Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results
Discussion
Materials and Methods
Supplementary Material
Acknowledgements
References
Introduction
Results
Discussion
Materials and Methods
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(21):5986-5994. doi:10.7150/thno.26650 This issue Cite
Research Paper
Chemoresistance Transmission via Exosome-Mediated EphA2 Transfer in Pancreatic Cancer
Jia Fan1*, Qian Wei2*, Eugene J Koay3, Yang Liu1,4, Bo Ning5, Paul W Bernard6, Ning Zhang4, Haiyong Han7, Matthew H Katz8, Zhen Zhao9,10 ![]() , Ye Hu1
, Ye Hu1 ![]()
1. School of Biological and Health Systems Engineering, Virginia G. Piper Biodesign Center for Personalized Diagnostics, The Biodesign Institute, Arizona State University, Tempe, AZ 85281, USA.
2. Tianjin Medical University Cancer Institute and Hospital, National Clinical Research Center for Cancer, Key Laboratory of Cancer Prevention and Therapy, Tianjin's Clinical Research Center for Cancer, Tianjin 300060, PR China.
3. Division of Radiation Oncology, University of Texas M.D. Anderson Cancer Center, Houston, Texas 77030, USA.
4. Research Center of Basic Medical Sciences & Cancer Institute and Hospital, National Clinical Research Center for Cancer, Tianjin Medical University, Tianjin 300070, PR China.
5. Center for Molecular Design and Biomimetics, The Biodesign Institute, Arizona State University, Tempe, AZ 85281, USA.
6. Department of Nanomedicine, Houston Methodist Research Institute, Houston, Texas 77030, USA.
7. Molecular Medicine Division, Translational Genomics Research Institute, Phoenix, AZ 85004, USA.
8. Department of Surgical Oncology, Division of Surgery, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA.
9. Department of Laboratory Medicine, Clinical Center, National Institutes of Health, Bethesda, MD 20892, USA.
10. Department of Pathology and Laboratory Medicine, Weill Cornell Medicine, New York, NY 10065, USA.
*These authors contributed equally to this work
Received 2018-4-12; Accepted 2018-10-4; Published 2018-11-13
Citation:
Fan J, Wei Q, Koay EJ, Liu Y, Ning B, Bernard PW, Zhang N, Han H, Katz MH, Zhao Z, Hu Y. Chemoresistance Transmission via Exosome-Mediated EphA2 Transfer in Pancreatic Cancer. Theranostics 2018; 8(21):5986-5994. doi:10.7150/thno.26650. https://www.thno.org/v08p5986.htm
Other stylesAbstract

Rationale: Exosomes are small extracellular vesicles secreted by most cells that are found in blood and other bodily fluids, and which contain cytoplasmic material and membrane factors corresponding to their cell type of origin. Exosome membrane factors and contents have been reported to alter adjacent and distant cell behavior in multiple studies, but the impact of cancer-derived exosomes on chemoresistance is less clear.
Methods: Exosomes isolated from three pancreatic cancer (PC) cell lines displaying variable gemcitabine (GEM) resistance (PANC-1, MIA PaCa-2, and BxPC-3) were tested for their capacity to transmit chemoresistance among these cell lines. Comparative proteomics was performed to identify key exosomal proteins that conferred chemoresistance. Cell survival was assessed in GEM responsive PC cell lines treated with recombinant Ephrin type-A receptor 2 (EphA2), a candidate chemoresistance transfer factor, or exosomes from a chemoresistant PC cell line treated with or without EphA2 shRNA.
Results: Exosomes from chemoresistant PANC-1 cells increased the GEM resistance of MIA PaCa-2 and BxPC-3 cell cultures. Comparative proteomics determined that PANC-1 exosomes overexpressed Ephrin type-A receptor 2 (EphA2) versus exosomes of less chemoresistant PC cell lines MIA PaCa-2 and BxPC-3. EphA2-knockdown in PANC-1 cells inhibited their ability to transmit exosome-mediated chemoresistance to MIA PaCa-2 and BxPC-3, while treatment of MIA PaCa-2 and BxPC-3 cells with soluble EphA2 did not promote chemoresistance, indicating that membrane carried EphA2 was important for the EphA2 chemoresistance effect.
Conclusion: Exosomal EphA2 expression could transmit chemoresistance and may potentially serve as a minimally-invasive predictive biomarker for PC treatment response. Further work should address whether additional exosomal factors regulate resistance to other cancer therapeutic agents for PC or other cancer types.
Keywords: Exosome, EphA2, Cytotoxic resistance, Pancreatic cancer, Gemcitabine
Introduction
Resistance to therapy is a primary cause of treatment failure in most human cancers [1, 2]. Pancreatic cancer (PC), one of the most challenging malignancies to treat, is characterized by aggressive local invasion, early metastasis, and a high degree of resistance to therapy. Patients with this lethal malignancy have an average 5-year survival rate of only 4% [3]. At present, surgical resection offers the only potential cure; however, 80%-85% of PC patients are diagnosed with advanced disease, which usually precludes complete resection [3]. Cytotoxic therapy is routinely used to treat both resectable and unresectable PC cases; however, only a small subset of patients responds to treatment, resulting in limited overall benefit, and despite intense research there has been little progress in the development of more effective therapies. Gemcitabine (GEM) and GEM-based chemotherapeutic combinations are standard treatments for PC, but exhibit a low response rate (24%) and a poor increase in median overall survival time (5.7 months) [4, 5]. FOLFIRINOX was recently adopted as a treatment option for patients with advanced PC, but it exhibits increased toxicity for a modest improvement in median overall survival time (11 months) [6]. Drug resistance is thought to be a major reason for treatment failure, indicating an urgent need to identify and apply predictive markers to segregate PC patients into personalized treatment regimens to minimize therapy resistance.
