Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(22):6386-6399. doi:10.7150/thno.29164 This issue Cite
Research Paper
Genomic Landscape and Endocrine-Resistant Subgroup in Estrogen Receptor-Positive, Progesterone Receptor-Negative, and HER2-Negative Breast Cancer
Xi-Yu Liu*, Ding Ma*, Xiao-En Xu, Xi Jin, Ke-Da Yu, Yi-Zhou Jiang ![]() , Zhi-Ming Shao
, Zhi-Ming Shao ![]()
Department of Breast Surgery, Fudan University Shanghai Cancer Center; Cancer Institute, Fudan University Shanghai Cancer Center; Department of Oncology, Shanghai Medical College, Fudan University, P.R. China
*These authors contributed equally to this work.
Received 2018-8-11; Accepted 2018-11-8; Published 2018-12-8
Abstract

Estrogen receptor-positive, progesterone receptor-negative, and human epidermal growth factor receptor 2 (HER2)-negative (ER+PR-HER2-) breast cancer comprise a special type of breast cancer that constitutes ~10% of all breast cancer patients. ER+PR-HER2- tumor benefits less from endocrine therapy, while its genomic features remain elusive. In this study, we systematically assessed the multiomic landscape and endocrine responsiveness of ER+PR-HER2- breast cancer.
Methods: This study incorporated five cohorts. The first and second cohorts were from the Surveillance, Epidemiology, and End Results database (n=130,856) and Molecular Taxonomy of Breast Cancer International Consortium (n=1,055) for analyzing survival outcomes and endocrine responsiveness. The third cohort was from The Cancer Genome Atlas (n=630) for multiomic analysis and endocrine-resistant subgroup exploration. The fourth cohort, from the MD Anderson database (n=92), was employed to assist gene selection. The fifth cohort was a prospective observational cohort from Fudan University Shanghai Cancer Center (n=245) that was utilized to validate the gene-defined subgroup by immunohistochemistry (IHC).
Results: Clinically, ER+PR-HER2- tumors showed lower endocrine responsiveness than did ER+PR+HER2- tumors. Genomically, copy number loss or promoter methylation of PR genes occurred in 75% of ER+PR-HER2- tumors, collectively explaining PR loss. ER+PR-HER2- tumors had higher TP53 (30.3% vs. 17.0%) and lower PIK3CA mutation rates (25.8% vs. 42.7%) and exhibited more ZNF703 (21.5% vs. 13.6%) and RPS6KB1 (18.5% vs. 7.8%) amplification events than ER+PR+HER2- tumors. Among ER+PR-HER2- tumors, nearly 20% were of the PAM50-defined non-luminal-like subgroup and manifested lower endocrine sensitivity scores and enriched biosynthesis, metabolism and DNA replication pathways. We further identified the non-luminal-like subgroup using three IHC markers, GATA3, CK5, and EGFR. These IHC-defined non-luminal-like (GATA3-negative, CK5-positive and/or EGFR-positive) tumors received limited benefit from adjuvant endocrine therapy.
Conclusion: ER+PR-HER2- breast cancer consists of clinically and genomically distinct groups that may require different treatment strategies. The non-luminal-like subgroup was associated with reduced benefit from endocrine therapy.
Keywords: ER+PR-HER2-, endocrine resistance, multiomic, non-luminal-like, breast cancer
Introduction
Progesterone receptor (PR), a downstream gene target of estrogen receptor (ER), is considered a biomarker of ER function and breast cancer prognosis [1]. Therefore, loss of PR in ER-positive breast cancer may be a surrogate indicator of an impaired ER pathway and a predictor of limited endocrine responsiveness. Breast cancer clinicians typically assess ER, PR, human epidermal growth factor receptor 2 (HER2) and Ki67 expression to define molecular subtypes and design treatment strategies [2]. Patients with ER+PR- breast cancer, which constitutes approximately 10%-15% of all breast cancers and is defined as “luminal-like” breast cancer, are recommended endocrine therapy indiscriminately [2-4]. Nevertheless, patients with ER+PR- status exhibit a higher recurrence rate and worse prognosis, which indicates that ER+PR- tumors are biologically and clinically more aggressive and may benefit from more escalated therapies [5-7]. Clinical data have suggested that ER+PR- tumors show less responsiveness to tamoxifen than do ER+PR+ tumors, while some clinical trials have revealed that ER+PR- tumors remain sensitive to aromatase inhibitors (AI) [8-12].
Efforts have been made to reveal the molecular features of ER+PR- breast cancer. For example, ER+PR- breast cancer has been associated with a significantly higher frequency of HER2 positivity than ER+PR+ cases [5, 7, 13]. Several reports have suggested that overexpression of the HER family of receptors, such as HER2, may activate the PI3K/Akt/mTOR signaling pathway and therefore contribute to the ER+PR- phenotype and tamoxifen resistance [1, 5, 14]. However, the HER2-positivity rate in ER+PR- breast cancer varies from 20%-30%, which indicates that the molecular nature of nearly 70%-80% of ER+PR- breast cancers may not be explained by the HER family-overexpression theory. The underlying mechanism of HER2-negative tumors in ER+PR- breast cancer await exploration. In addition, recent reports focusing on explaining PR loss in breast cancer suggested many possibilities, such as a malfunctioning ER pathway or hyperactive HER2 signaling pathways [1, 15, 16]. Although genomics changes, such as PR promoter hypermethylation or loss of heterozygosity at the PR gene locus, have received some attention [17, 18], an explicit illustration has not yet been clarified. Thus, utilizing multiomic data to explore the molecular features of ER+PR-HER2- is of great importance.
In this study, we combined five large and well-characterized cohorts of breast cancer patients with long-term follow-up data and multiomic data to (1) identify and characterize the clinical characteristics and molecular mechanisms of ER+PR-HER2- breast cancer and (2) establish a convenient immunohistochemistry (IHC)-based tool to distinguish subgroups within ER+PR-HER2- breast cancer for different treatment strategies. Clinicians can thereby identify distinctive tumor characteristics within the ER+PR-HER2- tumor group to design individualized treatment strategies.
Methods
Study cohorts
The first cohort was from the Surveillance, Epidemiology, and End Results (SEER) database and included 130,856 primary operable HER2-negative breast cancer patients (13,084 ER+PR-HER2- patients) with clinicopathologic characteristics and follow-up information for baseline and survival analysis. The outcome of interest was breast cancer-specific survival (BCSS), which was calculated from the date of diagnosis to the time of breast cancer death.
The second cohort was from the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) database. We included 1,055 cases of primary operable HER2-negative breast cancer (260 ER+PR-HER2- cases) with long-term follow-up, endocrine treatment and genomic information for survival analysis and endocrine response analysis. The primary endpoint was BCSS, which was calculated from the date of diagnosis to the time of breast cancer death.
For the third cohort, we included 630 tumors of primary operable breast cancer (66 ER+PR-HER2- tumors) from The Cancer Genome Atlas (TCGA) database. The data included gene expression, copy number alteration (CNA), and methylation for multiomic analysis and subgroup identification.
The fourth cohort was integrated from breast cancer data sets using a single U133A platform contributed by the MD Anderson Cancer Center (MDACC, Houston, TX, USA) (GSE25066, GSE20271, GSE20194) (92 ER+PR-HER2- breast tumors) [19-21]. Gene expression data were generated from biopsy before any therapy. Each of the hybridizations was uniformly renormalized by frozen robust multiarray analysis (fRMA) [22].
The fifth cohort was a prospective observational study cohort. A total of 245 consecutive operable patients treated in the Department of Breast Surgery at Fudan University Shanghai Cancer Center (FUSCC) from January 1, 2007, to December 31, 2014, were recruited according to the following criteria: (i) female patients diagnosed with unilateral disease; (ii) histologically confirmed invasive ductal carcinoma (IDC) or invasive lobular carcinoma (ILC) with the ER+PR-HER2- phenotype; and (iii) no metastatic loci at diagnosis. The exclusion criteria were as follows: (i) patients with breast carcinoma in situ and inflammatory breast cancer; and (ii) patients who received any type of treatment before surgery. Pathological examination of tumor specimens was performed in the Department of Pathology at FUSCC. ER, PR and HER2 status was reconfirmed by two experienced pathologists based on IHC and fluorescence in situ hybridization (FISH) [23-25]. The cutoff for ER-negative and PR-negative IHC status was less than 1% staining in the nuclei. HER2 status was considered negative when an IHC score was 0 or 1 or when HER2 amplification was absent (ratio <2.2) by FISH analysis. If any disagreements arose during the evaluation of the IHC and FISH results, a third pathologist was consulted. Patient follow-up was completed on March 1, 2018. The median length of follow-up was 49.9 months (interquartile range [IQR], 33.6 to 67.7 months). Recurrence-free survival (RFS) events included the following: the first recurrence of invasive disease at a local, regional, or distant site; contralateral breast cancer; and death from any cause. Patients without RFS events were censored at the last follow-up. Our study was approved by the independent Ethics Committee/Institutional Review Board of FUSCC (Shanghai Cancer Center Ethics Committee). All patients provided written informed consent before inclusion. Sufficient endocrine therapy was defined as receiving cumulative endocrine treatment for more than one year [26]. Insufficient treatment was defined as no more than one year or undergoing no endocrine therapy.
