Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Modulating antigen presentation...
Modulating cytotoxic T...
Modulating T helper cells (Th...
Modulating immunosuppressive...
Modulating soluble mediators in...
Modulating other components of...
Conclusions and perspectives
Abbreviations
Acknowledgements
References
Introduction
Modulating antigen presentation...
Modulating cytotoxic T...
Modulating T helper cells (Th...
Modulating immunosuppressive...
Modulating soluble mediators in...
Modulating other components of...
Conclusions and perspectives
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(1):126-151. doi:10.7150/thno.29431 This issue Cite
Review
Engineering Nanoparticles for Targeted Remodeling of the Tumor Microenvironment to Improve Cancer Immunotherapy
Shan Gao1, Dongjuan Yang1, Yan Fang1, Xiaojie Lin1, Xuechao Jin1, Qi Wang1, Xiyan Wang1, Liyuan Ke2, Kai Shi1 ![]()
1. Department of Pharmaceutics, School of Pharmacy, Shenyang Pharmaceutical University, Shenyang, Liaoning 117004, P. R. China
2. Liaoning Cancer Hospital & Institute, Shenyang, Liaoning 110042, P. R. China
Received 2018-8-23; Accepted 2018-11-1; Published 2019-1-1
Citation:
Gao S, Yang D, Fang Y, Lin X, Jin X, Wang Q, Wang X, Ke L, Shi K. Engineering Nanoparticles for Targeted Remodeling of the Tumor Microenvironment to Improve Cancer Immunotherapy. Theranostics 2019; 9(1):126-151. doi:10.7150/thno.29431. https://www.thno.org/v09p0126.htm
Other stylesAbstract

Owing to the fast-paced growth and cross-infiltration of oncology, immunology and molecular biology, tumor immunotherapy technology represented by immune checkpoint blockade and chimeric antigen receptor (CAR) T cell therapy has lately made remarkable advancements. In comparison with traditional chemotherapy, immunotherapy has the potential to elicit a stronger sustained antitumor immune response in those patients who have advanced malignant malignancies. In spite of the advancements made, a significant number of clinical research works have validated that an extensive proportion of cancer patients still manifest insensitivity to immunotherapy, primarily because of the immunomodulatory interactions between tumor cells and the immunosuppressive tumor microenvironment (TME), together mediating the immune tolerance of tumors and accordingly impacting the positive response to immunotherapy. The intricate immunosuppressive networks formed by stromal cells, inflammatory cells, vasculature, extracellular matrix (ECM), and their secreted cytokines in the TME, play a pivotal role in tumor immune escape. Specific blocking of inhibition pathways in the TME is expected to effectively prevent immune escape and tolerance of tumor cells in addition to their metastasis, accordingly improving the antitumor immune response at various phases of tumor growth. Emerging nanoscale targeted drug carriers truly suit this specific requirement due to their specificity, biocompatibility, and convenience of production. This review emphasizes recent attempts to remodel the tumor immune microenvironment using novel nanoparticles, which include specifically eliminating immunosuppressive cells, reprogramming immune regulatory cells, promoting inflammatory cytokines and blocking immune checkpoints. Targeted remodeling of the immunosuppressive TME using well-designed and fabricated nanoparticles provides a promising strategy for improving the effectiveness of current immunotherapy and is greatly significant.
Keywords: vaccines, tumor microenvironment, nanoparticles, cancer, immunotherapy
Introduction
Today, tumors are still counted among the most frequently found and serious diseases, directly damaging human life. Traditional treatments for malignant tumors include primarily surgical resection, radiotherapy, and chemotherapy, which show positive curative impacts on early tumors but fewer efficacies for advanced tumors and metastases, and have a higher risk of recurrence. In recent years, some advancement in cancer chemotherapy has been made, and the survival of cancer patients has been substantially extended, in particular for leukemia and malignant lymphoma [1]. Nevertheless, no satisfactory performance has been attained in the deadliest solid tumors. Thanks to the growth of molecular biology and associated disciplines, immunotherapy for cancer treatment has received increasing attention and has emerged as a more promising alternative to conventional surgery and radiochemotherapy [2].
In the early stages of tumor immunotherapy, substantial progress was made in using immune effector cells (e.g., tumor-infiltrating lymphocytes, cytokine-induced killer cells, lymphokine-activated killer cells, and NK cells) for adoptive treatment, which includes isolation of immunocompetent cells from cancer patients, amplification, and function identification in vitro, followed by transfusion back into the patient, resulting in killing of tumor cells directly or through stimulation of the immune response of the body. Nevertheless, owing to numerous issues—e.g., low amplification rate, difficulty in sourcing cells, lack of tumor antigen specificity—its application in the clinic is constrained [3]. Owing to deepening research addressing tumor molecular biology, a new technique based on modification of the T cell antigen receptor has been developed, namely chimeric antigen receptor T cell immunotherapy (CAR-T), which is capable of identifying tumor cells in the non-restrictive manner of the major histocompatibility complex (MHC). Today, CAR-T therapy has the potential of attaining a remission rate of 60-80% in patients with acute lymphoblastic leukemia, which is far superior to conventional chemotherapy [4]. Nevertheless, CAR-T continues to face huge challenges for treating solid tumors, primarily due to the inability of T cells to effectively infiltrate tumor tissues, off-target effects in the immunosuppressive TME, and the cytokine storm produced by their mechanism of killing tumor cells [5]. Besides using adoptively reintroduced tumor antigen-specific T cells to enhance the immune response towards tumors, encouraging progress has been made in the use of monoclonal antibodies (mAbs) to block immune checkpoints, accordingly enhancing the function of T cells. Co-stimulatory inhibitory molecules, which include mAbs of CTLA-4, PD-1 and PD-L1, have received approval by the FDA for treating advanced melanoma, small cell lung cancer, and metastatic bladder cancer [6]. Despite the fact that mAbs targeting co-suppressor molecules attain the objective of killing tumors through the activation of T cells, they do not have the potential for specifically activating tumor-specific T cell responses, which is expected to indispensably trigger polyclonal T cell activation and cause gravely adverse reactions such as autoimmune responses. Additionally, because mAbs poorly permeate tumor tissue, they are merely capable of relieving immunosuppression of T cells that are located at the edge of the tumors, and so still have a poor therapeutic effect on solid tumors [7]. Therapeutic tumor vaccines have emerged as a pivotal breakthrough in treating solid tumors since they have the ability to induce T cells to attack tumors with high specificity. The only epoch-making cancer vaccine product currently approved by the FDA is Provenge, adapted to the solid tumor of prostate cancer [8]. The therapeutic principle of this methodology works on the basis of introducing tumor-specific/-associated antigens (TSA/TAA) into dendritic cells (DCs), which are subsequently presented to tumor-specific T lymphocytes, which activates them and stimulates an effective immune response and killing of the tumor. To date, DCs-based therapeutic tumor vaccines have undergone phase III clinical trials in malignant melanoma, prostate cancer, non-small cell lung cancer (NSCLC) and prostate cancer (Table 1); nevertheless, the majority of their therapeutic effects are quite limited (<5%). The immunosuppressive TME plays a pivotal role in the ineffective immune response of antitumor vaccines, inhibiting not only antigen uptake and presentation but also the activation and infiltration of T lymphocytes by DCs in vivo.
Table 1
Typical nanoparticles-based cancer vaccines in clinical trials.
| Nanoparticles | Payloads | Clinical stages | Indications | Ref |
|---|---|---|---|---|
| Liposome (L-BLP25) | MUC-1, tecemotide monophosphoryl lipid A | Terminated after phase III | NSCLC | [9] |
| Liposome (AS15) | MAGE-A3, CpG 7909 monophosphoryl lipid A | Terminated after phase III | Melanoma, NSCLC | [10] |
| Liposome (ISCOMATRIX) | E7, saponin, | Terminated after Phase II | Melanoma | [11] |
| Liposome (DPX) | HLA-A2, Survivin polynucleotide | Phase I/II | Ovarian cancer | [12] |
| Liposome (Lipo-MERIT) | mRNA encoding four melanoma antigens (NY-ESO-1, MAGE-A3, tyrosinase, TPTE) | Phase I/II | Melanoma | [13] |
| Cholesteryl pullulan (CHP) | NY-ESO-1 protein | Phase I/II | Esophageal cancer | [14] |
| Autophagosomes (DPV) | HPV Imiquimod | Phase I/II | NSCLC Prostate cancer | [15] |
| Virus-like particles (VLPs) | Melan-A/MART-1, CpG | Phase I/II | Melanoma | [16] |
| Hybrid lipsome (Lipovaxin-MM) | Melanoma cell membrane, Antibody targeting DCs IFN-γ | Phase I/II | Melanoma | [17] |
In general, provided that a positive response to immunotherapy is dependent on immunomodulatory interactions between tumor cells and the TME, a comprehensive elucidation of the immunomodulatory mechanisms of the TME is expected to provide a new methodology for improving the effectiveness of current immunotherapy. The TME is an intricate environment, wherein tumors are dependent on a variety of extracellular matrices (for instance, collagen and laminin) and stromal cells, which include mesenchymal-derived fibroblasts, immune cells and vascular endothelial cells [18]. The TME provides support for cells in the matrix, in addition to secreting different kinds of cytokines and chemokines, constituting a bridge of information exchange between the TME and tumor cells. During tumor growth, the TME interacts with tumor cells by means of different kinds of immune cells (for instance, T lymphocytes, dendritic cells, macrophages, and myeloid-derived suppressor cells) to mediate their immune tolerance, accordingly impacting the clinical effect of immunotherapy. That is why blocking the immunosuppression of the TME benefits recovery and reconstruction of the normal antitumor immune defense potential of the human body, accordingly enhancing the comprehensive therapeutic effect of different types of tumor treatment methodologies, which include immunotherapy [19]. In recent years, the concept of the immune system regulating tumor progression is undergoing a new renaissance. By means of tumor-specific antigens expressed by tumor cells or cellular stress-induced molecules, the immune system is capable of specifically identifying cells and molecules and eliminating them prior to their causing damage, a process termed “tumor immune surveillance”. In spite of the presence of tumor immune surveillance, it is possible for tumors to occur within the normal immune system. Despite the fact that the immune system inhibits tumor proliferation, tumor cells have the ability to attenuate or evade immune stress in a manner similar to their evasion of classical tumor suppressor mechanisms [20].
Owing to the fast-paced growth of nanomaterials and the comprehensive understanding of TME-mediated immunotherapy in recent years, nanoparticles have been put to extensive use for regulating the TME and improvement of tumor immunotherapy: (1) Nanoparticles can be loaded with different types of adjuvants (e.g., peptides, proteins, and nucleic acids) and TSA/TAA in the same vector. These payloads can be precisely delivered to lymphoid organs (lymph nodes or spleen) or antigen-presenting cells by surface modification of the nanoparticles with targeting ligands as well as via their unique physicochemical attributes that facilitate recognition and uptake by DCs [21]. (2) Owing to the fact that the size of the nanoparticles is similar to that of pathogens, they have the ability to mimic the endocytosis of pathogens into endosomes, followed by promoting presentation of antigens by means of the MHC-I pathway in a "cross-presentation" manner. This is more likely to induce a long-term, effective, tumor-specific CTL response in comparison to free antigen. (3) Nanoparticles are also capable of safeguarding biomacromolecular immunotherapeutic components (e.g., peptide antigens, nucleic acid vaccines and DNA adjuvants) from premature degradation in the biological environment, accordingly enhancing their stability in vivo, while also being capable of sustained release of the antigen, helping to augment the intensity of the immune response [22]. (4) With the use of unique attributes of the TME, for instance, hypoxia, weakly acidic pH and tumor pressure gradient as well as the nature of extracellular matrix, it is possible to design and develop nanoparticles that have different types of environmental stimuli responses to precisely deliver the immunotherapeutic ingredients to particular cells or non-cellular components in the TME to improve the immune response [23]. (5) Nanoparticles can be easily subjected to functional chemical or biological modifications. Tumor stromal and immune cells typically overexpress or specifically express particular cellular surface molecules as well as secretory factors, providing a general idea for the functional design of nanoparticles. Targeting these cell markers has the potential to augment the uptake of nanoparticles by the TME and lower adverse effects on normal cells. (6) The efficacy of a single immunotherapy for primary solid tumors is usually constrained. Radio/ chemotherapy has the ability to kill some tumor cells in advance, leading to exposure of lots of tumor antigens in the TME, which can mobilize more immune effector cells for remodeling the immunosuppressive status of the TME. Nanoparticles are capable of combining immunomodulators with chemical drugs, photosensitizers and photothermal materials, which allows the combination of immunotherapy with chemotherapy, photodynamic therapy, and photothermal therapy to achieve synergistic antitumor effects [24].
The current review will summarize recent research progress addressing the synergistic effect of the TME on immunotherapy as the basis for engineering nanoparticles, and will analyze key challenges and prospects in the development of immunotherapy combined with nanotechnology.
Modulating antigen presentation with nanoparticles
Antigen presentation deals with the intricate mechanism in which antigens are taken up by antigen -presenting cells (APCs) and processed into immunogenic peptides, subsequently presented on the surface of APCs in the form of MHC peptide complexes, and finally identified by immune effector cells. Being specific, dendritic cells (DCs) are the most powerful professional APCs found in vivo, and constitute essential starting and regulating factors of the tumor immune response [25]. Nonetheless, antigen presentation and subsequent activation of immune effector cells by DCs is usually impaired by the immunosuppressive TME, which results in a poor immune response to tumors [26].
Targeting DCs
Dendritic cells (DCs) are regarded as effective APCs that have the most striking attribute of stimulating naïve T cells; accordingly, they are also key promoters of the immune response, in addition to playing a major role in inducting antitumor immunity. In accordance with their different abilities for stimulating T cell proliferation, DCs can be segregated into immature dendritic cells (imDCs) and mature dendritic cells (mDCs); mDCs are likely to induce Th1-type immune responses through the activation of TAAs-specific CTLs [27]. There are numerous antigenic peptides/MHC class II on the surface of DCs binding to TCRs that become the first signal of T cell activation; subsequently, co-stimulatory molecules like CD80, CD86, and CD40 are up-regulated for the purpose of providing the second signal for a sufficient T cell activation. DCs also self-secrete or induce other immune cells to synthesize and secrete cytokines (e.g., IL-12, TNF-α, and IFN-γ), aimed at providing the third signal, accordingly generating the effects of antigen-specific CTLs [28]. The immune impacts produced by tumor vaccines first require APCs, in particular DCs, for efficient uptake and presentation of tumor antigens. Soluble proteins are typically not conveniently absorbed by APCs, while antigen-loaded nanoparticles, being the same size as pathogens, are more conveniently recognized and ingested by APCs, accordingly increasing the immunogenicity of tumor vaccines [29]. In recent years, adoptive immunotherapy, which is based on DC vaccines for malignant tumors, has received increasing attention owing to the fast-paced growth of biotechnology and research addressing molecular mechanisms underlying cancer occurrence [30]. The basic principle behind this therapy involves separation of autologous DCs from the patients' bodies, followed by stimulation of their maturation by loading a proper source of tumor antigens in vitro. Following subcutaneous or intramuscular inoculation, the DC vaccines migrate to the immune organs (e.g., lymph nodes and spleen) and present TAAs to T cells for an effective immune response that is coupled with favorable clinical performances. Even though significant numbers of tumor vaccines have entered clinical trials for treating malignant tumors like melanoma, breast cancer and prostate cancer, extremely complicated operations and poor clinical effectiveness have constrained extensive application of this technology in the clinic [31].
