-ageing has been attributed to ageing pigmentation [24]. Both processes are cumulative, and the most noticeable age-related changes therefore occur in the superficial layer of the skin. In the present study, we show that cellular senescence is especially likely to occur in fibroblasts located in the upper dermis of pigmented skin. Senescent fibroblasts are expected to influence melanocytes via cross-talk that can readily occur through a damaged basement membrane [32]. We showed that senescent fibroblasts play a stimulatory role in pigmentation by upregulating the expression of the melanogenesis regulators MITF and tyrosinase in melanocytes, in agreement with previous results [13, 14]. Moreover, the impact of senescent fibroblasts on skin pigmentation was directly demonstrated when eliminating senescent cells with an intervention that reduced pigmentation. Together, these findings suggest that crosstalk occurs between melanocytes and senescent fibroblasts during the ageing process and that this crosstalk plays an important role in the stimulation of melanogenesis and subsequent ageing-related pigmentation.
In previous studies, facial SL showed flattened rather than elongated rete ridges [33, 34], and 10 of 25 examined samples contained p16INK4A-positive keratinocytes [34]. In the present study, p16INK4A-positive keratinocytes were observed in 5 of 17 samples. The number of p16INK4A-positive keratinocytes did not appear to be correlated with the number of p16INK4A-positive fibroblasts. It has been suggested that SL is most likely initiated by keratinocyte proliferation followed by quiescence in enlarged senescent keratinocytes with a higher melanin burden [34, 35]. According to the literature, the increased expression of pigmentary proteins is consistently observed regardless of the degree of progression in lentigo [35]. We therefore speculate that SL is a disorder that affects both melanocytes and keratinocytes in which melanocyte activation may be controlled by intricate interactions between melanocytes and senescent fibroblasts during the ageing process. Keratinocyte senescence may contribute to the increased epidermal thickness observed in SL cases. Nevertheless, it should be noted that we could not completely rule out the notion that senescent keratinocytes play a role in controlling pigmentation, and additional investigation will be required to resolve this issue.
An increasing amount of data in the literature are focused on the important role of melanogenic factors secreted by senescent phenotype fibroblasts in favouring hyperpigmentation linked to ageing [13, 14, 35, 36]. In particular, the ability of these cells to release high amounts of pro-pigmenting factors, such as hepatocyte growth factor (HGF), keratinocyte growth factor (KGF), and SCF could act in promoting pigmentation. Moreover, the crucial role of these factors in the development of age spots has been demonstrated in immunohistochemical studies showing positive reactivity for KGF and/or HGF [14, 35, 37]. However, in our RNA sequencing data, none of these factors appeared to have been significantly modulated in senescent cell models. The expression levels of the melanogenic factors HGF and KGF varied and were not consistent between replicative and UVA-induced senescent fibroblasts. Instead, only the LIF1 transcript was significantly upregulated in all samples. Moreover, many kinds of genes were differentially expressed in age spots, but melanocyte pigmentation-related genes were not among them (array data deposited in GSE109778), consistent with previous observations [38]. These results led us to hypothesize that the hyperpigmentation in age spots results from a decrease in the expression of the anti-melanogenic factor SDF1.
In the skin, dysregulation of SDF1 signalling was previously linked to autoimmune and chronic inflammatory dermatological disorders, such as psoriasis, atopic dermatitis and lupus [19]. Studies of vitiligo performed in animal models have shown that melanocyte-derived SDF1 plays an important role in the activation of melanocyte-specific autoimmunity and results in continuous loss of melanocytes [39]. The results of the present study suggest that beyond the case of vitiligo, in which an immune response is thought to be responsible for the destruction of normal melanocytes, SDF1 plays an inhibitory role in controlling cutaneous pigmentation in vivo. Among the many cytokines that affect melanocytes, several (TGFβ1, IFN-γ, DKK1, and clusterin) are known to have hypopigmentation effects and to be capable of independently modulating the expression levels of tyrosinase and related enzymes [40, 41]. We found that normal fibroblasts secrete considerable amounts of SDF1 and SDF1 inhibits pigmentation in melanocytes expressing CXCR4. In ageing skin, SDF1 levels were markedly reduced, and SDF1-deficient ageing skin showed increased pigmentation. Moreover, the restoration of SDF1 after RF treatment was accompanied by skin lightening. It is also possible that the ability to secrete SDF1 is altered in senescent cells (Figure 3A-B). The in vivo role of fibroblast-derived SDF1 in cutaneous pigmentation requires further investigation. Nevertheless, the inhibitory actions of SDF1 during the control of pigmentation suggest that SDF1 is a possible therapeutic target for the development of skin-lightening agents that can treat hyperpigmentation. As SDF1 expression was found to be epigenetically controlled and epigenetic modifications, such as DNA methylation, can be reversed, treatments for senile lentigo that use epigenetic “drugs” could be explored.
