Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Onco-immunology and Adoptive...
Cell sources and modality of the...
Gene engineering strategies in...
Combination of different...
Conclusions and Perspectives
Abbreviations
Acknowledgements
References
Introduction
Onco-immunology and Adoptive...
Cell sources and modality of the...
Gene engineering strategies in...
Combination of different...
Conclusions and Perspectives
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(20):5784-5800. doi:10.7150/thno.29035 This issue Cite
Review
Adoptive Cell Transfer: Is it a Promising Immunotherapy for Colorectal Cancer?
Jiaqiao Fan1 ![]() , Dong Shang1, Bing Han2, Jianxun Song3, Hailong Chen1
, Dong Shang1, Bing Han2, Jianxun Song3, Hailong Chen1 ![]() , Jin-Ming Yang4
, Jin-Ming Yang4 ![]()
1. Third General Surgery Department, The First Affiliated Hospital of Dalian Medical University, Dalian, China
2. Department of Pathology, College of Medicine, The Pennsylvania State University, Hershey, PA, USA
3. Department of Microbial Pathogenesis and Immunology, Texas A&M University Health Science Center, College Station, TX, USA
4. Department of Pharmacology, College of Medicine, The Pennsylvania State University, Hershey, PA, USA
Received 2018-8-6; Accepted 2018-10-4; Published 2018-11-10
Citation:
Fan J, Shang D, Han B, Song J, Chen H, Yang JM. Adoptive Cell Transfer: Is it a Promising Immunotherapy for Colorectal Cancer?. Theranostics 2018; 8(20):5784-5800. doi:10.7150/thno.29035. https://www.thno.org/v08p5784.htm
Other stylesAbstract

The last decade has witnessed significant advances in the adoptive cell transfer (ACT) technique, which has been appreciated as one of the most promising treatments for patients with cancer. Utilization of ACT can enhance the function of the immune system or improve the specificity and persistence of transferred cells. Various immune cells including T lymphocytes, natural killer cells, dendritic cells, and even stem cells can be used in the ACT despite their different functional mechanisms. Colorectal cancer (CRC) is among the most common malignancies and causes millions of deaths worldwide every year. In this review, we discuss the status and perspective of the ACT in the treatment of CRC.
Keywords: adoptive cell transfer, immunotherapy, colorectal cancer
Introduction
Colorectal cancer (CRC) is one of the most prevalent cancers worldwide and is the second most commonly diagnosed cancer in women and the third in men [1]. Also, CRC accounts for approximately 10% of cancer morbidity and is the fourth leading cause of cancer-related mortality. About 1.2 million people are diagnosed with CRC each year worldwide. Although multi-disciplinary efforts have led to significant progress and improvement in treating patients with CRC, half of the patients die from the disease eventually [2]. Surgery is currently the sole approach with the possibility to cure CRC and is generally the upfront option for patients with localized CRC significantly improving the overall survival (OS) [3]. Generally, chemotherapy and/or radiotherapy are prescribed following surgery to prevent local recurrence and distant organ metastases; however, liver metastases often occur among more than half of CRC patients, decreasing the survival rate [4, 5]. Thus, it is imperative to develop novel therapies for patients with CRC.
Onco-immunology and Adoptive Cell Transfer (ACT) for Cancer
The immune system can protect the host from tumorigenesis through immune surveillance mechanisms [6, 7]. The occurrence and development of cancer result from, at least partially, the deficiency of the immune system. The immune response is a highly coordinated process in healthy individuals for eliminating exogenous pathogen and mutated cells, but an excessive activity of this process may cause autoimmune damage. The schemes that the immune system uses to control the extent of immune reactivity such as upregulated expression of checkpoint proteins, activation of cell death programs, and accumulation of various immunosuppressive cells can also be exploited by cancer cells to escape the immune attack. Also, malignant cells may employ additional immune tolerance mechanisms including downregulation of MHC I molecules and specific antigens, reduced secretion of some immune suppressive molecules [8], depletion of the materials necessary for immune cells [9], and acquiring an acidic and hypoxic condition hostile to antitumor cells [10, 11]. The tumor entity contains a variety of stromal cells that can develop assorted mechanisms to prevent penetration of effector cells and to cause anergy or apoptosis [12]. It is known that the immune cells become deficient not only within tumor microenvironment but also during tumor initiation.
More than two decades ago, it was demonstrated that the expression of CD3ζ, a T cell receptor (TCR) intracellular domain, was downregulated in tumor-infiltrating lymphocytes (TILs) in a variety of cancers and caused a functional deficiency of immune effector cells [13-15]. It was also observed that the circulating lymphocytes of patients with malignant tumors had a downregulated expression of CD3ζ and an upregulated expression of the apoptosis-inducing molecule caspase3, which further inhibited the antitumor potential of the systemic immunity [16-18]. Although the existence of cytolytic effector cells within the tumor can elicit a naturally occurring antitumor response, the functional deficiency of the immune system suggests the necessity of therapeutic intervention to improve the immune function. The major objective of cancer immunotherapy is to reverse the immunosuppression and/or immune deficiency and to enhance the activity of immune effector cells.
A variety of strategies have been exploited to improve the immune function of cancer patients which include monoclonal antibodies, immune-activating cytokines, cancer vaccines, checkpoint inhibitors, and ACT. Soluble IL-2 was the first agent used to reverse the anergy of T cells [19, 20], but it had a moderate therapeutic activity and severe adverse effects. The first study of the ACT for cancer was reported in 1955 by Mitchison et al [21]. The therapeutic potential of the ACT, which is based on tumor-specific lymphocytes, was first tested by Fefer and Rosenberg about 40 years ago [22, 23] showing only limited efficacy. In 1998, Matsumoto showed that ACT could upregulate CD3ζ expression in peripheral blood lymphocytes and improve the immune function of cancer patients [24]. In recent years, due to the advancement of cellular and molecular biology techniques, such as T cell extraction, expansion and activation ex vivo, and genetic engineering techniques, ACT is becoming a viable therapeutic option for cancer patients who are refractory to conventional therapy. Indeed, ACT can lead to tumor regression and even eradication in some patients with advanced cancers. The ACT requires immune cells prepared from patients or donors or differentiated from stem cells. These immune cells are then activated and expanded in vitro, subjected to gene modification, and then infused into the patients through a peripheral vein or regional artery.
The ACT can also include dendritic cells (DCs) and/or immune effector cells, or combination of both. DCs are often used as vaccine carriers or antigen presenting cells (APCs) to prime naive T cells in vitro or in vivo. Cytotoxic T lymphocytes (CTL) and natural killer cells (NK) are the major effector cells in naturally occurring anti-tumor response and were exploited very early as tool cells for the ACT. An impressive success of ACT has been achieved in several types of cancers during the last two decades, and the most prominent progress was made in B lymphocyte leukemia and lymphoma [25, 26]. Rosenberg and his colleagues at the National Institute of Health (NIH) employed ACT in a hallmark study in solid tumors in 1988 [27] where 20 patients with advanced melanoma were treated with TILs and demonstrated therapeutic benefits. Since then, promising results were obtained in a variety of solid tumors including metastatic melanoma [28-31], renal cell carcinoma (RCC) [32, 33] , nasopharyngeal cancer [34], gastric cancer [35], hepatocellular carcinoma [36], and lung cancer [37]. ACT with TILs is yet the most effective immunotherapy strategy for patients with metastatic melanoma. Compared to other passive cancer immunotherapy strategies, ACT has several advantages: (1) tumor-specific lymphocytes can be selected, stimulated, and expanded to large numbers in vitro; (2) the infused cells can be genetically engineered with artificial genes to target desired tumor-associated antigens (TAA) selectively; (3) the host can be conditioned through lymphodepletion prior to ACT to yield a favorable condition for infused cell persistence. The ACT is now becoming one of the most promising strategies in cancer immunotherapy, and numerous clinical trials are in progress around the world which are listed in Table 1. In 2017, FDA approved two CD19-CAR (chimeric antigen receptor) T cells for acute refractory leukemia in children and B cell lymphoma in adults, indicating that ACT is now a treatment option in clinic.
Cell sources and modality of the ACT for treatment of CRC
Application of ACT for cancer treatment originated from the observation of TILs, which were the initiating cells used for the ACT. TILs can be extracted from freshly resected or biopsy tumor specimens, and the lymphocytes within the tumor-draining lymph nodes (lymph node lymphocytes, LNLs) have characteristics similar to TILs. However, it is often not easy to obtain fresh tumor specimens and lymph nodes from patients with advanced cancers. The peripheral blood mononuclear cells (PBMCs) are now used as the major alternative cell source for the ACT because of their easy extraction from the whole blood from patients or donors and expansion in vitro. More recently, stem cells have been used as a possible source for the ACT. Due to the urgent need for an industrial production of cells for the ACT, several strategies have been established for inducing differentiation of various immune cells from stem cells. Several cell types of ACT are listed in Table 2.
ACT with TILs and LNLs
TILs are the lymphocytes that accumulate at the tumor margins or infiltrate within the tumor. Use of TILs was first recommended by Klein and Vanky approximately four decades ago [38, 39]. TILs represent the host's naturally occurring antitumor response and can recognize and destroy cancer cells expressing TAAs, which are frequently mutated molecules and considered as foreign. TILs are not a homologous population of lymphocytes, whose function was first described in detail by Klein et al [40]. TILs can be isolated from single cell homogenates of fresh tumor specimens or cultured from tumor fragments. Several approaches for the preparation of TILs from fresh specimens have been developed which can expand TILs by thousands of folds in vitro for reinfusing back to the host (Figure 1). ACT using autologous TILs has achieved success in various malignancies, especially in metastatic melanoma. TILs can be produced massively from a variety of tumors including colon adenocarcinoma [41]. It was demonstrated that TILs had a protective role in rectal cancer and were positively correlated with patients' five-year survival rate [42]. More recently, Fridman et al performed a series of seminal basic studies on the role of TILs in CRC and demonstrated that TILs within CRC were beneficial to the patient's survival, suggesting that TILs can be used as a prognostic index [43-46]. Rosenberg et al conducted the first clinical trial of ACT using TILs at NIH in 1988 [27]. In this trial, 20 patients with advanced melanoma and renal cancer were treated with TILs followed by a high dose of IL-2 injection, and an objective response was observed in five patients.
