Impact Factor
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(3):747-760. doi:10.7150/thno.29989 This issue Cite
Research Paper
Lectin-Mediated pH-Sensitive Doxorubicin Prodrug for Pre-Targeted Chemotherapy of Colorectal Cancer with Enhanced Efficacy and Reduced Side Effects
Meinan Yao1, Xiaotu Ma2, Xin Zhang1, Linqing Shi1, Tianyu Liu1, Xiaolong Liang4, Huiyun Zhao1,3, Xiaoda Li1,3, Liqiang Li1, Hannan Gao1, Bing Jia1 ![]() , Fan Wang1,2
, Fan Wang1,2 ![]()
1. Medical Isotopes Research Center and Department of Radiation Medicine, School of Basic Medical Sciences, Peking University, Beijing 100191, P. R. China
2. Key Laboratory of Protein and Peptide Pharmaceuticals, CAS Center for Excellence in Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, P. R. China
3. Medical and Healthy Analytical Center, Peking University Health Science Center, Beijing 100191, P. R. China
4. Department of Ultrasonography, Peking University Third Hospital, Beijing 100191, P. R. China
Received 2018-9-16; Accepted 2018-12-12; Published 2019-1-24
Abstract

Doxorubicin (DOX) has been clinically used as a broad-spectrum chemotherapeutic agent for decades, but its clinical application is hindered by the lack of tumour specificity, severe cardiotoxicity and haematotoxicity. Pre-targeted strategies are highly tumour-specific, therapeutic approaches. Herein, a novel pre-targeted system was constructed, aiming to enhance anticancer efficacy of DOX and maximally reduce its side effects.
Methods: The DOX prodrug (bDOX) was first synthesized by conjugating DOX with mini-PEGylated (mPEGylated) biotin through a pH-sensitive bond. During the pre-targeted treatment, avidin was first administrated. After an optimized interval, bDOX was second administrated. The nontoxic prodrug bDOX was eventually transformed into the toxic anticancer form (DOX) by a pH-triggered cleavage specifically in tumour cells. The drug efficacy and side effect of the two-step, pre-targeted treatment were fully compared with free DOX in vitro and in vivo.
Results: The prodrug bDOX was quite stable under neutral conditions and nearly nontoxic, but was immediately transformed into the toxic anticancer form (DOX) under acidic conditions. Compared to free DOX, the pre-targeted bDOX exhibited a higher cellular uptake by human colorectal tumour cells (LS180 and HT-29 cells). In vivo evaluation performed on LS180 xenograft animal model demonstrated that the pre-targeted bDOX achieved a much more significant tumour inhibition than free DOX. The largely decreased, unwanted bystander toxicity was demonstrated by changes in body weight, cardiomyocyte apoptosis, blood routine examination and splenic pathological changes.
Conclusion: The high therapeutic efficacy, together with the minimal side effects, of this easily synthesized, pre-targeted system exhibited immense potentiality for the clinical application of DOX delivery.
Keywords: pre-targeted strategy, avidin, biotin, doxorubicin prodrug, chemotherapy
Introduction
Doxorubicin (DOX) has been clinically used as a chemotherapeutic agent for a wide range of cancers, such as acute leukaemia and breast carcinoma [1]. However, its clinical application is hindered by severe cardiotoxicity and haematotoxicity [2]. Drug efficacy is also limited by its lack of tumour-targeting ability and quick elimination from blood circulation [3, 4]. The enhancement of therapeutic efficiency and reduction of side effects remain severe challenges for DOX application. Many efforts have been made in previous studies to solve these two key challenges, including drug delivery systems (DDSs) and small molecular conjugates [5, 6]. The most representative formulae for DDSs are Myocet® and Doxil®/ Caelyx®, which are U.S. Food and Drug Administration (FDA)-approved DOX liposomes [7]. They can effectively reduce DOX cardiotoxicity and prolong blood circulation time [8]. However, their therapeutic efficacy is equal to free DOX and still needs to be improved. In addition to delivering DOX by DDSs, DOX can be covalently conjugated to certain compounds, such as antibodies [9] and peptides [10], to improve its tumour targeting ability. Although anticancer efficacy was improved somewhat, DOX uptake is inevitably high in the liver or kidney [11], where these compounds are metabolized or excreted. To further reduce the side effect of this one-step targeting strategy, pre-targeted treatments with multiple steps are emerging and have become a class of promising therapeutic approaches [12].
In previous reports, the first step of pre-targeted treatments primarily employs bispecific antibodies [13] or enzyme-conjugated antibodies, such as antibody-directed enzyme prodrug therapy (ADEPT) [14]. For the former strategy, a bispecific antibody was fabricated to offer a multivalent and simultaneous binding of two distinct antigens: tumour antigen and drug hapten [15]. For ADEPT, the antibody was conjugated with an enzyme to catalyse the nontoxic prodrug into an active, toxic form [14]. Compared to these two strategies, in which the modification of antibodies increases the complexity of synthesis and potential immunogenicity, the pre-targeted strategy utilizing avidin and biotin systems has specific advantages. Avidin has been shown to be biocompatible and safety referring to the results of phase II clinical trails [16]. Avidin can specifically bind to lectin, which is overexpressed on the surface of many types of cancer cells [17, 18]. Therefore, avidin itself, without any modification, can be employed in the first phase of pre-targeted treatment, making the pre-targeted strategy based on avidin and biotin systems more easily fabricated and more reliable than bispecific antibodies and ADEPT. At the same time, there is a high affinity between avidin and biotin, which is the highest non-covalent affinity (Kd = 10-15 M) in nature [19]. The excellent biocompatibility and low cost of avidin and biotin also encourage further clinical applications [20]. However, this pre-targeted strategy, based on a lectin-targeted avidin cooperated with biotin system, has thus far only been used for tumour imaging in published reports [18].
In this research, pre-targeted strategy based on a lectin-targeted avidin and biotin system was first utilized to deliver a chemotherapeutic drug with pH-sensitive drug release to enhance DOX's anticancer efficacy and reduce its side effects (Figure 1). DOX was conjugated to mini-PEGylated (mPEGylated) biotin via an acid-sensitive hydrazone bond [21, 22] to form the prodrug, biotinylated DOX (bDOX). The nearly nontoxic bDOX was quite stable under neutral conditions, but was quickly hydrolysed into cytotoxic DOX under acidic conditions. During the pre-targeted treatment, avidin was administered in the first step. Once concentrated within tumour mass by binding lectin and cleared from systemic circulation [23], bDOX was added in a second step. After binding to avidin, bDOX was endocytosed into lysosomes for acid-triggered DOX release. Due to the prodrug's fast-clearing pharmacokinetics and minimum toxicity, bDOX that was not bound to avidin was quickly eliminated from the blood circulation, with little harm to normal organs [24]. Thus, a strongly enhanced anticancer effect and largely reduced side effects were observed in a lectin-overexpressing colorectal tumour-bearing animal model.
Results
Preparation and characterization of bDOX
The synthesis of bDOX based on two steps was illustrated in Figure S1. First, mPEGylated biotin was conjugated with hydrazine hydrate to synthesize biotin-PEG4-hydrazide. The obtained biotin-hydrazide was verified by HPLC and MALDI-TOF mass spectrometry (Figure S2Aand Figure S3A). Then, DOX was conjugated to biotin-PEG4-hydrazide via pH-sensitive hydrazone bonds between the carbonyl group of DOX and the hydrazine group of biotin-hydrazide. The synthesized biotin-PEG4-hydrazone-DOX (i.e., bDOX) was characterized using HPLC, MALDI-TOF mass spectrometry and FTIR spectra (Figure S2B, Figure S3B, and Figure S4). As shown in FTIR spectra (Figure S4), the characteristic peak of DOX at 1729.83 cm-1 generated from the carbonyl stretch was replaced with a new peak at 1682.51 cm-1 generated from the C=N stretch, suggesting the formation of hydrazone bonds and the successful synthesis of bDOX.
