Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(22):6274-6290. doi:10.7150/thno.29580 This issue Cite
Research Paper
P-glycoprotein-targeted photodynamic therapy boosts cancer nanomedicine by priming tumor microenvironment
Chengqiong Mao1, Fang Li1, Yan Zhao1, Waldemar Debinski1,2, Xin Ming1,3 ![]()
1. Department of Cancer Biology and Comprehensive Cancer Center, Wake Forest University School of Medicine, Winston-Salem, NC 27157, USA;
2. Brain Tumor Center of Excellence, Thomas K Hearn Brain Tumor Research Center, Winston-Salem, NC 27157, USA;
3. Department of Biomedical Engineering, Wake Forest University School of Medicine, Winston-Salem, NC 27157, USA.
Received 2018-8-29; Accepted 2018-10-25; Published 2018-11-29
Citation:
Mao C, Li F, Zhao Y, Debinski W, Ming X. P-glycoprotein-targeted photodynamic therapy boosts cancer nanomedicine by priming tumor microenvironment. Theranostics 2018; 8(22):6274-6290. doi:10.7150/thno.29580. https://www.thno.org/v08p6274.htm
Other stylesAbstract

Cancer nanomedicines only modestly improve the overall survival of patients because their anticancer activity is limited by biological barriers posed by the tumor microenvironment. Currently, all the drugs in FDA-approved cancer nanomedicines are substrates for P-glycoprotein (Pgp), and thus, Pgp-mediated multidrug resistance (MDR) remains a hurdle for cancer nanomedicines.
Methods: In this study, Pgp-targeted photodynamic therapy (PDT) was developed to enhance the anticancer efficacy of nanomedicines by depleting MDR cancer cells as well as enhancing tumor penetration of nanomedicines. We first examined the Pgp specificity of our targeted PDT approach, and then tested combination therapy of PDT with Doxil in mixed tumor models of MDR cancer cells and stromal cells, mimicking human heterogeneous tumors.
Results: In vitro studies showed that the antibody-photosensitizer conjugates produced Pgp-specific cytotoxicity towards MDR cancer cells upon irradiation with a near-infrared light. The studies with a co-culture model of MDR cancer cells and stromal cells revealed synergistic effects in the combination therapy of PDT with Doxil. Using a mouse model of mixed tumors containing MDR cancer cells and stroma cells, we observed markedly enhanced tumor delivery of Doxil after PDT in vivo. We further examined the effects of the two modalities on individual cell populations and their synergism using an in vivo dual substrate bioluminescence assay. The results indicated that Pgp-targeted PDT specifically depleted MDR cancer cells and further enhanced Doxil's actions on both MDR cancer cells and stromal cells.
Conclusion: We conclude that our targeted PDT approach markedly enhances anticancer actions of nanomedicines by depleting MDR cancer cells and increasing their tumor penetration, and thereby, may provide an effective approach to facilitate translation of cancer nanomedicines.
Keywords: antibody conjugates, cancer multidrug resistance, cancer nanomedicine, cancer targeted therapy, P-glycoprotein, photodynamic therapy.
Introduction
Current cancer nanomedicines fail to improve the overall survival of cancer patients significantly because their efficacy is still limited by tissue and cellular barriers in tumors [1] (Scheme 1). Although nanoparticles (NPs) may take advantage of the enhanced permeability and retention (EPR) effect in tumor sites [2], the validity of this effect in human tumors is controversial because xenograft tumor models are fundamentally different from spontaneous human tumors, which are highly heterogeneous in terms of vascular leakage [1, 3, 4]. NPs may have better distribution in leaky sites of tumors; however, poor delivery to less leaky sites will result in incomplete eradication of the tumor and thereafter tumor relapse. On the other hand, cellular mechanisms leading to cancer resistance to conventional medicines pose the same challenges to cancer nanomedicines. P-glycoprotein (Pgp) is a primary cause for multidrug resistance (MDR) in cancers [5], and its overexpression has been found to be associated with treatment failure in more than half of human cancers, including ovary, colon and liver cancers, as well as leukemia and lymphoma [6-9]. Various strategies have been actively sought to overcome this resistance for more than three decades; however, they have yet to reach the oncology clinic [5].
Abundant nanoformulations have been developed to combat Pgp-medicated MDR [10], including micelles [11-14], liposomes [15, 16], carbon nanotubes [17-19], mesoporous silica NPs [20, 21], and gold NPs [22-24]. They are able to increase the accumulation of chemotherapy drugs at tumor sites through the EPR effects [25, 26], and further enhance intracellular drug concentration to compensate Pgp-medicated drug efflux [27-32]. However, the outcomes from this strategy are still limited due to poor penetration and low cellular internalization of the NPs [25, 33-35]. Therefore, none of these NPs have been approved by the FDA for treating MDR tumors. Liposomal doxorubicin (Doxil/Caelyx), daunorubicin (DaunoXome), vincristine (MARQIBO), irinotecan (ONIVYDE), and albumin NP of taxol (Abraxane) are currently the only FDA-approved cancer nanomedicines, and all the drugs in these NPs are substrates for Pgp [36] and Pgp-mediated drug efflux can decrease their anticancer activity, leading to MDR [37, 38]. Therefore, novel strategies are needed to overcome Pgp-mediated MDR for enhancing the therapeutic efficacy of nanomedicines, especially in patients that present MDR cancers.
Scheme 1
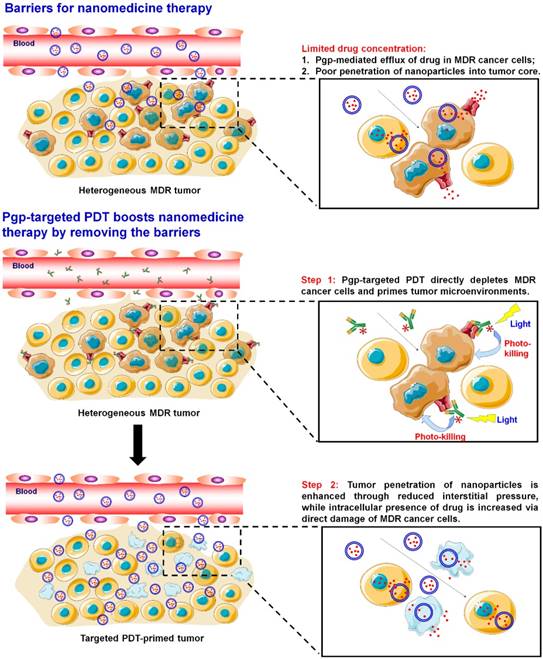
Pgp-targeted PDT boosts cancer nanomedicine. The barriers for cancer nanomedicine include tissue barriers preventing NP penetration and cellular barriers limiting intracellular delivery of drugs. Pgp-targeted PDT boosts nanomedicines therapy by depleting MDR cancer cells and enhancing tumor penetration of NPs.
Targeted photodynamic therapy (PDT) may provide an effective approach to overcome Pgp-mediated MDR towards cancer nanomedicines. PDT is a clinically approved cancer therapy [39], while targeted PDT is a highly cancer-specific approach to treat cancer by combining antibody-based cancer targeting and localized light activation of the photosensitizer (PS) [40]. Targeted PDT using cetuximab-IR700 to target epidermal growth factor receptor (EGFR) is currently being tested in a phase I/II trial for the treatment of head and neck cancers (NCT02422979) [41]. Further, EGFR-targeted PDT was reported to produce a “super EPR effect” at the tumor site that enhanced penetration of the NPs into the tumors [42].
In this study, we first examined the role of Pgp in chemoresistance to liposomal doxorubicin (Doxil) and albumin NP of taxol (Abraxane). Then, Pgp-targeted PDT was developed to enhance the anticancer efficacy of the nanomedicines by depleting MDR cancer cells as well as enhancing tumor penetration of the nanomedicines (Scheme 1). We prepared antibody-photosensitizer conjugates (APCs) using an anti-Pgp monoclonal antibody (Pab), and tested their specificity and phototoxicity using cell culture models. We further tested combination therapy of PDT with Doxil in a co-culture model of MDR cancer cells and stromal cells, which mimics human heterogeneous tumors. Then, their tumor delivery and anticancer efficacy were examined using a mouse model of mixed tumors containing MDR cancer cells and stromal cells, and we further examined the effects of the two modalities on individual cell populations and their synergism using an in vivo dual substrate bioluminescence assay.
Methods
Cell lines
3T3-MDR1, a mouse fibroblast cell line stably transfected with a cDNA coding for the human Pgp, was obtained from Dr. Michael Gottesman's laboratory at the National Cancer Institute (NCI). This cell line was maintained in DMEM cell culture medium (Corning Inc., Corning, NY, USA) supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich, St. Louis, USA), 400 IU/mL penicillin, 100 μg/mL streptomycin (Corning Inc.), and 60 ng/mL colchicine (Sigma-Aldrich). NCI-ADRRes is an adriamycin-resistant ovarian cancer cell line with high Pgp expression, and KB-8-5-11 is a MDR human KB carcinoma cell line independently selected with colchicine. Both of them were obtained from Dr. Gottesman's lab at NCI, and were maintained in the same condition as the 3T3-MDR1 cell line. OVCAR8 cells, the parental cell line of NCI-ADRRes cells, and 3T3 cells were from ATCC (Rockville, MD, USA). KB-3-1 cells, a subline of HeLa and the parental cell line of KB-8-5-11, were from Dr. Gottesman's lab. All these chemosensitive control cells were cultured in the same cell culture medium but without colchicine. GFP and/or firefly luciferase-expressing cells were constructed by transfection with reporter-encoding lentivirus (Biosettia, San Diego, CA, USA) according to a standard protocol provided by the vendor. The human cell lines were characterized by Genetica DNA Laboratories (Burlington, NC, USA) using short tandem repeat profiling.