Table 1
Clinical trials of the ACT in CRC
| Condition | Sponsor | Status | clinicaltrials.gov Identifier | Biological |
|---|---|---|---|---|
| Metastatic colorectal cancer | Gangham severance hospital, Seoul, The Republic of Korea | Enrolling by invitation | NCT03220984 | Immuncell-LC intravenous infusion using a CIK cell agent |
| Colorectal cancer | National center for tumor disease NCT Heidelberg, BW, Germany | recruiting | NCT02577588 | Re-activated T cells |
| Colorectal cancer | Jingzhou central hospital immunotherapy center, China | unknown | NCT02202928 | DC-CIK |
| Colorectal cancer | Guangxi Medical University Nanning, Guangxi, China | recruiting | NCT01839539 | DC-CIK |
| Colorectal cancer | Biotherapeutic Department of Chinese PLA General Hospital, China | unknown | NCT01801852 | NKT cells |
| Colorectal cancer | Envita Medical Centers Scottsdale, Arizona, United States | suspended | NCT00909558 | Autologous Natural Killer/Natural Killer T Cell Immunotherapy |
Table 2
Cell resource, types, management, and other characteristics of the ACT for CRC
| Cell modality | Cell resource | Phenotype of the cell pool | Handling method of cell | Cell working mechanism | Shortage of the cell | Perspective of the method |
|---|---|---|---|---|---|---|
| TILs | Fresh resected specimen | Mixed lymphocytes | Anti-CD3, anti-CD28, IL-2 | Including tumor-specific lymphocytes | Few and difficult to extract | Identification of tumor-specific TCR |
| LNLs | Regional lymph node | Mixed lymphocytes | Anti-CD3, anti-CD28, IL-2 | Including tumor-specific lymphocytes | Operation technique needed | Similar as above |
| CIKs | PBMCs | CD3+CD56+ | IFN γ, anti-CD3, IL-2 | APC independent natural killer | No tumor-specific | Gene engineered |
| NK | PBMCs | CD16+CD56+ | IL-2, IL-15 | APC independent natural killer | No tumor-specific | Gene engineered |
| Vγ9Vδ1+ | PBMCs | CD3+γδ TCR | Zoledronate, IL-2/ 2-methyl- 3-butenyl-1-pyrophosphate (2M3B1PP) | Innate and adaptive function | No tumor-specific | Exploring |
| αβ T cells | PBMCs | CD3+αβ TCR | CD3, CD28, IL-2 | Generally used to edit with TCR or CAR | No tumor-specific | Gene engineered |
| Differentiated lymphocytes | Stem cells | Variable phenotypes or gene engineered | Variable protocols | According to the final cells | At the initiation of development | Universal ACT cells resource |
Figure 1
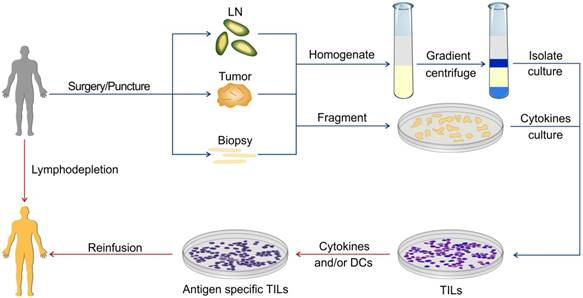
ACT using TILs. The specimens for preparing TILs can be obtained via surgery or puncture. These specimens can be homogenized or fragmented and then cultured. There are various protocols for the expansion of TILs in the presence of different cytokines or APC.
Although early trials of the ACT with TILs demonstrated efficacy for CRC patients, the results were also paradoxical. Gardini conducted a clinical trial in the 1990s in which 14 CRC patients with liver metastases were treated with TILs for the therapeutic effects of the ACT. TILs were extracted from the liver metastases of the radical resection specimens, stimulated, and expanded with IL-2. The TILs were then reinfused back to the patients. There was no significant difference in disease-free survival (DFS) between the TILs group and traditional chemotherapy [47]. In a later clinical study with patients with malignancies other than CRC, the investigators did not observe any encouraging objective response within heterogenous patients. Nevertheless, a moderate improvement in median survival was observed amongst patients receiving an intermediate or high dose of TILs compared with a low dose, suggesting that the high dose of TILs may be an effective approach [48]. The results suggested the need for improvement in procedures for TILs acquisition and expansion. TILs not only can be expanded directly from tumor specimens for the ACT in CRC but also be used to isolate TAA-specific CD8+ T cell clones or even identify tumor-specific TCRs.
In 2016, Rosenberg's team at NIH identified polyclonal CD8+ T cells against mutant KRAS G12D in TILs from metastatic lung lesions of a CRC patient. They expanded the KRAS G12D-specific CD8+ T cell clones and reinfused the TILs back to the patient and observed that 6 in 7 lung metastases were eradicated. Further, they resected the progressing lesion and found that it still expressed the mutated KRAS G12D but lost the gene encoding HLA-C*08:02 alleles. Subsequently, Tran et al. sequenced and synthesized the mutated KRAS G12D targeting TCRs, treated the in vitro expanded T cells with the TCRs and cocultured with pancreatic cells expressing the mutated KRAS G12D and observed a significant killing effect in the in vitro culture system [49]. Although there are different explanations for the results of this study [50], it demonstrated the existence of naturally occurring tumor-specific CTLs within TILs and showed the way to explore tumor-specific TCRs from millions of tumor-associated mutant epitopes [51]. More recently, various neoantigen-targeting CD8+ T cell clones and TCRs have been identified in patients with different types of cancers. However, several factors may hamper the successful application of TILs in CRC patients. It is difficult to harvest sufficient number of TILs from CRC specimens as relatively few effector cells infiltrate the CRC tumors [52, 53]. So far, sufficient TILs could only be obtained from patients with resectable melanoma and renal cancer. Several groups have exploited strategies to efficiently expand TILs in vitro along with improvements of TILs isolation and proliferation.
Another problem is of the presence of flora within the intestine which often contaminate the CRC specimens making it difficult to grow TILs from CRC tumors. To bypass this limitation, the tumor-draining lymph nodes were used to acquire the tumor-specific T cells. Lymph nodes are the major site for antigen presenting cells (APC) to process TAAs and present the determinant epitopes to TCR through MHC class II or I molecules. Tumor-draining lymph nodes receive lymph drainage from the tumor lesion and therefore LNLs contact the whole repertoires of TAAs and are primed naturally. It is hypothesized that tumor draining LNLs may have more tumor specificity than lymphocytes within general lymph nodes. Upon stimulation, LNLs displayed stronger proliferative potential than lymphocytes from uninvolved lymph nodes and may represent a reliable aseptic source of tumor-specific lymphocytes for ACT [54]. In the late 1990s, Satoh and Triozzi reported their individual studies on patients with metastatic CRC. They treated the patients with LNLs, which were expanded in vitro and observed positive response and significantly prolonged survival [55, 56], suggesting that ACT with LNLs can be safely used to treat malignant CRC. In a recent study, 16 middle and advanced stage CRC patients (seven with stage II-III and nine with stage IV CRCs) were treated with LNLs. The results showed that all patients with stage IV disease had an objective response and the survival of ACT group was significantly prolonged from 0.8 years to 2.6 years [57], further demonstrating the efficiency and feasibility of clinical application of LNLs in patients with CRC.
Based on these promising results, Jin et al. conducted a large phase I/II clinical study in 71 CRC patients (46 stage I-III patients underwent radical surgery and 25 stage IV patients underwent palliative surgery). The trial demonstrated that LNLs contained more activating CD3+CD69+ and CD4+CD69+ T lymphocytes than TILs. The major subtypes of expanded cells from LNLs were tumor-specific effector and central memory T cells. The OS of the patients in the LNL group improved significantly as compared to the control group (28 vs 14 months), and no side effects were observed [58]. Because the LNLs show similar or even better effects of ACT in CRC without the problem of contamination, they could be the major source of TIL-like cells for CRC adoptive therapy. Further investigations are warranted to address some hurdles for the use of TILs and LNLs as the ACT for CRC as these cell types occur naturally and have no immunogenicity and little adverse effects. More importantly, there exist tumor-specific TCRs against thousands of unknown TAAs. Liver metastasis is the most frequent complication in patients with advanced CRC ultimately leading to death. However, it is feasible to eliminate liver metastasis using the improved technique developed recently. Liver metastases may be an ideal source of TILs that can be harvested aseptically without contamination with the intestinal flora and used for the ACT to treat patients with CRC.
ACT with PBMCs
Acquisitions of TILs and LNLs require exquisite surgery techniques which are not feasible in patients with visceral cancers at a late stage. Most solid tumors are not as immunogenic as melanoma with too few TILs for harvesting and expansion to meet the requirements for the ACT. Also, the time required for ex vivo expansion of TILs is about 1 ~ 2 months which is too long for patients with the terminal disease. Therefore, it is necessary to explore an alternative source of TILs. PBMCs have been exploited as a source of ACT cells for three decades because of their large quantity and the simple isolation procedure. PBMCs can also be harvested from donors other than patients which is beneficial for the patients receiving standardized chemo/ radiotherapy. PBMCs can be stimulated in vitro with high doses of IL-2 to yield lymphokine-activated killer (LAK) cells. PBMCs are a heterogeneous cell population comprising lymphocytes (T cells, B cells, and NK cells), monocytes and dendritic cells, with lymphocytes typically in the range of 70-90%. PBMCs can not only be used as an integral heterogeneous cell pool but can also be purified as single phenotype cells. They can be used in their natural status after activation, expanded by different procedures, or genetically engineered with various artificial or selected genes to enhance their persistence and specificity (Figure 2).
Cytokine-induced killer (CIK) cells
The first antitumor effector cells generated from PBMCs are LAK cells, which can be isolated and activated in vitro by a high dose of IL-2. LAK cells are a heterogeneous cell pool consisting of NK cells and various T cells. This strategy had been a major method of cell transfer in cancer immunotherapy for almost two decades but was abandoned later because of its limited efficiency and severe adverse effects. CIK cells are the successor of LAK cells and can be obtained from PBMCs by sequentially adding variable cytokines instead of only IL-2 (Table 2). PBMCs acquire CD56 phenotype as well as nonspecific NK cell-like antitumor cytotoxicity in vitro. CIK cells were first characterized by Schmidt-Wolf in 1991 [59]. Compared to LAK cells, CIK cells can be produced more easily and display greater proliferation potential and stronger cytotoxicity against cancer cells [59-63]. The procedure for obtaining CIK cells in vitro has been well established [64-67].
The cytotoxic activity of CIK cells is mainly afforded by CD3+ CD56+ cells [68], which work in an MHC I molecule independent fashion, and is relevant for cancers that downregulate expression of MHC class I to escape immune reactivity. The cytotoxicity of CIK cells results from the release of granzyme B and perforin. However, the molecules involved in tumor recognition are not yet identified, although NKG2D signaling was considered as a candidate [69]. CIK cells do not display cytotoxicity against normal tissues particularly graft-versus-host disease. Several clinical protocols have been developed and demonstrated the feasibility of these passive transfer approaches with moderate toxicity [70].
Figure 2
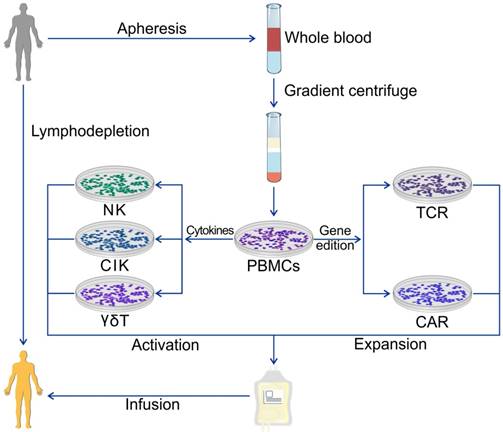
ACT using PBMCs. PBMCs are the most universal cell source of the ACT and are extracted from whole blood from patients or healthy donors. Various immune cells can be obtained for ACT using different protocols of cytokine management or gene edition.
The first clinical trial involving CRC patients with CIK cells was carried out by Schmidt-Wolf et al. in 1994 [71]. Ten patients with metastatic CRC, renal cell cancer, and lymphoma were included in the clinical trial of which five received CIK cells transfected with the IL-2 plasmid and another five received non-transfected CIK cells. Although the authors did not acquire any positive results, they showed, for the first time, that CIK cells could be safely administered in CRC patients. Since then, many clinical trials of ACT with CIK cells were carried out in patients with CRC. In a retrospective study, Zhang and his colleagues evaluated the effects of CIK cells on the prognosis of CRC patients and found that the ACT with CIK cells can decrease the recurrence and improve the OS [72]. CIK cells are not tumor type-specific enabling a broader application range. However, enhancing the antitumor specificity of CIK cells remains an unresolved issue.