Molecular and genomic research of PC suggests that varying responses to therapy may be attributable to tumor heterogeneity, implying that metastases may exhibit divergent responses due to clonal diversity [7]. Recent studies also indicate that cell-cell communications in the tumor microenvironment may be involved in therapy resistance [1, 8]. Tumor-derived exosomes contain proteins and nucleic acids that can serve as key mediators in cell-cell communications, increasing tumor progression and metastasis [9-15]. We hypothesized that exosomal transfer of a resistance factor from a chemoresistant PC tumors might also be able to increase chemoresistance of more susceptible PC clones. We therefore analyzed exosomes secreted by PC cell lines with variable GEM sensitivity to identify exosomal proteins that could transfer GEM resistance between PC cell lines. Our results indicate that Ephrin type-A receptor 2 (EphA2) expression was required for exosome-mediated transfer of GEM-resistance.
Results
Exosomes derived from chemoresistant PC cells confer GEM resistance
Studying mechanisms of tumor chemoresistance in vivo is challenging due to the heterogeneous nature of the tumor microenvironment. We therefore utilized in vitro cell culture to characterize the ability of PC-derived exosomes to increase resistance to GEM, a standard component of most PC treatment regimens, in a proof-of-concept study. We found that three well-characterized PC cell lines (PANC-1, MIA PaCa-2, and BxPC-3) displayed differential resistance to GEM, in agreement with previous observations [16-19], with PANC-1 cells exhibiting significantly greater GEM resistance than MIA PaCa-2 and BxPC-3 cells (Figure S1).
Exosomes were isolated from PANC-1 (GEM-resistant) and MIA PaCa-2 and BxPC-3 (GEM-sensitive) cell cultures by ultracentrifugation [20]. Transmission electron microscopy (TEM) analyses revealed characteristic cup-shaped exosomal morphology (Figure 1A and Figure S2) and dynamic light scattering studies found a typical size range of 30-100nm (Figure S3) [21], indicating that isolated vesicles were enriched in exosomes but not larger extracellular microvesicles. Western blot analyses detected the exosome markers CD63, CD9, and TSG101, while the Golgi protein marker GM130 was undetectable (Figure 1B), confirming the purity of these exosome preparations.
To determine if exosomes derived from GEM-resistant PC cells conferred resistance to GEM-sensitive PC cells, chemo-sensitive MIA PaCa-2 or BxPC-3 cultures were incubated with PANC-1 exosomes for 24 h (Figure 1C) and then challenged with GEM for 3 days, with fresh exosomes added every 24 h. PANC-1 exosome treatment significantly increased the GEM-resistance of MIA PaCa-2 and BxPC-3 cells (Figure 1D-E) corresponding to the exosome dose (Figure S4A-B). Further, fluorescent labeling revealed that PANC-1 exosomes were internalized by recipient cells (Figure 1F), suggesting that chemoresistance was mediated by delivery of exosome factors to the cell membrane and/or cytoplasm. In contrast, no change in viability was observed when MIA PaCa-2 cells were co-cultured with BxPC-3 exosomes or vice versa (Figure 1D-E and Figure S4C-D), demonstrating that exosomes of GEM-sensitive cells did not confer chemoresistance.
Identification of exosomal proteins associated with transfer of GEM resistance
Exosomes of each cell line were extracted for protein, which was size-fractionated by SDS-PAGE and silver-stained to detect candidate chemoresistance proteins overexpressed in PANC-1 vs. MIA PaCa-2 and BxPC-3 exosomes (Figure 2A-B). LC-MS/MS data from bands 1 and 2 used to query the UniProtKB/Swiss-Prot Homo sapiens databases identified 15 proteins, each with at least one unique peptide, that were differentially expressed with scores of >30 (Table 1). We selected the two highest scoring proteins, EphA2 and Erythrocyte band 7 integral membrane protein (stomatin), which were derived from band 1 and 2, respectively. EphA2 expression was ~5-fold higher in PANC-1 vs. MIA PaCa-2 and BxPC-3 exosomes, while stomatin expression was not differentially overexpressed in PANC-1 exosomes, leading us to focus on EphA2 (Figure 2C-D).
