Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
CDK/cyclin in cell cycle...
CKIs in cell cycle checkpoints
Selective autophagy
Autophagy regulation by cell...
Selective autophagy regulates...
Selective autophagy regulates...
Selective autophagy in cancer
Targeting general autophagy or...
Combined targeting of autophagy...
Conclusion
Abbreviations
Acknowledgements
References
CDK/cyclin in cell cycle...
CKIs in cell cycle checkpoints
Selective autophagy
Autophagy regulation by cell...
Selective autophagy regulates...
Selective autophagy regulates...
Selective autophagy in cancer
Targeting general autophagy or...
Combined targeting of autophagy...
Conclusion
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(1):104-125. doi:10.7150/thno.30308 This issue Cite
Review
Selective Autophagy Regulates Cell Cycle in Cancer Therapy
Kai Zheng1 ![]() , Zhendan He1, Kaio Kitazato2, Yifei Wang3
, Zhendan He1, Kaio Kitazato2, Yifei Wang3
1. School of Pharmaceutical Sciences, Health Science Center, Shenzhen University, Shenzhen, 518060, PR China;
2. Division of Molecular Pharmacology of Infectious Agents, Department of Molecular Microbiology and Immunology, Nagasaki University, Nagasaki, 852-8521, Japan
3. College of Life Science and Technology, Guangzhou Jinan Biomedicine Research and Development Center, Jinan University, Guangzhou 510632, PR China
Received 2018-9-30; Accepted 2018-10-30; Published 2019-1-1
Citation:
Zheng K, He Z, Kitazato K, Wang Y. Selective Autophagy Regulates Cell Cycle in Cancer Therapy. Theranostics 2019; 9(1):104-125. doi:10.7150/thno.30308. https://www.thno.org/v09p0104.htm
Other stylesAbstract

Aberrant function of cell cycle regulators results in uncontrolled cell proliferation, making them attractive therapeutic targets in cancer treatment. Indeed, survival of many cancers exclusively relies on these proteins, and several specific inhibitors are in clinical use. Although the ubiquitin-proteasome system is responsible for the periodic quality control of cell cycle proteins during cell cycle progression, increasing evidence clearly demonstrates the intimate interaction between cell cycle regulation and selective autophagy, important homeostasis maintenance machinery. However, these studies have often led to divergent rather than unifying explanations due to complexity of the autophagy signaling network, the inconsistent functions between general autophagy and selective autophagy, and the different characteristics of autophagic substrates. In this review, we highlight current data illustrating the contradictory and important role of cell cycle proteins in regulating autophagy. We also focus on how selective autophagy acts as a central mechanism to maintain orderly DNA repair and genome integrity by degrading specific cell cycle proteins, regulating cell division, and promoting DNA damage repair. We further discuss the ways in which selective autophagy may impact the cell cycle regulators, since failure to appropriately remove these can interfere with cell death-related processes, including senescence and autophagy-related cell death. Imbalanced cell proliferation is typically utilized by cancer cells to acquire resistance. Finally, we discuss the possibility of a potent anticancer therapeutic strategy that targets selective autophagy or autophagy and cell cycle together.
Keywords: selective autophagy, chemoresistance, senescence, DNA damage repair, cell cycle checkpoints
CDK/cyclin in cell cycle progression
The mammalian cell cycle is a well-organized and sophisticated regulation process, which is usually divided into G0/G1, S, G2, and M phases and is mainly controlled by different cyclin-dependent kinases (CDKs) and their functional cyclin partners [1]. Mitogenic signals activate signaling pathways required for proliferation, such as the RAS pathway. Cyclin then directly interacts with the corresponding CDK, which phosphorylates target proteins, activates transcriptional factors, and thus drives cell cycle progression (Figure 1). For instance, mitogenic signals trigger cells to exit quiescence (termed G0 phase) and re-enter the cell cycle (G1 into S phase) through activating the CDK4/6-cyclin D complex. Activated CDK4 and CDK6 can phosphorylate various cellular targets, among which the most important target is retinoblastoma tumor suppressor protein (RB). RB is a tumor suppressor that sequesters the transcription factor E2F to hinder cell cycle progression. Upon RB phosphorylation by CDK4/6-cyclin D, E2F is released and activates the expression of a set of genes critical for DNA replication and chromosome rearrangement (S phase). In addition to CDK4/6, CDK2 is another important G1/S transition regulator. In the late G1 phase, the combinatory effect of E2F-mediated transcriptional activation of cyclin E, CDK4/6-cyclin D-mediated removal of CDK inhibitor protein (CKI) p27 or p21, and cell division cycle 25A (CDC25A)-mediated dephosphorylation results in CDK2 activation to allow G1/S transition. Once entering S phase, CDK2 partner cyclin E undergoes promptly degradation through FBXW7-mediated ubiquitylation, and CDK2 forms an active complex with cyclin A2. Furthermore, CDK1 controls the cell cycle transition from G2 to M phase. Similar to CDK2, CDK1 activity is also inhibited by specific CDC2-inhibitory kinase MYT1-mediated phosphorylation at Thr14 and nuclear kinase WEE1-mediated Tyr15 phosphorylation, respectively, which are relieved by CDC25. During G2 phase, CDK1 is activated by binding to cyclin A2 and cyclin B. However, upon the initiation of mitosis, CDK1 interacts with cyclin B to form the so-called 'maturation promoting factor' to promote mitotic entry and several mitotic events. CDK1-cyclin B complex activates various enzymes critical for chromatin condensation, mitotic spindle formation, nuclear lamina depolymerisation and the reorganization of actin cytoskeleton. Finally, regulatory serine/threonine kinases, including Aurora kinases (Aurora A, Aurora B, and Aurora C) and Polo-like kinases (PLKs), are activated to control mitotic entry, mitotic exit, spindle formation, cytokinesis, and meiosis to ensure genetic stability in cell division.
CKIs in cell cycle checkpoints
Cell cycle checkpoints are control systems that ensure proper division of the cell and induce cell cycle arrest in response to intracellular DNA damage, exogenous cellular stress signals, and deficiencies in essential growth factors or nutrients. Cells that fail to proceed to the checkpoint initiate apoptosis, cell death, or permanent cell cycle arrest (referred to as “senescence”). By sensing defects that occur during essential processes such as DNA replication, checkpoints brake the cell cycle at a specific stage mainly through inhibiting the cell cycle-related kinase activity of CDKs (Figure 1). CKIs are an important class of tumor suppressor proteins that inhibit CDK activity through direct interaction with CDKs or cyclin-CDK holoenzymes. According to their functional mechanisms and targets, CKIs are classified into two types: the INK4 protein family, including p16, p15, p18 and p19, which binds to CDK4/6 and disrupts the binding of cyclin D, and the CIP/KIP protein family, including p21, p27, and p57, which binds both the cyclin and the CDK of a complex and primarily inhibits the kinase activity of CDK1 and CDK2. Upon DNA damage, the checkpoint ATM (Ataxia telangiectasia mutated)-CHK2 (cell cycle checkpoint kinase 2)-p53 signaling pathway transcriptionally induces the expression of p21, leading to the inhibition of CDK2-cyclin E complex and consequent G1 phase arrest. Additionally, activated ATR (Ataxia telangiectasia and Rad3-related) and ATM phosphorylate and activate CHK1 to cause S phase arrest. ATM/ATR-CHK1 pathway also induces cell cycle arrest at G2 phase by phosphorylating and inactiveting the CDC25 family of phosphatases which, in turn, maintain CDK1 and CDK2 in the phosphorylated and inactive status. In addition, WEE1 controls G2/M arrest and mitotic entry by inhibiting CDK1 through phosphorylating it at the amino acids Tyr15 and Thr14. Therefore, the coordinated activation of cell cycle checkpoints is especially important to maintain the stability of the genome and cell survival by specifically interrupting each cell cycle stage to provide sufficient time for the repair of various types of DNA damage. Chemoresistance caused by aberrant activities of cell cycle proteins has become a great obstacle in cancer therapy, providing a rationale for designing novel synthetic inhibitors [2-4].
Selective autophagy
Autophagy is a physiological “self-eating” degradative process within lysosomes to maintain metabolic homeostasis and cell survival under metabolic pressures, such as starvation and energy deficiency, as well as pathological processes, such as neurodegenerative diseases, infection, and cancer [5,6].
Different types of autophagy
Several types of autophagy, including microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy, have been documented (Figure 2) [6]. Macroautophagy is thus far the best characterized type of autophagy with major morphological changes in vesicular compartments.
CMA
CMA is one of the lysosomal proteolysis pathways that selectively degrades soluble cytosolic proteins recognized by a specific chaperone in lysosomes (Figure 2A). CMA does not require vesicles or membrane invaginations for delivery of targeted substrates to the lysosome. In the cytosol, soluble protein containing a KFERQ sequence is recognized by HSPA8/HSC70 and cochaperones, binds to lysosome-associated membrane protein type 2A (LAMP2A) on the cytosolic side of the lysosome surface, and subsequently stimulates the formation of the oligomeric LAMP2A translocation complex, and finally translocates into the lysosomal lumen for degradation. It has been estimated that approximately 30% of cytosolic proteins contain this motif and may be degraded by CMA. Nevertheless, CMA is maximally activated in response to oxidative, nitrosative, or metabolic stress and serves to balance dysregulation.
Figure 1
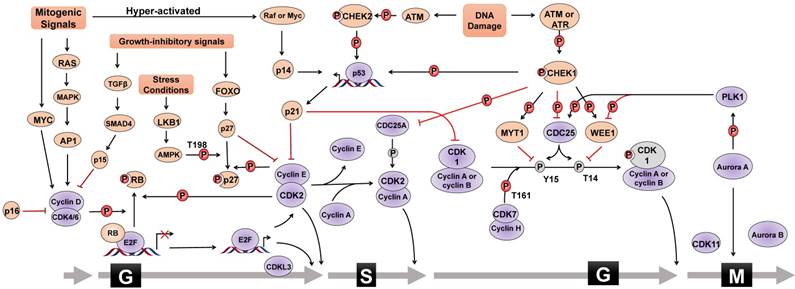
Cell cycle regulation. Cell cycle progression is regulated by the combinatory effect of several regulators including CDKs, cyclins and CKIs. Mitogenic signals activate cyclin-CDK complexes to promote the G1/S phase transition through phosphorylating (P) various substrates, such as RB, leading to the activation of E2F family of transcriptional factors. Cell cycle progression from S phase to G2 phase, as well as mitosis, is also under the control of the coordination of cyclin-CDK complexes, PLK1 and Aurora A kinases. In addition, CKIs is activated by different stimuli, such as DNA damage, growth inhibitory signals, or elevated mitogenic signals, to counteract the activity of cyclin-CDK complexes and to trigger cell cycle arrest. For instance, upon DNA damage, cell cycle halts at G1 phase via ATM-CHK2-p53 signal or S and G2 phase via ATM/ATR-CHK1 signal. Cell cycle regulators that promote cell cycle progress are denoted as Grey ovals whereas pink ovals represent regulators negatively modulating cell cycle transition. Grey circle with P means dephosphorylation. The form of “S323” means phosphorylation or dephosphorylation at amino acid residue Serine323. T=Threonine, S=Serine, Y=Tyrosine.
Microautophagy
Microautophagy (termed “endosomal microautophagy” in yeast cells), the least studied form of autophagy, engulfs cytoplasmic entities destined for degradation via direct membrane invagination at the surface of the lysosome or vacuole. The underlying molecular mechanism of the cargo delivery process remains largely unclear. Multiple endosomal-sorting complexes required for transport (ESCRT) systems have been shown to be critical for endosomal microautophagy, but not for microautophagy (Figure 2C). Additionally, endosomal microautophagy can also selectively degrade KFERQ-containing proteins recognized by HSC70. However, unlike CMA that exclusively relies on the function of LAMP2A, endosomal microautophagy requires the ESCRT systems for lysosome or endosome delivery.
Macroautophagy
Macroautophagy relies on the formation of autophagosomes (double-membraned vesicles) and their fusion with late endosomes or lysosomes to form amphisomes or autolysosomes, which are associated with the activity of two ubiquitin-like ATG conjugation systems (Figure 2B). Similar to microautophagy, cytoplasmic entities degraded by macroautophagy can occur either non-specifically or specifically via receptor-mediated recognition. A broad spectrum of regulators has been extensively investigated, among which are the particularly well-studied central element signals, the mechanistic target of rapamycin (mTOR) and the phosphatidylinositol 3-phosphate (PtdIns3P) Class III complex. As a sensor of cellular stress and growth factor signal, activated mTOR (Akt and MAPK signaling) suppresses macroautophagy by catalyzing the inactivating phosphorylation of the ULK1/ATG1 kinase complex that is critical for macroautophagy initiation and nucleation, whereas negative regulation of mTOR (AMPK and p53 signaling) promotes it. On the contrary, the PtdIns3P Class III complex, containing PIK3C3/VPS34, Beclin-1, p150, and Atg14L or UVRAG, promotes membrane trafficking and the appropriate localization of autophagic proteins to a pre-autophagosomal structure required for macroautophagy induction.
Each form of autophagy does not function in isolation or independently of other degradative systems, such as the ubiquitin proteasome system (UPS) [7,8]. For instance, constitutive activation of CMA is observed in cells deficient in macroautophagy or when the proteasome is chemically blocked [9,10].
Selective autophagy: selective removal of proteins and organelles
Autophagy can be divided into two types based on cargo specificity and delivery mechanism: non-selective autophagy, which involves disposing of cytoplasmic components in a relatively non-selective manner such as general microautophagy and macroautophagy, and selective autophagy, which requires the recognition of autophagy substrates by dedicated receptors [11,12]. CMA is the main form of selective autophagy which selectively degrades cytosolic proteins containing KFERQ-like motifs in a LAMP2A-dependent manner. In addition, specific autophagy receptor-mediated degradation of single proteins through macroautophagy and microautophagy are considered selective autophagy. Such selective microautophagy and macroautophagy are characterized by the enrichment of a precise autophagy substrate coupled to the requirement of specific LC3/GABARAP interacting region- (LIR) containing proteins or other scaffold proteins targeting for lysosomal degradation during the processes of lysosomal invagination and autophagosome formation. For instance, several LIR-containing proteins, such as SQSTM1/p62, NBR1, NDP52, and BNIP3, have been demonstrated to recognize and bind with specific substrates and tether them to the site of autophagosomal engulfment through their interaction with autophagosome-specific proteins, such as the members of the LC3 family [11,12]. Furthermore, the autophagy receptor-mediated selective removal of organelles under specific conditions, such as mitophagy (specific removal of mitochondria by micro- or macroautophagy) and pexophagy (macroautophagy-mediated preferential clearance of peroxisomes), can also be an alternative type of selective autophagy. In summary, selective autophagy seems to be instrumental in cleaning and remodeling specific metabolic regulators. This is significant because the dysfunction of these regulators leads to neoplasm development, neurodegeneration, or tumorigenesis.