While CIK cells express the NK cell marker CD56 and the T cell marker CD3, they may also work in an MHC-dependent fashion and eradicate cells presenting MHC molecules; this ability could be enhanced by TAA-bearing dendritic cells (DCs) [73, 74]. In recent years, the combination of CIK cells with DCs is emerging as an active immunotherapy research field. DCs can be pulsed with a specific antigen, whole tumor lysis, or even mutated RNAs to enhance the immune response and therapeutic effects of CIK cells by presenting specific TAAs to the cells. Gao et al. evaluated the effect of DC/CIK combination therapy in a group of patients following radical surgery for gastric cancers (GCs) and CRC. They found that ACT using DC/CIK could inhibit disease progression and increase OS [75]. In a series of clinical trials including hundreds of patients with CRC, several groups reported that DC/CIK-based immunotherapy could induce an antitumor response against CRC and increase PFS and OS [76-78]. Combination of the DC/CIK-based ACT with conventional chemotherapy was also tested and showed a significant improvement of OS in patients with CRC [79].
CIK cells could be engineered to express a chimeric antigen receptor (CAR) gene to enhance the specificity and penetration potential. Schmidt-Wolf et al. modified CIK cells with a CEA targeting CAR plasmid and demonstrated that the gene-engineered CIK cells displayed stronger cytotoxicity against autologous, primary CRC cells as compared with the unmodified CIK cells [67]. Thus, CIK cells may represent a new option in cancer immunotherapy. The perspectives and challenges of CIK in ACT remain in the exploitation of synergism with immunotherapy, other targeted therapies or, conventional chemotherapy [80].
NK cells
NK cells belong to the innate immunity system and constitute the first line of host defense against pathogens and mutated cells. The main characteristic of NK cells is the expression of CD56 and/or CD16 but without TCR-CD3 complex. NK cells can recognize and kill the pathogen-infected and mutated cells in an antigen presenting independent manner. Activation of NK cells is orchestrated through the integration of signals deriving from inhibitory and activating receptors expressed on their surface. Both activating and inhibitory receptors play key roles in tuning NK cell function, preventing NK cells from targeting “self” normal cells while allowing them to attack mutated or infected cells [81-83]. The most important activating receptors include natural killer group 2 (NKG2) and natural cytotoxicity receptors (NCR), which recognize ligands in cancer cells or infected cells. The inhibitory receptors of NK cells include killer immunoglobulin-like receptors (KIRs) that interact with their cognate MHC ligands. MHC class I molecules are aberrantly expressed in most CRC cells [84-86] which makes them susceptible to the NK cell-mediated cytotoxicity.
The antitumor activity of NK cells was first studied in hematopoietic malignancies prior to solid tumors. Miller and Burns reported in a series of clinical trials that autologous NK cells, although proliferating in vitro, did not show expansion and cytotoxicity in vivo after infusion [87, 88], but allogeneic NK cells could expand in vivo and showed cytotoxicity in certain hematopoietic malignancies and solid tumors [89]. It was reported that NK cells, isolated from PBMCs, activated and expanded in vitro, showed potent cytotoxic effects in various CRC cell lines and primary CRC cells, and the cytotoxic effect could be further enhanced by cetuximab, an EGFR monoclonal antibody [90]. NK cells can selectively infiltrate into and accumulate within tumors making them an ideal carrier of targeted therapy. It is believed that genetic engineering is an attractive approach to enhancing the potency and specificity of NK cells [91-94]. For instance, Yong et al. modified NK cells with a secretory TNF-related apoptosis-inducing ligand (TRAIL) gene and then injected the engineered NK cells into the peritoneal cavity of mice. They observed that the infused cells infiltrating into and accumulating in the tumor sites induced cancer cell apoptosis and delayed disease progression without significant adverse effects [95]. These studies underscored the therapeutic potential of the genetically engineered NK cells in treating CRC patients. Strandmann et al. tested a bispecific fused protein containing ULBP2 (a ligand of NKG2D) and a single chain of CEA antibody, which could bind with NKG2D on NK cells and CEA on CRC cells, and demonstrated that the bispecific ligand could induce the potent antitumor activity of NK cells against CRC cells [96]. More recently, Hans et al. [97] reported a protocol for obtaining NK cells from umbilical cord blood (UCB) stem cells (UCB-NK) and observed the potent antitumor activity of the UCB-NK cells. Despite the high rate of success in differentiating NK cells from UCB, it is recognized that the differentiated NK cells are easy to undergo apoptosis in the body. Stem cell-derived immune cells have their unique advantages in the ACT, which will be reviewed later.
Unselected or selected T lymphocytes
T lymphocytes, which are the major cell component of PBMCs and the critical effector cells in antitumor response, are the primary source of the ACT in cancer treatment. The phenotypic characteristic of T lymphocytes is the expression of TCR-CD3 complex, which can bind with the antigen epitope presented by MHC molecules and initiate the activation of signaling pathways with the help of co-receptors (CD4 and CD8) and costimulatory molecules (CD28, CD137). There are two major types of TCRs based upon the differences in the hypervariable chain and function, - αβ TCR and γδ TCR. The classical TCR is αβ TCR, which has hypervariable α and β chains. The frame of the TCR molecule is homogeneous, but the αβ chains are heterogeneous. Theoretically, there are approximately millions or even billions of TCRs, which can recognize millions of antigens epitopes. Despite the existence of cross-recognition, one TCR generally recognizes only one epitope. αβ T cells can be classified into CD4+ and CD8+ families according to their co-receptor molecules. CD8+ T cells are the main cytotoxic T lymphocytes, and CD4+ T cells are a heterogeneous T cell population with complicated “helping” functions as well as cytotoxic activity. Although CD8+ T cells are the main effector cells, purified CD8+ T cells have a poor persistence ability as they are not able to maintain their activity without the help of CD4+ T cells; therefore, mixed T cells are used in the ACT.
June et al. reported a seminal study in which they engineered the purified CD4+ and CD8+ T cells with different genes to improve specificity and potency [98]. Hasegawa et al. reported a series of clinical trials in which they used purified αβ T cells in combination with chemotherapy or monoclonal antibody to treat advanced or recurrent CRC patients. In these studies, they observed promising response rates and moderate adverse effects, and the CRC patients with liver metastasis could undergo surgical resection after chemo-immunotherapy [99-101]. As liver metastasis is the most common complication and the major reason for mortality in patients with advanced CRC, ACT using αβ T cells may be an effective way to improve the outcome of surgery. The major modality of adoptive infused cells, especially for gene engineering, are αβ T cells, unless specific lymphocytes from PBMCs, such as CIK, NK, and γδ T cells, are required. γδ T cells, a distinct subset of T cells that work in a unique fashion with αβ T cells, are comprised of about 0.5% of total CD3+ cells in human peripheral blood cells [102]. Although γδ T cells consist of a very low frequency of the CD3+ cell pool, they can be rapidly expanded both ex vivo and in vivo. γδ T cells can recognize and attack pathogens and mutated cells in an MHC class I molecule-unrestricted manner, displaying combined characteristics of both innate and adoptive immunity. They act as the upfront anticancer immune surveillance and possess cytotoxic activity and antigen-presenting capability [103, 104]. These features make γδ T cells an excellent candidate for the ACT. γδ T cells were previously recognized as homogeneous but are now considered heterogeneous. There are about 70 subsets of γδ T cells according to the hypervariable γ and δ chain but to date, only two of them, Vγ9Vδ1+ and Vγ9Vδ2+, have been exploited for ACT [105, 106]. Vγ9Vδ2+ T cells are the predominant subset of γδ T cells, but the Vγ9Vδ1+ subset may possess stronger antitumor activity. In 1996, Lotze reported that γδ T cells could recognize epithelium-derived malignant cells [107]. Kakimi and Ogawa independently reported in 2008 that γδ T cells could be isolated from PBMCs and expanded rapidly in vitro and secreted effector molecules such as IFN-γ, and showed cytotoxicity in a range of solid malignancy tumors including CRC [108, 109]. More recently, Capone and colleagues reported that systemic treatment of a CRC model with the human Vδ1+ γδ T cells not only controlled primary tumor growth but also prevented metastasis in the lung and liver [110]. Similarly, Jian et al. reported that the human Vδ1+ γδ T cells prepared from PBMCs showed more potent therapeutic activity than Vδ2+ γδ T cells in colon cancer both in vivo and in vitro, suggesting that Vδ1+ γδ T cells may be a promising candidate for the ACT for the treatment of CRC [111]. Further investigations are needed to improve the in vitro expansion strategies of γδ T cells and to better understand the mechanisms underlying the therapeutic efficacy of γδ T cells.
ACT using stem cell-derived immune cells
It is often difficult to get sufficient numbers of lymphocytes from patients, because: (a) most of the patients have been treated with chemotherapy and/or radiotherapy and have severe immune deficiency; (b) the immunogenicity of transferred allogeneic cells may induce autoimmunity; (c) a long waiting time-period for individual production; (d) high cost and unstable quantity of cell production. Therefore, the availability of “universal” immune cells for ACT becomes an overriding consideration. Pluripotent stem cells are believed to meet the requirements and can be easily passaged and expanded in vitro. The immune cells derived from pluripotent stem cells have the characteristics of low immunogenicity, potent persistence in vivo, and resistance to apoptosis. More importantly, pluripotent stem cells can be easily engineered in vitro with required genes and yield tumor-specific lymphocytes bearing TAA-specific receptor (TCR or CAR). Thus, pluripotent stem cells are suitable for generating homogenous cells for the ACT as well as reducing the cost (Figure 3).
Figure 3
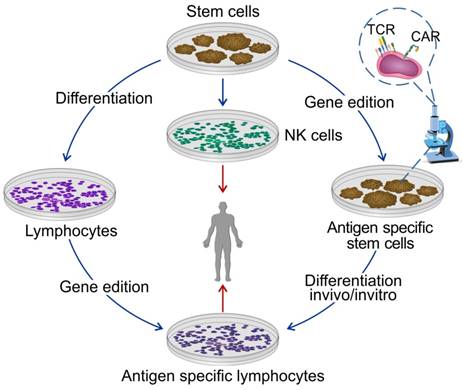
Differentiated immune cells from pluripotent stem cells for the ACT. Stem cells are considered as the most promising cell source for universal production of the ACT. Stem cells can differentiate into T cells or NK cells before or after gene edition.