Cytotoxicity of drugs in chemosensitive and chemoresistant cells
Dose-dependent cytotoxicity of doxorubicin (Sigma-Aldrich), Taxol (Sigma-Aldrich), Doxil (Johnson & Johnson), and Abraxane (Celgene) was quantified using Alamar Blue assay according to a method described previously [43, 44]. Briefly, five thousand cells were seeded in 96-well plates and were cultured overnight. Medium was replaced with the drugs in culture medium at a series of dilutions. Seventy-two hours post treatment, Alamar Blue reagent (Thermo Fisher Scientific, Waltham, MA, USA) was added and incubated for 2 h. The fluorescence of the samples was then measured on a CYTATION 5 imaging reader (BioTeK, Winooski, VT, USA) set at 540 nm excitation and 590 nm emission wavelengths. The mean drug concentrations required for 50% growth inhibition (IC50 values) were calculated using CompuSyn software (Version 1.0, ComboSyn Inc., Paramus, NJ, USA).
Measurement of cellular accumulation of doxorubicin and Doxil
Three pairs of chemosensitive (OVCAR8, KB-3-1, and 3T3) and chemoresistant (NCI-ADRRes, KB-8-5-11, and 3T3-MDR1) cell lines were incubated with 1 µM GF918 (Sigma-Aldrich), a Pgp inhibitor, for 30 min at 37 °C followed by adding 0.5 µg/mL doxorubicin or Doxil to the cells. After 24 h incubation, doxorubicin or Doxil accumulation was measured with flow cytometry on an LSR Fortessa flow cytometer (BD Bioscience, Franklin Lakes, NJ, USA). Twenty thousand events of cells were analyzed and only live cells were gated for quantitation of cellular accumulation. The data was processed using FlowJo software (FlowJo, Ashland, OR, USA).
Synthesis of Pab-IR700
Anti-Pgp monoclonal antibody 15D3 was produced in-house with a method described previously [45, 46]. Briefly, hybridoma cells were initially cultured in DMEM media containing 10% FBS. The serum content was reduced by serial dilution until culturing in serum-free hybridoma medium (Thermo Fisher Scientific). The media containing antibody was collected and the antibody was purified with a HiTrap Protein G HP column (GE Healthcare Life Sciences, Piscataway, NJ, USA). To prepare the APCs, Pab was incubated with IR700-NHS (LI-COR Biosciences, Lincoln, NE, USA) at a molar ratio of 1:4 in phosphate buffer (pH 8.0) for 1 h. The product of the conjugation was purified using a ZebaTM spin desalting column (40K MWCO, Thermo Fisher Scientific). The protein concentration of the antibody conjugates was determined with BCA protein assay kit (Thermo Fisher Scientific), and the IR700 concentration was quantified by measurement of the absorption at 689 nm in order to estimate the number of IR700 molecules conjugated to each antibody molecule. To monitor the synthesis process and characterize the final product, SEC-HPLC was performed using an UltiMate™ 3000 UHPLC system (Thermo Fisher Scientific) equipped with a diode-array detector. These molecules were first separated in the SEC column, and their UV-vis spectra were scanned with the inline diode-array detector to confirm their identities.
Cellular uptake of Pab-IR700
For flow cytometry experiments, cells were seeded on 24-well plates and were cultured overnight. Cells were treated with free IR700 or Pab-IR700 (both equivalent to 150 nM IR700) and incubated at 37 °C for 4 h. The cells were then trypsinized and suspended in PBS buffer. Fluorescence of the cells was acquired on a LSRFortessa flow cytometer. Ten thousand events of cells were analyzed and the data was processed using FlowJo software.
For confocal microscopy experiments, cells were seeded on 8-well Lab-Tek™ II Chambered Coverglass (Nalge Nunc, Rochester, NY, USA) and were cultured overnight. The cells were incubated with free IR700 or Pab-IR700 (both equivalent to 150 nM IR700) at 37 °C for 4 h. After washing with cold PBS twice, the cells were then fixed with 4% paraformaldehyde (Sigma-Aldrich), stained with DAPI (Thermo Fisher Scientific), and visualized with a ZEISS LSM 710 confocal microscope (Carl Zeiss AG, Oberkochen, Germany).
In vitro phototoxicity studies for Pab-IR700
The phototoxicity of free IR700 and Pab-IR700 was quantified using Alamar Blue assay [44, 47]. Briefly, five thousand cells were seeded in 96-well plates and cultured overnight. Medium was replaced with increasing concentrations of free IR700 or Pab-IR700. The cells were further incubated at 37 °C for 4 h. After washing, the cells were irradiated with a 690 nm LED light for 20 min to reach the light dose of 5 J/cm2. After 24 h, Alamar Blue reagent was added and incubated for 2 h. The fluorescence of the samples was then measured on a CYTATION 5 imaging reader. We also measured the phototoxicity of Pab-IR700 without the washing step after incubation.
The phototoxicity of Pab-IR700 was also examined with live/dead cell staining. Ten thousand cells were seeded in 96-well plates and were cultured overnight. Medium was replaced with the dose solution of Pab-IR700 (equivalent to 150 nM IR700). The cells were further incubated for 4 h at 37 °C. After washing with PBS, the cells were irradiated with LED light (5 J/cm2). An hour after NIR irradiation, the cells were co-stained with Calcein AM (2 µM) and PI (5 µg/mL) at room temperature for 30 min, rinsed with PBS, and then imaged with a Cytation 5 Imaging Reader.
Cellular singlet oxygen detection after targeted PDT
After being incubated with free IR700 or Pab-IR700 (equivalent to 150 nM IR700) overnight, KB-8-5-11 or KB-3-1 cells were treated with 10 μM CM-H2DCFDA (Thermo Fisher Scientific) and incubated for 30 min. Then, the cells were irradiated with LED light (5 J/cm2). The intracellular singlet oxygen generation was detected by observing the fluorescent product DCF using a Cytation 5 Imaging Reader.
In vitro combination therapy in a mixed cell culture model
A co-culture cell model was established to mimic spontaneous human tumors with heterogeneous expression of Pgp. One thousand chemosensitive 3T3 fibroblasts expressing Renilla luciferase (3T3-rLuc) and four thousand chemoresistant KB-8-5-11 cancer cells expressing firefly luciferase (KB-8-5-11-fLuc) were seeded in 96-well plates and were cultured overnight. For the combined treatment group, the cells were first treated with 1 μg/mL Pab-IR700 (equivalent to 15 nM IR700) at 37 °C overnight. After washing with PBS, the cells were irradiated with LED light at a light dose of 5 J/cm2, and then were treated with 100 μg/mL Doxil for 72 h. The monotherapies were performed according to the regimens in the combination therapy group. Dual luciferase activities after treatments were measured with a Dual-Luciferase® Reporter Assay System (Promega, Madison, WI, USA).
To study the synergistic anticancer effects of the combined therapy, one thousand chemosensitive 3T3 fibroblasts and four thousand KB-8-5-11 cancer cells were seeded in 96-well plates and were cultured overnight. Cells were treated as described above. Alamar Blue reagent was then added and incubated for 2 h to determine the cell viability. The combination index (CI) of the combination of targeted PDT and Doxil was calculated with the Chou and Talalay method using CompuSyn software.
Doxil accumulation was quantified after targeted PDT using the flow cytometry method. In this experiment, 100,000 cells were seeded in 12-well plates and were cultured overnight. The cells were first treated with 1 μg/mL Pab-IR700 at 37 °C overnight, and were then irradiated with LED light (5 J/cm2). After that, they were treated with 0.5 μg/mL Doxil for 24 h. Doxil accumulation was measured with flow cytometry on an LSR Fortessa flow cytometer.
Measurement of cellular ATP levels
To detect the intracellular and extracellular ATP concentrations, KB-8-5-11 and 3T3 cells were seeded in 12-well plates and were cultured overnight. Then, the cells were incubated with 1 μg/mL Pab-IR700 at 37 °C overnight. For intracellular ATP detection, cells were washed with PBS and incubated with fresh culture medium and were then irradiated with LED light at a light dose of 5 J/cm2. After washing with PBS thrice, cells were harvested with cell scrapers into 500 μL Tris-EDTA buffer (100 mM Tris, 4 mM EDTA, pH 7.5) at 30 min, 90 min and 240 min post light irradiation. After heating at 95 °C for 7 min and centrifuging at 18,544 × g for 3 min, the supernatants were used to determine the intracellular ATP concentration with an ATP determination kit (Thermo Fisher Scientific) according to the manufacturer's protocol. For extracellular ATP detection, cells were washed with PBS and incubated with the basal culture media containing 1% bovine serum albumin (Sigma-Aldrich) and 300 μM ARL67156 (Sigma-Aldrich), which inhibits ecto-nucleotidase activity and prevents degradation of extracellular ATP by FBS. Then, the cells were irradiated with LED light. The culture medium from each well was collected at 30 min, 90 min and 240 min post light irradiation and centrifuged at 2,365 × g for 5 min. The supernatants were used to determine the extracellular ATP concentration using an ATP determination kit.
Animals
All animal experiments were conducted in compliance with the Guide for the Care and Use of Laboratory Animal Resources (2011, US National Research Council), and the animal protocol was approved by the Wake Forest Institutional Animal Care and Use Committee. Female BALB/c nude mice (4-6 weeks old) that were purchased from Charles River (Wilmington, MA, USA) were used in the animal studies.
In vivo combination therapy in a mixed MDR tumor model
To evaluate the effects of targeted PDT on Doxil accumulation in tumors, a mixed MDR tumor model was established by inoculation of a mixture of 1×106 KB-8-5-11 cells and 5×104 3T3 cells in 0.1 mL PBS/Matrigel (1:1, v/v) into nude mice bilaterally. After 7 days, mice were i.v. injected with Pab-IR700 (200 µg). Fluorescence images were taken using an IVIS Imaging System (PerkinElmer, Inc., Waltham, MA, USA) for visualization of IR700 at 48 h post injection. After that, the tumors on the right site were exposed to 690 nm LED light at a total dose of 50 J/cm2. Thirty minutes after light irradiation, mice received Doxil (10 mg/kg) via i.v. injection. Fluorescence images were taken using an IVIS Imaging System for visualization of Doxil and IR700 at 15 min, 2 h, and 24 h after Doxil administration. Heart, liver, spleen, lung, kidney, and tumors were collected for ex vivo IVIS imaging to determine the distribution of Doxil and IR700 in the main organs at 24 h post injection of Doxil. Tumors were digested by AccumaxTM cell dissociation solution (STEMCELL Technologies Inc., Cambridge, MA, USA) for flow cytometry in order to determine intracellular Doxil levels in tumor cells.