Nanoparticles have exclusive potentials for enhancing antigen presentation efficiency in vivo and remodeling the immunosuppressive TME. Since tumor antigens share a good similarity with normal antigens, adjuvants are usually required to induce effective immune responses. Nanoscale drug delivery systems with uniform particle size and unique transport characteristics in vivo, are capable of substantially boosting the immunogenicity of tumor antigens by loading antigens and adjuvants individually in physically identical nanoparticles [32]. How to efficiently deliver the TAAs to DCs constitutes a pivotal prerequisite for nanocarrier systems for the promotion of tumor immunotherapy, and the particle size, shape and surface charge of nanovaccines are expected to play pivotal roles in antigen delivery. Among them, the particle size of the nanocarriers has a close association with the cellular uptake mechanism and endocytic pathway, determining the fate and overall biological impacts of the nanoparticles in cells [33]. Smaller nanoparticles, having a diameter of 25-40 nm, are able to reach draining lymph nodes through the tissue barrier faster than larger ones (>100 nm), which are typically retained at the injection site and transported to the lymph nodes by means of DCs; accordingly, smaller nanoparticles are believed to activate the immune response in a more effective manner. When the particle size of the nanocarrier is above 500 nm, they have a greater likelihood to be taken up by macrophages through macropinocytosis or phagocytosis. In addition to the effect of particle size, reports suggest that the shape of the nanocarriers also impacts the level of cellular uptake as well as biodistribution. It has been suggested that non-spherical nanoparticles are capable of avoiding non-specific cellular phagocytosis, accordingly prolonging systemic circulation. Nevertheless, non-spherical nanoparticles are not easily taken up by DCs in comparison with spherical nanoparticles [34]. Furthermore, the surface charge of nanoparticles also plays a pivotal role in the mechanism of particle internalization, even impacting the attributes of the immune response the nanoparticles induce. For instance, cationic nanoparticles can be taken up more rapidly by macrophages or DC cells, and, in turn, manifest a more robust potential for lysosomal escape. Nonetheless, they are prone to adsorption of serum proteins and react with negatively charged matrix components in the TME (e.g., collagen and hyaluronic acid), resulting in poor permeability of tumor tissues. Neutral particles (±10 mV) have been reported to possess the best long-circulation potential, and their penetration depth into tumors is three times that of charged particles [35].
Biodegradable nanoparticles are among the promising vehicles for cancer immunotherapy that have a proven efficacy for antigen presentation and cell stimulation. The most representative biodegradable polymer material is poly(lactic-co-glycolic acid) (PLGA), which is not only non-toxic but also possesses a good protection effect for antigens [36]. The size of PLGA nanoparticles is typically the same as that of pathogens, making them prone to absorption by APCs, accordingly enhancing the immune response. Recently, inorganic and metal nanoparticles have also been developed in the same manner [37]. Researchers have conjugated functional ligands to mesoporous silicon, calcium phosphate, gold, and upconversion nanoparticles for the preparation of nanovaccines that have the ability to induce CTLs-mediated antitumor immune responses [38-42]. Additionally, peptide micelles, dendrimers, oncolytic virus (OVs) and artificial exosomes have entered clinical trials as DCs nanovaccines, which have immense promise for antitumor immunotherapy [43-45]. Conversely, there are a variety of TSAs and surface functional molecules on tumor cell membranes, which are difficult to attain by conventional synthetic techniques. Despite the fact that patients' own tumors are regarded as the ideal means of producing antigenic materials, the expected effects have not yet been attained, most likely owing to dilution of TSAs by an extensive number of housekeeping proteins on tumor cells and their interference with the identification of tumor antigens [46]. Using nanoparticles covered with tumor cell membranes is likely to better address this issue (Figure 1). This novel drug delivery system is built around the basis of a polymeric nanocarrier, which carries a huge amount of adjuvant, together with a tumor cell-derived membrane layer coating containing different kinds of TSAs [47]. This nanoscale drug delivery system removes all intracellular housekeeping proteins, which allows for better recognition of TSAs by the immune system [48]. Other investigations have made use of erythrocyte membrane antigens to improve DCs targeting and antigen presentation efficiency [49].
Figure 1
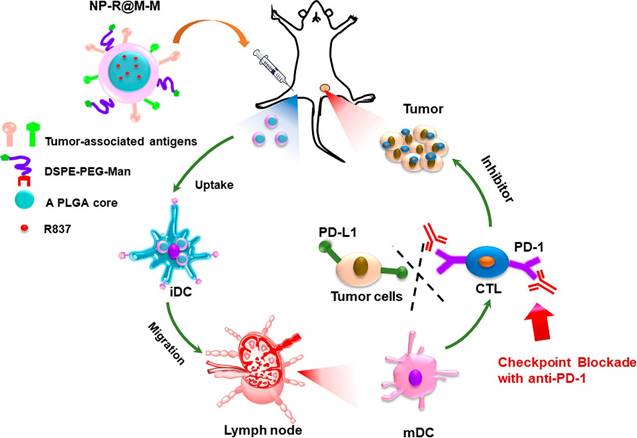
Schematic illustration of tumor cell membrane-coated PLGA nanoparticles that act as nanovaccines to induce antitumor immunity. The PLGA nanoparticles were first loaded with a toll-like receptor 7 agonist of imiquimod (R837) and then coated with a cancer cell membrane whose surface proteins were capable of acting as tumor-specific antigens (NP-R@M). By further surface modification with mannose as a ligand (NP-R@M-M), the obtained nanovaccines showed enhanced cellular uptake capacity by APCs, such as DCs, which are expected to be stimulated to the mature state to trigger an antitumor immune response. Reproduced with permission from [47], copyright 2018 American Chemical Society.
Engineering aAPCs
Adoptive immune therapy using autologous or allogeneic immune cells has been extensively tested over the past few decades and has made immense progress in antitumor clinical applications. In spite of these advances, treatment with innate immune effector cells is constrained by the cost and complexity of generating tumor-specific T cells. Alternatively, a strategy making use of biomimetic materials for the development of artificial antigen presentation cells (aAPCs) has attracted significant attention [50]. This concept receives inspiration from the natural antigen presentation mechanism, wherein specific MHC-peptides and co-stimulatory molecules (e.g., CD3, CD28) are conjugated onto synthetic nanoparticles for efficiently amplifying antigen-specific T cells rather than APCs [51]. Engineering multifunctional nanoparticles allows implementation of this new immunotherapy strategy [52]. Dextran-coated superparamagnetic iron oxide nanoparticles have been designed as a type of aAPCs for the amplification of T cells, on which both the MHC-Ig dimer and anti-CD28 antibody were conjugated for the provision of both the antigen-specific signal and co-stimulatory signal, correspondingly. When the nanoscale aAPCs are bound to T cell receptors, they accumulate in the magnetic field, leading to enrichment of T cells and accordingly, improving the activation of T cells together with their antitumor activity [53]. Magnetic field-induced aAPCs have the potential of stimulating both the activation and proliferation of antigen-specific T lymphocytes, which provides a new direction for tumor immunotherapy. Iron oxide nanoparticles-derived biomimetic magnetosomes were developed as multifunctional aAPCs in which the leucocyte membrane was camouflaged and modified using MHC-I molecules and anti-CD28 antibody, correspondingly [54]. Upon effective amplification and stimulation ex vivo by nanoscale aAPCs, the re-infused antigen-specific CD8+ T cells were visually guided with the magnetosomes to tumors tissues with the help of magnetic resonance imaging (MRI). The results suggested that aAPCs had the potential of retarding growth of a lymphoma model in vivo without significant systemic toxicity. Accordingly, we expect that aAPCs are going to serve as powerful artificial antigen-presenting constructs for both the stimulation and amplification of T cells.
Modulating cytotoxic T lymphocytes with nanoparticles
Cytotoxic T lymphocytes (CTLs) are a class of T cells that have CD8+ surface markers and are restricted by MHC class I molecules; they are responsible for eliminating cancer cells in the adaptive immune system [55]. Upon activation following recognition of tumor antigens presented by APCs coupled with the simultaneous acquisition of synergistic stimulation signals provided by costimulatory molecules such as B7/CD28 and CD40/CD40L, CD8+ T cells will proliferate and differentiate into functional CTLs. Following identification of tumor antigens, CTLs perform their tumor killing function by secreting perforin, granzymes, and IFN-γ [56]. Overall, tumor cell evasion of immune surveillance primarily occurs when CD8+ CTLs are ineffectively activated. Several investigations have confirmed that the greater the number of infiltrating CTLs in tumor tissues, the better the patient's prognosis [57]. Nonetheless, tumor cells are still not eradicated despite sufficient CTL infiltration in the tumor tissue. The mechanisms involved in the immune escape of tumor cells include a weakened antigen presentation ability of DCs owing to interference by the TME during their maturation mechanism, a lack of co-stimulatory molecules in APCs, and decreased expression of MHC-I antigens on the surface of tumor cells, which are capable of indirectly undermining CTLs' response in the TME. For instance, there are a number of cytokines in the tumor immune microenvironment that are capable of inhibiting the functions of CTLs, with IL-10 and TGF-β being the most obvious [58, 59]. IL-10 blocks the transformation of T cells into CTLs, while TGF-β inhibits the proliferation, differentiation, and immune activity of CTLs and NK cells [60]. This is why the activity of CTLs is usually inhibited and they are unable to effectively exert an antitumor impact subjected to the co-regulation of many immune factors in the TME [61].
Modulating engineered T cells
Aimed at improving the reactivity and specificity of T cells against the tumor, a new chimeric antigen receptor T cell immunotherapy (CAR-T), which is based on the principle of antibody recognition, has been successfully developed [62]. This technique holds the potential of producing a large number of specific T lymphocytes against tumor antigens, selectively targeting and killing tumor cells with the help of the non-MHC restriction. The principle of CAR-T technology deals with combining the high affinity of antibodies against tumor antigens with the killing effect of T lymphocytes, in addition to using genetic engineering technology to link the variable region fragments of single-chain antibodies (scFv), costimulatory molecules, and signal-transducing peptides together. Subsequent to transfection into lymphocytes by means of retrovirus or lentivirus packaging, the recombinant chimeric receptor specifically binds to the corresponding antigen expressed by the tumor cells, such as a monoclonal antibody, accordingly exerting a tumor killing impact that is subject to activation of the signal transduction peptide [63]. Despite the fact that CAR-T technology has attained outstanding performances in treating acute lymphocytic leukemia and non-Hodgkin's lymphoma and is regarded as one of the most promising therapies for cancers, there are still some issues and limitations that must be addressed [64]. Firstly, the course of treatment is quite intricate, requiring extraction of T lymphocytes from the peripheral blood of patients and amplification in vitro, typically for more than 2 to 3 weeks, prior to adoptive reinfusion [65]. With the above considerations, in situ construction of CAR-T in vivo provides a new direction for improving CAR-T therapy. Poly (β-amino ester) nanoparticles with a payload of plasmid DNA encoding leukemia-specific chimeric antigen receptors (CAR) were designed and prepared for antitumor immunotherapy [66]. Upon anti-CD3 antibodies-mediated endocytosis by lymphocytes, the polymeric nanoparticles selectively transfected CAR genes into the nuclei of host T cells, subject to guidance from both a microtubule-associated sequence and nuclear localization signals coupled on the surface of the nanocarriers. T cells programmed by the synthetic nanoparticles were observed to express CAR within 24 to 48 hours in vitro. Following systematic administration in vivo, the nanoparticles were rapidly identified and bound to peripherally circulating T cells, and were highly distributed in the spleen, lymph nodes and bone marrow of mice. Additionally, they manifested comparable therapeutic efficacy to conventional CAR-T in a murine lymphoblastic leukemia framework. The survival of mice with the use of both therapies increased by an average of 58 days compared to the control group. Despite the fact that this in situ production of CAR-T cells in vivo is a representation of an innovative approach for overcoming both the time and cost of current autologous reinfusion techniques, it has yet to be verified whether this methodology is capable of effectively producing CAR-T cells as well as a durable immune response in the human body, and whether there are toxicity issues caused by off-target effects.
Another factor that limits the clinical application of CAR-T therapy is its poor efficacy against solid tumors due to the suppressive TME that inactivates tumor infiltrating lymphocytes (TILs) through the production of different kinds of immunosuppressive molecules. Additionally, the treatment usually releases a large number of inflammatory cytokines (e.g., IFN-γ, TNF-α) in the body, which is attributed to the systemic administration, accordingly leading to multiple organ failure [67]. This is the reason that nanoparticles-based tumor-targeted therapy is required for the purpose of remodeling the TME without giving rise to the systemic toxicity. Owing to the fact that the small molecule adenosine has the ability to inhibit the function of both CD4+ and CD8+ T cells by binding to and activating the A2a adenosine receptor (A2aR) expressed on the surface of T cells, small molecule antagonists specific to A2aR can be put to use for blocking this inhibitory pathway. Nevertheless, their clinical application is constrained due to the difficulty in delivering them to immune cells within the TME. In a bid to overcome this limitation, CD19 CAR-engineered T cells were employed as active chaperones for successfully delivering antagonist-loaded cross-linked liposomes to TILs deep in the immunosuppressive TME. No significant reduction in the tumor size of mice bearing SKOV3.CD19 tumor with CAR-T cells treatment or a mixture of CAR-T cells and liposomes was observed. In contrast, five of the six tumor-bearing mice treated with CAR-T cell-conjugated liposomes manifested a decline in tumor size of more than 50% and one mouse manifested a 44% decline. Ex vivo analysis shed light on the fact that CAR-T cell-conjugated liposomes are capable of inducing 10.8% of activated T cells, in addition to high levels of IFN-γ secretion in tumor tissues [68].
Enhancing immune checkpoints blockade
It is fully known that T cells constitute the key recognition and effector cells of the acquired immune response, and their activation requires the simultaneous presence of two signals: one is the antigen-specific signal, the first signal mediated by the T cell receptor (TCR) and MHC; the second is the co-stimulatory signal that is mediated by the membrane protein molecules expressed on the surface of T cells as well as their ligands. The complete activation of T cells subsequent to antigen recognition must be dependent on this signal pathway. On the basis of the regulatory functions of co-stimulatory molecules on T cell activation signals, positive co-stimulatory molecules (including CD28/CD80/ CD86, 4-1BB/4-1BBL, OX40/OX40L) improve TCR signaling-mediated immune responses; on the other hand, negative co-stimulatory molecules (including PD-1/PD-L1, CTLA-4/CD80/CD86, Tim-3/Galectin-9) suppress the immune response mediated by TCR signals, which are also termed immune checkpoints [69]. These co-stimulatory molecules initiate, stimulate, amplify, and enhance the immune response at different stages, together with precisely mediating their extent and duration. In tumor tissues, the negative regulatory checkpoints occupy a dominant position while inhibiting the process of T cell activation. Tumor cells resort to immune checkpoints for the purpose of evading the attack of immune cells, accordingly they are one of the key reasons for tumor immune tolerance. Accordingly, immune checkpoints constitute key factors in the maintenance of self-tolerance and modulate signaling pathways of immune responses, playing a pivotal role in the protection of the body from autoimmunity and inflammation by impacting and interfering with the immune response of CTLs [70].
Figure 2
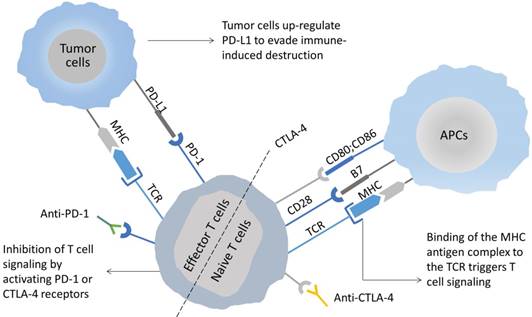
The mechanism of immune checkpoint blockade using anti-CTLA-4 and anti-PD-1/PD-L1 mAbs. CTLA-4 and PD-1/PD-L1 represent two T cell-inhibitory receptors with independent mechanisms of action. (1) Effective T cell activation requires at least two signals: First, T cells recognize antigen peptide-MHC complex on the APC surface by TCR. Second, co-stimulation through combining CD28 with CD80/CD86. Since CTLA-4 has a greater affinity for CD80/CD86 than CD28, it preferentially binds to the ligand of CD28 and, at sufficient levels, inhibits immune activation. (2) PD-1 is expressed by T cells, while PD-L1 is expressed in tumor cells and tumor-infiltrating immune cells. Inhibition of the interaction between PD-1 and its ligands is expected to significantly enhance the function of T cells and lead to antitumor activity. Accordingly, therapeutic blockade of immunosuppressive checkpoints provides a potential means of boosting antitumor immunity.
