Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(6):1777-1793. doi:10.7150/thno.31581 This issue Cite
Research Paper
Therapeutic Targeting of BRD4 in Head Neck Squamous Cell Carcinoma
Yaping Wu1,2*, Yanling Wang1*, Pengfei Diao1, Wei Zhang1,3, Jin Li1,2,3, Han Ge1, Yue Song1, Zhongwu Li2, Dongmiao Wang2, Laikui Liu3, Hongbing Jiang2, Jie Cheng1,2 ![]()
1. Jiangsu Key Laboratory of Oral Disease, Nanjing Medical University, Jiangsu 210029, China PRC
2. Department of Oral and Maxillofacial Surgery, Affiliated Stomatological Hospital, Nanjing Medical University, Nanjing, 210029, China PRC
3. Department of Oral Pathology, Affiliated Stomatological Hospital, Nanjing Medical University, Nanjing, 210029, China PRC
*These two authors contributed equally to this study.
Received 2018-11-17; Accepted 2019-1-27; Published 2019-2-28
Citation:
Wu Y, Wang Y, Diao P, Zhang W, Li J, Ge H, Song Y, Li Z, Wang D, Liu L, Jiang H, Cheng J. Therapeutic Targeting of BRD4 in Head Neck Squamous Cell Carcinoma. Theranostics 2019; 9(6):1777-1793. doi:10.7150/thno.31581. https://www.thno.org/v09p1777.htm
Other stylesAbstract

The bromodomain and extraterminal family members are epigenetic readers and transcriptional coactivators which are critically involved in various biological processes including tumorigenesis. BRD4 has been increasingly appreciated as a key oncogene and promising anticancer target. Here, we sought to characterize the expression of BRD4 and its tumorigenic roles as well as therapeutic targeting in HNSCC.
Methods: Expression of BRD4 mRNA and protein was determined by bioinformatics interrogation of publically available databases, primary HNSCC samples and 4NQO-induced HNSCC animal model. The tumorigenic roles of BRD4 in HNSCC were evaluated by genetic and pharmacological approach in vitro and in vivo. Therapeutic efficiency of BRD4 targeting by JQ1 was assessed in three preclinical models including xenograft model, 4NQO-induced model and patients-derived xenograft model. Gene candidates responsible for therapeutic effects of JQ1 were identified by transcriptional profiling in HNSCC cells after JQ1 exposure.
Results: Significant upregulation of BRD4 was found in primary HNSCC samples and 4NQO-induced HNSCC model. Its overexpression associated with aggressive clinicopathological features and inferior overall and disease-free survival. BRD4 depletion by genetic silencing or pharmacological inhibition impaired cell proliferation, migration and invasion and reduced tumor growth and metastasis in vivo. Transcriptional profiling of HNSCC cells following JQ1 exposure identified hundreds of genes which might mediated its antitumor effects and enriched in cancer-relevant pathways. A novel prognostic risk score derived from JQ1-regulated genes was developed to stratify patients into subgroups with favorable or inferior prognosis.
Conclusions: Our findings reveal that BRD4 serves as a novel and critical mediator underlying tumorigenesis and a robust prognostic biomarker in HNSCC. Therapeutic targeting of BRD4 represents a potent and promising strategy against HNSCC.
Keywords: BRD4, bromodomain and extraterminal domain, JQ1, head neck squamous cell carcinoma
Introduction
Head neck squamous cell carcinoma (HNSCC) represents the sixth most common malignancy worldwide with approximately 500,000 new cases diagnosed each year and remains as one leading cause of cancer-related death [1]. Currently, comprehensive and multimodality therapeutic approaches including surgical resection, chemotherapy and radiotherapy against HNSCC have been established. Nevertheless, the long-term survival rates for patients with HNSCC, especially for those with advanced diseases, have not been significantly improved largely due to high incidence of locoregional recurrence, cervical lymph node metastasis and therapeutic resistance [2, 3]. Despite previous many efforts have been devoted to unravelling the cellular and molecular mechanisms underlying HNSCC tumorigenesis, our understanding regarding this malignancy remains incomplete known until now [4]. Therefore, identifying novel prognostic biomarkers and druggable targets has been an imperative need for better patient stratification, treatment-decision guiding and discovery of effective therapies for HNSCC, which ultimately improve patients' survival and quality of life [5].
