Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(5):1247-1263. doi:10.7150/thno.29884 This issue Cite
Research Paper
Therapeutic effects of human monoclonal PSMA antibody-mediated TRIM24 siRNA delivery in PSMA-positive castration-resistant prostate cancer
Sheng-Jia Shi1#, Li-Juan Wang2#, Dong-Hui Han1#, Jie-Heng Wu3#, Dian Jiao4, Kai-Liang Zhang5, Jiang-Wei Chen6, Yu Li1, Fa Yang1, Jing-Liang Zhang1, Guo-Xu Zheng7, An-Gang Yang3, Ai-Zhi Zhao8 ![]() , Wei-Jun Qin1
, Wei-Jun Qin1 ![]() , Wei-Hong Wen3
, Wei-Hong Wen3 ![]()
1. Department of Urology, Xijing Hospital, Fourth Military Medical University, 710032 Xi'an, P.R. China
2. Department of Dermatology, First Affiliated Hospital of Xi'an Jiaotong University, 710061 Xi'an, P.R. China
3. State Key Laboratory of Cancer Biology, Department of Immunology, Fourth Military Medical University, 710032 Xi'an, P.R. China
4. Department of Urology, Tangdu Hospital, Fourth Military Medical University, 710038 Xi'an, P.R. China
5. Department of Orthopedics, Tangdu Hospital, Fourth Military Medical University, 710038 Xi'an, P.R. China
6. Department of Cardiology, Xijing Hospital, Fourth Military Medical University, 710032 Xi'an, P.R. China.
7. Department of Physiology and Pathophysiology, Fourth Military Medical University, 710032 Xi'an, P.R. China
8. OriMAbs Ltd. Science center, Room 544. 3624 Market Street, PA 19104, USA
# These authors contributed equally to this work.
Received 2018-9-12; Accepted 2019-1-14; Published 2019-2-7
Citation:
Shi SJ, Wang LJ, Han DH, Wu JH, Jiao D, Zhang KL, Chen JW, Li Y, Yang F, Zhang JL, Zheng GX, Yang AG, Zhao AZ, Qin WJ, Wen WH. Therapeutic effects of human monoclonal PSMA antibody-mediated TRIM24 siRNA delivery in PSMA-positive castration-resistant prostate cancer. Theranostics 2019; 9(5):1247-1263. doi:10.7150/thno.29884. https://www.thno.org/v09p1247.htm
Other stylesAbstract

Background and Aims: Prostate specific membrane antigen (PSMA) is specifically expressed on prostate epithelial cells and markedly overexpressed in almost all prostate cancers. TRIM24 is also up-regulated from localized prostate cancer to metastatic castration-resistant prostate cancer (CRPC). Because of the high relevance of TRIM24 for cancer development and the universal expression of PSMA in CPRC, we investigated the efficacy of human monoclonal PSMA antibody (PSMAb)-based platform for the targeted TRIM24 siRNA delivery and its therapeutic efficacy in CRPC in vivo and in vitro.
Methods: The therapeutic complexes were constructed by conjugating PSMAb and sulfo-SMCC-protamine, and encapsulating TRIM24 siRNA. Flow cytometry, immunofluorescence, and fluorescence imaging were performed to detect the receptor-binding, internalization, and targeted delivery of PSMAb-sulfo-SMCC-protamine (PSP)-FAM-siRNA complex (PSPS) in vitro and in vivo. CCK-8, plate-colony formation, apoptosis, cell cycle, and Transwell assays were performed to evaluate the therapeutic potential of the PSP-TRIM24 siRNA complex in vitro, whereas the in vivo therapeutic efficacy was monitored by small animal imaging, radiography, and micro CT.
Results: We confirmed that PSP could efficiently protect siRNA from enzymatic digestion, enable targeted delivery of siRNA, and internalize and release siRNA into PSMA-positive (PSMA+) prostate cancer cells in vitro and in vivo. Silencing TRIM24 expression by the PSP-TRIM24 siRNA complex could dramatically suppress proliferation, colony-formation, and invasion of PSMA+ CRPC cells in vitro, and inhibit tumor growth of PSMA+ CRPC xenografts and bone loss in PSMA+ CRPC bone metastasis model without obvious toxicity at therapeutic doses in vivo.
Conclusion: PSMAb mediated TRIM24 siRNA delivery platform could significantly inhibit cell proliferation, colony-formation, and invasion in PSMA+ CRPC in vitro and suppressed tumor growth and bone loss in PSMA+ CRPC xenograft and bone metastasis model.
Keywords: CRPC, PSMA, TRIM24, RNA interference
Introduction
An estimated 16490 new prostate cancer cases and 29430 deaths were recorded in the USA in 2018 so far [1]. Androgen deprivation therapy is one of the most widely used management strategies for primary prostate cancer and the major component of systemic treatments for recurrent or metastatic prostate cancer [2]. However, due to amplification or point mutations in the androgen receptor (AR) gene [3, 4] and other underlying mechanisms yet not fully understood, nearly all primary prostate cancers eventually become androgen resistance and readily progress to castration-resistant prostate cancer (CRPC) stage [5]. Unfortunately, because almost all CRPC patients are refractory to chemotherapy, it is currently an incurable disease [6]. Therefore, new treatment strategies for CRPC patients are urgently needed.
Prostate-specific membrane antigen (PSMA) is specifically up-regulated on prostate epithelial cells in almost all prostate cancers, especially in metastatic CRPC cases [7, 8]. Thus, PSMA is considered an excellent therapeutic target for CRPC [9]. In our previous study, we had obtained a PSMA specific single-chain antibody fragment (scFv) (termed gy1) from a large yeast-display naive human scFv library which could specifically recognize the extracellular domain of PSMA [10]. Furthermore, we reconstructed this scFv into a human monoclonal PSMA antibody (PSMAb) and provided evidence that PSMAb could specifically bind with and internalize into PSMA+ prostate cancer cells with high binding affinity in vitro and in vivo. In addition, we also confirmed that PSMAb could inhibit tumor growth through antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) in PSMA+ CRPC cell xenografts in vivo (Wu et al. unpublished data). Thus, it was essential to explore new effective therapies for CRPC patients based on PSMAb.
Tripartite motif-containing protein 24 (TRIM24) (originally transcriptional intermediary factor 1α) was reported to be positively correlated with carcinogenesis and cancer development in multiple cancers, such as glioblastoma [11], gastric cancer [12], and cervical cancer [13]. More importantly, TRIM24 could function as a chromatin-associated epigenetic reader protein and an oncogenic transcriptional activator by interacting with several nuclear receptors such as androgen receptor [14] or estrogen receptor [15] via its tandem PHD-bromodomain. Furthermore, it was reported that TRIM24, whose expression was increased from primary prostate cancer to CRPC, could promote the proliferation of CRPC cells under low androgen conditions by augmenting AR signaling [16]. These observations indicated that TRIM24 could be an ideal therapeutic target in CRPC.
Protecting siRNAs from enzymatic digestion and facilitating their internalization into tumor cells in vivo remain a challenge in RNA interference (RNAi) [17]. Compared to the techniques involving fusion proteins, monoclonal antibody-based targeted delivery systems have the advantage of using clinically used and readily available monoclonal antibodies. Furthermore, monoclonal antibody-based siRNA delivery system has been shown to be more efficient and safer than liposome- or nanoparticle-based siRNA delivery system which lacks specific targeting ability [18, 19]. Nicole et.al reported a reliable method which could deliver siRNA in stable and cell type-specific manner by a monoclonal antibody-based siRNA delivery system [20]. In the present study, we investigated the efficacy of the PSMAb-based platform for the targeted delivery of TRIM24 siRNA and its therapeutic effects in CRPC.
Materials and Methods
Plasmid construction, expression, and purification of human PSMAb in CHO-S cells
The coding sequences for the viable region of the heavy and light chains (gy1) were joined with corresponding constant regions of human IgG1 and synthesized and subsequently incorporated into the bicistronic eukaryotic expression vector Lh1. FreeStyle MAX transfection reagent was used to transiently transfect PSMAb-expressing vector into CHO-S cells (Invitrogen, Life Technologies, Paisley, Scotland, UK). At day 7 after transfection, the supernatants were collected, centrifuged at 4°C at 5000rpm for 20min and filtered through 0.45 μm filter. The same volume of binding buffer (20 mM sodium phosphate, pH 7.0) was then mixed with the supernatant. PSMAb was then purified using HiTrap rProtein A Fast Flow 5ml column (GE Healthcare, USA) and AKTA FPLC system (GE Healthcare, USA).
Coupling of PSMAb with protamine sulfate
Coupling of PSMAb with protamine sulfate was performed as previously reported [20]. Briefly, 3 mM protamine solution (cat. no. 539122. Merck, Darmstadt, Germany), 50 mM sulfo-SMCC (Sulfosuccinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate) solution (Thermo scientific, Waltham, MA, USA), and 370 μL ddH2O were incubated for 2h at 37°C with shaking at 700 rpm. Subsequently, sulfo-SMCC-protamine complexes were incubated with 1mg PSMAb overnight at 4°C. After using gel filtration chromatography to desalt the PSP sulfate in Zeba spin desalting columns (Thermo Scientific), the protein content was measured by using the bicinchoninic acid assays and the PSP complexes were stored at 4°C.
Encapsulation of siRNA in PSP
FAM-siRNA was synthesized by Shanghai Genepharma Co., Ltd and chemically modified by 2'-O-methylation to reduce off-target effects and enhance its stability. Encapsulating siRNAs in PSP complexes was performed as previously reported [20]. Briefly, 60 nM PSP and 300 nM FAM-siRNA were subjected to shaking at 1000 rpm for 3h at room temperature. Subsequently, gel shift and serum stability assays were performed for visualizing the estimated siRNA load capacity of PSP and serum stability of PSPS. For estimation of siRNA load capacity, PSPS (molar ratio: 1:20) complexes were subjected to 0.6% agarose gel electrophoresis followed by ethidium bromide staining. Following electrophoretic separation, the agarose gel was exposed to UV transillumination followed by imaging. The unbound siRNA duplex was of 20 bp size, and PSPS was immobile because of its large size. For serum stability of PSPS, 90 μL PSPS was incubated with 10 μL FBS or SFM for 1-24 h at 37°C. Subsequently, 6x gel loading dye was added and the samples were applied to 0.6% agarose gel, and fluorescence spectrophotometer was used to measure the FAM-siRNA fluorescence intensity.