Figure 2
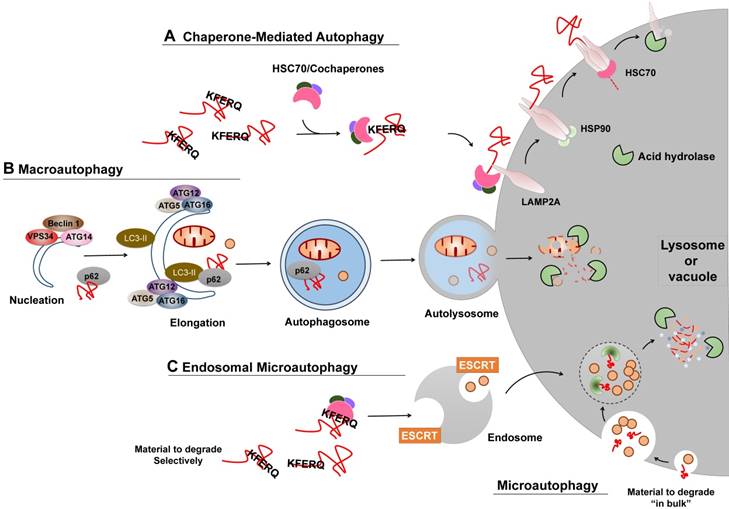
Different types of autophagy. (A) CMA. Proteins degraded by CMA are identified in the cytosol by HSC70/cochaperons complexes. Upon binding to the KFERQ motif, HSC70/cochaperons complexes bring them to the surface of lysosomes and bind to the receptor protein LAMP2A, which in turn promotes LAMP2A multimerization to form a translocation complex within the help of Heat shock protein 90 (Hsp90). Upon unfolding, substrate proteins cross the lysosomal membrane and undergo complete degradation. (B) Macroautophagy. Efficient macroautophagy involves the sequestration of cellular material, the formation of autophagosomes, their fusion with lysosomes, and lysosomal degradation of the autophagolysosomal content. An unidentified membrane source delivers lipid bi-layers for the formation of preautophagosomal membranes, the so-called 'nucleation process', which recruits the PtdIns3P Class III complex consisting of at least BECN1, VPS34, ATG14 or UVRAG. Besides, two ubiquitin-like ATG conjugation systems are activated to allow for autophagosomes formation and cytosolic cargo incorporation (either specifically via receptors or nonspecifically). For instance, the ATG12-ATG5:ATG16L1 complex conjugates phosphoethanolamine to LC3 to form lapidated LC3 (LC3-II), which may promote substrate uptake upon binding to different receptors, such as p62. After the completed autophagosomes have enclosed substrates, they can fuse with endosomes or lysosomes to form amphisomes or autolysosomes, the molecular machinery of which is shared with the endocytic pathway. Finally the inner content gets released into the lysosome/autolysosome and is being degraded by lysosomal hydrolases. (C) Microautophagy and Endosomal microautophagy. Endosomes engulf cytosolic materials within multivesicular bodies that contain other cytosolic components, and degrade these materials when fusion with lysosomes. The endosomal invagination relies on the ESCRT systems. Microautophagy can also selectivity degrade proteins bearing the KFERQ motif. Those KFERQ-containing cargos are targeted by HSC70 and unlike CMA, delivered to lysosome or endosome through the binding of HSC70 to endosomal membrane phosphatidylserines, independent of LAMP2A.
At present, there have been many remarkable insights into the refined function of autophagy, primarily macroautophagy, involved in the replication stress response and DNA repair pathways [13-15]. Although increasing evidence clearly demonstrates how specific cell cycle proteins, such as CDKs, CKIs, and checkpoints, regulate autophagy and vice visa, these studies have often led to divergent rather than unifying explanations due to the complexity of autophagy signaling network, the inconsistent functions between general autophagy and selective autophagy, and the different characteristics of autophagic substrates. Therefore, determining the mechanisms by which selective autophagy and the cell cycle are intertwined will provide novel insight into chemoresistance and improved therapeutic strategies for cancer.
Autophagy regulation by cell cycle regulators
The critical question of autophagy regulation and function in the orchestration of cell division remains unclear. Many studies suggest that autophagy and the cell cycle are coordinated and reciprocally regulated. However, different autophagic statuses during mitosis and interphase are shown, and contradictory findings are presented, possibly due to the results of varied experimental evaluations [16-22]. Additionally, the specificity of autophagy is not addressed, as investigators in most studies focus on the accumulation of LC3 puncta and the transformation from LC3-I to LC3-II, the typical biomarkers used so far to monitor macroautophagic responses. Whether microautophagy and CMA are also discriminately regulated in cell cycle progression remains to be clarified.
In general, the most common conclusion that can be drawn from several studies is that basal macroautophagy is detected in all cell cycle phases. Especially in response to DNA damage, cell cycle regulators may directly regulate macroautophagy to remove hazardous substances, to provide emergency substrates and energy, and to promote the mobilization and activity of DNA damage repair complexes before entering into mitosis. However, whether macroautophagy occurs at a higher or lower level during mitosis remains controversial. A few studies showed that macroautophagy is inhibited during mitosis in asynchronously proliferating human glioblastoma cells or in mitotic arrest cells synchronized by nocodazole treatment [17-19]. Macroautophagy may function as a protective mechanism to prevent unintended loss of organelles and chromosomes during mitosis. There is also evidence indicating that macroautophagy actively persists during mitosis [20-22]. Macroautophagy may play some important roles in mitotic chromosome congression and outer kinetochore assembly, and macroautophagy deficiency causes centrosome amplification, leading to abnormal cell division and genome instability. Further work is required to elucidate the underlying mechanism by which macroautophagy is activated during mitosis.
Interestingly, a growing body of evidence demonstrates that Aurora kinase and the majority of CDKs inhibit macroautophagy to keep its level under a threshold in normal condition, whereas CKIs, activated by DNA damage and other growth-inhibitory signals, activate macroautophagy (Table 1). It seems that cooperation between CDKs and CKIs maintains a low basal macroautophagic activity during cell cycle progression and provides a way to meet fitness with fast macroautophagic responsiveness, adapting quickly to changes in the situational environment.
Macroautophagy regulation in cell cycle progression
Increasing evidence suggests that physiological CDK inhibition is a critical macroautophagy regulation system and those cell cycle regulators, including CDKs, cyclins, Aurora kinases, and PLKs, directly link the macroautophagic responses or its associated pathway to cell stress.
Macroautophagy regulated by G0/G1 to S transition proteins
Recently, CDK4 and CDK6 have been implicated in macroautophagy inhibition, and inhibiting CDK4/6 by chemical inhibitors or siRNA induces macroautophagy in multiple cancer cells [23-27]. It has been shown that CDK4/6-cyclin D1 complex is able to phosphorylate LKB1 at Ser325, leading to AMPK inactivation and macroautophagy inhibition [26,27]. Additionally, results of a chemoproteomics assay show that CDK4/6 inhibitor palbociclib engages several lipid kinases that are critical for PtdIns3P generation and subsequent autophagososome formation, modulates PI3K/ AKT-AMPK signaling, and interacts with several subunits of the PtdIns3P Class III complex, such as Beclin-1, UVRAG and VPS34, all of which indicate the involvement of AMPK and the Beclin-1 signaling axis in CDK4/ 6-mediated macroautophagy inhibition [25]. CDK2 also has an inhibitory effect on macroautophagy. Knockdown of CDK2 alone induces a modest increase in macroautophagy [23], and CDK2 may inhibit macroautophagy by activating mTOR directly [28] or indirectly through AKT activation [29]. The CDK2- cyclin A2 complex can phosphorylate C-terminal AKT at Ser477 and Thr479, leading to AKT hyperactivation and aberrant cell cycle progression [29]. Furthermore, cyclin-dependent kinase-like 3 (CDKL3), a CDK3 homolog that is involved in G1 to S phase transition, has also been demonstrated to inhibit macroautophagy, probably by stimulating mTOR activity through its MAPK-like kinase activity [30].
CDK5 is predominantly expressed in nondividing neurons to promote microtubule depolymerizetion and mitosis and has not been considered as a potential cell-cycle regulator [31]. Most recently, CDK5 has been demonstrated to genetically interact with and phosphorylate acinus at Ser437 [32]. Acinus is primarily a nuclear protein that promotes starvation-independent basal macroautophagy to suppress neurodegeneration. However, in an Alzheimer's disease model, CDK5 is hyperactivated by Amyloid β1-42 (Aβ) to modulate the RB/E2F pathway to directly affect the function of tumor suppressors and transcription factors, resulting in G1 phase re-entry, aberrant cell cycle progression, and ultimately neurodegeneration and oncogenesis [33]. Aβ-mediated activation of CDK5 triggers aberrant activation of macroautophagy via phosphorylating RB and subsequently promoting E2F-mediated transcriptionally enhanced expression of macroautophagy genes [34,35]. In addition, CDK5 can induce macroautophagy through phosphorylating endophilin B1 (EndoB1, a member of the endophilin family that was initially implicated in the regulation of mitochondrial morphology and apoptosis) at Thr145, promoting its dimerization and recruiting the UVRAG-Beclin 1 complex to initiate macroautophagy [36]. Notably, CDK5-mediated phosphorylation of EndoB1 is selectively required for stressor-induced macroautophagy, it but has negligible effect on basal macroautophagy, indicating that different machinery may control basal and induced macroautophagy under pathological conditions [37,38].
Macroautophagy regulated by G2 to M transition and mitosis proteins
Controversial results regarding the activity of macroautophagy during mitosis exist [17-22]. Additionally, some mitotic cell cycle regulators exhibit opposing effects on macroautophagy regulation, creating more uncertainty.
CDKs-mediated regulation
One noticeable example that suppresses macroautophagy during the mitotic stage of the cell cycle is the mitotic kinase CDK1-mediated regulation [17,18]. CDK1 can directly phosphorylate VPS34 at Thr159 in their common substrate recognition motif, thereby interrupting the interaction between VPS34 and Beclin-1 and resulting in decreased macroautophagy in mitotic cells [18].
Table 1
Macroautophagy regulated by cell cycle proteins
| Cell-cycle proteins | Autophagy Regulation | Possible Mechanism | Ref |
|---|---|---|---|
| G0/G1 to S transition | |||
| CDK4/6-Cyclin D1 | Inhibition | Affect lipid kinases and PtdIns3P production; Modulate the PI3K/AKT-AMPK signaling; Interact with subunits of the PtdIns3P class III complexes | 24,25 |
| Inhibition | Phosphorylate LKB1 at Ser325, leading to AMPK inactivation | 26,27 | |
| CDK2 | Inhibition | Activate mTOR directly or indirectly through phosphorylating AKT at Ser477 and Thr479 | 28,29 |
| CDKL3 | Inhibition | Stimulate mTOR activity through its MAPKs-liked kinase activity | 30 |
| CDK5 | Induction | Phosphorylate Acinus at Ser437 | 32 |
| Promote E2F-mediated transcriptionally activation of autophagy genes; | 33- 35 | ||
| Phosphorylate EndoB1 at Thr145 and subsequent promote the recruitment of the PtdIns3P class III complexes | 36-38 | ||
| G2 to M transition | |||
| CDK1 | Inhibition | Phosphorylate VPS34 at Thr159, resulting in FBXL20-mediated degradation and impaired interaction between Beclin-1 and VPS34 | 18,39 |
| CDK5 | Inhibition | Phosphorylate VPS34 at Thr668 to inhibit its lipid enzymatic activity and decrease PtdIns3P production | 18 |
| CDK11 | Inhibition | Affect autophagic flux | 40 |
| Cyclin H-CDK7 | Induction | Promote autophagy genes expression and assembly | 41 |
| Mitosis | |||
| Aurora A | Inhibition | Activate mTOR signaling | 43- 48 |
| PLK1 | Induction | Phosphorylate the mTOR component RPTOR/RAPTOR | 52,53 |
| Inhibition | Activate mTOR signaling | 51 | |
| CKIs | |||
| p16 | Induction | - | 54 |
| p14 | Induction | Release Beclin 1 from the inhibitory binding of Bcl-XL | 55 |
| p27 | Induction | Promote the expression of autophagy-related gene in p53-CD95-activation dependent manner | 56- 58 |
| p21 | Induction | - | 54,60 |
| Inhibition | - | 61 | |
| p53 (nuclear) | Induction | Transcriptional activating members of the autophagy-lysosome core machinery and the AMPK-mTOR signaling; activating genes involved in releasing Beclin 1 from the inhibitory interactions with Bcl-2 and Bcl-XL | 62,63 |
| p53 (cytoplasmic) | Inhibition | Modulate the AMPK/mTOR pathway | 64- 66 |
Importantly, phosphorylation of Thr159 also triggers the ubiquitination and degradation of VPS34 by FBXL20, an F-box protein controlled by p53-dependent transcription [39]. In addition to CDK1, CDK5 can also phosphorylate VPS34 at Thr159 and Thr668 [18]. Such CDK5-mediated Thr668 phosphorylation independently inhibits VPS34 lipid enzymatic activity and thus decreases PtdIns3P production and autophagososome formation. Furthermore, CDK11 plays a more complex role in regulating macroautophagy. CDK11 function is linked to the regulation of mitosis and is similar to that of CDK1. CDK11 is required for successful macroautophagy, and loss of CDK11 results in increased basal macroautophagy initiation and reduced autophagosome turnover [40]. Finally, the yeast kinases Ccl1-Kin28 complex, equivalent to cyclin H-CDK7 complex in mammalian which phosphorylates and activates CDK1 at Thr161, is newly identified as an initiator of macroautophagy through maintaining the expression of Atg29 and Atg31 and the assembly of Atg1 complex [41]. Therefore, it will be interesting to examine whether mammalian cyclin H-CDK7 exerts a similar effect on macroautophagy induction.
Aurora A-mediated inhibition
Aurora A mainly regulates centrosome maturation and separation as well as bipolar spindle assembly and orientation [42]. Other than its functions in mitosis, Aurora A can regulate macroautophagy through modulating the mTOR signaling pathway, as evident by that inhibition of Aurora A by inhibitor Alisertib induces macroautophagy in various human cancer cells to cause cell death or higher chemosensitivity [43-46]. Aurora A enhances the activity of MAPK, which subsequently promotes the dissociation of TSC1/2 complex and increases the ability of RheB to upregulate mTOR activation [47,48]. Such Aurora A-mediated macroautophagy inhibition may also act as an explanation for the low autophagic activity in mitosis.