As revealed experimentally, suppressive immune checkpoint pathways are always activated in the inflammatory TME, which allow tumor cells to evade immune surveillance, in addition to eradicating their immune response to TILs [71]. This has resulted into the growth of different types of immune checkpoint inhibitors (ICIs) for reinvigorating dysfunctional or exhausted T cells through the restoration of immunity that are currently being tested in clinical trials or have been approved for a variety of advanced metastatic cancers. Cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) is a leukocyte differentiation antigen and a transmembrane receptor on T cells, sharing the B7 ligand with its co-stimulatory molecule receptor (CD28). Upon binding to the B7 molecule, it stimulates T cell anergy, accordingly becoming involved in the negative regulation of immune response [72]. CTLA-4 on the surface of T cells is highly homologous to CD28 and is mediated by interacting with primary ligands (B7-1/CD80 and B7-2/CD86) on the surface of APCs (Figure 2). Contrary to the functions of CD28, CTLA-4 primarily inhibits T cell activation, in addition to inducing their unresponsiveness. The antitumor mechanisms of CTLA-4 antibody primarily include the following two types: (1) modulation of tumor-specific immune effector cells, for instance, CD8+ T cells for promotion of their clonal proliferation; (2) removal of Tregs to relieve the inhibition of the tumor-associated immune response [73]. Likewise, programmed death receptor-1 (PD-1) and its ligand (PD-L1) constitute an innovative group of ICIs limiting the excessive immune response to antigens and preventing autoimmunity [74]. Among them, PD-1 can be expressed on a variety of immune cells, which include NK cells, B lymphocytes, T lymphocytes, DCs, and activated monocytes. PD-L1 is overexpressed on most tumor cells and promotes cancer evasion of immune surveillance via inhibiting functions of CTLs. Accordingly; an elevated expression of PD-L1 in tumors is linked to incapability or depletion of T cells, accordingly leading to avoidance of immune surveillance. The PD-1/PD-L1 pathway modulates immunosuppression primarily with the help of the following mechanisms: (1) binding of PD-L1 on the surface of tumor cells and myeloid-derived suppressor cells (e.g., TMAs, MDSCs, etc.) with PD-1 on the surface of tumor-specific T cells has the potential to induce apoptosis and depletion of TILs in TME. The involved mechanisms include blocked expression of anti-apoptotic factor Bcl-xL and phosphorylation of PI3K, which are deemed essential for the maintenance and survival of CD8+ T cells by costimulatory signals provided by CD28 ligation. (2) The activated PD-1 prevents T cells from proliferating by means of selectively inhibiting the signaling pathways of RAS/ MEK/ERK and PI3K/AKT, accordingly blocking cell cycle-related gene transcription as well as protein expression. (3) The expression of PD-L1 on the surface of APCs is capable of promoting the transformation of CD4+ T cells into induced Tregs (iTregs), together with maintaining their immunosuppressive function by down-regulating the phosphorylation levels of mTOR, AKT, S6 and ERK2, and up-regulating PTEN expression in CD4+ T cells. This is why blocking the PD-1/PD-L1 signaling pathway is expected to restore the function of effector CD8+ T cells, meanwhile suppressing the function of Tregs and MDSCs, accordingly enhancing the antitumor effect of the immune system [75].
Figure 3
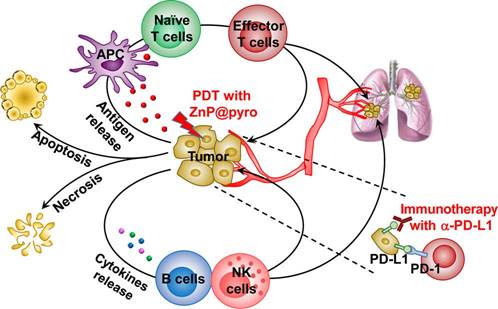
Illustration of the mechanisms underlying the combination of PDT with immunogenic ZnP@pyro that enhances the sensitivity of metastatic tumors to PD-L1 blockade immunotherapy. PDT with ZnP@pyro induced ICD and lead to release of TAAs, which were then presented to naïve T cells to stimulate the production and proliferation of tumor-specific effector T cells. In addition, PDT with ZnP@pyro also elicited an inflammatory environment that enhanced infiltration of effector T cells and other immune cells (such as B cells and NK cells) into primary and metastatic tumors. When combined with ICIs targeting PD-L1, PDT with ZnP@pyr not only eradicated the primary tumors, but also rejected the metastatic tumors through a systemic antitumor immune response. Reproduced with permission from [79], copyright 2016 American Chemical Society.
Despite the substantial progress that ICIs have made in clinical applications in recent years, some issues requiring solutions have started to emerge. One is that monotherapy that makes use of ICIs has limited durable clinical responses in just a small fraction of patients because of the occurrence of primary and adaptive resistances to ICIs therapy [76]. Accordingly, it is essential to combine ICIs with other treatments, for instance, chemotherapy, radiotherapy and photothermal therapy, to maximize the clinical benefit of immune checkpoint blockade therapy [77]. A multifunctional PLGA nanoparticle was developed that co-encapsulated the near-infrared (NIR) dye indocyanine green for enabling photothermal therapy and the TLR7 agonist imiquimod for activating immune responses [78]. Upon NIR-induced photothermal ablation of the primary tumor through administration of the nanoparticles, the released TAAs worked with the nanoparticulate adjuvant to carry out a vaccine-like function. In combination with the ICI therapy, a robust immune response was generated that suppressed immunosuppressive Tregs and cleared distant remaining tumor cells in a murine breast cancer model. A similar investigation made use of zinc pyrophosphate (ZnP) nanoparticles loaded with the photosensitizer pyrolipid (ZnP@pyro) for PDT to improve the sensitivity of tumor to PD-L1 blockade immunotherapy [79]. Upon irradiation with a light-emitting diode at 670 nm, apoptosis and necrosis of tumor cells was directly induced by generating of singlet oxygen. Additionally, PDT with immunogenic ZnP@pyro induced immunogenic cell death (ICD) and released TAAs, which were subsequently presented to naïve T cells to stimulate both the production and proliferation of tumor-specific effector T cells. In combination with PD-L1 checkpoint blockade immunotherapy in a mouse 4T1 mammary cancer model, PDT with ZnP@pyr eliminated the primary tumors and rejected metastatic tumors via a systemic antitumor immune response (Figure 3). On the basis of the understanding of the role of MEK and PI3K pathways in tumorigenesis, where the antiapoptotic protein expression mediates tolerance to T cell immune responses, a nanoscale supramolecular therapeutic was designed and synthetized from the active molecular subunits of selumetinib and PI103 for the purpose of targeting both signals, respectively [80]. Contrary to conventional lipid nanocarriers, the supramolecular nanostructures provided higher stability as well as drug loading capacity in vitro. In combination with PD-1/PD-L1 inhibitors, the nanoscale supramolecular targeted therapeutic improved the antitumor performance in models of melanoma and breast cancers in vivo.
Another factor that limits the clinical performance of ICIs is that a substantial number of cancers do not respond to PD-1/PD-L1 immunotherapy. In particular, microsatellite stable (MSS) or mismatch repair (MMR)-proficient solid tumors, for instance, pancreatic ductal epithelial adenocarcinoma and colorectal carcinoma, are non-responsive. In addition, systemic administration of these antibody inhibitors inevitably leads to adverse effects, termed immune-related adverse events (irAEs), which limit their therapeutic utility in clinical trials. Aimed at addressing these issues, an engineered plasmid DNA coding a protein antagonist of PD-L1 called “trap” was designed and loaded in lipid-protamine-DNA (LPD) nanoparticles. Employment of targeted LPD nanoparticles enabled in situ expression of the protein antagonist locally and transiently in the tumor tissue, accordingly lowering the toxicity of the systemic administration. In combination with oxaliplatin (OxP)-mediated ICD, the PD-L1 trap enhanced the response of MSS tumors to the immunotherapy, manifesting substantial antitumor efficacy coupled with the prolonged survival [81]. In a significant aspect, the OxP treatment substantially augmented infiltration of CD8+ T cells and CD4+ T cells, activated DCs in tumors, and increased levels of Th1-type cytokines (which included IFN-γ and TNF-α); on the other hand, the changed levels of IL-4 and IL-10 were not significant. As suggested by these findings, the combination of OxP and PD-L1 traps in nanoparticles revoked the immunosuppressive TME of orthotopic MMR-proficient colorectal tumors, leading to activation of T cells and accordingly, boosting the efficacy of the immunotherapy. A similar investigation made use of a binary synergistic TME-activated prodrug nanoparticle in a bid to trigger ICD and elicit antitumor immunity. The nanoparticles were constructed from a homodimer of oxaliplatin (OXA) and indoleamine-2,3-dioxygenase (IDO) inhibitor with a dual responsiveness to tumor acidity and reduction. The activated OXA promoted intratumoral accumulation of CTLs by triggering ICD in TME, while the IDO inhibitor down-regulated IDO-mediated immunosuppression and suppressed regulatory T cells [82].
Modulating T helper cells (Th cells) with nanoparticles
Th cells, which are also termed CD4+ cells, are considered to be pivotal organizers of cell-mediated immunity, besides having involvement in various stages of the immune response. There are antigen receptors on their surface, which identify antigenic fragments presented by the MHC class II molecules of APCs [83]. When activated, immature CD4+ T cells differentiate into various subtypes, which include Th1, Th2 and Th17 cells. Th1 cells are documented to produce IFN-γ, IL-2 and TNF-α, which are involved in inducing macrophage activation, assisting in cytotoxic T cell differentiation, and mediating cellular immune responses (Figure 4). On the other hand, the primary roles of Th2 cells deal with assisting B cell activation, proliferation, differentiation, and maturation, and induction of the production of specific antibodies [84]. Subject to co-induction of TGF-β and IL-6, immature CD4+ cells are capable of differentiating into the Th17 subpopulation, mediating the occurrence and progress of inflammatory responses, autoimmune diseases, cancers and transplant rejection by means of secreting IL-17 and IL-6. As revealed experimentally, Th17 cells are capable of inducing the activation of tumor-specific CD8+ T cells, promote the recruitment of dendritic cells into tumor tissues, and increase tumor-specific CD8+ T cells in draining lymph nodes [85]. Together, the immature CD4+ cells also differentiate into Tregs, expressing Foxp3 under induction of TGF-β alone, which is involved in immune regulation by means of secreting TGF-β. The balance between Tregs and Th17 cells has emerged as a prominent factor in the regulation of tumor immunity. Investigations have revealed that they are important members of the immunosuppressive TME, and, subject to specific conditions, can be converted into each other, together playing a pivotal role in the maintenance of the body's own immune stability and antitumor immunity. When the number of Tregs in TME is substantially increased, the antitumor immune response of the body can be inhibited; on the other hand, removal of Tregs has the ability to substantially increase TH17 cells in the tumor tissue, accordingly reconstructing antitumor immunity [86].
Figure 4
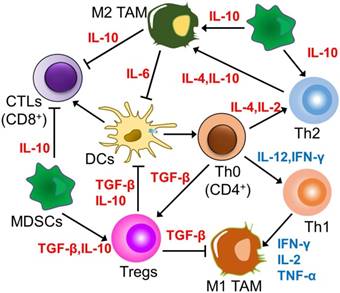
The main interaction between Th cells and other immune cells in TME. Th2 cells, M2 polarized TAMs, and MDSCs enhance each other's proliferation and phenotypes, in addition to maintaining the immunosuppressive effects of tumors. Together with Tregs, these cells suppress the activity and proliferation of immune effector cells, including Th1, M1 polarized TAMs, and CTLs.
In the immunosuppressive TME, tumor cells always induce elevated levels of Th2 cytokines, for instance, IL-4, IL-6 and IL-10, inhibiting the production of Th1 cytokines like IL-2, IL-12 and IFN-γ, and accordingly, preventing the activation of CD8+ CTLs precursors [87]. In the same manner, the key strategy for modulating Th cells deals with regulating the Th1/Th2 balance of CD4+ T cell response with nanoparticles. Despite the fact that both Th1 and Th2 subtypes are able to mediate antitumor immune responses, Th1 cells secreting IFN-γ have more effectiveness in this role. Research has shed light on the fact that this balance is primarily impacted by the processing and presentation mechanism of antigens by APCs [88]. There have been reported various factors impacting the Th1/Th2 immune response elicited by APCs, which include their maturation status and the uptake pathway of antigens, where the shape and size of nanoparticles crucially contribute towards this mechanism. The role of particle morphological features in the modulation of the immune response intensity and subtypes has been evaluated with the use of OVA as a model antigen. In vivo immunization research has revealed that smaller spherical polyethylene particles (193 nm) give rise to a stronger Th1-biased immune response, while larger rod-shaped particles (1530 nm) are inclined to produce a Th2 immune response [89]. The size-dependent immunomodulatory effects of gold nanoparticles could be ascribed to different mechanisms of their internalization, accumulation levels, and intracellular distribution within DCs, resulting in various modulations of mature signaling. That is why both the shape and size of nanoparticles are expected to constitute key parameters requiring consideration for inducing specific immunity in vaccine development. The precise design of the nanoparticle morphology for vaccination is likely to drive the Th1/Th2 balance following immunization, which is expected to help design a productive vaccine delivery system against different kinds of tumors. Furthermore, the redox properties of nanoparticles also shift this balance. Two inorganic nanoparticles, TiO2 and CeO2, have been presented as having approximately opposite effects on human DCs and Ths. TiO2 nanoparticles, having oxidative characteristics, enhanced DCs maturation, leading to Th1-biased responses; on the other hand, antioxidant CeO2 nanoparticles induced APCs to secrete the anti-inflammatory cytokine IL-10, resulting in Th2-dominated cell subtypes [90]. The opposite results are owing to the different potential of the nanoparticles to modulate reactive oxygen species (ROS), inducing an inflammatory response with the help of activation of the NLRP3 inflammatory complex coupled with expression of the pro-inflammatory cytokine IL-1β downstream of the ROS pathway.
Modulating immunosuppressive cells in the TME with nanoparticles
Tumor-associated immunosuppressive cells constitute a cohort of negative regulatory cells inhibiting the immune function in TME, which include regulatory T cells (Tregs), tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs). Once employed by tumor cells in the TME, they are expected to inhibit the body's antitumor immune response by means of paracrine effects, which include inhibiting the function and differentiation of APCs and T cell depletion and dysfunction, accordingly letting tumor cells escape the body's immune surveillance. Accordingly, precise regulation of these tumor-associated immunosuppressive cells requires rational design of a nanoparticle carrier for targeted delivery of immunomodulators into the TME for in vivo applications (Figure 5).
Figure 5
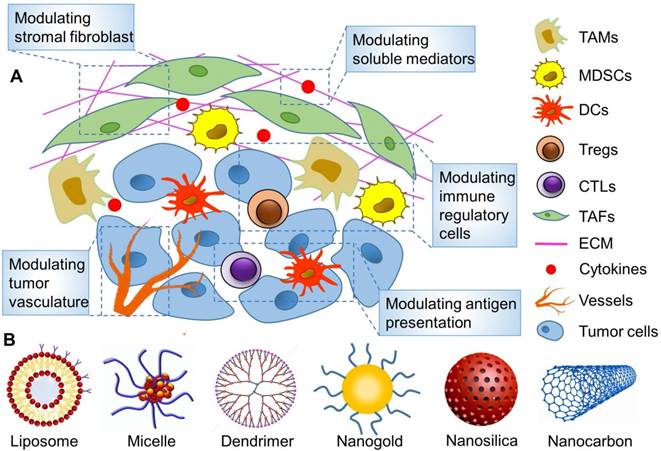
Targeted remodeling of immunosuppressive cells in the TME with nanoparticles to improve cancer immunotherapy. (A) The main strategies for modulating TME on the basis of nanoparticles. (B) Various reported nanoparticles used to improve cancer immunotherapy by remodeling TME.
Targeting Tregs
Tregs are a group of lymphocytes that negatively regulate the body's immune response. They typically play a pivotal role in the maintenance of autoimmune tolerance, in addition to being considered immunosuppressive [91]. Tregs negatively regulate the immune response primarily by three mechanisms: (1) induction of T cell apoptosis through cell-cell interactions; (2) suppression of immune responses through secretion of cytokines, for instance, TGF-β and IL-10; (3) release of perforin and granzymes in order to kill CTLs, monocytes and DCs directly [92]. Investigations have revealed that high levels of Tregs are observed in a variety of malignant tumors; these infiltrate lymph nodes, peripheral blood, and tumor-draining lymph nodes and are closely associated with the occurrence and progression of tumors, having a negative correlation with therapeutic outcomes. Accordingly, removal of Tregs or blocking of their immunosuppressive functions is expected to restore the antitumor effects of immunotherapies [93].