Patient selection, data preparation, bioinformatic analysis methods, and supplementary tables and figures are included in Supplementary Material.
Statistical analyses
Statistical analyses were performed using Stata/MP version 14.1 (StataCorp., College Station, TX, USA), GraphPad Prism version 7.00 for Mac (GraphPad Software, La Jolla California, USA) and R version 3.4.1 (R Core Team, Vienna, Austria) statistical software. Pearson's chi-square test and Fisher's exact test were employed for comparison of categorical clinicopathologic variables. Survival analysis was constructed by Kaplan-Meier plots and log-rank test. A Cox proportional hazard regression model adjusted for other prognostic covariates was used to calculate hazard ratios (HRs) and 95% confidence intervals (CIs). Student's t-test, the Mann-Whitney Wilcoxon test and the Kruskal-Wallis test were utilized to compare continuous variables and ordered categorical variables. A two-sided P-value less than 0.05 was considered significant unless otherwise stated.
Results
ER+PR-HER2- tumors possess specific clinicopathologic characteristics and patients exhibit worse survival outcomes
In the current study, five cohorts were utilized (Table S1). We summarize the clinicopathologic characteristics of ER+PR-HER2- breast cancer from the SEER and METABRIC cohorts in Table 1. The majority of patients in the ER+PR-HER2- group were older (≥50: SEER/METABRIC 81.9%/91.9%) than those in the ER+PR+HER2- group (≥50: SEER/ METABRIC 76.3%/82%) and the triple-negative breast cancer (TNBC) group (≥50: SEER/METABRIC 67.9%/69%). There were more black people in the ER+PR-HER2- group than in the ER+PR+HER2- group (SEER: 14.4% vs. 8.7%). Grade 3 tumors were more frequent in the ER+PR-HER2- group than in the ER+PR+HER2- group (SEER/METABRIC 40%/43.9% vs. 17.3%/29.9%) but less common than in the TNBC group (SEER/ METABRIC 78%/ 84.7%). ER+PR-HER2- tumors presented higher T stage, more positive lymph nodes (LN) and, thus, later stage than ER+PR+HER2- tumors (P<0.05). ER+PR-HER2- patients appeared to receive more treatment than ER+PR+HER2- patients, including chemotherapy (SEER: 50.6% vs. 30.5%), endocrine therapy (METABRIC: 79.2% vs. 67.7%) and mastectomy (SEER/METABRIC 40.3%/59.6% vs. 36.0%/53.2%). There was no difference between the PR- and PR+ phenotypes with regard to radiotherapy.
Clinicopathological characteristics of ER+PR-HER2- breast cancer cases from SEER and METABRIC cohorts compared with ER+PR+HER2- cases.
| Cohort 1: SEER | CCa | P-valueb | Cohort 2: METABRIC | P-valueb | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ER+PR+HER2- | ER+PR-HER2- | TNBC | ER+PR+HER2- | ER+PR-HER2- | TNBC | |||||
| N=99,058 (%) | N=13,084 (%) | N=18,714 (%) | N=626 (%) | N=260 (%) | N=169 (%) | |||||
| Median Follow-up (IQR) (mo) | 26 (11-41) | 125.8 (75.3-194.0) | ||||||||
| Age | 18-49 | 23,446 (23.7) | 2,375 (18.2) | 6,007 (32.1) | 0.04 | <0.001 | 113 (18.1) | 21 (8.1) | 52 (31.0) | <0.001 |
| ≥50 | 75,612 (76.3) | 10,709 (81.9) | 12,707 (67.9) | 513 (82.0) | 239 (91.9) | 117 (69.0) | ||||
| Race | White | 79,932 (80.7) | 9,827 (75.1) | 13,250 (70.8) | 0.06* | <0.001 | - | - | - | |
| Black | 8,653 (8.7) | 1,883 (14.4) | 3,892 (20.8) | - | - | - | ||||
| AS/AI/AP | 9,762 (9.9) | 1,278 (9.8) | 1,440 (7.7) | - | - | - | ||||
| N/A | 711 (0.7) | 96 (0.7) | 112 (0.6) | - | - | - | ||||
| Histologic type | IDC | 82,193 (83.0) | 10,787 (82.4) | 16,318 (87.2) | 0.01 | <0.001 | 626 (100.0) | 260 (100.0) | 169 (100.0) | - |
| ILC | 12,183 (12.3) | 1,747 (13.4) | 169 (0.9) | 0 | 0 | 0 | ||||
| Others and N/A | 4,682 (4.7) | 550 (4.2) | 2,227 (11.9) | 0 | 0 | 0 | ||||
| Grade | 1 | 30,398 (30.7) | 2,541 (19.4) | 356 (1.9) | 0.36* | <0.001 | 78 (12.5) | 24 (9.2) | 3 (2.0) | 0.001 |
| 2 | 48,530 (49.0) | 4,814 (36.8) | 2,939 (15.7) | 329 (52.6) | 113 (43.5) | 21 (12.3) | ||||
| 3 | 17,119 (17.3) | 5,228 (40.0) | 14,597 (78.0) | 187 (29.9) | 114 (43.9) | 143 (84.7) | ||||
| Others and N/A | 3,011 (3.0) | 501 (3.8) | 842 (4.5) | 32 (5.1) | 9 (3.5) | 2 (1.0) | ||||
| T stage | T0-1 | 67,342 (68.0) | 7,433 (56.8) | 8,290 (44.3) | 0.22* | <0.001 | 297 (47.4) | 108 (41.5) | 71 (41.9) | 0.014 |
| T2 | 25,747 (26.0) | 4,365 (33.4) | 7,991 (42.7) | 310 (49.5) | 133 (51.2) | 86 (50.7) | ||||
| T3-T4 | 5,908 (6.0) | 1,269 (9.7) | 2,396 (12.8) | 19 (3.0) | 18 (6.9) | 12 (7.4) | ||||
| N/A | 61 (0.1) | 17 (0.1) | 37 (0.2) | 0 (0) | 1 (0.4) | 0 (0.0) | ||||
| LN status | Negative | 70,356 (71.0) | 8,809 (67.3) | 12,332 (65.9) | 0.03 | <0.001 | 378 (60.4) | 129 (49.6) | 90 (53.2) | 0.003 |
| Positive | 28,679 (29.0) | 4,268 (32.6) | 6,363 (34.0) | 248 (39.6) | 131 (50.4) | 79 (46.8) | ||||
| N/A | 23 (0.0) | 7 (0.1) | 19 (0.1) | - | - | - | ||||
| Stage | I | 58,663 (59.2) | 6,383 (48.8) | 6,943 (37.1) | 0.07 | <0.001 | 245 (39.1) | 83 (31.9) | 48 (28.6) | 0.005 |
| II | 30,973 (31.3) | 4,878 (37.3) | 8,759 (46.8) | 354 (56.6) | 153 (58.9) | 104 (61.1) | ||||
| III | 9,411 (9.5) | 1,823 (13.9) | 3,012 (16.1) | 27 (4.3) | 24 (9.2) | 17 (10.3) | ||||
| Chemotherapy | Yes | 30,178 (30.5) | 6,614 (50.6) | 14,766 (78.9) | 0.14 | <0.001 | 55 (8.8) | 27 (10.4) | 88 (52.2) | 0.455 |
| No/ Unknown | 68,880 (69.5) | 6,470 (49.5) | 3,948 (21.1) | 571 (91.2) | 233 (89.6) | 81 (47.8) | ||||
| Radiation | Yes | 57,901 (58.5) | 7,313 (55.9) | 9,581 (51.2) | 0.02 | <0.001 | 395 (63.1) | 163 (62.7) | 128 (75.9) | 0.909 |
| No/Unknown | 41,157 (41.6) | 5,771 (44.1) | 9,133 (48.8) | 231 (36.9) | 97 (37.3) | 41 (24.1) | ||||
| Endocrine therapy | Yes | - | - | - | - | - | 424 (67.7) | 206 (79.2) | 51 (30.1) | 0.001 |
| No/Unknown | - | - | - | 202 (32.3) | 54 (20.8) | 118 (69.9) | ||||
| Surgery | BCS | 61,055 (61.6) | 7,343 (56.1) | 9,451 (50.5) | 0.04 | <0.001 | 290 (46.3) | 101 (38.9) | 81 (47.8) | 0.034 |
| Mastectomy | 35,618 (36.0) | 5,275 (40.3) | 8,346 (44.6) | 333 (53.2) | 155 (59.6) | 86 (50.7) | ||||
| Others and N/A | 2,385 (2.4) | 466 (3.6) | 917 (4.9) | 3 (0.5) | 4 (1.5) | 2 (1.5) | ||||
AS/AI/AP: Alaskan native/American Indian, and Asian/Pacific Islander, and others-unspecified; CC: contingency coefficient; ER: estrogen receptor; HER2: human epidermal growth factor receptor 2; IDC: invasive ductal carcinoma; ILC: invasive lobular carcinoma; IQR: interquartile range; LN: lymph node; METABRIC: Molecular Taxonomy of Breast Cancer International Consortium; N/A: not available; PR: progesterone receptor; SEER: Surveillance, Epidemiology, and End Results; TNBC: triple negative breast cancer.
a Contingency coefficient >0.02 is defined as significant. * is gamma coefficient.
b Pearson's chi-square test was performed between the ER+PR+HER2- and ER+PR-HER2- groups.