PLK1-mediated modulation
Polo-like kinase 1 (PLK1), the most frequently investigated PLK protein, can phosphorylate and activate CDC25C to promote the activation of CDK1-Cyclin B complex and the initiation of G2/M transition [49]. Interestingly, several studies exhibit the controversial effect of PLK1 on macroautophagy modulation. For instance, PLK1 is highly expressed in cancer cells and pharmacological inhibition of PLK1 by specific inhibitors RO3280 or BI2536 induces macroautophagy partly via mTOR pathway dephosphorylation [50,51]. Additionally, long-term treatment with BI2536 leads to mitotic arrest of prostate adenocarcinoma LNCaP cells, correlating with macroautophagy activation, which indicates a macroautophagy inhibition function of PLK1 [50]. In contrast, through quantitative proteomic analysis, PLK1 has been confirmed to induce macroautophagy in quiescence and interphase cells [52,53]. PLK1 resides with mTOR at lysosomes and decreases its activity by directly interacting with and phosphorylating the mTOR component RPTOR/RAPTOR [52]. Overexpression of active PLK1 decreases lysosomal association of the PLK1-mTOR complexes, whereas PLK1 inhibition promotes lysosomal localization of mTOR. One explanation for those controversial results may be that the methodology measuring autophagic activity or retarding cells in mitosis, as well as the length of cell cycle arrest and cell types, may add variations to the complex functions of PLK1 in mitotic macroautophagy regulation.
Macroautophagy regulation by cell cycle checkpoints
In response to different cell cycle stresses, several cell cycle checkpoints exist and two major signals, ATM-CHEK2 and ATR-CHEK1, activate p53 and different CKIs, which in turn modulate the function of specific CDK/cyclin complex to control the on-off switch for cell death via modulating macroautophagy.
Macroautophagy regulated by CKIs
In contrast to CDKs, CKIs have been demonstrated to stimulate macroautophagy in CDK-dependent or -independent ways, resulting in promotion of tumor growth. For instance, overexpression of p16, an inhibitor of the CDK4/6-cyclin D complex, is able to induce macroautophagy at baseline or after starvation [54]. p14, an activator of p53-mediated G1 cell cycle perturbation, can bind to Bcl-XL and release Beclin 1 from the inhibitory binding of Bcl-XL, leading to increased macroautophagy [55]. Another example of enhanced macroautophagy by CKIs is provided by p27, which restricts the cell cycle at G1 phase through binding to and inhibiting CDK2-cyclin E and CDK4-cyclin D complex. Beyond braking cell cycle, p27 controls the balance between macroautophagy and cell death upon different stressful stimulations [23,56-58]. Both low energy status and amino acid starvation stabilize p27 through the energy-sensing LKB1-AMPK pathway-mediated phosphorylation at Thr198, leading to the induction of macroautophagy that senses nutrient concentration and promotes bioenergetics rather than cell death [23]. Consistently, ectopic expression of p27 is sufficient to induce macroautophagy and the evasion of apoptosis. This macroautophagy-inducing effect of p27 partially relies on its CDK inhibitory function, as depletion of cyclin-binding region or CDK2 and CDK4 affecting p27-induced effects on macroautophagy [23]. In addition, p27 can promote the expression of macroautophagy-related gene in a p53-CD95-activation-dependent manner [59]. Furthermore, there is increasingly evidence of the connection between p21 and macroautophagy [54,60,61]. Regarding the modulation of macroautophagy, different results have emerged, and it seems that p21 promotes cell death upon death stimuli via inducing macroautophagy in cancer cells or inhibiting macroautophagy in normal cells, respectively. For instance, overexpressing p21 is sufficient to induce macroautophagy and senescence in breast cancer cells [54]. Quinacrine, an antimalarial agent, presents anticancer activity and induces p21-dependent macroautophagy in colon cancer HCT-116 cells [60]. In p21-/- HCT-116 cells, macroautophagy, as well as apoptosis, is decreased when compared to the parental cells, implying that p21 activates macroautophagy to accelerate cell death. In contrast, p21 exerts an inhibitory effect on macroautophagy in normal cells. Ceramide is an apoptosis inducer and induces apoptosis in p21+/+ mouse embryonic fibroblasts (MEFs) [61]. However, interference of p21 expression with siRNA initiates macroautophagy and hinders the occurrence of apoptosis. In ceramide-treated p21-/- MEFs, there is a high macroautophagic activity and low apoptosis, while exogenous expression of p21 decreases the number of macroautophagic cells and enhances apoptosis, suggesting that p21 inhibits macroautophagy to switch macroautophagy to apoptosis under death stimuli.
p53-mediated Regulation
The particularly well-studied connection between macroautophagy and stress-induced cell cycle responses is the function of p53, one of the most extensively characterized tumor suppressor proteins. Under various stimuli, cytoplasmic p53 undergoes rapid post-translational modifications that allow for the evasion of Mdm2-mediated ubiquitination and proteasomal degradation and translocates into the nucleus to initiate appropriate cellular responses, such as apoptosis, macroautophagy, or cell cycle arrest. Upon DNA damage, nuclear p53 induces macroautophagy by transactivating a wide range of macroautophagy-related genes. Those genes can be divided into several clusters based on function [reviewed in 62,63], including activating the AMPK-mTOR signaling (e.g., AMPK subunits β1 and β2, Sestrin 1/2, TSC2, Ddit4, PTEN), releasing Beclin 1 from the inhibitory interactions with Bcl-2 and Bcl-XL (e.g., DRAM, DAPK1), and promoting the expression of members of the macroautophagy-lysosome core machinery (e.g., ULK1, ULK2, Atg7). It is reasonable that induction of macroautophagy by p53 may assist the evasion of tumor cells from DNA damage-mediated cell death, probably via removing pro-apoptotic mitochondria and reducing oxidative phosphorylation. Surprisingly, the cytoplasmic pool of p53 executes an opposite role that suppresses macroautophagy, presumably through modulating the AMPK/mTOR pathway [64]. It is important to note that this cytoplasmic p53-mediated macroautophagy induction is a cell cycle-dependent phenomenon that occurs only at the G1 and S phases [65]. In addition, nutrient deprivation and mTOR inhibition induces Mdm2-dependent proteasomal degradation of cytoplasmic p53, indicating that efficient macroautophagy activation requires degradation of the cytoplasmic, non-nuclear pool of p53 [64]. Therefore, further work is needed to understand how the cell determines whether to induce macroautophagy with nuclear p53 or inhibit macroautophagy with cytoplasmic p53 and how this process is regulated.
Because cytoplasmic p53 induces mitochondrial membrane permeabilization, apoptosis, and senescence, cancers may acquire resistance through inactivating or functionally down-regulating p53 (frequently inactivated by mutations). It is reasonable that inactivation of the p53 system in the cytoplasm may result in an enhanced constitutive baseline level of macroautophagy to provide advantage for cancer cells bearing metabolic stress caused by hypoxia and nutrition depletion in tumor microenvironment. Unexpectedly, many tumors are found to have high levels of mutated p53, and interestingly, one third of those p53 mutants in tumors exclusively localize to the cytoplasm to repress macroautophagy, whereas p53 mutants fail to inhibit macroautophagy when the protein accumulates in the nucleus [66]. Therefore, macroautophagy inhibition caused by some cancer-associated p53 variants might be “corrected” by additional mutations or epigenetic changes, for instance, activating mutations in H-Ras or K-Ras, to elicit excessive macroautophagy.
Taken together, any perturbation of p53 system, either activation or inhibition by different stimuli, may modulate macroautophagy to fuel the survival of stressed cells.
Selective autophagy regulates cell cycle
In response to different stimuli, UPS-mediated proteolysis modulates cell cycle progression, DNA repair machinery, and cellular death rates [67]. Selective autophagy, including CMA and selective macroautophagy, has been confirmed to function both separately or in conjunction with the UPS to regulate cell cycle progression by removing key cell cycle regulators and other signaling adaptors (Table 2).
Selective macroautophagy-mediated degradation of cell cycle proteins
Unlike the non-specific uptake of cytosolic entities associated with starvation-mediated macroautophagy, selective macroautophagy identifies and clears autophagic substrates that are recognized and tethered to the nascent autophagic membrane by specific autophagy receptors, such as p62 and NBR1. Accumulating evidence illustrates that selective macroautophagy acts as an intrinsic and alternative antagonism against uncontrolled CDK-cyclin complex activity and aberrant cell cycle regulation (Figure 3). Although there are studies demonstrating that other proteins, including cyclin E, CDC25 and CDK2, are degraded in macroautophagy-dependent way [68,69], it is not yet known whether their degradation are non-selective or selective.
Figure 3
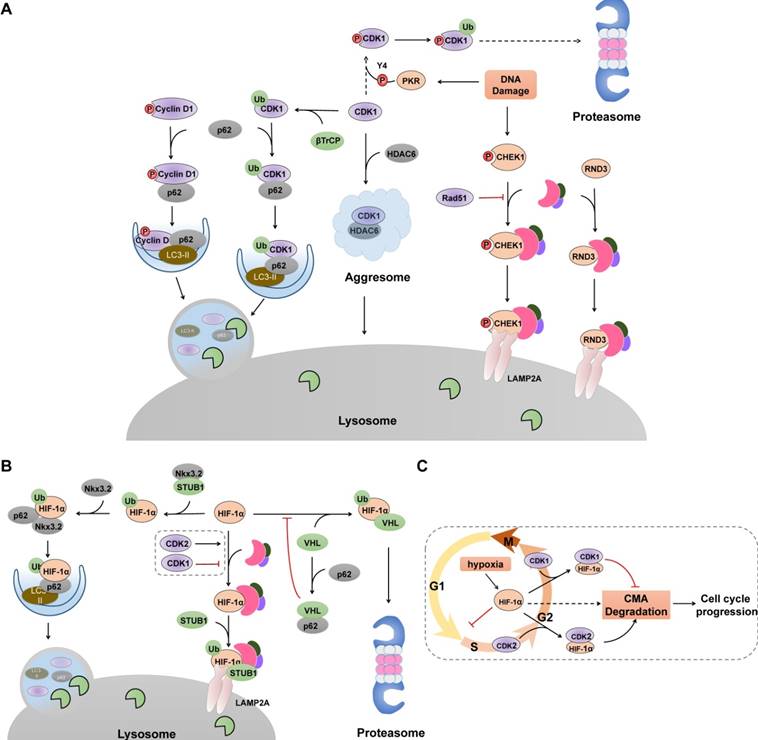
Selective Autophagy degrades cell cycle proteins. (A) Selective autophagy, cooperated with UPS, acts as an on-off switch of cell cycle progression via controlling the stability of several cell cycle regulators. Selective macroautophagy receptor p62 selectively recognizes CDK1, cyclin D1 and p27 (not shown), recruits them into autophagosome through its interaction with LC3. In contrast, CHK1 and RND3 are degraded via CMA pathway. Green ovals denote ubiquitin ligases. (B) Hif-1α undergoes lysosomal degradation through its interaction with HSC70, LAMP2 and STUB1. p62-mediated recognition also promotes Hif-1α degradation in selective macroautophagy. (C) Cell cycle-related degradation of Hif-1α. Hypoxia prevents the VHL-mediated proteasomal degradation of Hif-1α, resulting elevated levels of Hif-1α and subsequent cell cycle arrest at G1/S phase. CDK2 physically interacts with Hif-1α and promotes its degradation whereas CDK1 protects HIF-1α from lysosomal degradation through unclarified mechanism.
Table 2
Selective autophagy-mediated degradation of cell cycle proteins
| Cell-cycle proteins | Autophagy type | Autophagy Receptor | Physiological Relevance | Ref |
|---|---|---|---|---|
| Cell cycle progression | ||||
| Cyclin D1 | Selective Macroautophagy | SQSTM1, LC3 | Cell cycle arrest and reduced tumorigenesis | 77-79 |
| CDK1 | Selective Macroautophagy | SQSTM1, HDAC6 | Tumor cell death | 80,81 |
| Cyclin A2 | Selective Macroautophagy | SQSTM1 | Prevent G2/M arrest, Cell cycle progression | 22 |
| Cell division | ||||
| RhoA | Selective Macroautophagy | p62 | Cytokinesis failure and aneuploidy | 112 |
| KIF23 | Selective Macroautophagy | p62, NBR1 | Midbody ring derivatives degradation | 115 |
| CEP55 | Selective Macroautophagy | NBR1 | Midbody ring derivatives degradation | 116 |
| CHMP4B | Selective Macroautophagy | p62, LC3 | Micronuclei elimination | 119 |
| Cell cycle checkpoint | ||||
| CHEK1 | CMA | HSC70, LAMP2A | Maintain genome integrity and cell cycle progression | 132 |
| p27 | Selective Macroautophagy | SQSTM1 | Maintain T cell homeostasis and function | 87 |
| RND3 | CMA | HSC70, LAMP2A | Tumor cell proliferation | 108 |
| DNA Damage Repair | ||||
| RNF168 | Selective Macroautophagy | p62 | Impair DSB repair | 127 |
| USP14 | Selective Macroautophagy | p62 | Suppress RNF168-mediated DSB repair | 130 |
| HP1α | Selective Macroautophagy | p62 | Promote homologous recombination DSB repair process | 131 |
| Senescence | ||||
| GATA4 | Selective Macroautophagy | p62 | Inhibit SASP program and cellular senescence | 123 |
| Lamin B1 | Selective Macroautophagy | LC3 | Drives senescence upon oncogenic and genotoxic insults | 124 |
| ∆133p53α | Selective Macroautophagy | p62, LC3 | Promote activate p53-mediated cellular senescence | 125 |
| Hypoxia | ||||
| Hif-1α | CMA | HSC70, LAMP2A | cell cycle progression and cell proliferation | 97-99 |
Degradation of Cyclin D1
Cyclin D1 has a relevant role in the initiation and progression of cancer, and high cyclin D1 expression correlates with poor overall survival rate [70]. It is not surprising that macroautophagic activity is inversely correlated with cyclin D1 expression, as cyclin D1 promotes macroautophagy inhibition [26,27]. Both the UPS and autophagy can degrade cyclin D1. For instance, cyclin D1 undergoes Skp1-Cul1-F box (SCF) E3 ligase-mediated proteasomal degradation in normal actively cycling cells [71-73], or in cancer cells treated with kinase inhibitors in an ERK1/2- dependent or GSK3β-dependent phosphorylation of Thr286 [74-76]. However, in some specific types of cancers treated with radiotherapy or DNA-damage agents, selective macroautophagy is activated to degrade cyclin D1 to inhibit tumorigenesis (Figure 3A). Cyclin D1 co-localizes with Beclin 1 and after ubiquitination, is selectively degraded in autolysosome through its interaction with autophagy receptor p62 [77-79]. Besides, ubiquitination of cyclin D1 requires GSK3β-mediated phosphorylation at Thr286 [79]. Those results indicate that cancer cells preferentially activate selective autophagy to control cyclin D1 to adjust cell cycle progression under DNA damage.