The four PEG monomers in bDOX were employed to increase the solubility of hydrophobic DOX and avoid possible steric hindrance to the binding of biotin and avidin. The affinity between bDOX and avidin was verified in vitro. Three block agents (biotin, IgG, or vitamin C) were adopted to attempt to block bDOX from binding to avidin. Avidin was pre-fixed on the well bottoms of ELISA plates, and excess block agents were added and incubated before the addition of bDOX. The bDOX binding to avidin was detected. As shown in Figure S5, without the addition of a blocking agent, ~ 50% bDOX was bound to avidin. In contrast, the binding amount of bDOX greatly decreased to ~ 5% when biotin was pre-added. However, the addition of IgG and vitamin C had no significant influence on the binding of bDOX and avidin, showing that the binding between bDOX and avidin was competitively inhibited by biotin and that bDOX retained its specific affinity for avidin.
pH-sensitive DOX release
The pH-sensitivity of bDOX was evaluated by dissolving bDOX in a PB (Phosphate Buffer) solution with varying pH (5.0, 6.0, and 7.4). At various time intervals, the released DOX was detected by HPLC. As Figure 2 shows, in a neutral condition (pH 7.4), only a slight proportion (~ 15%) of bDOX was hydrolysed to DOX even over long periods (30 h). In stark contrast, as pH decreased, the amount of hydrolysed bDOX greatly increased. The accumulated hydrolysed bDOX reached ~ 64% (pH 6.0) and ~ 97% (pH 5.0) for 30 h.
Illustration of lectin-mediated pre-targeted bDOX for cancer treatment. (A) The distribution and elimination of avidin and bDOX. (B) The cellular entry process of pre-targeted bDOX. (C) The structure of bDOX.
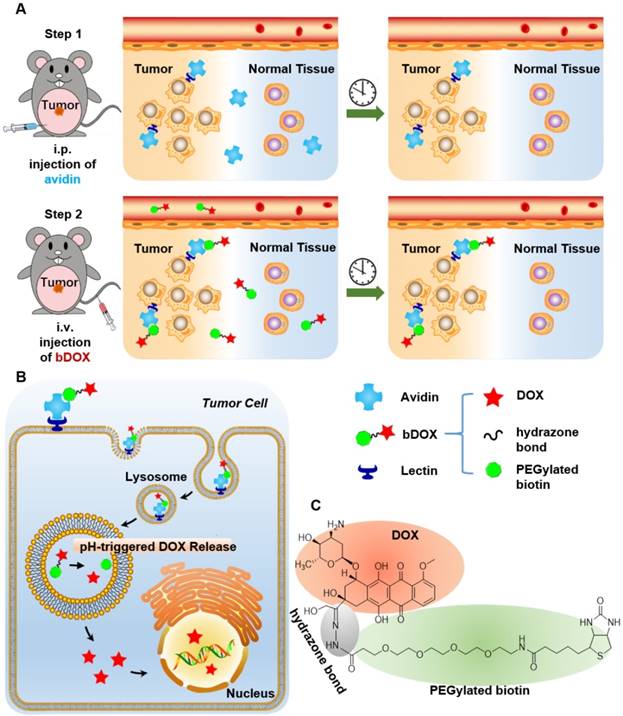
Acid-sensitive DOX release from bDOX. (A-C) HPLC characterization of bDOX hydrolysis after dissolving bDOX in PB buffer solutions with varying pH for different times. Retention time was ~ 23 min for DOX and ~ 25 min for bDOX. (D) Quantification of released DOX from bDOX with varying pH for different times. The values represent mean ± SD (n = 3).
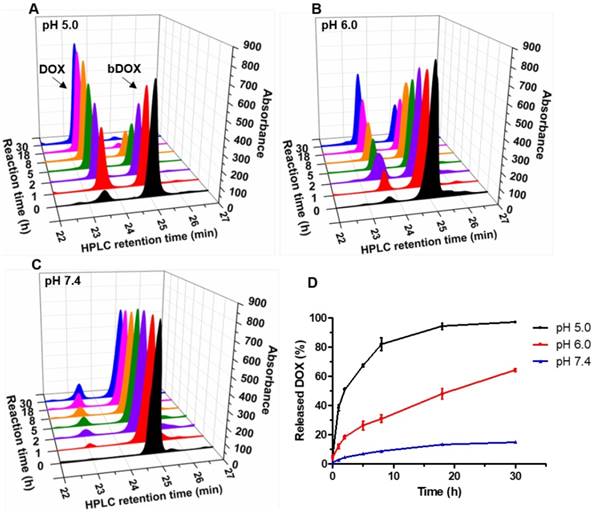
Chemotherapeutic effect of bDOX in vitro. Relative cell viability of (A) HT-29 , (B) LS180 and (C) HEK293 after incubating with un-pre-targeted bDOX (bDOX in graph), DOX or pre-targeted bDOX (pre-bDOX in graph) at different concentrations for 48 h. Log M represents the logarithmic concentration of DOX (mmol/L). *** p < 0.001. The values represent mean ± SD (n = 3). (pre-bDOX: pre-targeted bDOX).
In vitro anticancer efficacy
The chemotherapeutic effect of pre-targeted bDOX was evaluated on HT-29, LS180 and HEK293 cells in vitro. Cells were seeded in 96-well plates and incubated with different concentrations of pre-targeted bDOX for 48 h. Un-pre-targeted bDOX and free DOX were used as control drugs. For all three cell lines, un-pre-targeted bDOX showed minimized cytotoxicity compared to pre-targeted bDOX or free DOX (Figure 3). For both human colorectal cancer cell lines (HT-29 and LS180 cells), pre-targeted bDOX achieved significantly better chemotherapeutic effects than free DOX (Figure 3A-B). After incubating for 48 h, the IC50 value of the pre-targeted bDOX for HT-29 cells was only ~ 20% of the free DOX; for LS180 cells, it was only ~ 25% (Table S1). However, for the normal human cell line (HEK293 cells), the cytotoxicity of pre-targeted bDOX was obviously lower than that of free DOX (Figure 3C).
To explore the inner mechanism of cytotoxicity of pre-targeted bDOX, cellcycle-dependent changes were analysed by flow cytometry before and after incubating HT-29 cells with pre-targeted bDOX. As Figure S6 shows, before incubation, there were 67.45% cells in G1 phase. However, it increased to 69.54% and 76.76% after incubating for 12 and 24 h, respectively. Thus, pre-targeted bDOX can cause S arrest of the cell cycle.
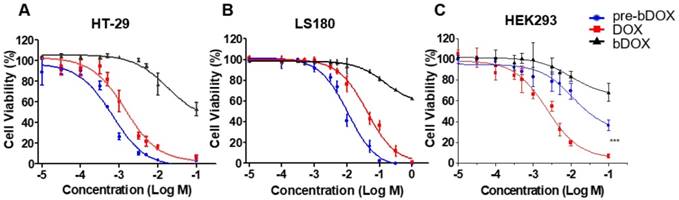
In vitro cellular uptake. (A) Confocal laser scanning microscopy (CLSM) images of cellular uptake of bDOX, free DOX and pre-targeted bDOX in HEK293 , LS180 , and HT-29 cells. The scale bar represents 25 μm. (B) Fluorescence intensity percentages of the treated cells. The values represent mean ± SD (n = 7). * p < 0.05; ** p < 0.01. (pre-bDOX: pre-targeted bDOX).