Cell culture
Human PSMA-positive (PSMA+) prostate cancer cell lines LNCaP, C4-2, PC-3-PSMA+, and PSMA-negative (PSMA-) prostate cancer cell line DU-145, PC-3 cells were maintained in F-12K, DMEM, RPMI-1640, or EMEM medium (Gibco Life Technologies, Paisley, Scotland, UK) supplemented with 1% penicillin-streptomycin (Invitrogen Life Technologies, USA) and10% fetal bovine serum (Gibco Life Technologies, USA). The cells were cultured at 37°C with 5% CO2 in a humidified incubator.
Flow cytometry (FC) for binding and internalization assays
C4-2, DU-145, PC-3, LNCaP, and PC-3-PSMA+ were harvested, washed, and re-suspended in FACS buffer (BD, CA, USA) at the density of 5×105-1×106 cells/mL. For binding assays, cells were stained with 200μL 50nM PSMAb (positive control) or PSP for 30 min at 4°C in darkness, followed by washing and staining with 100 μL PE-conjugated anti-human IgG Fc antibody (HP6017, Biolegend, CA, USA) for 30 min at 4°C in the dark. For PSP internalization assay, PE-PSMAb and PE-PSP conjugate was produced by incubating PSMAb and PSP with PE-labeled-anti-IgG-Fc 2nd antibody at the ratio of 1:1 for 1h at 4 °C. Then, the bound antibody was removed by adding trypsin to C4-2/PC-3 cells incubated PE-PSMAb or PE-PSP at 4°C (negative control) or 37°C for 1 h. After washing with FACS buffer, cells were analyzed by FC (Beckman Coulter, Brea, CA, USA). For the PSPS internalization assay, C4-2/PC-3 were incubated with 100nM PSPS or IgG-sulfo-SMCC-protamine (IgG-P)-FAM-siRNA (negative control) for 3 h at 37°C. C4-2/PC-3 cells transfected with FAM-siRNA using Lipofectamine 2000 was applied as a positive control. After washing with FACS buffer, cells were analyzed by Beckman Coulter.
Immunofluorescence staining (IF) for binding and internalization assays
PC-3 and C4-2 cells were plated at the density of 5×104 cells per well in 24-well plates with coverslips and cultured overnight. For PSP binding and internalization assays, cells were incubated with 200μl 100nM PSP, PSMAb (Positive control) or IgG (Negative control) for 1h at 4°C (binding assay) or 3 h at 37°C (internalization assay) before washing and fixing with 4% paraformaldehyde for 25 min. Cells were then incubated with 0.5% Triton X-100 and 5% BSA for 1 h at 37°C for blocking followed by incubation with FITC-conjugated goat anti-human IgG for 1 h at 4°C in darkness. For the PSPS internalization assay, cells were incubated with 200 μL 100nM PSPS or IgG-P-FAM-siRNA (negative control) for 1h at 4°C (binding assay) or 3 h at 37°C (internalization assay) before washing and fixing with 4% paraformaldehyde for 20 min. DAPI and phalloidin were used to visualize the nuclei and cytoskeleton, respectively. Finally, cells were washed and observed under FluoView FV1000 (Olympus, Japan), and images were captured.
IF staining for TUNEL, Ki67 and TRIM24 expression
Slides were deparaffinized and rehydrated. The tissue sections were then incubated with Proteinase K working solution for 30 min. After washing twice, 50μL TUNEL (Lewes, UK) reaction mixture was added and incubated for 60 min. For the Ki-67 or TRIM24 expression, slides were deparaffinized and rehydrated before incubating with 3% H2O2. The primary antibody for Ki-67 (1:400, Cell Signaling Technology, Inc., MA, USA) or TRIM24 (1:100, Abcam, Cambridge, UK) was added to the slides which were incubated overnight. Next day 100μL PE- IgG Fc antibody (Biolegend) was added and incubated for 1 h at 4 °C in darkness, and subsequently stained with 4',6-diamidino-2-phenylindole (DAPI). The slides were observed under FluoView FV1000 (Olympus), and images were captured.
Western Blots, CCK-8, colony formation, cell apoptosis, cell cycle, cell migration and invasion assays, qRT-PCR, and cellular ELISA
These experiments were performed following our previously published procedures [10, 21, 22].
Immunohistochemistry (IHC)
IHC for TRIM24 was carried out as previously described [23]. Briefly, slides were deparaffinized and rehydrated before incubating with 3% H2O2. The primary antibody for TRIM24 (1:100, Abcam) was added to the slides which were incubated overnight. Slides were incubated with biotinylated secondary antibody for 30 min. Subsequently, DAB chromogen was used for visualization.
Animal models and in vivo fluorescence imaging (FLI) of PSP or PSPS
All animals were treated in accordance with the Guide for the Care and Use of Laboratory Animals, 1996, by the National Research Council (US) Institute for Laboratory Animal Research [24]. Animal experiments were approved by the Ethics Committee of the Fourth military medical University (Xi'an, China).
5×106 PC-3-PSMA+ or PC-3 cells were subcutaneously injected in the right flank. Imaging study was performed when the tumor volume reached 100 mm3. PSP was labeled with indocyanine green ICG to analyze its enrichment in PSMA+ cells. Extra dye was removed through dialysis. 0.2 μmol/kg of the ICG-labeled PSP, PSMAb, or IgG was intravenously injected. For in vivo enrichment and internalization assays of PSPS, each mouse was intravenously injected with 0.2 μmol/kg of PSPS, IgG-P-FAM-siRNA, or 1 μmol/kg FAM-siRNA. Subsequently, ICG or FAM fluorescence was monitored in a real-time manner using the Xenogen IVIS Kinetic imaging system.
PSP-TRIM24 treatment in vivo
When the tumor volume reached 200 mm3, PSP-TRIM24 siRNA (5mg/kg), PSP-NC (5mg/kg), IgG-P-TRIM24 siRNA (5mg/kg) or PBS was injected into the tail veins of the xenograft mice. Tumor growth was also observed using BLI every week. Tumors and important organs were collected to prepare paraffin sections for IHC, TUNEL, Ki-67 staining, and H.E. staining. Furthermore, RNA and protein were extracted from each tumor. Serum was withdrawn to examine liver toxicity by checking the ALT and AST level, and kidney toxicity by checking the BUN and Cr level.
For treatments of CRPC bone metastasis models, 20 nude mice were anesthetized with ketamine/xylazine and injected with 1×106 firefly luciferase-expressing PC-3-PSMA+ cells in the left tibia. The mice were randomly divided into four groups (PBS, IgG-P-TRIM24 siRNA 5mg/kg, PSP-NC 5mg/kg, and PSP-TRIM24 siRNA 5mg/kg). Two weeks later, PSP-TRIM24 siRNA, PSP-NC, IgG-P-TRIM24 siRNA, or PBS was injected into xenograft mice, and each mouse was treated twice a week, for five times. Luciferase imaging was utilized every week and X-rays were taken every two weeks. When several mice developed lesions that began to break through the cortical bone, became paraplegic, or began to lose weight due to increased tumor burden, mice were anesthetized and sacrificed. Left tibia was excised for further evaluation of the PSP-TRIM24 siRNA therapeutic effects by using a Scanco μCT vivaCT40 (SCANCO Medical AG, Swiss) using 12 μM resolution.
Statistical analysis
IBM SPSS statistical software (version 20.0) was used to perform statistical analysis. Student's t-test was used for data analysis and P values were determined using 2-sided tests. P values < 0.05 were considered to have statistical significance.
Results
Characterization of PSP and PSPS
Atomic force microscopic (ATM) images of PSP and PSPS were shown in Figures 1A and 1B. Size range analysis showed that PSP (Figure 1C) display mono-dispersion with an average size of 16 nm. Differential scanning calorimetry (DSC) assay showed that the Tm (midpoint transition temperature) of PSP was 73.48 °C, indicating good thermostability (Figure 1D). Size exclusion chromatography-high performance liquid chromatography (SEC-HPLC) results indicated molecular weight of PSP to be approximately 165.8 kilodaltons (KD), and PSP mainly existed as monomers (Figure 1E). Gel shift assay showed that PSP encapsulated siRNA duplexes (Figure 1F) and released them in the cytoplasm (Figure 1G).
Cellular ELISA revealed that the binding affinity of PSP or PSPS for C4-2 cells was high with the Kd (equilibrium dissociation constant) values of 0.40 nM and 0.32 nM respectively, which was similar as PSMAb (Figure 2A and 2B). In the Gel shift assay, FAM-siRNA was encapsulated by PSP at a molar ratio of 1:1-1:4. When the molar ratio was beyond 1:8, unbound siRNA could be detected (Figure 2C). Fluorometric biding assay showed that 1 mol PSP molecule bound to approximately 5 mol FAM-siRNA (Figure 2D). Only minor intensity changes were detected between fetal bovine serum (FBS) and serum-free medium (SFM) by the gel shift assay (Figure 2E) and fluorometric assay (Figure 2F). We further investigated the stability of PSPS (PSP/FAM-siRNA: 1/5) in the physiological fluid. There were only minor changes in siRNA concentration or fluoresce intensity when incubated with human serum for 1h-72h (Figure S1A and S1B).
PSP and PSPS bind to and internalize into PSMA+ prostate cancer cells
FCM (Figure 3A) and IF analyses (Figure 3B and Figure S2A) revealed that PSP could specifically bind to PSMA+ prostate cancer cells, such as C4-2, LNCaP, and PC-3-PSMA+ (Figure 3A and Figure 3B) whereas it did not bind to PSMA- prostate cancer cells such as DU-145 and PC-3 (Figure 3A and Figure S2A). Internalization of PSP and the presence of the fluorescent signal in C4-2 (Figure 3C and 3D) but not PC-3 (Figure S2B) cell was obvious by FCM and IF assays as displayed in Figure 3C-3D and Figure S2B, respectively. Furthermore, both FCM (Figure 3E) and IF (Figure 3F and Figure S3) assays demonstrated that PSPS could specifically deliver siRNA to PSMA+ cells but not PSMA- cells.