Figure 1
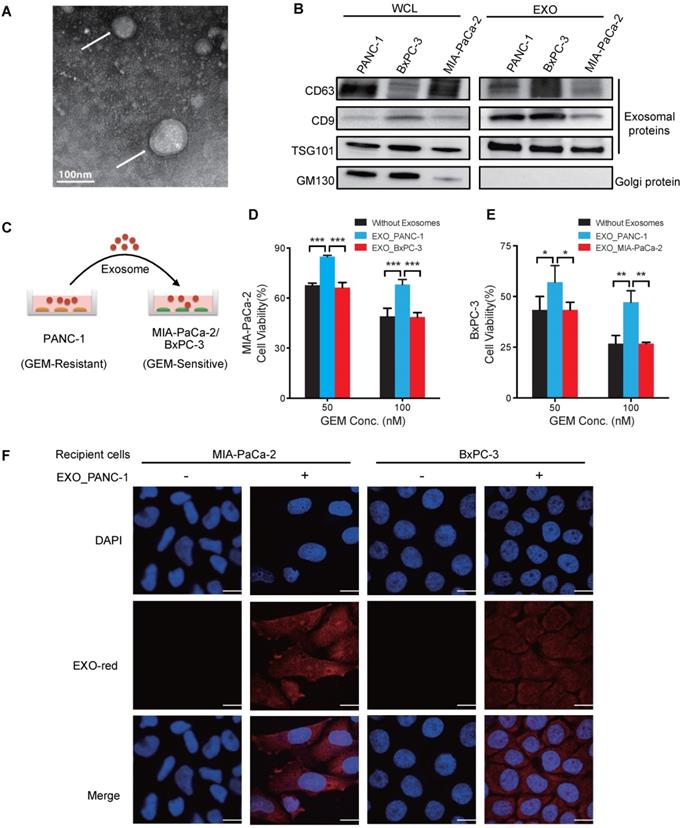
Exosomes of GEM-resistant PC cells can transfer chemoresistance. (A) TEM image of exosomes isolated from PANC-1 cells and negatively stained by uranyl acetate (arrows). (B) Western blot analysis of exosomes (EXOs) and whole-cell lysates (WCLs) of PANC-1, MIA PaCa-2, and BxPC-3 cells for exosome (CD63, CD9, and TSG101) and golgi (GM130) protein markers. (C) Experimental design of exosome uptake studies. (D-E) GEM cytotoxicity in (D) MIA PaCa-2 cells or (E) BxPC-3 after 24 h pre-treatment with 20 µg/mL exosomes from PANC-1 (EXO_PANC-1), BxPC-3 (EXO_BxPC-3) or MIA PaCa-2 (EXO_MIA PaCa-2) cells, with fresh exosomes added every 24 h of the 3 day GEM treatment. Cell viability is presented as a percentage of control (no drug or exosomes) viability. (F) Exosome internalization in MIA PaCa-2 and BxPC-3 cells incubated for 2 h with or without EXO-Red-stained exosomes and then stained with DAPI for nuclear visualization. Bar indicates 10 µm; Data indicate mean±SD; n=6; *p<0.05; **p<0.01, ***p<0.001.
Figure 2
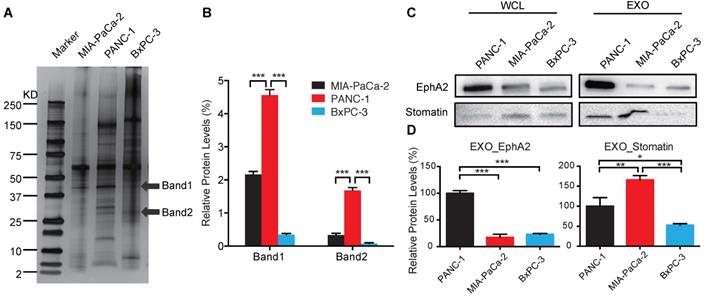
Identification of candidate exosome chemoresistance proteins. (A) Western blot of exosomal proteins of MIA PaCa-2, PANC-1, and BxPC-3 cells (arrows indicate protein bands subjected to LC-MS/MS sequence analysis). (B) Relative expression of selected exosomal protein bands normalized to the sum of the band densities in each lane. (C) Western blot of candidate proteins in WCLs and EXOs of PANC-1, MIA PaCa-2, and BxPC-3 cells. (D) Relative exosome EphA2 and Stomatin expression. Data indicate mean±SD (n=3); *p<0.05, **p<0.01, ***p<0.001.
Table 1
Proteins identified by LC MS/MS sequence from exosomal bands 1 and 2.
| No. | Protein name | ID | Score | No. of peptides | Band |
|---|---|---|---|---|---|
| 1 | Ephrin type-A receptor 2 | P29317 | 124 | 6 | 1 |
| 2 | Erythrocyte band 7 integral membrane protein | P27105 | 115 | 2 | 2 |
| 3 | MARCKS-related protein | P49006 | 90 | 4 | 1 |
| 4 | Protein eva-1 homolog B | P58658 | 88 | 2 | 2 |
| 5 | 14-3-3 protein eta | Q04917 | 79 | 4 | 2 |
| 6 | Lactadherin | Q08431 | 70 | 4 | 1 |
| 7 | 14-3-3 protein sigma | P31947 | 53 | 2 | 2 |
| 8 | CD44 antigen | P16070 | 45 | 1 | 2 |
| 9 | Cytochrome b-c1 complex subunit 2 | P22695 | 43 | 1 | 1 |
| 10 | Inter-alpha-trypsin inhibitor heavy chain H2 | P19823 | 43 | 1 | 1 |
| 11 | Ribonuclease inhibitor | P13489 | 40 | 1 | 1 |
| 12 | Retinoic acid-induced protein 3 | Q8NFJ5 | 40 | 1 | 1 |
| 13 | Rho GDP-dissociation inhibitor 1 | P52565 | 38 | 1 | 2 |
| 14 | Integrin alpha-6 | P23229 | 38 | 1 | 2 |
| 15 | Perilipin-3 | O60664 | 38 | 1 | 2 |
Exosomal EphA2 mediates transmission of GEM resistance to GEM-sensitive PC cells
To determine whether exosomal EphA2 (EXO_EphA2) was responsible for transmission of GEM resistance to GEM-sensitive cells, we generated PANC-1 cultures where >90% of cells stably expressed EphA2 shRNA (Figure S5). PANC-1 cells expressing EphA2-shRNA-1 (PANC-1EphA2-) revealed a ~80% decrease in EphA2 expression and a ~25% decrease in resistance to 100 nM GEM (Figure 3A-C), and their exosomes failed to transfer chemoresistance to GEM-sensitive MIA PaCa-2 and BxPC-3 cells (Figure 3D-E). To address whether exosomal delivery is required for the EphA2-mediated chemoresistance effect, MIA PaCa-2 and BxPC-3 cells were incubated with bioactive recombinant EphA2, both before and during GEM treatment. MIA PaCa-2 or BxPC-3 cells treated with recombinant EphA2 protein did not exhibit altered responses to GEM, and failed to demonstrate EphA2 protein uptake (Figure S6), unlike THP-1 macrophages that exhibited marked accumulation of free EphA2 during this incubation period. These results strongly suggest that exosome protein delivery played a critical role in EphA2-mediated GEM resistance.