In vitro cellular uptake
From the cytotoxicity results shown above, pre-targeted bDOX exhibited a much higher anticancer effectiveness than free DOX. To determine whether the enhanced effectiveness resulted from enhanced drug cellular uptake, cells were incubated with pre-targeted bDOX, un-pre-targeted bDOX and free DOX and the intracellular bDOX and DOX were traced and quantified by fluorescence using a confocal laser scanning microscope (CLSM). DOX and bDOX exhibited almost the same property and ability of fluorescence excitation and emission (Figure S7), which made it possible to compare their intracellular concentrations by fluorescence intensity. As shown in Figure 4, when incubating cells with bDOX alone (un-pre-targeted bDOX group), all three types of cells exhibited minimized drug uptake that was much lower than that seen when incubating with free DOX, coincident with the minimized cytotoxicity of bDOX. The reduced cytotoxicity of bDOX itself might result from biotin-PEG4 conjugation, which may make it more difficult for bDOX to diffuse through the cell membrane compared to free DOX.
At the same time, the drug uptake of the pre-targeted bDOX group was higher than that of the free DOX group for these two colorectal cancer cell lines (HT-29 and LS180), which was coincident with the enhanced anticancer efficacy of pre-targeted bDOX. Free DOX exhibited no obvious distinction between tumour and normal cell lines, and there was a high drug uptake in HEK293 cells when incubating with free DOX. In stark contrast, pre-targeted bDOX endowed DOX with the ability to select and specifically target tumour cells, and thus the drug uptake of the pre-targeted bDOX was quite weaker than free DOX in the normal cell line (HEK293).
Besides, for the pre-targeted bDOX group, the drug uptake of HT-29 cells was nearly two-fold higher than that of LS180 cells, and it was much lower for HEK293 cells. This difference was coincident with the level of lectin expression of the three cell lines. As Figure S8 shows, the lectin expression efficiency of the HT-29 and LS180 cells was 95.4% and 39.7%, respectively, as measured by flow cytometry; for HEK293 cells, it was 1.38%. Higher expression of lectin would recruit more avidin, leading to more binding and endocytosis of bDOX [18, 25].
Subcellular localization of pre-targeted bDOX
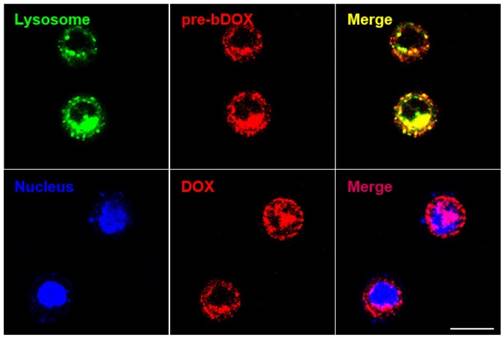
In order to confirm whether bDOX could be transported into the lysosome through avidin-mediated endocytosis to realize pH-sensitive hydrolysis into DOX, the subcellular distribution of avidin and bDOX was tracked by CLSM. First, intracellular colocalization between avidin and lysosome was performed on HT-29 cells to explore whether avidin could be endocytosed into the lysosome as reported previously [17]. As Figure S9 shows, after incubating cells with Cy3-labelled avidin, the labelled avidin was largely colocalized with the LysoTrackerTM-labelled lysosome. Colocalization between pre-targeted bDOX and lysosome was then examined. HT-29 cells were first incubated with avidin, followed by incubation with bDOX 4 h later. There was a large overlay between the fluorescence of bDOX and the labelled lysosome (Figure 5), showing bDOX's successful transportation into the lysosome. Pearson's correlation between lysosome and bDOX was as high as 0.7415 (Figure S10and Table S2). The same fluorescence of bDOX seen in lysosome was also largely observed in the nucleus (Figure 5and Figure S10); this fluorescence was emitted from DOX that was hydrolysed from bDOX in the lysosomes and transported into the nucleus [26]. The three-dimensional image of colocalization between bDOX and lysosomesas well as bDOX and nuclei was shown in Movie S1. These colocalization examinations were also performed on LS180 cells, and the results were coincident with HT-29 cells (Figure S11).
Subcellular localization of pre-targeted bDOX performed on HT-29 cells. Lysosomes were stained by LysoTrackerTM Deep Red and colored in green; nuclei were stained by Hoechst 33342 and colored in blue. The pretargeted bDOX was colored in red. The scale bar represents 25 μm. (pre-bDOX: pre-targeted bDOX).
Timing of pre-targeting protocol
Optimizing the efficacy of pre-targeted chemotherapy requires optimization of the timing of pre-targeting step to allow for a thorough elimination of avidin in off-target organs. The probe 99mTc-biotin was synthesized and characterized (Figure S12). The selected dose of 15 mg/kg avidin and the timing of radioactivity detection at 2 h after 99mTc-biotin administration were based on literature data [27]. The delay between the avidin chase and 99mTc-biotin injection was examined from 1 h to 8 h (Figure S13 and Figure 6A). There was a striking increase in the radioactivity of tumours with a time lapse after avidin injection and a peak at 5 h, followed by a rapid decrease from the sixth hour. The signal from tumours at 5 h post injection (p.i.) was higher than other pre-targeting intervals, with statistical significance (p < 0.05). The gradual decrease in normal tissue signal was accompanied by an increase of the tracer's signal at the tumour site; at 5 h p.i., major organ radioactivity dropped by > 80%. As shown in Figure 6A, the uptake of 99mTc-biotin in LS180 tumours was 20.6 ± 5.7%ID/g at 5 h p.i., which was above 109-fold higher in heart, 3-fold higher in liver, 8-fold higher in spleen, 2-fold higher in kidney, and 7-fold higher in colon. Obviously, the 5-h pre-targeting time exhibited adequate systemic clearance, resulting in excellent tumour-to-normal tissue contrast.
However, in this work, 99mTc-biotin served as a tracer of avidin distributed in vivo, so that Figure 6A shows an integrated biodistribution of both avidin and 99mTc-biotin. The influence of 99mTc-biotin on avidin was examined by experiments measuring 99mTc-biotin biodistribution. 99mTc-biotin injection was performed without avidin, and 1 h, 2 h and 4 h later, radioactivity detection was employed (Figure S14A). The signal decreased rapidly and was quite low (< 0.6%ID/g) at 2 h p.i. in the tissues except for kidney, which showed the highest 99mTc uptake, in accordance with the reported elimination of 99mTc-biotin largely via the renal pathway [27, 28]. 99mTc-biotin injection was also performed after 5 h of avidin chase, followed by radioactivity detection 1 h, 2 h and 4 h p.i. (Figure S14B). Pre-targeted 99mTc-biotin showed similar biodistribution profiles in tissues other than the liver and kidney. Some lectin expressed in liver and kidney resulted in the accumulation of avidin [29]. Higher 99mTc uptake by the liver and kidney indicated that the biodistribution of 99mTc-biotin was significantly dependent on avidin. Therefore, 5 h after avidin chase was the optimal pre-targeting time, and subsequent animal studies will follow this timing sequence.
Biodistribution of pre-targeted bDOX
After confirming the optimal interval between the injection of avidin and bDOX, the biodistribution of pre-targeted bDOX was explored and compared to that of free DOX. LS180-bearing BALB/c nude mice were randomly divided into two groups (n = 3), the pre-targeted bDOX group and free DOX group, and respectively i.v. injected with bDOX and DOX at equivalent doses (avidin was pre-injected into mouse abdomen 5 h before bDOX injection). Four hours after dosing, major organs and tumours were isolated, and the fluorescence from bDOX or DOX was detected (Figure 6B). Drug accumulation in the tumours of pre-targeted bDOX was much higher than that of free DOX with a statistically significant difference (p < 0.05). However, for normal organs, especially the heart and liver (Figure 6C), the drug fluorescence of the pre-targeted bDOX group was much weaker than the free DOX group, showing successful and enhanced drug accumulation in tumours with reduced drug accumulation in normal organs.