Figure 1
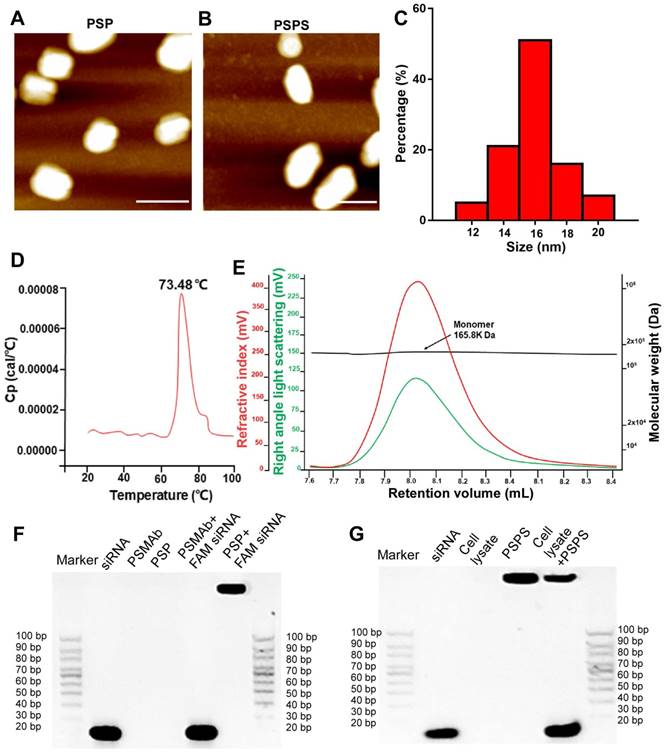
Characterization of PSP and PSPS. ATM micrograph of (A) PSP and (B) PSPS. (C). Size distribution of PSP. (D). Thermostability of PSP. (E). Characterization of PSP by SEC-HPLC showing the molecular weight of PSP is 165.8 KD and it mainly exists as monomers. (F). Characterization of siRNA payload: gel shift assay of free siRNA, PSMAb, PSP, a mixture of PSMAb and FAM-siRNA (molar ratio: PSP/FAM-siRNA:1/1), and a mixture of PSP and FAM-siRNA (molar ratio: PSP/FAM-siRNA:1/1). (G). Characterization of siRNA release: gel shift assay of free siRNA, cell lysate, PSPS, and a mixture of PSPS and cell lysate. Scale bars = 20 nm. Representative results of 3 independent experiments are shown.
Figure 2
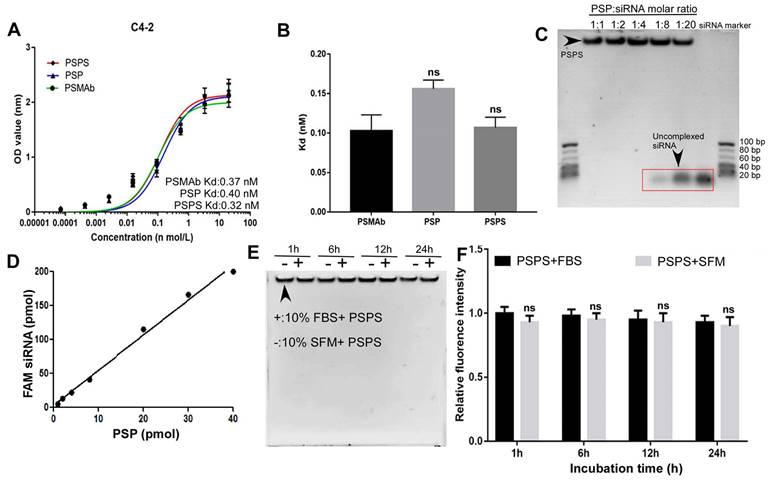
Binding affinity and quality control of PSPS. (A) and (B). Detection and statistical analysis of the binding affinity of PSMAb, PSP, and PSPS by ELISA. (C). Specific siRNA payload estimation of PSP. Different molar ratios of PSP complexes to FAM-siRNA were incubated for 2h and gel shift assays were performed. (D). Specific FAM-siRNA payload of PSP was analyzed by the fluorescent intensity of FAM-siRNA bound to PSP captured on protein A/G agarose beads. (E). Protective effect of PSP for FAM-siRNA (PSP/FAM-siRNA:1/5) against enzymatic digestion. (F). Fluorescence intensity assay ns: not significant vs control group. Data are shown as the mean ± S.E. ns: not significant. Representative results of 3 independent experiments are shown.
PSP and PSPS specifically enrich in PSMA+ CRPC xenografts in vivo
Both ICG-labeled-PSP and PSPS diffused throughout the entire body including the tumor tissues 6 h after the injection as detected by FLI. ICG-labeled-PSP and PSPS were then steadily cleared but were still retained in PSMA+ tumor tissues even 96 h post-injection (Figure 4A&4C). Specific accumulation was not observed in PSMA- tumor tissues (Figure S4A and S4C). By comparison, FAM-siRNA alone rapidly dissipated from the whole body and couldn't be detected after 24 h (Upper panel, Figure 4C) whereas, IgG-P-FAM-siRNA was gradually cleared from the body, but not retained specifically in PC-3-PSMA+ tumor tissues (Middle panel, Figure 4C).
Ex vivo fluorescence imaging confirmed the presence of ICG-labeled-PSP and PSPS in PC-3-PSMA+ tumor tissues (Right panel, Figure 4B and 4D) but not in PC-3 tumor tissues (Figure S4B and S4D). Also, if the PC-3-PSMA+ xenografted nude mice were injected with IgG-P-FAM-siRNA, no significant fluorescent signal could be found in tumor tissues (Middle panel, Figure 4D). Upon injection with ICG-labeled-PSP and PSPS, fluorescent signals could also be detected in metabolic organs, such as liver and kidney, and organs which are rich in blood supply, such as lung and spleen (Right panel, Figure 4B&4D and Figure S4B and S4D). However, no fluorescent signal could be detected in brain, intestine, and heart (Right panel, Figure 4B and 4D and Figure S4B and S4D). Similarly, no fluorescent signal could be detected in major organs or tumor tissues when the nude mice injected with FAM-siRNA alone (Left panel, Figure 4D).
Finally, FAM fluorescence signals in cryosections of tumors could be detected in PC-3-PSMA+-xenografted nude mice injected with PSPS (Right panel, Figure 4E). Neither the PC-3-PSMA+ xenografted nude mice injected with IgG-FAM-siRNA or FAM-siRNA alone nor PC-3 xenografted nude mice injected with PSPS showed FAM fluorescence signals in the tumor rim (Left and middle panel, Figure 4E and Figure S4E). Consistent with our previous results, we could also detect FAM fluorescence signals in the metabolic organs (Figure 4F and Figure S4F).
PSP-TRIM24 siRNA complexes effectively silence TRIM24 expression, and inhibit cell proliferation, colony formation, and invasion in PSMA+ CRPC cells in vitro
TRIM24 expression was significantly downregulated in C4-2 cells incubated with PSP-TRIM24 siRNA (P<0.05, Figure 5A-5C). However, in PC-3 cells, TRIM24 expression in PSP-TRIM24 siRNA-treated group was similar to that of controls (P>0.05, Figure S5D-5F). As determined by the CCK-8 and plate colony formation assays, the proliferation rate (P<0.05, Figure 5D) and colony-forming ability (P<0.05, Figure 5E and 5F) of C4-2 cells incubated with PSP-TRIM24 siRNA was significantly decreased than those of the control group.
Figure 3
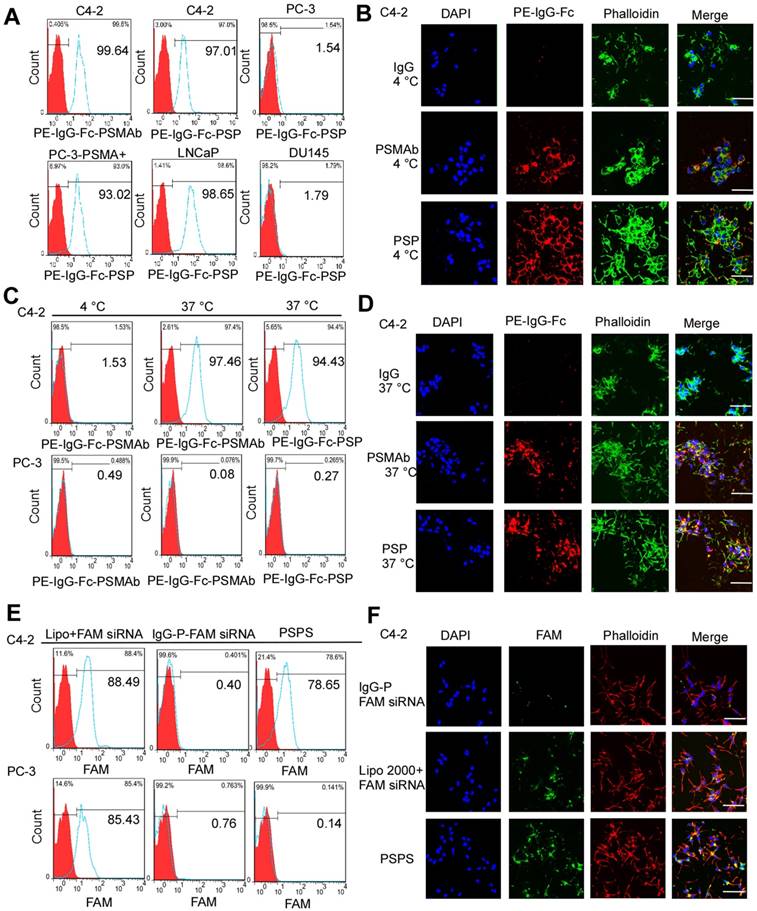
Binding and internalization of PSP and PSPS in PSMA+ or PSMA- CRPC cells. Specific binding of PSP with PSMA+ prostate cancer cell lines analyzed by (A) FCM assays and (B) IF assays (400x). PSMA+ prostate cancer cell lines C4-2 (FCM and IF), PC-3-PSMA+ (FC), LNCaP (FCM) and PSMA- prostate cancer cell lines PC-3 (FCM) and DU145 (FCM)were incubated with 50nM PSP, PSMAb (positive control) or human IgG (negative control) followed by PE-IgG-Fc antibody. Specific internalization of PSP into PSMA+ CRPC cells analyzed by (C) FCM and (D) IF assays (x 400). For the FCM assay, C4-2 or PC-3 cells were incubated with 100nM PE-PSP and PE-PSMAb for 1 h at 4°C or 37°C followed by trypsin digestion. For IF assay, C4-2 cells were incubated with 100nM PSP, PSMAb (positive control), or human IgG (negative control) for 3 h at 37°C followed by PE-conjugated anti-human IgG Fc antibody. Internalization of FAM-siRNA into PSMA+ CRPC cells analyzed by (E) FCM assay and (F) IF assay (x 400). C4-2 (FCM and IF) or PC-3 (FCM) cells were incubated with 100 nM PSPS. DAPI was used to visualize the nuclei. Scale bars = 20 μm. Representative results of 3 independent experiments are shown.