Overall, there are two strategies for targeting Tregs in tumors with nanoparticles: The first involves combination with anti-CTLA4 checkpoint blockade. Glucocorticoid-induced TNFR-related receptor (GITR) has been shown to be overexpressed predominantly by intratumoral CD4+ Tregs. A single-walled carbon nanotube (SWCNT) with ligands against GITR was firstly reported for Tregs targeting [94]. Ou et al. performed a combined treatment to melanoma with tLyp1 peptide-modified hybrid nanoparticles (hNPs) and anti-CTLA4 immune checkpoint inhibitor [95]. The conjugated moiety of tLyp1 peptide was capable of mediating efficient hNPs targeting to Nrp1 receptor, which is expressed in intratumoral Tregs. The combination with anti-CTLA4 antibody allowed the authors to augment tumor immunotherapy by suppressing intratumoral Treg function and elevating TILs in the TME. Another viable strategy employs tyrosine kinase inhibitors. As revealed experimenttally, activation of STAT3, a family of proteins that plays a regulatory role in signal transduction and transcription, mediates the expression of inflammatory factors as well as their corresponding immune responses [96]. Being more specific, it has been revealed that STAT3 of immune cells infiltrated in the TME is usually aberrantly activated, promoting secretion and expression of Th2 cytokines, accordingly suppressing Th1 responses as well as facilitating survival of Tregs. Frequent STAT3 activation in tumor cells is primarily owing to the convergence of STAT3 by a number of tyrosine kinases, which include VEGFR, PDGFR, EGFR and Src [97]. Moreover, sunitinib, which is a tyrosine kinase inhibitor targeting epidermal growth factor receptor (EGFR), was encapsulated in PLGA-based micelles to improve the effectiveness of Trp2 nanovaccine in treating melanoma. The delivery system enhanced infiltration of CD8+ T cells in tumor tissues and remodeled the stroma and angiogenesis in the ECM. The proportion of immunosuppressive cells, for instance, Tregs and MDSCs, was also lowered in the tumor suppressive microenvironment, which was accompanied by a tendency towards the Th1-dominant cytokine phenotype. Owing to the inhibition of STAT3 signaling pathways, the nanoscale micelles were able to substantially improve the efficacy of the vaccine against proliferation and immune escape of tumor cells [98].
Targeting TAMs
TAMs are important immunoregulatory cells, likely accounting for 50% of solid tumor tissues. TAMs are macrophages that infiltrate in the tumor stroma, performing the same kinds of functions as alternatively activated M2 macrophages, for instance, secreting immunosuppressive cytokines as well as growth factors that inhibit T cell proliferation and activation, promoting tumor cell growth, participating in tumor angiogenesis, and facilitating tumor invasion and metastasis [99]. In the early phase of tumor progression, TAMs with M2 phenotype are capable of promoting neovascularization and improving tumor cell invasion ability and the metastatic behavior of tumor cells. During tumor metastasis, M2-polarized macrophages promote formation of the tumor "pre-metastasis microenvironment" and the extravasation, survival, and sustained growth of tumor cells [100]. Additionally, TAMs have the ability to release different kinds of growth factors, for instance, VEGF to induce angiogenesis, and secrete Th2 cytokines in order to suppress tumor immunity [101].
On the basis of the origin and functions of TAMs, one viable strategy for targeting TAMs with nanoparticles deals with inhibiting recruitment of inflammatory monocytes to the TME, which can be attained through preventing tumor cells from secreting chemokines that attract macrophages, or by blocking macrophage surface receptors to prevent signal transduction [102]. Lipid nanoparticles with siRNA payload have been developed to modulate the expression of chemokine receptor CCR2 in inflammatory monocytes [103]. When administered in a systematic manner, the nanoparticles rapidly accumulated in the spleen and bone marrow, followed by high cellular localization of siRNA in monocytes. Subsequent to silencing of CCR2 mRNA in the inflammatory monocyte subset, the nanoparticles selectively inhibited recruitment of these cells as well as TAMs into the xenografts, accordingly adjusting the tumor progression in vivo. The second viable strategy involves reshaping the TAM phenotype by reprogramming the signaling pathway of TAM differentiation [104]. Despite the fact that TAMs typically manifest alternatively activated M2 features associated with Th2 immune response, research has revealed that they can be reprogrammed towards the classically activated M1 phenotype that holds the responsibility for promoting inflammation and tumor inhibition in the presence of Th1 cytokines. It has been reported that iron oxide nanoparticles possess the intrinsic potential of polarizing immunosuppressive TAMs into the pro-inflammatory Th1 type. The reprogrammed macrophages substantially inhibited the growth of inoculated adenocarcinomas, in addition to preventing the development of liver metastasis in mice [105]. Poly(β-amino ester) nanoparticles with IL-12 payload were designed and synthesized for targeted immunotherapy [106]. The nanoparticles promotes systemic administration of IL-12 and released IL-12 in a TME-responsive manner, which allowed subsequent local reversal of the TAMs phenotype in the TME, coupled with enhanced therapeutic efficacy against solid tumors with negligible cytotoxicity. Furthermore, direct deletion of infiltrated TAMs constitutes another possible strategy. On the basis of the knowledge that sialic acid receptors are typically overexpressed on TAMs, liposomes with a payload of epirubicin and surface modification with sialic acid were synthesized, which increased cellular uptake and drug accumulation in TAMs in vitro [107]. Owing to the depletion of TAMs in the TME, the liposomes manifested a robust antitumor efficacy in a S180 murine sarcoma model in vivo.
Targeting MDSCs
MDSCs represent a group of undifferentiated bone marrow-derived heterogeneous cell populations that are identified by the co-expression of Gr-1 and CD11b [108]. These cells constitute substantial barriers to natural antitumor immunity and immunotherapy, in addition to having the potential for impairing differentiation of innate immune cells, for instance, macrophages and DCs. Upon induction with chemokines secreted by tumors, they migrate into the TME and take part in the immunosuppression, together with promoting vascular growth, tumor invasion, and metastasis [109]. Furthermore, MDSCs are also able to inhibit the functions of T cells through up-regulating the expression of arginine and nitric oxide synthase in TME as well as inducing production of Tregs [110].
Like macrophages, MDSCs can also be polarized to the M1 or M2 phenotype, characterized by secretion of Th1 (e.g., IL-12, IFN-γ, TNF-α) or Th2 (e.g., IL-10, TGF-β) cytokines. That is why reprogramming MDSCs in order to shift their polarization from M2 to M1 has the ability to provide new insights into the improvement of the efficacy of cancer immunotherapy. Two cationic polymers, cationic dextran and polyethyleneimine, have been demonstrated to have the ability to repolarize MDSCs by means of modulating toll-like receptors (TLRs) signaling in MDSCs, accordingly allowing reactivation of tumor immune surveillance as well as improved efficacy of immunotherapy [111]. Furthermore, another feasible strategy for modulating MDSCs deals with inhibiting their function and development. On the basis of the high expression of scavenger receptor type B-1 (SCARB1) on MDSCs, a receptor with high affinity for high-density lipoprotein (HDL), synthetic HDL biomimetic nanoparticles were prepared for inhibiting the functions of MDSCs [112]. As revealed by in vitro T cell proliferation assays, the HDL nanoparticles inhibited the activity of MDSCs by means of specifically binding to SCARB1. Additionally, HDL nanoparticles-mediated suppression of MDSCs substantially retarded tumor growth, in addition to enhancing the number of CD8+ T cells and depleting Tregs in the metastatic TME of murine B16F10 melanoma models. Furthermore, direct depletion of infiltrated MDSCs provides a third viable strategy. Pluronic-stabilized propylene sulfide micelles were developed carrying 6-thioguanine, a cytotoxic drug put to use in treating myeloid leukemia. In B16F10 melanoma and E.G7-OVA thymoma models, the micelles maintained a reduction in the numbers of circulating monocytic and granulocytic MDSCs for a period of seven consecutive days. Moreover, removal of MDSCs in the TME improved the efficacy of adoptively transferred tumor-specific CD8+ T cells substantially in vivo, accordingly affording a new perspective for cancer immunotherapy [113].
Modulating soluble mediators in the TME with nanoparticles
Targeting cytokines
Cytokines are small-molecular proteins secreted by immune cells that transmit information among immune cells, and include interleukins (ILs), interferons (IFNs), tumor necrosis factors (TNFs), colony stimulating factors (CSFs), and chemokines. These cytokines in the TME give rise to vasodilation and recruitment of immune cells to tumor sites, in addition to promoting the growth and metastasis of tumor cells, together with stimulation of vascular lymphatic vessels. It has now been identified that many of the immunosuppressive effects produced in the TME are ascribed to a disequilibrium of cytokines derived from pro-inflammatory Th1 (secreting IL-2, IL-6, IFN-γ and TNF-β) and anti-inflammatory Th2 (secreting IL-4, IL-5 and IL-10) cells [114]. Despite the fact that dosing of exogenous Th1 cytokines has manifested substantial efficacy in stimulating DCs and CTLs in vivo, these preclinical results derived from animal models have not been completely successful in clinical practice owing to poor tissue specificity coupled with the high cytotoxicity of the exogenous therapeutic agents [115].
In recent years, it has been discovered that members of the interleukin (IL) cytokine family play a pivotal role in tumor immunotherapy, in particular IL-2, IL-12 and IL-27. IL-2, also termed the T cell growth factor, is a cytokine with broad biological activity primarily produced by activated CD4+ T cells and CD8+ T cells. Being a pivotal factor in the regulation of immune response, IL-2 constitutes the only cytokine drug approved by the FDA for treating advanced malignant melanoma [116]. Nevertheless, as a kind of small secreted protein, it usually undergoes degradation and rapid clearance from systemic circulation, accordingly requiring repeated high-dose injections and inevitably resulting in serious side effects. Despite the fact that adenovirus-mediated IL-2 gene therapy has manifested substantial antitumor efficacy, biosafety concerns still inhibit its clinical application. Synthetic nanoparticles are likely to provide protective and local accumulation of exogenous cytokines by means of the enhanced permeability and retention (EPR) effect, in addition to receptor-mediated active targeting. Conversely, the ineffectiveness of traditional cytokine therapy is primarily ascribed to the presence of numerous immunosuppressive cytokines and chemokines in the TME, among which TGF-β plays a key role in the depletion of TILs and increase in the number of Tregs. Accordingly, nanoscale liposomal polymeric gels (nLGs) with a payload of cyclodextrin inclusion complex were designed and synthesized for the purpose of simultaneously delivering IL-2 and TGF-β antagonist to the TME [117]. The nLGs substantially delayed tumor growth, enhanced infiltration of activated CD8+ T cells in tumors, and improved the survival of metastatic melanoma-bearing mice. In the same manner, IL-12 is regarded as a pivotal player in cytokine immunotherapy as it is also considered to be a NK cell-stimulating factor and is primarily produced by DCs, macrophages, B lymphocytes, as well as other APCs [118]. Its primary immunomodulatory effect involves inducing early Th native cells to differentiate into Th1 cells and promoting their proliferation and activation. IL-12 also indirectly inhibits the formation of tumor blood vessels by inhibiting MMP9, VEGF, and TGF-β. That is why it constitutes a productive target for modulating the TME and enhancing antitumor immune responses. Nonetheless, application of IL-12 in clinical trials has been suspended owing to severe systemic nonspecific toxicity coupled with unsatisfactory therapeutic outcomes. Alternatively, administration of IL-12 by targeted gene delivery is likely to provide a sustained low level of IL-12 expression throughout the treatment period. A nanoscale self-assembly derived from PEGylated polylactic acid and cationic phospholipid was fabricated for targeted delivery of plasmid DNA [119]. Murine Ct26 colon carcinoma cells transfected with the pIL12-loaded self-assembled complex stably expressed and secreted IL-12, accordingly enhancing the T cell-mediated antitumor immune response in vivo.
Transforming growth factor-β (TGF-β) is a pleiotropic and multifunctional regulatory cytokine belonging to the Th2 cytokine family that regulates a variety of biological effects that include inhibition of inflammatory cells proliferation and lymphocyte differentiation, and promotion of extracellular matrix expression, for instance, collagen and fibronectin [120]. In the TME, TGF-β is primarily secreted by tumor cells and stromal cells, which include infiltrating immune cells and fibroblasts. When bound to its receptor TβRII, TGF-β commences the classical Smad and non-Smad signaling pathways (which include PI3K-Akt, RhoA and MAPK), in addition to taking part in several processes, for instance, tumor growth, angiogenesis, metastasis, and invasion [121]. TGF-β is also capable of converting effector T cells, which normally attack tumor cells by means of the inflammatory response, into regulatory T cells, which suppress immune surveillance. The presence of TGF-β in the TME gives rise to the loss of MHC class I molecules in malignant cells, accordingly allowing tumor cells to escape immune surveillance. That is why TGF-β is also able to inhibit the activation of NK cells, owing to the fact that they eradicate tumor cells by monitoring the expression of MHC class I molecules and activating receptor ligands on the cell surface [122]. In this manner, antagonizing the functions of TGF-β and its receptor provides a productive strategy for blocking the immunosuppressive effects of the cytokine, accordingly increasing the antitumor immune response. Nanoparticles have been reported for delivering the tumor antigen of Trp-2 peptide and CpG oligonucleotide adjuvant to DCs, to elicit a productive systemic immune response [123]. Nevertheless, this vaccine was less effective for late-stage B16F10 melanoma in a subcutaneous homologous model, which was primarily a result of the elevated levels of immunosuppressive cytokines in the TME, for instance, TGF-β. Moreover, targeted delivery of siRNA against TGF-β with the use of liposome-protamine-hyaluronic acid (LPH) nanoparticles lead to ~5% knockdown of TGF-β in the advanced TME. Down-regulation of TGF-β increased the tumor-infiltrating CD8+ T cell ratio and decreased regulatory T cell levels in comparison with the vaccine treatment alone, accordingly increasing the vaccine efficacy and attaining a tumor growth inhibition rate amounting to 52% [124]. Most recently, a nanoemulsion of the antifibrotic natural active compound, fraxinellone, was developed for the purpose of reversing the immunosuppressive TME of desmoplastic melanoma. Following systemic administration, the nanoemulsion with a particle size of ~145 nm deposited in the stroma and removed tumor-associated fibroblasts (TAFs) in an effective manner. Treatment with the nanosized antifibrotic compound increased the level of the Th1 cytokine IFN-γ, which elicited antitumor immunity, simultaneously lowering the levels of immunosuppressive TGF-β, CCL2, and IL-6. A combination of a tumor-specific BRAF peptide vaccine with the nano-emulsified fraxinellone led to enhanced tumor-specific T cell infiltration, coupled with activation of death receptors on the surface of tumor cells, and induction of increased apoptotic tumor cell death [125].