It should be noted that the cell culture model of senescent fibroblasts used in our study may not necessarily represent the cells that actually impact melanogenesis in the facial area as the fibroblasts were obtained from the foreskins of neonates or young adults with skin phototype III or IV. To induce replicative senescence, dermal fibroblasts were cultured at a high serum concentration to induce cell proliferation. This would not normally be observed in vivo, in which these cells are in general quiescent [42]. Indeed, the ageing process dermal fibroblasts undergo in their physiological tissue environment has only been partially defined in in vitro aged dermal fibroblasts [43]. Therefore, the specific and detailed roles of known melanogenic factors in promoting pigmentation independent of the expression of SDF1 during the development of SL should be considered, and further investigations are necessary.
In conclusion, the results of this study provide an integrated view of how senescent fibroblasts contribute to skin pigmentation. Our findings suggest that senescent fibroblasts and their phenotype switching are promising therapeutic targets for delaying or preventing ageing-associated pigmentation. These data may provide a new therapeutic paradigm, such as a stroma-targeting therapies that involve energy-based devices that incorporate intense-pulse light (IPL), pulsed-diode lasers, and pico lasers, to treat pigmentary disorders.
Abbreviations
5-Aza: 5-aza 2-deoxycytidine; FSP1: fibroblasts-specific protein 1; MITF: microphthalmia-associated transcription factor; PA/EA: pigmented area/epidermal area; rhSDF1: recombinant human SDF1; RS: replicative senescence; SA-β-Gal: senescence-associated β-galactosidase; SASP: senescence-associated secretory phenotype; SDF1: stromal-derived factor 1; SL: senile lentigo; TYR: tyrosinase; UVAiS: UVA-induced senescence.
Supplementary Material
Supplementary figures.
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2016R1D1A1B03932749, NRF-2012R1A5A2048183, and NRF-2017R1A2B4006665) and by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare of the Republic of Korea (HN14C0094).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bastonini E, Kovacs D, Picardo M. Skin pigmentation and pigmentary disorders: Focus on epidermal/dermal cross-talk. Ann Dermatol. 2016;28:279-89
2. Kim M, Han JH, Kim JH, Park TJ, Kang HY. Secreted frizzled-related protein 2 (sFRP2) functions as a melanogenic stimulator; The role of sFRP2 in UV-induced hyperpigmentary disorders. J Invest Dermatol. 2016;136:236-44
3. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu Rev Pathol. 2010;5:99-118
4. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J Clin Invest. 2013;123:966-72
5. Kim YH, Choi YW, Lee JH, Soh EY, Kim JH, Park TJ. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat Commun. 2017;8:15208
6. Mine S, Fortunel NO, Pageon H, Asselineau D. Aging alters functionally human dermal papillary fibroblasts but not reticular fibroblasts: A new view of skin morphogenesis and aging. PLoS One. 2008;3:e4066
7. Coppé JP, Patil CK, Rodier F, Krtolica A, Beauséjour CM, Parrinello S. et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS One. 2010;5:e9188
8. Velarde MC, Demaria M. Targeting senescent cells: Possible implications for delaying skin aging. Gerontology. 2016;62:513-8
9. Ghosh K, Capell BC. The senescence-associated secretory phenotype: Critical effector in skin cancer and aging. J Invest Dermatol. 2016;136:2133-9