Table 3
TAAs exploited for CRC ACT
| TAAs | Interventions | Sponsor | Status | clinicaltrials.gov Identifier |
|---|---|---|---|---|
| CEA | CAR-T | Shanghai Tumor Hospital | Recruiting | NCT02959151 |
| CEA | CAR-T | Christie Hospital Manchester | Terminated (due to safety concerns and lack of efficacy) | NCT01212887 |
| CEA | CAR-T | Roger Williams Medical Center Providence | Suspended (Funding) | NCT01723306 |
| CEA | CAR-T | Southwest Hospital of Third Military Medical University | Recruiting | NCT02349724 |
| MUC1 | CAR-T | PersonGen Biomedicine (Suzhou) Co. Ltd | Recruiting | NCT02617134 |
| MUC1 | CAR-pNK | PersonGen Bio Therapeutics (Suzhou) Co. Ltd. | Recruiting | NCT02839954 |
| HER2 | CAR-pNK | Southwest Hospital of Third Military Medical University | Recruiting | NCT02713984 |
| CD133 | CAR-T | Chinese PLA General Hospital | Recruiting | NCT02541370 |
| EpCAM | CAR-T | IEC of Chengdu Medical College | Recruiting | NCT03013712 |
| EGFR | CAR-T | The Second People's Hospital of Shenzhen | recruiting | NCT03152435 |
| Mutant KRAS G12D | Young TILs | National institutes of health clinical center | Recruiting | NCT01174121 |
| P53 | Soluble TCR with IL2 gene | University of Colorado MD Anderson Cancer Center Orlando H.Lee Moffitt Cancer Center & Research Institute Tampa University of Washington, Seattle Cancer Care Center Seattle | Completed | NCT00496860 |
| KRAS G12 V | HLA-A*1101 restricted mTCR | National Institutes of Health Clinical Center | recruiting | NCT03190941 |
| Anti CEA | Murine derived Ig TCR | Beth Israel Deaconess Medical Center Boston, Massachusetts, United States | completed | NCT00004178 |
There are several types of stem cells with the potential to differentiate into lymphocytes, including embryonic stem cells (ESCs), hematopoietic stem cells (HSCs), mesenchymal stem cells (MSCs), umbilical cord blood stem cells (UCB-SCs), and induced pluripotent stem cells (iPSCs). Several studies have reported the ability of pluripotent stem cells to differentiate into T lineage cells in vitro and in vivo [112-114]. ESCs are the most potent stem cells that can produce lymphocytes, but, due to ethical issues, it is difficult to obtain ESCs. iPSCs provide another homogenous starting cell platform for the ACT because they have characteristics similar to ESCs. Both ESCs and iPSCs can be genetically engineered in vitro to produce specific cells to serve as a “universal” cell source for cancer killing lymphocytes. By contrast, genetic modification is often difficult with PBMCs-derived NK and T cells. In 2011, Song et al. reported a proof-of-concept study of obtaining Ag-specific T lymphocytes from iPSC for the ACT. In this study, they demonstrated that the genetically modified iPSCs with ovalbumin (OVA)-specific TCR could develop OVA-specific CD8+ T cells in vivo and control tumor growth [115]. More recently, the same group reported another seminal study in which OVA-specific CTLs were developed through a combination of in vitro and in vivo differentiation. The investigators demonstrated that adoptive transfer of iPSC-derived CTLs could successfully suppress tumor growth and that the integration of BCL-xL and surviving genes could enhance the persistence and anti-tumor effect of the CTLs [116]. Kawamoto et al. reported a study in which they generated iPSCs from mature CTLs specific for MART-1 (a melanoma antigen epitope) and regenerated the iPSCs back to T lymphocytes. They showed that the iPSCs could efficiently generate TCRβ+CD4+CD8+ cells expressing MART-1-specific TCR, and the regenerated T cells displayed specific reactivity to MART-1 epitope [117]. UCB-SCs are another important cell source to produce cells for the ACT and reliable to yield NK cells from UCB-SCs. Hans et al. demonstrated the antitumor activity of UCB-NK cells [97], but, compared with the ESC-derived NK cells, UCB-NK cells displayed less cytotoxicity against tumor cells because of their functional immaturity [118]. Although stem cell-derived immune cells display several advantages for the ACT, there are still hurdles to their successful application in clinical trials. Since it is difficult to develop human-derived feeder cells, murine feeder cells are used for propagating T lymphocytes in vitro which may result in immunogenicity in patients. Another major obstacle is that the differentiation of T lymphocytes from stem cells is not stable, and it is a major challenge to increase the stability and standardize this approach.
Gene engineering strategies in ACT
The major advance of the ACT during the past decades is of the gene editing technique, which makes it possible to generate TAA-specific effector cells for the tumors that lack immunogenicity or to reprogram the adoptively transferred cells to improve their persistence and/or anti-apoptosis potential. Table 3 lists a variety of TAAs that are being tested in CRC. The approaches to modify effector cell's genome can be classified into two categories: gene insertion and gene knockout. As the gene knockout technique has been used in several experimental therapies, here we mainly focus on the gene insertion techniques that have been used in the treatment of CRC. Gene insertion is a strategy to insert an artificial or selected gene into the immune cell's genome to enhance the specificity, persistence, and anti-tumor activity. The goal can be achieved mainly through viral vectors, such as retroviruses and the lentivirus system, or transposons and electroporation technique. TCR and CAR are the star genes that have been tested in numerous experiments and clinical trials. Other genes with the ability to improve the antitumor effects of effector cells, such as synthetic Notch receptor, are being tested.
TCR engineering
T lymphocytes work in a TCR-dependent manner, as immune surveillance and mutated cell eradication depend on recognition of abnormal antigens by TCR on the surface of T lymphocytes. However, most cancer cells lack specific antigen expression and/or have a downregulated expression of MHC class I molecule and therefore cannot be recognized by effector T cells. There may be naturally occurring antitumor response caused by tumor-specific TILs, but they may be in small numbers or inhibited by the immunosuppressive mechanism. To overcome this problem, a possible scheme is to exploit artificial TCRs or screen naturally occurring TCRs targeting the antigens overexpressed by cancer cells but not expressed or expressed at a low level by normal cells and engineer infused cells with these TCR genes. T cells with engineered TCRs have demonstrated therapeutic potential in immunotherapy, although they only have a limited antigen repertoire and still need antigen presentation.
An artificial TCR is generally produced from immunized murine, because the TCRs bypass the self-tolerance process of the natural TCRs in the thymus, and theoretically they can be designed to target self-antigens [119]. Although artificial TCRs can elicit a potent antitumor effect, they may cause severe adverse effects and immunogenicity in humans. In a reported phase I study, three patients with advanced CRC refractory to conventional treatments were infused with autologous T cells genetically engineered with murine-derived CEA-targeted TCRs. All three patients showed a profound decrease in the serum CEA level and one patient experienced tumor regression; nevertheless, all patients had severe autoimmune colitis. The results of the study indicated the risk of using the antigens expressed by cancer cells as well as normal tissues as the target of TCRs [120]. Due to this risk, further investigations of the CEA-specific TCRs have been suspended. On the other hand, use of naturally occurring TCRs in cancer treatment appears to be promising. In 1991, van der Bruggen identified the first human TAA-MAGEA1 antigen in a melanoma patient and cloned the gene which was a hallmark in ACT therapy using TCRs [121]. Since then, many other antigen genes have been cloned [122, 123]. NY-NEO-1, a cancer testis antigen (CTA), is an attractive target antigen for immunotherapy, although it is only expressed in a minority of CRC patients [124] but can be induced by DNA methylation inhibitor, 5-aza-2'-deoxycytidine (DAC). The approach has been used to enhance the sensitivity of tumor cells to NY-ESO-1 TCR-transduced T cells. Richard and Edus reported a study combining epigenetic modulation of cancer cells with DAC and ACT based on NY-ESO-1-targeting TCRs [125, 126]. They induced NY-ESO-1 expression in CRC cells both in vitro and in vivo, then treated the cells and tumor-bearing animals with the NY-ESO-1 specific TCR-engineered T cells. They observed that the epigenetic modulation could sensitize CRC cells to NY-ESO-1-specific T cells. Gustav et al. isolated a naturally occurring TCR from a long-term surviving CRC patient who received treatment with a synthetic TGFβRII peptide vaccine, and then engineered the autologous T cells with the TCR gene. They demonstrated that the genetically modified cells could produce several cytotoxic cytokines following a short incubation with the target cells [127].
In a xenograft mouse model of colon cancer, significant suppression of tumor growth and improved survival rate were achieved following infusion of the T cells [125]. This pilot study paved a new way in the development of ACT through use of TCRs from successfully vaccinated patients. In a recent study reported by Rosenberg's group mentioned previously, four different TCR subsets specific to mutant KRAS G12D were identified, synthesized, and transduced to autologous T cells. The transduced T cells could recognize the allogeneic pancreatic tumor cells expressing a mutant KRAS G12D [49]. The results indicated the possibility that we can clone TCRs targeting some mutant epitopes from one patient and modify T cells with the TCRs to target allogeneic patients bearing different tumors but with the same mutant epitopes. However, screening a TAA-specific TCR among millions or even billions of naturally occurring TCRs remains a technical challenge. Besides, there are several other problems associated with the application of the TCR-engineered T cells. For instance, TCR-antigen recognition requires the antigen presented by MHC class I molecule to be expressed on the cancer cell surface, but most cancer cells have a down-regulation of MHC class I expression [128, 129]. Although one TCR recognizes one antigen epitope theoretically, the on-target off-tumor effect can cause lethal side effects and must be considered carefully as some TCRs not only recognize the epitope on the targeting antigen but also target similar epitopes on unknown antigens. Therefore, exploring and developing new more powerful and specific approaches with less/minimal side effects are imperative. Also, the immunogenicity of most types of cancers to host lymphocytes is low because of the positive and negative selection through the immune system during the development of malignancy. This suggests that with the exception of some mutant peptides, there is a lack of exclusive tumor-specific antigens, which are not expressed on normal tissues. However, the existence of naturally occurring antitumor response and the complete regression of some tumors under nonspecific immune activation therapy, such as IL-2 and checkpoint inhibitors, indicate that combined targeting of the TCR may eliminate malignant cells.
CAR engineering
TCR initiates T cell activation and the activated T cells exert antitumor effects. Since it is difficult to utilize tumor-specific TCR and TCR-specific antigens are rare, efforts have been made to exploit synthetic molecules to mimic TCR signaling that targets selected antigens. These efforts began three decades ago when Gross et al. used an artificial molecule containing a double CD3 ζ chain as the intercellular signaling domain and heavy and light chains of an antibody as the extracellular binding domain [130]. As described by Irving [131] and Romeo [132], the first-generation CAR was exploited by Eshhar et al. in 1992 with a chimeric single chain comprising an antibody-binding domain and a CD3 ζ chain [133]. The basic structure of CAR consists of a costimulatory molecule signal and a transmembrane domain. Indeed, CARs are a series of artificial genes that comprise an extracellular antigen-binding domain, an extracellular spacer, a transmembrane hinge domain, and an intracellular signaling domain with costimulatory molecules such as CD28, and/or 4-1BB, and/or OX40, and a T cell activating signaling domain-CD3ζ chain [134, 135]. The extracellular component derived from an antibody single chain variable fragment (scFv) binds with a specific TAA in an MHC-independent manner. The hinge domain, generally derived from CD4, CD8, CD28, or IgG1 molecule, connects scFv with the intracellular domain derived from a costimulatory molecule and then CD3ζ molecule. The intracellular domain of the first-generation CAR was designed by using a CD3ζ domain. The second and third generation of CARs has one or two costimulatory domains with a CD3ζ segment. The gene complex is typically inserted into the T cell genome using γ-retroviral or lentiviral transduction systems. Unlike TCR-recognizing peptide epitopes, the variable region of CAR may recognize protein, lipid, or carbohydrate motifs, i.e., CAR may recognize a wider range of antigens in an MHC-unrestricted fashion [126]. The affinity and avidity of CAR with its targeting antigen can be tuned by designing the molecular structure, and one TAA can be selected by different epitopes to be exploited as a CAR target.
Compared to TCR, CAR is considered as more feasible in both experimental and clinical therapies. The clinical success of CD19-targeted CAR-T cells in hematological malignancies has attracted great attention in the last decade. Although the success of ACT in treating solid tumors is limited, progress is on the way, and several CAR-T-based ACT clinical trials for solid tumors are currently ongoing [136-138]. CEA might also be an important target of CAR in CRC, because it showed effectiveness in some late-stage CRC patients. In 2006, Robert and Takashi independently developed a protocol to transduce CEA-targeted CAR genes into PBMCs by using lentivirus and retrovirus systems, and successfully edited PBMCs with the CAR genes and demonstrated its potent cytotoxic effect against CEA+ tumor cells in vitro and in vivo [139, 140]. More recently, Qian et al. reported a phase I clinical trial of the ACT with CAR against CEA in CRC patients with CEA+ metastases. They observed that after infusion with the CAR-T cells, 70% of the patients refractory to conventional treatments experienced stable disease with two patients showing tumor shrinkage, and no severe adverse effects occurred. Furthermore, most of the patients had a long-term reduction of serum CEA. The persistent CAR-T cells in peripheral blood could be detected for a long period of time, especially in the patients who received a second CAR-T cell infusion [141].