For the tumor response experiments, 1×106 KB-8-5-11-GFP-fLuc cells and 5×104 3T3-RFP-rLuc cells were suspended in 0.1 mL PBS/Matrigel (1:1, v/v) and injected into the floor of the mouth via an extra-oral approach. After 7 days, mice were randomly allocated into four groups (n = 8): PBS, targeted PDT, Doxil, and targeted PDT + Doxil. The mice in the PDT and PDT + Doxil groups received i.v. injection of 200 µg Pab-IR700 at day 7. Fluorescence images were taken using an IVIS Imaging System for visualization of IR700 at 48 h post-injection. After that, the mice in the PDT and PDT + Doxil groups were exposed to the 690 nm LED light at a total dose of 50 J/cm2. The mice in the Doxil and PDT + Doxil groups received Doxil (10 mg/kg) via i.v. injection 30 min after light irradiation. Tumor growth was measured with in vivo dual substrate bioluminescence imaging (BLI) and by a caliper twice per week. Mice were euthanized if any tumor volume exceeded 1,500 mm3. The body weight of the mice was recorded to evaluate in vivo toxicity.
Immunohistochemical analysis
Five days after the light irradiation, some mice were sacrificed and tumor tissues were excised for immunohistochemical analyses. Briefly, tumors were collected and fixed in freshly prepared 4% paraformaldehyde for 1 day. Tumor samples were paraffin-embedded, sectioned and stained with hematoxylin and eosin (H&E, Thermo Fisher Scientific). Nuclei of tumor cells were stained with DAPI (Thermo Fisher Scientific). Images of the stained sections were taken with a ZEISS LSM 710 confocal microscope.
Statistical analysis
Quantitative data are expressed as mean ± SD. Means were compared using Student's t-test for two-sample comparison or one-way ANOVA followed by Tukey's post-hoc analysis for multiple comparisons using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA). P values < 0.05 were considered statistically significant. Survival analysis was conducted with Kaplan-Meier curves, and their comparison was performed with the log-rank (Mantel-Cox) test.
Results
Efficacies of Doxil and Abraxane are limited by Pgp
Doxil (liposomal formulation of doxorubicin) and Abraxane (albumin NP of taxol) are the most commonly used cancer nanomedicines. We first characterized both NPs with transmission electron microscopy (TEM) and dynamic light scattering (DLS). As shown Figure S1, Doxil showed a particle size of 85.3 ± 0.9 nm, whereas Abraxane showed a particle size of 102.6 ± 1.1 nm. The results are consistent with previous reports. In order to examine if their therapeutic efficacies are limited by Pgp-mediated drug resistance, we first measured the dose-dependent cytotoxicity of doxorubicin (Dox), Doxil, Taxol, and Abraxane in three pairs of chemosensitive/chemoresistant cell lines: KB-3-1 (Pgp negative and chemosensitive) and KB-8-5-11 (Pgp positive and chemosensitive); OVCAR8 (Pgp negative and chemosensitive) and NCI-ADRRes (Pgp positive and chemoresistant); 3T3 (Pgp negative and chemosensitive) and 3T3-MDR1 (Pgp positive and chemoresistant). The cell viability curves are shown in Figure 1A-C, and the IC50 values of their cytotoxicity were calculated as shown in Table 1. The cytotoxicity of Taxol was similar to that of Abraxane in both chemosensitive and chemoresistant sublines (Figure 1 and Table 1). However, Doxil was less toxic than doxorubicin in these cells (Figure 1 and Table 1), likely due to delayed drug release from the liposome and slow endosomal escape. We also calculated the ratios of the IC50 values in chemoresistant cells to those in chemosensitive cells in each pair of cell lines in order to examine if the agent's cytotoxicity is limited by Pgp-mediated drug resistance. Ratios larger than 1 indicate strong effects by MDR, whereas small ratios indicate weak effects. As shown in Table 1, the ratios of Doxil are > 3.6 for the 3T3-MDR1/3T3 pair, > 29 for NCI-ADRRes/OVCAR8, and > 28.2 for KB-8-5-11/KB-3-1, indicating that the anticancer activity of Doxil was limited by Pgp-mediated MDR. Abraxane exhibits even larger effects than Doxil, and the ratios are 10.4 for 3T3-MDR1/3T3, 1000.0 for NCI-ADRRes/OVCAR8, and 785.7 for KB-8-5-11/KB-3-1. Overall, when compared to the drugs in free forms (doxorubicin and Taxol), their corresponding nanomedicines did not exhibit any advantages in terms of overcoming Pgp-mediated MDR.
Table 1
IC50 values (µg/mL) for cytotoxicity assay of doxorubicin, Doxil, Taxol, and Abraxane in chemosensitive and chemoresistant cells. The ratios of the IC50 values of chemoresistant cells to those of chemosensitive cells in each pair of cell lines are shown in blue.
| Dox | Doxil | Taxol | Abraxane | |
|---|---|---|---|---|
| 3T3 | 7.1 × 10-2 | 5.6 × 101 | 1.1 × 10-1 | 3.6 × 102 |
| 3T3-MDR1 | 6.2 × 10-1 | >2.0 × 102 | 2.7 × 100 | 3.8 × 103 |
| 3T3-MDR1/3T3 Ratio | 8.7 × 100 | >3.6 × 100 | 2.5 × 101 | 1.1 × 101 |
| OVCAR8 | 4.1 × 10-1 | 6.9 × 100 | 4.6 × 10-2 | 3.3 × 10-3 |
| NCI-ADRRes | 4.5 × 101 | >2.0 × 102 | >2.5 × 100 | 3.3 × 100 |
| NCI-ADR/OVCAR8 Ratio | 1.1 × 102 | >2.9 × 101 | >5.4 × 101 | 1.0 × 103 |
| KB-3-1 | 2.0 × 10-1 | 7.1 × 100 | 2.5 × 10-3 | 1.4 × 10-3 |
| KB-8-5-11 | 1.0 × 101 | >2.0 × 102 | 9.8 × 10-1 | 1.1 × 100 |
| KB-8-5-11/KB-3-1 Ratio | 5.0 × 102 | >2.8 × 101 | 3.9 × 102 | 7.9 × 102 |
In order to confirm that the drug resistance to Doxil and Abraxane is caused by Pgp-medicated drug efflux, three pairs of cell lines were incubated with Pgp inhibitor GF918 first and then 0.5 µg/mL doxorubicin or Doxil was added to the cells. After that, doxorubicin and Doxil accumulation in different cancer cells was evaluated by flow cytometry after 24 h incubation. As shown in Figure 2, the intracellular levels of both doxorubicin and Doxil in the chemoresistant sublines (NCI-ADRRes, KB-8-5-11, and 3T3-MDR1) were significantly lower than those in their respective control cell lines, at about 10% levels, indicating drug efflux of both doxorubicin and Doxil in these chemoresistant cells. However, when chemoresistant cells were incubated with the Pgp inhibitor GF918, the accumulation of both doxorubicin and Doxil was reversed to similar levels as in the chemosensitive cells. These results indicate that drug resistance to Doxil and doxorubicin is caused by Pgp-mediated efflux. We conclude that the therapeutic efficacies of both the FDA-approved cancer nanomedicines are reduced by Pgp-medicated drug efflux, and thus, the resultant MDR remains a hurdle for cancer nanomedicines.
Figure 1
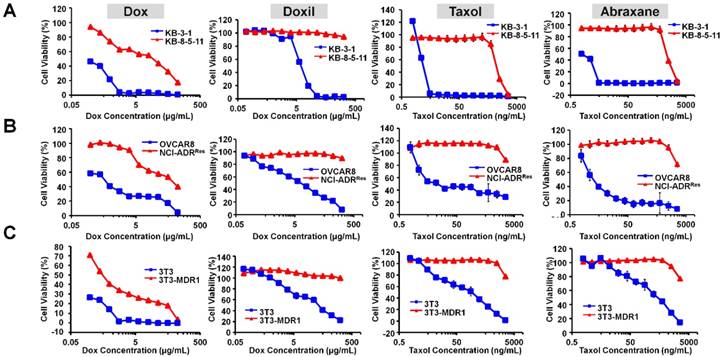
Therapeutic efficacies of Doxil and Abraxane are limited by Pgp. (A) Dose-dependent toxicity of doxorubicin, Doxil, Taxol, and Abraxane in KB-3-1 (Pgp negative and chemosensitive) and KB-8-5-11 (Pgp positive and chemoresistant) cells after 72 h incubation. (B) Dose-dependent toxicity of doxorubicin, Doxil, Taxol, and Abraxane in OVCAR8 (Pgp negative and chemosensitive) and NCI-ADRRes (Pgp positive and chemoresistant) cells after 72 h incubation. (C) Dose-dependent toxicity of doxorubicin, Doxil, Taxol, and Abraxane in 3T3 (Pgp negative and chemosensitive) and 3T3-MDR1 (Pgp positive and chemoresistant) cells after 72 h incubation. Data are mean ± SD (n = 3).