Exosome uptake by chemosensitive PC cells is independent of EXO_EphA2 expression
Exosome fusion with a plasma membrane transfers soluble and membrane-associated factors to recipient cells and blocking this uptake can inhibit exosome-mediated phenotypes. EXO-Red-labeled PANC-1EphA2- and PANC-1Ctrl_shRNA exosomes were both efficiently internalized by MIA PaCa-2 and BxPC-3 cells (Figure 4A), which revealed similar uptake of exosomes derived from PANC-1, PANC-1EphA2- or PANC-1Ctrl_shRNA cells (Figure 4B-C), indicating that exosome uptake was independent of EXO_EphA2 expression.
EphA2 expression in MIA PaCa-2 and BxPC-3 cells increased dose-dependently after incubation with PANC-1 exosomes (Figure 5A-B) revealing a 1.5-fold increase at the highest dose, while PANC1EphA2- exosomes failed to increase cellular EphA2 expression, even after incubation with a 20 µg/mL exosome dose (Figure 5C-D).
Discussion
Resistance to therapy contributes to poor PC patient outcomes [22, 23]. Exosomes present in tumor microenvironments can be internalized by adjacent cells and modify the phenotype of the recipient cell to reflect the regulatory functions of the exosome cargo. Our study results now indicate that PC tumor exosomes can transmit chemoresistance, potentially allowing chemoresistant PC cells to transfer resistance to sensitive PC cells within the same tumor or at other anatomical sites. Chemoresistance transfer from GEM-resistant to GEM-sensitive PC cells was found to correspond with and require EXO_EphA2 expression.
Figure 3
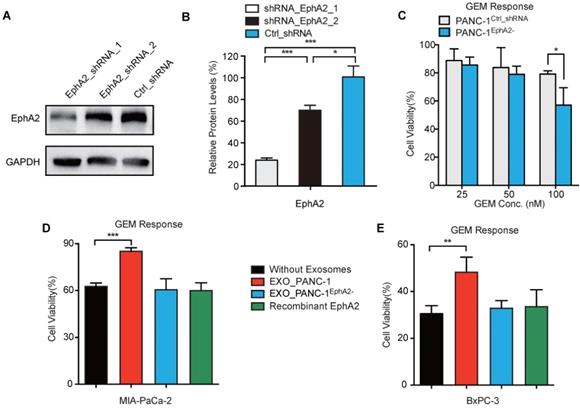
Exosomal EphA2 confers GEM resistance. (A) Western blot and (B) GAPDH-normalized EphA2 levels in PANC-1 cells transduced with lentiviruses expressing EphA2_shRNA_1, EphA2_shRNA_2, or Ctrl_shRNA. (C) Viability of PANC-1Ctrl_shRNA and PANC-1EphA2- (EphA2_shRNA_1 transduced) cells after 72h GEM treatment, normalized to untreated cell viability. (D-E) GEM cytotoxicity (100nM for 72 h) in (D) MIA PaCa-2 and (E) BxPC-3 cells after exposure to 20 µg/mL EXO_PANC-1 or EXO_PANC-1EphaA2- or recombinant EphA2 (0.5 µg/mL). Data represent mean±SD; Western: n=3; MTT assay: n=6; *p<0.05, **p<0.01, ***p<0.001.
Multiple proteomic and metabolomic studies have attempted to define the intracellular circuitry governing PC chemoresistance [24-29], but few have focused on the role of PC-derived exosomes, which our results now indicate can transfer GEM-resistance via an EphA2-dependent mechanism. Most previous reports have focused on exosome-mediated affects to promote rather than transfer resistance to chemotoxic agents or radiation exposure, and tend to identify relatively non-specific mechanisms. For example, one study has proposed that exosomes can function as drug exporters to promote chemoresistance, finding that doxorubicin accumulates in shed vesicles and that chemoresistance corresponds with vesicle shedding rate across a variety of cell lines [30]. Exosome transfer from stromal cells to breast cancer cells has also been reported to promote STAT-1-dependent radiation resistance by exosomal RNA-mediated activation of the RIG-I pattern recognition receptor [13]. Few studies have examined the ability of exosomes of chemoresistant cancer cells to transfer drug resistance. One study has proposed that exosome-mediated miRNA transfer from docetaxel-resistant to docetaxel-sensitive breast cancer cells may enhance drug resistance through an affect to decrease expression of the tumor suppressor protein PTEN [31]. We believe our results, however, provide the first example of direct exosomal transfer of a chemoresistance factor altering the chemoresistance phenotype of the recipient cell.
EphA2 is overexpressed and mediates therapy resistance in breast cancer, cervical cancer, and melanoma [32-36]. Recent studies have revealed that EphA2 repression attenuates PC invasiveness and increases melanoma sensitivity to vemurafenib and breast cancer sensitivity to tamoxifen [36-38], suggesting that EphA2 represents a promising target for future cancer therapeutics. Our results reveal a previously unknown ability of EphA2-enriched exosomes to transfer chemoresistance. Improving the predictability of a patient's response to therapy is urgently required to customize therapy and improve overall survival, particularly for PC patients, who have poor therapy response rates that lead to rapid patient mortality. Despite great effort expended on biomarker development, effective predictive markers for PC treatment remain elusive [39]. Characterization of exosomal factors that regulate chemoresistance may serve as a robust source of functional biomarkers. Our findings suggest that EXO_EphA2 may play an important role in PC drug resistance, and implies that it may be useful to analyze other specific exosome sub-populations as biomarkers of PC drug resistance. Future studies need to be performed in PC patient populations with well-defined treatment regimens to validate the clinical utility of EXO_EphA2, and other potential exosome factors, as a biomarker of PC drug resistance. Validation of EXO_EphA2 level as a biomarker of PC drug resistance would allow affected patients to be assigned to EphA2-targeted therapeutic regimens, several which are currently under study, to offer a targeted approach to individual therapy.