Figure 4
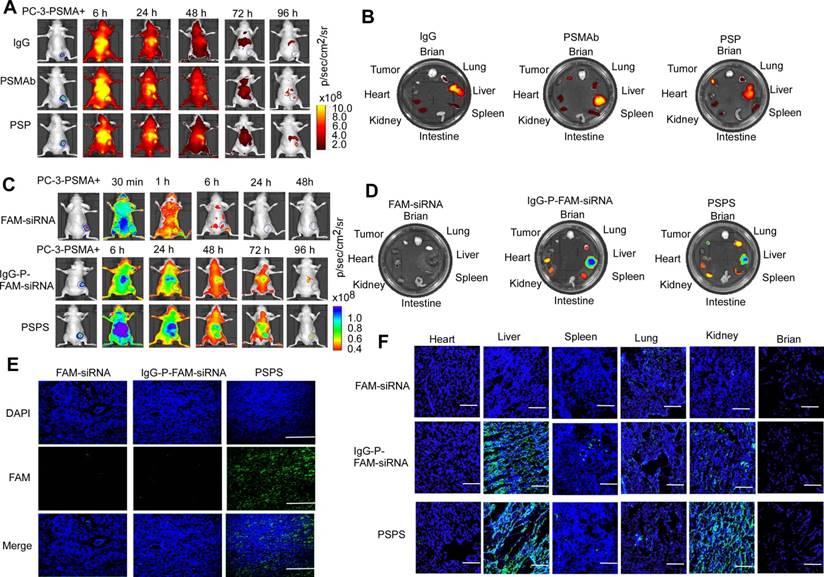
Specific enrichment of PSP and PSPS in PSMA+ CRPC xenografts in vivo. (A) Dynamic whole-body and (B) major organs distribution of ICG-labeled PSP in vivo. ICG-labeled PSP was injected into PC-3-PSMA+ xenografted nude mice. The whole-body distribution of ICG-labeled PSP was monitored at indicated time points, and the resected major organs distribution was monitored at 96 h after ICG-labeled PSP injection by fluorescence imaging excited at 745 nm. BLI was acquired to identify the CRPC tumor tissues. (C) Dynamic whole-body and (D) major organs distribution of PSPS in vivo. PSPS was injected into PC-3-PSMA+ xenograft nude mice. The whole-body distribution of PSPS was monitored at indicated time points, and the resected major organ distribution was monitored at 96 h after PSPS injection by fluorescence imaging excited at 488 nm. BLI was acquired to identify the CRPC tumor tissues. IgG-P-FAM-siRNA and FAM-siRNA were used as negative controls. Tumors and major organs were resected 96h after PSPS injection. Fluorescence images of (E) tumor and (F) major organs cryosections were taken by laser scanning confocal microscopy. IgG-P-FAM-siRNA and FAM-siRNA were used as negative controls. Scale bars = 50 μm. Data are shown as the mean ± S.E. Representative results of 3 independent experiments are shown.
Furthermore, TRIM24 siRNA delivered by PSP induced early apoptosis (P<0.05, Figure 6A and 6B) and cell cycle arrest at G1 stage (P<0.05, Figure 6C and 6D) in C4-2 cells as shown by flow cytometry. Wound healing and Transwell assays indicated a significant decrease in migration distance (P<0.05, Figure 7A and 7B), migrative ability (P<0.05, Figure 7C and 7D), and invasive ability (P<0.05, Figure 7E and 7F) of C4-2 cells incubated with PSP-TRIM24 siRNA compared to the other groups. But, there was no obvious change in proliferation (P>0.05, Figure S6A), colony-forming ability(P>0.05, Figure S6B and S6C), cell apoptosis (P>0.05, Figure S6D and S6E), cell cycle (P>0.05, Figure S5F and S5G), migration distance (P>0.05, Figure S7A and S7B), migrative ability (P>0.05, Figure S7C and S7D), and invasive ability (P>0.05, Figure S7E and S7F) of PC-3 cells incubated with PSP-TRIM24 siRNA.
PSP-TRIM24 siRNA complexes inhibit tumor growth in PSMA+ CRPC xenografts in vivo
As shown by the bioluminescence imaging (BLI) data and growth curves, slower tumor growth, smaller tumor volume, and lighter tumor weight were observed in PC-3-PSMA+ xenografts in nude mice receiving PSP-NC and PSP-TRIM24 siRNA intravenously than in the mice receiving IgG-P-TRIM24 siRNA or PBS. The PSP-TRIM24 siRNA group showed better results in all three categories than the PSP-NC group: better growth inhibition (P<0.05, PSP-NC vs PBS or IgG-P-TRIM24 siRNA; P<0.01, PSP-TRIM24 siRNA vs PBS or IgG-P-TRIM24 siRNA; Figure. 8A and 8B); smaller tumor volume and lighter tumor weight (P<0.05, PSP-NC vs PBS or IgG-P-TRIM24 siRNA; P<0.01, PSP-TRIM24 siRNA vs PBS or IgG-P-TRIM24 siRNA; Figure 8C and 8D). Furthermore, H&E staining revealed morphological abnormalities, such as necrotic foci, in PC-3-PSMA+ xenografted tumors in nude mice receiving PSP-TRIM24 siRNA treatment but not in control groups (Figure 8E). There was no obvious difference in tumor growth speed (P>0.05, Figure S8A and S8B), tumor volume (Figure S8C), tumor weight (P>0.05, Figure S8D), and morphology (Figure S8E) among PC-3 xenografts of nude mice receiving PSP-TRIM24 siRNA and control treatments.
Figure 5
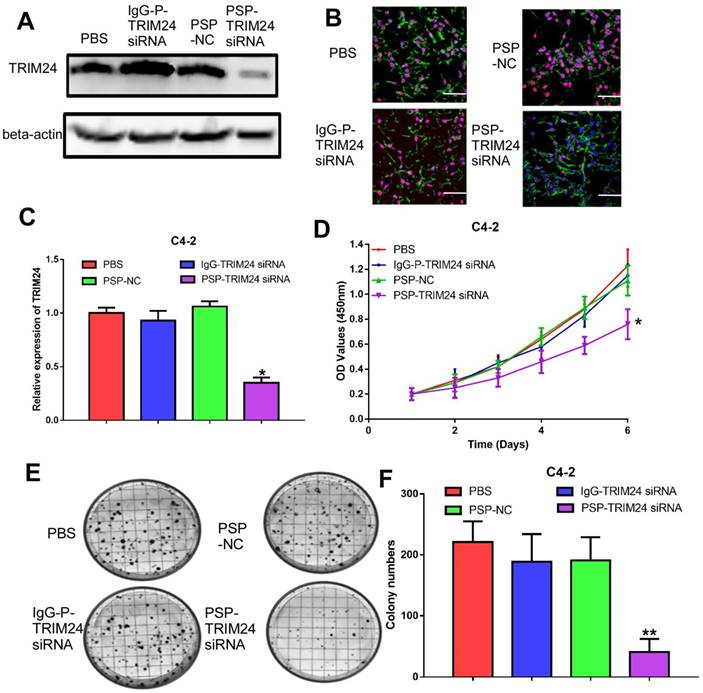
Inhibition of cell proliferation in PSMA+ CRPC cells in vitro by PSP-mediated delivery of TRIM24 siRNA. PSMAb-mediated RNAi reduced TRIM24 target gene expression in the PSMA+ CRPC cells. C4-2 cells were exposed to 100 nM of PSP-TRIM24-siRNA. Analysis of TRIM24 by (A) Western blot analysis, (B) IF (x400; Cells were fixed and stained with TRIM24 antibody (Red), phalloidin (Green) to visualize the cytoskeleton, and with DAPI (Blue) to visualize the nuclei.), and (C) qRT-PCR were performed for TRIM24. Proliferation of C4-2 cells, treated as indicated were analyzed by (D) CCK-8 assay (E) and (F) plate colony formation assay. Scale bars = 20 μm. Data are shown as the mean ± S.E. *P<0.05, **P<0.01. Representative results of 3 independent experiments are shown.
PSP-TRIM24 siRNA complexes effectively silence TRIM24 expression, suppress cell proliferation and induce apoptosis in PSMA+ CRPC xenografts in vivo
TRIM24 expression was significantly downregulated in the PSP-TRIM24 siRNA treatment group of PC-3-PSMA+ xenografts (P<0.05, Figure 9A-9C). However, in the PC-3 xenografts, the expression of TRIM24 in the PSP-TRIM24 siRNA group was similar to the other groups (P>0.05, Figure S9A-S9C). TUNEL assay revealed a more intense fluorescent signal indicative of apoptosis in PC-3-PSMA+ xenografts in nude mice receiving PSP-TRIM24 siRNA treatment than in the PSP-NC group (P<0.05, PSP-NC vs PBS or IgG-P-TRIM24 siRNA; P<0.01, PSP-TRIM24 siRNA vs PBS or IgG-P-TRIM24 siRNA; Figure 9D and 9F). The fluorescent signal was not detected in PC-3-PSMA+ xenografts in nude mice receiving PBS or control IgG-P-TRIM24 siRNA (Figure 9D and 9F). Also, no obvious fluorescent signal was found in PC-3 xenografts of nude mice receiving PSP-TRIM24 siRNA or the control treatments (P>0.05, Figure S9D and S9F). Ki-67 assay also showed that percent of positive Ki-67 cells was significantly down-regulated in PC-3-PSMA+ xenografts of nude mice receiving PSP-NC or PSP-TRIM24 siRNA treatments compared with those receiving PBS or IgG-P-TRIM24 siRNA (P<0.05, PSP-NC vs PBS or IgG-P-TRIM24 siRNA; P<0.01, PSP-TRIM24 siRNA vs PBS or IgG-P-TRIM24 siRNA; Figure 9E and 9G). No obvious change in the percent of positive Ki-67 cells was found in PC-3 xenografts of nude mice receiving PSP-TRIM24 siRNA and control treatments (P>0.05, Figure S9D and S9F).