Chemokines constitute a class of low molecular weight (8-10 kDa) cytokines, which are chemotactic for immune cells. When specifically bound to G protein-coupled receptors, they exert an extensive array of biological effects and play a pivotal role in generating, differentiating, and developing immune cells, and regulating immune responses [126]. Chemokines play a dual role in the biological behavior of tumors. Owing to the expression of various Th1-type chemokine receptors on the surface of CD8+ T cells, CD4+ T cells, and NK cells, they attract these immune effector cells to the tumor site, localizing them to the specific microenvironment, and in turn induce their degranulation and release of perforin and granzymes to kill tumors. Additionally, because mature DCs express chemokine receptors in large quantities, for instance, CCR4, CCR7, and CXCR4, chemokines also recruit APCs to the TME, accordingly improving the immune response of T cells to tumor antigens [127]. Conversely, tumor cells and mesenchymal cells are likely to secrete and release chemokines, for instance, CXCL12, specifically activating and attracting immunosuppressive cells, for instance, Tregs, MDSCs, and TAMs into TME, accordingly substantially attenuating the antitumor immune response of immune effector cells. Furthermore, chemokines are capable of stratifying vascular endothelial cells and stimulating macrophages for the production of VEGF, accordingly leading to tumor angiogenesis. Considering the bidirectional regulation of chemokines on the TME, a feasible strategy for targeting chemokines involves linking the chemokine gene and the TAA gene as DNA vaccines, and transfecting them into the body by targeted nanocarriers. A fusion protein, which consists of a chemokine adjuvant and a tumor antigen, is expected to improve the host's immune response to tumor antigens. As an example, folic acid-modified chitosan nanoparticles were employed for encapsulating the gene expression plasmid of interferon-inducible protein-10 (i.e., CXCL10). In combination with a vaccine generated from the fusion of DCs and hepatocellular carcinoma cells (HCC), they were able to effectively inhibit the growth of implanted HCC tumors and prolonged the survival of mice. The combination therapy also substantially lowered MDSCs in mouse spleens, local tumors, and bone marrow, meanwhile augmenting tumor-specific IFN-γ responses [128]. Another feasible strategy involves blocking the binding of chemokines to immune effector cells with the help of targeted nanocarriers, accordingly relieving their immunosuppressive effects in the TME. As investigations suggest, the limited T cell infiltration by the inhibitory TME constitutes a pivotal cause of failure in the treatment of ICIs, and CXCL12 constitutes a key chemokine inhibiting T cell infiltration. Small molecule inhibitors (for instance, AMD3100) and monoclonal antibodies directed against the CXCL12/CXCR4 axis have been put to use for the purpose of blocking this immunosuppressive effect; nevertheless, systemic toxicity and poor tumor tissue permeability limit the therapeutic efficacy of both regimens. It was accordingly put forward to encapsulate a plasmid that encodes a fusion protein (called trap) specifically binding CXCL12 and PD-L1 into LPD nanoparticles for treating pancreatic cancer (Figure 6). The LPD nanocarriers allowed for local and transient expression of the trap, accordingly lowering the systemic toxicity while accumulating primarily around the blood vessels. MDSCs were decreased following the PD-L1 trapping alone in an allograft model, but Tregs still remained at high levels. Upon combination with CXCL12 capture, both Tregs and MDSCs were substantially lowered, significantly increasing the infiltration of CD3+ T cells into the tumor mass and enhancing the immune killing effect of PD-L1 trapping on tumors [129].
Targeting immunosuppressive enzymes
Indoleamine-2,3-dioxygenase (IDO) catalyzes the reaction of tryptophan in cells for the production of canine urea, which leads to depletion of tryptophan as well as inhibition of T cells and NK cells proliferation [130]. Being an immunomodulatory enzyme, IDO is primarily produced by numerous alternately activated macrophages and other immunoregulatory cells. It is also capable of inhibiting the functions of T cells and NK cells, and participates in immune tolerance through the generation and activation of Tregs and MDSCs. The mechanism by which overexpressed IDO remodels TME and favors immune tolerance is primarily by means of the production of kynurenine, a metabolite of L-tryptophan produced by tryptophan dioxygenase, which can activate the aryl hydrocarbon receptor for promoting the differentiation of Foxp3+ Tregs and suppressing antitumor immune responses [131]. Furthermore, emerging evidence indicates that activation of the IDO pathway is likely to serve as a preferred nodal modification pathway for tumor immune escape during cancer progression, for instance, via a survival-like PI3K pathway or the angiogenic VEGF pathway [132]. Considering the primary role of IDO in tumor immune tolerance, immunotherapy strategies that make use of IDO as a target have been put forward using 1-methyltryptophan (1-MT) as a more effective inhibitor that exerts an enzyme inhibition function through competition with the substrate tryptophan to bind IDO [133]. Nonetheless, the key findings of clinical trials have suggested that the performance of monotherapy with IDO inhibitors in cancer immunotherapy is limited and requires mandatory combination with other treatments [134]. A synergistic immunotherapeutic strategy for local targeting of immunosuppressive PD-1 and IDO has been developed for the treatment of melanoma, wherein 1-MT-modified hyaluronic acid was self-assembled into nanocapsules along with an anti-PD-1 antibody payload, followed by further embedding in transdermal microneedles [135]. The resulting delivery device elicited sustained release of the antibody and enhanced retention of ICIs in the TME. The combined use of IDO inhibitors augmented the number of CTLs and reduced the immunosuppression of the TME, accordingly attaining potent antitumor efficacy in a B16F10 mouse melanoma model (Figure 7). In another research work, mesoporous silica nanoparticles layered with IDO inhibitor (indoximod)-conjugated phospholipid bilayers were prepared, which were loaded with oxaliplatin in a bid to induce ICD [136]. Upon intratumoral administration in an orthotopic pancreatic ductal adenocarcinoma (PDAC) model, the nanoparticles effectively induced an adaptive immune response against PDAC. Additionally, they induced substantial tumor regression by means of recruiting and expanding TILs, accompanied by down-regulating Foxp3+ Tregs.
Figure 6
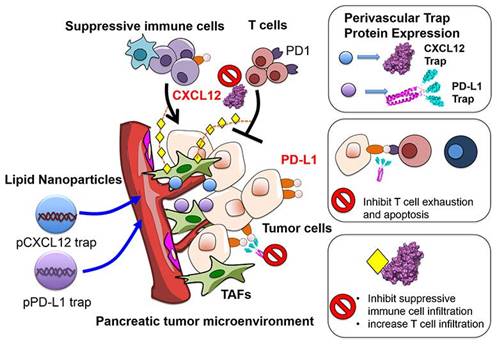
Illustration of the mechanisms underlying LPD nanoparticles-mediated combination immunotherapy with PD-L1 and CXCL12 trap for the treatment of pancreatic cancer. Plasmids encoding PD-L1 and CXCL12 trap were encapsulated into nanoparticles. Local and transient delivery of the encapsulated plasmids reduced their systemic toxicity and allowed accumulation in perivascular cells. The CXCL12 capture protein secreted from perivascular cells promoted effective capture of CXCL12 chemokines, which not only directly reduced infiltration of immunosuppressive cells (such as MDSCs and Tregs) through the CXCL12 / CXCR4 axis, but also inhibited the expression of PD-L1 by regulating the MAPK pathway. Reproduced with permission from [129], copyright 2017 American Chemical Society.
Matrix metalloproteinases (MMPs) are a family of zinc-dependent endopeptidases that degrade different kinds of tissue extracellular matrices including bone. Owing to the fact that the primary conditions for tumor cell invasion and metastasis are degradation of the ECM and destruction of the basement membrane, MMPs are regarded as the primary protease in this mechanism, as they not only degrade various protein components in the ECM, but also deteriorate histological barriers for tumor cell invasion [137]. Considering the fact that MMPs constitute the key factors in the promotion of the tumor invasion and metastasis, inhibition of MMPs has emerged as a new strategy for clinically preventing and treating tumors; nevertheless, the efficacy of most inhibitors has not been confirmed owing to poor specificity [138]. Overexpression of MMP-2 and MMP-9 in the ECM of tumor tissues can serve as a trigger for release of payloads from nanocarriers. Various surface-functionalized PEGylated nanoparticles targeting TME have been highlighted in which specific ligands of nucleosome or p32/gC1qR receptors on tumor cells were conjugated to the long-chain of PEG by means of a MMP-cleavable linker [139, 140]. Upon the responsive removal of the PEG shield, exposed surface-attached cell-penetrating peptide (TATp) triggers intracellular delivery of the system. Most recently, a peptide substrate of MMP-2 was coupled to the D-peptide antagonist of PD-L1 (DEAP- DPPA-1) and assembled with IDO inhibitor (NLG919) for the purpose of constructing a dual-targeted immunotherapeutic nanoparticle [141]. Specific cleavage of the substrate by MMP-2 in the TME allowed local release of both antagonists and simultaneous blockade of immune checkpoint and tryptophan metabolism, accordingly facilitating recruitment and proliferation of tumor-specific CD8+ T cells and substantially retarding tumor growth in a melanoma model.
Figure 7
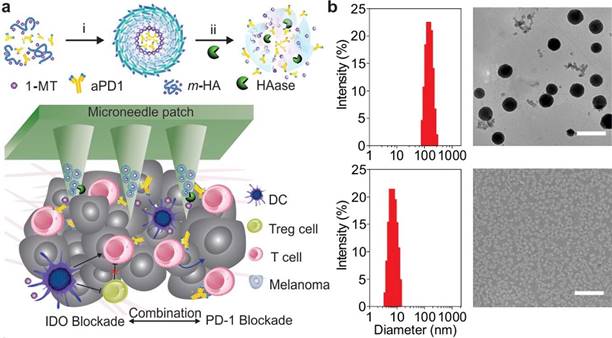
Schematic illustration of a microneedle-based transcutaneous drug delivery system loaded with self-assembled immunotherapeutic nanoparticles. (A) The IDO inhibitor 1-MT and hyaluronic acid (HA) were covalently conjugated to form an amphiphilic structure (m-HA), which then self-assembled into nanoparticles to encapsulate the anti-PD-1 antibody (aPD1). Drug release was activated via digestion by hyaluronidase (HAase), which is overexpressed in the TME. The subsequently triggered release of 1-MT blocked the IDO-mediated immunosuppressive pathway in TME, thereby enhancing the ability of aPD1 to block immune checkpoints. (B) The obtained nanoparticles were characterized by an average hydrodynamic size of 151 nm, which was consistent with transmission electron microscopy observation (upper). After 24 h of continuous incubation with HAase, the particles gradually dissociated and the size was decreased to 8 nm (lower). Reproduced with permission from [135], copyright 2016 American Chemical Society.
Modulating other components of the TME with nanoparticles
Targeting extracellular matrix and stromal fibroblasts
Extracellular matrix (ECM) is a collection of extracellular macromolecules constituting structural proteins and proteoglycans, which are synthesized and secreted by supporting cells [142]. Therein, fibroblasts constitute a key constituent of ECM, making important contributions towards the occurrence, proliferation, invasion, and metastasis of tumors. Tumor-associated fibroblasts (TAFs) are activated fibroblast populations isolated from tumor tissues, and play a pivotal role in the regulation of the balance of the system by means of direct cell-cell contact and secretion of soluble factors, for instance, fibroblast-specific protein (FSP-1), growth factors (for instance, VEGF), fibroblast activation protein (FAP), and smooth-muscle actin (SMA) [143]. Recently, growing evidence has suggested that the emergence of multidrug resistance is dependent not only on the genetic behavior of the tumor cells themselves, but also on their interaction with the TME [144]. TAFs are regarded as the key stromal cells in the TME, accounting for more than 50% of solid tumor tissues, e.g., PDAC [145]. In comparison with normal fibroblasts, activated TAFs have substantial changes in morphological attributes and functional gene expression [146]. On the one hand, abundant collagen fibers exist in the tumor stroma that are derived from the ECM proteins secreted by TAFs, which confine nanomedicines in the tumor stroma, significantly lowering the concentration of therapeutic agents in tumor cells and promoting drug resistance[147]. On the other hand, TAFs are capable of secreting a variety of soluble cytokines (for instance, TGF-β, SDF-1, and HGF) in order to act in a paracrine manner on adjacent tumor cells, accordingly activating their proliferation or anti-apoptotic signaling pathways and lowering their sensitivity to the chemotherapy [148]. In the same manner, targeting TAFs in the TME provides a pivotal strategy for the reversal of stroma-mediated multidrug resistance through inhibition of their activated signaling pathway. It has been observed that Wnt16 is overexpressed in cytotoxic agents-injured TAFs, which constitutes a key contributor towards drug resistance in cancer therapy. Nanoparticles assembled with LPH were employed for the purpose of encapsulating siRNA against Wnt16, wherein, anisamide ligand was coupled to the vectors to target σ receptor, which is overexpressed on both TAFs and tumor cells [149]. The results suggested the efficacy of Wnt16 knockdown in the impaired TAFs, and indicated that this is a promising combination strategy with cisplatin nanoparticles in a stroma-rich bladder carcinoma model. Likewise, an anti-fibrotic natural chemical quercetin substantially inhibited the expression of Wnt16 in activated TAFs [150]. Moreover, systemic administration of quercetin nanoparticles resulted in substantial down-regulation of Wnt16 expression and a synergistic antitumor effect with cisplatin nanoparticles in a bladder cancer model. α-SMA positive fibroblasts and collagen in the TME were also substantially lowered following the combined therapy. More recently, micelles self-assembled from telmisartan-grafted glycolipid were developed for targeting angiotensin II type I receptor (AT1R) overexpressed on TAFs as well as tumor cells. Elimination of TAFs by the micelles resulted in apoptosis of the ECM coupled with enhanced vulnerability of the tumor cells to chemotherapeutics [151].
Targeting tumor vasculature
Tumor neovascularization is the mechanism wherein vascular endothelial cells in the TME change from a comparatively more static to fast-paced growth under the action of related stimulating angiogenic signals, which is a pivotal prerequisite for tumor growth [152]. Tumor neovascularization caters to the growth and metabolism requirements of tumor tissue, in addition to becoming a pivotal means for tumor cell invasion and distant metastasis. In comparison with conventional tumor treatment, anti-angiogenesis strategies have the advantages of not giving rise to drug resistance, convenient access of drug to target cells, inhibition of tumor metastasis, and small adverse reactions; accordingly, it has emerged as one of the pivotal methodologies for the comprehensive treatment of malignant tumors [153]. Tumor angiogenesis is dependent on the dynamic equilibrium between angiogenic factors, for instance, VEGF, and antiangiogenic factors like angiostatin; therefore, current investigations targeting tumor vasculature for antitumor progression have emphasized activation of endogenous angiogenesis inhibitors and inhibition of pro-angiogenic factors as well as direct repair of vascular endothelial cells [154]. For example, an NIR laser-triggered hollow CuS nanoparticle with a payload of vinyl azide and surface functional modification of c(RGDfE) peptide was put forward as a targeted and noninvasive physical therapy strategy aimed at effectively destroying the tumor neovasculature. NIR irradiation resulted in a local temperature rise and triggered rapid release of nitrogen bubbles from the vinyl azide, which are likely to immediately burst, leading to destruction of the tumor neovasculature as well as surrounding carcinoma cells [155]. Monoclonal antibodies (for instance, bevacizumab), tyrosine kinase receptor inhibitors (TKIs), and a variety of protein inhibitors targeting VEGF-associated pathways have been employed for remodeling the tumor vasculature to enhance cancer therapies; nevertheless, only modest clinical outcomes have been attained [156]. Morphological and functional normalization of tumor vasculature are expected to reconstruct the pressure gradients between intravascular and interstitial tissues, accordingly facilitating intratumoral delivery of nanoparticles. In this regard, it has been reported that nanoparticle-mediated transport of antiangiogenic agents enhanced tumor blood flow by means of vascular normalization, which, in turn, is expected to benefit from the enhanced perfusion and oxygenation of tumor cells. In the same manner, targeted gold nanoparticles (AuNPs) have been developed with a payload of endostatin as neovascularization blocker for tumor blood perfusion improvement, local hypoxia mitigation, and tumor vasculature normalization. In combination with neovascularization blockade, the nanoparticles substantially enhanced the efficacy of 5-fluorouracil with significant down-regulation of both VEGF and HIF-1α [157].