Biodistribution of avidin and pre-targeted bDOX. (A) Biodistribution of 99mTc-biotin injected at different times (1 h, 2 h, 4 h, 5 h and 6 h) after the injection of avidin. The values represent mean ± SD (n = 4). (B) Bright-field (upper) and DOX fluorescence (below) images of tumour and major organs ex vivo after injecting free DOX or pre-targeted bDOX. The scale bar represents 1 cm. (C) Quantification of Figure 6B, exhibiting average relative DOX fluorescence intensity of tumour, heart and liver. The values represent mean ± SD (n = 3). (D) CLSM images of DAPI-stained frozen sections showing drug accumulation in tumour and heart tissues after injecting PBS, un-pre-targeted bDOX, free DOX or pre-targeted bDOX. Blue colour represents DAPI-stained nuclei, and pseudo-red colour represents fluorescence from DOX or bDOX. The scale bar represents 50 μm. * p < 0.05. (pre-bDOX: pre-targeted bDOX).
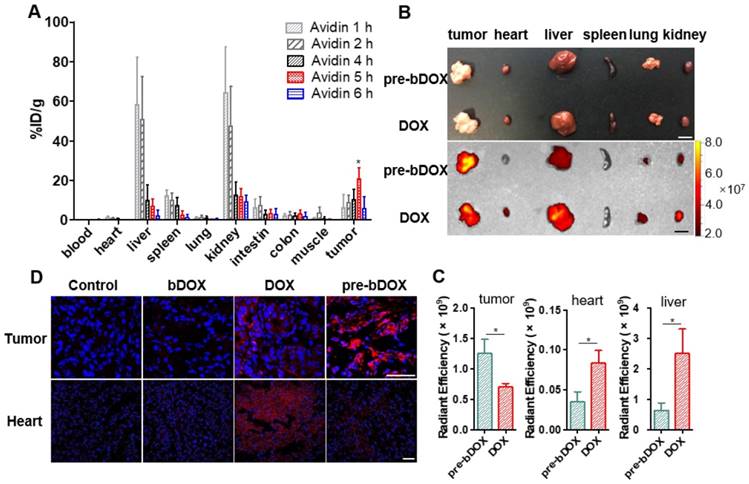
As cardiotoxicity is the most severe side effect of DOX, drug accumulation in tumour and heart tissues was further confirmed by CLSM using the aforementioned animal model. After administering pre-targeted bDOX, un-pre-targeted bDOX (no avidin given before bDOX injection), free DOX, and PBS every two days for a total of 5 administrations, mouse tumour and heart tissues were collected, frozen, sectioned and stained with DAPI. The bDOX and DOX fluorescence signals were observed under CLSM. As Figure 6D shows, there was more drug accumulation in tumours after injecting pre-targeted bDOX compared to free DOX injection. At the same time, drug accumulation in tumours following an injection with un-pre-targeted bDOX was significantly lower than that with free DOX injection, which is consistent with the minimum cellular uptake of individual bDOX. Both pre-targeted and un-pre-targeted bDOX showed a lower drug accumulation in the heart than free DOX due to the lack of lectin expression in the heart tissue and the low level of cellular uptake of bDOX. In summary, pre-targeted bDOX can achieve a higher drug accumulation in tumours and less accumulation in the heart compared to free DOX.
In vivo anticancer efficacy and evaluation of side effects
In vivo anticancer efficacy was evaluated in 5-week-old BALB/c nude mice. Four days after implantating LS180 cells into the abdomen, mice were divided into four groups (n = 5): free DOX, pre-targeted bDOX, un-pre-targeted bDOX, and control. The groups received different treatments: (1) for the free DOX group, 5 mg/kg free DOX was i.v. injected; (2) for the pre-targeted bDOX group, mice were first i.p. injected with 15 mg/kg avidin and bDOX was i.v. injected 5 h later. The dose of bDOX was equivalent to the molar quantity of free DOX; (3) for the un-pre-targeted bDOX group, PBS, rather than avidin, was given before bDOX injection; (4) for the control group, after injecting avidin, biotin was given instead of bDOX. The first treatment was designated day 0, and treatments were repeated every two days for a total of five administrations. Tumour growth was monitored by bioluminescence imaging (BLI) every two days, utilizing the transfected luciferase of LS180 cells. As Figure 7A-B and Figure S15 show, tumours in the un-pre-targeted bDOX and control groups grew quickly, with no significant difference between these two groups in tumour growth due to the minimal toxicity of bDOX. However, compared to free DOX, a stronger anticancer effect was observed for pre-targeted bDOX. At the end of therapy (day 10), tumours in the pre-targeted bDOX group were much smaller than that of the free DOX group, with a statistically significant difference (p < 0.01). After therapy, mice were sacrificed and tumours were collected, counted and weighed (Figure 7C-D). The tumour weight per mouse in the pre-targeted bDOX group was much lower than that in the free DOX group, in accordance with BLI results. For the pre-targeted bDOX group, the tumour number per mouse was also much lower than that in the free DOX group (Figure S16). The number of tumours per mouse represented a part of tendency of tumour metastasis.
In vivo evaluation of chemotherapeutic effects and side effects. (A) Representative bioluminescence images of LS180-bearing BALB/c nude mice on different days after various treatments, showing the rate of tumour growth. The mice in each group are shown in Figure S15. (B) Quantification of the relative tumour fluorescence intensity in Figure 7A and Figure S15. The values represent mean ± SD (n = 5). (C) A photograph of tumours collected from different groups after therapy (day 10). Note that tumours from one mouse were stacked into a pile. The scale bar represents 2 cm. (D) Average tumour weight per mouse for different groups, weighed and calculated from Figure 7C. The values represent mean ± SD (n = 5). (E) Relative mouse body weight of various groups during therapy. The values represent mean ± SD (n = 5). Changes in blood routine examination parameters: (F) WBC and (G) MO of various groups during therapy. The values represent mean ± SD (n = 5). TUNEL-stained frozen section of (H) tumour and (I) heart tissues after the treatments. Compared to free DOX, increased tumour cellular apoptosis and decreased cardiomyocyte apoptosis were exhibited after a treatment with pre-targeted bDOX. The scale bar represents 25 μm. (J) H&E-stained tumour and spleen tissue sections after the treatments. Compared to free DOX, increased tumour necrosis and decreased spleen damage were exhibited after a treatment with pre-targeted bDOX. The scale bar represents 100 μm. * p < 0.05; ** p < 0.01; *** p < 0.001. (MO: monocytes; ns: no significance; pre-bDOX: pre-targeted bDOX; WBC: white blood cells).