Figure 2
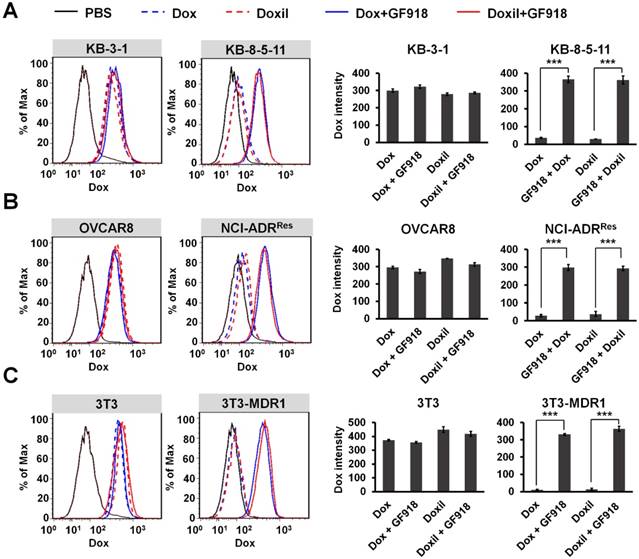
Efflux of doxorubicin and Doxil by Pgp. Three pairs of chemosensitive and chemoresistant cell lines, KB-3-1 and KB-8-5-11 (A), OVCAR8 and NCI-ADRRes (B), and 3T3 and 3T3-MDR1 (C), were incubated with 1 µM Pgp inhibitor GF918 and then 0.5 µg/mL doxorubicin or Doxil were added. Doxorubicin or Doxil accumulation within 24 h was determined by flow cytometry. Data are presented as mean ± SD (n = 3, ***p < 0.001).
Figure 3
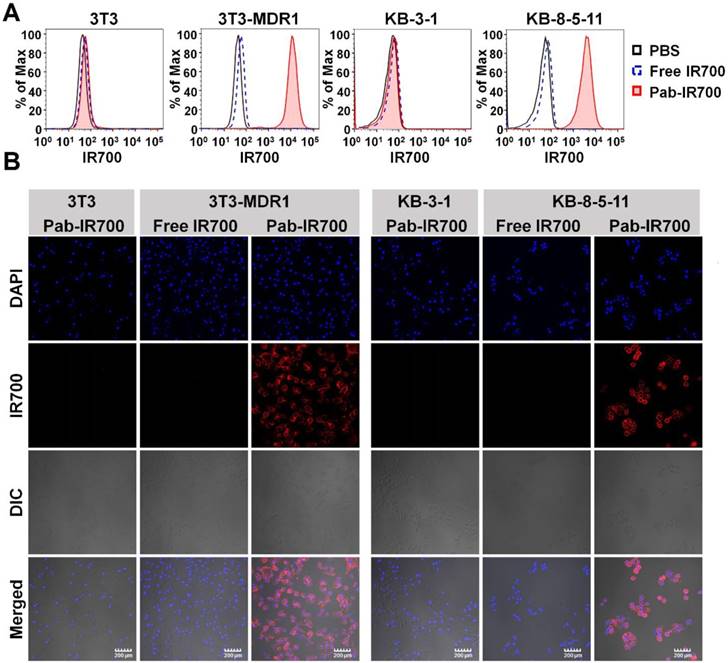
Pgp-IR700 specifically binds to Pgp-expressing chemoresistant cells. (A) Flow cytometry analysis of cellular uptake of free IR700 and Pab-IR700 in the chemosensitive cells 3T3 and KB-3-1 as well as the chemoresistant cells 3T3-MDR1 and KB-8-5-11 after 4 h incubation. (B) Confocal images of cellular uptake of free IR700 or Pab-IR700 in the chemosensitive cells 3T3 and KB-3-1 as well as the chemoresistant cells 3T3-MDR1 and KB-8-5-11 after 4 h incubation. Nuclei were labelled with DAPI (blue). Scale bar, 200 µm.
Pgp-targeted PDT specifically depletes chemoresistant cancer cells
In order to overcome Pgp-mediated chemoresistance, we prepared Pgp-specific APCs, Pab-IR700, with an anti-Pgp monoclonal antibody described previously [46]. We characterized the final product with an SEC-HPLC system equipped with a diode-array detector. As shown in Figure S2, Pab-IR700 eluted slightly earlier than Pab, and both eluted much earlier than free IR700, indicating successful conjugation and purification of Pab-IR700. Further, from the UV-vis spectra of individual peaks in the HPLC graphs, Pab-IR700 shows absorption at 280 nm and 689 nm, whereas Pab and free IR700 only show single absorption at 280 nm and 689 nm, respectively. These results indicated that Pab-IR700 was prepared in excellent purity.
We then examined the cellular uptake of Pab-IR700 in Pgp-positive cell lines, 3T3-MDR1 and KB-8-5-11, as well as Pgp-negative cell lines, 3T3 and KB-3-1, and used free IR700 as control. The results from both flow cytometry and confocal microscopy showed that free IR700 did not bind to any cell line, whereas Pab-IR700 specifically bound to the cell membrane of Pgp-expressing chemoresistant cells 3T3-MDR1 and KB-8-5-11 (Figure 3A-B). These results demonstrate the high Pgp specificity of Pab-IR700.
Phototoxicity of free IR700 and Pab-IR700 was measured with Alamar Blue assay after 4 h incubation with the PSs followed by light irradiation. As shown in Figure 4A, free IR700 did not cause phototoxicity to any cells, whereas Pab-IR700 produced dose-dependent cell death only in Pgp-expressing and chemoresistant 3T3-MDR1 and KB-8-5-11 cells, but not in Pgp-negative 3T3 and KB-3-1 cells, indicating that Pab-IR700-mediated photokilling is highly specific to Pgp-expressing MDR cells. In addition, the washing step after the Pab-IR700 treatment did not change the phototoxicity profiles (Figure 4A), indicating that the antibody conjugates produce phototoxicity only when they bind to target cells specifically. Similar results of phototoxicity were obtain with live/dead cell staining experiments. As shown in Figure S3, Pab-IR700 caused dramatic cytotoxicity towards 3T3-MDR1, KB-8-5-11, and NCI-ADRRes cells upon NIR irradiation; whereas, the same treatments did not affect cell viability of the control cells including 3T3, KB-3-1, and OVCAR8 cells. These results confirmed that Pab-IR700-mediated PDT is highly specific to Pgp.
To study the mechanism of the photokilling of Pab-IR700, we examined singlet oxygen generation in KB-8-5-11 and KB-3-1 cells after targeted PDT with an oxidative stress indicator CM-H2DCFDA. As shown in Figure 4B, bright green fluorescence was only observed in KB-8-5-11 cells after Pab-IR700 treatment followed by light irradiation. But in the other treatments, the singlet oxygen generation was barely detectable, suggesting that photoirradiation of cell-bound Pab-IR700 produces singlet oxygen and further triggers cell death events in Pgp-expressing chemoresistant cells. In light of the result that IR700 lacks photothermal activity (Figure S4), we concluded that the cell death from light irradiation of Pab-IR700 was mediated by photodynamic effects.
Figure 4
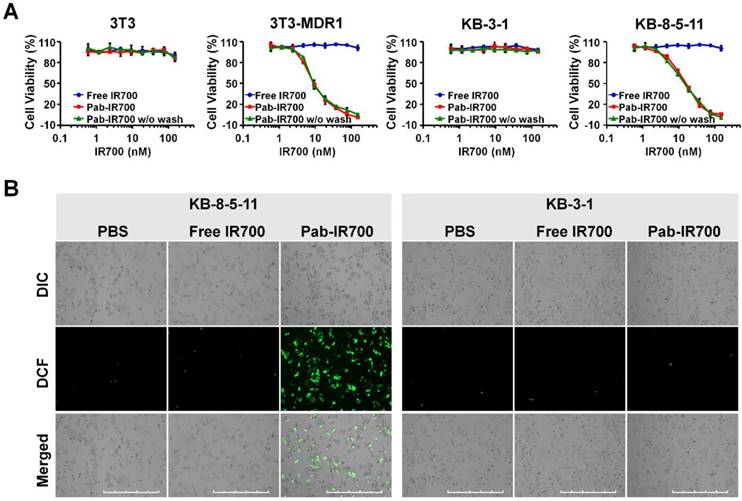
Pgp-targeted PDT specifically kills Pgp-expressing chemoresistant cells. (A) Dose-dependent phototoxicity of free IR700 and Pab-IR700 in 3T3, 3T3-MDR1, KB-3-1, and KB-8-5-11 cells at a light dose of 5 J/cm2 was measured with Alamar Blue assay. The phototoxicity of Pab-IR700 without the washing step was also measured. Data are mean ± SD (n = 3, *** p < 0.001). (B) Singlet oxygen generation of targeted PDT determined with the probe CM-H2DCFDA in KB-8-5-11 and KB-3-1 cells after NIR irradiation at 5 J/cm2. Scale bar, 1000 µm.
Pgp-targeted PDT overcomes chemoresistance towards Doxil in cell culture
To test if Pgp-targeted PDT can overcome drug resistance towards cancer nanomedicines, we combined our Pgp-targeted PDT with Doxil and tested this combination approach in a mixed cell culture model in which only some cells express Pgp. This combination therapy addresses a potential limitation of our targeted PDT in human tumors with heterogeneous Pgp expression. The co-cultured cell model is composed of Renilla luciferase-expressing chemosensitive 3T3 fibroblasts (3T3-rLuc), mimicking stromal cells in tumors, and firefly luciferase-expressing chemoresistant KB-8-5-11 cancer cells (KB-8-5-11-fLuc) (Figure 5A). When the mixed cells were treated with targeted PDT only, the firefly luciferase level decreased to 36% of that of the PBS group (Figure 5A); however, the Renilla luciferase was not affected by PDT, indicating that Pgp-targeted PDT only caused death of chemoresistant cancer cells (Figure 5A). When the mixed cells were treated with Doxil only, the firefly luciferase level did not change significantly but the Renilla luciferase level decreased to 8% of that of the PBS group (Figure 5A), indicating that Doxil could only inhibit growth of chemosensitive cells. The combination therapy of targeted PDT and Doxil produced significantly higher toxicity towards KB-8-5-11-fLuc cells compared to single PDT treatment, with a further 26% decrease in the firefly luciferase level from the single PDT treatment (Figure 5A).