Survival analysis showed that the ER+PR-HER2- group had intermediate outcomes between those of the ER+PR+HER2- (best outcomes) and TNBC (worst outcomes) groups (Figure 1A-D and Table S2, P<0.001). In addition, the ER+PR-HER2- group had an outcome similar to that of the ER-PR-HER2+ group in the SEER cohort (Figure S1A-B). With extended follow-up time, the survival curve of the ER+PR-HER2- group moved closer to that of the TNBC curve (Figure 1C-D) and finally exhibited no significant difference in 10-year overall survival (OS) (log-rank test, P=0.241, Tables S2-S3) or even worse OS after long-term follow-up (Figure S1C-D). A multivariate analysis among these three groups that adjusted for age, race, grade, stage, histology, chemotherapy and surgery showed that ER+PR-HER2- status was an independent predictor of recurrence and shorter survival (Table 2 and Table S4). This outcome disadvantage of the ER+PR-HER2- group was more apparent in the young, white, invasive ductal carcinoma (IDC), and LN-positive subgroups (Figure S1E). In the endocrine therapy subgroup, the ER+PR-HER2- group had a poorer outcome than the ER+PR+HER2- group (BCSS: P=0.008, Figure 1E), while in the subgroup without endocrine therapy, no survival difference was detected between these two groups (BCSS: P=0.587, Figure 1F).
Survival analysis between ER+PR-HER2-, ER+PR+HER2- and TNBC breast cancer. Kaplan-Meier curves of (A) breast cancer-specific survival from the Surveillance, Epidemiology, and End Results (SEER) database; (B) overall survival from the SEER database; (C) breast cancer-specific survival from the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) database; (D) overall survival from the METABRIC database; (E) breast cancer-specific survival within the hormone-received group from the METABRIC database; and (F) breast cancer-specific survival within the no-hormone group from the METABRIC database. The log-rank test was used to calculate the P-value.
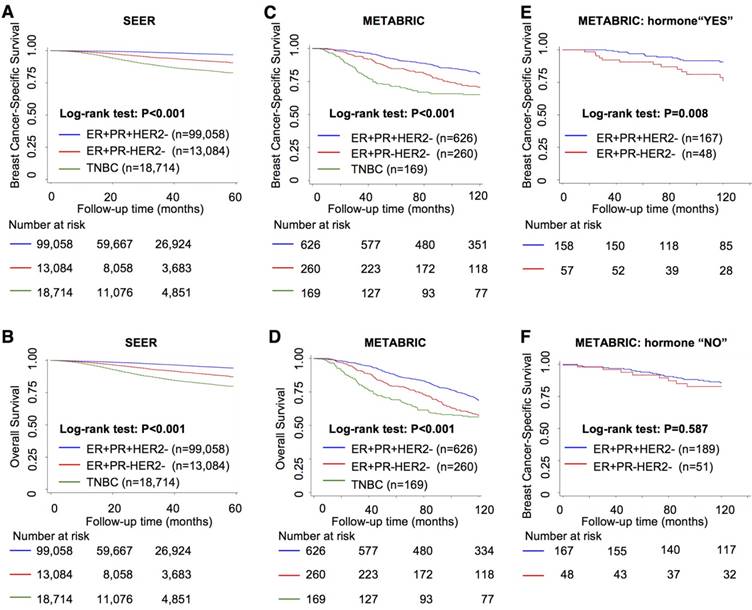
Univariate and multivariate analysis by Cox proportional hazards models in SEER and METABRIC cohorts.
| Cohort 1: SEER BCSS | Cohort 2: METABRIC BCSS | |||||
|---|---|---|---|---|---|---|
| Univariate | Univariate | |||||
| HR | 95% CI | P | HR | 95% CI | P | |
| ER+PR+HER2- | 1 | - | - | 1 | - | - |
| ER+PR-HER2- | 3.67 | 3.31-4.06 | <0.001 | 1.70 | 1.26-2.30 | 0.001 |
| ER-PR-HER2- | 7.58 | 7.06-8.15 | <0.001 | 2.41 | 1.78-3.27 | <0.001 |
| Multivariatea | Multivariateb | |||||
| HR | 95% CI | P | HR | 95% CI | P | |
| ER+PR+HER2- | 1 | - | - | 1 | - | - |
| ER+PR-HER2- | 2.85 | 2.58-3.14 | <0.001 | 1.54 | 1.14-2.09 | 0.005 |
| TNBC | 5.43 | 5.03-5.86 | <0.001 | 2.02 | 1.43-2.84 | <0.001 |
BCSS: breast cancer-specific survival; CI: confidence intervals; ER: estrogen receptor; HR: hazard ratio; HER2: human epidermal growth factor receptor 2; METABRIC: Molecular Taxonomy of Breast Cancer International Consortium; PR: progesterone receptor; SEER: Surveillance, Epidemiology, and End Results; TNBC: triple negative breast cancer.
a Adjusted by Cox proportional hazards models including age, race, stage, grade, histology, chemotherapy, and surgery.
b Adjusted by Cox proportional hazards models including age, grade, stage, chemotherapy and surgery.
Overall, the survival outcomes of the ER+PR-HER2- group fell between those of the ER+PR+HER2- and TNBC groups. In addition, ER+PR-HER2- tumors benefited less from endocrine therapy than did ER+PR+HER2- tumors.
Genomic and epigenetic alterations contribute to PR loss in ER+PR-HER2- breast cancer
To obtain deeper insights into the genomic and epigenetic characteristics of ER+PR-HER2- breast cancer, we explored potential mechanisms that contribute to PR loss in the TCGA cohort. First, we assessed the messenger RNA (mRNA) and protein expression levels of ER and PR (Figure 2A-B). As demonstrated in Figure 2A-B, there was no difference in ER expression level between ER+PR-HER2- and ER+PR+HER2- breast cancer. However, the ER+PR-HER2- group had significantly lower PR expression levels than the ER+PR+HER2- group (P<0.001). This result indicated that PR loss happened at or even before the level of transcription, rather than during posttranscriptional regulation.
Genomic and epigenomic causes of progesterone receptor (PR) loss within ER+HER2- breast cancer from the TCGA cohort. (A) Log2-transformed mRNA expression levels (RSEM) of ESR1, PR and MKI67. P-value was calculated by the Mann-Whitney test. Median with 95% confidence intervals (CI) is displayed. (B) Protein expression levels by reverse phase protein array (RPPA) of ER-alpha and PR. P-value was calculated by the Mann-Whitney test. Median with 95% CI is displayed. (C) Somatic copy number alteration (SCNA) status of PR among the ER+PR-HER2-, ER+PR+HER2- and TNBC groups. Loss: hemizygous or homozygous deletion; amplification: high-level amplification. The rate of PR copy number loss in each group is displayed (P=0.23 between ER+PR+HER2- and ER+PR-HER2- groups). (D) PR mRNA expression level by different PR copy number status within ER+PR- breast cancer. The mRNA expression value was calculated as log2(RSEM+1). The P-value was calculated by the Mann-Whitney test. Median with 95% CI is displayed. (E) PR promoter methylation levels between ER+PR-HER2- and ER+PR+HER2- breast cancer. The promoter methylation probe for PR was cg01671895. The P-value was calculated by the Mann-Whitney test. Median with 95% CI is displayed. (F) Correlation analysis between PR promoter methylation levels and mRNA expression levels by Pearson's correlation test (r=-0.6, R2=0.36, P<0.05). (G) PR promoter methylation levels between the PR copy loss group and the neutral/gain group in ER+PR-HER2- breast cancer. The P-value was calculated by the Mann-Whitney test. Median with 95% CI is displayed. (H) Pie chart summary of PR loss causes from the ER+PR-HER2- breast cancer group. Promoter hypermethylation was defined as “β-value > 0.2”.