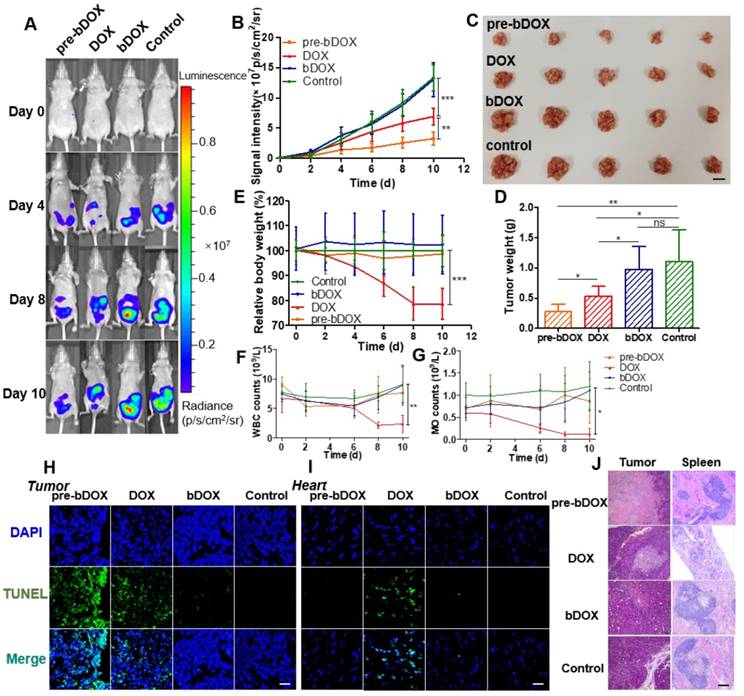
Cellular apoptosis in tumour and heart cells was examined using the TUNEL assay. After therapy, mice from each group (n = 3) were sacrificed and tumour and heart tissues were collected, frozen, sectioned and assayed using the TUNEL method [30]. As Figure 7H shows, the most severe tumour cellular apoptosis was observed in mice treated with pre-targeted bDOX, which was more severe than mice treated with free DOX, showing the enhanced anticancer effect achieved by pre-targeted bDOX. Un-pre-targeted bDOX only caused a slight tumour cellular apoptosis. For heart tissue, obvious cellular apoptosis was caused by the injection of free DOX (Figure 7I); however, there was only a negligible apoptosis with the pre-targeted bDOX or un-pre-targeted bDOX treatments. The results of cellular apoptosis in tumours and the heart after treatment were consistent with the biodistribution evaluation results. Anticancer effect was enhanced by pre-targeted bDOX, while side effects on the heart were greatly reduced.
Changes in body weight are often adopted as an indicator of possible side effects of a chemotherapeutic drug. The average body weight of each group was monitored every two days during therapy. As Figure 7E shows, an obvious decline in body weight was observed for mice treated with free DOX compared to the control group (p < 0.001), indicating the severe side effects of free DOX. In stark contrast, neither the un-pre-targeted bDOX group nor the pre-targeted bDOX group significantly differed from the control group in body weight.
Blood routine examination was adopted to further evaluate the side effects of various treatments. For mice treated with free DOX, the level of white blood cells (WBC) and monocytes (MO) showed obvious changes during therapy (Figure 7F-G), further confirming the severe side effects of free DOX. However, there was no significant difference between the pre-targeted bDOX group and control group, fully showing its minimal side effects.
Tumours and major organs (heart, liver, spleen, lung and kidney) from different groups were collected after therapy, sectioned, and stained with H&E to evaluate possible side effects (Figure 7J). In accordance with the rate of tumour growth (Figure 7A) and the level of cellular apoptosis, tumour sections of the pre-targeted bDOX group had much more necrosis than the free DOX group. Although most organs showed no obvious changes among the treatment groups (Figure S17), there were great morphological and pathological changes in the spleen with free DOX. The spleen in the free DOX group shrank significantly (Figure S18), and splenic corpuscles decreased in size and number, with nearly incomplete structures. Furthermore, there were massive immature red blood cells in the red pulp, indicating possible toxicity of myelosuppression. However, the severe toxicity of free DOX to the spleen was not observed in the pre-targeted bDOX group, showing its overwhelming advantage in terms of reduced side effects and enhanced anticancer efficacy.
Discussion
In this study, a novel pre-targeted system based on avidin and bDOX was successfully constructed and applied for the treatment of lectin-overexpressing colorectal cancer. The biotinylation of archetype DOX cooperated with the application of a pre-targeted strategy achieved enhanced anticancer efficacy and less unwanted side effects compared to the DOX used currently in clinic.
While entering into the body, avidin concentrated on lectin-overexpressing tumours and eliminated from normal organs with little lectin expression after a proper interval. Then bDOX was added and targeted to the avidin in tissues (Figure 1). Afterwards, avidin/bDOX complex was endocytosed into lysosomes. The quick response to the acidic environment could enable bDOX to realize robust DOX release in lysosomes and then DOX would enter cell nuclei. The anticancer effect of pre-targeted bDOX relies on the free form of DOX hydrolysed from bDOX, which mainly operates by interacting with DNA and inhibiting topoisomerase II in order to inhibit DNA synthesis [2]. For free bDOX (i.e., uncombined with avidin), the stability maintained in the neutral condition, the difficulty in entering cells, and the quick elimination kinetics could minimize premature DOX release during systemic blood circulation, thereby reducing side effects.
The insufficiency of the selectivity and targeting towards tumour cells is a severe problem faced by most chemotherapeutic drugs, which promotes their side effects. The pre-targeted strategy can effectively overcome this insufficiency, however. The tumour selectivity of pre-targeted bDOX could reduce drug uptake in normal cells with low lectin expression, and thus decrease damage to normal organs. The targeting ability of pre-targeted bDOX led to higher cellular uptake than the free diffusion of free DOX [31], thereby promoting enhanced anticancer efficacy.
A dose of 15 mg/kg avidin and 5 h pre-targeting time were the optimum conditions for pre-targeted therapy, allowing adequate systemic clearance and excellent tumour-to-normal tissue contrast. Under this situation, pre-targeted bDOX exhibited a more favourable tumour biodistribution than free DOX (i.e., significantly higher drug accumulation in tumours with a much lower concentration in normal organs, especially the heart and liver). The chemotherapy results in tumour-bearing mice were in accordance with the biodistribution evaluation. Tumours in the pre-targeted bDOX group were much smaller in volume, less in number, and lighter in weight, showing that pre-targeted bDOX had a stronger effect on inhibiting tumour growth and a greater potentiallity to prevent tumour metastasis than free DOX. Signs of harmful side effects, which include an apparent decline in body weight, obvious changes in blood routine examination, abnormal morphological and pathological changes in the spleen, and widespread cardiomyocyte apoptosis, indicated the severe side effects of free DOX. Therefore, compared with free DOX, pre-targeted bDOX achieved a more powerful tumour inhibition with minimal damage to normal tissues following an equimolar administration; therefore, pre-targeted bDOX might break the restriction of the current DOX toxicity-limited dosage.
Pre-targeted bDOX showed overwhelming advantages over free DOX in many aspects. Firstly, an organic combination of the pre-targeted system and DOX was achieved, which endowed the free DOX with a highly specific tumour targeting ability. Secondly, lectin-mediated endocytosis significantly increased the cellular uptake of DOX and may reduce the quantity of DOX pumped out. Similar to most small-molecule chemotherapeutic drugs, DOX enters tumour cells through free diffusion; however, significant quantities may be pumped out of cells by glycoproteins [32]. Pre-targeted bDOX, on the other hand, was endocytosed into lysosomes. Therefore, drug resistance was likely to be avoided to some extent [33]. Thirdly, the hydrolysis of the pH-sensitive bond of bDOX was only triggered in an acidic environment to release the active anticancer drug (i.e., free DOX); therefore, DOX toxicity towards normal tissues could be largely reduced.
Currently, many chemotherapeutic drugs associated with DOX are approved for clinical use or in clinical trials. However, there are still some problems with these drugs. For example, Doxil® is the first FDA-approved DOX nano-liposome. After its administration, Doxil accumulates in tumours via the enhanced permeability and retention (EPR) effect; up to 98% of the drug in plasma is liposome-associated DOX, which does not release DOX until clearance under the long blood circulation half-life [7], thereby reducing cardiotoxicity. Similarly, bDOX in our research is stable and non-toxic in neutral conditions (e.g., plasma). However, although Doxil concentration in tumour is 4 to 16 times higher than that of DOX [34], liposomes release DOX slowly and inadequately, which is limited by the tumour microenvironment [35]. The insufficient drug release as well as the heterogeneity of the EPR effect in tumors hinder the efficacy of Doxil. For instance, Doxil was approved by the FDA and European Medicines Evsluation Agency (EMA) for the treatment of metastatic breast cancer in 2003 and multiple myeloma in 2007 with its reduced cardiotoxicity and improved safety profile, but its efficacy was equivalent to DOX [7]. In this study, we aimed at overcoming these problems utilizing a pre-targeted bDOX. Avidin endowed bDOX the ability to target tumours and the effective release of DOX, which was triggered in the acidic environment of lysosomes.