PSP-TRIM24 siRNA complex inhibits tumor growth in bone metastasis model of PSMA+ CRPC cells in vivo
As is evident by BLI (Figure 10A), small animal X-ray (Figure 10B), and micro 3D micro-computed tomography (Figure 10C&10D and Supplementary video 1-4), the progression of cancer-induced bone loss in PC-3-PSMA+ xenografts of nude mice receiving PSP-NC and PSP-TRIM24 siRNA treatments was inhibited compared with the IgG-P-TRIM24 siRNA or PBS treatment groups. The PSP-TRIM24 siRNA group showed better inhibition of bone loss than the PSP-NC group.
In vivo toxicity evaluation of multiple dosing of PSP-TRIM24 siRNA complex
After receiving an intravenous injection of PSP-TRIM24 siRNA 5 times (5mg/kg), the body weight of PSP-TRIM24 treatment and control groups did not show any descending trend both in PC-3-PSMA+ (P>0.05, Figure 11A) and PC-3 (P>0.05, Figure S10A) xenografted nude mice. The liver and kidney function analyses also did not show significant changes in AST (glutamic oxaloacetic transaminase), ALT (glutamic pyruvic transaminase), Cr (creatinine), and BUN (blood urea nitrogen) (P>0.05, Figure 11B and Figure S10B) between the two groups. More importantly, no morphological abnormalities could be detected by H&E staining in the major organs of PC-3-PSMA+ (Figure 11C) or PC-3 (Figure S10C) xenografted nude mice receiving 5 intravenous injections (5mg/kg) of PSP-TRIM24 siRNA.
Figure 6
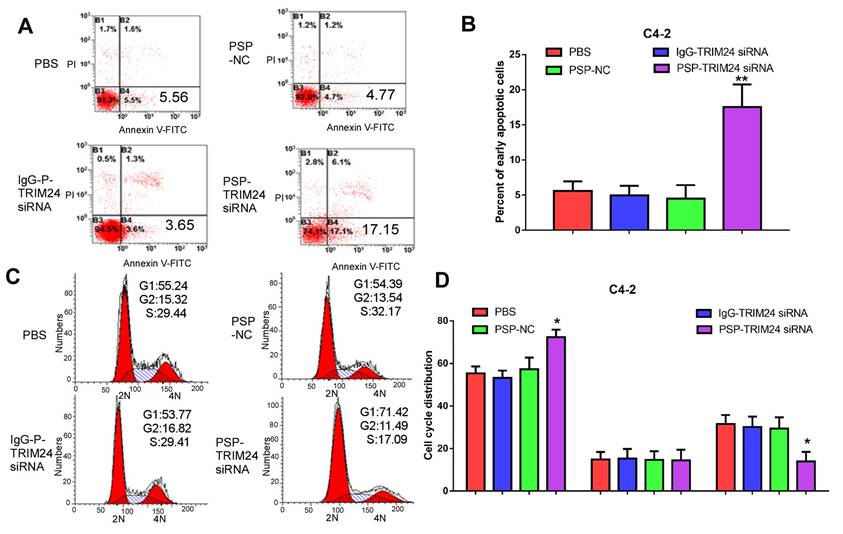
Cell apoptosis in PSMA+ CRPC cells in vitro by PSP-mediated delivery of TRIM24 siRNA. Flow cytometry assays of (A) apoptosis and (C) cell cycle of C4-2 cells treated as indicated. Statistical analysis of (B) results shown in A and (D) results shown in C from 3 independent experiments. Data are shown as the mean ± S.E. *P<0.05, **P<0.01. The representative results of 3 independent experiments are shown.
Figure 7
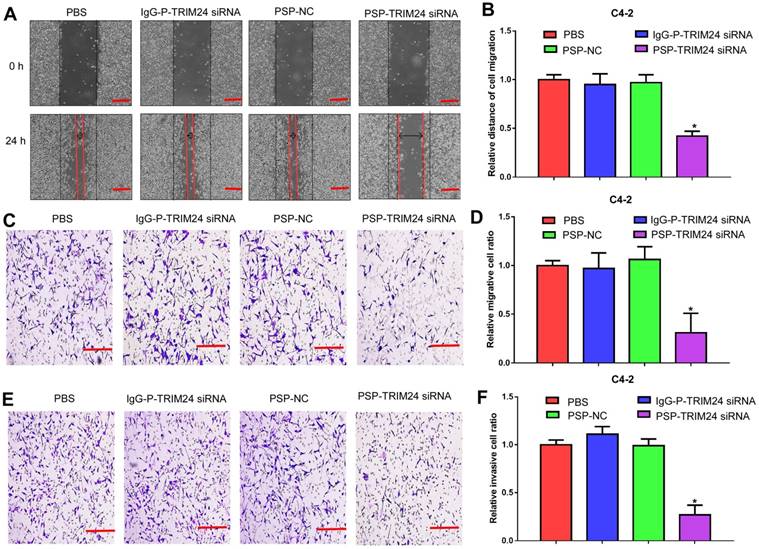
Inhibition of cell migration and invasion in PSMA+ CRPC cells in vitro by PSP-mediated delivery of TRIM24 siRNA. (A). Migrative distance of C4-2 cells treated as indicated was analyzed by wound healing assays. Numbers of (C) migrative and (E) invasive C4-2 cells treated as indicated were analyzed by the Transwell assay. (B), (D), and (F). Statistical analyses of A, C, and E, respectively. Scale bars = 100 μm. Data are shown as the mean ± S.E. *P<0.05. Representative results of 3 independent experiments are shown.
Discussion
In the present study, we provided evidence that PSPS could protect siRNA from enzymatic digestion and specifically bind to and internalized by PSMA+ prostate cancer cells in vitro and in vivo. Furthermore, silencing the expression of TRIM24 by PSP-TRIM24 siRNA complex suppressed cell proliferation, colony formation, migration, and invasion, and induced apoptosis and cell cycle arrest in PSMA+ CRPC cells in vitro. We also investigated the therapeutic effects of PSP-TRIM24 siRNA complex in PSMA+ and PSMA- CRPC cell-xenografted nude mice and found that PSP-TRIM24 siRNA complex suppressed tumor growth of PSMA+ CRPC xenografts by inducing apoptosis and inhibiting cell proliferation. More significantly, PSP-TRIM24 siRNA complex inhibited bone loss in the bone metastasis model of PSMA+ CRPC.
To efficiently and safely deliver siRNA to prostate cancer cells, we had previously developed fusion proteins consisting of anti-PSMA scFv, truncated protamine (tP), and HA2 fragment (FH) sequence. Our results demonstrated that the fusion proteins enabled targeted delivery of siRNA which was internalized into PSMA+ prostate cancer cells [25]. Although, the anti-PSMA scFv-based siRNA delivery platform exhibited several advantages compared to nanoparticles, lipid spheres, or even an exosomal siRNA delivery system, it still had some unavoidable drawbacks, such as easy availability, potential immunogenicity, rapid renal clearance, short circulation half-life, and failure in activating complement system-mediated cellular immunity [26, 27]. To overcome these obstacles in the clinical application of siRNA delivery systems, we adopted a fast and reliable method first described by Bäumer et.al [20, 28] and established stable and efficient transfer carriers for siRNA by PSMA dependent, human monoclonal antibody-mediated delivery platform. Furthermore, to effectively silence TRIM24 expression and reduce off-target effects, siRNAs chemically modified by 2'-O-methylation were loaded in our PSPS delivery platform. Consistent with the previous study, we demonstrated that, after conjugating with sulfo-SMCC-protamine and encapsulating siRNA, PSP, and PSPS had similar binding affinities as PSMAb alone indicating that protamine and siRNAs did not decrease the affinity of PSMAb for PSMA+ prostate cancer cells. Also, PSP complexes could efficiently bind with siRNA and increase the serum stability of siRNA. Furthermore, PSP and PSPS could also specifically bind and internalize into PSMA+ prostate cancer cells in vitro and specifically enrich in PSMA+ CRPC xenografts in vivo. Thus, we proved the effectiveness of targeted siRNA delivery platform based on PSP complexes.
We examined TRIM24 and PSMA expression in the tissues of 492 patients of prostate adenocarcinoma (PRAD) (red box) and 152 healthy controls (Black box) by using GEPIA website (http://gepia.cancer-pku.cn/) to analyze TCGA data. We found both TRIM24 (P>0.05, Figure S5A) and PSMA (P<0.05, Figure S5B) expression to be upregulated in PRAD compared to healthy controls. More importantly, we detected a positive association between TRIM24 expression and PSMA expression in PRAD (r=0.34, P<0.05, Figure S5C). The simultaneous high expression of both antibody-targeted antigen and siRNA-targeted tumor-associated transcript is the most important criterion for antibody-mediated siRNA delivery in cancer [18]. PSMA is known to be upregulated in CRPC cells which were also widely reported to overexpress TRIM24 [16]. In our study, we also found TRIM24 expression to be positively associated with PSMA expression in PRAD suggesting TRIM24 to be an ideal target of PSMAb-based siRNA delivery platform.