Targeting tumor hypoxia
During tumor progression, the vascular network cannot be timely established, and the neovascularization becomes abnormal in structure owing to the rapid proliferation of tumor cells, which results in a decline in oxygen content, nutrient deficiency, and accumulation of acidic substances in the TME [158]. In the hypoxic microenvironment, tumor cells activate an array of adaptive genes (for instance, VEGF, TAp73, and GLUT1) and immunosuppressive cells (e.g., Tregs, MDSCs) for obtaining immune escape potential by means of the hypoxia-inducible factors (HIFs) pathway, accordingly facilitating invasion, metastasis, and drug resistance. Furthermore, the hypoxic TME secretes a variety of immunosuppressive cytokines, for instance, TGF-β, that induce apoptosis of CTLs as well as production of CD4+CD25+ Tregs [159]. It also inhibits the expression of not only antigen-presenting molecules, but also co-stimulatory factors and chemokine receptors on the surface of DCs, rendering it impossible for DCs to present tumor antigens to T cells. This is why hypoxia poses a key challenge for the delivery of therapeutic drugs to solid tumors, in particular while delivering nanoparticles with a large size. Interventional treatment strategies for tumor hypoxia are likely to effectively augment the blood perfusion capacity of the tumor, in addition to lowering the level of hypoxia in the TME and also enhancing the tumor immune response [160]. For instance, considering the fact that the typical hypoxic attributes of solid tumors substantially limit the efficiency of PDT, nanoscale carbon dots with surface modification of protoporphyrin photosensitizer and targeting motif were synthesized for light-driven water decomposition, aimed at producing oxygen. Doping the carbon dots with carbon nitride (C3N4) enhanced their absorption of red light, efficiently leading to splitting of water in vivo. In this manner, the attained multifunctional nanocomposites augmented the intracellular oxygen concentration when subject to light irradiation, in addition to enhancing the production of reactive oxygen in the tumor hypoxic environment, accordingly reversing hypoxia-induced PDT resistance in tumor cells [161]. Recently, a variety of nanoparticles based on inorganic materials like manganese dioxide (MnO2) have been put to use for relieving tumor hypoxia through the catalysis of hydrogen peroxide (H2O2) in the TME for the generation of oxygen, accordingly enhancing the outcome of cancer therapy [162, 163]. MnO2 nanoparticles were attained with the help of in situ biomineralization on a template of bovine serum albumin, which were further surface-coated with a coordination polymer developed by the assembly of high-Z element hafnium ions and cisplatin prodrug [164]. In this system, the MnO2 core triggered decomposition of tumor endogenous H2O2 for the production of oxygen in situ, accordingly overcoming the radiotherapy tolerance caused by hypoxia. Simultaneously, the coordination polymers were used as radiosensitizers to improve radiotherapy due to the strong absorption capacity of hafnium ions to X-rays. Additionally, the nanoparticles were degraded to divalent manganese ions when subjected to the acidic conditions of the tumor, achieving magnetic resonance imaging in vivo. The intelligent multifunctional nanoparticles were dual responsive to both acidic and hypoxic conditions in the TME, and successfully achieved a synergistic therapeutic effect to chemotherapy and radiotherapy, which is expected to become a new degradable integrated diagnosis and treatment preparation. More recently, multifunctional biomimetic core-shell nanoparticles were reported for synergistic chemotherapy and immunotherapy. A pH-sensitive zeolitic imidazolate framework 8 was integrated with catalase and doxorubicin as oxygen generator and drug, respectively, which in turn were coated with mouse melanoma cell membrane for tumor targeting ability together with the ability to elicit an immune response because of enrichment of antigens. The biomimetic core-shell nanoparticles with oxygen generation down-regulated the expression of HIF-1α in the TME, accordingly lowering the expression of PD-L1. When combined with PD-1 mAbs to blockade immune checkpoints, the dual inhibition of the PD-1/PD-L1 axis by the multifunctional nanoparticles elicited a substantial immune response and was robustly potent in retarding tumor recurrence and metastasis [165].
Targeting nitric oxide
Nitric oxide (NO) is a ubiquitous bioactive small molecule with unpaired electrons in mammals generated by the catalysis of L- arginine with nitric oxide synthase (NOS). Being an intercellular messenger, NO is not only involved in normal physiological activities (which include vascular growth, smooth muscle relaxation, immune response, apoptosis, and synaptic transmission), but also have close association with occurrence and development of tumors [166]. NO is not only an effector molecule of tumor immunity but is also a regulatory molecule of different kinds of immune cells. When macrophages or neutrophils are activated, large amounts of NOs are usually expressed, followed by the production of high concentrations of NO, which directly impair DNA and lead to cytotoxicity by binding with intracellular superoxide anions to produce nitroxide radicals. Furthermore, excess NO also promotes NK cell activity, activates monocytes, and regulates T lymphocytes for the enhancement of the immune response [167]. Contrarily, continuous low concentration of NO is expected to inhibit tumor cell apoptosis by inhibiting T lymphocyte proliferation, modulating mast cell response, and participating in tumor angiogenesis. In accordance with the dual roles of NO in various phases of tumor initiation, progression, and metastasis, increasing the NO level beyond the critical concentration that favors tumor progression activates tumor cell growth inhibition and apoptosis pathways, providing us with a new strategy to improve the sensitivity of refractory tumor cells to radiochemotherapy and immunotherapy.
There are different strategies to manipulate the production and exogenous transport of NO in vivo, which include iNOS gene transfection and induction, and administration of NO donor drugs [168]. The NO donors can be encapsulated or linked to a chemical moiety for the formation of a functional polymer, which mimics the production of endogenous NO for targeted therapy. A polymeric micelle-based NO delivery system was introduced for the first time, with the aim to overcome MDR effects, wherein, S-nitrosothiols, a NO donor comprising thiol and nitric oxide, was conjugated to the hydrophobic segment of the diblock copolymer. Upon endocytosis of the micelles, redox-responsive intracellular release of NO was initiated for exerting cytotoxicity. In comparison with the monotherapy, pretreatment with the micelles reduced the IC50 of cisplatin by 5-fold in neuroblastoma cells [169]. Inspired by this investigation, an innovative mesoporous silica-coated upconversion nanoparticle (UCNP) was designed and synthesized chemically, in which Roussin's black salt was loaded as a NO donor for cancer therapy. Upon NIR irradiation, UCNP up-converted NIR photons into higher energy UV photons for photolyzing the NO precursors. An on-demand supply of NO was attained through simple adjustment of the illumination intensity, wherein higher NIR irradiation resulted in cytotoxicity by producing a high concentration of NO, while low NIR irradiation produced a low concentration of NO that suppressed P-glycoprotein expression in cancer cells, accordingly overcoming multidrug resistance [170]. Another viable strategy deals with harnessing living cells with innate tumor homing ability as “hitchhikers” of nanomedicines, conferring biomimetic features on them in terms of crossing the vascular barrier and reaching the inner poorly perfused regions of tumor tissues [171]. Macrophages carrying particles with photoactivated theranostic payloads into tumors overcame the challenges usually encountered with the therapeutic administration of NO, and in addition provided the potential for multiple treatment strategies with the use of a combination system. The macrophages carrying particles penetrated deeply into tumor sphere models of NIH-3T3/4T1. Upon irradiation by NIR light, NO was released from the photoactivated NO-releasing moiety of manganese nitrosyl; in addition, emission from Nd3+-doped up-converting nanoparticles imaged the particle location [172].
Table 2
Various strategies for nanoparticles-based TME modulation in cancer immunotherapy.
| Targeting sites | Nanoparticles (NPs) | Payloads | Tumor models | Ref |
|---|---|---|---|---|
| APCs | Liposome /3-methylglutarylated poly(glycidol) | OVA | E.G7-OVA | [32] |
| PLGA NPs | STAT3 siRNA, OVA, ICG, R837 | E.G7-OVA | [173] | |
| Lipid-calcium-phosphate NPs | Trp2, CpG, | B16F10 | [123] | |
| Dendrimer / peptide | pcDNA3-TRP2/gp70 | B16F10 | [174] | |
| Gold NPs / PEG | OVA, CpG | B16-OVA | [46] | |
| Upconversion NPs /PEG-PEI | OVA | B16-OVA | [41] | |
| Micelle / PEG-PLL-PLLeu | Poly I:C, OVA, STAT3 siRNA | B16-OVA | [42] | |
| TAMs | Lipid NPs / C12-200, cholesterol, PEG-DMG | CCR2 siRNA | EL4, CT26 | [103] |
| Poly(β-amino ester) | IL-12 | B16F10 | [106] | |
| Liposome / sialic acid | Epirubicin | S180 sarcoma | [107] | |
| Tregs | Carbon nanotubes / anti-GITR mAb | Fluorochrome | B16 | [94] |
| Lipid-PLGA / tLyp1 | Imatinib, anti-CTLA-4 mAb | B16 | [95] | |
| Lipid-calcium phosphate | Sunitinib; Trp2, CpG, | B16F10 | [98] | |
| MDSCs | High-density lipoprotein-like NPs | DiD | B16F10 | [112] |
| Micelles of propylene sulfide | 6-thioguanine | B16F10, E.G7-OVA | [113] | |
| Liposome / DSPE-PEG-PDP | Complement C3 | 4T1 | [175] | |
| Iron oxide / dextran | Ferumoxytol | KP1 | [105] | |
| Cytokines | Liposome containing anti-CD137/IL-2 | OVA | B16F10 | [176] |
| Liposome encapsulated cyclodextrin | TGF-β inhibitor, IL-2 | B16 melanoma | [117] | |
| MPEG-PLA hybrid DOTAP | IL-12 plasmid | Colorectal Ct26 | [119] | |
| CTLA-4/ PD-1/PD-L1 | PLGA NPs / anti-CTLA4 mAb | Indocyanine green, Imiquimod | 4T1 | [78] |
| Micelles / PC, DSPE-PEG/ anti-PD-L1 mAb | Selumetinib, PI103 | B16F10, 4T1 | [80] | |
| Lipid-protamine-DNA | PD-L1/CXCL12 trap | KPC98027 | [129] | |
| Lipid-protamine-DNA | PD-L1 trap, OXP | CT26 | [81] | |
| TAFs | Lipid-cisplatin, Lipid-protamine | Wnt16 siRNA | UMUC3/NIH 3T3 | [149] |
| Lipid-cisplatin, Lipid-calcium phosphate | Quercetin | UMUC3/ NIH 3T3 | [150] | |
| Micelle / telmisartan - glycolipid | Doxorubicin | MCF-7 | [151] | |
| Neovascular | CuS NPs / c(RGDfE) | Vinyl azide | HeLa | [155] |
| Gold NPs | Endostatin | H22 | [157] | |
| Hypoxia | Carbon dots doped with carbon nitride | Protoporphyrin | 4T1 | [161] |
| MnO2 NPs / mannan-hyaluronic acid | Doxorubicin | 4T1 | [162] | |
| MnO2 NPs / mesoporous silica | Doxorubicin | MCF-7/ADR | [163] | |
| MnO2 NPs / BSA / hafnium | Cisplatin | 4T1 | [164] | |
| Nitric oxide | Micelle /oligo (ethyleneglycol-methacrylate) / S-nitrosothiol | Cisplatin | Neuroblastoma | [169] |
| Upconversion NPs / mesoporous SiO2 | Roussin's black salt, Doxorubicin | MCF-7/ADR | [170] |
Conclusions and perspectives
With the collaboration of immunology and materials science, an array of new tumor immunotherapies based on nanoparticles is being developed. Nanoparticles provide an attractive delivery mode to target the tumor immune microenvironment, capable of effectively safeguarding vaccines and immunomodulatory molecules, improving their biodistribution and release kinetics in vivo, and increasing their accumulation in tumor sites. In addition, they are capable of targeting stromal cells, immune cells, and tumor cells together with performing local immune regulation in the TME, accordingly enhancing the effects of tumor immunotherapy and lowering systemic immunotoxicity. Nevertheless, there are still a number of challenges to making nanoparticles that enhance the efficacy of immunotherapy and translating them to the clinic for extensive applications.
The tumor immune microenvironment is an extremely complex system, with cross-talk among immune cells, stromal cells, cytokines, and tumor cells. We currently lack a comprehensive understandding of the immune regulation mechanisms of tumors, which is why we are encountering a number of difficulties and confusions when attempting to solve antitumor issues from the perspective of immunity. Further investigations dealing with the mechanisms of how tumor cells escape from immune surveillance and immune killing effects can provide more immunotherapeutic targets for nanoparticles to modulate the TME. An enhanced understanding of the biochemical attributes of cells and matrix components infiltrated in the TME is expected to constitute a pivotal basis for constructing intelligent nanomedicine delivery systems responding to the TME. Further elucidation of the TME-mediated molecular regulation of tumor cells as well as immune tolerance mechanisms is expected to help guide nanoparticles to exercise new TME immune regulation strategies, in addition to more effectively relieving the suppressive effects of the TME on immunotherapy.
In the development of nanoparticles for conventional drug delivery, it is typically deemed essential to take into consideration how to avoid elimination of the immune system. Nonetheless, with regard to tumor immunotherapy, it is essential to construct nanoparticles that have the potential to strongly interact with various pathways of the immune system. Accordingly, in designing and preparing nanoparticles, corresponding adjustments require being incorporated in accordance with their clinical applications aimed at attaining specific therapeutic outcomes. For instance, the size and surface charge of nanoparticles are factors requiring special consideration in the precise delivery of immunomodulators in order to target the tumor immune microenvironment. In spite of the fact that small-sized nanoparticles are more likely to penetrate into tumors than larger ones, nanoparticles that are too small (e.g. less than 20 nm) tend to be rapidly cleared by the kidneys and therefore cannot be effectively accumulated in tumor tissues. The promising way to solve this dilemma is to keep the nanoparticles in a relatively large size (such as about 100 nm) during the systematic circulation process. Once reaching the tumor sites, they are expected to undergo particle size reduction caused by TME stimulation, such as enzymatic degradation and redox decomposition, thereby facilitating tumor penetration. Similarly, the surface charge of nanoparticles can also be kept neutral before cell uptake, which is conducive to the accumulation and penetration of tumor tissue. Upon entering TME, the nanoparticles will be triggered by TME to expose positively charged groups to facilitate their cellular endocytosis, lysosome escape and nuclear entry.
Despite the fact that nanoparticles for immunotherapy have manifested attractive potentials in preclinical investigations, there are still numerous difficulties to be solved in their clinical translation. Firstly, the safety of nanomaterials limits their successful translation to the clinical field [177]. The selection of material compositions should be carried out using the FDA's list of certifications, and these ingredients are also preferably biodegradable without leading to biological aggregation and resistance. Secondly, some inherent attributes of nanoparticles, for instance, high cation density and surface activity or leakage of toxic metal ions, are all issues requiring careful consideration. Thirdly, controllable industrial production of nanoparticles also constitutes a major factor that restricts their successful clinical translation; in particular the polydispersity of nanoparticles is crucial to the quality and clinical efficacy of the final products [178].
It is also worth mentioning that, subsequent to entering systemic circulation, nanoparticles will adsorb serum proteins and opsonins onto their surface, forming a dynamic nanoparticle-protein complex, also known as the protein corona, which impacts numerous physicochemical attributes of the nanoparticles, including their size, surface chemistry, hydrophobicity, and aggregation state, accordingly determining their biological fate in vivo, which includes cellular uptake, intracellular transport, pharmacokinetics, biodistribution as well as toxicity [179]. Additionally, the protein corona is likely to mask targeting ligands, causing off-target effects. When target cells interact with surface-functionalized nanoparticles in vivo, they identify and process the protein-complexed nanoparticles instead of the naked ones; therefore, the targeting efficiency, in addition to clearance by the mononuclear phagocyte system, immunogenicity, as well as therapeutic effects of the nanoparticles, are primarily independent of their composition and surface functionalization, but dependent rather on the attributes, alignment, and residence time of adsorbed proteins on the surface of the nanoparticles. Taking into consideration the fact that protein corona formation of nanomaterials in vivo is unavoidable, this phenomenon is expected to be employed as a positive means for targeted drug delivery by designing synthetic nanoparticles that capture plasma proteins that are particularly identified by the receptors of target cells. This targeting strategy, developed on the basis of the protein corona effect, requires identification of a composition of adsorbed plasma proteins having the highest affinity to nanoparticles with various compositions and surface attributes. Recent investigations have revealed that patients are likely to have unique protein coronas, as suggested by the fact that the same nanomaterials incubated with plasma proteins from patients with different pathologies adsorb protein coronas with different compositions [180]. Therefore, extensive work is required to better understand the interactions between nanoparticles and organisms.
In prospective research work on tumor immunotherapy, nanoparticles with multiple TME stimuli responsiveness, for instance, extracellular acidity, hypoxia, and matrix enzymes, and external stimuli responsiveness, e.g., photothermal, will emerge as key research directions, which are expected to overcome multiple physiological barriers encountered while delivering immunomodulators in vivo. Nevertheless, multiple stimuli-responsive nanoparticles also have issues related to their intricate design and the synthesis of carrier materials, such as difficult quality control. It is believed that with the continued development of materials science, tumor immunology, molecular and cell biology, these issues will be effectively solved, and intelligent nanoparticles will be developed towards more precise directions. That is why cancer immunology in combination with nanotechnology is an emerging field.