This lectin-mediated avidin and bDOX system has special advantages for clinical translation. As a natural protein, the non-toxicity of avidin has already been clinically verified [36]. This easily obtained, low-cost protein (extracted from egg whites) is only found in birds, amphibians and reptiles, meaning that it is particularly suitable for the treatment of mammals, as there is no endogenous avidin that can interfere with the pre-targeted treatment [37]. Also, the interaction between avidin and biotin is not obviously affected by pH from 2 to 13, enzymes, or temperature. The incidence and mortality rates of colorectal cancer are the third and fourth highest globally [38, 39]. Patients with stage II colorectal cancer are at high risk of recurrence; those with stage III disease are recommended for adjuvant chemotherapy [40]. The pre-targeted bDOX may potentially be a choice in the adjuvant chemotherapy of colorectal cancer as well as a replacement of free DOX or Doxil in clinical practice.
Due to the modular nature of the designed pre-targeted system, the following applications can be developed: (a) Other small-molecule chemotherapeutic agents can be delivered by this pre-targeted system; (b) by utilizing single-photon emission computed tomography (SPECT), 99mTc-biotin can be employed for in vivo molecular imaging to non-invasively monitor tumour growth situations. 99mTc-biotin can also detect the expression level of lectin, thereby predicting whether patients are suitable for this pre-targeted treatment before therapy.
In conclusion, a pre-targeted system based on lectin-mediated avidin and biotin was applied for the first time to cancer chemotherapy. The pre-targeted bDOX exhibited a great potential for clinical translation as a simple and effective anticancer therapeutic agent to replace the traditional free DOX, or even Doxil, with improved drug efficacy and minimal toxicity.
Methods
Preparation and characterization of prodrug bDOX
Synthesis of biotin-hydrazide: 7.37 mg of biotin-PEG4-C2H5COOH was dissolved in methyl alcohol (150 µL); then, 16 mg EDC dissolved in distilled water (25 µL) and 10 mg NHS dissolved in methyl alcohol (25 µL) were added to the biotin-PEG4-C2H5COOH solution. The mixture was stirred at room temperature for 0.5 h, and the 75 µL hydrazine monohydrate was added and stirred overnight. The product, biotin-PEG4-C2H5CON2H3 (biotin-hydrazide), was purified with a semi-preparative HPLC chromatography YMC C-18 column (5 μm, 250 × 4.5 mm) (YMC Co., Ltd., Kyoto, Japan) using a 0-33% gradient elution system. The aqueous phase was aqueous NH4OAc buffer (26 mM, pH 7.0) and the organic phase was acetonitrile. The biotin-hydrazide was collected by freeze-drying (7.2 mg, 92.3%).
Synthesis of bDOX: Doxorubicin hydrochloride (4 mg) and biotin-hydrazide (4 mg) were dissolved in extra dry DMF (2.8 mL) in the presence of 4 μL trifluoroacetic acid. After stirring in the dark at room temperature overnight, the product biotin-PEG4-C2H5CON2H2-DOX (bDOX) was purified with a semi-preparative HPLC column using a 0-80% gradient elution system with aqueous NH4OAc buffer (26 mM, pH 7.0) (solvent A) and MeCN (solvent B), and the red powder was collected by lyophilization (3.8 mg, 52.6%).
The bDOX was determined by mass spectrometer (Bruker Co., Ettlingen, Germany), infrared spectrometer (PerkinElmer, Inc., Spokane, WA, USA) and 1H-NMR spectroscopy (Bruker spectrometer, 300 MHz, Ettlingen, Germany), and the optical properties of DOX and bDOX were analysed by a fluorescence spectrophotometer (PerkinElmer, Inc., Spokane, WA, USA) with a slit width of 5 nm.
In vitro drug release
Triplicate bDOX was dissolved in 0.2 M PB buffer at pH 5.0, 6.0, and 7.4 to a final concentration of 0.485 mM. The mixed solutions were shaken at 120 rpm at 37 °C. The concentration of released DOX from bDOX was analysed by HPLC at various time intervals (0, 1, 2, 5, 8, 18, and 30 h) with a reference to the DOX standard calibration curve. The assay was conducted in triplicate.
In vitro chemotherapy and cytotoxicity
Cell proliferation inhibition was examined in HT-29 and LS180 cells, and cytotoxicity was evaluated in HEK293 cells. Cells were plated at 3.63 × 104 cells/well in 100 μL of media in 96-well plates the night before treatment with pre-targeted bDOX, bDOX, or free DOX. Their concentrations varied from 0.01 to 103 µM DOX. The avidin of the pre-targeted bDOX group had been incubated for 4 h in advance, and the dose of avidin in each well was 100 μM. After 48 h, the cellular viability was detected using an enzyme-linked immune detector at 450 nm absorbance with a CCK-8 kit (Cell Counting Kit-8, Dojindo Laboratories, Tokyo, Japan) and the half-growth inhibition concentration (IC50) was calculated with the Prism software. The assay was performed in triplicate and repeated twice.
Drug uptake and cellular distribution of bDOX
HT-29 cells were seeded into glass-bottom dishes (1.5 × 105 cells per dish). After incubating for 12 h, the medium was removed. Cy3-labelled avidin was purified by the PD-10 column. Cells were incubated with 2.2656 μM Cy3-labelled avidin for 4 h; then, nuclei were further labelled with the nuclear dye Hoechst 33342 for 1 h and lysosomes were labelled with LysoTrackerTM Deep Red (Thermo Fisher Scientific, Rockford, IL) for 0.5 h.
Three cell lines, HT-29, LS180 and HEK293, which had different lectin expression levels, were seeded into glass-bottom dishes (1.5 × 105 cells per dish). After incubating for 12 h, the medium was removed and pre-targeted bDOX, free DOX, and bDOX were added to the cells for 4 h at an equivalent concentration of 9.0625 µM DOX. The avidin of the pre-targeted bDOX group had been incubated for 4 h in advance, and the dose of avidin in each dish was 2.2656 μM. Following incubation, the cells were washed with PBS and they were further labelled with the nuclear dye Hoechst 33342 for 1 h and the lysosomal dye LysoTrackerTM Deep Red for 0.5 h. Avidin-labelled Cy3 was added to HT-29 cells and incubated for 4 h, and Hoechst 33342 and LysoTrackerTM Deep Red were added as before. The cells were incubated at 37 °C for all steps. Images were taken using a confocal laser scanning microscope (CLSM), and stacks were recorded with the confocal laser scanning system TCS STED (Leica Microsystems, Wetzlar, Germany).
In vivo drug biodistribution
All animal experiments were performed in accordance with the Guidelines of Peking University Animal Care and Use Committee.
Forty-four BALB/c mice were divided into eleven groups (n = 4 per group) at random. Five groups separately received an intraperitoneal injection of avidin (300 μg in 100 μL PBS per mouse [41]) at 1, 2, 4, 6 and 8 h ahead of time, and 99mTc-biotin (8 μCi per mouse) was injected intravenously (i.v.). Euthanasia was administered after 2 h. Another three groups of mice received intraperitoneal injection of avidin (300 μg in 100 μL PBS per mouse) 5 h ahead of time, and 99mTc-biotin (8 μCi per mouse) was injected into the tail vein. Euthanasia was administered after 1, 2, and 4 h, respectively. The last three groups were administered a single dose of 99mTc-biotin (8 μCi per mouse) i.v., and euthanasia was administered after 1, 2, and 4 h, respectively. Blood, heart, liver, spleen, lung, kidney, intestine, colon, and muscle were harvested and weighed. The radioactivity of the tissues was measured using a γ counter (Packard, Meriden, CT, USA). The results are expressed as a percentage of injected dose per gram of tissue (%ID/g tissue).