Degradation of CDK1-Cyclin A2
Similar to cyclin D1, several studies have reported that after DNA damage, CDK1 is ubiquitinated by E3 ligase SCF (βTrCP) and subsequently interacts directly with p62 targeting for lysosomal degradation [80]. When the autophagosomal pathway is overloaded (for example, when the proteasome is blocked), CDK1 interacts with HDAC6 and accumulates into aggresomes for later degradation [81]. However, dsRNA-activated protein kinase (PKR) has also been demonstrated to phosphorylate CDK1 at Tyr4 and trigger its subsequent polyubiquitination and proteasomal degradation after doxorubicin treatment [82]. One possible explanation is that such PKR-mediated phosphorylation may induce some types of conformational changes in CDK1 that originally allow for E3 ubiquitin ligase, such as βTrCP, to access the ubiquitination sites on CDK1 [83]. In light of this, different tumor types, depending on their pathogenic spectrum mutations, may display different sensitivities to CDK1 degradation and whether PKR activation by p53 determines the degradation pathway of CDK1 in the context of genotoxic stress. Moreover, cyclin A2, one partner of CDK1 that accumulates from late G1 to M phase, is degraded by the proteasome system before metaphase, whereas selective macroautophagy acts as a complementary mechanism to prevent the accumulation of cyclin A2 at the end of mitosis and the G2/M arrest [22]. The two pathways function in parallel and macroautophagy inhibition induces a mitotic delay that is a consequence of the partial stabilization of cyclin A2.
Degradation of p27
Numerous studies have determined that p27 is degraded by the proteasome system upon cell cycle entry through CDK2-cyclin E-mediated phosphorylation and ubiquitin ligase SCF-SKP2-mediated recognition [84,85]. Another study indicates that p27 is downregulated through endolysosomal degradation, mediated by an interaction with endosomal vesicular-trafficking regulator SNX6 [86], indicating an alternative degradation strategy usurped by cells to respond to different stimuli and modulate cell cycle progression. In support of this hypothesis, a recent study identifies that in both native and activated T lymphocytes, the degradation of p27 relies on macroautophagy [87], which is consistent with previous reports that macroautophagy-deficient conventional T and invariant NKT lymphocytes fail to proliferate efficiently [88-90]. p27 forms large polymers and is ubiquitinated and physiologically associated with p62 targeting for lysosomes [87]. Further work is needed to determine whether other cell cycle regulators are degraded by selective macroautophagy to establish an elaborate regulating network linking aberrant cell cycle control system to autophagy dysfunction in T lymphocytes.
CMA-mediated degradation of cell cycle proteins
CMA exerts an protumorigenic effect in several tumors though selectively degrading tumor suppressors, pro-apoptotic and anti-proliferation proteins [91,92]. Growing evidence suggests CMA is a mechanism for maintaining genome integrity and forms a complex regulatory network to balance cell cycle progression and arrest. Consequently, blocking CMA may be a broadly useful anti-oncogenic therapy.
Degradation of HIF-1α
Cell cycle arrest and proliferation inhibition are fundamental adaptive responses to hypoxia and are partially induced through the actions of HIF-1α (hypoxia inducible factor 1) [93]. HIF-1α can interrupt cell cycle at G1 phase and inhibit DNA replication by binding to the minichromosome maintenance complex components, promoting their loading on chromatin but blocking their phosphorylation and activation [94]. Under hypoxic conditions, HIF-1α degradation in proteasomes is inhibited, and elevated HIF-1α levels cause cell cycle arrest. Therefore, cancer cells must be able to mobilize other degradation systems, such as CMA, to selectively degrade HIF-1α for DNA replication to proceed during hypoxia. Indeed, both the UPS and CMA are utilized by cells to degrade HIF-1α [95-98] (Figure 3B). Upon proline hydroxylation, HIF-1α is ubiquitinated and subsequent degraded in proteasomes by the von Hippel-Lindau (VHL) ubiquitin ligase complex in an oxygen-dependent manner [95,96], whereas in both normoxic and hypoxic conditions, CMA components HSC70 and LAMP2A interact with HIF-1α and promote the recruitment of an E3 ubiquitin ligase STUB1/CHIP for lysosomal translocation and degradation [97-99]. Additionally, selective macroautophagy has been demonstrated to degrade HIF-1α via p62-mediated recognition and binding to the STUB1-HIF-1α complex, which can be further enhanced by chondrogenic factor Nkx3.2 [100]. Furthermore, p62 can interact directly with the VHL E3 ligase complex and block the VHL-mediated ubiquitylation and proteasomal degradation of HIF-1α [101], indicating an alternative crosstalk that switches HIF-1α degradation between these two systems. Recently, CDK1 and CDK2 have been demonstrated to regulate CMA-mediated HIF-1α degradation in an opposite manner [99] (Figure 3C). Prior to M phase, CDK1 is activated with its partner cyclin B, which protects HIF-1α from degradation and enhances its transcriptional activity. By contrast, CDK2-cyclin A or CDK2-cyclin E complex promotes HIF-1α degradation to allow cell cycle progression from G1 to S phase transition. The mechanisms by which CMA recognizes the different interactions between CDK1 and CDK2 and how HIF-1α is targeted for lysosomal degradation are unclear. Considering that phosphorylation of HSC70 affects its binding affinity to its substrate [102], and CDK1 has been described to promote the phosphorylation of HSC70 to control the abundance of G1 cyclins and cell cycle progression [103], it is plausible that CDK1 may phosphorylate HSC70 and impair its interaction with HIF-1α, thereby impairing CMA-mediated degradation.
Degradation of RND3
RND3/RhoE, one member of the RHO family, can inhibit cell proliferation and cause cell cycle arrest at G1 phase through the increase of PTEN and p27 as well as through the decrease of activated AKT and cyclin D1 [104-106]. Thus, the rapid proliferation of several cancers depends on its constant degradation. Beyond the proteasomal degradation of RND3 by SKP2 to orchestrate cellular proliferation [107], CMA is overactivated in cancer cells and acts as an alternative pathway for RND3 degradation. CMA components HSC70 and LAMP2A interact with RND3, which in turn, are engulfed and degraded by lysosomes [108]. Importantly, such boosted CMA-mediated quality control of RND3 is only detectable in cancer cells whereas in normal cells, the CMA-RND3-proliferation axis is absent or very weak.
Selective autophagy in cell division
The UPS, mainly via anaphase-promoting complex/ cyclosome-mediated proteasomal degradation of mitotic regulators, has been considered a major control system for sequential and successful cell division. However, increasing evidence suggests selective autophagy has a supplemental role in regulating the progression of cell division and maintenance of the genome. In addition to the previously discussed intricate relationships between selective autophagy and specific cell cycle proteins, specialized contributions in mitosis and cytokinesis have also emerged.
Selective macroautophagy regulates cytokinesis
Cytokinesis is a process in which the cytoplasm of a single eukaryotic cell divides into two daughter cells after chromosome segregation. Although many studies have shown the failure of cytokinesis following knock-down of several PtdIns3P Class III complex genes important for macroautophagy, such as Beclin 1, VPS34 and UVRAG [109-111], the underlying causality remains undefined and also seems to be macroautophagy-independent. However, p62-mediated selective macroautophagy has been clearly shown to orchestrate cytokinesis via restricting activation of RhoA, a small GTPase that regulates cell shape and completion of cytokinesis via F-actin reticulation at the midbody [112]. p62 acts as the molecular adapter to recruit ubiquitinated active RhoA to autolysosomes for degradation. Consequently, defects in macroautophagy (knockdown or gene deletion of macroautophagy core genes, such as Atg5, Atg7 and Sqstm1, or the usage of inhibitors) drives cytokinesis failure and aneuploidy. Therefore, selective autophagy appears to function as an integral part of the checkpoint machinery at the cytokinesis midzone by mediating RhoA and Cyclin A2 (discussed above) degradation in late mitosis.
Selective macroautophagy removes midbody ring
The midbody ring is the central region of the midbody characterized by a phase-dense circular structure where several cytokinesis-coupled events converge, such as cell cycle regulators degradation and cytoskeleton reorganization. After cytokinesis, the midbody ring randomly enters into one daughter cell and is degraded in proteasomes, or eliminated by extrusion to the extracellular space [113,114]. The remnant midbody ring derivatives can also be degraded by selective autophagy [115-117]. Autophagy factors, such as p62 and Atg8, associate with the midbody during abscission and are moved together into the daughter cell. Notably, upon cytokinesis, a complex consist of ubiquitin E3 ligase TRAF6, p62, NBR1 and the interacting adaptor protein WDFY3, transports to the midbody region. Several midbody ring proteins, including KIF23, are then ubiquitinated by TRAF6, leading to their recognition by p62 and NBR1. After completion of cytokinesis, core autophagic machinery is recruited by p62 and NBR1 to form an autophagic membrane around the midbody ring derivatives and contribute to its subsequent degradation [115]. NBR1 can also bind with the midbody centrosomal protein 55 (CEP55) to promote the delivery of the midbody ring to the lysosome for degradation [116].
Selective autophagy in micronuclei elimination
The micronucleus is a small nucleus that forms when some chromosomes or chromosome fragments are not properly incorporated into the newly formed daughter cell nuclei during cell division. Since micronuclei are usually destroyed by cytoplasmic nucleases, they are likely removed through selective autophagy [118]. Part of the micronuclei colocalizes with autophagy proteins, such as LC3, Ulk1, LAMP2, and p62, as well as charged multivesicular body protein 4B (CHMP4B), a member of the ESCRT machinery, indicating the involvement of selective autophagy in micronuclei clearance [118,119]. In addition, deprivation of LC3 and p62 increases the frequency of micronuclei [120]. p62 interacts with RAD51 and PARP-1, two proteins involved in DNA repair and genomic stability which accumulate at micronuclei, thus demonstrating a connection between the autophagic pathway and the nuclear stability. Further studies are needed to resolve the detailed mechanisms and determine the extent to which selective autophagy contributes to micronuclei elimination versus other mechanisms.
Selective autophagy regulates cell cycle stress response
Radiation and genotoxic drugs, such as alkylating agents, mainly cause DNA fracture and interfere with DNA replication and transcription to inhibit tumor cell division. This usually triggers the DNA damage repair response (DDR), a complicated signaling network integrating cell cycle checkpoints, DNA damage repair, and DNA damage tolerance pathways to promote orderly completion of genome duplication and cell cycle progression. Damaged DNA is repaired due to the involvement of DDR sensors, such as the BRCA1 complex and the Mre11-Rad50-Nbs1 (MRN) complex, and the activation of checkpoint pathways ATM-CHK2-p53 and ATR-CHK1 [121,122]. Persistent DNA damage also triggers cellular senescence, a stress-activated genetic program that permanently prevents cells from further proliferation and is an irreversible state of cell-cycle arrest. Senescence also leads to secretion of diverse cytokines, chemokines, various growth factors and proteases/inhibitors, which constitute the senescence-associated secretory phenotype (SASP). Notably, growing evidence also highlights the importance of selective autophagy in modulating DNA damage repair and senescence [123-126].
Selective autophagy promotes DNA damage repair
General autophagy has been demonstrated to promote the DDR process, deficiency of which always results in genomic instability and aneuploidy [126]. In addition, selective autophagy may promote efficient DNA double-strand breaks (DSBs) repair by regulating the levels of nuclear components of homologous recombination (HR) or non-homologous end-joining (NHEJ) (Figure 4A). For instance, following DNA damage, E3 ligase RNF168 is recruited to the sites of DSBs, where it facilitates the ubiquitination of histone H2A and the subsequent recruitment of DNA repair proteins. However, in autophagy-defective cells, RNF168 binds to nuclear-accumulated p62 and its activity is inhibited by such interaction, leading to the failure of recruitment of DNA repair proteins BRCA1, RAP80, and RAD51 [127]. In addition, nuclear p62 interacts with FLNA (filamin A) and RAD51, two essential regulators of HR, and promotes their proteasomal degradation within the nucleus, resulting in impaired DSB repair [128]. Furthermore, p62 recruits nuclear polyubiquitinated proteins that are involved in multiple DDR, such as BLM/WRN DNA helicases, the Mre11 complex, or TopBP1, to promyelocytic leukemia bodies where they aid in the proteasomal degradation in the nucleus [129]. Alternatively, USP14, a deubiquitnase that negatively regulates levels of RNF168 and suppresses RNF168-dependent ubiquitination signaling, is selectively recognized by p62 targeting for lysosomal degradation [130]. When autophagy function is compromised, accumulated p62 aggregates sequester USP14 and promote its translocation to DSB sites, thus impairing DDR. Furthermore, DSB repair in heterochromatin requires moving outside heterochromatin protein 1 (HP1) to complete the recombination. During the efficient HR DSB repair process, HP1α physically interacts with and is ubiquitinated at residue K154 by RAD6, an E2 ubiquitin-conjugating enzyme important for the repair of UV-induced DNA damage. It is ultimately degraded by p62-mediated selective autophagy, leading to a loosen chromatin structure that facilitates the formation of RAD51 nucleoprotein filaments and successful HR [131].
In addition, selective autophagy, particularly CMA, participates in the cytosolic quality control of CHK1 [132-134] (Figure 3A). Upon DNA damage, CHK1 is activated by kinase ATR or ATM to cause cell cycle arrest and thereby provides sufficient time for DNA repair. Cogent evidence demonstrates that after completion of repair, CHK1 is selectively recognized by HSC70, the core component of the CMA machinery, and degraded in lysosomes (Figure 4B). Correspondingly, disruption of lysosomal function leads to the accumulation of phosphorylated CHK1 in the nucleus and the destabilization of the hyper-phosphorylated DNA repair MRN complex, resulting in insufficient DNA repair and permanent cell cycle arrest [132]. Furthermore, RAD51, a key component of the DNA repair complex and a protein highly expressed in cancer cells, can stabilize CHK1 by negatively modulating macroautophagy, suggesting that selective macroautophagy is also involved in CHK1 degradation [135]. RAD51 interacts with CHK1 via its DMC1 domain to positively regulate the expression of CHK1 and promote tumor cells growth.
Collectively, these data support a positive function of selective autophagy in DDR signal transduction and DSB repair.