The past decades have witnessed tremendous progress in epigenetic profiling, functional interrogation and therapeutic targeting in human cancer [6]. Cancer cells are characterized by dysregulated epigenetic landscape and exploited the chromatin regulatory machinery to enforce oncogenic transcriptional programs [7]. Of particular interests, bromodomain and extraterminal domain (BET) proteins are key epigenetic readers characterized by two tandem bromodomains (BD1 and 2) and mainly recognize acetylated lysine of histone H3 and H4 on chromatin, sometimes the acetylated non-histone proteins, to influence gene expression [8]. BRD4, the well-studied member of the BET family, usually recruits the transcription elongation factor P-TEFb, Mediator, transcriptional factors or select histone modifiers to facilitate transcriptional activation of target genes [9, 10]. Compelling evidence has revealed that BRD4 is critically involved in various cellular processes including cell cycle progression, cell growth control, apoptosis and tumorigenesis when it goes awry [11-13]. BRD4 is frequently overexpressed and clinically linked to multiple types of human cancer by enhancing oncogenic functions of major cancer- drivers by elevating their expression and functions in cancer, such as c-Myc in leukemia, ERG and AR in prostate cancer as well as ALDH in ovarian cancer [14-17].
Given the essential roles of BRD4 underlying transcriptional activation and tumorigenesis, selective inhibition of BRD4 has therefore been pursued as an attractive therapeutic strategy against cancer [18, 19]. Until now, several selective small-molecule inhibitors like JQ1 and I-BET762 have been developed to target the amino-terminal bromodomains of BRD4, which exhibits potent anti-proliferative effects with promising translational significance in a growing range of malignancies [15, 20, 21]. JQ1, the first-in class thienodiazepine small-molecule, binds the bromodomains of BET protein with exquisite shape complementarity and nanomolar affinity resulting in potent, transient and competitive displacement of BRD4 from acetylated chromatin, thus preferentially blocks transactivation of specific sets of genes in a dose- or context-dependent manner [21, 22]. Collectively, these aforementioned findings established a compelling rationale for therapeutic targeting BRD4 in cancer.
However, its expression pattern, detailed tumorigenic roles as well as translational potential as a novel therapeutic target in HNSCC remain largely undefined yet. In this study, we sought to assess BRD4 expression and functions by integrating bioinformatics', genetic and pharmacological approaches by using online databases, clinical samples and preclinical animal models. Our findings further highlight that BRD4 is critically involved in HNSCC tumorigenesis and hold great potentials as a novel prognostic marker and therapeutic target for HNSCC.
Methods
Cell lines, chemicals and treatments
Human Oral Keratinocytes (HOK), HEK293T and a panel of HNSCC cell lines including Cal27, Fadu, SCC4, SCC25, HN4 and HN6 were used. HOK, HEK293T, Cal27, Fadu, SCC4 and SCC25 were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and authenticated by short tandem repeat profiling at regular intervals. HN4 and HN6 were generous gifts from Dr. Wantao Chen (Shanghai Jiaotong University). HNSCC cell lines were grown in DMEN/F12 (Invitrogen) supplemented with 10% FBS (Gibco) and 100 units/ml penicillin and streptomycin, and maintained at 37℃ in a 5% CO2-humidified incubator. Mycoplasma testing was routinely performed during the course of this study. JQ1 (SML1524) and OTX015 (SML1605) were purchased from Sigma-Aldrich and dissolved in DMSO. Cells were treated with JQ1 or OTX015 with varied concentrations and time course as indicated.
DNA constructs, viral production and transfection/infection
Two independent short hairpin RNAs (shRNA) targeting human BRD4 mRNA (NM_058243.2) (shRNA-1: TRCN0000196576-GCCAAATGTCTACACAGTATA; shRNA-2: TRCN0000199427 CAGTGACAGTTCGACTGATGA) were subcloned into pLKO.1 puro lentiviral vector and verified by direct sequencing. The shRNA vector containing sequence without targeting any known human gene was used as negative control (shNC). The human BRD4 overexpression construct tagged with single FLAG was generated by inserting the BRD4 full-length cDNA template into lentiviral plasmid pLenti CMV-GFP-Puro and then verified by direct sequencing. Lentiviral particles were prepared by transiently co-transfecting HEK293T cells with individual lentiviral constructs or controls together with packaging and envelope plasmids (pCMV-VSV-G and pCMV-Δ8.2) using calcium-phosphate method. These viral supernatants were filtered, concentrated and stored until use. The efficiencies of BRD4 knockdown or overexpression constructs were confirmed by western blot following cell infection in vitro. The stable cell clones were selected by appropriate antibiotics (puromycin, 2-5μg/ml, Sigma) for at least one week after virus infection or plasmids transfection.