Although a part of the KB-8-5-11-fLuc cell population survived targeted PDT, some of these cells might be partially damaged, and the function of Pgp-mediated drug efflux might be compromised. Thus, further Doxil treatment caused greater cytotoxicity. To examine this potential mechanism for the synergism between the two modalities, we measured cellular uptake of Doxil after targeted PDT. Thus, chemosensitive and chemoresistant cell lines underwent the targeted PDT procedure, which was followed by incubation with Doxil for 24 h. Then, live cells that survived the PDT procedure were gated and their Doxil levels were quantified with flow cytometry. The results in Figure 5B showed that the targeted PDT procedure significantly increased cellular accumulation of Doxil in the chemoresistant cells that survived the PDT procedure, and this increase was not observed in chemosensitive cells. We then concluded that targeted PDT can compromise Pgp-mediated efflux function and assist intracellular delivery of Doxil, even if the PDT procedure does not kill the cells.
Figure 5
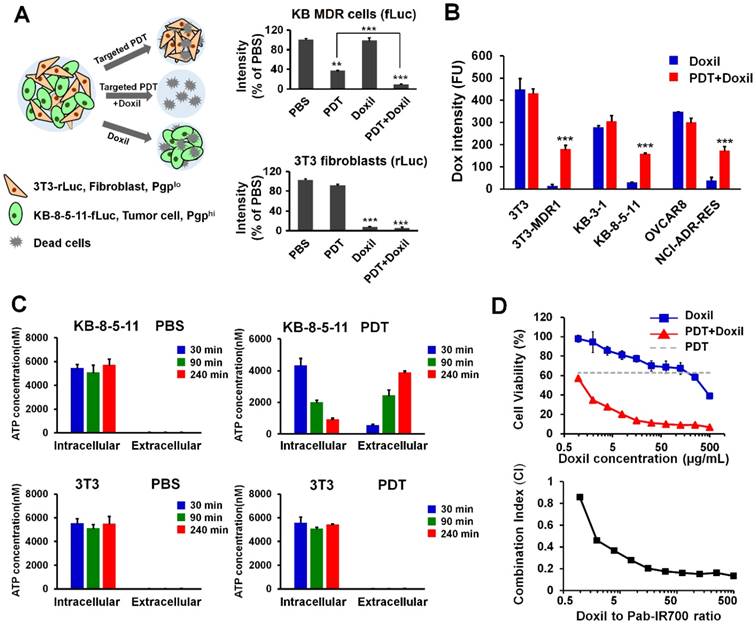
Synergistic combination therapy in an in vitro co-culture model. (A) Chemosensitive 3T3-rLuc fibroblasts and chemoresistant KB-8-5-11-fLuc cancer cells were co-cultured and received different treatments of PBS, targeted PDT, Doxil (100 µg/mL), and targeted PDT followed by Doxil treatment. Relative firefly and Renilla luciferases' activities were measured to indicate the viability of chemoresistant KB-8-5-11 and chemosensitive 3T3 cells. Data are presented as mean ± SD (n = 3, *** p < 0.001; ** p < 0.01). (B) Doxil accumulation in chemoresistant/chemosensitive cells after targeted PDT was measured by flow cytometry. Data are presented as mean ± SD (n = 3, *** p < 0.001 between the Doxil and combination groups). (C) Intracellular and extracellular ATP concentrations in KB-8-5-11 and 3T3 cells were measured at 30 min, 90 min or 240 min after the targeted PDT procedure. Data are presented as mean ± SD. (D) Dose-dependent cytotoxicity of Doxil in KB-8-5-11/3T3 mixed cells after the targeted PDT procedure was measured by Alamar Blue assay (upper). The combination index (CI) of combination therapy was calculated over the ratios of Doxil to Pab-IR700 (lower).
Pgp confers drug resistance by mediating the ATP-dependent efflux of chemotherapy drugs, and Pgp-mediated drug efflux highly depends on intracellular ATP concentration [48]. We speculated that partial damage of MDR cells caused leakage of intracellular ATP, and thereby reduced the efflux activity of Pgp. In order to examine the mechanism for the synergism, we measured the intracellular and extracellular ATP levels in the chemoresistant cell line KB-8-5-11 before and after the targeted PDT procedures. As shown in Figure 5C, KB-8-5-11 cells in the control conditions demonstrated low extracellular ATP level (~20 nM) and high intracellular ATP level (~6000 nM), which drives the Pgp's action of mediating drug efflux against the concentration gradient. Pgp-targeted PDT dramatically decreased the intracellular ATP level in KB-8-5-11 cells by 85%, but did not change the level in Pgp-negative 3T3 cells (Figure 5C). The dramatic decrease of the intracellular ATP level could reduce Pgp's efflux activity, which led to increased Doxil accumulation as shown in Figure 5B.
To further validate if there is synergism between targeted PDT and Doxil, we examined the dose-dependent cytotoxicity of Doxil with or without targeted PDT in the mixed cell model with Alamar Blue assay. The results in Figure 5D showed that Pgp-targeted PDT enhanced the Doxil-induced cell death. The CI was further calculated with the Chou-Talalay method. As shown in Figure 5D, combination therapy showed an overall CI value < 1 within the concentrations tested, indicating a clear synergy of targeted PDT and Doxil in this heterogeneous cell model.
Pgp-targeted PDT increased Doxil accumulation in mixed tumors in vivo
We further tested our combination therapy in a mouse model of mixed MDR tumors. Poor drug penetration through tumor stroma causes insufficient drug access to cancer cells, leading to drug resistance [49], and thus a mixed tumor model that contains both cancer cells and tumor stroma cells more closely mimics human tumors that use multiple mechanisms to resist therapeutics [50]. As shown in Figure 6A, we inoculated a mixture of KB-8-5-11 cells (Pgp-expressing MDR cancer cells) and 3T3 cells (Pgp-negative stromal cells) into nude mice bilaterally. At day 7, mice received Pab-IR700 via i.v. injection. We observed that both left-flank tumor and right-flank tumor showed Pab-IR700 accumulation 48 h post-injection (Figure 6A). Then, we gave a light irradiation only on the right-flank tumors and the tumors in the left flank served as control. After 30 min, mice received a single dose of Doxil (10 mg/kg) via i.v. injection. IVIS fluorescence imaging showed that Doxil started to accumulate in the right-flank tumor as soon as 15 min post injection, and Doxil accumulation in the right-flank tumor increased up to 24 h post injection (Figure 6A). On the other hand, left-flank tumors that were not irradiated showed little Doxil accumulation (Figure 6A), indicating that our targeted PDT enhanced Doxil accumulation in the tumors. In the meantime, IR700 signal disappeared on the right-flank tumor due to photo-bleaching (Figure 6A). We harvested heart, liver, spleen, lung, kidney, and tumors for ex vivo IVIS imaging to determine Doxil distribution in the main organs 24 h post Doxil injection (Figure 6B and Figure S5). The right-flank tumors showed 6.5-fold higher fluorescence intensity than the left-flank tumors (Figure 6B), confirming the enhancement of tumor delivery of Doxil by targeted PDT. Tumors were then digested into single cells for flow cytometry analysis in order to determine intracellular levels of Doxil in tumor cells. As shown in Figure 6C, the intracellular level of Doxil in the irradiated tumors was 5.2-fold higher than that in the unirradiated tumors. We further investigated the patterns of tumoral Doxil penetration after light irradiation by staining CD31 as a marker of blood vessel. As shown in Figure 6D-E and Figures S6-7, the irradiated tumors showed a higher fluorescence intensity of Doxil than un-irradiated tumors, indicating that the higher accumulation of Doxil was caused by targeted PDT. Moreover, the irradiated tumors showed a more even distribution of Doxil than the un-irradiated tumors (Figure 6D-E and Figures S6-7). It showed a distance of penetration up to 320 µm in the irradiated tumors but only 50 µm in the control tumors (Figure 6E). These results further confirm that the Pgp-targeted PDT increased overall delivery and penetration of Doxil in the mixed tumors.
In vivo combination therapy in mixed tumor model
To examine the tumor response of combination therapy with targeted PDT and Doxil, a mixed tumor model was established by inoculating nude mice with a mixture of KB-8-5-11-fLuc-GFP cells plus 3T3-rLuc-RFP cells. MDR cancer cells and stromal cells express different reporters, and so the effects of the combination therapy on individual cell types and their interplay can be examined by tracking individual reporters using IVIS imaging. At Day 7, xenograft-bearing mice were allocated into four groups (n = 8) and were treated with PBS, Doxil, PDT, or PDT followed by Doxil. The mice in the PDT and PDT + Doxil groups received 200 µg Pab-IR700 via i.v. injection. Then, the mice in these two groups received light irradiation at tumor sites at a total dose of 50 J/cm2. Half an hour after light irradiation, the mice in groups Doxil and PDT + Doxil received Doxil (10 mg/kg). Growth of KB MDR cancer cells and 3T3 stromal cells in the tumors were monitored individually using dual substrate BLI. As shown in Figure 7A, BLI data in the mixed tumors showed that targeted PDT reduced tumoral firefly luciferase level by 82% but Doxil did not change it, indicating that KB MDR cells were resistant to Doxil but sensitive to PDT treatment. In vivo observation of the resistance of KB MDR cells towards Doxil in Figure 6A is consistent with the in vitro toxicity data, which indicated that Pgp-positive cells are resistant to both free and liposomal doxorubicin as it is a substrate for Pgp (Figure 1-2). In contrast, 3T3 fibroblasts in the mixed tumors only responded to chemotherapy with Doxil, which reduced Renilla luciferease by 59% (Figure 7A). PDT followed by Doxil treatment reduced KB MDR cell growth by 92%, which is significantly higher than single PDT treatment (p < 0.01) (Figure 7A). On the other hand, combination therapy reduced stromal cell growth by 76%, which is significantly greater than single Doxil treatment (p < 0.05) (Figure 7A). Thus, combination therapy suppressed growth of both cell populations in mixed tumors with an efficacy that is superior to single treatments, indicating that targeted PDT enhanced Doxil's actions on both MDR cancer cells and stromal cells.