10. Waldera Lupa DM, Kalfalah F, Safferling K, Boukamp P, Poschmann G, Volpi E. et al. Characterization of skin aging-associated secreted proteins (SAASP) produced by dermal fibroblasts isolated from intrinsically aged human skin. J Invest Dermatol. 2015;135:1954-68
11. Fisher GJ, Kang S, Varani J, Bata-Csorgo Z, Wan Y, Datta S. et al. Mechanisms of photoaging and chronological skin aging. Arch Dermatol. 2002;138:1462-70
12. Quan T, He T, Voorhees JJ, Fisher GJ. UV irradiation blocks cellular responses to transforming growth factor beta by downregulating its type II receptor and inducing Smad7. J Biol Chem. 2001;276:26349-56
13. Salducci M, André N, Guéré C, Martin M, Fitoussi R, Vié K, Cario-André M. et al. Factors secreted by irradiated aged fibroblasts induce solar lentigo in pigmented reconstructed epidermis. Pigment Cell Melanoma Res. 2014;27:502-4
14. Kovacs D, Cardinali G, Aspite N, Cota C, Luzi F, Bellei B. et al. Role of fibroblast-derived growth factors in regulating hyperpigmentation of solar lentigo. Br J Dermatol. 2010;163:1020-7
15. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C. et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci. 1995;92:9363-7
16. Kim HS, Song MC, Kwak IH, Park TJ, Lim IK. Constitutive induction of p-Erk1/2 accompanied by reduced activities of protein phosphatases 1 and 2A and MKP3 due to reactive oxygen species during cellular senescence. J Biol Chem. 2003;278:37497-510
17. Brenner M, Coelho SG, Beer JZ, Miller SA, Wolber R, Smuda C. et al. Long lasting molecular changes in human skin after repetitive in situ UV irradiation. J Invest Dermatol. 2009;129:1002-11
18. Teixidó J, Martínez-Moreno M, Díaz-Martínez M, Sevilla-Movilla S. The good and bad faces of the CXCR4 chemokine receptor. Int J Biochem Cell Biol. 2017;95:121-31
19. Zgraggen S, Huggenberger R, Kerl K, Michael D. An important role of the SDF-1/CXCR4 axis in chronic skin inflammation. PLoS One. 2014;9:e93665
20. Ruiz EJ, Oeztuerk-Winder F, Ventura JJ. A paracrine network regulates the cross-talk between human lung stem cells and the stroma. Nat Commun. 2014;5:3175
21. Grönniger E, Weber B, Heil O, Peters N, Stäb F, Wenck H. et al. Aging and chronic sun exposure cause distinct epigenetic change in human skin. PLoS Genet. 2010;6:e1000971
22. Koch CM, Suschek CV, Lin Q, Bork S, Goergens M, Joussen S. et al. Specific age-associated DNA methylation changes in human dermal fibroblasts. PLoS One. 2011;6:e16679
23. Qian H, Xu X. Reduction in DNA methyltransferases and alteration of DNA methylation pattern associate with mouse skin ageing. Exp Dermatol. 2014;23:357-9
24. Moulin L, Cenizo V, Antu AN, André V, Pain S, Sommer P. et al. Methylation of LOXL1 promoter by DNMT3A in aged human skin fibroblasts. Rejuvenation Res. 2017;20:103-10
25. Wendt MK, Johanesen PA, Kang-Decker N, Binion DG, Shah V, Dwinell MB. Silencing of epithelial CXCL12 expression by DNA hypermethylation promotes colonic carcinoma metastasis. Oncogene. 2006;25:4986-97
26. Moskovits N, Kalinkovich A, Bar J, Lapidot T, Oren M. p53 attenuates cancer cell migration and invasion through repression of SDF-1/CXCL12 expression in stromal fibroblasts. Cancer Res. 2006;66:10671-6
27. Maroni P, Bendinelli P, Matteucci E, Desiderio MA. HGF induces CXCR4 and CXCL12-mediated tumor invasion through Ets1 and NF-kappaB. Carcinogenesis. 2007;28:267-79
28. el-Domyati M, el-Ammawi TS, Medhat W, Moawad O, Brennan D, Mahoney MG. et al. Radio frequency facial rejuvenation: Evidence-based effect. J Am Acad Dermatol. 2011;64:524-35