Abbreviations
APCs: antigen presenting cells; AuNPs: gold nanoparticles; CAR-T: chimeric antigen receptor T-cell immunotherapy; CSFs: colony stimulating factors; COX: cyclooxygenase; CTLA-4: cytotoxic T lymphocyte-associated antigen-4; CTLs: cytotoxic T lymphocytes; DCs: dendritic cells; ECM: extracellular matrix; EGFR: epidermal growth factor receptor; EPR: enhanced permeability and retention; ERBB2IP: erbb2 interacting protein; FAP: fibroblast activation protein; FSP-1: fibroblast-specific protein-1; GITR: glucocorticoid-induced TNFR-related receptor; HA: hyaluronic acid; HCC: hepatocellular carcinoma cells; HDL: high-density lipoprotein; hNPs: hybrid nanoparticles; ICD: immunogenic cell death; ICIs: immune checkpoint inhibitors; IDO: indoleamine-2,3-dioxygenase; IFNs: interferons; ILs: interleukins; imDCs: immature dendritic cells; irAEs: immune-related adverse events; iTregs: induced T cells; LPD: lipid-protamine-DNA; mAbs: monoclonal antibodies; mDCs: mature dendritic cells; MDSCs: myeloid-derived suppressor cells; MHC: major histocompatibility complex; MMPs: matrix metalloproteinases; MRI: magnetic resonance imaging; MSS: microsatellite stability; 1-MT: 1-methyltryptophan; NIR: near-infrared; NK: natural killer; NSCLC: non-small cell lung cancer; NOS: nitric oxide synthase; OxP: oxaliplatin; PDAC: pancreatic ductal adenocarcinoma; PD-1: programmed death receptor-1; PD-L1: programmed death receptor-1 ligand; PDT: photodynamic therapy; PLGA: poly(lactic-co-glycolic acid); SCARB1: scavenger receptor type B-1; SMA: smooth-muscle actin; STAT3: signal transducer and activator of transcription 3; SWCNT: single-walled carbon nanotube; TAA: tumor-associated antigen; TAMs: tumor-associated macrophages; TAFs: tumor-associated fibroblasts; TCR: T cell receptor; Th cells: T helper cells; TILs: tumor infiltrating lymphocytes; TKIs: tyrosine kinase receptor inhibitors; TLRs: toll-like receptors; TME: tumor microenvironment; TGF-β: Transforming growth factor-β; TNFs: tumor necrosis factors; Tregs: regulatory T cells; TSA: tumor specific antigen; UCNP: upconversion nanoparticles.
Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 31671020) for financial support.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen YL, Chang MC, Cheng WF. Metronomic chemotherapy and immunotherapy in cancer treatment. Cancer Lett. 2017;400:282-92
2. Del Paggio JC. Immunotherapy: Cancer immunotherapy and the value of cure. Nat Rev Clin Oncol. 2018;15:268-70
3. Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269-81
4. Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013;39:49-60
5. D'Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis. 2018;9:282
6. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350-5
7. Garber K. Driving T-cell immunotherapy to solid tumors. Nat Biotechnol. 2018;36:215-9
8. Cheever MA, Higano CS. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res. 2011;17:3520-6
9. Wurz GT, Kao CJ, Wolf M, DeGregorio MW. Tecemotide: an antigen-specific cancer immunotherapy. Hum Vaccin Immunother. 2014;10:3383-93
10. Vansteenkiste JF, Cho BC, Vanakesa T, De Pas T, Zielinski M, Kim MS. et al. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17:822-35
11. Thomas R, Al-Khadairi G, Roelands J, Hendrickx W, Dermime S, Bedognetti D. et al. NY-ESO-1 based immunotherapy of cancer: current perspectives. Front Immunol. 2018;9:947
12. Karkada M, Berinstein NL, Mansour M. Therapeutic vaccines and cancer: focus on DPX-0907. Biologics. 2014;8:27-38
13. Grippin AJ, Sayour EJ, Mitchell DA. Translational nanoparticle engineering for cancer vaccines. Oncoimmunology. 2017;6:e1290036
14. Kageyama S, Wada H, Muro K, Niwa Y, Ueda S, Miyata H. et al. Dose-dependent effects of NY-ESO-1 protein vaccine complexed with cholesteryl pullulan (CHP-NY-ESO-1) on immune responses and survival benefits of esophageal cancer patients. J Transl Med. 2013;11:246
15. Wang H, Mooney DJ. Biomaterial-assisted targeted modulation of immune cells in cancer treatment. Nat Mater. 2018;17:761-72
16. Sander AF, Lollini PL. Virus-like antigen display for cancer vaccine development, what is the potential? Expert Rev Vaccines. 2018;17:285-8
17. Gargett T, Abbas MN, Rolan P, Price JD, Gosling KM, Ferrante A. et al. Phase I trial of Lipovaxin-MM, a novel dendritic cell-targeted liposomal vaccine for malignant melanoma. Cancer Immunol Immun. 2018;67:1461-72
18. Karlou M, Tzelepi V, Efstathiou E. Therapeutic targeting of the prostate cancer microenvironment. Nat Rev Urol. 2010;7:494-509
19. Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263-74
20. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E. et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35(Suppl):S185-S98
21. Zhao L, Seth A, Wibowo N, Zhao CX, Mitter N, Yu C. et al. Nanoparticle vaccines. Vaccine. 2014;32:327-37
22. Leleux J, Roy K. Micro and nanoparticle-based delivery systems for vaccine immunotherapy: an immunological and materials perspective. Adv Healthc Mater. 2013;2:72-94
23. Du J, Lane LA, Nie S. Stimuli-responsive nanoparticles for targeting the tumor microenvironment. J Control Release. 2015;219:205-14
24. Sau S, Alsaab HO, Bhise K, Alzhrani R, Nabil G, Iyer AK. Multifunctional nanoparticles for cancer immunotherapy: A groundbreaking approach for reprogramming malfunctioned tumor environment. J Control Release. 2018;274:24-34
25. Zhu G, Zhang F, Ni Q, Niu G, Chen X. Efficient nanovaccine delivery in cancer immunotherapy. ACS Nano. 2017;11:2387-92
26. Wang C, Ye Y, Hu Q, Bellotti A, Gu Z. Tailoring biomaterials for cancer immunotherapy: emerging trends and future outlook. Adv Mater. 2017;29:1606036
27. Liu Y, Cao X. Intratumoral dendritic cells in the anti-tumor immune response. Cell Mol Immunol. 2015;12:387-90
28. Le Gall CM, Weiden J, Eggermont LJ, Figdor CG. Dendritic cells in cancer immunotherapy. Nat Mater. 2018;17:474-5
29. Zupančič E, Curato C, Paisana M, Rodrigues C, Porat Z, Viana AS. et al. Rational design of nanoparticles towards targeting antigen-presenting cells and improved T cell priming. J Control Release. 2017;258:182-95
30. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12:265-77
31. Bol KF, Schreibelt G, Gerritsen WR, de Vries IJ, Figdor CG. Dendritic cell-based immunotherapy: state of the art and beyond. Clin Cancer Res. 2016;22:1897-906
32. Yuba E, Harada A, Sakanishi Y, Watarai S, Kono K. A liposome-based antigen delivery system using pH-sensitive fusogenic polymers for cancer immunotherapy. Biomaterials. 2013;34:3042-52
33. Moon JJ, Huang B, Irvine DJ. Engineering nano- and microparticles to tune immunity. Adv Mater. 2012;24:3724-46
34. Decuzzi P, Godin B, Tanaka T, Lee SY, Chiappini C, Liu X. et al. Size and shape effects in the biodistribution of intravascularly injected particles. J Control Release. 2010;141:320-7
35. He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31:3657-66
36. MB H, YT L. Programmed nanoparticles for combined immunomodulation, antigen presentation and tracking of immunotherapeutic cells. Biomaterials. 2014;35:590-600
37. Irvine DJ, Hanson MC, Rakhra K, Tokatlian T. Synthetic nanoparticles for vaccines and immunotherapy. Chem Rev. 2015;115:11109-46
38. Mody KT, Popat A, Mahony D, Cavallaro AS, Yu C, Mitter N. Mesoporous silica nanoparticles as antigen carriers and adjuvants for vaccine delivery. Nanoscale. 2013;5:5167-79
39. Zhao Y, Huo M, Xu Z, Wang Y, Huang L. Nanoparticle delivery of CDDO-Me remodels the tumor microenvironment and enhances vaccine therapy for melanoma. Biomaterials. 2015;68:54-66
40. Niikura K, Matsunaga T, Suzuki T, Kobayashi S, Yamaguchi H, Orba Y. et al. Gold nanoparticles as a vaccine platform: influence of size and shape on immunological responses in vitro and in vivo. ACS Nano. 2013;7:3926-38
41. Xiang J, Xu L, Gong H, Zhu W, Wang C, Xu J. et al. Antigen-loaded upconversion nanoparticles for dendritic cell stimulation, tracking, and vaccination in dendritic cell-based immunotherapy. ACS Nano. 2015;9:6401-11
42. Luo Z, Wang C, Yi H, Li P, Pan H, Liu L. et al. Nanovaccine loaded with poly I:C and STAT3 siRNA robustly elicits anti-tumor immune responses through modulating tumor-associated dendritic cells in vivo. Biomaterials. 2015;38:50-60
43. Vacas-Cordoba E, Climent N, De La Mata FJ, Plana M, Gomez R, Pion M. et al. Dendrimers as nonviral vectors in dendritic cell-based immunotherapies against human immunodeficiency virus: steps toward their clinical evaluation. Nanomedicine-UK. 2014;9:2683-702
44. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2016;15:143
45. Bell BM, Kirk ID, Stefanie Hiltbrunner, Susanne Gabrielsson, Jarred J. Bultema. Designer exosomes as next-generation cancer immunotherapy. Nanomed-Nanotechnol. 2016;12:163-9
46. Almeida JPM, Lin AY, Figueroa ER, Foster AE, Drezek RA. In vivo gold nanoparticle delivery of peptide vaccine induces anti-tumor immune response in prophylactic and therapeutic tumor models. Small. 2015;11:1453-9
47. Yang R, Xu J, Xu L, Sun X, Chen Q, Zhao Y. et al. Cancer cell membrane-coated adjuvant nanoparticles with mannose modification for effective anticancer vaccination. ACS Nano. 2018;12:5121-9
48. Fang RH, Hu CMJ, Luk BT, Gao W, Copp JA, Tai Y. et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14:2181-8
49. Guo Y, Wang D, Song Q, Wu T, Zhuang X, Bao Y. et al. Erythrocyte membrane-enveloped polymeric nanoparticles as nanovaccine for induction of antitumor immunity against melanoma. ACS Nano. 2015;9:6918-33
50. Eggermont LJ, Paulis LE, Tel J, Figdor CG. Towards efficient cancer immunotherapy: advances in developing artificial antigen-presenting cells. Trends Biotechnol. 2014;32:456-65
51. Perica K, De León Medero A, Durai M, Chiu YL, Bieler JG, Sibener L. et al. Nanoscale artificial antigen presenting cells for T cell immunotherapy. Nanomed-Nanotechnol. 2014;10:119-29
52. Hickey JW, Vicente FP, Howard GP, Mao HQ, Schneck JP. Biologically inspired design of nanoparticle artificial antigen-presenting cells for immunomodulation. Nano Lett. 2017;17:7045-54
53. Perica K, Bieler JG, Schutz C, Varela JC, Douglass J, Skora A. et al. Enrichment and expansion with nanoscale artificial antigen presenting cells for adoptive immunotherapy. ACS Nano. 2015;9:6861-71
54. Zhang Q, Wei W, Wang P, Zuo L, Li F, Xu J. et al. Biomimetic magnetosomes as versatile artificial antigen-presenting cells to potentiate T-cell-based anticancer therapy. ACS Nano. 2017;11:10724-32
55. Rahir G, Moser M. Tumor microenvironment and lymphocyte infiltration. Cancer Immunol Immun. 2012;61:751-9
56. Speiser DE, Ho PC, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol. 2016;16:599-611
57. Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W. et al. Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer Cell. 2016;29:285-96
58. Sun Z, Fourcade J, Pagliano O, Chauvin JM, Sander C, Kirkwood JM. et al. IL10 and PD-1 cooperate to limit the activity of tumor-specific CD8+ T cells. Cancer Res. 2015;75:1635-44
59. Chen W, Ten Dijke P. Immunoregulation by members of the TGFbeta superfamily. Nat Rev Immunol. 2016;16:723-40
60. Ravi R, Noonan KA, Pham V, Bedi R, Zhavoronkov A, Ozerov IV. et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFbeta enhance the efficacy of cancer immunotherapy. Nat Commun. 2018;9:741
61. Anderson KG, Stromnes IM, Greenberg PD. Obstacles posed by the tumor microenvironment to T cell activity: a case for synergistic therapies. Cancer Cell. 2017;31:311-25
62. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361-5
63. Lim WA, June CH. The principles of engineering immune cells to treat cancer. Cell. 2017;168:724-40
64. Ledford H. Safety concerns blight promising cancer therapy. Nature. 2016;538:150-1
65. Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16:566-81
66. Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D. et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nanotechnol. 2017;12:813-20
67. Baitsch L, Fuertes-Marraco SA, Legat A, Meyer C, Speiser DE. The three main stumbling blocks for anticancer T cells. Trends Immunol. 2012;33:364-72
68. Siriwon N, Kim YJ, Siegler E, Chen X, Rohrs JA, Liu Y. et al. CAR-T cells surface-engineered with drug-encapsulated nanoparticles can ameliorate intratumoral T-cell hypofunction. Cancer Immunol Res. 2018;6:812-24
69. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56-61
70. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-64
71. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205-14
72. Walker LS, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol. 2011;11:852-63
73. Du X, Tang F, Liu M, Su J, Zhang Y, Wu W. et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. 2018;28:416-32
74. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328-32
75. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568-71
76. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14:561-84
77. Melero I, Berman DM, Aznar MA, Korman AJ, Perez Gracia JL, Haanen J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer. 2015;15:457-72
78. Chen Q, Xu L, Liang C, Wang C, Peng R, Liu Z. Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Nat Commun. 2016;7:13193
79. Duan X, Chan C, Guo N, Han W, Weichselbaum RR, Lin W. Photodynamic therapy mediated by nontoxic core-shell nanoparticles synergizes with immune checkpoint blockade to elicit antitumor immunity and antimetastatic effect on breast cancer. J Am Chem Soc. 2016;138:16686-95
80. Kulkarni A, Natarajan SK, Chandrasekar V, Pandey PR, Sengupta S. Combining immune checkpoint inhibitors and kinase-inhibiting supramolecular therapeutics for enhanced anticancer efficacy. ACS Nano. 2016;10:9227-42
81. Song W, Shen L, Wang Y, Liu Q, Goodwin TJ, Li J. et al. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat Commun. 2018;9:2237
82. Feng B, Zhou F, Hou B, Wang D, Wang T, Fu Y. et al. Binary cooperative prodrug nanoparticles improve immunotherapy by synergistically modulating immune tumor microenvironment. Adv Mater. 2018;30:e1803001
83. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME. et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344:641-5
84. Haabeth OA, Lorvik KB, Hammarstrom C, Donaldson IM, Haraldsen G, Bogen B. et al. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat Commun. 2011;2:240
85. Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S. et al. T Helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. 2009;31:787-98
86. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y. et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772-84
87. Borst J, Ahrends T, Babala N, Melief CJM, Kastenmuller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18:635-47
88. Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immun. 2005;54:721-8
89. Kumar S, Anselmo AC, Banerjee A, Zakrewsky M, Mitragotri S. Shape and size-dependent immune response to antigen-carrying nanoparticles. J Control Release. 2015;220:141-8
90. Schanen BC, Das S, Reilly CM, Warren WL, Self WT, Seal S. et al. Immunomodulation and T helper TH(1)/TH(2) response polarization by CeO(2) and TiO(2) nanoparticles. PLoS One. 2013;8:e62816
91. Darrasse-Jeze G, Podsypanina K. How numbers, nature, and immune status of foxp3(+) regulatory T-cells shape the early immunological events in tumor development. Front Immunol. 2013;4:292
92. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S. et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol. 2013;14:307-8
93. Su S, Liao J, Liu J, Huang D, He C, Chen F. et al. Blocking the recruitment of naive CD4(+) T cells reverses immunosuppression in breast cancer. Cell Res. 2017;27:461-82