Figure 6
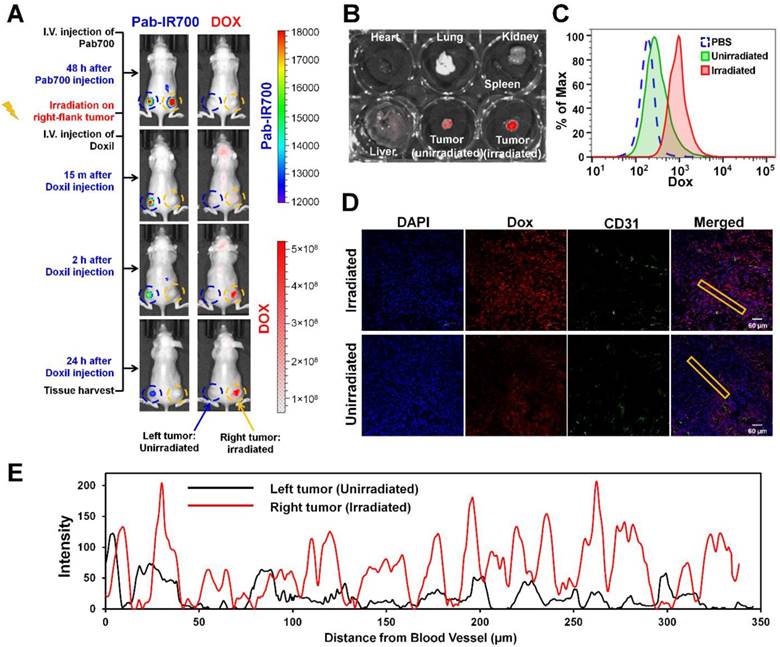
Pgp-targeted PDT enhances Doxil accumulation and penetration in mixed tumor. (A) KB/3T3 mixed tumor xenograft-bearing mice were i.v. injected with Pab-IR700, followed by light treatment only on the right-flank tumors. After 0.5 h, mice were injected with Doxil (10 mg/kg). Fluorescence imaging at 24 h post injection showed marked uptake of Doxil in light-treated tumors. (B) Biodistribution of Doxil in heart, lung, spleen, kidney, liver, left-flank and right-flank tumors at 24 h post injection. (C) After fluorescence imaging, tumors were harvested and digested into single cells for flow cytometry to track the intracellular levels of Doxil in the irradiated tumors and unirradiated tumors. (D) Confocal image of intratumoral distribution of Doxil in light-irradiated or un-irradiated tumors 24 h post injection. CD31-stained blood vessels are shown in green, Doxil is shown in red, and DAPI-stained cell nuclei are shown in blue. Scale bar, 60 μm. (E) Distribution of Doxil in the tumors with or without light irradiation. Distance was calculated from CD31-stained blood vessels. A region marked with the rectangular frame in (D) was selected for this analysis.
Figure 7
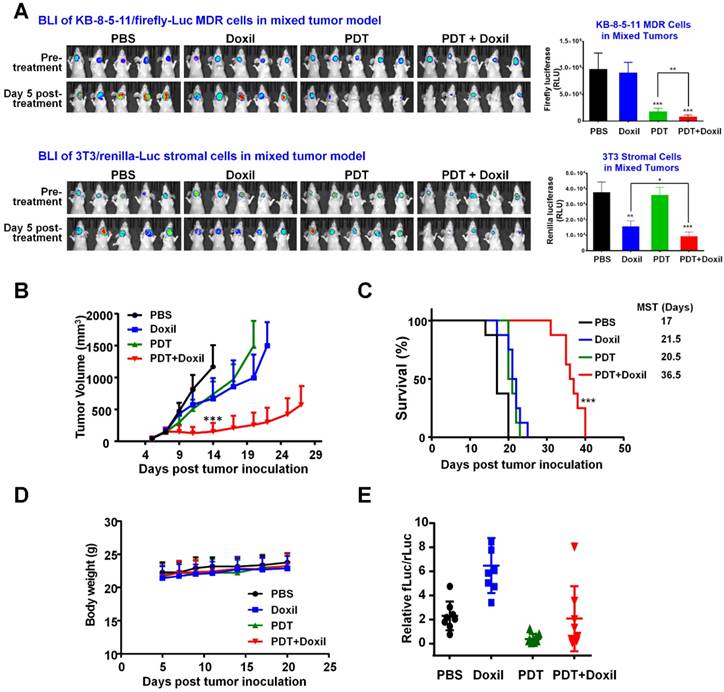
Tumor responses to combination therapy of targeted PDT and Doxil in a mouse mixed tumor model. A mixed tumor model was established by inoculating nude mice with KB-8-5-11-fLuc-GFP plus 3T3-rLuc-RFP cells. Xenograft-bearing mice were treated with Doxil (10 mg/kg), PDT, or PDT followed by Doxil. Growth of KB MDR cancer cells and 3T3 stromal cells in the tumors were monitored individually using an in vivo dual substrate BLI assay. (A) BLI and quantitative data indicated that KB MDR cells were resistant to Doxil, but sensitive to targeted PDT treatment. On the contrary, 3T3 cells only responded to chemotherapy with Doxil. Targeted PDT followed by Doxil treatment produced superior suppression of both cell populations to single treatments in the mixed tumors. Data are presented as mean ± SD (n = 8, *** p < 0.001, ** p < 0.01, and * p < 0.05). (B) Tumor growth curves showed that the combination of PDT with Doxil outperformed the two single treatments in suppressing mixed tumor growth. Data are presented as mean ± SD (n = 8, ***p < 0.001). (C) Kaplan-Meier survival curves confirmed that the combination therapy achieved superior tumor survival to the monotherapies (n = 8, *** p < 0.001). (D) Body weights of mice after different treatments were recorded from day 4 to day 20. Data are mean ± SD, n=8. (E) In vitro dual-reporter assay was performed to quantitate the firefly luciferase and Renilla luciferase activities in the tumor tissues that were harvested at the end point.
Tumor growth curves (Figure 7B) show that the combination of PDT with nanomedicine outperformed the two single therapeutics in suppressing growth of the mixed tumors. The median survival time of the PBS group was 17 days, and those of the PDT and Doxil groups were 21.5 days and 20.5 days, respectively, whereas that of the PDT + Doxil group was 36.5 days (Figure 7C), indicating that targeted PDT plus Doxil dramatically extended the survival of the mixed tumor-bearing mice. All treatments did not reduce the body weight of mice (Figure 7D), indicating that no significant side effects of these treatments were observed in vivo. We also performed an in vitro dual-reporter assay to quantitate the firefly luciferase ( in tumor cells) and Renilla luciferase (in stromal fibroblast cells) activities in the tumor tissues harvested at the end of the experiment (Figure 7E), which showed consistent results with the BLI data in Figure 7A.
We collected tumor tissues at day 14, five days post treatments, for histological analyses. As shown in Figure 8, fibroblasts and tumor cells demonstrated different cell morphologies in H&E images. Fibroblasts were spindle shaped and elongated (yellow dotted circles), whereas tumor cells were irregularly round shaped with enlarged cell nuclei and formed tumor nests in the tumor tissues (white dotted circles). In the PBS group, the sections showed tumor nests uniformly grew in the fibroblasts, and both cells exhibited typical cell morphology. In the Doxil group, the sections were full of tumor nest while fibroblasts were crowded in small and elongated regions with dead cells and cavities, indicating that Doxil preferentially killed fibroblasts over chemoresistant tumor cells. In the targeted PDT group, the tumor nests were full of dead cells and the cavities inside were smaller than those in the PBS group, whereas the fibroblasts grew towards the tumor nests, indicating specific killing of tumor cells by Pgp-targeted PDT. Further, in the combination therapy group, both tumor cells and fibroblasts showed pyknotic nuclei and cytoplasmic concentration, indicating that the combination therapy killed both cell populations. The fluorescence images in Figure 8 showed that Doxil only reduced the 3T3-RFP fibroblast population and targeted PDT depleted the KB-8-5-11-GFP cell population specifically, whereas the combination therapy dramatically decreased both cell populations. These images further confirmed the results in Figure 7.
Figure 8
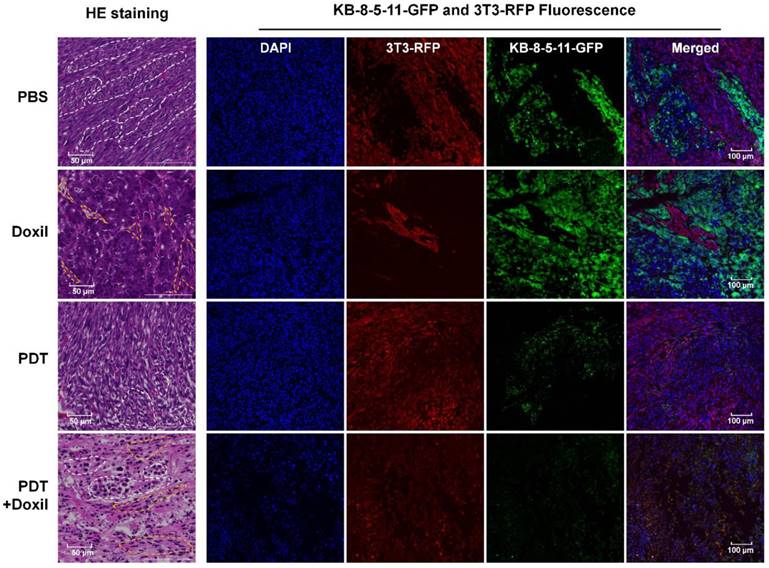
Histological analyses of the mixed tumors after combination therapy. Xenograft-bearing mice were treated with Doxil, PDT, or PDT followed by Doxil. Tumor tissues were collected for histological analyses 5 days post treatments. HE staining of the tumor sections was performed. Cell nuclei were stained with DAPI (blue). KB-8-5-11-fLuc-GFP (green) and 3T3-rLuc-RFP (red) cells were observed by fluorescence microscopy. The tumor nests are marked with white dotted circles, whereas the fibroblasts are marked with yellow dotted circles.