CEA-targeted CAR-T cells can also be used regionally to treat the peritoneal and liver metastases, the deadliest complications of the advanced CRC and the major cause of death. A group at Boston University reported a series of pre-clinical and clinical trials using the CEA-targeted CAR-T cells infused via the hepatic artery to treat CRC liver metastasis. They showed the safety and effectiveness of CAR-T cells in the regional treatment as compared to systemic delivery [142-144]. More recently, this team reported a pre-clinical study demonstrating that intraperitoneal infusion of CAR-T cells could more effectively reduce the peritoneal tumor burden than systemic delivery. They also observed that regional delivery of CAR-T cells into the intraperitoneal cavity not only provided durable protection against intraperitoneal metastases but also restrained the growth of extra-abdominal tumor [145]. Guanylate cyclase C (GUCY2C) is a membrane-bound cyclase selectively expressed on the apical surfaces of the normal intestinal epithelium from the duodenum to the rectum and has limited expression in extra-intestinal tissues [146]. Primary and metastatic CRC, esophageal, and gastric cancers universally overexpress GUCY2C [147-150]. Magee et al. found in a murine model with CRC metastases that the GUCY2C-targeted CAR-T cells showed an immense potential to treat CRC metastases without autoimmune adverse effect which is likely due to GUCY2C expression on the apical surfaces of intestinal epithelial cells. This study established the first proof-of-principle for the GUCY2C CAR-T cells as a candidate for the ACT in treating CRC patients [151].
Tumor-associated glycoprotein-72 (TAG-72) is an oncofetal mucin-like glycoprotein overexpressed by most human epithelial adenocarcinomas including CRC but rarely in benign or normal cells [152-154]. TAG-72 was first used in immunotherapy as a target of monoclonal antibody but did not show any significant effect. Moghimi et al. developed TAG-72 as a target of CAR-T cells and generated four TAG-72-specific CARs with the same antigen binding site but with different spacers and signaling domains [155]. Their results suggested that TAG-72-targeting CARs might be a promising tool for the ACT. Two Phase I clinical trials using the first-generation TAG-72 CAR-T cells were conducted two decades ago. In these studies, the investigators infused the TAG-72 CAR-T cells through intravenous or hepatic artery injection in patients with advanced CRC and observed infiltration of TAG-72-specific engineered CAR-T cells into the tumor [156].
Despite the promising results of the CAR-based ACT, further studies are needed to validate this approach for CRC especially because an extremely adverse reaction was observed in a patient with advanced CRC after treatment with the ERBB2-targeting CAR-T cells. This patient received adoptive transfer of the T cells engineered to express ERBB2-targeted CARs containing CD28, 4-1BB and CD3 ζ signaling moieties; within 15 minutes after cell infusion, the patient experienced respiratory distress and died 5 days later. This case alerts us to the risk of ACT using CAR-T cells and the importance of the target antigen epitope of CAR [157]. Because the T cells have to be activated and expanded for a long time before and after gene engineering, persistence is another important problem that needs to be resolved for CAR-T therapy.
Combination of different strategies for the ACT
As promising immunotherapy for cancer, ACT has its advantages in expanding the cohort of in vivo tumor-specific effector lymphocytes. However, this therapy also has disadvantages, which mainly include the lack of tumor-specific antigens and poor stability and trafficking of the transferred cells. A variety of strategies have been explored to overcome these shortcomings and to sensitize tumor cells to ACT. As mentioned above, modification of artificial genes for engineering cells to be transferred is an active field. ACT, in combination with other therapies, is another attractive approach to enhancing the effectiveness of the ACT. Chemotherapy and/or radiotherapy, or lymphodepletion to yield space for transferred cell expansion are often used before ACT as a general treatment. In a pilot study, Hodge et al. demonstrated that irradiation could enhance Fas expression in the MC38 cell line (a colorectal cancer cell line) and improve the in vivo as well as in vitro antitumor activity of the infused CEA-targeting CD8+ lymphocytes [158]. Monoclonal antibodies have been extensively used to treat malignant tumors as a single regimen and can also be utilized to enhance the effect of NK cells likely by binding with Fc receptors (CD16) and mediating ADCC (antibody-dependent cell-mediated cytotoxicity). Also, it was reported that an EGFR monoclonal antibody, cetuximab, can enhance the antitumor ability of NK cells [90].
The feasibility and safety of the combination of chemotherapy or monoclonal antibody with transferred αβ T cells were demonstrated in a phase I clinical study in patients with stage IV CRC [100-102]. Surgery in combination with ACT has been tested in clinical trials in patients with hepatic metastases. Promising results were obtained after injecting the CAR-T cells through the liver artery during radical liver resection. Regional delivery can enhance the trafficking of the transferred cells within the tumor site and alleviate or avoid the adverse effects of the gene-modified cells. Cytoreduction by surgery can enhance the penetration of infused cells by decreasing the mass of tumor which is beneficial for the ACT. Thus, a combination of different immunotherapies may hold the most promising prospect for the ACT because they can modulate the immune surveillance with different targets or mechanisms, produce synergistic effects, and/or neutralize the adverse effects.
Immune checkpoint inhibitors are an important component of immunotherapy and have shown impressive results in several malignant diseases. Checkpoint molecules belong to B7 family proteins, a group of important negative modulators of T cell activation which play important roles in avoiding an autoimmune reaction. Most of the checkpoint molecules can be exploited by cancer cells and cancer stroma cells (MDSC and Treg cells) to induce immune tolerance and T cells anergy. Increased expression of immune checkpoint proteins is one of the most important characteristics of cancer cells and activated T cells, and checkpoint signals are the leading cause of anergy and apoptosis of transferred T cells. The combination with a checkpoint inhibitor may improve the trafficking of infused cells and reverse their anergy. In a pilot study published in 2013, it was reported that PD-1 blockade could increase the activation and proliferation of the Her-2-targeting CAR-T cells in vitro and regress the growth of established tumors in vivo. The PD-1 inhibitor did not increase the number of CAR-T cells in the tumor, but enhanced their antitumor effect and was associated with a reduction of MDSCs at the tumor site, suggesting that PD-1 blockade can improve the immune microenvironment of the tumor [159].
Costimulatory molecules may be another promising target that can be exploited to improve the ACT. It was demonstrated that 4-1 BB was expressed in activated T cells especially TILs [160, 161], and the agonistic 4-1 BB monoclonal antibody could increase the antitumor response through directly activating CD8+ T cells [162, 163]. More recently, it was reported that in Her2 + colon cancer, the Her-2-targeting CAR-T cells had an increased 4-1 BB expression upon contact with targeted cells, and treatment with an anti-4-1 BB enhanced the IFN γ secretion by CAR-T cells in vitro. Further, CAR-T in combination with the 4-1 BB mAb could completely regress the established tumors in mice without causing autoimmunity [164]. This study also reported reductions of MDSCs and Tregs, and mature NK cells within the tumor site, which might be the underlying mechanism for the increased antitumor activity of the combination of CAR-T with the 4-1 BB antibody [164].
Immunotherapy for malignant cancers utilizing vaccines can also be exploited to enhance the specificity of transferred cells. Tumor vaccines consist of whole tumor cell lysates, DC vaccines, peptide vaccines, and mRNA vaccines. DCs as an antigen presenting cells can prime and activate naïve T cells through MHC-peptide complex and co-stimulatory molecules pathway and are often used together with effector T cells especially CIK cells. Two clinical trials combining DC vaccines and CIK cells for CRC patients showed the elevation of serum levels of cytokines and OS benefit without severe adverse effects except for fever [75, 76]. The potential deficiency of the DC-CIK therapy is that the percent of the T cells with corresponding TCR that can bind with the peptide presented by the DCs may be low in the whole pool of transferred CIK cells, and the efficiency of the antigen presentation is not high. Currently, studies are exploring the personal neoantigen vaccine and RNA mutanome vaccines for enhancement of the specificity and efficiency of antigen presentation [165, 166].
Conclusions and Perspectives
Although the therapeutic potential of the ACT in the treatment of cancers has been increasingly appreciated, there are still numerous impediments to the effectiveness of ACT in the treatment of CRC and other solid tumors. These drawbacks include brief persistence and poor trafficking of immune cells within the immunosuppressive tumor microenvironment, anergy, and weak and inadequate proliferation of immune cells as well as removal of the gene-engineered T cells upon tumor regression. Some of these problems can be resolved by better manipulating the engineered T cells. However, most of the obstacles mentioned above can only be overcome by a combination of other treatment methods, such as chemo and/or radiotherapy, surgery, and use of check-point inhibitors, monoclonal antibodies, DC vaccines, and even some small molecules that can modulate the metabolism and immune microenvironment of cancer cells. As solid tumors are extremely heterogeneous and may possess multiple immunosuppressive mechanisms, it is imperative to explore more effective and personalized strategies to strengthen the efficacy of ACT.
Most of the preclinical and clinical studies of ACT used unselected T cells including cytotoxic, helper, and even immune suppressive Treg cells, as cytotoxic T cells cannot proliferate without helper T cells. Recently, CD4+ and CD8+ T cells were modified with various CAR genes. CD4+ and CD8+ T cells could be redirected with a CAR gene containing ICOS signaling domain and a CAR gene containing CD28 or 4-1BB signaling domain, respectively. The ICOS signaling dramatically enhanced the in vivo stability of CD4+ CAR T cells thereby increasing the persistence of CD8+ T cells, because of the high dependence of CD8+ CAR T cells on the helper effect provided by the redirected CD4+ CAR T cells [98]. These studies may pave the way for further investigating the cooperation of CD4+ and CD8+ T cells. Another critical issue of the ACT lies in the genes used to edit transferred cells. The naturally occurring in vivo antitumor response suggests the existence of tumor-specific epitopes and TCRs. However, despite considerable advances in the techniques, it is difficult to identify such epitopes or TCRs among the repertoire of billions of candidates. Designing of CAR molecules may present another challenge to the successful application of the ACT. CD3 and CD28 signaling pathways are important for the initiation of T cell priming but adversely affect T cell activation and proliferation if they are stimulated repeatedly. This may be one of the reasons for the anergy of transferred cells since the CAR is designed to activate CD3 and CD28 signaling pathways forcibly. A recent study reported a novel CAR that stimulated IL-2R intracellular signaling pathway JAK-STAT and demonstrated the advantage of the novel CAR T cells with antitumor effects in blood cancers and solid tumors as compared with conventional second generation CAR with CD28 or 4-1BB co-stimulatory domain [167]. Since the IL2R signaling pathway is the main mechanism for maintaining the function and proliferation of activated T cells, this approach may represent another promising strategy of ACT gene design-for stimulating the IL2R signaling pathway. It is believed that ACT, alone or in combination with other therapies, may hold great promise for the treatment of CRC and other cancers.
Abbreviations
CRC, colorectal cancer; ACT, adoptive cell therapy/adoptive cell transfer; OS, overall survival; NK, natural killer; CTL, cytotoxicity T lymphocytes; DC, dendritic cells; APC, antigen-presenting cells; TAA, tumor-associated antigen; TIL, tumor infiltrateing lymphocytes; LNL, lymph node lymphocyte; PBMCs, peripheral blood mononuclear cells; iPSC, induced pluripotent stem cell; LAK, lymphokine-activated killer; CIK, cytokine-induced killer; TCR, T cell receptor; CAR, chimeric antigen receptor.