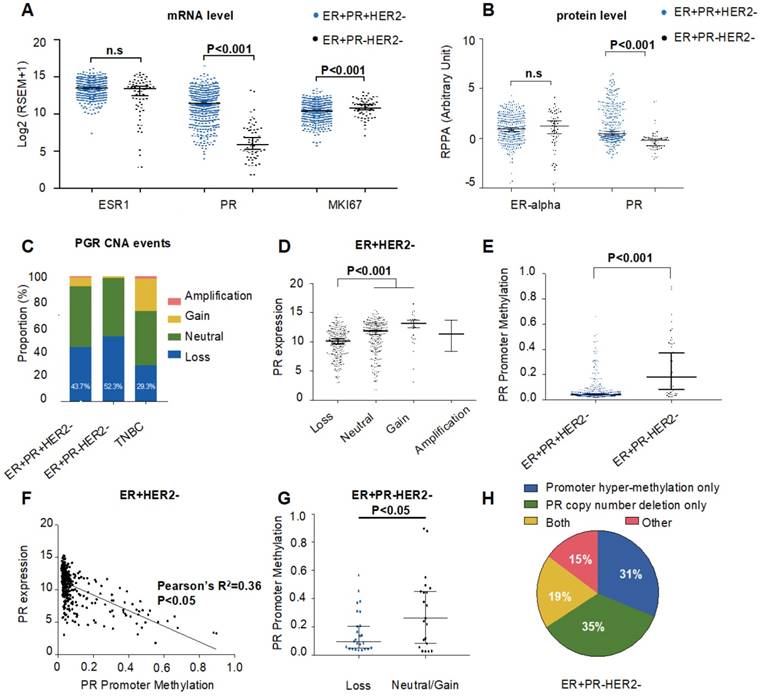
We further evaluated CNA events. We found that ER+PR-HER2- breast cancers possessed more copy number (CN) losses (hemizygous or homozygous deletion) in the PR genomic locus than did ER+PR+HER2- cases (Figure 2C, TCGA: 52.3% vs. 43.7%, P=0.23; Figure S2A, METABRIC: 27.5% vs. 17.5%, P=0.008). Within those ER+HER2- cases, tumors with PR CN loss displayed significantly lower PR mRNA expression (P<0.001, Figure 2D), suggesting that PR CN loss contributed to PR negativity. Nonetheless, a difference in PR expression was still present between the PR- and PR+ groups within the PR CN loss subgroup (Figure S2B), which indicated that other mechanisms participated in PR negativity.
Given previous studies on gene methylation and expression downregulation, we next investigated the relationship between PR expression and PR methylation. Figure 2E shows that the methylation level of the PR promoter was significantly higher in ER+PR-HER2- tumors than in ER+PR+HER2- tumors (P<0.001). This inverse correlation between PR expression and PR promoter methylation suggested that PR promoter methylation contributed to PR negativity in ER+HER2- tumors (Figure 2F). In addition, the methylation level of PR enhancer also correlated with PR negativity (Figure S2C-D). Interestingly, the promoter methylation level within the PR CN loss subgroup was significantly lower in ER+PR-HER2- cases (P<0.05, Figure 2G). Figure 2H summarizes the finding that only 19% of tumors had promoter hypermethylation and CN loss events at the same time.
Overall, genomic alterations such as PR CNA loss and PR methylation appear to be relatively independent mechanisms downregulating PR expression, which may consequently lead to PR negativity in ER+HER2- tumors.
Intrinsic genomic features of ER+PR-HER2- breast cancer were revealed
To extend our findings to the driver events of ER+PR-HER2- breast cancer, we explored specific somatic mutation and CNA events in ER+PR-HER2- tumors. Overall, ER+PR-HER2- tumors presented an intermediate somatic mutation load between those of ER+PR+HER2- and TNBC tumors (Table S5, P=0.007). Specifically, PR- tumors featured significantly more TP53 mutation events (30.3% vs. 17.0%, P=0.010) and fewer PIK3CA mutation events (25.8% vs. 42.7%, P=0.009) than did PR+ tumors, after adjusting for age, stage, race, and histology by logistic regression (Figure 3A and Table S5).
Genomic driver events in ER+PR-HER2- breast cancer. (A) TP53 and PIK3CA mutation rates in ER+PR-HER2- breast cancer compared with ER+PR+HER2- breast cancer and TNBC. Pearson's chi-square test was performed between each two groups. *: P<0.05; **: P<0.01; ***: P<0.001. Mutation rate of TP53 and PIK3CA is shown. (B) Frequency of significant somatic copy number alteration events in ER+PR-HER2- breast cancer (>0: copy number amplification; <0: copy number loss) from the TCGA cohort. (C) Kaplan-Meier curves of breast cancer-specific survival by Zinc Finger Protein 703 (ZNF703) amplification events or (D) by RPS6KB1 amplification events from the METABRIC database. (E) Enriched pathways (NOM P<0.01) related to Zinc Finger Protein 703 (ZNF703) amplification; and (F) related to Ribosomal Protein S6 Kinase B1 (RPS6KB1) amplification within ER+HER2- breast cancer from the TCGA cohort by gene set enrichment analysis (GSEA). amp: amplification.
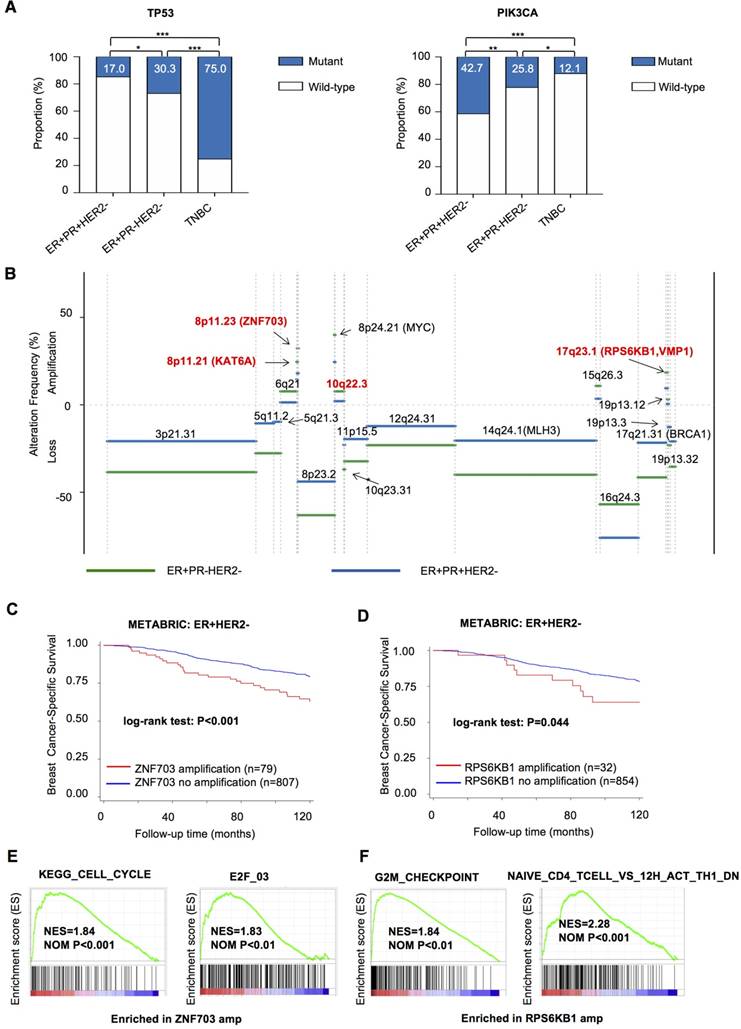
We then summarized significant focal somatic CNA events in the ER+PR-HER2- group after logistic regression adjustment (Figure 3B and Tables S6-S7). In particular, Chr8p11.21, Chr8p11.23, Chr10q22.3, and Chr17q23.1 were the four most frequent events occurring in the ER+PR-HER2- group (P<0.05, Table S8). Notably, ZNF703 on Chr8p11.23 (ER+PR-HER2- vs. ER+PR-HER2- vs. TNBC in TCGA: 21.5% vs. 13.6% vs. 7.3%; in METABRIC: 16.4% vs. 5.8% vs. 2.0%) and RPS6KB1 on Chr17q23.1 (ER+PR-HER2- vs. ER+PR-HER2- vs. TNBC in TCGA: 18.5% vs. 7.8% vs. 6.5%; in METABRIC: 5.6% vs. 2.8% vs. 0.0%) are two highly amplified oncogenes in ER+PR-HER2- breast cancer (Table S9).
In the METABRIC ER+HER2- cohort, ZNF703/ RPS6KB1 amplification was an indicator of poor breast cancer-specific survival (BCSS) (Figure 3C-D). The ZNF703 amplification group also showed a significantly poor OS (P<0.001), while the RPS6KB1 amplification group did not (P=0.318) (Figure S3A-B). Univariate and multivariate analyses showed that ZNF703 amplification was an independent prognostic factor (Table S10), while RPS6KB1 amplification showed a trend towards predicting worse BCSS independently (P=0.060). In addition, clinicopathologic analysis showed that ZNF703/RPS6KB1 amplification was related to a higher grade (Table S11). In summary, ZNF703/RPS6KB1 amplification was an unfavorable prognostic factor for ER+HER2- breast cancer. Furthermore, the ZNF703/RPS6KB1 high-expression group exhibited a poor BCSS (Figure S3C-F). Univariate and multivariate analysis showed that high RPS6KB1 expression was an independent prognostic factor (Table S12). The correlation between clinicopathologic features and ZNF703/ RPS6KB1 expression is summarized in Table S13. To summarize, high ZNF703/RPS6KB1 expression also indicated a poor prognosis. Surprisingly, tumors with ZNF703 amplification benefited more from endocrine therapy (Figure S3G-H). Furthermore, an increase in ZNF703/RPS6KB1 gene expression following copy number amplification was noted, indicating that these two genes were both cis-regulated by gene copy number alteration (Figure S4A-D). Thus, amplification events in both genes were functional.