Materials and Methods
Materials
The PC cell lines PANC-1, MIA PaCa-2, and BxPC-3 were purchased from the American Type Culture Collection (Manassas, VA). Bovine growth serum, RPMI 1640, and DMEM medium were purchased from HyClone (Logan, UT). GEM was purchased from the pharmacy at Houston Methodist Research Institute and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was obtained from Life Technologies (Grand Island, NY). Puromycin was purchased from Invitrogen (Carlsbad, CA). Mouse monoclonal anti-CD9, rabbit polyclonal anti-CD63, goat polyclonal anti-Stomatin, and mouse monoclonal anti-GAPDH antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Mouse monoclonal anti-TSG101 and rabbit monoclonal anti-GM130 antibodies were purchased from Abcam (Cambridge, MA). Mouse monoclonal anti-EphA2 antibody was purchased from Millipore (Billerica, MA). GIPZ Lentiviral EphA2 shRNAs were purchased from GE Dharmacon (Lafayette, CO).
Figure 4
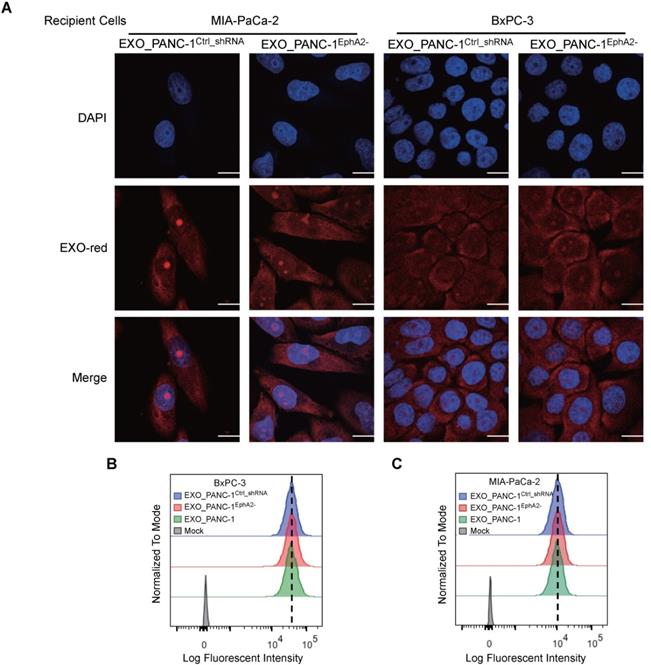
Exosomal EphA2 expression does not affect exosome internalization. (A) Confocal images of MIA PaCa-2 and BxPC-3 cells mock-treated or incubated for 2 h with the indicated EXO-Red-stained exosomes, then fixed, permeabilized, and stained with DAPI (blue). The scale bar represents 10 µm. (B-C) Flow cytometric analysis of the fluorescence intensity of (B) BxPC-3 and (C) MIA PaCa-2 cells after 2h co-culture with the indicated EXO-Red-labeled exosomes.
Figure 5
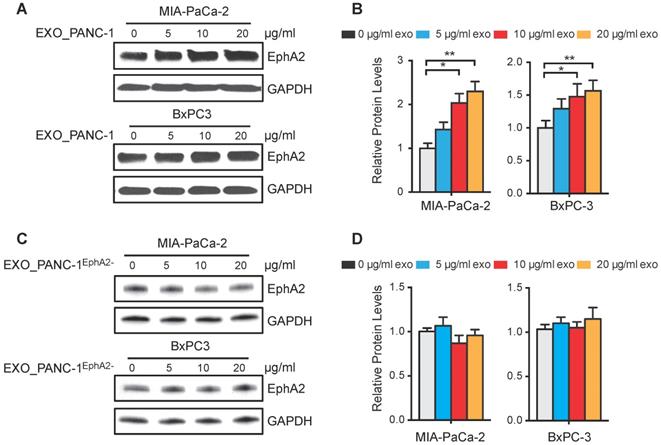
EphA2 uptake by recipient PC cells. (A and C) Western blots of MIA PaCa-2 and BxPC3 WCLs after 96 h culture with (A) PANC-1 or (C) PANC-1EphA2- exosomes. (B and D) GAPDH-normalized EphA2 expression. Data represent mean±SD; n=3; *p<0.05, **p<0.01, ***p<0.001.
Cell culture
PANC-1 and BxPC-3 cells were cultured in RPMI 1640 containing 10% fetal bovine serum (FBS), and MIA PaCa-2 cells in DMEM with 10% FBS for growth or maintenance or serum-free medium for exosome collection. Cultures were incubated at 37°C in a humidified 5% CO2 incubator and supplemented with penicillin (100 U) and streptomycin (100µg/mL).
Exosome isolation and analyses
Exosomes were isolated using a slight modification of an established protocol [20, 40, 41]. Briefly, cells were cultured with 10% FBS until reaching 70% confluence, washed 3x with PBS, then cultured for 48h in serum-free medium, after which supernatants were collected and centrifuged for 10 min at 300×g at 4°C to remove dead cells, then transferred and centrifuged for 30 min at 9000×g to remove cell debris. Supernatant was centrifuged at 100,000×g for 2 h to generate exosome pellets, which were resuspended in PBS and then centrifuged at 100,000×g for 2 h at 4°C, and resulting exosome pellets were resuspended in PBS for characterization or use in cell culture experiments or dissolved in M-PER mammalian protein extraction reagent (Pierce, Rockford, IL) for Western blot analyses.