Twenty-five BALB/c nude mice bearing LS180 xenografts were divided into five groups (n = 5 per group) at random. For five groups, 99mTc-biotin (8 μCi per mouse) was injected via the tail vein at 1, 2, 4, 5 and 6 h post-avidin chase (300 μg in 100 μL PBS per mouse, intraperitoneally), and the animals were euthanized 2 h later. Tumour tissues, blood samples and various organs were collected as described above and counted for 99mTc to calculate the %ID/g (mean ± SD).
DOX biodistribution
The biodistribution profiles of the pre-targeted bDOX and free DOX were tested in nude mice bearing diffuse LS180 tumours. Pre-targeted bDOX and DOX (10 mg/kg body weight of DOX) were injected into the tail vein of mice into which avidin and saline had been pre-injected i.p. 5 h earlier (n = 3). Euthanasia was administered 4 h later [42], and major organs and tumours were collected and rinsed with saline. The content of DOX or bDOX distributed in isolated tissues was imaged using a near-infrared fluorescence imaging system (IVIS Spectrum, Waltham, Massachusetts, USA).
DOX biodistribution in the tumour and heart was also examined by CLSM. After an i.v. administration of pre-targeted bDOX (5 mg DOX/kg), free DOX (5 mg/kg), bDOX (5 mg DOX/kg), and PBS every two days for a total of five times, mouse tumour and heart tissues were excised and embedded in O.C.T. (Optimum Cutting Temperature compound) medium (SAKURA Tissue-Tek, Torrance, USA) and sectioned into 0.2-μm slices using a freezing microtome (Leica, Solms, Germany). The sections were stained with DAPI and observed under CLSM. The fluorescence of DOX (λex = 488 nm, λem = 580 nm) and DAPI were measured for observation.
In vivo tumour chemotherapy and monitoring
Mice were implanted with the LS180 tumour as described above. Briefly, female BALB/c nude mice (5 w, 15-19 g) were ordered, and 5 × 106 LS180 cells in 100 μL PBS per mouse were implanted intraperitoneally. Due to rapid LS180 tumour growth, mice were randomly assigned to four groups (n = 6 per group) and treated on day 5 after implantation, when tumours were just palpable. Mice received five doses of pre-targeted bDOX (5 mg DOX/kg), free DOX (5 mg/kg), bDOX (5 mg DOX/kg), and empty vehicles at the same concentration as pre-targeted bDOX. For the pre-targeted group, avidin was administered at 300 μg in 100 μL PBS per dose 5 h in advance. The treatment was administered every two days and tumour growth was monitored via bioluminescence imaging (BLI) by an IVIS spectrum. Prior to analysis, animals were injected i.p. with D-luciferin (150 mg/kg), anaesthetized with isoflurane, and imaged 7 min postinjection. Images were acquired with a 1-s exposure time and minimum binning factor. After ten days, mice were euthanized, and tumours were dissected, weighed, and photographed. Tumours and major organs were collected for paraffin sectioning and stained with haematoxylin and eosin (H&E) for histopathological analysis. In addition, tumour and heart tissues were excised for embedding in O.C.T. medium, frozen in liquid nitrogen, and sectioned into 0.2-μm slices using a freezing microtome. Cell apoptosis of freezing sections was detected using a TUNEL assay kit (Roche Co., Ltd.) according to the manufacturer's instruction.
In vivo toxicity evaluation
Tumour-bearing mice were prepared as in the animal therapy experiment described above and were randomly divided into four groups (n = 6). Mice received i.v. administrations of pre-targeted bDOX (5 mg DOX/kg), free DOX (5 mg/kg), bDOX (5 mg DOX/kg), and empty vehicle. Body weight fluctuations were recorded. The blood routine examination was monitored every two days. Blood samples were collected from the mice eye socket vein before every drug administration and two days later after the last dose, and examined using the blood cell automatic analysis machinery.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 5 (GraphPad Software Inc., La Jolla, CA). All values were presented as mean ± SD. Statistical analysis was done using a 1-way ANOVA for multiple groups and an unpaired Student's t-test. A p-value of < 0.05 was considered statistically significant. For all tests, symbols represent the following: * p < 0.05; ** p < 0.01; *** p < 0.001; ns, not significant.
Abbreviations
ADEPT: antibody-directed enzyme prodrug therapy; BLI: bioluminescence imaging; CLSM: confocal laser scanning microscope; DDSs: drug delivery systems; DOX: doxorubicin; EMA: European Medicines Evsluation Agency; EPR: enhanced permeability and retention; FDA: Food and Drug Administration; H&E: haematoxylin and eosin; IC50: half-growth inhibition concentration; MO: monocytes; SPECT: single photon emission computed tomography; WBC: white blood cells.
Supplementary Material
Supplementary methods, figures, tables and movie legend.
Supplementary movie 1.
Acknowledgements
The authors would like to acknowledge the contributions of the following individuals to this work: Yihui Xu of Key Laboratory of Infection and Immunity, Institute of Biophysics, Chinese Academy of Sciences for her support in bioluminescence imaging, and Junfeng Hao of Institute of Biophysics, Chinese Academy of Sciences for her help in histopathological analysis.