Figure 8
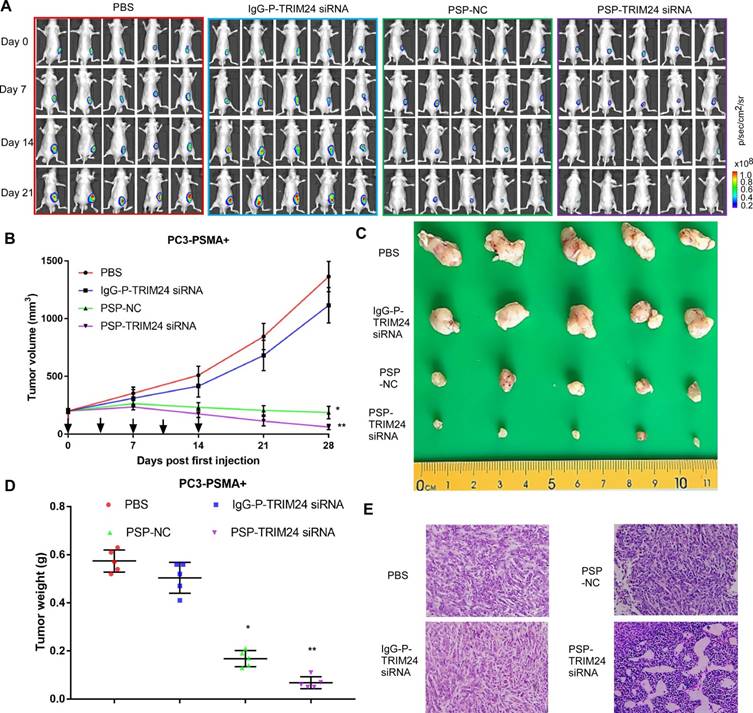
Inhibition of tumor growth in PC-3-PSMA+-xenografted mice by PSP-mediated delivery of TRIM24 siRNA. (A). Tumor growth of different PC-3-PSMA+ groups treated as indicated was observed using BLI every week after the first treatment. (B). Tumor volume analysis showing growth of PC-3-PSMA+ tumors after receiving indicated treatments. The arrows indicate the five treatments. (C). Digital pictures of resected tumors after receiving indicated treatments (D). Weight analysis of PC-3-PSMA+ tumors after receiving indicated treatments (E). Paraffin-embedded sections of resected PC-3-PSMA+ tumors from each group were stained with H&E. Data are shown as the mean ± S.E. (n=5). *P < 0.05, **P<0.01. Representative results of 3 independent experiments are shown.
Figure 9
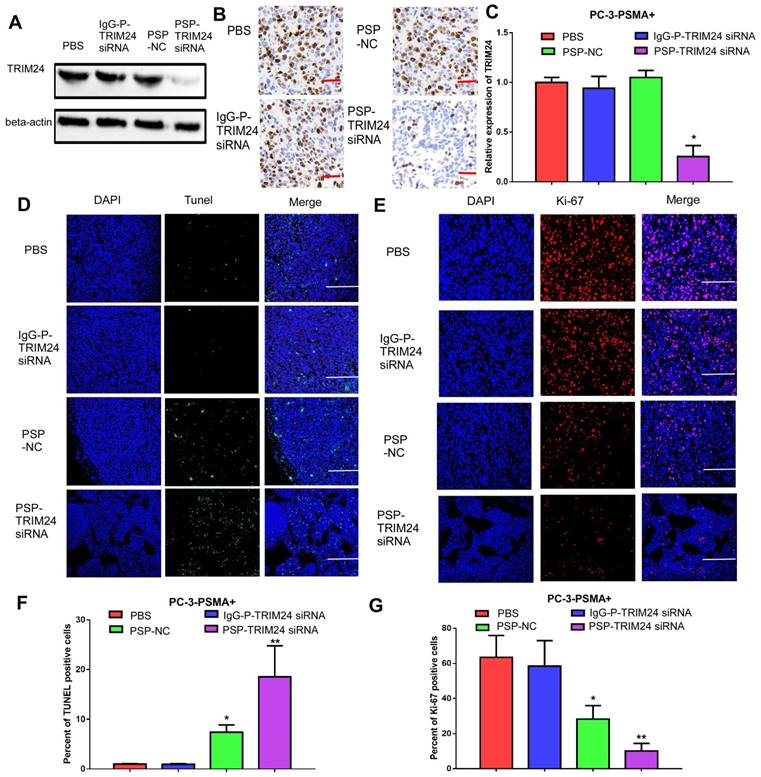
Inhibition of TRIM24 expression and promotion of apoptosis in PC-3-PSMA+ xenografts by PSP-mediated delivery of TRIM24 siRNA. PSMAb-mediated RNAi reduced TRIM24 target gene expression in the PSMA+ PC-3 cells in vivo. Nude mice bearing PC-3-PSMA+ xenografts received five treatments as indicated, and paraffin-embedded sections of resected tumors from each group were analyzed as follows: (A) Western blot analysis, (B) IHC, and (C) qRT-PCR (D) TUNEL (x100, Scale bars = 50 μm) and (E) Ki67 (x200, Scale bars = 20 μm). (F). Statistical analysis of D. (G). Statistical analysis of E. Data are shown as the mean ± S.E. *P<0.05, **P<0.01. Representative results of 3 independent experiments are shown.
It has been demonstrated that genetic alterations in TP53, AR, and PTEN, which is the suppressor of PI3K/AKT signaling, are associated with development and progression of prostate cancer [29]. Mechanistic studies have shown that TRIM24 could activate PIK3CA transcription and enhance PI3K/AKT signaling pathway in low-grade gliomas, glioblastomas, and in prostate cancer [16, 30, 31]. Furthermore, TRIM24 could function as an ubiquitin ligase and negatively regulate p53 expression through proteasome-mediated degradation [32]. It was also reported TRIM24 could promote the proliferation of CRPC cells under low androgen condition by augmenting AR signaling [16]. Consistent with the previous study, silencing the expression of TRIM24 in PSMA+ CRPC by PSP-TRIM24 siRNA complex inhibited the proliferation, migration, and invasion and simultaneously promoted cell apoptosis both in vivo and in vitro. Thus, our results demonstrated TRIM24, the key regulator of PI3K/AKT, AR, and p53, promoted the proliferation, colony-formation, and invasion of CRPC.
Figure 10
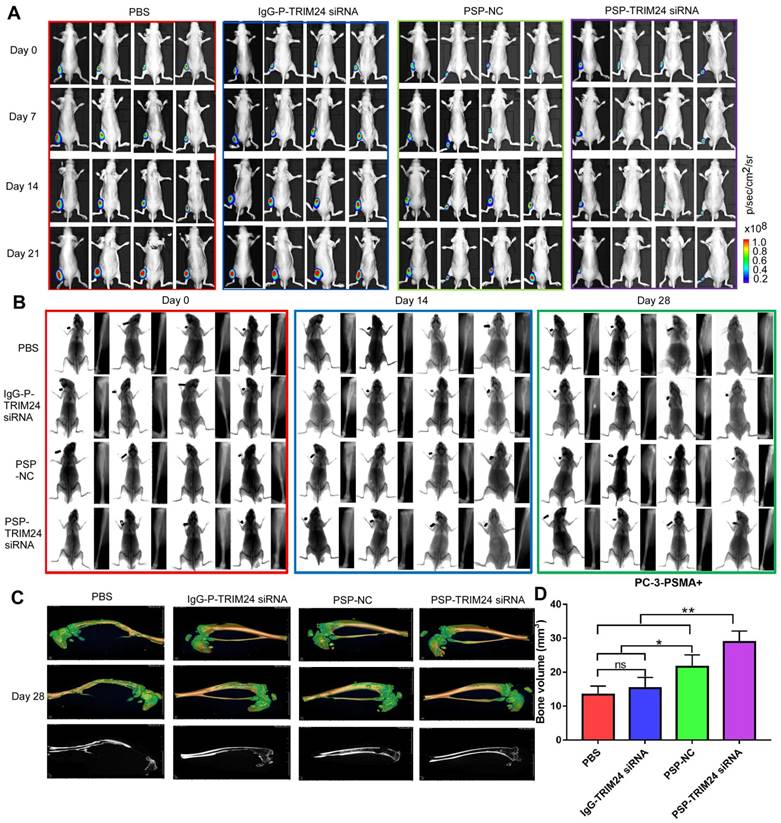
TRIM24 siRNA effectively delivered by PSP Inhibition of bone loss in PC-3-PSMA+ bone metastasis model by PSP-mediated delivery of TRIM24 siRNA. (A). Tumor growth of various PC-3-PSMA+ groups treated as indicated was observed using BLI every week after the first treatment. (B). Bone loss of various PC-3-PSMA+ groups treated as indicated was observed using X-ray every two weeks after the first treatment. (C) Bone loss and (D) Bone volume analysis of various PC-3-PSMA+ groups treated as indicated was observed using micro-CT after the mice were anesthetized and sacrificed. Data are shown as the mean ± S.E. ns: not significant, *P<0.05, **P<0.01. Representative results of 3 independent experiments are shown.
While evaluating the therapeutic effects of PSP-TRIM24 siRNA complex in vitro and in vivo, we observed an interesting phenomenon. The PSP-NC complex, which had no significant effects on cell proliferation, colony formation, and invasion in vitro, exerted obvious anti-tumor effects in vivo which could be explained by the fact that PSP contained PSMAb with the conventional IgG1 constant region and could produce ADCC and CDC effects in vivo [33, 34]. Our previous study had also found that in the presence of PSMAb, both human and mouse NK cells could induce cytotoxicity in C4-2 cells which was stronger in the groups with higher E/T ratios (Wu et al. unpublished data). However, by performing HE staining we observed morphological abnormalities and necrotic foci in tumors of PC-3-PSMA+-xenografted nude mice receiving PSP-TRIM24 siRNA treatment but not in control groups. This could be due to the synergistic effects of silencing TRIM24 expression in CRPC cells and CDC and ADCC effects mediated by PSMAb. Therefore, PSP-TRIM24 siRNA complex could not only mediate ADCC or CDC effects in vivo but also suppress cell proliferation, colony formation, and invasion both in vitro and in vivo.