RNA extraction and real time RT-PCR
Total RNA was extracted from fresh tissues or cells with Trizol reagent (Invitrogen) and then subjected to reverse transcription and PCR reactions using PrimeScriptTM RT-PCR kit (Takara) as described previously [23]. The detailed gene-specific primers for genes of interests were listed in Table S7. Relative mRNA expression was quantified as compared to internal control GAPDH or 18sRNA using comparative CT method.
Protein extraction and western blot analysis
Cells were lysed with ice-cold RIPA buffer containing protease inhibitor cocktail (Roche). Lysates were then separated by SDS-PAGE and transferred onto PVDF membranes (Bio-Rad). After 5% non-fat dry milk blocking and overnight incubation with primary antibodies, the blots were detected using appropriate horseradish peroxidase-conjugated secondary antibodies (Invitrogen). Immunoreactive bands on the blots were detected by ECL chemiluminescence kit (Millpore). Detailed information for primary antibodies were listed in Table S8. The relative levels of each protein were quantified with ImageJ software and GAPDH or β-Actin served as loading control.
Cell viability, proliferation and apoptosis assays
Cell proliferation and viability were monitored by absorbance using the MTT assay. Approximately 1000-3000 cells/well were seeded in the 96-well plates. At the indicated time points, 5mg/ml MTT (Sigma) was added to the cells and incubated at 37℃ for another 4h. Absorbance at 490 nm was measured with an automatic enzyme-linked immunosorbent assay reader (Molecular Devices). Cell apoptosis were determined by propidium iodide (PI) or Annexin V: PI Apoptosis Detection Kit (BD Bioscience) in a FACS Caliber BD flow cytometer. Data were processed using BD FACSuite analysis software.
Cell migration and invasion assay
Cell migration and invasion assays were performed using wound healing and transwell chambers (8-μm pore size, Corning) with Matrigel (BD Pharmingen) pre-coating as we previously reported [23, 24].
Colony formation and tumorsphere assay
For colony formation assay, 103 cells were placed into 6-well plates or dishes, allowed to grow for two weeks. After fixation and staining with crystal violet, these colonies (50μm in size) were visualized and counted. For tumorsphere formation assay, the disassociated single cells (104/ml) were cultured in serum-free DMEM/F-12 supplemented with B27, 20ng/ml EGF (BD Bioscience) and 10ng/ml bFGF (BD Bioscience), and grown in ultra-low-attachment plates (Corning) for 10-14 days. For in vitro serial passages, these tumorspheres were harvested and dissociated into single cells by 0.1% trypsin and gentle pipette, and then filtered, re-plated to form secondary sphere in aforementioned serum-free media. Tumorspheres with diameter larger than 50μm were counted under microscope.
Patients and tissue specimens
A total number of 103 patients with primary HNSCC receiving surgical treatment at the Department of oral and maxillofacial surgery, Affiliated Hospital of Stomatology, Nanjing Medical University between Jan. 2008 and Dec. 2015 were retrospectively enrolled. Patient inclusion criteria were described as follows: primary HNSCC without any prior history of chemotherapy or radiotherapy; patients underwent radical tumor resection and elective or therapeutic neck dissection as required; detailed demographic, clinical, pathological and follow-up data. The archived sample blocks and haematoxylin-eosin staining slides of each patient were retrieved and analyzed to confirm the previous histological diagnosis according to the established histological criteria. Twenty-four samples of healthy tongue and buccal mucosa were obtained from no-tumor surgery for histopathological examination. In addition, 65 pairs of primary HNSCC samples and adjacent non-tumor mucosa were freshly collected (Jan.2016-Dec.2017) upon surgical resection of primary lesions within 15 min and snap-frozen in liquid nitrogen, stored in -80℃ until use. Written informed consent was obtained from these patients or donors. This study protocol was reviewed and approved by the Research Ethic Committee of Nanjing Medical University.