94. Sacchetti C, Rapini N, Magrini A, Cirelli E, Bellucci S, Mattei M. et al. In vivo targeting of intratumor regulatory T cells using PEG-modified single-walled carbon nanotubes. Bioconjug Chem. 2013;24:852-8
95. Ou W, Thapa RK, Jiang L, Soe ZC, Gautam M, Chang JH. et al. Regulatory T cell-targeted hybrid nanoparticles combined with immuno-checkpoint blockage for cancer immunotherapy. J Control Release. 2018;281:84-96
96. Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15:234-48
97. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14:736-46
98. Huo M, Zhao Y, Satterlee AB, Wang Y, Xu Y, Huang L. Tumor-targeted delivery of sunitinib base enhances vaccine therapy for advanced melanoma by remodeling the tumor microenvironment. J Control Release. 2017;245:81-94
99. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K. et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344:921-5
100. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559-63
101. Ruffell B, Affara N, Coussens L. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012;33:119-26
102. Silva VL, Al-Jamal WT. Exploiting the cancer niche: Tumor-associated macrophages and hypoxia as promising synergistic targets for nano-based therapy. J Control Release. 2017;253:82-96
103. Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G. et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29:1005-10
104. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399-416
105. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A. et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. 2016;11:986-94
106. Wang Y, Lin YX, Qiao SL, An HW, Ma Y, Qiao ZY. et al. Polymeric nanoparticles promote macrophage reversal from M2 to M1 phenotypes in the tumor microenvironment. Biomaterials. 2017;112:153-63
107. Zhou S, Zhang T, Peng B, Luo X, Liu X, Hu L. et al. Targeted delivery of epirubicin to tumor-associated macrophages by sialic acid-cholesterol conjugate modified liposomes with improved antitumor activity. Int J Pharm. 2017;523:203-16
108. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19:108-19
109. Ouzounova M, Lee E, Piranlioglu R, El Andaloussi A, Kolhe R, Demirci MF. et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat Commun. 2017;8:14979
110. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37:208-20
111. He W, Liang P, Guo G, Huang Z, Niu Y, Dong L. et al. Re-polarizing myeloid-derived suppressor cells (MDSCs) with cationic polymers for cancer immunotherapy. Sci Rep. 2016;6:24506
112. Plebanek MP, Bhaumik D, Bryce PJ, Thaxton CS. Scavenger receptor type B1 and lipoprotein nanoparticle inhibit myeloid-derived suppressor cells. Mol Cancer Ther. 2018;17:686-97
113. L. Jeanbart, I. C. Kourtis, A. J. van der Vlies, M. A. Swartz, J. A. Hubbell. 6-Thioguanine-loaded polymeric micelles deplete myeloid-derived suppressor cells and enhance the efficacy of T cell immunotherapy in tumor-bearing mice. Cancer Immunol Immun. 2015;64:1033-46
114. Protti MP, De Monte L. Cross-talk within the tumor microenvironment mediates Th2-type inflammation in pancreatic cancer. Oncoimmunology. 2012;1:89-91
115. Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol. 2015;15:271-82
116. A delicate balance. tweaking IL-2 immunotherapy. Nat Med. 2012;18:208-9
117. Park J, Wrzesinski SH, Stern E, Look M, Criscione J, Ragheb R. et al. Combination delivery of TGF-β inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat Mater. 2012;11:895-905
118. Tugues S, Burkhard SH, Ohs I, Vrohlings M, Nussbaum K, vom Berg J. et al. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015;22:237-46
119. Liu X, Gao X, Zheng S, Wang B, Li Y, Zhao C. et al. Modified nanoparticle mediated IL-12 immunogene therapy for colon cancer. Nanomedicine-UK. 2017;13:1993-2004
120. Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31:220-7
121. Pickup M, Novitskiy S, Moses HL. The roles of TGFbeta in the tumour microenvironment. Nat Rev Cancer. 2013;13:788-99
122. Araki K, Shimura T, Suzuki H, Tsutsumi S, Wada W, Yajima T. et al. E/N-cadherin switch mediates cancer progression via TGF-beta-induced epithelial-to-mesenchymal transition in extrahepatic cholangiocarcinoma. Br J Cancer. 2011;105:1885-93
123. Xu Z, Ramishetti S, Tseng Y-C, Guo S, Wang Y, Huang L. Multifunctional nanoparticles co-delivering Trp2 peptide and CpG adjuvant induce potent cytotoxic T-lymphocyte response against melanoma and its lung metastasis. J Control Release. 2013;172:259-65
124. Xu Z, Wang Y, .Zhang L, Huang L. Nanoparticle-delivered transforming growth factor-β siRNA enhances vaccination against advanced melanoma by modifying tumor microenvironment. ACS Nano. 2014;8:3636-45
125. Hou L, Liu Q, Shen L, Liu Y, Zhang X, Chen F. et al. Nano-delivery of fraxinellone remodels tumor microenvironment and facilitates therapeutic vaccination in desmoplastic melanoma. Theranostics. 2018;8:3781-96
126. Franciszkiewicz K, Boissonnas A, Boutet M, Combadiere C, Mami-Chouaib F. Role of chemokines and chemokine receptors in shaping the effector phase of the antitumor immune response. Cancer Res. 2012;72:6325-32
127. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17:559-72
128. Hu Z, Chen J, Zhou S, Yang N, Duan S, Zhang Z. et al. Mouse IP-10 Gene delivered by folate-modified chitosan nanoparticles and dendritic/tumor cells fusion vaccine effectively inhibit the growth of hepatocellular carcinoma in mice. Theranostics. 2017;7:1942-52
129. Miao L, Li J, Liu Q, Feng R, Das M, Lin CM. et al. Transient and local expression of chemokine and immune checkpoint traps to treat pancreatic cancer. ACS Nano. 2017;11:8690-706
130. Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R. et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol. 2011;12:870-8
131. Salazar F, Awuah D, Negm OH, Shakib F, Ghaemmaghami AM. The role of indoleamine 2,3-dioxygenase-aryl hydrocarbon receptor pathway in the TLR4-induced tolerogenic phenotype in human DCs. Sci Rep. 2017;7:43337
132. Munn DH, Mellor AL. IDO in the tumor microenvironment: Inflammation, counter-regulation, and tolerance. Trends Immunol. 2016;37:193-207
133. Hornyak L, Dobos N, Koncz G, Karanyi Z, Pall D, Szabo Z. et al. The role of indoleamine-2,3-dioxygenase in cancer development, diagnostics, and therapy. Front Immunol. 2018;9:151
134. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33:321-2
135. Ye Y, Wang J, Hu Q, Hochu GM, Xin H, Wang C. et al. Synergistic transcutaneous immunotherapy enhances antitumor immune responses through delivery of checkpoint Inhibitors. ACS Nano. 2016;10:8956-63
136. Lu J, Liu X, Liao YP, Salazar F, Sun B, Jiang W. et al. Nano-enabled pancreas cancer immunotherapy using immunogenic cell death and reversing immunosuppression. Nat Commun. 2017;8:1811
137. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52-67
138. Vandenbroucke RE, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov. 2014;13:904-27
139. Zhu L, Kate P, Torchilin VP. Matrix metalloprotease 2-responsive multifunctional liposomal nanocarrier for enhanced tumor targeting. ACS Nano. 2012;6:3491-8
140. Sun Z, Li R, Sun J, Peng Y, Xiao L, Zhang X. et al. Matrix metalloproteinase cleavable nanoparticles for tumor microenvironment and tumor cell dual-targeting drug delivery. ACS Appl Mater Interfaces. 2017;9:40614-27
141. Cheng K, Ding Y, Zhao Y, Ye S, Zhao X, Zhang Y. et al. Sequentially responsive therapeutic peptide assembling nanoparticles for dual-targeted cancer immunotherapy. Nano Lett. 2018;18:3250-8
142. Minton K. Extracellular matrix: Preconditioning the ECM for fibrosis. Nat Rev Mol Cell Bio. 2014;15:766-7
143. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 (+) T cells to protect tumour cells. Nat Commun. 2018;9:948
144. Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin Oncol. 2018;15:366-81
145. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582-98
146. Miao L, Newby JM, Lin CM, Zhang L, Xu F, Kim WY. et al. The binding site barrier elicited by tumor-associated fibroblasts interferes disposition of nanoparticles in stroma-vessel type tumors. ACS Nano. 2016;10:9234-58
147. Miao L, Lin CM, Huang L. Stromal barriers and strategies for the delivery of nanomedicine to desmoplastic tumors. J Control Release. 2015;219:192-204
148. Pure E, Lo A. Can targeting stroma pave the way to enhanced antitumor immunity and immunotherapy of solid tumors? Cancer Immunol Res. 2016;4:269-78
149. Miao L, Wang Y, Lin CM, Xiong Y, Chen N, Zhang L. et al. Nanoparticle modulation of the tumor microenvironment enhances therapeutic efficacy of cisplatin. J Control Release. 2015;217:27-41
150. Hu K, Miao L, Goodwin TJ, Li J, Liu Q, Huang L. Quercetin remodels the tumor microenvironment to improve the permeation, retention, and antitumor effects of nanoparticles. ACS Nano. 2017;11:4916-25
151. Zhu Y, Wen L, Shao S, Tan Y, Meng T, Yang X. et al. Inhibition of tumor-promoting stroma to enforce subsequently targeting AT1R on tumor cells by pathological inspired micelles. Biomaterials. 2018;161:33-46
152. Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T. et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature. 2017;544:250-4
153. Ojha T, Pathak V, Shi Y, Hennink WE, Moonen CTW, Storm G. et al. Pharmacological and physical vessel modulation strategies to improve EPR-mediated drug targeting to tumors. Adv Drug Deliver Rev. 2017;119:44-60
154. Chen F, Cai W. Tumor vasculature targeting: a generally applicable approach for functionalized nanomaterials. Small. 2014;10:1887-93
155. Gao W, Li S, Liu Z, Sun Y, Cao W, Tong L. et al. Targeting and destroying tumor vasculature with a near-infrared laser-activated "nanobomb" for efficient tumor ablation. Biomaterials. 2017;139:1-11
156. Ferrara N, Adamis AP. Ten years of anti-vascular endothelial growth factor therapy. Nat Rev Drug Discov. 2016;15:385-403
157. Li W, Zhao X, Du B, Li X, Liu S, Yang XY. et al. Gold nanoparticle-mediated targeted delivery of recombinant human endostatin normalizes tumour vasculature and improves cancer therapy. Sci Rep. 2016;6:30619
158. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393-410
159. Patel A, Sant S. Hypoxic tumor microenvironment: Opportunities to develop targeted therapies. Biotechnol Adv. 2016;34:803-12
160. Qiu GZ, Jin MZ, Dai JX, Sun W, Feng JH, Jin WL. Reprogramming of the tumor in the hypoxic niche: The emerging concept and associated therapeutic strategies. Trends Pharmacol Sci. 2017;38:669-86
161. Zheng DW, Li B, Li CX, Fan JX, Lei Q, Li C. et al. Carbon-dot-decorated carbon nitride nanoparticles for enhanced photodynamic therapy against hypoxic tumor via water splitting. ACS Nano. 2016;10:8715-22
162. Song M, Liu T, Shi C, Zhang X, Chen X. Bioconjugated manganese dioxide nanoparticles enhance chemotherapy response by priming tumor-associated macrophages toward M1-like phenotype and attenuating tumor hypoxia. ACS Nano. 2016;10:633-47
163. Chen Y, Yin Q, Ji X, Zhang S, Chen H, Zheng Y. et al. Manganese oxide-based multifunctionalized mesoporous silica nanoparticles for pH-responsive MRI, ultrasonography and circumvention of MDR in cancer cells. Biomaterials. 2012;33:7126-37
164. Chen Q, Feng L, Liu J, Zhu W, Dong Z, Wu Y. et al. Intelligent albumin-MnO2 nanoparticles as pH-/H2 O2 -responsive dissociable nanocarriers to modulate tumor hypoxia for effective combination therapy. Adv Mater. 2016;28:7129-36
165. Zou MZ, Liu WL, Li CX, Zheng DW, Zeng JY, Gao F. et al. A multifunctional biomimetic nanoplatform for relieving hypoxia to enhance chemotherapy and inhibit the PD-1/PD-L1 axis. Small. 2018;14:e1801120
166. Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nat Rev Cancer. 2006;6:521-34
167. Xu W, Liu LZ, Loizidou M, Ahmed M, Charles IG. The role of nitric oxide in cancer. Cell Res. 2002;12:311-20
168. Kim J, Yung BC, Kim WJ, Chen X. Combination of nitric oxide and drug delivery systems: tools for overcoming drug resistance in chemotherapy. J Control Release. 2017;263:223-30
169. Duong HT, Kamarudin ZM, Erlich RB, Li Y, Jones MW, Kavallaris M. et al. Intracellular nitric oxide delivery from stable NO-polymeric nanoparticle carriers. Chem Commun. 2013;49:4190-2
170. Zhang X, Tian G, Yin W, Wang L, Zheng X, Yan L. et al. Controllable generation of nitric oxide by near-infrared-sensitized upconversion nanoparticles for tumor therapy. Adv Funct Mater. 2015;25:3049-56
171. Anselmo AC, Mitragotri S. Cell-mediated delivery of nanoparticles: taking advantage of circulatory cells to target nanoparticles. J Control Release. 2014;190:531-41
172. Evans MA, Huang PJ, Iwamoto Y, Ibsen KN, Chan EM, Hitomi Y. et al. Macrophage-mediated delivery of light activated nitric oxide prodrugs with spatial, temporal and concentration control. Chem Sci. 2018;9:3729-41
173. Heo MB, Lim YT. Programmed nanoparticles for combined immunomodulation, antigen presentation and tracking of immunotherapeutic cells. Biomaterials. 2014;35:590-600
174. Daftarian P, Kaifer AE, Li W, Blomberg BB, Frasca D, Roth F. et al. Peptide-conjugated PAMAM dendrimer as a universal DNA vaccine platform to target antigen-presenting cells. Cancer Res. 2011;71:7452-62
175. Kullberg M, Martinson H, Mann K, Anchordoquy TJ. Complement C3 mediated targeting of liposomes to granulocytic myeloid derived suppressor cells. Nanomedicine-UK. 2015;11:1355-63
176. Zhang Y, Li N, Suh H, Irvine DJ. Nanoparticle anchoring targets immune agonists to tumors enabling anti-cancer immunity without systemic toxicity. Nat Commun. 2018;9:6
177. Mahmoudi M. Debugging nano-bio interfaces: systematic strategies to accelerate clinical translation of nanotechnologies. Trends Biotechnol. 2018;36:755-69
178. Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF. et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1:16014
179. Nguyen VH, Lee BJ. Protein corona: a new approach for nanomedicine design. Int J Nanomedicine. 2017;12:3137-51
180. Corbo C, Molinaro R, Tabatabaei M, Farokhzad OC, Mahmoudi M. Personalized protein corona on nanoparticles and its clinical implications. Biomater Sci. 2017;5:378-87
Author contact
![]() Corresponding author: Dr. Kai Shi, Department of Pharmaceutics, School of Pharmaceutical Science, Shenyang Pharmaceutical University, Shenyang 117004, China. Tel: +86-24-43520557. Email address: shikaiedu.cn.
Corresponding author: Dr. Kai Shi, Department of Pharmaceutics, School of Pharmaceutical Science, Shenyang Pharmaceutical University, Shenyang 117004, China. Tel: +86-24-43520557. Email address: shikaiedu.cn.
Citation styles
APA
Gao, S., Yang, D., Fang, Y., Lin, X., Jin, X., Wang, Q., Wang, X., Ke, L., Shi, K. (2019). Engineering Nanoparticles for Targeted Remodeling of the Tumor Microenvironment to Improve Cancer Immunotherapy. Theranostics, 9(1), 126-151. https://doi.org/10.7150/thno.29431.
ACS
Gao, S.; Yang, D.; Fang, Y.; Lin, X.; Jin, X.; Wang, Q.; Wang, X.; Ke, L.; Shi, K. Engineering Nanoparticles for Targeted Remodeling of the Tumor Microenvironment to Improve Cancer Immunotherapy. Theranostics 2019, 9 (1), 126-151. DOI: 10.7150/thno.29431.
NLM
Gao S, Yang D, Fang Y, Lin X, Jin X, Wang Q, Wang X, Ke L, Shi K. Engineering Nanoparticles for Targeted Remodeling of the Tumor Microenvironment to Improve Cancer Immunotherapy. Theranostics 2019; 9(1):126-151. doi:10.7150/thno.29431. https://www.thno.org/v09p0126.htm
CSE
Gao S, Yang D, Fang Y, Lin X, Jin X, Wang Q, Wang X, Ke L, Shi K. 2019. Engineering Nanoparticles for Targeted Remodeling of the Tumor Microenvironment to Improve Cancer Immunotherapy. Theranostics. 9(1):126-151.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.