Figure 11
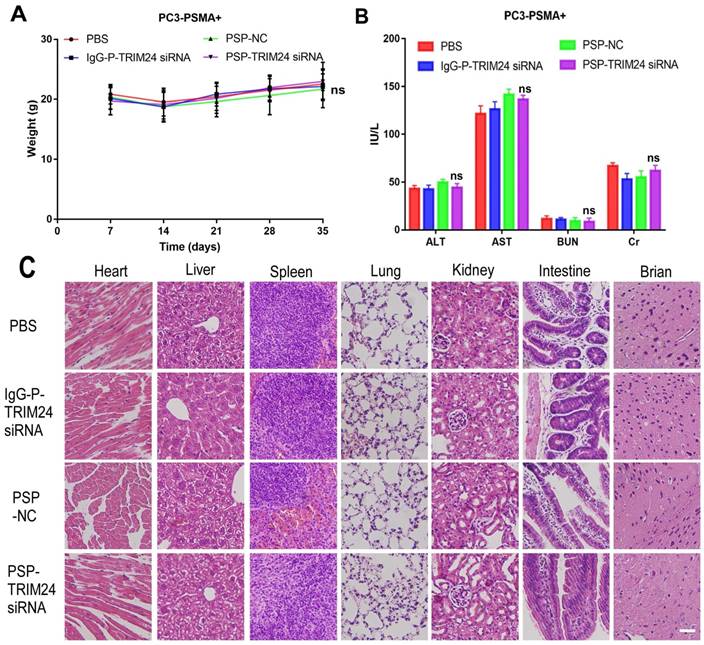
In vivo toxicity of TRIM24 siRNA delivered by PSP in the PC-3-PSMA+ group. (A). Body weight analysis of each PC-3-PSMA+ group after receiving treatments as indicated. (B). Liver and kidney functions of each PC-3-PSMA+ group receiving five times the indicated treatments. (C). Paraffin-embedded sections of resected major organs from each PC-3-PSMA+ group receiving five times the indicated treatments were stained with H&E and documented by microscopy (x200). Scale bars = 20 μm. Data are shown as the mean ± S.E for each group of mice (n=5). ns: not significant. Representative results of 3 independent experiments are shown.
Once prostate cancer progresses to metastatic CRPC (mCRPC), the treatment options are limited to chemotherapy or alternative hormonal therapy with enzalutamide. However, all current treatments for mCRPC have little effect on micrometastatic foci [35]. Only the PSMA targeted therapy could effectively recognize and eradicate the PSMA+ CRPC cells in micrometastatic foci [9]. Compared to previous studies of the targeted delivery of siRNA in CRPC, our PSMAb-mediated TRIM24 siRNA delivery platform for CRPC therapy had several advantages. First, PSP-TRIM24 siRNA effectively inhibited the bone loss in the bone metastasis model of PSMA+ CRPC. Second, besides silencing TRIM24 expression in CRPC, PSMAb containing the constant region of human IgG1 induced additional therapeutic effects of ADCC and CDC. The vast majority of targeted therapy for CRPC, on the other hand, used receptor ligands, peptides, or scFv that could only identify and bind with antigens and lacked additional therapeutic effects [36, 37]. Third, PSMAb-based TRIM24 siRNA delivery had high affinity with PSMA, which is approximately ten times higher than that of the extensively studied J591 PSMA antibody (3 nM) [38], and was helpful in avoiding the off-target effects during therapy. Fourth, PSP-TRIM24 siRNA with a relatively high molecular weight and long half-life compared to low molecular weight agents that are currently in preclinical studies or clinical trials [9, 39-41] could provide effective therapy during a relatively long time. And finally, previously reported PSMA antibodies were either mouse or humanized monoclonal antibodies, which were obtained after immunization of mice with either PSMA+ prostate cancer cells, cell lysates, or purified PSMA protein [42-44]. Thus, the murine origin of these antibodies may cause serious immunogenic side effects when applied in humans severely restraining their application in the clinic [45, 46]. However, PSMAb used in our study was a fully human antibody with a higher bio-safety profile and lower immunogenicity than the currently used humanized PSMA antibodies.
Conclusion
We have confirmed that PSP complexes could efficiently protect siRNA from enzymatic digestion and enable targeted delivery of siRNA to PSMA+ prostate cancer cells in vitro as well as in vivo. Silencing TRIM24 expression by the PSP-TRIM24 siRNA complex could remarkably inhibit cell proliferation, colony formation, and invasion of PSMA+ CRPC cells in vitro and suppressed tumor growth and bone loss in PSMA+ CRPC xenografts and bone metastasis model without obvious toxicity in vivo. Thus, PSMAb-mediated TRIM24 siRNA delivery platform provides a valuable alternative for CRPC patients when the conventional treatments are of little curative value.
Abbreviations
AR: Androgen receptor; CRPC: Castrate-resistant prostate cancer; Metastatic CRPC: mCRPC; GBM: Glioblastoma; PRAD: Prostate adenocarcinoma; Specific single-chain antibody fragment: scFv; PSMA: Prostate-specific membrane antigen; PSMA+: PSMA-positive; PSMA-: PSMA-negative; PSMAb: Human monoclonal PSMA antibody; PSP: PSMAb-sulfo-SMCC-protamine; PSPS: PSP-FAM-siRNA complexes; TRIM24: Tripartite Motif-containing protein 24; Kd: Equilibrium dissociation constant; KD: kilodalton; Tm: Midpoint transition temperature; RNA i: RNA interference; IgG-P: IgG-sulfo-SMCC-protamine; Sulfo-SMCC: Sulfo succinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate; Truncated protamine: tP; HA2 fragment: FH; PSP-NC: PSMAb-sulfo-SMCC-protamine-Normal control scrambled siRNA; FCM: Flow cytometry; ATM: Atomic force microscope; DSC: Differential Scanning Calorimetry; SEC-HPLC: Size exclusion chromatography-High Performance Liquid Chromatography; ICG: Indocyanine green; IF: Immunofluorescence; FLI: Fluorescence imaging; BLI: Bioluminescence imaging; ADCC: Antibody-dependent cell-mediated cytotoxicity; CDC: Complement-dependent cytotoxicity; FBS: Fetal bovine serum; SFM: Serum-free medium; TMB: 3, 3ˊ, 5, 5ˊ-tetramethylbenzidine; ALT: Glutamic pyruvic transaminase; AST: Glutamic oxaloacetic transaminase; BUN: Blood urea nitrogen; Cr: Creatinine; ns: not significant.
Supplementary Material
Supplementary figure legends, figures and video legends.
Supplementary video 1.
Supplementary video 2.
Supplementary video 3.
Supplementary video 4.
Acknowledgements
We would like to express our gratitude to Dr. Changhong Shi (Laboratory Animal Center, Airforce Military Medical University, China) for the xenograft model experiment. We thank Mrs. Yunxin Cao and Mr. Jintao Hu (Department of Immunology, Airforce Military Medical University, China) for flow cytometry analysis.
Financial support
This work was partly supported by grants from the National Key Basic Research Development Program (973 Program) (No. 2013CB530500) and the National Natural Science Foundation of China (No. 81372225, 81372771, 81772734, 81802935).
Author Contributions
Conceptualization: Wen WH, Qin WJ, and Zhao AZ; Methodology, Wu JH, Han DH, and Zhao AZ; Investigation: Shi SJ, Wang LJ, Zhang KL, Chen JW, Jiao D, Li Y, Zhang JL, and Yang F; Writing-Original Draft: Shi SJ and Wang LJ; Writing: Review and Editing, Wen WH, Zhao AZ and Yang AG; Funding Acquisition: Wen WH, Qin WJ and Yang AG; Supervision: Yang AG and Zheng GX.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7-30
2. Sonnenburg DW, Morgans AK. Emerging Therapies in Metastatic Prostate Cancer. Curr Oncol Rep. 2018;20:46
3. Waltering KK, Helenius MA, Sahu B, Manni V, Linja MJ, Janne OA. et al. Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Res. 2009;69:8141-9
4. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP. et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239-43
5. Tilki D, Schaeffer EM, Evans CP. Understanding Mechanisms of Resistance in Metastatic Castration-resistant Prostate Cancer: The Role of the Androgen Receptor. Eur Urol Focus. 2016;2:499-505
6. Heidenreich A, Bastian PJ, Bellmunt J, Bolla M, Joniau S, van der Kwast T. et al. EAU guidelines on prostate cancer. Part II: Treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur Urol. 2014;65:467-79
7. Ross JS, Sheehan CE, Fisher HA, Kaufman RP Jr, Kaur P, Gray K. et al. Correlation of primary tumor prostate-specific membrane antigen expression with disease recurrence in prostate cancer. Clin Cancer Res. 2003;9:6357-62
8. Sweat SD, Pacelli A, Murphy GP, Bostwick DG. Prostate-specific membrane antigen expression is greatest in prostate adenocarcinoma and lymph node metastases. Urology. 1998;52:637-40
9. Wustemann T, Haberkorn U, Babich J, Mier W. Targeting prostate cancer: Prostate-specific membrane antigen based diagnosis and therapy. Med Res Rev. 2018
10. Han D, Wu J, Han Y, Wei M, Han S, Lin R. et al. A novel anti-PSMA human scFv has the potential to be used as a diagnostic tool in prostate cancer. Oncotarget. 2016;7:59471-81
11. Lv D, Li Y, Zhang W, Alvarez AA, Song L, Tang J. et al. TRIM24 is an oncogenic transcriptional co-activator of STAT3 in glioblastoma. Nat Commun. 2017;8:1454
12. Miao ZF, Wang ZN, Zhao TT, Xu YY, Wu JH, Liu XY. et al. TRIM24 is upregulated in human gastric cancer and promotes gastric cancer cell growth and chemoresistance. Virchows Arch. 2015;466:525-32
13. Fang Z, Zhang L, Liao Q, Wang Y, Yu F, Feng M. et al. Regulation of TRIM24 by miR-511 modulates cell proliferation in gastric cancer. J Exp Clin Cancer Res. 2017;36:17
14. Kikuchi M, Okumura F, Tsukiyama T, Watanabe M, Miyajima N, Tanaka J. et al. TRIM24 mediates ligand-dependent activation of androgen receptor and is repressed by a bromodomain-containing protein, BRD7, in prostate cancer cells. Biochim Biophys Acta. 2009;1793:1828-36
15. Tsai WW, Wang Z, Yiu TT, Akdemir KC, Xia W, Winter S. et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature. 2010;468:927-32