Immunohistochemical staining and scoring
Immunohistochemical staining was performed on 4μm-thick sections from formalin-fixed paraffin-embedded clinical samples. The staining procedure was performed as we reported previously [25, 26]. Negative controls (without primary antibody incubation) were included in each staining run. Immunoreactivity was semi-quantitatively evaluated according to staining intensity and distribution using the immunoreactive score which was calculated as intensity score × proportion score. Intensity score was defined as 0, negative; 1, weak; 2, moderate; 3, strong. The proportion score was defined as 0, negative; 1, <10%; 2, 11-50%; 3, 51-80%; 4, >80% positive cells. The total score ranged from 0 to 12. Accordingly, the immunoreactivity of each slide was categorized into three subgroups based on the final score: 0, negative; 1-4, low expression; 4-12, high expression as we reported previously [25, 26].
HNSCC xenograft model and JQ1 treatment
All experiments involving animal subjects were in accordance with the institutional animal welfare guidelines and approved by Institutional Animal Care and Use Committee of Nanjing Medical University. Six-week-old female nu/nu mice were obtained and maintained in a specific pathologic-free environment. Cancer cells suspended in total 100μL PBS and Matrigel (1:1) were inoculated subcutaneously on the single or both flanks (at least 6 animals per experimental group). Tumor incidence and growth were monitored after inoculation and tumor diameters were measured by calipers every 3 days after tumor masses were identified. For drug treatment animal experiments, 2×106 viable Cal27/Fadu cells were inoculated subcutaneously in nude mice. Four weeks later, mice bearing tumors with approximately 100 mm3 were randomly divided into two subgroups (6 mice per group) which were scheduled to receive the following treatments: 50 mg/kg JQ1 (dissolved in 10% cyclodextrine), once every day by intraperitoneal injection or vehicle only in controls for consecutive 15 days. Tumor volume was calculated as follows: volume=a×b2/2, where a and b were defined as the longest and shortest diameter, respectively. Final tumor weights were also measured upon animals were sacrificed. The tumor samples were processed for H&E staining and immunohistochemical staining.
4-nitroquinoline 1-oxide (4NQO)-induced HNSCC animal model and JQ1 treatment
For 4NQO-induced HNSCC animal model, 6-week-old C57BL/6 mice were treated with drinking water containing 50μg/mL 4NQO for consecutive 16 weeks and then given with normal water for another 8 weeks [27]. Animals with normal drinking water were used as negative control. Lesions in tongue were visually inspected twice every week. Samples were harvested at 16, 20 and 24 weeks after chemical administration and subjected to histopathological analyses. To detected the effects of JQ1 treatment in this chemical-induced HNSCC animal model, 20 mice with prominent tongue lesions at 24th week were randomly divided (n=10 per group) and treated daily with either JQ1 at 50 mg/kg by intraperitoneal injection or vehicle control. After three consecutive weeks with JQ1 treatment, the samples were harvested and collected for histopathological analyses.
Establishment of HNSCC patient-derived xenograft (PDX) and JQ1 treatment
Fresh samples upon surgery within 15 min were trimmed to remove necrotic tissues and transferred to a clean 50mL centrifuge tube containing DMEM/F12 with 1:100 penicillin/streptomycin (Gibco). After thorough washing, the tumor samples were cut into approximately 3-mm pieces and directly transplanted into flanks 4-6 week female NOD/SCID mice. Subsequently, the sites of implantation were checked twice each week for possible tumor formation. Tumor masses usually appeared about 2-4 months after inoculation. Tumor masses were harvested, dissected and implanted in another group of NOD/SCID mice using the same procedure until they reached approximately 1.0 cm in diameter. The passage harboring the original patient-derived tissue was termed P0 with subsequent generations labelled consecutively (P1, P2, P3, and so on). In this study, the third generation (P2) was expanded for HNSCC treatment. When the volume of PDX tumors reached about 100mm3, these mice were randomly divided into two groups (at least n=6 per group). Mice were treated daily with either JQ1 at 50 mg/kg IP or vehicle control. The samples were harvested after three weeks JQ1 treatment.
Data mining and interrogation of BRD2, BRD3 and BRD4 in HNSCC via publically available database
The original data concerning mutational landscape and expression of BRD2, BRD3 and BRD4 in HNSCC were retrieved from publically available databases Oncomine (https://www.oncomine.org/) and TCGA (https://cancergenome.nih.gov/). The mRNA expression levels of BRD2-4 (log2- transformed) in HNSCC and normal counterparts were retrieved from diverse studies and compared. The associations between expression status of BRD2-4 (high or low using median value as cutoff) and patient survival were determined by Kaplan-Meir analysis.