This research was supported by the National Key R&D Program of China (2017YFA0205600), National Natural Science Foundation of China (NSFC) projects (81371614, 81630045, 81420108019, 81621063, 81427802), and Strategic Priority Research Program of the Chinese Academy of Sciences (XDA12020216).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jehn CF, Hemmati P, Lehenbauer-Dehm S, Kummel S, Flath B, Schmid P. Biweekly pegylated liposomal doxorubicin (Caelyx) in heavily pretreated metastatic breast cancer: a phase 2 study. Clin Breast Cancer. 2016;16:514-9
2. Damiani RM, Moura DJ, Viau CM, Caceres RA, Henriques JAP, Saffi J. Pathways of cardiac toxicity: comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch Toxicol. 2016;90:2063-76
3. Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185-229
4. Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219-34
5. Bildstein L, Dubernet C, Couvreur P. Prodrug-based intracellular delivery of anticancer agents. Adv Drug Deliv Rev. 2011;63:3-23
6. Jiang K, Song X, Yang L, Li L, Wan Z, Sun X. et al. Enhanced antitumor and anti-metastasis efficacy against aggressive breast cancer with a fibronectin-targeting liposomal doxorubicin. J Control Release. 2017
7. Barenholz Y. Doxil (R) - The first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160:117-34
8. Chan S, Davidson N, Juozaityte E, Erdkamp F, Pluzanska A, Azarnia N. et al. Phase III trial of liposomal doxorubicin and cyclophosphamide compared with epirubicin and cyclophosphamide as first-line therapy for metastatic breast cancer. Ann Oncol. 2004;15:1527-34
9. Ma ZX, He H, Sun FM, Xu Y, Huang XQ, Ma YX. et al. Selective targeted delivery of doxorubicin via conjugating to anti-CD24 antibody results in enhanced antitumor potency for hepatocellular carcinoma both in vitro and in vivo. J Cancer Res Clin. 2017;143:1929-40
10. Pallerla S, Gauthier T, Sable R, Jois SD. Design of a doxorubicin-peptidomimetic conjugate that targets HER2-positive cancer cells. Eur J Med Chem. 2017;125:914-24
11. Wibroe PP, Ahmadvand D, Oghabian MA, Yaghmur A, Moghimi SM. An integrated assessment of morphology, size, and complement activation of the PEGylated liposomal doxorubicin products Doxil (R), Caelyx (R), DOXOrubicin, and SinaDoxosome. J Control Release. 2016;221:1-8
12. Patra M, Zarschler K, Pietzsch HJ, Stephan H, Gasser G. New insights into the pretargeting approach to image and treat tumours. Chem Soc Rev. 2016;45:6415-31
13. Green DJ, O'Steen S, Lin Y, Comstock ML, Kenoyer AL, Hamlin DK. et al. CD38 bispecific antibody pretargeted radioimmunotherapy for multiple myeloma and other B cell malignancies. Blood. 2017
14. Sharma SK, Bagshawe KD. Antibody directed enzyme prodrug therapy (ADEPT): trials and tribulations. Adv Drug Deliv Rev. 2017;118:2-7
15. Khaw BA, Gada KS, Patil V, Panwar R, Mandapati S, Hatefi A. et al. Bispecific antibody complex pre-targeting and targeted delivery of polymer drug conjugates for imaging and therapy in dual human mammary cancer xenografts: targeted polymer drug conjugates for cancer diagnosis and therapy. Eur J Nucl Med Mol Imaging. 2014;41:1603-16
16. Paganelli G, De Cicco C, Ferrari ME, Carbone G, Pagani G, Leonardi MC. et al. Intraoperative avidination for radionuclide treatment as a radiotherapy boost in breast cancer: results of a phase II study with Y-90-labeled biotin. Eur J Nucl Med Mol Imaging. 2010;37:203-11
17. Hama Y, Urano Y, Koyama Y, Kamiya M, Bernardo M, Paik RS. et al. A target cell-specific activatable fluorescence probe for in vivo molecular imaging of cancer based on a self-quenched avidin-rhodamine conjugate. Cancer Res. 2007;67:2791-9
18. Yao Z, Zhang M, Sakahara H, Saga T, Arano Y, Konishi J. Avidin targeting of intraperitoneal tumor xenografts. J Natl Cancer Inst. 1998;90:25-9
19. Green NM. Avidin and streptavidin. Methods Enzymol. 1990;184:51-67
20. Prakash S, Hazari PP, Meena VK, Jaswal A, Khurana H, Kukreti S. et al. Biotinidase resistant (68)Gallium-radioligand based on biotin/avidin interaction for pretargeting: synthesis and preclinical evaluation. Bioconjugate Chem. 2016;27:2780-90
21. Du C, Qian J, Zhou L, Su Y, Zhang R, Dong CM. Biopolymer-drug conjugate nanotheranostics for multimodal imaging-guided synergistic cancer photothermal-chemotherapy. ACS Appl Mater Interfaces. 2017;9:31576-88
22. Luo WX, Wen G, Yang L, Tang J, Wang JG, Wang JH. et al. Dual-targeted and pH-sensitive doxorubicin prodrug-microbubble complex with ultrasound for tumor treatment. Theranostics. 2017;7:452-65
23. Sakahara H, Saga T. Avidin-biotin system for delivery of diagnostic agents. Adv Drug Deliv Rev. 1999;37:89-101
24. Sharkey RM, Goldenberg DM. Cancer radioimmunotherapy. Immunotherapy. 2011;3:349-70
25. Ren WX, Han J, Uhm S, Jang YJ, Kang C, Kim JH. et al. Recent development of biotin conjugation in biological imaging, sensing, and target delivery. Chem Commun. 2015;51:10403-18
26. Dutertre M, Chakrama FZ, Combe E, Desmet FO, Mortada H, Polay Espinoza M. et al. A recently evolved class of alternative 3'-terminal exons involved in cell cycle regulation by topoisomerase inhibitors. Nat Commun. 2014;5:3395
27. Dong C, Yang S, Shi J, Zhao H, Zhong L, Liu Z. et al. SPECT/NIRF dual modality imaging for detection of intraperitoneal colon tumor with an avidin/biotin pretargeting system. Sci Rep. 2016;6:18905
28. Jia B, Shi J, Yang Z, Xu B, Liu Z, Zhao H. et al. 99mTc-labeled cyclic RGDfK dimer: initial evaluation for SPECT imaging of glioma integrin alphavbeta3 expression. Bioconjug Chem. 2006;17:1069-76
29. Sharkey RM, Karacay H, Cardillo TM, Chang CH, McBride WJ, Rossi EA. et al. Improving the delivery of radionuclides for imaging and therapy of cancer using pretargeting methods. Clin Cancer Res. 2005;11:7109s-21s
30. Rami A, Benz A. Exclusive activation of caspase-3 in mossy fibers and altered dynamics of autophagy markers in the mice hippocampus upon status epilepticus induced by kainic acid. Mol Neurobiol. 2018;55:4492-503
31. Guo Y, Luo J, Tan S, Otieno BO, Zhang Z. The applications of Vitamin E TPGS in drug delivery. Eur J Pharm Sci. 2013;49:175-86
32. Li R, Wu R, Zhao L, Wu M, Yang L, Zou H. P-glycoprotein antibody functionalized carbon nanotube overcomes the multidrug resistance of human leukemia cells. ACS Nano. 2010;4:1399-408
33. Wei T, Chen C, Liu J, Liu C, Posocco P, Liu X. et al. Anticancer drug nanomicelles formed by self-assembling amphiphilic dendrimer to combat cancer drug resistance. Proc Natl Acad Sci U S A. 2015;112:2978-83
34. Soloman R, Gabizon AA. Clinical pharmacology of liposomal anthracyclines: focus on pegylated liposomal Doxorubicin. Clin Lymphoma Myeloma. 2008;8:21-32
35. Alibolandi M, Abnous K, Mohammadi M, Hadizadeh F, Sadeghi F, Taghavi S. et al. Extensive preclinical investigation of polymersomal formulation of doxorubicin versus Doxil-mimic formulation. J Control Release. 2017;264:228-36
36. Lesch HP, Kaikkonen MU, Pikkarainen JT, Yla-Herttuala S. Avidin-biotin technology in targeted therapy. Expert Opin Drug Del. 2010;7:551-64
37. Kovacs-Nolan J, Phillips M, Mine Y. Advances in the value of eggs and egg components for human health. J Agric Food Chem. 2005;53:8421-31
38. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115-32
39. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-86
40. Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383:1490-502
41. Yao ZS, Zhang ML, Sakahara H, Saga T, Arano Y, Konishi J. Avidin targeting of intraperitoneal tumour xenografts. J Natl Cancer Inst. 1998;90:25-9
42. Duan X, Xiao J, Yin Q, Zhang Z, Yu H, Mao S. et al. Smart pH-sensitive and temporal-controlled polymeric micelles for effective combination therapy of doxorubicin and disulfiram. ACS Nano. 2013;7:5858-69
Author contact
![]() Corresponding authors: Dr. Bing Jia, Medical Isotopes Research Center, Peking University, Beijing, China, 100191; Phone: (+86-10) 8280-2871; Fax: (+86-10) 8280-1145; E-mail: jiabingedu.cn and Prof. Fan Wang, Medical Isotopes Research Center, Peking University, Beijing, China, 100191; Phone: (+86-10) 8280-2871; Fax: (+86-10) 8280-1145; E-mail: wangfanedu.cn
Corresponding authors: Dr. Bing Jia, Medical Isotopes Research Center, Peking University, Beijing, China, 100191; Phone: (+86-10) 8280-2871; Fax: (+86-10) 8280-1145; E-mail: jiabingedu.cn and Prof. Fan Wang, Medical Isotopes Research Center, Peking University, Beijing, China, 100191; Phone: (+86-10) 8280-2871; Fax: (+86-10) 8280-1145; E-mail: wangfanedu.cn