Discussion
FDA-approved cancer nanomedicines, including liposomal doxorubicin (Doxil) and albumin NP of taxol (Abraxane), only modestly improve the overall survival of cancer patients compared with conventional chemotherapeutics, though they reduce toxicity by changing the biodistribution and pharmacokinetics of the agents [51]. Thus, clinically used NPs fail to achieve superior tumor delivery to conventional agents to enhance survival of cancer patients significantly. One biological barrier in tumor microenvironments limiting nanomedicine delivery is drug efflux mediated by ABC transporters, primarily Pgp. All the drugs in cancer nanomedicines, including doxorubicin, daunorubicin, taxol, vincristine, and irinotecan, are substrates for Pgp and/or other ABC transporters and thus, these transporters may reduce the therapeutic efficacy of these nanomedicines [36]. We selected liposomal doxorubicin, Doxil, and albumin NP of taxol, Abraxane, to examine Pgp-mediated drug resistance towards these nanomedicines. Cytotoxicity results in Figure 1 showed that two Pgp-expressing MDR cells and Pgp-transfected 3T3 cells are highly resistant to both nanomedicines, with resistance levels that are similar or even greater than those of the chemotherapy agents in their free forms. Utilizing the fluorescence of doxorubicin, we studied Pgp-mediated efflux of Doxil by flow cytometry. As shown in Figure 2, intracellular doxorubicin levels after Doxil treatment were substantially lower in Pgp-expressing MDR cells and 3T3-MDR1 cells than their respective control cells, and co-incubation with the Pgp inhibitor GF918 increased Doxil uptake in Pgp-expressing cells to the levels in their control cells. These results strongly indicated that Pgp pumps the chemotherapy agents out of Pgp-expressing cancer cells after nanomedicine uptake, reduces their anticancer activities, and causes drug resistance. Our results are consistent with previous studies that indicated Pgp causes cancer MDR towards Abraxane [37, 38]. Although NPs enter cancer cells via endocytosis, the drugs need to be released from the NPs to be pharmacologically active. After release, the drugs can bind to Pgp inside the cells to be pumped out. Hypoxia-inducible factor-1 upregulates Pgp expression and so its expression level in the hypoxic core of tumors is even higher [52]. This may pose a greater challenge to NPs than conventional chemotherapy agents, because penetration of NPs to the core of tumors is hindered by other biological barriers in tumors, including the presence of extracellular matrix, low blood flow, and high interstitial pressure [51].
There are currently no clinical solutions to overcome Pgp-mediated MDR in cancers [5]. Three generations of small molecule inhibitors has been developed to overcome Pgp-mediated drug resistance [7], yet none of them have been approved by the FDA [53]. The main reason for this failure is a lack of cancer specificity of these inhibitors, which could cause side effects in normal tissues [53]. For example, Pgp is expressed in the blood-brain barrier to prevent toxic compounds from entering the brain. Small molecule Pgp inhibitors could block this protection mechanism and cause neurotoxicity [54]. Other novel strategies, including NP-based co-delivery of Pgp inhibitors [55] and siRNA [56], generated some positive outcomes in preclinical models. However, none of these have advanced to clinical studies due to inefficient efficacy and safety concerns.
In this study, we tested if Pgp-targeted PDT can overcome Pgp-mediated MDR and improve therapy with cancer nanomedicines. The results shown in Figure 3 and Figure 4 indicated that our approach achieves high cancer specificity towards Pgp-expressing MDR cells through combining antibody-based cancer targeting and locoregional tumor ablation with laser irradiation. Thus, it overcomes the main limitation of small-molecule Pgp inhibitor approaches and thus is expected to have great translational importance in targeted cancer therapy. In addition, this approach is highly synergistic with cancer nanomedicines through dual mechanisms: depletion of MDR cancer cells and enhancement of tumor penetration of NPs. In in vitro studies with a co-culture model of MDR cancer cells and stromal 3T3 cells, we observed that targeted PDT specifically killed Pgp-expressing MDR cells, while Doxil was only effective in stromal cells (Figure 5A). Further, partial damage of MDR cells by PDT compromised Pgp-mediated efflux function by releasing intracellular ATP, and thus, further enhanced Doxil uptake and the sensitivity of these cells (Figure 5A-C). Overall, the combination of targeted PDT followed by Doxil treatment produced synergistic actions in this co-culture model (Figure 5D). We further tested if this synergism can be translated into animal models. In tumor delivery studies, we observed over 6-fold higher overall Doxil uptake in KB/3T3-mixed tumors (Figure 6A-B), and over 5-fold higher intracellular Doxil level in the cells of the mixed tumors after targeted PDT procedures compared to the control tumors (Figure 6C). In addition, Doxil distributed more evenly in the tumors after targeted PDT than in the control tumors (Figure 6D-E and Figures S6-7). All these data indicated that targeted PDT enhanced Doxil delivery and microdistribution in MDR tumors. In tumor response studies with the KB/3T3-mixed tumors, we further examined the effects of the two modalities on individual cell populations and their synergism using an in vivo dual substrate BLI. The results indicated that Pgp-targeted PDT specifically depleted MDR cancer cells whereas Doxil preferentially killed 3T3 stromal cells (Figure 7A). Combination therapy produced greater killing than the monotherapies on both cell populations (Figure 7A), indicating targeted PDT enhanced Doxil's actions on both MDR cancer cells and stromal cells in vivo. Thus, the synergism may occur by two mechanisms. On one hand, targeted PDT partially damages some MDR cells and compromises Pgp-mediated efflux function, resulting in greater Doxil delivery in MDR cells. This mechanism is also supported by the in vitro data (Figure 5). On the other hand, targeted PDT enhances overall tumor delivery of Doxil, promoting its actions in both MDR and stromal cells. Overall, combination therapy achieved greater tumor growth suppression and increased the survival of xenograft-bearing mice without causing significant toxicity (Figure 7B-D).
In conclusion, all drugs in FDA-approved cancer nanomedicines are substrates for Pgp [36], and their anticancer efficacy is limited by tumoral Pgp based on the results in this study and those in previous studies [37, 38]. Our targeted PDT approach is highly cancer specific through combining antibody-based cancer targeting and locoregional tumor ablation, and it markedly enhanced anticancer actions of nanomedicines by depleting MDR cancer cells and increasing their tumor penetration. PDT is a clinically approved cancer therapy [39], and targeted PDT is currently in clinical testing [41]. Thus, our targeted PDT approach may provide a clinically effective approach to facilitate translation of cancer nanomedicines in the near future.
Abbreviations
APCs: antibody-photosensitizer conjugates; BLI: bioluminescence imaging; Dox: doxorubicin; EGFR: epidermal growth factor receptor; EPR: enhanced permeability and retention; FBS: fetal bovine serum; LED: Light-emitting diode; MDR: multidrug resistance; NPs: nanoparticles; Pab: anti-Pgp monoclonal antibody; PDT: photodynamic therapy; Pgp: P-glycoprotein; PSs: photosensitizers.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by NIH grants 5R01CA194064 and UL1TR001420. The authors would like to thank Dr. Michael Gottesman (NCI) for providing 3T3-MDR1, NCI-ADRRes, and KB-8-5-11 cells, and Dr. Michael Miley (UNC Antibody Core Facility) for assistance in producing anti-Pgp antibody. The authors would also like to acknowledge the support of the Wake Forest Baptist Comprehensive Cancer Center Tumor Tissue and Pathology Shared Resource and Flow Cytometry Shared Resource, supported by the National Cancer Institute's Cancer Center Support Grant award number P30CA012197.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chauhan VP, Jain RK. Strategies for advancing cancer nanomedicine. Nat Mater. 2013;12:958-62
2. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387-92
3. Kwon IK, Lee SC, Han B, Park K. Analysis on the current status of targeted drug delivery to tumors. J Control Release. 2012;164:108-14
4. Ruenraroengsak P, Cook JM, Florence AT. Nanosystem drug targeting: Facing up to complex realities. J Control Release. 2010;141:265-76
5. Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer. 2018;18:452-64
6. Fu D. Where is it and how does it get there - intracellular localization and traffic of P-glycoprotein. Front Oncol. 2013;3:321
7. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714-26
8. Hu T, Li Z, Gao CY, Cho CH. Mechanisms of drug resistance in colon cancer and its therapeutic strategies. World J Gastroenterol. 2016;22:6876-89
9. Tang Y, Wang Y, Kiani MF, Wang B. Classification, treatment strategy, and associated drug resistance in breast cancer. Clin Breast Cancer. 2016;16:335-43
10. Patel NR, Pattni BS, Abouzeid AH, Torchilin VP. Nanopreparations to overcome multidrug resistance in cancer. Adv Drug Deliv Rev. 2013;65:1748-62