We further evaluated enrichment pathways in the ZNF703/RPS6KB1 amplification group by gene set enrichment analysis (GSEA) within ER+HER2- breast cancer (detailed data not shown). Enriched cell cycle-related gene sets, such as KEGG_CELL_CYCLE, correlated with ZNF703 amplification (Figure 3E and Table S14). In addition, representative cell cycle-related genes (CCND1, CCNE2 and MKI67) were upregulated in the ZNF703-amplification group (Figure S5A). Significantly enriched E2F family gene sets, such as E2F_03, were related to ZNF703 amplification (Figure 3E and Table S15). Within those E2F family genes, E2F1, E2F2, E2F5, E2F7 and E2F8 were significantly upregulated genes in the ZNF703-amplification group (Figure S5B). Pathways enriched in RPS6KB1-amplified tumors were involved in cell cycle-related and immune response-related gene sets (Figure 3F). These results will provide clues for further research on these two genes.
Refining the “non-luminal-like” subgroup to deprioritize endocrine therapy within ER+PR-HER2- breast cancer by IHC
Evidence has emerged that intrinsic molecular subtypes (e.g., PAM50) could help classify patients with distinct biological features and responses to certain treatments even within a predefined IHC-based subtype [27, 28]. We therefore investigated PAM50 subtypes within the ER+PR-HER2- group from the TCGA cohort. In the ER+PR-HER2- group, 75% belonged to the luminal-like subtype, while 20% were non-luminal-like (normal-like excluded) (Figure 4A). This result indicated that tumors with the ER+PR-HER2- phenotype possessed a higher likelyhood of being the non-luminal-like subtype than did tumors with the ER+PR-HER2- phenotype (19% vs. 3%, P<0.001).
Identification of “non-luminal-like” subgroup from ER+PR-HER2- breast cancer. (A) PAM50 subtype distributions among the ER+PR-HER-, ER+PR+HER2- and TNBC groups from the TCGA cohort. (B) Enriched pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG) collection of the non-luminal-like subgroup compared to the luminal-like subgroup of the TCGA cohort by Pathifier algorithm. Heatmap showing the pathway scores of each ER+PR-HER2- tumor. (C) Endocrine sensitivity scores of different subtypes of ER+PR-HER2- breast cancer from the TCGA cohort. Endocrine sensitivity score = (0.8×ESR1 + 1.2×PGR + BCL2 + SCUBE2) / 4. The P-value was calculated by the Mann-Whitney test. (D) Differentially expressed candidate genes between luminal-like and non-luminal-like subgroups of ER+PR-HER2- breast cancer from the TCGA cohort. P-value was calculated by the Mann-Whitney test. (E) Criteria of subtyping based on the immunohistochemistry (IHC) status of GATA3, CK5 and EGFR in ER+PR-HER2- breast cancer from the Fudan University Shanghai Cancer Center (FUSCC) cohort. (F) Kaplan-Meier curves of recurrence-free survival within the “luminal-like” group and (G) within the “non-luminal-like” group from the FUSCC cohort treated with either sufficient endocrine therapy or not. The P-value was calculated by the log-rank test. (H) Clinical response to endocrine therapy or chemotherapy in non-luminal-like patients with advanced ER+PR-HER2- breast cancer. Responses were analyzed according to RECIST version 1.1. ALND: axillary lymph node dissection; CEF: CTX+Epi+5-FU plan.
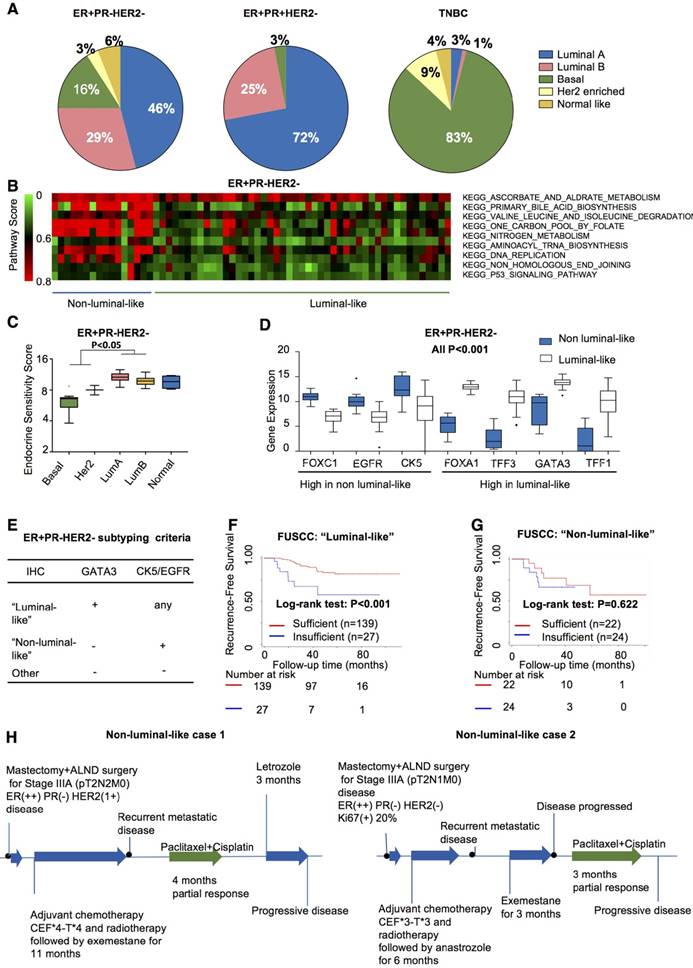
To explore whether these non-luminals in ER+PR-HER2- tumors differed from non-luminals in ER+PR+HER2-, TNBC and the other general population (all breast cancer patients from the TCGA dataset excluding patients with ER+PR-HER2-, ER+PR+HER2- or triple-negative breast cancer). PCA analysis and hierarchical clustering were performed based on gene expression profiles of 5000 genes with top-ranked standard deviations (Figure S6). Figure S6A-B shows that the non-luminals from ER+PR-HER2- breast cancer clustered together with non-luminals in the general population, mainly with the non-luminals in TNBCs. In other words, non-luminals in the ER+PR-HER2- population showed a similar expression pattern with non-luminals in the general population, especially in TNBCs. The non-luminal-like subtype in the ER+PR-HER2- group presented higher Pathifier [29] scores in biosynthesis, metabolism and DNA replication (Figure 4B) and lower endocrine sensitivity scores (P<0.05, Figure 4C and Figure S7A).
We next performed a differential expression analysis to select characterized genes. Figure 4D shows that higher expressions of FOXC1, EGFR, and CK5 were significantly associated with the non-luminal-like subtype, while FOXA1, TFF3, GATA3, and TFF1 tended to associate with the luminal-like subtype (all P<0.001, Figure 4D). We chose three genes based on the differential expression results, published research, and clinical routines. The detailed selection procedures are summarized in Supplementary Material. The combination of GATA3, CK5 and EGFR significantly discriminated luminal-like and non-luminal-like subtypes in the TCGA cohort and the MDACC cohort (AUC>0.9, Figure S7B-C). We further validated these three genes in the FUSCC cohort to determine individual cases as “non-luminal-like” or “luminal-like” by a feasible IHC method following these criteria: (a) a tumor was defined as “luminal-like” if it was GATA3-positive; (b) a tumor was placed into the “non-luminal-like” subgroup if it was GATA3-negative and positive for at least one of CK5 (coded by KRT5) or EGFR; and (c) a tumor was placed into the “other” subgroup if all three markers were negative (Figure 4E). Following these criteria, 67.8% (166/245) luminal-like and 18.7% (46/245) non-luminal-like subtypes were identified from 245 ER+PR-HER2- cases. Non-luminal-like cases exhibited a poorer prognosis than did luminal-like cases (Figure S7D), and non-luminal-like status was an independent prognostic factor for recurrence after adjusting for age at diagnosis, tumor size, node status, grade, chemotherapy and radiation (Table S16, HR=3.12, 95% CI 1.61-6.03, P=0.001). Interestingly, ER+PR-HER2- cases with a luminal-like subtype benefited more from sufficient endocrine therapy (log-rank P<0.001, Figure 4F). In contrast, the non-luminal-like subtype did not benefit from sufficient endocrine therapy (log-rank P=0.622, Figure 4G; interaction P<0.001, Table S17). According to the IHC-based criteria, we summarized the treatment efficacy of non-luminal-like (n=10) and luminal-like (n=10) cases (Table S18). For those non-luminal-like cases, the median recurrence-free time during adjuvant hormone therapy was significantly shorter than that of luminal-like cases (P=0.017). During salvage endocrine treatment procedure, the median progression-free time of non-luminal-like cases was also significantly shorter (P=0.034). Two representative non-luminal-like cases from the FUSCC cohort are presented in Figure 4H. The first case encountered a recurrence metastatic event during endocrine therapy, which responded partially to chemotherapy but progressed after changing to endocrine therapy (Figure 4H and Figure S8A). The second patient had a recurrence metastatic event during endocrine therapy and did not respond to another endocrine drug but did respond partially to chemotherapy (Figure 4H and Figure S8B). The detailed treatment procedures of other non-luminal-like cases are shown in Figure S9. The data indicate that hormone therapy might not effectively prevent recurrence or delay disease progression for non-luminal-like cases. Thus, it may not be appropriate to utilize hormone therapy as a primary method to control disease progression for non-luminal-like cases.