Dynamic light scattering was performed with 10 µg of PBS-resuspended exosomes to generate size distribution data that was analyzed using Zetasizer Software. For TEM analyses, 10 µL of purified exosome fraction (100 µg/mL) was placed on non-glow-discharged carbon-coated grids for 10 min and then negatively stained with 10 µL of 2% uranyl acetate for 1 min, after which negative stain solution was removed by wicking onto filter paper, and the dried grids were viewed using a Philips/FEI CM-12 transmission electron microscope operated at 80 KeV [20].
Determination of protein concentration
Whole cells or purified exosomes were lysed in M-PER mammalian protein extraction reagent in the presence of a protease inhibitor cocktail (Pierce, Rockford, IL). The protein concentration was determined using a bicinchoninic acid (BCA) assay kit (Pierce, Rockford, IL) according to the manufacturer's instructions.
Western blot analysis
Western blot assays were performed with precast Tris-Bis 4%-15% gradient gels and nitrocellulose membranes (Bio-Rad, Richmond, CA) using standard methods. Specific protein signals in loaded whole-cell lysates (WCLs, 20 µg) or exosome lysates (EXOs, 10 µg) were analyzed by densitometric analyses normalized to a loading control or an internal standard.
Isolation and identification of exosomal proteins
Exosomal proteins were isolated and identified using a previously described method [42, 43]. Briefly, silver-stained SDS-PAGE gel slices were digested with trypsin, and the digests were separated using Ultimate 3000 nano-LC (Dionex Corporation, USA) with a nC18 enrichment column (Dionex C18 Pepmap 100, 5 µm particle, 100 Å pore, 300 µm i.d. ×5 mm) and a Dionex nC18 analytical column (C18 Pepmap 100, 3 µm particle, 100 Å pore, 75 µm i.d. ×150 mm). Each 10 µL sample was dried by vacuum centrifugation and resuspended to ~20 µL with 0.1% formic acid/2% acetonitrile before injection. Flow rates of 20 µL/min and 300 nL/min were used for the loading and analytical columns, respectively. Eluted peptides were analyzed on a linear ion trap LTQ Velos Pro mass spectrometer (Thermo Scientific, San Jose, CA). One MS scan was followed by eight MS/MS scans, and MS/MS spectra were queried against the UniProt database (www.uniprot.org) using in-house Mascot 2.3.0 (www.matrixscience.com) software and a mass tolerance of 0.5 Da.
EphA2 shRNA knockdown
PANC-1 cells were transduced with lentiviral vectors expressing Eph2A (EphA2_shRNA_1: 5'-GAACTTCAACACAGCCTGG-3' or EphA2_shRNA_2: 5'-AGAGGTTGAAAGTCTCCTT-3') or control (Ctrl_shRNA) shRNA, then incubated with 10 µg/mL puromycin (Invitrogen, Carlsbad, CA) for 2 days to select for transduced cells, with EphA2 expression knockdown confirmed by Western blot.
GEM sensitivity assays
Cells were seeded in 96-well plates at a density of 2×103 cells/well (MIA PaCa-2 cells) or 3×103 cells/well (PANC-1 and BxPC-3 cells) and allowed to attach for 24 h, then cultured for 72 h with 1-100 nM GEM, as indicated. MTT (5 mg/mL) was then added to each well, and after 4 h the media was aspirated and 100 µL of DMSO was added to dissolve formazan crystals. Plates were gently agitated for 5 min and absorbance was measured at 570 nm and 630 nm to determine cell viability.
For EphA2-mediated viability studies, MIA PaCa-2 and BxPC-3 cells were seeded in 24-well plates (1×105 cells/well), cultured for 24 h in serum-containing media supplemented with 0, 5, 10, or 20 µg/mL PANC-1 or PANC-1EphA2- exosomes, or 0.04, 0.2 or 0.5µg/mL recombinant EphA2, then re-seeded into 96-well plates (2 × 103 cells/well MIA PaCa-2; 3 × 103 cells/well BxPC-3) and cultured for 3 days with 0, 25, 50, or 100 nM GEM, with fresh exosomes added every 24 h, and then analyzed by MTT cell viability assays according to the manufacturer's instructions.
Exosome internalization
Exosomes were labeled with EXO-Red (System Biosciences, Mountain View, CA) according to the manufacturer's recommendations. MIA PaCa-2, BxPC-3 cells and THP-1 cells were plated at 2.5×104 cells/well in 8 well chamber slides (BD Falcon, Bedford, MA), allowed to adhere for 24 h in serum-containing medium, washed 3x with PBS, cultured for 2 h at 37°C in serum-containing medium supplemented with 20 µg/mL EXO-Red-labeled PANC-1 exosomes or 0.5 µg/mL recombinant fluorescent-labeled EphA2 protein. THP-1 monocytes were treated with 50 ng/mL phorbol 12-myristate 13-acetate (PMA) to differentiate into macrophage. Recombinant EphA2 was labeled with Alexa Fluor 488 dyes according to the manufacturer's instructions. For microscopy studies, cells were washed 3x with PBS, incubated for 15 min at 25°C with 4% paraformaldehyde, and incubated for 5 min at 25°C in 1:1000 4'6-diamidino-2-phenylindole (DAPI)/PBS solution, and exosomes were visualized using a laser scanning confocal microscope with a 40× objective (FV-100; Olympus). For flow cytometry studies, cells were PBS washed, detached, PBS washed, diluted in PBS with 10% BSA, and then analyzed on a BD FACS AriaII (BD Biosciences, San Jose, CA) to measure exosome uptake by florescence intensity [44].