29. Seo KY, Yoon MS, Kim DH, Lee HJ. Skin rejuvenation by microneedle fractional radiofrequency treatment in Asian skin: Clinical and histological analysis. Lasers Surg Med. 2012;44:631-6
30. Na J, Zheng Z, Dannaker C, Lee SE, Kang JS, Cho SB. Electromagnetic initiation and propagation of bipolar radiofrequency tissue reactions via invasive non-insulated microneedle electrodes. Sci Rep. 2015;5:16735
31. Spandau DF, Lewis DA, Somani AK, Travers JB. Fractionated laser resurfacing corrects the inappropriate UVB response in geriatric skin. J Invest Dermatol. 2012;132:1591-6
32. Goyarts E, Muizzuddin N, Maes D, Giacomoni PU. Morphological changes associated with aging: Age spots and the microinflammatory model of skin aging. Ann N Y Acad Sci. 2007;1119:32-9
33. Yonei N, Kaminaka C, Kimura A, Furukawa F, Yamamoto Y. Two patterns of solar lentigines: A histopathological analysis of 40 Japanese women. J Dermatol. 2012;39:829-32
34. Shin J, Park JY, Kim SJ, Kang HY. Characteristics of keratinocytes in facial solar lentigo with flattened rete ridges: Comparison with melasma. Clin Exp Dermatol. 2015;40:489-94
35. Chen N, Hu Y, Li WH, Eisinger M, Seiberg M, Lin CB. The role of keratinocyte growth factor in melanogenesis: A possible mechanism for the initiation of solar lentigines. Exp Dermatol. 2010;19:865-72
36. Goorochurn R, Viennet C, Tissot M, Locatelli F, Granger C, Varin-Blank N. et al. Differential morphological and functional features of fibroblasts explanted from solar lentigo. Br J Dermatol. 2017;177:109-11
37. Hasegawa K, Fujiwara R, Sato K, Shin J, Kim SJ, Kim M. et al. Possible involvement of keratinocyte growth factor in the persistence of hyperpigmentation in both human facial solar lentigines and melasma. Ann Dermatol. 2015;27:626-9
38. Choi W, Yin L, Smuda C, Batzer J, Hearing VJ, Kolbe L. Molecular and histological characterization of age spots. Exp Dermatol. 2017;26:242-8
39. Speeckaert R, Ongenae K, van Geel N. Alterations of CXCL12 in serum of patients with vitiligo. J Invest Dermatol. 2017;137:1586-8
40. Lee J, Kim M, Park TJ, Kang HY. Fibroblast-derived clusterin negatively regulates pigmentation. J Invest Dermatol. 2017;137:1812-5
41. Natarajan VT, Ganju P, Singh A, Vijayan V, Kirty K, Yadav S. et al. IFN-γ signaling maintains skin pigmentation homeostasis through regulation of melanosome maturation. Proc Natl Acad Sci USA. 2014;111:2301-10
42. Mignon C, Uzunbajakava NE, Raafs B, Botchkareva NV, Tobin DJ. Photobiomodulation of human dermal fibroblasts in vitro: Decisive role of cell culture conditions and treatment protocols on experimental outcome. Sci Rep. 2017;7:2797
43. Tigges J, Krutmann J, Fritsche E, Haendeler J, Schaal H, Fischer JW. et al. The hallmarks of fibroblast ageing. Mech Ageing Dev. 2014;138:26-44
Author contact
![]() Corresponding authors: Tae Jun Park M.D., Ph.D., Department of Biochemistry and Molecular Biology, Ajou University School of Medicine, Suwon, 443-721, Korea. Tel: +82-31-219-5055; Fax: +82-31-219-5059; E-mail: park64ac.kr and Hee Young Kang M.D., Ph.D., Department of Dermatology, Ajou University School of Medicine, Suwon, 443-721, Korea. Tel: +82-31-219-5188; Fax: +82-31-219-5189; E-mail: hykangac.kr
Corresponding authors: Tae Jun Park M.D., Ph.D., Department of Biochemistry and Molecular Biology, Ajou University School of Medicine, Suwon, 443-721, Korea. Tel: +82-31-219-5055; Fax: +82-31-219-5059; E-mail: park64ac.kr and Hee Young Kang M.D., Ph.D., Department of Dermatology, Ajou University School of Medicine, Suwon, 443-721, Korea. Tel: +82-31-219-5188; Fax: +82-31-219-5189; E-mail: hykangac.kr