Overall, we defined a “non-luminal-like” subgroup within ER+PR-HER2- breast cancer that exhibited poorer outcome and received less benefit from endocrine therapy. In addition, we developed an easy-to-use IHC-based classifier to identify these cases, which is of great clinical significance.
Discussion
Precision treatment is becoming the ultimate goal for treating breast cancer. Although hormone receptor-positive breast cancer benefits greatly from endocrine therapy, a proportion of ER-positive patients still suffer from recurrence. Identification of those distinct populations remains of great clinical significance. It has been debated whether the presence of PR, an indicator of an intact ER pathway, plays a role in endocrine responsiveness [ 5, 8, 30]. In the current study, we explored the clinical characteristics and genomic landscapes of ER+PR-HER2- breast cancer. Our data revealed that nearly 20% of ER+PR-HER2- tumors belong to non-luminal-like subtypes by PAM50. Therefore, we developed a workable 3-gene IHC classifier to define tumor subtypes within ER+PR-HER2- breast cancer. Evaluation of distinct subtypes using such a feasible tool is very valuable for predicting responses to endocrine drugs and developing individualized therapies in clinical practice.
ER+PR- tumors have been reported to occur more frequently in elderly and postmenopausal women and to have worse survival [3, 5, 7, 8, 30, 31]. However, the role of PR loss in predicting endocrine therapy sensitivity is controversial [8, 10, 30, 32, 33]. In addition, ER+PR- tumors presented two to three times higher rates of HER2 positivity, which was reported to be associated with endocrine resistance, than did ER+PR+ tumors [5, 13].
Given this information, we first took advantage of the large SEER database and long-term METABRIC database for investigating the clinicopathological features of ER+PR-HER2- breast cancer, and we validated that PR loss in ER+HER2- breast cancer was not only a prognostic factor but also a predictor of response to endocrine therapy. We next investigated multiomic events promoting PR loss in ER+HER2- tumors. Several theories can explain PR loss in ER-positive breast tumors, including the nonfunctional ER theory [16]. However, our results showed that PR expression decreased at both the mRNA and protein levels while ER was transcriptionally active (Figure 2A-B). This phenomenon also indicated that PR loss reflected the downregulation of PR expression rather than posttranscriptional regulation. Other reported potential mechanisms for PR loss included growth signaling activation, hypermethylation of the PR promoter, or LOH at the PR gene locus [1, 17, 18]. In our analysis, we found that both PR CN loss and hypermethylation in PR regulatory regions correlated with reduced PR expression (Figure 2C-F and Figure S2A-C). Interestingly, PR promoter methylation and PR CN loss occurred independently (Figure 2G) and covered 75% of ER+PR-HER2- tumors (Figure 2H), which suggested that both genomic and epigenomic aberrance at the PR gene locus were quite common in ER+PR-HER2- tumors.
Genomically, ER+PR-HER2- tumors had driver events distinct from those of ER+PR+HER2- tumors, including a higher TP53 mutation rate, a lower PIK3CA mutation rate (Figure 3A) and a series of CN gain or loss events (Figure 3B), some of which have not previously been reported [34, 35]. Among these events, ZNF703 on Chr8p11.23 and RPS6KB1 on Chr17q23.1 were found more frequently in ER+PR-HER2- tumors. ZNF703 is a member of the NET/NlZ family of zinc finger transcription factors. It was initially recognized as a candidate oncogene based on a frequent amplicon 8p11-12 in human breast cancer [36-39]. Amplification of the ZNF703 gene preferentially occurs in luminal B breast cancer, usually coamplified with CCND1 [38, 39]. A high level of ZNF703 expression or amplification has been associated with worse survival in breast cancer [39-41]. ZNF703 is also associated with tumor progression in many other types of human cancers, such as lung cancer [42, 43], colorectal cancer [44] and head and neck squamous cell carcinoma [45]. Tumors with ZNF703 overexpression have a distinctive expression signature. Holland et al. reported that a lipid metabolism and detoxification pathway was altered in tumors with high ZNF703 levels [39]. In vitro studies revealed that ZNF703 overexpression in breast cancer cell lines could promote cell proliferation and increase stem cells through WNT, NOTCH, Akt/mTOR, or TGFβ signaling [39, 40]. It has also been identified that ZNF703 overexpression regulates ER and E2F1 transcriptional activity, downregulating ER expression [40]. Our analysis showed that ZNF703 amplification correlated with poor outcome in ER+HER2- breast cancer and participated in cell cycle progression through the E2F family of regulators. Only one study has reported that high ZNF703 levels may lead to tamoxifen resistance through Akt/mTOR signaling [46]. ER+HER2- tumors with ZNF703 amplification appeared to benefit more from endocrine therapy (Figure S3G-H); therefore, further investigation of the effects of ZNF703 on hormone sensitivity is needed. RPS6KB1, situated in the commonly amplified 17q21-23, encodes a member of the ribosomal S6 kinase family of serine/threonine kinases [47]. The encoded protein S6K1 acts downstream of mTOR signaling to promote protein synthesis, cell growth and cell cycle progression. This protein plays different roles in cancer through multiple mechanisms: 1) by phosphorylating EIF4B, RPS6 and EEF2K, S6K1 induces protein synthesis at the ribosome; 2) by repressing the pro-apoptotic function of BAD, S6K1 contributes to cell survival; 3) as a downstream target of mTORC1 signaling, S6K1 transmits the PI3K/AKT1 signal; and 4) phosphorylation of S6K1 has been correlated with autophagy inhibition or activation [32]. Although S6K1 is overexpressed in many tumors, high-level amplification of RPS6KB1 is limited to breast cancer [48]. Overexpression of this protein has previously been found to be associated with worse survival outcomes, chemotherapy resistance and radiotherapy resistance in breast cancer [49-51]. However, S6K1 is also implicated in regulating ERα transcriptional activity by phosphorylating ER-Ser 167 [52] and increasing tamoxifen sensitivity [53]. There is actually a positive regulatory loop between S6K1 and ERα in control of breast cancer cell proliferation [54]. S6K1 may be utilized as a prognostic marker and a therapeutic target.
ER+PR-HER2- breast cancer is a heterogeneous entity. Ethier et al. previously reported that ER+PR-HER2- tumors were composed predominantly of luminal B and HER2-enriched molecular subtypes [55]. In contrast, our analysis found that nearly 20% of ER+PR-HER2- tumors were non-luminal-like (mainly basal-like) with a significantly lower endocrine resistance score. Treatment of those cases with endocrine therapy for 5 years or even more is questionable. Thus, identifying the non-luminal-like subgroup within the ER+PR-HER2- phenotype is important. This study developed a feasible IHC-based tool (GATA3, CK5, and EGFR) to distinguish non-luminal-like from luminal-like subtypes within the ER+PR-HER2- phenotype. Of the three genes selected in this article, the transcription factor GATA3 has previously been reported to bind to the cis-regulatory elements upstream of the ERα gene and promote ER expression [56]. Thus, GATA3 is defined as an ER-related gene and a marker of the luminal subtype of breast cancer [57, 58]. In addition, several studies have provided evidence that a high GATA3 mRNA expression level correlates with hormone therapy benefits in ER+ breast cancer patients [59, 60]. CK5 and EGFR are two commonly used markers for basal-like breast cancer [61-63]. Evidence has shown that CK5-expressing cells exhibit increased cancer stem cell and therapy-resistant properties [64]. EGFR protein plays an important role in promoting cell proliferation and migration [65]. More importantly, EGFR expression in ER+PR-HER2- tumors may serve as a potential therapeutic target for treatment [66, 67].
Importantly, the identified non-luminal-like ER+PR-HER2- cases obtained limited benefit from endocrine therapy, while luminal-like cases benefited despite PR loss. We defined two non-luminal-like cases using this tool from the FUSCC cohort. Both cases encountered recurrence and metastasis during a short period of endocrine therapy, which indicates primary resistance to endocrine drugs. Instead, chemotherapy was more effective in controlling their disease (Figure 4H and Figure S9).