Statistical analysis
Statistical analysis was performed using one-way or two-way ANOVA with post hoc Bonferroni tests for each individual comparison. GraphPad Prism software (San Diego, CA) was used to perform the calculations. Differences with a p-value <0.05 were considered statistically significant. Data were expressed as the mean±SD (n≥3).
Supplementary Material
Supplementary figures.
Acknowledgements
We thank the Intramural Research Programs of the Clinical Center of the National Institutes of Health. We thank Christopher J. Lyon and Vollter Anastas for scientific editing. Grant Support: The work was primarily supported by research funding provided by NIH (U01CA214254, R01HD090927, R01AI122932, R01AI113725, R21Al126361-01 and 5P50CA126752-08), Arizona Biomedical Research Commission (ABRC) young investigator award, Fred Hutchinson Cancer Research Center (0000917241), Intramural Research Program of the NIH, PANCAN-AACR Career Development Award (14-20-25-KOAY), Radiological Society of North America seed grant (RSD1429), National Natural Science Foundation of China (81702996), and the Shiekh Ahmed Center for Pancreatic Cancer Research and the Center for Radiation Oncology Research at The University of Texas M.D. Anderson Cancer Center.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99:1441-54
2. Swartz MA, Iida N, Roberts EW, Sangaletti S, Wong MH, Yull FE. et al. Tumor microenvironment complexity: emerging roles in cancer therapy. Cancer Res. 2012;72:2473-80
3. Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011;378:607-20
4. Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR. et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403-13
5. Ryan DP. Chemotherapy for advanced exocrine pancreatic cancer. UpToDate. 2015
6. Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y. et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817-25
7. Samuel N, Hudson TJ. The molecular and cellular heterogeneity of pancreatic ductal adenocarcinoma. Nat Rev Gastroenterol Hepatol. 2012;9:77-87
8. Koay EJ, Truty MJ, Cristini V, Thomas RM, Chen R, Chatterjee D. et al. Transport properties of pancreatic cancer describe gemcitabine delivery and response. J Clin Invest. 2014;124:1525-36
9. Kahlert C, Melo SA, Protopopov A, Tang J, Seth S, Koch M. et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J Biol Chem. 2014;289:3869-75
10. Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G. et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883-91
11. Taylor DD, Gercel-Taylor C. Exosomes/microvesicles: mediators of cancer-associated immunosuppressive microenvironments. Semin Immunopathol. 2011;33:441-54
12. Corrado C, Raimondo S, Chiesi A, Ciccia F, De Leo G, Alessandro R. Exosomes as intercellular signaling organelles involved in health and disease: basic science and clinical applications. Int J Mol Sci. 2013;14:5338-66
13. Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T. et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell. 2014;159:499-513
14. Azmi AS, Bao B, Sarkar FH. Exosomes in cancer development, metastasis, and drug resistance: a comprehensive review. Cancer Metastasis Rev. 2013;32:623-42
15. Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK. et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17:816-26
16. Bold RJ, Chandra J, McConkey DJ. Gemcitabine-induced programmed cell death (apoptosis) of human pancreatic carcinoma is determined by Bcl-2 content. Ann Surg Oncol. 1999;6:279-85
17. Giovannetti E, Mey V, Danesi R, Mosca I, Del Tacca M. Synergistic cytotoxicity and pharmacogenetics of gemcitabine and pemetrexed combination in pancreatic cancer cell lines. Clin Cancer Res. 2004;10:2936-43
18. Hong SP, Wen J, Bang S, Park S, Song SY. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int J Cancer. 2009;125:2323-31
19. Huanwen W, Zhiyong L, Xiaohua S, Xinyu R, Kai W, Tonghua L. Intrinsic chemoresistance to gemcitabine is associated with constitutive and laminin-induced phosphorylation of FAK in pancreatic cancer cell lines. Mol Cancer. 2009;8:125
20. Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006 Chapter 3: Unit 3 22
21. Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569-79
22. Farrell JJ, Elsaleh H, Garcia M, Lai R, Ammar A, Regine WF. et al. Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology. 2009;136:187-95
23. Kim MP, Gallick GE. Gemcitabine resistance in pancreatic cancer: picking the key players. Clin Cancer Res. 2008;14:1284-5
24. Kim Y, Han D, Min H, Jin J, Yi EC. Comparative proteomic profiling of pancreatic ductal adenocarcinoma cell lines. Mol Cells. 2014;37:888-98
25. Suenaga S, Kuramitsu Y, Wang Y, Baron B, Kitagawa T, Akada J. et al. Human pancreatic cancer cells with acquired gemcitabine resistance exhibit significant up-regulation of peroxiredoxin-2 compared to sensitive parental cells. Anticancer Res. 2013;33:4821-6
26. Zhou J, Du Y. Acquisition of resistance of pancreatic cancer cells to 2-methoxyestradiol is associated with the upregulation of manganese superoxide dismutase. Mol Cancer Res. 2012;10:768-77