Acknowledgements
This study was supported by a grant from the National Natural Science Foundation of China (No. 81573751).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2015;65:5-29
2. Torre LA, Bray F, Sieqel RL, Siegel RL, Ferlay J. et al. Global cancer statistics 2012. CA Cancer J Clin. 2015;65:87-108
3. Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordinger B. et al. Colorectal cancer. Lancet. 2010;375:1030-47
4. Fong Y, Cohen AM, Fortner JG, Enker WE, Turnbull AD, Coit DG. et al. Liver resection for colorectal metastases. J Cancer Clin Oncol. 1997;15:938-46
5. Van Cutsem E, Nordlinger B, Adam R, Kohne CH, Pozzo C, Poston G. et al. Towards a pan-European consensus on the treatmsent of patients with colorectal liver metastases. Eur J Cancer. 2006;42:2212-21
6. Mlecnik B, Tosolini M, Charoentong P, Kirilovsky A, Bindea G, Berger A. et al. Biomolecular network reconstruction identifies T-cell homing factors associated with survival in colorectal cancer. Gastroenterology. 2010;138:1429-40
7. Stoll G, Bindea G, Mlecnik B, Galon J, Zitovgel L, Kroemer G. Meta-analysis of organ-specific differences in the structure of the immune infiltrate in major malignancies. Oncotarget. 2015;6:11894-909
8. Mukherjee P, Glinardi AR, Madsen CS, Tinder TL, Jacobs F, Parker J. et al. MUC1-specific CTLs are non-functional within a pancreatic tumor microenvironment. Glycoconj J. 2001;18:931-942
9. Soliman H, Mediavilla-Varela M, Antonia S. Indoleamine 2.3-dioxygenase: is it an immune suppressor. Cancer J. 2010;16:354-9
10. Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res. 2011;71:6921-25
11. Corbet C, Feron O. Tumour acidosis: from the passenger to the driver's seat. Nat Rev Cancer. 2017;17:577-93
12. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74-80
13. Nakagomi H, Petersson M, Magnusson I, Juhlin C, Matsuda M, Mellstedt H. et al. Decreased expression of the signal-transducing ζ chains in tumor-infiltrating T-cells and NK cells of patients with colorectal carcinoma. Cancer Res. 1993;53:5610-5612
14. Finke JH, Zea AH, Stanley J, Longo DL, Mizoguchi H, Tubbs RR. et al. Loss of T-cell receptor ζ chain and p56lck in T-cells infiltrating human renal cell carcinoma. Cancer Res. 1993;53:5613-16
15. Yoong K, Adams D. Interleukin 2 restores CD3-ζ chain expression but fails to generate tumour-specific lytic activity in tumour-infiltrating lymphocytes derived from human colorectal hepatic metastases. Br J Cancer. 1998;77:1072-81
16. Frydecka I, Kaczmarek P, Bocko D, Kosmaczewska A, Morilla R, Catovsky D. Expression of signal-transducing zeta chain in peripheral blood T cells and natural killer cells in patients with Hodgkin's disease in different phases of the disease. Leuk Lymphoma. 1999;35:545-54
17. Takahashi A, Kono K, Amemiya H, Iizuka H, Fujii H, Matsumoto Y. Elevated caspase-3 activity in peripheral blood T cells coexists with increased degree of T-cell apoptosis and down-regulation of TCR zeta molecules in patients with gastric cancer. Clin Cancer Res. 2001;7:74-80
18. Dworacki G, Meidenbauer N, Kuss I, Hoffmann TK, Gooding W, Lotze M. et al. Decreased ζ chain expression and apoptosis in CD3+ peripheral blood T lymphocytes of patients with melanoma. Clin Cancer Res. 2001;7:947-57
19. Rosenberg SA, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR. et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271:907-13
20. Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol. 2014;192:5451-58
21. Mitchison N. Studies on the immunological response to foreign tumor transplants in the mouse. J Exp Med. 1955;102:157-77
22. Fefer A. Immunotherapy and chemotherapy of Moloney sarcoma virus-induced tumors in mice. Cancer Res. 1969;29:2177-83
23. Rosenberg SA, Terry WD. Passive immunotherapy of cancer in animals and man. Adv Cancer Res. 1977;25:323-88
24. Kono K, Ichihara F, Iizuka H, Sekikawa T, Matsumoto Y. Expression of signal transducing T-cell receptor ζ molecules after adoptive immunotherapy in patients with gastric and colon cancer. Int J Cancer. 1998;78:301-05
25. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rherngold SR. et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509-18
26. Avanzi MP, Brentjens RJ. Emerging role of CAR T cells in non-Hodgkin's lymphoma. J Natl Compr Canc Netw. 2017;15:1429-37
27. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST. et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. N Engl J Med. 1988;319:1676-80
28. Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299-08
29. Hershkovitz L, Schachter J, Treves AJ, Besser MJ. Focus on adoptive T cell transfer trials in melanoma. Clin Dev Immunol. 2010;2010:260267
30. Robbins PF, Morgan RA, Feldman SA, Yang JC, Shermy RM, Dudley ME. et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Cancer Clin Oncol. 2011;29:917-24
31. Maio M. Melanoma as a model tumour for immuno-oncology. Ann Oncol. 2012;23(suppl_8):viii10-4
32. Kurokawa T, Oelke M, Mackensen A. Induction and clonal expansion of tumor-specific cytotoxic T lymphocytes from renal cell carcinoma patients after stimulation with autologous dendritic cells loaded with tumor cells. Int J Cancer. 2001;91:749-56
33. Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C. et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21:904-12
34. Secondino S, Zecca M, Licitra L, Gurrado A, Schiavetto I, Bossi P. et al. T-cell therapy for EBV-associated nasopharyngeal carcinoma: preparative lymphodepleting chemotherapy does not improve clinical results. Ann Oncol. 2011;23:435-41
35. Kono K, Takahashi A, Sugai H, Fuji H, Choudhury AR, Kiessling R, Matsumoto Y. Dendritic cells pulsed with HER-2/neu-derived peptides can induce specific T-cell responses in patients with gastric cancer. Clin Cancer Res. 2002;8:3394-00
36. Takayama T, Sekine T, Makuuchi M, Yamasaki S, Kosuge T, Yamamoto J. et al. Adoptive immunotherapy to lower postsurgical recurrence rates of hepatocellular carcinoma: a randomised trial. Lancet. 2000;356:802-07
37. Ratto GB, Zino P, Mirabelli S, Minuti P, Aguilina R, Fantino G. et al. A randomized trial of adoptive immunotherapy with tumor-infiltrating lymphocytes and interleukin-2 versus standard therapy in the postoperative treatment of resected nonsmall cell lung carcinoma. Cancer. 1996;78:244-51
38. Vose BM, Vanky F, Argov S, Klein E. Natural cytotoxicity in man: activity of lymph node and tumor-infiltrating lymphocytes. Eur J Immunol. 1977;7:753-57
39. Klein E, Svedmyr E, Jondal M, Vanky F. Functional studies on tumor-infiltrating lymphocytes in man. Isr J Med Sci. 1977;13:747-52
40. Klein E, Vanky F, Galili U, Vose BM, Foop M. Separation and characteristics of tumor-infiltrating lymphocytes in man. Contemp Top Immunobiol. 1980;10:79-107
41. Rosenberg S, Spiess AP, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233:1318-21
42. Jass J. Lymphocytic infiltration and survival in rectal cancer. J Clin Pathol. 1986;39:585-89
43. Fridman WH, Galon J, Dieu-Nosjean MC, Cremer I, Fisson S, Damotte D. et al. Immune infiltration in human cancer: prognostic significance and disease control. Curr Top Microbiol Immunol. 2011;344:1-24
44. Pages F, Galon J, Dieu-Nosjean MC, Tatour E, Sautes-Fridman C, Fridman WH. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene. 2010;29:1093-02
45. Pagès F, Galon J, Fridman WH. The essential role of the in situ immune reaction in human colorectal cancer. J Leukocyte Biol. 2008;84:981-87
46. Galon J, Fridman WH, Pagès F. The adaptive immunologic microenvironment in colorectal cancer: a novel perspective. Cancer Res. 2007;67:1883-86
47. Gardini A, Erolani G, Riccobon A, Ravaioli M, Ridofi L, Flamini E. et al. Adjuvant, adoptive immunotherapy with tumor infiltrating lymphocytes plus interleukin-2 after radical hepatic resection for colorectal liver metastases: 5-year analysis. J Surg Oncol. 2004;87:46-52
48. Dillman R, Schiltz P, DePriest C, Barth N, Beutel L, de Leon C. et al. Tumor-infiltrating lymphocytes and interleukin-2: dose and schedules of administration in the treatment of metastatic cancer. Cancer Biother Radiopharm. 2004;19:730-37
49. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L. et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. 2016;375:2255-62
50. Rech AJ, Vonderheide RH. T-Cell transfer therapy targeting mutant KRAS. N Engl J Med. 2017;376:e11
51. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV. et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415-21
52. Yee C, Riddell SR, Greenberg PD. Prospects for adoptive T cell therapy. Curr Opin Immunol. 1997;9:702-08
53. Frost P, Caliliw R, Belldegrun A, Bonavida Bl. Immunosensitization of resistant human tumor cells to cytotoxicity by tumor infiltrating lymphocytes. Int J Oncol. 2003;22:431-37
54. Marits P, Karlsson M, Dahi K, Larsson P, Wanders A, Thorn M. et al. Sentinel node lymphocytes: tumour reactive lymphocytes identified intraoperatively for the use in immunotherapy of colon cancer. Br J Cancer. 2006;94:1478-84
55. Satoh K, Kan N, Okino T, Mise K, Yamsaki S, Harada T. et al. The therapeutic effect of OK-432-combined adoptive immunotherapy against liver metastases from gastric or colorectal cancers. Biotherapy. 1993;6:41-49
56. Kim JA, Bresler HS, Martin EW Jr, Aldrich W, Heffelfinger M, Triozzi PL. Cellular immunotherapy for patients with metastatic colorectal carcinoma using lymph node lymphocytes localized in vivo by radiolabeled monoclonal antibody. Cancer. 1999;86:22-30
57. Karlsson M, Martis P, Dahi K, Dagoo T, Enerback S, Thorn M. et al. Pilot study of sentinel-node-based adoptive immunotherapy in advanced colorectal cancer. Ann Surg Oncol. 2010;17:1747-57
58. Zhen YH, Liu XH, Yang Y, Li B, Tang JL, Zeng QX, e t al. Phase I/II study of adjuvant immunotherapy with sentinel lymph node T lymphocytes in patients with colorectal cancer. Cancer Immunol Immunother. 2015;64:1083-93
59. Schmidt-Wolf IG, Negrin RS, Kiem HP, Blume KG, Weissman IL. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J Exp Med. 1991;174:139-49
60. Scheffold C, Brandt K, Johnston V, Lefterova P, Degen B, Schontube M. et al. Potential of autologous immunologic effector cells for bone marrow purging in patients with chronic myeloid leukemia. Bone Marrow Transplant. 1995;15:33-39
61. Schmidt-Wolf IG, Lefterova P, Johnston V, Scheffold C, Csipai M, Mehta BA. et al. Sensitivity of multidrug-resistant tumor cell lines to immunologic effector cells. Cell Immunol. 1996;169:85-90
62. Ren X, Yu J, Liu H, Zhang P, An X, Zhang N. et al. Th1 bias in PBMC induced by multicycles of auto-CIKs infusion in malignant solid tumor patients. Cancer Biother Radiopharm. 2006;21:22-33
63. Lu PH, Negrin RS. A novel population of expanded human CD3+ CD56+ cells derived from T cells with potent in vivo antitumor activity in mice with severe combined immunodeficiency. J Immunol. 1994;153:1687-96