16. Groner AC, Cato L, de Tribolet-Hardy J, Bernasocchi T, Janouskova H, Melchers D. et al. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell. 2016;29:846-58
17. Jiang K, Li J, Yin J, Ma Q, Yan B, Zhang X. et al. Targeted delivery of CXCR4-siRNA by scFv for HER2(+) breast cancer therapy. Biomaterials. 2015;59:77-87
18. Tushir-Singh J. Antibody-siRNA conjugates: drugging the undruggable for anti-leukemic therapy. Expert Opin Biol Ther. 2017;17:325-38
19. Juliano RL. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016;44:6518-48
20. Baumer N, Appel N, Terheyden L, Buchholz F, Rossig C, Muller-Tidow C. et al. Antibody-coupled siRNA as an efficient method for in vivo mRNA knockdown. Nat Protoc. 2016;11:22-36
21. Shi SJ, Wang LJ, Yu B, Li YH, Jin Y, Bai XZ. LncRNA-ATB promotes trastuzumab resistance and invasion-metastasis cascade in breast cancer. Oncotarget. 2015;6:11652-63
22. Zheng J, Qin W, Jiao D, Ren J, Wei M, Shi S. et al. Knockdown of COUP-TFII inhibits cell proliferation and induces apoptosis through upregulating BRCA1 in renal cell carcinoma cells. Int J Cancer. 2016;139:1574-85
23. Shi SJ, Wang LJ, Wang GD, Guo ZY, Wei M, Meng YL. et al. B7-H1 expression is associated with poor prognosis in colorectal carcinoma and regulates the proliferation and invasion of HCT116 colorectal cancer cells. PLoS One. 2013;8:e76012
24. National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals. Washington (DC): National Academies Press (US). 1996
25. Su Y, Yu L, Liu N, Guo Z, Wang G, Zheng J. et al. PSMA specific single chain antibody-mediated targeted knockdown of Notch1 inhibits human prostate cancer cell proliferation and tumor growth. Cancer Lett. 2013;338:282-91
26. Padayachee ER, Biteghe FAN, Malindi Z, Bauerschlag D, Barth S. Human Antibody Fusion Proteins/Antibody Drug Conjugates in Breast and Ovarian Cancer. Transfus Med Hemother. 2017;44:303-10
27. Farajnia S, Ahmadzadeh V, Tanomand A, Veisi K, Khosroshahi SA, Rahbarnia L. Development trends for generation of single-chain antibody fragments. Immunopharmacol Immunotoxicol. 2014;36:297-308
28. Baumer S, Baumer N, Appel N, Terheyden L, Fremerey J, Schelhaas S. et al. Antibody-mediated delivery of anti-KRAS-siRNA in vivo overcomes therapy resistance in colon cancer. Clin Cancer Res. 2015;21:1383-94
29. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM. et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215-28
30. Zhang LH, Yin AA, Cheng JX, Huang HY, Li XM, Zhang YQ. et al. TRIM24 promotes glioma progression and enhances chemoresistance through activation of the PI3K/Akt signaling pathway. Oncogene. 2015;34:600-10
31. Lv D, Jia F, Hou Y, Sang Y, Alvarez AA, Zhang W. et al. Histone Acetyltransferase KAT6A Upregulates PI3K/AKT Signaling through TRIM24 Binding. Cancer Res. 2017;77:6190-201
32. Allton K, Jain AK, Herz HM, Tsai WW, Jung SY, Qin J. et al. Trim24 targets endogenous p53 for degradation. Proc Natl Acad Sci U S A. 2009;106:11612-6
33. Ponath P, Menezes D, Pan C, Chen B, Oyasu M, Strachan D. et al. A Novel, Fully Human Anti-fucosyl-GM1 Antibody Demonstrates Potent In Vitro and In Vivo Antitumor Activity in Preclinical Models of Small Cell Lung Cancer. Clin Cancer Res. 2018
34. Kashyap MK, Amaya-Chanaga CI, Kumar D, Simmons B, Huser N, Gu Y. et al. Targeting the CXCR4 pathway using a novel anti-CXCR4 IgG1 antibody (PF-06747143) in chronic lymphocytic leukemia. J Hematol Oncol. 2017;10:112
35. Nuhn P, De Bono JS, Fizazi K, Freedland SJ, Grilli M, Kantoff PW. et al. Update on Systemic Prostate Cancer Therapies: Management of Metastatic Castration-resistant Prostate Cancer in the Era of Precision Oncology. Eur Urol. 2018
36. Michalska M, Schultze-Seemann S, Kuckuck I, Wolf P. In Vitro Evaluation of Humanized/De-immunized Anti-PSMA Immunotoxins for the Treatment of Prostate Cancer. Anticancer Res. 2018;38:61-9
37. Tai W, Li J, Corey E, Gao X. A ribonucleoprotein octamer for targeted siRNA delivery. Nat Biomed Eng. 2018;2:326-37
38. McDevitt MR, Barendswaard E, Ma D, Lai L, Curcio MJ, Sgouros G. et al. An alpha-particle emitting antibody ([213Bi]J591) for radioimmunotherapy of prostate cancer. Cancer Res. 2000;60:6095-100
39. Zhou J, Neale JH, Pomper MG, Kozikowski AP. NAAG peptidase inhibitors and their potential for diagnosis and therapy. Nat Rev Drug Discov. 2005;4:1015-26
40. Afshar-Oromieh A, Babich JW, Kratochwil C, Giesel FL, Eisenhut M, Kopka K. et al. The Rise of PSMA Ligands for Diagnosis and Therapy of Prostate Cancer. J Nucl Med. 2016;57:79S-89S
41. Vallabhajosula S, Nikolopoulou A, Babich JW, Osborne JR, Tagawa ST, Lipai I. et al. 99mTc-labeled small-molecule inhibitors of prostate-specific membrane antigen: pharmacokinetics and biodistribution studies in healthy subjects and patients with metastatic prostate cancer. J Nucl Med. 2014;55:1791-8
42. Nakajima T, Mitsunaga M, Bander NH, Heston WD, Choyke PL, Kobayashi H. Targeted, activatable, in vivo fluorescence imaging of prostate-specific membrane antigen (PSMA) positive tumors using the quenched humanized J591 antibody-indocyanine green (ICG) conjugate. Bioconjug Chem. 2011;22:1700-5
43. Ruggiero A, Holland JP, Hudolin T, Shenker L, Koulova A, Bander NH. et al. Targeting the internal epitope of prostate-specific membrane antigen with 89Zr-7E11 immuno-PET. J Nucl Med. 2011;52:1608-15
44. Chang SS, Reuter VE, Heston WD, Bander NH, Grauer LS, Gaudin PB. Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm PSMA expression in tumor-associated neovasculature. Cancer Res. 1999;59:3192-8
45. Bander NH, Milowsky MI, Nanus DM, Kostakoglu L, Vallabhajosula S, Goldsmith SJ. Phase I trial of 177lutetium-labeled J591, a monoclonal antibody to prostate-specific membrane antigen, in patients with androgen-independent prostate cancer. J Clin Oncol. 2005;23:4591-601
46. Tagawa ST, Milowsky MI, Morris M, Vallabhajosula S, Christos P, Akhtar NH. et al. Phase II study of Lutetium-177-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 for metastatic castration-resistant prostate cancer. Clin Cancer Res. 2013;19:5182-91
Author contact
![]() Corresponding authors: Weihong Wen, Department of Immunology, Fourth Military Medical University, 169 Changle West Road, Xi'an, China 710032. Tel.: +86 29 84774532; Fax: +86 29 83253816; Email: wenweihedu.cn; Weijun Qin, Department of Urology, Xijing Hospital, Fourth Military Medical University, 127 Changle West Road, Xi'an, China 710032. Tel.: +86 29 84775321; Email: qinweijedu.cn; Aizhi Zhao, OriMAbs Ltd. Science center, Room 544. 3624 Market Street, PA 19104, USA. Tel: 215-966-6286; Email: zhaoaizhicom.
Corresponding authors: Weihong Wen, Department of Immunology, Fourth Military Medical University, 169 Changle West Road, Xi'an, China 710032. Tel.: +86 29 84774532; Fax: +86 29 83253816; Email: wenweihedu.cn; Weijun Qin, Department of Urology, Xijing Hospital, Fourth Military Medical University, 127 Changle West Road, Xi'an, China 710032. Tel.: +86 29 84775321; Email: qinweijedu.cn; Aizhi Zhao, OriMAbs Ltd. Science center, Room 544. 3624 Market Street, PA 19104, USA. Tel: 215-966-6286; Email: zhaoaizhicom.
Citation styles
APA
Shi, S.J., Wang, L.J., Han, D.H., Wu, J.H., Jiao, D., Zhang, K.L., Chen, J.W., Li, Y., Yang, F., Zhang, J.L., Zheng, G.X., Yang, A.G., Zhao, A.Z., Qin, W.J., Wen, W.H. (2019). Therapeutic effects of human monoclonal PSMA antibody-mediated TRIM24 siRNA delivery in PSMA-positive castration-resistant prostate cancer. Theranostics, 9(5), 1247-1263. https://doi.org/10.7150/thno.29884.
ACS
Shi, S.J.; Wang, L.J.; Han, D.H.; Wu, J.H.; Jiao, D.; Zhang, K.L.; Chen, J.W.; Li, Y.; Yang, F.; Zhang, J.L.; Zheng, G.X.; Yang, A.G.; Zhao, A.Z.; Qin, W.J.; Wen, W.H. Therapeutic effects of human monoclonal PSMA antibody-mediated TRIM24 siRNA delivery in PSMA-positive castration-resistant prostate cancer. Theranostics 2019, 9 (5), 1247-1263. DOI: 10.7150/thno.29884.
NLM
Shi SJ, Wang LJ, Han DH, Wu JH, Jiao D, Zhang KL, Chen JW, Li Y, Yang F, Zhang JL, Zheng GX, Yang AG, Zhao AZ, Qin WJ, Wen WH. Therapeutic effects of human monoclonal PSMA antibody-mediated TRIM24 siRNA delivery in PSMA-positive castration-resistant prostate cancer. Theranostics 2019; 9(5):1247-1263. doi:10.7150/thno.29884. https://www.thno.org/v09p1247.htm
CSE
Shi SJ, Wang LJ, Han DH, Wu JH, Jiao D, Zhang KL, Chen JW, Li Y, Yang F, Zhang JL, Zheng GX, Yang AG, Zhao AZ, Qin WJ, Wen WH. 2019. Therapeutic effects of human monoclonal PSMA antibody-mediated TRIM24 siRNA delivery in PSMA-positive castration-resistant prostate cancer. Theranostics. 9(5):1247-1263.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.