27. Ohmine K, Kawaguchi K, Ohtsuki S, Motoi F, Egawa S, Unno M. et al. Attenuation of phosphorylation by deoxycytidine kinase is key to acquired gemcitabine resistance in a pancreatic cancer cell line: targeted proteomic and metabolomic analyses in PK9 cells. Pharm Res. 2012;29:2006-16
28. Chen YW, Liu JY, Lin ST, Li JM, Huang SH, Chen JY. et al. Proteomic analysis of gemcitabine-induced drug resistance in pancreatic cancer cells. Mol Biosyst. 2011;7:3065-74
29. Fujimura Y, Ikenaga N, Ohuchida K, Setoyama D, Irie M, Miura D. et al. Mass spectrometry-based metabolic profiling of gemcitabine-sensitive and gemcitabine-resistant pancreatic cancer cells. Pancreas. 2014;43:311-8
30. Shedden K, Xie XT, Chandaroy P, Chang YT, Rosania GR. Expulsion of small molecules in vesicles shed by cancer cells: association with gene expression and chemosensitivity profiles. Cancer Res. 2003;63:4331-7
31. Chen WX, Liu XM, Lv MM, Chen L, Zhao JH, Zhong SL. et al. Exosomes from drug-resistant breast cancer cells transmit chemoresistance by a horizontal transfer of microRNAs. PLoS One. 2014;9:e95240
32. Zhuang G, Brantley-Sieders DM, Vaught D, Yu J, Xie L, Wells S. et al. Elevation of receptor tyrosine kinase EphA2 mediates resistance to trastuzumab therapy. Cancer Res. 2010;70:299-308
33. Lu M, Miller KD, Gokmen-Polar Y, Jeng MH, Kinch MS. EphA2 overexpression decreases estrogen dependence and tamoxifen sensitivity. Cancer Res. 2003;63:3425-9
34. Zhou C, Zhong Q, Rhodes LV, Townley I, Bratton MR, Zhang Q. et al. Proteomic analysis of acquired tamoxifen resistance in MCF-7 cells reveals expression signatures associated with enhanced migration. Breast Cancer Res. 2012;14:R45
35. Harada K, Hiramoto-Yamaki N, Negishi M, Katoh H. Ephexin4 and EphA2 mediate resistance to anoikis through RhoG and phosphatidylinositol 3-kinase. Exp Cell Res. 2011;317:1701-13
36. Miao B, Ji Z, Tan L, Taylor M, Zhang J, Choi HG. et al. EPHA2 is a mediator of vemurafenib resistance and a novel therapeutic target in melanoma. Cancer Discov. 2015;5:274-87
37. Gokmen-Polar Y, Toroni RA, Hocevar BA, Badve S, Zhao Q, Shen C. et al. Dual targeting of EphA2 and ER restores tamoxifen sensitivity in ER/EphA2-positive breast cancer. Breast Cancer Res Treat. 2011;127:375-84
38. Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. Ligation of EphA2 by Ephrin A1-Fc inhibits pancreatic adenocarcinoma cellular invasiveness. Biochem Biophys Res Commun. 2004;320:1096-102
39. Winter JM, Yeo CJ, Brody JR. Diagnostic, prognostic, and predictive biomarkers in pancreatic cancer. J Surg Oncol. 2013;107:15-22
40. Logozzi M, De Milito A, Lugini L, Borghi M, Calabro L, Spada M. et al. High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients. PLoS One. 2009;4:e5219
41. Li J, Sherman-Baust CA, Tsai-Turton M, Bristow RE, Roden RB, Morin PJ. Claudin-containing exosomes in the peripheral circulation of women with ovarian cancer. BMC Cancer. 2009;9:244
42. Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452:181-6
43. Ishihama Y. Proteomic LC-MS systems using nanoscale liquid chromatography with tandem mass spectrometry. J Chromatogr A. 2005;1067:73-83
44. Christianson HC, Svensson KJ, van Kuppevelt TH, Li JP, Belting M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc Natl Acad Sci U S A. 2013;110:17380-5
Author contact
![]() Corresponding authors: Zhen Zhao, Department of Pathology and Laboratory Medicine, Weill Cornell Medicine. Ye Hu, School of Biological and Health Systems Engineering, Virginia G. Piper Biodesign Center for Personalized Diagnostics, The Biodesign Institute, Arizona State University, Tempe, AZ 85281, USA
Corresponding authors: Zhen Zhao, Department of Pathology and Laboratory Medicine, Weill Cornell Medicine. Ye Hu, School of Biological and Health Systems Engineering, Virginia G. Piper Biodesign Center for Personalized Diagnostics, The Biodesign Institute, Arizona State University, Tempe, AZ 85281, USA
Citation styles
APA
Fan, J., Wei, Q., Koay, E.J., Liu, Y., Ning, B., Bernard, P.W., Zhang, N., Han, H., Katz, M.H., Zhao, Z., Hu, Y. (2018). Chemoresistance Transmission via Exosome-Mediated EphA2 Transfer in Pancreatic Cancer. Theranostics, 8(21), 5986-5994. https://doi.org/10.7150/thno.26650.
ACS
Fan, J.; Wei, Q.; Koay, E.J.; Liu, Y.; Ning, B.; Bernard, P.W.; Zhang, N.; Han, H.; Katz, M.H.; Zhao, Z.; Hu, Y. Chemoresistance Transmission via Exosome-Mediated EphA2 Transfer in Pancreatic Cancer. Theranostics 2018, 8 (21), 5986-5994. DOI: 10.7150/thno.26650.
NLM
Fan J, Wei Q, Koay EJ, Liu Y, Ning B, Bernard PW, Zhang N, Han H, Katz MH, Zhao Z, Hu Y. Chemoresistance Transmission via Exosome-Mediated EphA2 Transfer in Pancreatic Cancer. Theranostics 2018; 8(21):5986-5994. doi:10.7150/thno.26650. https://www.thno.org/v08p5986.htm
CSE
Fan J, Wei Q, Koay EJ, Liu Y, Ning B, Bernard PW, Zhang N, Han H, Katz MH, Zhao Z, Hu Y. 2018. Chemoresistance Transmission via Exosome-Mediated EphA2 Transfer in Pancreatic Cancer. Theranostics. 8(21):5986-5994.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.