64. Zoll B, Lefterova P, Csipai M, Finke S, Trojaneck B, Ebert O. et al. Generation of cytokine-induced killer cells using exogenous interleukin-2,-7 or-12. Cancer Immunol Immunother. 1998;47:221-26
65. Sangiolo D, Martinuzzi E, Todorovic M, Vitaggio K, Vallario A, Jordaney N. et al. Alloreactivity and anti-tumor activity segregate within two distinct subsets of cytokine-induced killer (CIK) cells: implications for their infusion across major HLA barriers. Int Immunol. 2008;20:841-48
66. Wang Y, Dai H, Li H, Lv H, Wang T, Fu X. et al. Growth of human colorectal cancer SW1116 cells is inhibited by cytokine-induced killer cells. Clin Dev Immunol. 2011;2011:621414
67. Schlimper C, Hombach AA, Abken H, Schmidt-Wolf IG. Improved activation toward primary colorectal cancer cells by antigen-specific targeting autologous cytokine-induced killer cells. Clin Dev Immunol. 2012;2012:238924
68. Pievani A, Borleri G, Pende D, Moretta L, Rambaldi A, Golay J. et al. Dual-functional capability of CD3+ CD56+ CIK cells.a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood. 2011;118:3301-10
69. Verneris MR, Karimi M, Baker J, Jayaswai A, Negrin RN. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood. 2004;103:3065-72
70. Introna M. CIK as therapeutic agents against tumors. J Autoimmun. 2017;85:32-44
71. Schmidt-Wolf IG, Finke S, Trojaneck B, Denkena A, Lefterova P, Schwella N. et al. Phase I clinical study applying autologous immunological effector cells transfected with the interleukin-2 gene in patients with metastatic renal cancer.colorectal cancer and lymphoma. Br J Cancer. 1999;81:1009-16
72. Zhang J, Zhu L, Zhang Q, He X, Yin Y, Gu Y. et al. Effects of cytokine-induced killer cell treatment in colorectal cancer patients: A retrospective study. Biomed Pharmacother. 2014;68:715-20
73. Wang K, Gao X, Pang J, Liu X, Cai Y, Zhang Y. et al. Dendritic cells transduced with a PSMA-encoding adenovirus and cocultured with autologous cytokine-induced lymphocytes induce a specific and strong immune response against prostate cancer cells. Urol Oncol. 2009;27:26-32
74. Mosińska P, Gabryelska A, Zasada M, Fichna J. Dual functional capability of dendritic cells-cytokine-induced killer cells in improving side effects of colorectal cancer therapy. Front Pharmacol. 2017;8:126
75. Gao D, Li C, Xie X, Zhao P, Wei X, Sun W. et al. Autologous tumor lysate-pulsed dendritic cell immunotherapy with cytokine-induced killer cells improves survival in gastric and colorectal cancer patients. PLoS One. 2014;9:e93886
76. Zhu H, Yang X, Li J, Ren Y, Zhang T, Zhang C. et al. Immune response, safety, and survival and quality of life outcomes for advanced colorectal cancer patients treated with dendritic cell vaccine and cytokine-induced killer cell therapy. Biomed Res Int. 2014;2014:603871
77. Niu J, Ren Y, Zhang T, Yang X, Zhu W, Zhu H. et al. Retrospective comparative study of the effects of dendritic cell vaccine and cytokine-induced killer cell immunotherapy with that of chemotherapy alone and in combination for colorectal cancer. Biomed Res Int. 2014;2014:214727
78. Lin T, S ong C, Chou DY, Zhang H, Zhao J. Clinical effects of autologous dendritic cells combined with cytokine-induced killer cells followed by chemotherapy in treating patients with advanced colorectal cancer: a prospective study. Tumor Biol. 2016;37:4367-72
79. Mosińska P, Gabryelska A, Zasada M, Fichna J. Dual functional capability of dendritic cells-cytokine-induced killer cells in improving side effects of colorectal cancer therapy. Front Pharmacol. 2017;8(e963424):126-133
80. Gao X, Mi Y, Guo N, Xu H, Xu L, Gou X. et al. Cytokine-induced killer cells as pharmacological tools for cancer immunotherapy. Front Immunol. 2017;8:774
81. Uhrberg M, Valiante NM, Shum BP, Shilling HG, Lienert-Weidenbach K, Corliss B. et al. Human diversity in killer cell inhibitory receptor genes. Immunity. 1997;7:753-63
82. Braud VM, Allan DS, O'Callaghan CA, Soderstrom K, D'Andrea A, Ogg GS. et al. HLA-E binds to natural killer cell receptors CD94/NKG2A.B and C. Nature. 1998;391:795-9
83. Ljunggren HG, Kärre K. In search of the 'missing self': MHC molecules and NK cell recognition. Immunol Today. 1990;11:237-44
84. Sandel M, Speetjens FM, Menon AG, Albertsson PA, Basse PH, Hokland M. et al. Natural killer cells infiltrating colorectal cancer and MHC class I expression. Mol Immunol. 2005;42:541-46
85. Maleno l, Aptsiauri N, Cabrera T, Gallego A, Paschen A, Lopez-Nevot MA. et al. Frequent loss of heterozygosity in the β2-microglobulin region of chromosome 15 in primary human tumors. Immunogenetics. 2011;63:65-71
86. Menon AG, Morreau H, Tollenaar RA, Alphenaar E, Van Puijenbroek M, Putter H. et al. Down-regulation of HLA-A expression correlates with a better prognosis in colorectal cancer patients. Lab Invest. 2002;82:1725-33
87. Miller JS, Tessmer-Tuck J, Pierson BA, Weisdorf D, McGlave P, Blazar BR. et al. Low dose subcutaneous interleukin-2 after autologous transplantation generates sustained in vivo natural killer cell activity. Biol Blood Marrow Transplant. 1997;3:34-44
88. Burns LJ, Weisdorf DJ, DeFor TE, Vesole DH, Repka TL, Blazar BR. et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: a phase I/II trial. Bone Marrow Transplant. 2003;32:177-86
89. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK. et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051-57
90. Veluchamy JP, Spanholtz J, Tordoir M, Thijssen VL, Heideman DA, Verheul HM. et al. Combination of NK cells and cetuximab to enhance anti-tumor responses in RAS mutant metastatic colorectal cancer. PLoS One. 2016;11:e0157830
91. Yang Q, Hokland ME, Bryant JL, Zhang Y, Nannmark U, Watkins SC. et al. Tumor-localization by adoptively transferred,interleukin-2-activated NK cells leads to destruction of well-established lung metastases. Int J Cancer. 2003;105:512-519
92. Edsparr K, Basse PH, Goldfarb RH, Albertsson P. Matrix metalloproteinases in cytotoxic lymphocytes impact on tumour infiltration and immunomodulation. Cancer Microenviron. 2011;4:351-60
93. Goding SR, Yang Q, Knudsen KB, Potter DM, Basse PH. Cytokine gene therapy using adenovirally transduced.tumor-seeking activated natural killer cells. Hum Gene Ther. 2007;18:701-11
94. Goding S, Yang Q, Mi Z, Robbins PD, Basse PH. Targeting of products of genes to tumor sites using adoptively transferred A-NK and T-LAK cells. Cancer Gene Ther. 2007;14:441-50
95. Song X, Hong SH, Kwon WT, Bailey LM, Basse P, Bartlett DL. et al. Secretory TRAIL-armed natural killer cell-based therapy: in vitro and in vivo colorectal peritoneal carcinomatosis xenograft. Mol Cancer Ther. 2016;15:1591-601
96. Rothe A, Jachimowicz RD, Borchmannn S, Madlener M, Kebler J, Reiners KS. et al. The bispecific immunoligand ULBP2-aCEA redirects natural killer cells to tumor cells and reveals potent anti-tumor activity against colon carcinoma. Int J Cancer. 2014;134:2829-2840
97. Veluchamy JP, Lopez-Lastra S, Spanholtz J, Bohme F, Kok N, Heideman DA. et al. In vivo efficacy of umbilical cord blood stem cell-derived NK cells in the treatment of metastatic colorectal cancer. Front Immunol. 2017;8:87
98. Guedan S, Posey AD Jr, Shaw C, Wing A, Da T, Patel PR. et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI insight. 2018:3
99. Yoshida Y, Naito M, Yamada T, Aisu N, Daibo K, Mera T. et al. Adoptive chemoimmunotherapy using activated αβ T cells for stage IV colorectal cancer. Anticancer Res. 2016;36:3741-46
100. Ishii F, Yoshida Y, Yamauchi Y, Aisu N, Kojima D, Mera T. et al. Hepatectomy for liver metastases of colorectal cancer after adoptive chemoimmunotherapy using activated αβ T-cells. Anticancer Res. 2017;37:3933-39
101. Yoshida Y, Naito M, Yamada T, Aisu N, Kojima D, Mera T. et al. Clinical study on the medical value of combination therapy involving adoptive immunotherapy and chemotherapy for stage IV colorectal cancer (COMVI Study). Anticancer Res. 2017;37:3941-46
102. Chien Y, Meyer C, Bonneville M. γδ T cells: first line of defense and beyond. Annu Rev Immunol. 2014;32:121-55
103. Brandes M, Willimann K, Moser B. Professional antigen-presentation function by human γδ T cells. Science. 2005;309:264-8
104. Gertner-Dardenne J, Bonnafous C, Bezombes C, Capietto AH, Scaglione V, Ingoure S. et al. Bromohydrin pyrophosphate enhances antibody-dependent cell-mediated cytotoxicity induced by therapeutic antibodies. Blood. 2009;113:4875-84
105. Davis MM, Bjorkman PJ. T-cell antigen receptor genes and T-cell recognition. Nature. 1988;334:395
106. Rock EP, Sibbald PR, Davis MM, Chien YH. CDR3 length in antigen-specific immune receptors. J Exp Med. 1994;179:323-28
107. Maeurer MJ, Martin D, Walter W, Liu K, Zitvogel L, Halusczcak H. et al. Human intestinal Vdelta1+ lymphocytes recognize tumor cells of epithelial origin. J Exp Med. 1996;183:1681-96
108. Kondo M, Sakuta K, Noguchi A, Ariyoshi N, Sato K, Sato S. et al. Zoledronate facilitates large-scale ex vivo expansion of functional γδ T cells from cancer patients for use in adoptive immunotherapy. Cytotherapy. 2008;10:842-56
109. Murayama M, Tanaka Y, Yagi J, Uchiyama T, Ogawa K. Antitumor activity and some immunological properties of γδ T-cells from patients with gastrointestinal carcinomas. Anticancer Res. 2008;28:2921-31
110. Devaud C, Rousseau B, Netzer S, Pitard V, Paroissin C, Khairallah C. et al. Anti-metastatic potential of human Vδ1+ γδ T cells in an orthotopic mouse xenograft model of colon carcinoma. Cancer Immunol Immunother. 2013;62:1199-210
111. Wu D, Wu P, Wu X, Ye J, Wang Z, Zhao S. et al. Ex vivo expanded human circulating Vδ1 γδT cells exhibit favorable therapeutic potential for colon cancer. Oncoimmunology. 2015;4:e992749
112. Schmitt TM, de Pooter RF, Gronski MA, Cho SK, Ohashi PS, Zuniga-Pflucker JC. Induction of T cell development and establishment of T cell competence from embryonic stem cells differentiated in vitro. Nat Immunol. 2004;5:410
113. La Motte-Mohs RN, Herer E, Zúñiga-Pflücker JC. Induction of T-cell development from human cord blood hematopoietic stem cells by Delta-like 1 in vitro. Blood. 2005;105:1431-39
114. Lei F, Haque R, Weiler L, Vrana KE, Song J. T lineage differentiation from induced pluripotent stem cells. Cell Immunol. 2009;260:1-5
115. Lei F, Zhao B, Haque R, Xiong X, Budgeon L, Christensen ND. et al. In vivo programming of tumor antigen-specific T lymphocytes from pluripotent stem cells to promote cancer immunosurveillance. Cancer Res. 2011;71:4742-47
116. Haque M, Song J, Fino K, Sandhu P, Wang Y, Ni B. et al. Melanoma immunotherapy in mice using genetically engineered pluripotent stem cells. Cell Transplant. 2016;25:811-27