The strengths of our study are summarized as follows. This study evaluated the multiomic features of ER+PR-HER2- tumors collaboratively for the first time and identified some driver events that may help explore significant treatment targets. Furthermore, this study discovered non-luminal-like subtypes in ER+PR-HER2- tumors and established an important and feasible tool for identifying this subtype, which benefits less from endocrine therapy.
Inevitably, there are some limitations in our study. First, the FUSCC cohort was a prospective observational study that did not include intervention randomization. Thus, this study is susceptible to the inherent biases of this type of study design. To minimize the bias, we included multiple cohorts to validate our conclusions. To further validate the IHC subtyping criteria in a prospective cohort, we recently designed an open-label, multicenter, randomized clinical trial. In this trial, we aim to compare the efficacy and safety of chemotherapy versus hormone therapy in the group determined as non-luminal-like patients based on the IHC subtyping criteria. We hypothesize that patients defined as non-luminal-like will benefit less from hormone therapy and might be more sensitive to chemotherapy. In the future, we will continue to focus on treatment strategies for ER+PR-HER2- breast cancer, the details of which will be included in a future article. In addition, different cohorts provided various end-points that may warrant more caution in interpreting some conclusions. Finally, for some cases, the “insufficient” endocrine therapy was a result of early tumor recurrence; therefore, prospective studies are needed.
Conclusions
In conclusion, ER+PR-HER2- breast tumors are more malignant than ER+PR+HER2- tumors clinically, biologically and genomically. The identifycation of non-luminal-like subtypes by a three-marker IHC method (GATA3, CK5, and EGFR) is a clinically feasible method of guiding systemic endocrine therapy in ER+PR-HER2- tumors.
Abbreviations
BCSS: breast cancer-specific survival; Chr: chromosome; CNA: copy number alteration; ER: estrogen receptor; fRMA: frozen robust multiarray analysis; FUSCC: Fudan University Shanghai Cancer Center; GSEA: gene set enrichment analysis; HER2: human epidermal growth factor receptor 2; IHC: immunohistochemistry; MDACC: MD Anderson Cancer Center; METABRIC: Molecular Taxonomy of Breast Cancer International Consortium; PR: progesterone receptor; RFS: recurrence-free survival; SEER: Surveillance, Epidemiology, and End Results; TCGA: the Cancer Genome Atlas.
Acknowledgements
This study was funded by grants from the National Natural Science Foundation of China (81874112, 81502278, 81572583, and 81372848), the Training Plan of Excellent Talents in Shanghai Municipality Health System (2017YQ038), the “Chen Guang” project supported by Shanghai Municipal Education Commission and Shanghai Education Development Foundation (17CG01), Shanghai Pujiang Program (18PJD007), and the Training Plan of Excellent Talents of Fudan University Shanghai Cancer Center (YJYQ201602). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Authors' contributions
YZJ and ZMS outlined the manuscript; all authors contributed to the literature search, data collection, data analysis, and data interpretation. XYL and DM provided the figures and drafted the manuscript, with additional input from all authors. All authors approved the final manuscript. XYL and DM contributed equally to this work.
Ethics approval and consent to participate
Our study was approved by the independent Ethics Committee/Institutional Review Board of FUSCC. The data released by the SEER, METABRIC, TCGA and MDACC databases are publicly available and deidentified and, therefore, do not require informed patient consent.
Supplementary Material
Supplementary methods, figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cui X, Schiff R, Arpino G, Osborne CK, Lee AV. Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy. J Clin Oncol. 2005;23:7721-35
2. Goldhirsch A, Wood WC, Coates AS, Gelber RD, Thurlimann B, Senn HJ. et al. Strategies for subtypes—dealing with the diversity of breast cancer: highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann Oncol. 2011;22:1736-47
3. Purdie CA, Quinlan P, Jordan LB, Ashfield A, Ogston S, Dewar JA. et al. Progesterone receptor expression is an independent prognostic variable in early breast cancer: a population-based study. Br J Cancer. 2014;110:565-72
4. Sun JY, Wu SG, Li FY, Lin HX, He ZY. Progesterone receptor loss identifies hormone receptor-positive and HER2-negative breast cancer subgroups at higher risk of relapse: a retrospective cohort study. Onco Targets Ther. 2016;9:1707-13
5. Arpino G, Weiss H, Lee AV, Schiff R, De Placido S, Osborne CK. et al. Estrogen receptor-positive, progesterone receptor-negative breast cancer: association with growth factor receptor expression and tamoxifen resistance. J Natl Cancer Inst. 2005;97:1254-61
6. Dunnwald LK, Rossing MA, Li CI. Hormone receptor status, tumor characteristics, and prognosis: a prospective cohort of breast cancer patients. Breast Cancer Res. 2007;9:R6
7. Bae SY, Kim S, Lee JH, Lee HC, Lee SK, Kil WH. et al. Poor prognosis of single hormone receptor- positive breast cancer: similar outcome as triple-negative breast cancer. BMC Cancer. 2015;15:138
8. Bardou VJ, Arpino G, Elledge RM, Osborne CK, Clark GM. Progesterone receptor status significantly improves outcome prediction over estrogen receptor status alone for adjuvant endocrine therapy in two large breast cancer databases. J Clin Oncol. 2003;21:1973-9
9. Rakha EA, El-Sayed ME, Green AR, Lee AH, Robertson JF, Ellis IO. Prognostic markers in triple-negative breast cancer. Cancer. 2007;109:25-32
10. Viale G, Regan MM, Maiorano E, Mastropasqua MG, Dell'Orto P, Rasmussen BB. et al. Prognostic and predictive value of centrally reviewed expression of estrogen and progesterone receptors in a randomized trial comparing letrozole and tamoxifen adjuvant therapy for postmenopausal early breast cancer: BIG 1-98. J Clin Oncol. 2007;25:3846-52
11. Yu KD, Liu GY, Di GH, Wu J, Lu JS, Shen KW. et al. Progesterone receptor status provides predictive value for adjuvant endocrine therapy in older estrogen receptor-positive breast cancer patients. Breast. 2007;16:307-15
12. Cuzick J, Sestak I, Baum M, Buzdar A, Howell A, Dowsett M. et al. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 10-year analysis of the ATAC trial. Lancet Oncol. 2010;11:1135-41
13. Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233-47
14. Ellis MJ, Ma C. Letrozole in the neoadjuvant setting: the P024 trial. Breast Cancer Res Treat. 2007;105(Suppl 1):33-43
15. Dowsett M, Harper-Wynne C, Boeddinghaus I, Salter J, Hills M, Dixon M. et al. HER-2 amplification impedes the antiproliferative effects of hormone therapy in estrogen receptor-positive primary breast cancer. Cancer Res. 2001;61:8452-8
16. Horwitz KB, McGuire WL. Estrogen control of progesterone receptor in human breast cancer. Correlation with nuclear processing of estrogen receptor. J Biol Chem. 1978;253:2223-8
17. Lapidus RG, Nass SJ, Davidson NE. The loss of estrogen and progesterone receptor gene expression in human breast cancer. J Mammary Gland Biol Neoplasia. 1998;3:85-94
18. Winqvist R, Hampton GM, Mannermaa A, Blanco G, Alavaikko M, Kiviniemi H. et al. Loss of heterozygosity for chromosome 11 in primary human breast tumors is associated with poor survival after metastasis. Cancer Res. 1995;55:2660-4
19. Hatzis C, Pusztai L, Valero V, Booser DJ, Esserman L, Lluch A. et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. Jama. 2011;305:1873-81
20. Popovici V, Chen W, Gallas BG, Hatzis C, Shi W, Samuelson FW. et al. Effect of training-sample size and classification difficulty on the accuracy of genomic predictors. Breast Cancer Res. 2010;12:R5
21. Tabchy A, Valero V, Vidaurre T, Lluch A, Gomez H, Martin M. et al. Evaluation of a 30-gene paclitaxel, fluorouracil, doxorubicin, and cyclophosphamide chemotherapy response predictor in a multicenter randomized trial in breast cancer. Clin Cancer Res. 2010;16:5351-61
22. McCall MN, Bolstad BM, Irizarry RA. Frozen robust multiarray analysis (fRMA). Biostatistics. 2010;11:242-53
23. Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S. et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28:2784-95
24. Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH. et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31:3997-4013
25. Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ. et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol. 2007;25:118-45
26. Yu KD, Jiang YZ, Hao S, Shao ZM. Molecular essence and endocrine responsiveness of estrogen receptor-negative, progesterone receptor-positive, and HER2-negative breast cancer. BMC Med. 2015;13:254
27. Perou CM, Jeffrey SS, van de Rijn M, Rees CA, Eisen MB, Ross DT. et al. Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci U S A. 1999;96:9212-7
28. Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A. et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003;100:8418-23
29. Drier Y, Sheffer M, Domany E. Pathway-based personalized analysis of cancer. Proc Natl Acad Sci U S A. 2013;110:6388-93
30. Rakha EA, El-Sayed ME, Green AR, Paish EC, Powe DG, Gee J. et al. Biologic and clinical characteristics of breast cancer with single hormone receptor positive phenotype. J Clin Oncol. 2007;25:4772-8
31. Braun L, Mietzsch F, Seibold P, Schneeweiss A, Schirmacher P, Chang-Claude J. et al. Intrinsic breast cancer subtypes defined by estrogen receptor signalling-prognostic relevance of progesterone receptor loss. Mod Pathol. 2013;26:1161-71
32. Fenton TR, Gout IT. Functions and regulation of the 70kDa ribosomal S6 kinases. Int J Biochem Cell Biol. 2011;43:47-59
33. Early Breast Cancer Trialists' Collaborative G. Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. Lancet. 2015;386:1341-52
34. Creighton CJ, Kent Osborne C, van de Vijver MJ, Foekens JA, Klijn JG, Horlings HM. et al. Molecular profiles of progesterone receptor loss in human breast tumors. Breast Cancer Res Treat. 2009;114:287-99
35. Carracedo A, Salido M, Corominas JM, Rojo F, Ferreira BI, Suela J. et al. Are ER+PR+ and ER+PR- breast tumors genetically different? A CGH array study. Cancer Genet. 2012;205:138-46
36. Garcia MJ, Pole JC, Chin SF, Teschendorff A, Naderi A, Ozdag H. et al. A 1 Mb minimal amplicon at 8p11-12 in breast cancer identifies new candidate oncogenes. Oncogene. 2005;24:5235-45
37. Adelaide J, Finetti P, Bekhouche I, Repellini L, Geneix J, Sircoulomb F. et al. Integrated profiling of basal and luminal breast cancers. Cancer Res. 2007;67:11565-75
38. Kwek SS, Roy R, Zhou H, Climent J, Martinez-Climent JA, Fridlyand J. et al. Co-amplified genes at 8p12 and 11q13 in breast tumors cooperate with two major pathways in oncogenesis. Oncogene. 2009;28:1892-903
39. Holland DG, Burleigh A, Git A, Goldgraben MA, Perez-Mancera PA, Chin SF. et al. ZNF703 is a common Luminal B breast cancer oncogene that differentially regulates luminal and basal progenitors in human mammary epithelium. EMBO Mol Med. 2011;3:167-80
40. Sircoulomb F, Nicolas N, Ferrari A, Finetti P, Bekhouche I, Rousselet E. et al. ZNF703 gene amplification at 8p12 specifies luminal B breast cancer. EMBO Mol Med. 2011;3:153-66
41. Reynisdottir I, Arason A, Einarsdottir BO, Gunnarsson H, Staaf J, Vallon-Christersson J. et al. High expression of ZNF703 independent of amplification indicates worse prognosis in patients with luminal B breast cancer. Cancer Med. 2013;2:437-46
42. Baykara O, Bakir B, Buyru N, Kaynak K, Dalay N. Amplification of chromosome 8 genes in lung cancer. J Cancer. 2015;6:270-5
43. Baykara O, Dalay N, Kaynak K, Buyru N. ZNF703 Overexpression may act as an oncogene in non-small cell lung cancer. Cancer Med. 2016;5:2873-8
44. Ma F, Bi L, Yang G, Zhang M, Liu C, Zhao Y. et al. ZNF703 promotes tumor cell proliferation and invasion and predicts poor prognosis in patients with colorectal cancer. Oncol Rep. 2014;32:1071-7
45. Yang H, Jiang WQ, Cao Y, Sun YA, Wei J, An X. et al. Elevated ZNF703 protein expression is an independent unfavorable prognostic factor for survival of the patients with head and neck squamous cell carcinoma. Dis Markers. 2015;2015:640263
46. Zhang X, Mu X, Huang O, Xie Z, Jiang M, Geng M. et al. Luminal breast cancer cell lines overexpressing ZNF703 are resistant to tamoxifen through activation of Akt/mTOR signaling. PLoS One. 2013;8:e72053
47. Sunami T, Byrne N, Diehl RE, Funabashi K, Hall DL, Ikuta M. et al. Structural basis of human p70 ribosomal S6 kinase-1 regulation by activation loop phosphorylation. J Biol Chem. 2010;285:4587-94
48. Sinclair CS, Rowley M, Naderi A, Couch FJ. The 17q23 amplicon and breast cancer. Breast Cancer Res Treat. 2003;78:313-22
49. Perez-Tenorio G, Karlsson E, Waltersson MA, Olsson B, Holmlund B, Nordenskjold B. et al. Clinical potential of the mTOR targets S6K1 and S6K2 in breast cancer. Breast Cancer Res Treat. 2011;128:713-23
50. Barlund M, Forozan F, Kononen J, Bubendorf L, Chen Y, Bittner ML. et al. Detecting activation of ribosomal protein S6 kinase by complementary DNA and tissue microarray analysis. J Natl Cancer Inst. 2000;92:1252-9
51. van der Hage JA, van den Broek LJ, Legrand C, Clahsen PC, Bosch CJ, Robanus-Maandag EC. et al. Overexpression of P70 S6 kinase protein is associated with increased risk of locoregional recurrence in node-negative premenopausal early breast cancer patients. Br J Cancer. 2004;90:1543-50
52. Yamnik RL, Digilova A, Davis DC, Brodt ZN, Murphy CJ, Holz MK. S6 kinase 1 regulates estrogen receptor alpha in control of breast cancer cell proliferation. J Biol Chem. 2009;284:6361-9
53. Yamashita H, Nishio M, Kobayashi S, Ando Y, Sugiura H, Zhang Z. et al. Phosphorylation of estrogen receptor alpha serine 167 is predictive of response to endocrine therapy and increases postrelapse survival in metastatic breast cancer. Breast Cancer Res. 2005;7:R753-64
54. Maruani DM, Spiegel TN, Harris EN, Shachter AS, Unger HA, Herrero-Gonzalez S. et al. Estrogenic regulation of S6K1 expression creates a positive regulatory loop in control of breast cancer cell proliferation. Oncogene. 2012;31:5073-80
55. Ethier JL, Ocana A, Rodriguez Lescure A, Ruiz A, Alba E, Calvo L. et al. Outcomes of single versus double hormone receptor-positive breast cancer. A GEICAM/9906 sub-study. Eur J Cancer. 2018;94:199-205
56. Asch-Kendrick R, Cimino-Mathews A. The role of GATA3 in breast carcinomas: a review. Hum Pathol. 2016;48:37-47
57. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA. et al. Molecular portraits of human breast tumours. Nature. 2000;406:747-52
58. Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H. et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869-74
59. Ciocca V, Daskalakis C, Ciocca RM, Ruiz-Orrico A, Palazzo JP. The significance of GATA3 expression in breast cancer: a 10-year follow-up study. Hum Pathol. 2009;40:489-95
60. Liu J, Prager-van der Smissen WJ, Look MP, Sieuwerts AM, Smid M, Meijer-van Gelder ME. et al. GATA3 mRNA expression, but not mutation, associates with longer progression-free survival in ER-positive breast cancer patients treated with first-line tamoxifen for recurrent disease. Cancer Lett. 2016;376:104-9
61. Sood N, Nigam JS. Correlation of CK5 and EGFR with clinicopathological profile of triple-negative breast cancer. Patholog Res Int. 2014;2014:141864
62. Rao C, Shetty J, Prasad KH. Immunohistochemical profile and morphology in triple - negative breast cancers. J Clin Diagn Res. 2013;7:1361-5
63. Sutton LM, Han JS, Molberg KH, Sarode VR, Cao D, Rakheja D. et al. Intratumoral expression level of epidermal growth factor receptor and cytokeratin 5/6 is significantly associated with nodal and distant metastases in patients with basal-like triple-negative breast carcinoma. Am J Clin Pathol. 2010;134:782-7
64. Fettig LM, McGinn O, Finlay-Schultz J, LaBarbera DV, Nordeen SK, Sartorius CA. Cross talk between progesterone receptors and retinoic acid receptors in regulation of cytokeratin 5-positive breast cancer cells. Oncogene. 2017;36:6074-84
65. Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol. 2018;12:3-20
66. Jamdade VS, Sethi N, Mundhe NA, Kumar P, Lahkar M, Sinha N. Therapeutic targets of triple-negative breast cancer: a review. Br J Pharmacol. 2015;172:4228-37
67. Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z. et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10:5367-74
Author contact
![]() Corresponding authors: Dr. Yi-Zhou Jiang, Fudan University Shanghai Cancer Center, No. 270 Dong'an Road, Shanghai, 200032, P.R. China. Email: yizhoujiangedu.cn and Dr. Zhi-Ming Shao, Fudan University Shanghai Cancer Center, No. 270 Dong'an Road, Shanghai, 200032, P.R. China. Email: zhimingshaocom
Corresponding authors: Dr. Yi-Zhou Jiang, Fudan University Shanghai Cancer Center, No. 270 Dong'an Road, Shanghai, 200032, P.R. China. Email: yizhoujiangedu.cn and Dr. Zhi-Ming Shao, Fudan University Shanghai Cancer Center, No. 270 Dong'an Road, Shanghai, 200032, P.R. China. Email: zhimingshaocom