Selective autophagy regulates senescence
Many stresses, including DNA damage, oxidative stress, and oncogenic stress, can activate cellular senescence. Although increasing evidence demonstrates the intimate connection and dual roles (anti- or pro-senescence) of autophagy in regulating cellular senescence [136,137], the potential mechanistic aspects of this complex relationship and the discriminate functions between general autophagy and selective autophagy remain to be explained. In addition, autophagy and senescence occur in parallel and their relationship is more subtle that senescence can occur independently of autophagy [138,139]. Importantly, two recent works describe the opposing effects of selective autophagy in the regulation of cellular senescence. For example, p62-dependent selective autophagy of GATA4 acts as a mechanism for anti-senescence [123], whereas LC3B-lamin B1-dependent selective autophagy of nuclear lamina acts as a mechanism of pro-senescence [124]. GATA4 is a transcriptional factor that, under normal conditions, binds to the specialized autophagic receptor p62 for degradation by selective autophagy [123]. Upon senescence-inducing stimuli, DDR kinases ATM and ATR disable the interaction between p62 and GATA4, releasing GATA4 to activate the SASP program and cellular senescence. In contrast to GATA4, nuclear lamina protein Lamin B1 exerts an anti-senescence function and increases cell proliferation rate. In the basal cellular state, autophagy protein LC3 is present in the nucleus and directly binds to the lamin-associated domains of Lamin B1 on chromatin [124]. Upon oncogenic and genotoxic stresses, but not starvation, the LC3-Lamin B1 interaction promotes Lamin B1 degradation by selective autophagy and drives senescence. Additionally, autophagy is found to selectively degrade ∆133p53α, a p53 isoform that inhibits full length p53 and results in activating p53-mediated cellular senescence [125]. Under normal conditions, STUB1, a chaperone-associated E3 ubiquitin ligase, ubiquitinates and maintains ∆133p53α expression. During replicative senescence, STUB1 is downregulated, and ∆133p53α interacts with p62 and LC3 and is ubiquitinated for autophagic degradation. It seems that STUB1 competes with an unidentified E3 ligase to ubiquitinate ∆133p53α with different conjugation types (e.g. K48-linked or K63-linked) STUB1 protects ∆133p53α whereas unidentified E3 ligase promotes its degradation. Together, the contribution of selective autophagy to senescence seems to depend on the autophagic substrates and receptors that allow autophagy to have specificity. Future work is needed to find additional autophagy receptor-substrate pairs that act in the senescence regulatory network.
Figure 4
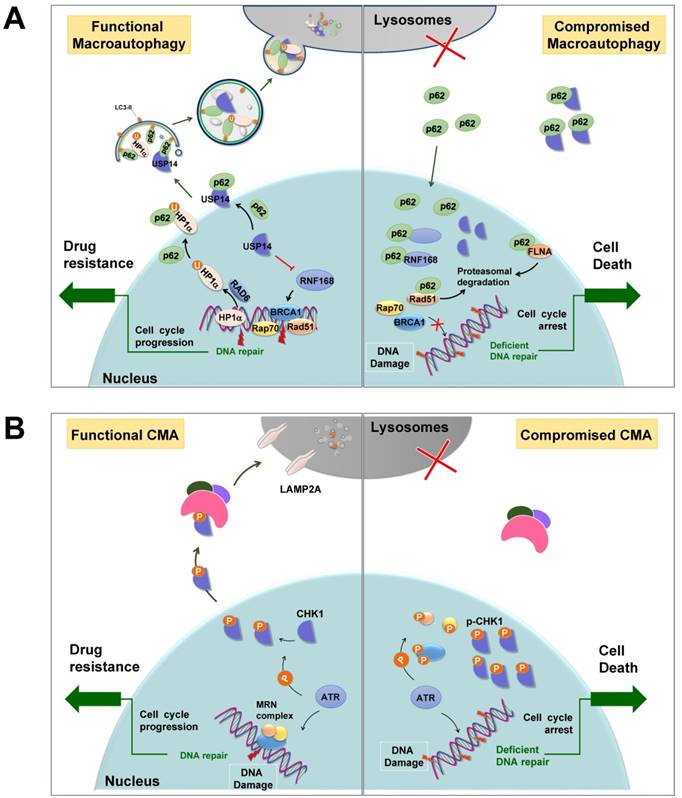
Selective autophagy promotes DNA damage repair. (A) p62-mediated selective autophagic degradation of USP14 promotes RNF168-dependent ubiquitination signaling and facilitates DNA repair proteins transporting to DSB sites. Besides, selective degradation of HP1α results in an open chromatin structure that facilitates HR. When autophagy is deficient, p62 accumulates in nuclei, where it binds to and inhibits RNF168. p62 also sequesters USP14 and promotes its translocation to DSB sites, resulting impaired DDR. In addition, p62 promotes the proteasomal degradation of DNA repair protein RAD51 and FLNA. U: ubiquitin. (B) CMA mediates the degradation of CHK1, dysfunction of which leads to the accumulation of activated CHK1 in nuclei to inhibit DNA repair and cause prolonged cell cycle arrest. Modified from Ref.132.
Selective autophagy in cancer
It is well known that autophagy plays complex and contradictory role in cancer [140-142]. Briefly, on one hand, autophagy is initially considered a mechanism that suppresses tumor initiation. Direct evidence comes from mouse genetic studies [143-146]. Tumor initiation increases when autophagy is impaired by the deletion of core autophagy genes, such as ATG5, ATG7, and Beclin-1, indicating suppression of spontaneous tumorigenesis by autophagy through an intrinsic cellular mechanism. Besides, autophagy defects due to constitutive activation of the autophagy-suppressing mTOR pathway or monoallelical loss of Beclin-1 (40% to 75%) are common in human tumors, such as prostate, breast, and ovarian cancers [142,147]. In addition, autophagy-defective tumor cells also display elevated genome damage under stress and a dysregulated cell cycle [148,149]. Considering the important role of general autophagy in energy homeostasis, cell cycle control, and DNA damage repair, a possible mechanistic explanation may be that autophagy deficiency causes the accumulation of reactive oxygen species, the prolonged DNA damage, and dysfunctional mitochondria, which are all implicated in tumorigenesis [149]. Indeed, deficiency in autophagy leads to the accumulation of p62 and endoplasmic reticulum chaperones, which may in turn, alter NF-kB regulation and gene expression to promote tumorigenesis [150].
On the other hand, autophagy promotes the survival and proliferation of established tumor cells. Due to inherent deficiencies in the microenvironment, cancer cells rely on autophagy more than normal cells and activated autophagy is able to satisfy the requirement for aberrant proliferation of cancer cells, which is associated with increased metabolic and biosynthetic utilization [140-145]. For instance, autophagy sustains growth of fully formed tumors, including lung cancers driven by B-Raf oncogene [151], pancreatic ductal adenocarcinoma [152], CNS malignancies [153], as well as multiple cancers driven by Ras oncogene [154-157]. As a result, blocking autophagy is an appealing therapeutic target. Indeed, genetic inhibition or pharmacological inhibition of autophagy by chloroquine or its derivative hydroxychloroquine (HCQ) in vitro or in vivo has demonstrated significant therapeutic responses in cancers, such as K-Ras-driven lung and pancreatic cancer [158-160]. Together, it seems that autophagy exerts its tumor-suppressive or protumorigenic roles depending on specific factors including tumor stage, cellular microenviroment, and the origin of tissue.
In contrast to general autophagy's contradictory roles in cancer development, most of the works suggest selective autophagy, such as CMA, p62-mediated selective macroautophagy, mitophagy, and pexophagy, as protumorigenic mechanisms [161,162]. CMA activity, as well as the protein levels of CMA components, is markedly elevated in most tumors [91,161-163]. Upregulated CMA exerts its protumorigenic effects though selectively degrading tumor suppressors, degrading pro-apoptotic and anti-proliferation proteins, stabilizing pro-survival proteins, maintaining the Warburg effect, and protecting against cytotoxic agents, radiation, and hypoxia (selectively degrading CHK1 and Hif-1α, Figure 4B). As a result, blocking CMA decreases the survival and tumorigenicity of cancer cells, causes tumor shrinkage, and reduces metastasis in preformed xenografts [91]. Selective macroautophagy also has a protumorigenic function via regulating the cell cycle stress response. For instance, autophagy receptor p62-mediated selective macroautophagy promotes DNA damage repair and proliferation of cancer cells via selectively degrading RNA168, USP14 and HP1α (Figure 4A). Additionally, p62-mediated selective degradation of GATA4 acts as an anti-senescence mechanism to promote tumorigenesis [123]. Another survival-promoting function of selective autophagy is maintaining signaling complexes at an appropriate level critical for cancer cell proliferation. For instance, the invasion and survival of cancer cells require focal adhesion kinase (FAK)-mediated appropriate activation of Src kinases [164]. Following loss of FAK signaling, Src is overactivated to reduce cancer cell viability. In this circumstance, the selective autophagic pathway is stimulated to selectively degrade overactive Src with the help of autophagy receptor c-Cbl, an E3 ubiquitin ligase binding LC3 via its LIR-motif [165]. Furthermore, Ret, a receptor tyrosine kinase involved in oncogenic activation of multiple cancers, is similarly degraded in selective autophagy dependent manner upon FAK signaling disruption [166]. In summary, failure in selective autophagy is prone to induce accumulation of damaged organelles and dysregulated protein quality control to facilitate neoplastic transformation. In contrast, selective autophagy may degrade misfolded proteins and dysfunctional organelles to build a relative stable intracellular context to ensure survival of tumor cells and guarantee the development of already established tumors.
Targeting general autophagy or selective autophagy
Autophagy modulation is an exciting area for anticancer therapy. Ideally, autophagy induction could prevent tumor formation at the initial stage and enhance anticancer immune responses. In addition, drug-induced overactivated autophagy could trigger autophagic cell death in cancer cells, particularly those in which the apoptotic machinery is compromised. Alternatively, most relevant studies have implicated that autophagy is a protective mechanism conferring to chemoresistance. Autophagy inhibition could thereby sensitize resistant cancer cells to chemotherapies and specifically target autophagy-addicted tumors [142,167,168]. The activation of autophagy protects cancer cell from chemotherapy and contributes to the inherit resistance involving several signaling pathways, such as EGFR signaling, p53 signaling, MAPK/p38 signaling and microRNAs [168]. Currently, all autophagy modulating drugs in clinical trials are autophagy inhibitors, primarily seeking to increase tumor sensitivity to conventional therapies [168].
Chloroquine and its derivative HCQ are currently the only FDA-approved modulators of autophagy [169,170]. HCQ mainly inhibits lysosomal acidification to impair autophagosome maturation and cargo degradation. HCQ is used as a single agent in phase I and II clinical trials to treat breast cancer and prostate cancer. In addition, HCQ is combined with several standard anticancer agents to treat small cell lung cancer, pancreatic carcinoma, colorectal cancer, advanced adenocarcinoma, advanced cancers, malignant solid tumor, and others [169,170]. However, due to the unclear pharmacodynamic and poor pharmacokinetics properties of HCQ in clinical settings, additional autophagy-linked protein targets are required.
The effect of HCQ as explored in the current clinical trials for various cancers is still unsatisfactory. Furthermore, targeting the general autophagic machinery by HCQ has proven risky, provoking severe side effects in the patient, such as retinal toxicity [170]. Therefore, the requirement for more specific autophagy modulation tailored to different cancer phenotypes remains when considering the pro-survival role of autophagy and the complexity of each tumor entity. There are studies demonstrating a possible conversion from tumor suppression to tumor promotion when general autophagy inhibition is used as an anti-tumor inflammatory response strategy [171,172]. When inhibiting general autophagy is used as an anti-cancer strategy, the interaction between autophagy and tumor microenvironment should be paid more attention because such crosstalk determines the effective immune response to chemotherapy [167].
It is possible that a superior strategy used in tumor suppression would be targeting specific types of selective autophagy rather than general autophagy, the latter of which would indifferently affect all types of autophagy and other cellular processes. For instance, the lysosome is critical for extracellular secretion, plasma membrane remodeling, cell signaling, and energy metabolism. As a result, lysosomal destruction by HCQ could significantly affect other aspects of cellular homeostasis and thereby produce unpredictable side effects. Moreover, most present studies clearly demonstrate the protumorigenic function of selective autophagy, compared with the contradictory effect of general autophagy in tumor development [161,162]. Therefore, it is reasonable that targeting specific components of selective autophagy rather than targeting general autophagy will be the most effective approach in cancer therapy. Additionally, due to the different functions of selective autophagy substrates and receptors, selective autophagy modulation may impact tumorigenesis in varied ways, implying specific approaches that will ultimately allow for personalized medication. Such therapeutic tactics will also inspire the development of novel drugs that would be appropriately combined with conventional chemotherapy or radiotherapy.
Combined targeting of autophagy and the cell cycle in cancer treatment
Cancers are characterized by uncontrolled proliferation, and increasing evidence clearly demonstrates that cell cycle stress responses, including DNA repair and senescence acquisition are associated with the efficacy of chemotherapy and/or radio-therapy [121,122]. The above results indicate a clear crosstalk between autophagy and cell cycle regulation, and malfunction of one leads to deregulation of the other (Figure 5A). Therefore, the relevance of selective autophagy in executing these cell-cycle stress responses and the role of autophagy in determining cellular life and death decisions in these contexts are of considerable interest. More importantly, cell cycle inhibitors, such as genotoxic drugs that cause DNA damage and cell cycle arrest to inhibit tumor cell division, always activate autophagy, which delays cell death and may therefore lead to chemoresistance [15,126,173]. It is important to evaluate the ability of specific autophagy to affect the efficacy and selectivity of cell cycle inhibitors in clinic. As a result, strategies that target both autophagy and the cell cycle stress response are considered novel and potent therapeutic routes.
Selective autophagy/cell cycle interaction in cancer treatment
Selective autophagy is preferred to remove specific cell cycle proteins to strengthen the fitness of cancer cells under genotoxic stress, and the complicated reciprocal inhibition of cell cycle regulators and selective autophagy links the cell cycle to the changed replicative stress. Upon DNA damage, selective autophagy is stimulated as a result of CKI activation and CDK degradation, which in turn provides sufficient time to compromise the integrity of the genome. After DNA repair, timely degradation of checkpoint proteins and DNA repair proteins allows for cycle progression and the survival of cancer cells (Figure 5B). As a result, modulation of selective autophagy can sensitize cancer cells to anticancer genotoxic agents and DNA repair inhibition by triggering the accumulation and prolonged clearance of DNA damage and finally causing permanent cell cycle arrest or cell necrotic death (Table 2) (Figure 5C). Furthermore, various potential anticancer drugs, rather than DNA-damaging agents, have been demonstrated to cause overactivated and irreversible autophagy. This impinges on the dynamic stability of cell cycle proteins to trigger cycle arrest, senescence, and autophagy-related cell death [167] (Figure 5D). Therefore, targeting selective autophagy, combined with already available chemotherapeutic genotoxic drugs, DNA repair inhibitors, and cell cycle-targeted agents, emerges as a promising strategy for anticancer treatment. Indeed, there are several examples that demonstrate the potent anticancer efficacy of combinatory targeting of autophagy and cell cycle.