11. Muddineti OS, Kumari P, Ray E, Ghosh B, Biswas S. Curcumin-loaded chitosan-cholesterol micelles: evaluation in monolayers and 3D cancer spheroid model. Nanomedicine (Lond). 2017;12:1435-53
12. Huang SY LJ, Zhu H, Hussain A, Liu Q, Li J. et al. PEGylated doxorubicin micelles loaded with curcumin exerting synergic effects on multidrug resistant tumor cells. J Nanosci Nanotechnol. 2017;17:2873-80
13. Wei X, Liu L, Guo X, Wang Y, Zhao J, Zhou S. Light-activated ROS-responsive nanoplatform codelivering apatinib and doxorubicin for enhanced chemo-photodynamic therapy of multidrug-resistant tumors. ACS Appl Mater Interfaces. 2018;10:17672-84
14. Guo X, Wei X, Jing Y, Zhou S. Size changeable nanocarriers with nuclear targeting for effectively overcoming multidrug resistance in cancer therapy. Adv Mater. 2015;27:6450-6
15. Alamoudi K, Martins P, Croissant JG, Patil S, Omar H, Khashab NM. Thermoresponsive pegylated bubble liposome nanovectors for efficient siRNA delivery via endosomal escape. Nanomedicine (Lond). 2017;12:1421-33
16. Gao M, Xu Y, Qiu L. Sensitization of multidrug-resistant malignant cells by liposomes co-encapsulating doxorubicin and chloroquine through autophagic inhibition. J Liposome Res. 2017;27:151-60
17. Li R, Wu R, Zhao L, Wu M, Yang L, Zou H. P-glycoprotein antibody functionalized carbon nanotube overcomes the multidrug resistance of human leukemia cells. ACS Nano. 2010;4:1399-408
18. Li Y, Gao X, Yu Z, Liu B, Pan W, Li N. et al. Reversing multidrug resistance by multiplexed gene silencing for enhanced breast cancer chemotherapy. ACS Appl Mater Interfaces. 2018;10:15461-6
19. Pai CL, Chen YC, Hsu CY, Su HL, Lai PS. Carbon nanotube-mediated photothermal disruption of endosomes/lysosomes reverses doxorubicin resistance in MCF-7/ADR Cells. J Biomed Nanotechnol. 2016;12:619-29
20. Cheng W, Liang C, Xu L, Liu G, Gao N, Tao W. et al. TPGS-functionalized polydopamine-modified mesoporous silica as drug nanocarriers for enhanced lung cancer chemotherapy against multidrug resistance. Small. 2017;13:1700623
21. Sun L, Wang D, Chen Y, Wang L, Huang P, Li Y. et al. Core-shell hierarchical mesostructured silica nanoparticles for gene/chemo-synergetic stepwise therapy of multidrug-resistant cancer. Biomaterials. 2017;133:219-28
22. Wang F, Wang YC, Dou S, Xiong MH, Sun TM, Wang J. Doxorubicin-tethered responsive gold nanoparticles facilitate intracellular drug delivery for overcoming multidrug resistance in cancer cells. ACS Nano. 2011;5:3679-92
23. Gurav D, Varghese OP, Hamad OA, Nilsson B, Hilborn J, Oommen OP. Chondroitin sulfate coated gold nanoparticles: a new strategy to resolve multidrug resistance and thromboinflammation. Chem Commun (Camb). 2016;52:966-9
24. Chen M, Hou C, Huo D, Bao J, Fa H, Shen C. An electrochemical DNA biosensor based on nitrogen-doped graphene/Au nanoparticles for human multidrug resistance gene detection. Biosens Bioelectron. 2016;85:684-91
25. Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine (Lond). 2012;7:597-615
26. Yuan Y, Cai T, Xia X, Zhang R, Chiba P, Cai Y. Nanoparticle delivery of anticancer drugs overcomes multidrug resistance in breast cancer. Drug Deliv. 2016;23:3350-7
27. Katiyar SS, Muntimadugu E, Rafeeqi TA, Domb AJ, Khan W. Co-delivery of rapamycin- and piperine-loaded polymeric nanoparticles for breast cancer treatment. Drug Deliv. 2016;23:2608-16
28. Song XR, Cai Z, Zheng Y, He G, Cui FY, Gong DQ. et al. Reversion of multidrug resistance by co-encapsulation of vincristine and verapamil in PLGA nanoparticles. Eur J Pharm Sci. 2009;37:300-5
29. Owatari S, Akune S, Komatsu M, Ikeda R, Firth SD, Che XF. et al. Copper-transporting P-type ATPase, ATP7A, confers multidrug resistance and its expression is related to resistance to SN-38 in clinical colon cancer. Cancer Res. 2007;67:4860-8
30. van Vlerken LE, Duan Z, Seiden MV, Amiji MM. Modulation of intracellular ceramide using polymeric nanoparticles to overcome multidrug resistance in cancer. Cancer Res. 2007;67:4843-50
31. Huang IP, Sun SP, Cheng SH, Lee CH, Wu CY, Yang CS. et al. Enhanced chemotherapy of cancer using pH-sensitive mesoporous silica nanoparticles to antagonize P-glycoprotein-mediated drug resistance. Mol Cancer Ther. 2011;10:761-9
32. Gandhi NS, Tekade RK, Chougule MB. Nanocarrier mediated delivery of siRNA/miRNA in combination with chemotherapeutic agents for cancer therapy: current progress and advances. J Control Release. 2014;194:238-56
33. Sun Q, Ojha T, Kiessling F, Lammers T, Shi Y. Enhancing tumor penetration of nanomedicines. Biomacromolecules. 2017;18:1449-59
34. Zhang Y, Chen W, Yang C, Fan Q, Wu W, Jiang X. Enhancing tumor penetration and targeting using size-minimized and zwitterionic nanomedicines. J Control Release. 2016;237:115-24
35. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20-37
36. Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48-58
37. Vallo S, Kopp R, Michaelis M, Rothweiler F, Bartsch G, Brandt MP. et al. Resistance to nanoparticle albumin-bound paclitaxel is mediated by ABCB1 in urothelial cancer cells. Oncology Letters. 2017;13:4085-92
38. Zhao MZ, Lei CN, Yang YD, Bu XL, Ma HL, Gong H. et al. Abraxane, the nanoparticle formulation of paclitaxel can induce drug resistance by up-regulation of P-gp. Plos One. 2015:10
39. Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO. et al. Photodynamic therapy of cancer: an update. CA Cancer J Clin. 2011;61:250-81
40. Mew D, Wat CK, Towers GH, Levy JG. Photoimmunotherapy: treatment of animal tumors with tumor-specific monoclonal antibody-hematoporphyrin conjugates. J Immunol. 1983;130:1473-7
41. Mitsunaga M, Ogawa M, Kosaka N, Rosenblum LT, Choyke PL, Kobayashi H. Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat Med. 2011;17:1685-91
42. Sano K, Nakajima T, Choyke PL, Kobayashi H. Markedly enhanced permeability and retention effects induced by photo-immunotherapy of tumors. ACS Nano. 2013;7:717-24
43. Ming X, Carver K, Wu L. Albumin-based nanoconjugates for targeted delivery of therapeutic oligonucleotides. Biomaterials. 2013;34:7939-49
44. Yuan A, Yang B, Wu J, Hu Y, Ming X. Dendritic nanoconjugates of photosensitizer for targeted photodynamic therapy. Acta Biomater. 2015;21:63-73
45. Mao C, Zhao Y, Li F, Li Z, Tian S, Debinski W. et al. P-glycoprotein targeted and near-infrared light-guided depletion of chemoresistant tumors. J Control Release. 2018;286:289-300
46. Wang M, Mao C, Wang H, Ling X, Wu Z, Li Z. et al. Molecular imaging of P-glycoprotein in chemoresistant tumors using a dual-modality PET/fluorescence probe. Mol Pharm. 2017;14:3391-8
47. Li F, Zhao Y, Mao C, Kong Y, Ming X. RGD-modified albumin nanoconjugates for targeted delivery of a porphyrin photosensitizer. Mol Pharm. 2017;14:2793-804
48. Bansal T, Akhtar N, Jaggi M, Khar RK, Talegaonkar S. Novel formulation approaches for optimising delivery of anticancer drugs based on P-glycoprotein modulation. Drug Discov Today. 2009;14:1067-74
49. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583-92
50. Gottesman MM, Lavi O, Hall MD, Gillet JP. Toward a better understanding of the complexity of cancer drug resistance. Annu Rev Pharmacol Toxicol. 2016;56:85-102
51. Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653-64
52. Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002;62:3387-94
53. Yu M, Ocana A, Tannock IF. Reversal of ATP-binding cassette drug transporter activity to modulate chemoresistance: why has it failed to provide clinical benefit? Cancer Metastasis Rev. 2013;32:211-27
54. Fox E, Bates SE. Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther. 2007;7:447-59
55. Nieto Montesinos R, Beduneau A, Pellequer Y, Lamprecht A. Delivery of P-glycoprotein substrates using chemosensitizers and nanotechnology for selective and efficient therapeutic outcomes. J Control Release. 2012;161:50-61
56. Chen Y, Bathula SR, Li J, Huang L. Multifunctional nanoparticles delivering small interfering RNA and doxorubicin overcome drug resistance in cancer. J Biol Chem. 2010;285:22639-50
Author contact
![]() Corresponding author: Xin Ming, Department of Cancer Biology and Comprehensive Cancer Center, Wake Forest University School of Medicine, Winston-Salem, NC 27157, USA. E-mail: xmingedu
Corresponding author: Xin Ming, Department of Cancer Biology and Comprehensive Cancer Center, Wake Forest University School of Medicine, Winston-Salem, NC 27157, USA. E-mail: xmingedu
Citation styles
APA
Mao, C., Li, F., Zhao, Y., Debinski, W., Ming, X. (2018). P-glycoprotein-targeted photodynamic therapy boosts cancer nanomedicine by priming tumor microenvironment. Theranostics, 8(22), 6274-6290. https://doi.org/10.7150/thno.29580.
ACS
Mao, C.; Li, F.; Zhao, Y.; Debinski, W.; Ming, X. P-glycoprotein-targeted photodynamic therapy boosts cancer nanomedicine by priming tumor microenvironment. Theranostics 2018, 8 (22), 6274-6290. DOI: 10.7150/thno.29580.
NLM
Mao C, Li F, Zhao Y, Debinski W, Ming X. P-glycoprotein-targeted photodynamic therapy boosts cancer nanomedicine by priming tumor microenvironment. Theranostics 2018; 8(22):6274-6290. doi:10.7150/thno.29580. https://www.thno.org/v08p6274.htm
CSE
Mao C, Li F, Zhao Y, Debinski W, Ming X. 2018. P-glycoprotein-targeted photodynamic therapy boosts cancer nanomedicine by priming tumor microenvironment. Theranostics. 8(22):6274-6290.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.