117. Vizcardo R, Masuda K, Yamada D, Ikawa T, Shimizu K, Fujii S. et al. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8+ T cells. Cell stem cell. 2013;12:31-36
118. Woll PS, Grzywacz B, Tian X, Marcus RK, Knorr DA, Verneris MR. et al. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood. 2009;113:6094-101
119. Schmitt TM, Aggen DH, Stromnes IM, Dossett ML, Richman SA, Kranz DM. et al. Enhanced-affinity murine T-cell receptors for tumor/self-antigens can be safe in gene therapy despite surpassing the threshold for thymic selection. Blood. 2013;122:348-56
120. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA. et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19:620-6
121. van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B. et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643-47
122. Sahin U, Tureci O, Schmitt H, Cochlovius B, Johannes T, Schmits R. et al. Human neoplasms elicit multiple specific immune responses in the autologous host. Pro Natl Acad Sci U S A. 1995;92:11810-13
123. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14:135
124. Scanlan MJ, Welt S, Gordon CM, Chen YT, Gure AO, Stockert E. et al. Cancer-related serological recognition of human colon cancer. Cancer Res. 2002;62:4041-47
125. Wargo JA, Robbins PF, Li Y, Zhao Y, EI-Gamil M, Caragacianu D. et al. Recognition of NY-ESO-1+ tumor cells by engineered lymphocytes is enhanced by improved vector design and epigenetic modulation of tumor antigen expression. Cancer Immunol Immunother. 2009;58:383-94
126. Chou J, Voong LN, Mortales CL, Towlerton AM, Pollack SM, Chen X. et al. Epigenetic modulation to enable antigen-specific T cell therapy of colorectal cancer. J Immunother. 2012;35:131-41
127. Inderberg EM, Walchli S, Myhre MR, Trachsel S, Almasbak H, Kvalheim G. et al. T cell therapy targeting a public neoantigen in microsatellite instable colon cancer reduces in vivo tumor growth. Oncoimmunology. 2017;6:e1302631
128. Bubeník J. MHC class I down-regulation: tumour escape from immune surveillance? Int J Oncol. 2004;25:487-91
129. Leone P, Shin EC, Perosa F, Vacca A, Dammacco F, Racanelli V. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst. 2013;105:1172-87
130. Gross G, Gorochov G, Waks T, Eshhar Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant Proc. 1989;21:127-30
131. Irving BA, Weiss A. The cytoplasmic domain of the T cell receptor ζ chain is sufficient to couple to receptor-associated signal transduction pathways. Cell. 1991;64:891-901
132. Romeo C, Seed B. Cellular immunity to HIV activated by CD4 fused to T cell or Fc receptor polypeptides. Cell. 1991;64:1037-46
133. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Pro Natl Acad Sci U S A. 1993;90:720-4
134. Sadelain MR. et al. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388-98
135. Sheen AJ, Sherlock DJ, Irlam J, Hawkins RE, Giham DE. T lymphocytes isolated from patients with advanced colorectal cancer are suitable for gene immunotherapy approaches. Br J Cancer. 2003;88:1119-27
136. Lo AS, Ma Q, Liu DL, Junghans RP. Anti-GD3 chimeric sFv-CD28/T-cell receptor ζ designer T cells for treatment of metastatic melanoma and other neuroectodermal tumors. Clin Cancer Res. 2010;16:2769-80
137. Loskog A, Giandomenico V, Rossig C, Pule M, Dotti G, Brenner MK. Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia. 2006;20:1819-28
138. Eshhar Z. Adoptive cancer immunotherapy using genetically engineered designer T-cells: First steps into the clinic. Curr Opin Mol Ther. 2010;12:55-63
139. Sasaki T, Ikeda H, Sato M, Ohkuri T, Abe H, Kuroki M. et al. Antitumor activity of chimeric immunoreceptor gene-modified Tc1 and Th1 cells against autologous carcinoembryonic antigen-expressing colon cancer cells. Cancer Sci. 2006;97:920-27
140. Simmons A, Whitehead RP, Kolokoltsov AA, Davey RA. Use of recombinant lentivirus pseudotyped with vesicular stomatitis virus glycoprotein G for efficient generation of human anti-cancer chimeric T cells by transduction of human peripheral blood lymphocytes in vitro. Virol J. 2006;3:8
141. Zhang C, Wang Z, Yang Z, Wang M, Li S, Li Y. et al. Phase I escalating-eose trial of CAR-T therapy targeting CEA+ metastatic colorectal cancers. Mol Ther. 2017;25:1248-58
142. Saied A, Licata L, Burga RA, Thorn M, McCormack E, Stainken BF. et al. Neutrophil: lymphocyte ratios and serum cytokine changes after hepatic artery chimeric antigen receptor-modified T-cell infusions for liver metastases. Cancer Gene Ther. 2014;21:457-62
143. Burga RA, Thorn M, Point GR, Guha P, Nguyen CT, Licata LA. et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother. 2015;64:817-29
144. Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR. et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res. 2015;21:3149-59
145. Katz S, Point GR, Cunetta M, Thorn M, Guha P, Espat NJ. et al. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther. 2016;23:142-8
146. Frick GS, Pitari GM, Weinberg DS, Hyslop T, Schulz S, Waldman SA. Guanylyl cyclase C: a molecular marker for staging and postoperative surveillance of patients with colorectal cancer. Expert Rev Mol Diagn. 2005;5:701-13
147. Snook AE, Eisenlohr LC, Rothstein JL, Waldman SA. Cancer mucosa antigens as a novel immunotherapeutic class of tumor-associated antigen. Clin Pharmacol Ther. 2007;82:734-39
148. Carrithers SL, Barber MT, Biswas S, Parkinson SJ, Park PK, Goldstein SD. et al. Guanylyl cyclase C is a selective marker for metastatic colorectal tumors in human extraintestinal tissues. Pro Natl Acad Sci U S A. 1996;93:14827-832
149. Birbe R, Palazzo JP, Walters R, Weinberg D, Schulz S, Waldman SA. Guanylyl cyclase C is a marker of intestinal metaplasia.dysplasia.and adenocarcinoma of the gastrointestinal tract. Hum Pathol. 2005;36:170-9
150. Schulz S, Hyslop T, Haaf J, Bonaccorso C, Nielsen K, Witek ME. et al. A validated quantitative assay to detect occult micrometastases by reverse transcriptase-polymerase chain reaction of guanylyl cyclase C in patients with colorectal cancer. Clin Cancer Res. 2006;12:4545-52
151. Magee MS, Kraft CL, Abraham TS, Baybutt TR, Marszalowicz GP, Li P. et al. GUCY2C-directed CAR-T cells oppose colorectal cancer metastases without autoimmunity. Oncoimmunology. 2016;5:e1227897
152. Thor A, Ohuchi N, Szpak CA, Johnson WW, Schlom J. Distribution of oncofetal antigen tumor-associated glycoprotein-72 defined by monoclonal antibody B72. 3. Cancer Res. 1986;46:3118-24
153. Wolf BC, D'Emillia JC, Salem RR, DeCoste D, Sears HF, Gottlieb LS. et al. Detection of the tumor-associated glycoprotein antigen (TAG-72) in premalignant lesions of the colon. J Natl Cancer Inst. 1989;81:1913-17
154. Guadagni F, Roselli M, Amato T, Cosimelli M, Perri P, Casali V. et al. CA 72-4 measurement of tumor-associated glycoprotein 72 (TAG-72) as a serum marker in the management of gastric carcinoma. Cancer Res. 1992;52:1222-27
155. Sharifzadeh Z, Rahbarizadeh F, Shokrgozar MA, Ahmadvand D, Mahboudi F, Jamnani FR. et al. Genetically engineered T cells bearing chimeric nanoconstructed receptors harboring TAG-72-specific camelid single domain antibodies as targeting agents. Cancer Lett. 2013;334:237-44
156. Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG. et al. Safety tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer. 2017;5:22
157. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843-51
158. Chakraborty M, Abrams SI, Camphausen K, Liu K, Scott T, Coleman CN. et al. Irradiation of tumor cells up-regulates Fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J Immunol. 2003;170:6338-47
159. John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, e t al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19:5636-46
160. Vinay DS, Kwon BS. 4-1BB (CD137) an inducible costimulatory receptor as a specific target for cancer therapy. BMB Rep. 2014;47:122-9
161. Ye Q, Song DG, Poussin M, Yamamoto T, Best A, Li C. et al. CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin Cancer Res. 2014;20:44-55
162. Miller RE, Jones J, Le T, Whitmore J, Boiani N, Gliniak B. et al. 4-1BB-specific monoclonal antibody promotes the generation of tumor-specific immune responses by direct activation of CD8 T cells in a CD40-dependent manner. J Immunol. 2002;169:1792-800
163. Gauttier V, Judor JP, Le Guen V, Cany J, Ferry N, Conchon S. Agonistic anti-CD137 antibody treatment leads to antitumor response in mice with liver cancer. Int J Cancer. 2014;135:2857-67
164. Mardiana S, John LB, Henderson MA, Slaney CY, von Scheidt B, Giuffrida L. et al. A Multifunctional Role for Adjuvant Anti-4-1BB Therapy in Augmenting Antitumor Response by Chimeric Antigen Receptor T Cells. Cancer Res. 2017;77:1296-1309
165. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217-21
166. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222-6
167. Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K. et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat Med. 2018;24:352-59
Author contact
![]() Corresponding authors: Jin-Ming Yang, M.D, Ph. D, Department of Pharmacology, Penn State Cancer Institute, The Penn State University or Hailong Chen, Ph. D, Third General Surgery Department, The First Affiliated Hospital of Dalian Medical University, Zhongshan Road 222#, Dalian, China or Jiaqiao Fan, Ph. D, Third General Surgery Department, The First Affiliated Hospital of Dalian Medical University, Zhongshan Road 222#, Dalian, China
Corresponding authors: Jin-Ming Yang, M.D, Ph. D, Department of Pharmacology, Penn State Cancer Institute, The Penn State University or Hailong Chen, Ph. D, Third General Surgery Department, The First Affiliated Hospital of Dalian Medical University, Zhongshan Road 222#, Dalian, China or Jiaqiao Fan, Ph. D, Third General Surgery Department, The First Affiliated Hospital of Dalian Medical University, Zhongshan Road 222#, Dalian, China
Citation styles
APA
Fan, J., Shang, D., Han, B., Song, J., Chen, H., Yang, J.M. (2018). Adoptive Cell Transfer: Is it a Promising Immunotherapy for Colorectal Cancer?. Theranostics, 8(20), 5784-5800. https://doi.org/10.7150/thno.29035.
ACS
Fan, J.; Shang, D.; Han, B.; Song, J.; Chen, H.; Yang, J.M. Adoptive Cell Transfer: Is it a Promising Immunotherapy for Colorectal Cancer?. Theranostics 2018, 8 (20), 5784-5800. DOI: 10.7150/thno.29035.
NLM
Fan J, Shang D, Han B, Song J, Chen H, Yang JM. Adoptive Cell Transfer: Is it a Promising Immunotherapy for Colorectal Cancer?. Theranostics 2018; 8(20):5784-5800. doi:10.7150/thno.29035. https://www.thno.org/v08p5784.htm
CSE
Fan J, Shang D, Han B, Song J, Chen H, Yang JM. 2018. Adoptive Cell Transfer: Is it a Promising Immunotherapy for Colorectal Cancer?. Theranostics. 8(20):5784-5800.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.