Figure 5
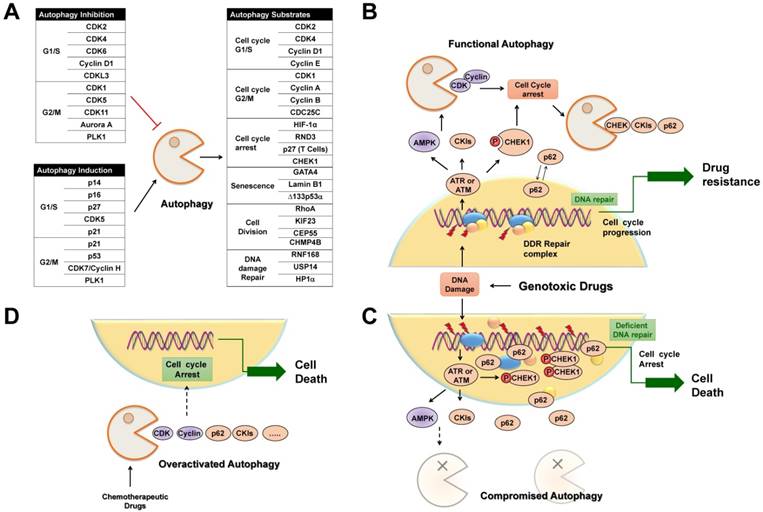
Selective autophagy regulates cell cycle arrest in cancer treatment. (A) Tables list cell cycle regulators that inhibit or activate autophagy, respectively. Also possible regulators belonging to autophagic substrates are summarized. (B) Upon DNA damage caused by genotoxic agents, selective autophagy is activated to facilitate cell cycle arrest, DNA repair and subsequent cell cycle progression through the selective degradation of cell cycle regulators. (C) When autophagy activity is compromised, abnormally high levels of active CHK1 and p62 persist in the nucleus, resulting in prolonged DNA damage and defective DNA maintenance and repair, and finally permanent cell cycle arrest or senescence. (D) Chemotherapeutic agents induce overactivated autophagy to degrade cell cycle regulators to cause permanent cell cycle arrest and autophagy-related cell death.
Examples of combinatory targeting of autophagy and CDK/cyclin
Aberrant function of cell cycle regulators results in uncontrolled cell proliferation, making them attractive therapeutic targets in cancer treatment. Recently, several pan-CDK and Aurora A inhibitors, and promising selective CDK4/6 inhibitors palbociclib and ribociclib, have enter the clinical setting for the treatment of cancer cells that always depend on specific CDKs or cyclins [1,70,174]. However, only relatively low percentage of patients receiving such targeted therapies shows long-term response and satisfactory outcome, leading to the requirement of combination therapies that provide additional therapeutic benefits [1]. Interestingly, the reciprocal regulation between autophagy and cell cycle proteins makes autophagy a promising co-target. One example is the usage of CDK4/6 inhibitors. Preclinical studies indicate that hyperactivated CDK4/6-cyclin D complex may drive the aberrant cell cycle progression of particular oncogene-driven tumors, which may be accompanied by inhibited macroautophagy in an AMPK-mTOR dependent manner [24,25]. As expected, inhibition of CDK4/6 by palbociclib causes cell cycle arrest at G1 phase and inhibits growth of tumor xenografts [54,175-177]. However, inevitable adverse events exist, and only some patients showed satisfactory and long-term responses. One possible explanation is that palbociclib may remove the inhibitory effect of the CDK4/6-cyclin D complex on autophagy, leading to activated autophagy and enhanced drug resistance in particular cancers. Several studies have consistently demonstrated that CDK4/6 inhibitors induce autophagy in different cancer cells, and the simultaneous blockade of CDK4/6 and autophagy significantly exacerbates cancer cell death [24,178-181]. For instance, autophagy prevents palbociclib-induced senescence and the combinatory usage of inhibitor HCQ with low-dose palbociclib synergistically induces irreversible growth inhibition, significant increment of reactive oxygen species, and finally cellular senescence in cancers possessing functional G1/S checkpoint [180]. Furthermore, overexpression of Aurora A has been reported in various cancer types, making Aurora A a promising molecular target for cancer therapy [182]. Notably, Aurora A inhibits macroautophagy via mTOR activation and, similarly to CDK4/6, suppresses macroautophagy and increases the sensitivity of cancer cells to apoptotic cell death induced by Aurora A inhibition [43,183]. Taken together, it is important to evaluate the contribution of specific autophagy to cell death and autophagy modulators can be utilized to remove the acquired resistance and enhance the efficacy of targeting cell cycle progression regulators.
Examples of combinatory targeting of autophagy and cell cycle checkpoints
Two cell cycle checkpoint signals, ATM-CHK2-p53 and ATR/ATM-CHK1, are activated upon genotoxic and replicative stress to arrest cell cycle progression until the damaged DNA is fixed. Defects in one or more checkpoints render cancer cells more dependent on the remaining pathways, inhibition of which may cause simultaneous perturbation of both DDR signals, resulting in dramatically genomic instability and cell death, the so-called 'synthetic lethality' process. For instance, ATR-CHK1 pathway is particularly necessary for the survival of cancers with the frequently inactivation of p53. In this respect, ATR-CHK1 inhibitors will thereby enhance killing of tumor cells through blocking cell cycle arrest and causing mitotic catastrophe [184,185]. Notably, ATM/ATR-CHK1 inhibitors have been demonstrated to increase radiosensitivity through inhibiting autophagy [186-188]. Besides, combinatory inhibitions of autophagy and ATM/ATR-CHK1 signals also corporately enhance cancer cell death [189,190]. One possible explanation is that inhibition of selective autophagy may interrupt the prompt autophagic degradation of CHK1, leading to the accumulation of phosphorylated CHK1 in the nucleus and consequently the prolonged cell cycle arrest, along with delayed DNA damage clearance and ultimately cell death [132,134]. In summary, autophagy modulation can be a promising pharmacological intervention strategy to bypass chemoresistance induced by DNA damage agents or cell cycle-targeted inhibitors. Future work is needed to identify reliable biomarkers indicating susceptible cancer subsets that respond best to the combinatory therapy targeting the cell cycle and selective autophagy.
Conclusion
Autophagy plays a key role in regulating various important biological phenomena, especially in coordinating cell responses, death, or survival, in response to stressful stimuli.
Information on the involvement of cell cycle regulators in autophagy regulation has increased. However, the specificity of autophagy (microautophagy, macroautophagy, or CMA) in cell cycle regulation and whether the degradation of cell cycle proteins is selective or non-selective should be clarified. In addition, a comprehensive understanding of how specific cell cycle arrests and selective autophagy are intertwined is still needed. For instance, the relevant cell cycle regulators for preferred autophagic degradation, the spectrum and the underlying mechanism by which autophagy receptors selectively recognize cell cycle proteins, and the signals that trigger the initial ubiquitination of relevant substrates for lysosomal delivery are not completely known. In addition, the different ubiquitin patterns of selective autophagy, K26-, K63-, or K48-type ubiquitination, emphasizes the need for identification of the involved large unknown E3 ubiquitin ligases, the coordination of which during autophagy induction and degradation remains to be elucidated. Moreover, due to the interconnectivity of the three types of autophagy and the shared usage of some components by macro- and microautophagy, further studies should not only focus on the degradation of single proteins but also on the extent to which other stress response factors are degraded simultaneously in order to discriminate the precise role of selective autophagy in cell cycle control.
Although there are fundamental molecular interactions between the pathways of cell cycle stress response and autophagy modulation, it is difficult to parse how specific interactions contribute to cross-regulation in the face of the overarching effect of death stimuli on cancer cell cycle progression and proliferation. The ability of selective autophagy to identify and degrade single cell cycle proteins or organelles makes it the preferred route to overcome chemoresistance of cancer cells [91,92,162]. However, sequential crosstalk among the different autophagy pathways exists [7-9], and it will be challenging to explore specific ways to compromise selective autophagy activity without affecting general autophagy and without disrupting the major pathways between autophagy and the cell cycle. Furthermore, beyond established paradigms, additional signals and mechanisms of selective autophagic removal of cell cycle arrest must be delineated. Finally, substantial efforts to develop novel CDK-selective inhibitors and selective autophagy inhibitors, as well as combinatory therapeutic strategies under active clinical investigation can be anticipated.
Abbreviations
ATM: Ataxia telangiectasia mutated; ATR: Ataxia telangiectasia and Rad3-related; CDC25: cell division cycle 25; CDK: cyclin-dependent kinase; CEP55: midbody centrosomal protein 55; CHK: cell cycle checkpoint kinase; CHMP4B: charged multivesicular body protein 4B; CKI: CDK inhibitor protein; CMA: chaperone-mediated autophagy; DDR: DNA damage response; DSBs: DNA double-strand breaks; EndoB1: endophilin B1; ESCRT: endosomal-sorting complexes required for transport; FAK: focal adhesion kinase; HCQ: hydroxychloroquine; HIF-1α: hypoxia inducible factor 1; HP1: heterochromatin protein 1; HR: homologous recombination; LAMP2A: lysosome-associated membrane protein type 2A; LIR: LC3/GABARAP interacting region; MEFs: mouse embryonic fibroblasts; MRN: Mre11-Rad50-Nbs1 complex; NHEJ: non-homologous end-joining; mTOR: mechanistic target of rapamycin; PKR: dsRNA-activated protein kinase; PLK: Polo-like kinase; PtdIns3P: phosphatidylinositol 3-phosphate; RB: retinoblastoma tumor suppressor protein; SASP: senescence-associated secretory phenotype; SCF: Skp1-Cul1-F box; UPS: ubiquitin-proteasome system; VHL: von Hippel-Lindau ubiquitin ligase complex.
Acknowledgements
This review was funded by the National Natural Science Foundation of China (No. 81603341, 81573471 and 31670360), the International Cooperation Project of Guangdong Province (No. 2015A050502028), the Marine and Fishery Bureau Industrialization Project of Guangdong Province (No. GD2013-B02-003), and the New Teacher Natural Science Research Project of Shenzhen University (No. 2018021).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. 2017;17:93-115
2. Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12:801-17
3. Qiu Z, Oleinick NL, Zhang J. ATR/CHK1 inhibitors and cancer therapy. Radiother Oncol. 2018;126:450-464
4. Rundle S, Bradbury A, Drew Y, Curtin NJ. Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers (Basel). 2017:9 pii: E41
5. Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65-75
6. Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F. et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36:1811-1836
7. Park C, Cuervo AM. Selective autophagy: talking with the UPS. Cell Biochem Biophys. 2013;67:3-13
8. Wong E, Cuervo AM. Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol. 2010;2:a006734
9. Kaushik S, Massey A, Mizushima N, Cuervo AM. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell. 2008;19:2179-2192
10. Koga H, Martinez-Vicente M, Macian F, Verkhusha VV, Cuervo AM. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat Commun. 2011;2:386
11. Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279-96
12. Zaffagnini G, Martens S. Mechanisms of Selective Autophagy. J Mol Biol. 2016;428:1714-24
13. Eliopoulos AG, Havaki S, Gorgoulis VG. DNA Damage Response and Autophagy: A Meaningful Partnership. Front Genet. 2016;7:204
14. Hewitt G, Korolchuk VI. Repair, Reuse, Recycle: The Expanding Role of Autophagy in Genome Maintenance. Trends Cell Biol. 2017;27:340-351
15. Bordin DL, Lima M, Lenz G, Saffi J, Meira LB, Mésange P. et al. DNA alkylation damage and autophagy induction. Mutat Res. 2013;753:91-9
16. Kaminskyy V, Abdi A, Zhivotovsky B. A quantitative assay for the monitoring of autophagosome accumulation in different phases of the cell cycle. Autophagy. 2011;7:83-90
17. Eskelinen EL, Prescott AR, Cooper J, Brachmann SM, Wang L, Tang X. et al. Inhibition of autophagy in mitotic animal cells. Traffic. 2002;3:878-93
18. Furuya T, Kim M, Lipinski M, Li J, Kim D, Lu T. et al. Negative regulation of Vps34 by Cdk mediated phosphorylation. Mol Cell. 2010;38:500-11
19. Sit KH, Paramanantham R, Bay BH, Chan HL, Wong KP, Thong P. et al. Sequestration of mitotic (M-phase) chromosomes in autophagosomes: mitotic programmed cell death in human Chang liver cells induced by an OH* burst from vanadyl(4). Anat Rec. 1996;245:1-8
20. Liu L, Xie R, Nguyen S, Ye M, McKeehan WL. Robust autophagy/mitophagy persists during mitosis. Cell Cycle. 2009;8:1616-20
21. Li Z, Ji X, Wang D, Liu J, Zhang X. Autophagic flux is highly active in early mitosis and differentially regulated throughout the cell cycle. Oncotarget. 2016;7:39705-39718
22. Loukil A, Zonca M, Rebouissou C, Baldin V, Coux O, Biard-Piechaczyk M. et al. High-resolution live-cell imaging reveals novel cyclin A2 degradation foci involving autophagy. J Cell Sci. 2014;127:2145-50
23. Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M. et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218-24
24. Iriyama N, Hino H, Moriya S, Hiramoto M, Hatta Y, Takei M. et al. The cyclin-dependent kinase 4/6 inhibitor, abemaciclib, exerts dose-dependent cytostatic and cytocidal effects and induces autophagy in multiple myeloma cells. Leuk Lymphoma. 2017;18:1-12
25. Sumi NJ, Kuenzi BM, Knezevic CE, Remsing Rix LL, Rix U. Chemoproteomics reveals novel protein and lipid kinase targets of clinical CDK4/6 inhibitors in lung cancer. ACS Chem Biol. 2015;10:2680-6
26. Casimiro MC, Di Sante G, Di Rocco A, Loro E, Pupo C, Pestell TG. et al. Cyclin D1 restrains oncogene-induced autophagy by regulating the AMPK-LKB1 signaling Axis. Cancer Res. 2017;77:3391-3405
27. Brown NE, Jeselsohn R, Bihani T, Hu MG, Foltopoulou P, Kuperwasser C. et al. Cyclin D1 activity regulates autophagy and senescence in the mammary epithelium. Cancer Res. 2012;72:6477-89
28. Su M, Wang J, Wang C, Wang X, Dong W, Qiu W. et al. MicroRNA-221 inhibits autophagy and promotes heart failure by modulating the p27/CDK2/mTOR axis. Cell Death Differ. 2015;22:986-99
29. Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D. et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature. 2014;508:541-5
30. Szyniarowski P, Corcelle-Termeau E, Farkas T, Høyer-Hansen M, Nylandsted J, Kallunki T. et al. A comprehensive siRNA screen for kinases that suppress macroautophagy in optimal growth conditions. Autophagy. 2011;7:892-903
31. Pozo K, Bibb JA. The Emerging Role of Cdk5 in Cancer. Trends Cancer. 2016;2:606-618
32. Nandi N, Tyra LK, Stenesen D, Krämer H. Stress-induced Cdk5 activity enhances cytoprotective basal autophagy in Drosophila melanogaster by phosphorylating acinus at serine437. Elife. 2017;6:e30760
33. Lopes JP, Agostinho P. Cdk5: multitasking between physiological and pathological conditions. Prog Neurobiol. 2011;94:49-63
34. Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene. 2008;27:4860-4
35. Jiang H, Martin V, Gomez-Manzano C, Johnson DG, Alonso M, White E. et al. The RB-E2F1 pathway regulates autophagy. Cancer Res. 2010;70:7882-93
36. Wong AS, Lee RH, Cheung AY, Yeung PK, Chung SK, Cheung ZH. et al. Cdk5-mediated phosphorylation of endophilin B1 is required for induced autophagy in models of Parkinson's disease. Nat Cell Biol. 2011;13:568-79
37. Wen Z, Shu Y, Gao C, Wang X, Qi G, Zhang P. et al. CDK5-mediated phosphorylation and autophagy of RKIP regulate neuronal death in Parkinson's disease. Neurobiol Aging. 2014;35:2870-80
38. Su LY, Li H, Lv L, Feng YM, Li GD, Luo R. et al. Melatonin attenuates MPTP-induced neurotoxicity via preventing CDK5-mediated autophagy and SNCA/α-synuclein aggregation. Autophagy. 2015;11:1745-59
39. Xiao J, Zhang T, Xu D, Wang H, Cai Y, Jin T. et al. FBXL20-mediated Vps34 ubiquitination as a p53 controlled checkpoint in regulating autophagy and receptor degradation. Genes Dev. 2015;29:184-96
40. Wilkinson S, Croft DR, O'Prey J, Meedendorp A, O'Prey M, Dufès C. et al. The cyclin-dependent kinase PITSLRE/CDK11 is required for successful autophagy. Autophagy. 2011;7:1295-301
41. Zhu J, Deng S, Lu P, Bu W, Li T, Yu L. et al. The Ccl1-Kin28 kinase complex regulates autophagy under nitrogen starvation. J Cell Sci. 2016;129:135-44
42. Borisa AC, Bhatt HG. A comprehensive review on Aurora kinase: Small molecule inhibitors and clinical trial studies. Eur J Med Chem. 2017;140:1-19
43. Zou Z, Yuan Z, Zhang Q, Long Z, Chen J, Tang Z. et al. Aurora kinase A inhibition-induced autophagy triggers drug resistance in breast cancer cells. Autophagy. 2012;8:1798-810
44. Xu LZ, Long ZJ, Peng F, Liu Y, Xu J, Wang C. et al. Aurora kinase A suppresses metabolic stress-induced autophagic cell death by activating mTOR signaling in breast cancer cells. Oncotarget. 2014;5:7498-511
45. Zhu Q, Yu X, Zhou ZW, Zhou C, Chen XW, Zhou SF. Inhibition of aurora a kinase by alisertib induces autophagy and cell cycle arrest and increases chemosensitivity in human hepatocellular carcinoma hepg2 cells. Curr Cancer Drug Targets. 2017;17:386-401
46. Liu Z, Wang F, Zhou ZW, Xia HC, Wang XY, Yang YX. et al. Alisertib induces G2/M arrest, apoptosis, and autophagy via PI3K/Akt/mTOR- and p38 MAPK-mediated pathways in human glioblastoma cells. Am J Transl Res. 2017;9:845-73
47. Wan XB, Long ZJ, Yan M, Xu J, Xia LP, Liu L. et al. Inhibition of Aurora-A suppresses epithelial-mesenchymal transition and invasion by downregulating MAPK in nasopharyngeal carcinoma cells. Carcinogenesis. 2008;29:1930-7
48. Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179-93
49. van de Weerdt BC, Medema RH. Polo-like kinases: a team in control of the division. Cell Cycle. 2006;5:853-64
50. Deeraksa A, Pan J, Sha Y, Liu XD, Eissa NT, Lin SH. et al. Plk1 is upregulated in androgen-insensitive prostate cancer cells and its inhibition leads to necroptosis. Oncogene. 2013;32:2973-83
51. Tao YF, Li ZH, Du WW, Xu LX, Ren JL, Li XL. et al. Inhibiting PLK1 induces autophagy of acute myeloid leukemia cells via mammalian target of rapamycin pathway dephosphorylation. Oncol Rep. 2017;37:1419-1429
52. Ruf S, Heberle AM, Langelaar-Makkinje M, Gelino S, Wilkinson D, Gerbeth C. et al. PLK1 (polo like kinase 1) inhibits MTOR complex 1 and promotes autophagy. Autophagy. 2017;13:486-505
53. Chen LL, Wang YB, Song JX, Deng WK, Lu JH, Ma LL. et al. Phosphoproteome-based kinase activity profiling reveals the critical role of MAP2K2 and PLK1 in neuronal autophagy. Autophagy. 2017;21:1-12
54. Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J. et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, "fueling" tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle. 2012;11:3599-610
55. Pimkina J, Humbey O, Zilfou JT, Jarnik M, Murphy ME. ARF induces autophagy by virtue of interaction with Bcl-xl. J Biol Chem. 2009;284:2803-10
56. Coats S, Flanagan WM, Nourse J, Roberts JM. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science. 1996;272:877-80
57. Campos T, Ziehe J, Palma M, Escobar D, Tapia JC, Pincheira R. et al. Rheb promotes cancer cell survival through p27Kip1-dependent activation of autophagy. Mol Carcinog. 2016;55:220-9
58. Zada S, Noh HS, Baek SM, Ha JH, Hahm JR, Kim DR. Depletion of p18/LAMTOR1 promotes cell survival via activation of p27(kip1) -dependent autophagy under starvation. Cell Biol Int. 2015;39:1242-50
59. Zhang G, Park MA, Mitchell C, Walker T, Hamed H, Studer E. et al. Multiple cyclin kinase inhibitors promote bile acid-induced apoptosis and autophagy in primary hepatocytes via p53-CD95-dependent signaling. J Biol Chem. 2008;283:24343-58
60. Mohapatra P, Preet R, Das D, Satapathy SR, Choudhuri T, Wyatt MD. et al. Quinacrine-mediated autophagy and apoptosis in colon cancer cells is through a p53- and p21-dependent mechanism. Oncol Res. 2012;20:81-91
61. Fujiwara K, Daido S, Yamamoto A, Kobayashi R, Yokoyama T, Aoki H. et al. Pivotal role of the cyclin-dependent kinase inhibitor p21WAF1/CIP1 in apoptosis and autophagy. J Biol Chem. 2008;283:388-97
62. Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22:181-5
63. Mathiassen SG, De Zio D, Cecconi F. Autophagy and the Cell Cycle: A Complex Landscape. Front Oncol. 2017;7:51
64. Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M. et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676-87
65. Tasdemir E, Maiuri C, Tajeddine N, Criollo A, Hickman G, Geneste O. et al. Cell cycle-dependent induction of autophagy, mitophagy and reticulophagy. Cell Cycle. 2007;6:2263-7
66. Morselli E, Tasdemir E, Maiuri MC, Galluzzi L, Kepp O, Criollo A. et al. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle. 2008;7:3056-61
67. Gilberto S, Peter M. Dynamic ubiquitin signaling in cell cycle regulation. J Cell Biol. 2017;216:2259-2271
68. Lee KH, Lee SJ, Lee HJ, Choi GE, Jung YH, Kim DI. et al. Amyloid β1-42 (Aβ1-42) Induces the CDK2-Mediated Phosphorylation of Tau through the Activation of the mTORC1 signaling pathway while promoting neuronal cell death. Front Mol Neurosci. 2017;10:229
69. Pathania AS, Guru SK, Kumar S, Kumar A, Ahmad M, Bhushan S. et al. Interplay between cell cycle and autophagy induced by boswellic acid analog. Sci Rep. 2016;6:33146
70. VanArsdale T, Boshoff C, Arndt KT, Abraham RT. Molecular Pathways: Targeting the Cyclin D-CDK4/6 Axis for Cancer Treatment. Clin Cancer Res. 2015;21:2905-10
71. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3b regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499-3511
72. Lin DI, Barbash O, Kumar KG, Weber JD, Harper JW, Klein-Szanto AJ. et al. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Mol Cell. 2006;24:355-66
73. Santra MK, Wajapeyee N, Green MR. F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature. 2009;459:722-5
74. Park GH, Song HM, Kim YS, Jeon Y, Koo JS, Jeong HJ. et al. Anti-cancer activity of Bacillus amyloliquefaciens AK-0 through cyclin D1 proteasomal degradation via GSK3β-dependent phosphorylation of threonine-286. Pharmazie. 2017;72:348-354
75. Liu SL, Liu Z, Zhang LD, Zhu HQ, Guo JH, Zhao M. et al. GSK3β-dependent cyclin D1 and cyclin E1 degradation is indispensable for NVP-BEZ235 induced G0/G1 arrest in neuroblastoma cells. Cell Cycle. 2017;16:2386-2395
76. Kim MK, Park GH, Eo HJ, Song HM, Lee JW, Kwon MJ. et al. Tanshinone I induces cyclin D1 proteasomal degradation in an ERK1/2 dependent way in human colorectal cancer cells. Fitoterapia. 2015;101:162-8
77. Pirtoli L, Belmonte G, Toscano M, Tini P, Miracco C. Cyclin D1 Co-localizes with Beclin-1 in Glioblastoma Recurrences: A clue to a therapy-induced, autophagy-mediated degradative mechanism? Anticancer Res. 2016;36:4057-62
78. Wu SY, Lan SH, Lin XZ, Su IJ, Tsai TF, Yen CJ. et al. Autophagy regulates human hepatocellular carcinoma tumorigenesis through selective degradation of cyclin D1. Biomark Genomic Med. 2014;6:187
79. Wu SY, Lan SH, Wu SR, Chiu YC, Lin XZ, Su IJ. et al. Hepatocellular carcinoma-related cyclin D1 is selectively regulated by autophagy degradation system. Hepatology. 2018;68:141-154
80. Herrero-Ruiz J, Mora-Santos M, Giráldez S, Sáez C, Japón MA, Tortolero M. et al. βTrCP controls the lysosome-mediated degradation of CDK1, whose accumulation correlates with tumor malignancy. Oncotarget. 2014;5:7563-74
81. Galindo-Moreno M, Giráldez S, Sáez C, Japón MÁ, Tortolero M, Romero F. Both p62/SQSTM1-HDAC6-dependent autophagy and the aggresome pathway mediate CDK1 degradation in human breast cancer. Sci Rep. 2017;7:10078
82. Yoon CH, Miah MA, Kim KP, Bae YS. New Cdc2 Tyr 4 phosphorylation by dsRNA-activated protein kinase triggers Cdc2 polyubiquitination and G2 arrest under genotoxic stresses. EMBO reports. 2010;11:393-399
83. Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T. et al. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFb-TrCP. Proc Natl Acad Sci USA. 2004;101:4419-4424
84. Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V. et al. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682-5
85. Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A. et al. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev. 1999;13:1181-9
86. Fuster JJ, Gonzalez JM, Edo MD, Viana R, Boya P, Cervera J. et al. Tumor suppressor p27(Kip1) undergoes endolysosomal degradation through its interaction with sorting nexin 6. FASEB J. 2010;24:2998-3009
87. Jia W, He MX, McLeod IX, Guo J, Ji D, He YW. Autophagy regulates T lymphocyte proliferation through selective degradation of the cell-cycle inhibitor CDKN1B/p27Kip1. Autophagy. 2015;11:2335-45
88. Pei B, Zhao M, Miller BC, Vela JL, Bruinsma MW, Virgin HW. et al. Invariant NKT cells require autophagy to coordinate proliferation and survival signals during differentiation. J Immunol. 2015;194:5872-84
89. Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204:25-31
90. Dowling SD, Macian F. Autophagy and T cell metabolism. Cancer Lett. 2018;419:20-26
91. Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, Cuervo AM. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med. 2011;3:109ra117
92. Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014;24:92-104
93. Hackenbeck T, Knaup KX, Schietke R, Schödel J, Willam C, Wu X. et al. HIF-1 or HIF-2 induction is sufficient to achieve cell cycle arrest in NIH3T3 mouse fibroblasts independent from hypoxia. Cell Cycle. 2009;8:1386-95
94. Hubbi ME. et al. A nontranscriptional role for HIF-1α as a direct inhibitor of DNA replication. Sci Signal. 2013;6:ra10
95. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME. et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271-5
96. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ. et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468-72
97. Hubbi ME, Hu H. et al. Chaperone-mediated autophagy targets hypoxia-inducible factor-1α (HIF-1α) for lysosomal degradation. J Biol Chem. 2013;288:10703-14
98. Ferreira JV, Fôfo H, Bejarano E, Bento CF, Ramalho JS, Girão H. et al. STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy. 2013;9:1349-66
99. Hubbi ME, Gilkes DM, Hu H. et al. Cyclin-dependent kinases regulate lysosomal degradation of hypoxia-inducible factor 1α to promote cell-cycle progression. Proc Natl Acad Sci U S A. 2014;111:E3325-34
100. Im S, Kim DW. Nkx3.2 induces oxygen concentration-independent and lysosome-dependent degradation of HIF-1α to modulate hypoxic responses in chondrocytes. Cell Signal. 2017;36:127-138
101. Chen K, Zeng J, Xiao H, Huang C, Hu J, Yao W. et al. Regulation of glucose metabolism by p62/SQSTM1 through HIF1α. J Cell Sci. 2016;129:817-30
102. Liu T, Singh R, Rios Z, Bhushan A, Li M, Sheridan PP. et al. Tyrosine phosphorylation of HSC70 and its interaction with RFC mediates methotrexate resistance in murine L1210 leukemia cells. Cancer Lett. 2015;357:231-41
103. Truman AW, Kristjansdottir K, Wolfgeher D, Hasin N, Polier S, Zhang H. et al. CDK-dependent Hsp70 Phosphorylation controls G1 cyclin abundance and cell-cycle progression. Cell. 2012;151:1308-18
104. Zhao H, Yang J, Fan T, Li S, Ren X. RhoE functions as a tumor suppressor in esophageal squamous cell carcinoma and modulates the PTEN/PI3K/Akt signaling pathway. Tumour Biol. 2012;33:1363-74
105. Villalonga P, Fernandez de Mattos S, Ridley AJ. RhoE inhibits 4EBP1 phosphorylation and eIF4E function impairing cap-dependent translation. J Biol Chem. 2009;284:35287-96
106. Poch E, Minambres R, Mocholi E, Ivorra C, Perez-Arago A, Guerri C. et al. RhoE interferes with Rb inactivation and regulates the proliferation and survival of the U87 human glioblastoma cell line. Exp Cell Res. 2007;313:719-31
107. Lonjedo M, Poch E, Mocholí E, Hernández-Sánchez M, Ivorra C, Franke TF. et al. The Rho family member RhoE interacts with Skp2 and is degraded at the proteasome during cell cycle progression. J Biol Chem. 2013;288:30872-82
108. Zhou J, Yang J, Fan X, Hu S, Zhou F, Dong J. et al. Chaperone-mediated autophagy regulates proliferation by targeting RND3 in gastric cancer. Autophagy. 2016;12:515-28
109. Sagona AP, Nezis IP, Pedersen NM, Liestøl K, Poulton J, Rusten TE. et al. PtdIns(3)P controls cytokinesis through KIF13A-mediated recruitment of FYVE-CENT to the midbody. Nat Cell Biol. 2010;12:362-71
110. Thoresen SB, Pedersen NM, Liestøl K, Stenmark H. A phosphatidylinositol 3-kinase class III sub-complex containing VPS15, VPS34, Beclin 1, UVRAG and BIF-1 regulates cytokinesis and degradative endocytic traffic. Exp Cell Res. 2010;316:3368-78
111. You SY, Park YS, Jeon HJ, Cho DH, Jeon HB, Kim SH. et al. Beclin-1 knockdown shows abscission failure but not autophagy defect during oocyte meiotic maturation. Cell Cycle. 2016;15:1611-9
112. Belaid A, Cerezo M, Chargui A, Corcelle-Termeau E, Pedeutour F, Giuliano S. et al. Autophagy plays a critical role in the degradation of active RHOA, the control of cell cytokinesis, and genomic stability. Cancer Res. 2013;73:4311-22
113. Dubreuil V, Marzesco AM, Corbeil D, Huttner WB, Wilsch-Bräuninger M. Midbody and primary cilium of neural progenitors release extracellular membrane particles enriched in the stem cell marker prominin-1. J Cell Biol. 2007;176:483-95
114. Kieserman EK, Glotzer M, Wallingford JB. Developmental regulation of central spindle assembly and cytokinesis during vertebrate embryogenesis. Curr Biol. 2008;18:116-23
115. Isakson P, Lystad AH, Breen K, Koster G, Stenmark H, Simonsen A. TRAF6 mediates ubiquitination of KIF23/MKLP1 and is required for midbody ring degradation by selective autophagy. Autophagy. 2013;9:1955-64
116. Kuo TC, Chen CT, Baron D, Onder TT, Loewer S, Almeida S. et al. Midbody accumulation through evasion of autophagy contributes to cellular reprogramming and tumorigenicity. Nat Cell Biol. 2011;13:1214-23
117. Pohl C, Jentsch S. Midbody ring disposal by autophagy is a post-abscission event of cytokinesis. Nat Cell Biol. 2009;11:65-70
118. Rello-Varona S, Lissa D, Shen S, Niso-Santano M, Senovilla L, Mariño G. et al. Autophagic removal of micronuclei. Cell Cycle. 2012;11:170-6
119. Sagona AP, Nezis IP, Stenmark H. Association of CHMP4B and autophagy with micronuclei: implications for cataract formation. Biomed Res Int. 2014;2014:974393
120. Yan S, Liu L, Ren F, Gao Q, Xu S, Hou B. et al. Sunitinib induces genomic instability of renal carcinoma cells through affecting the interaction of LC3-II and PARP-1. Cell Death Dis. 2017;8:e2988
121. O'Connor MJ. Targeting the DNA Damage Response in Cancer. Mol Cell. 2015;60:547-60
122. Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12:587-98
123. Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L. et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349:aaa5612
124. Dou Z, Xu C, Donahue G, Shimi T, Pan JA, Zhu J. et al. Autophagy mediates degradation of nuclear lamina. Nature. 2015;527:105-109
125. Horikawa I, Fujita K, Jenkins LM, Hiyoshi Y, Mondal AM, Vojtesek B. et al. Autophagic degradation of the inhibitory p53 isoform Delta133p53alpha as a regulatory mechanism for p53-mediated senescence. Nat Commun. 2014;5:4706
126. Gomes LR, Menck CFM, Leandro GS. Autophagy Roles in the Modulation of DNA Repair Pathways. Int J Mol Sci. 2017;18:pii E2351
127. Wang Y, Zhang N, Zhang L, Li R, Fu W, Ma K. et al. Autophagy Regulates Chromatin Ubiquitination in DNA Damage Response through Elimination of SQSTM1/p62. Mol Cell. 2016;63:34-48
128. Hewitt G, Carroll B, Sarallah R, Correia-Melo C, Ogrodnik M, Nelson G. et al. SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy. 2016;12:1917-1930
129. Pankiv S, Lamark T, Bruun JA, Øvervatn A, Bjørkøy G, Johansen T. Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. J Biol Chem. 2010;285:5941-53
130. Sharma A, Alswillah T, Singh K, Chatterjee P, Willard B, Venere M. et al. USP14 regulates DNA damage repair by targeting RNF168-dependent ubiquitination. Autophagy. 2018;14:1976-1990
131. Chen S, Wang C, Sun L, Wang DL, Chen L, Huang Z. et al. RAD6 promotes homologous recombination repair by activating the autophagy-mediated degradation of heterochromatin protein HP1. Mol Cell Biol. 2015;35:406-416
132. Park C, Suh Y, Cuervo AM. Regulated degradation of chk1 by chaperone-mediated autophagy in response to DNA damage. Nat Commun. 2015;6:6823
133. Liu EY, Xu N, O'Prey J, Lao LY, Joshi S, Long JS. et al. Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc Natl Acad Sci U S A. 2015;112:773-8
134. Zheng K, Li Y, Wang S, Wang X, Liao C, Hu X. et al. Inhibition of autophagosome-lysosome fusion by ginsenoside Ro via the ESR2-NCF1-ROS pathway sensitizes esophageal cancer cells to 5-fluorouracil-induced cell death via the CHEK1-mediated DNA damage checkpoint. Autophagy. 2016;12:1593-613
135. Zhu X, Pan Q, Huang N, Wu J, Zhen N, Sun F. et al. RAD51 regulates CHK1 stability via autophagy to promote cell growth in esophageal squamous carcinoma cells. Tumour Biol. 2016 [Epub ahead of print]
136. Gewirtz DA. Autophagy and senescence: a partnership in search of definition. Autophagy. 2013;9:808-12
137. White E, Lowe SW. Eating to exit: autophagy-enabled senescence revealed. Genes Dev. 2009;23:784-7
138. Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF. et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798-803
139. Goehe RW, Di X, Sharma K, Bristol ML, Henderson SC, Valerie K. et al. The autophagy-senescence connection in chemotherapy: must tumor cells (self) eat before they sleep? J Pharmacol Exp Ther. 2012;343:763-78
140. White E. The role for autophagy in cancer. J Clin Invest. 2015;125:42-46
141. Kenific CM, Debnath J. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol. 2015;25:37-45
142. Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F. et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34:856-880
143. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A. et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809-20
144. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077-82
145. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S. et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795-800
146. Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O. et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275-84
147. Levine B, Kroemer G. Autophagy in the Pathogenesis of Disease. Cell. 2008;132:27-42
148. Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K. et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367-81
149. Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S. et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621-35
150. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY. et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062-75
151. Levy JM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK. et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov. 2014;4:773-80
152. Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H. et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717-29
153. White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401-10
154. Kimmelman AC. Metabolic Dependencies in RAS-Driven Cancers. Clin Cancer Res. 2015;21:1828-34
155. Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM. et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011;22:165-78
156. Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G. et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460-70
157. Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, Choi YH. et al. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem. 2011;286:12924-32
158. Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S. et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4:914-27
159. Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM. et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27:1447-61
160. Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC. et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014;4:905-13
161. Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018;19:365-381
162. Daskalaki I, Gkikas I, Tavernarakis N. Hypoxia and Selective Autophagy in Cancer Development and Therapy. Front Cell Dev Biol. 2018;6:104
163. Li L, Fang R, Liu B, Shi H, Wang Y, Zhang W. et al. Deacetylation of tumor-suppressor MST1 in Hippo pathway induces its degradation through HBXIP-elevated HDAC6 in promotion of breast cancer growth. Oncogene. 2016;35:4048-57
164. Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513-609
165. Sandilands E, Serrels B, Wilkinson S, Frame MC. Src-dependent autophagic degradation of Ret in FAK-signalling-defective cancer cells. EMBO Rep. 2012;13:733-40
166. Sandilands E, Serrels B, McEwan DG, Morton JP, Macagno JP, McLeod K. et al. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat Cell Biol. 2011;14:51-60
167. Maes H, Rubio N, Garg AD, Agostinis P. Autophagy: shaping the tumor microenvironment and therapeutic response. Trends Mol Med. 2013;19:428-46
168. Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M. et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:e838
169. Chude CI, Amaravadi RK. Targeting autophagy in cancer: update on clinical trials and novel inhibitors. Int J Mol Sci. 2017:18 pii: E1279
170. Shi TT, Yu XX, Yan LJ, Xiao HT. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother Pharmacol. 2017;79:287-294
171. Rao S, Yang H, Penninger JM, Kroemer G. Autophagy in non-small cell lung carcinogenesis: A positive regulator of antitumor immunosurveillance. Autophagy. 2014;10:529-31
172. Townsend KN, Hughson LR, Schlie K, Poon VI, Westerback A, Lum JJ. Autophagy inhibition in cancer therapy: metabolic considerations for antitumor immunity. Immunol Rev. 2012;249:176-94
173. Bhat P, Kriel J, Shubha Priya B. et al. Modulating autophagy in cancer therapy: Advancements and challenges for cancer cell death sensitization. Biochem Pharmacol. 2018;147:170-182
174. O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13:417-30
175. Acevedo M, Vernier M, Mignacca L, Lessard F, Huot G, Moiseeva O. et al. A CDK4/6-dependent epigenetic mechanism protects cancer cells from PML-induced senescence. Cancer Res. 2016;76:3252-64
176. Bourdeau V, Ferbeyre G. CDK4-CDK6 inhibitors induce autophagy-mediated degradation of DNMT1 and facilitate the senescence antitumor response. Autophagy. 2016;12:1965-1966
177. Iriyama N, Hino H, Moriya S, Hiramoto M, Hatta Y, Takei M. et al. The cyclin-dependent kinase 4/6 inhibitor, abemaciclib, exerts dose-dependent cytostatic and cytocidal effects and induces autophagy in multiple myeloma cells. Leuk Lymphoma. 2018;59:1439-1450
178. Valenzuela CA, Vargas L, Martinez V, Bravo S, Brown NE. Palbociclib-induced autophagy and senescence in gastric cancer cells. Exp Cell Res. 2017;360:390-396
179. Okada Y, Kato S, Sakamoto Y, Oishi T, Ishioka C. Synthetic lethal interaction of CDK inhibition and autophagy inhibition in human solid cancer cell lines. Oncol Rep. 2017;38:31-42
180. Vijayaraghavan S, Karakas C, Doostan I, Chen X, Bui T, Yi M. et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun. 2017;8:15916
181. Small J, Washburn E, Millington K, Zhu J, Holder SL. The addition of abemaciclib to sunitinib induces regression of renal cell carcinoma xenograft tumors. Oncotarget. 2017;8:95116-95134
182. Mountzios G, Terpos E, Dimopoulos MA. Aurora kinases as targets for cancer therapy. Cancer Treat Rev. 2008;34:175-82
183. Shang YY, Yao M, Zhou ZW, Cui J, Xia L, Hu RY. et al. Alisertib promotes apoptosis and autophagy in melanoma through p38 MAPK-mediated aurora a signaling. Oncotarget. 2017;8:107076-88
184. Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol Med. 2011;17:88-96
185. Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825-37
186. Zhou ZR, Yang ZZ, Wang SJ, Zhang L, Luo JR, Feng Y. et al. The Chk1 inhibitor MK-8776 increases the radiosensitivity of human triple-negative breast cancer by inhibiting autophagy. Acta Pharmacol Sin. 2017;38:513-523
187. Liou JS, Wu YC, Yen WY, Tang YS, Kakadiya RB, Su TL. et al. Inhibition of autophagy enhances DNA damage-induced apoptosis by disrupting CHK1-dependent S phase arrest. Toxicol Appl Pharmacol. 2014;278:249-58
188. Cui Y, Palii SS, Innes CL, Paules RS. Depletion of ATR selectively sensitizes ATM-deficient human mammary epithelial cells to ionizing radiation and DNA-damaging agents. Cell Cycle. 2014;13:3541-50
189. Wang FZ, Fei HR, Cui YJ, Sun YK, Li ZM, Wang XY. et al. The checkpoint 1 kinase inhibitor LY2603618 induces cell cycle arrest, DNA damage response and autophagy in cancer cells. Apoptosis. 2014;19:1389-98
190. Lin CS, Wang YC, Huang JL, Hung CC, Chen JY. Autophagy and reactive oxygen species modulate cytotoxicity induced by suppression of ATM kinase activity in head and neck cancer cells. Oral Oncol. 2012;48:1152-8
Author contact
![]() Corresponding author: zhengkedu.cn or y.k.zhengcom (Kai Zheng). Address: School of Pharmaceutical Sciences, Health Science Center, Shenzhen University, Nanhai Rd 3688, Shenzhen 518060, Guangdong, PR China. Tel: +86 755 26917542
Corresponding author: zhengkedu.cn or y.k.zhengcom (Kai Zheng). Address: School of Pharmaceutical Sciences, Health Science Center, Shenzhen University, Nanhai Rd 3688, Shenzhen 518060, Guangdong, PR China. Tel: +86 755 26917542
Citation styles
APA
Zheng, K., He, Z., Kitazato, K., Wang, Y. (2019). Selective Autophagy Regulates Cell Cycle in Cancer Therapy. Theranostics, 9(1), 104-125. https://doi.org/10.7150/thno.30308.
ACS
Zheng, K.; He, Z.; Kitazato, K.; Wang, Y. Selective Autophagy Regulates Cell Cycle in Cancer Therapy. Theranostics 2019, 9 (1), 104-125. DOI: 10.7150/thno.30308.
NLM
Zheng K, He Z, Kitazato K, Wang Y. Selective Autophagy Regulates Cell Cycle in Cancer Therapy. Theranostics 2019; 9(1):104-125. doi:10.7150/thno.30308. https://www.thno.org/v09p0104.htm
CSE
Zheng K, He Z, Kitazato K, Wang Y. 2019. Selective Autophagy Regulates Cell Cycle in Cancer Therapy. Theranostics. 9(1):104-125.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.