Transcriptional profiling and gene set enrichment analysis (GSEA)
Genome-wide transcriptional profiling was performed in cells treated with JQ1 for indicated time. RNA samples were sent to transcriptional profiling using Agilent SurePrint G3 Human Expression v3 platform at the core facility, Shanghai Biotechnology corporation. Raw data were normalized by Quantile algorithm, limma packages in R and deposited in the Gene Expression Omnibus under the accession number GSE122522. Pairwise comparisons between treatment group and control were conducted to detect differentially expressed genes using Bioconductor package DESeq2. Gene set enrichment analysis was performed via GSEA (version v3.0) to identify gene ontology and biological process enrichments following the official user guide of GSEA [28].
Identification and validation of prognostic significance of JQ1-regulated gene signature in HNSCC
To identify JQ1-regulated gene signature with prognostic significance in HNSCC, we initially profiled the differentially expressed genes upon JQ1 exposure in Cal27 and Fadu cells by microarray and identified the overlapped genes as potential candidates responsible JQ1's anti-cancer effects. Then, univariate regression assay was utilized to identify candidates which were significantly associated with overall survival (with P value of less than 0.05) in TCGA-HNSCC datasets which were further filtered using Robust likelihood-based modelling for 1000 times via R environment with Rbsurv package and multivariate Cox regression analysis with top statistical significance [29, 30]. A risk score formula based on the expression level and coefficient of these selected candidates was generated and its optimal cut-off point was selected at the maximal sensitivity and specificity by receiver operating characteristics (ROC) curve. Subsequently, the prognostic values of this risk score were validated in TCGA-HNSCC dataset and other online available HNSCC cohorts (GSE41613 and GSE42743) [31].
Statistical analysis
All quantitative data in the present study was shown as mean±SD of two or three independent experiments and compared with Student's t-test or ANOVA with Bonferroni post hoc test unless otherwise specified. The potential associations between BRD4 expression and various clinicopathological parameters were evaluated by Chi-square or Fisher exact test. The overall survival (OS) and disease-free survival (DFS) rates of patients were estimated using Kaplan-Meier method and compared with Log-rank test. The prognostic analyses were performed by univariate and multivariate Cox regression models to determine the individual clinicopathological variables with patient overall survival. P values less than 0.05 (two-sided) were considered statistically significant. All statistical analyses were performed using GraphPad Prism 7, SPSS 22.0 software and R 3.5.1 with Survival package, ROC package and Rbsurv package.
Results
Overexpression of BRD4 mRNA associates with tumor aggressiveness in HNSCC
Previous studies have reported that BRD4 is aberrantly overexpressed in a broad spectrum of human cancer and significantly associated with malignant features and poor prognosis [13, 16, 32, 33]. To explore the expression pattern of BRD4 in HNSCC, we initially evaluated the mRNA expression patterns of BRD2, BRD3 and BRD4 using the publically available datasets including TCGA-HNSCC and Oncomine. As shown in Figure 1A and Figure S1A, all BRD2, BRD3 and BRD4 mRNAs were significantly upregulated in TCGA-HNSCC samples as compared to their normal counterparts. In addition, as shown in Figure 1B-D, data mining and interrogation from Oncomine database indicated marked overexpression of BRD4 mRNA in HNSCC samples from Ginos' [34], Peng's [35] and Ye's [36] cohorts, whereas BRD2 and BRD3 mRNAs weren't consistently elevated in HNSCC but largely comparable with normal counterparts in these three independent cohorts (Figure S1A).
Moreover, our data from qRT-PCR in 65 freshly collected and paired HNSCC samples and adjacent not-tumor mucosa indicated that only BRD4 mRNA was significantly increased in HNSCC samples compared to the paired normal epithelial (Figure 1E, Figure S1B-C). Status of BRD4 mRNA expression categorized with median value as cutoff was found to be positively associated with tumor size and cervical node metastasis with P-value 0.0414 and 0.0225, respectively (Fisher exact test, Table S1). Only high BRD4 mRNA expression correlated with advanced pathological grade in TCGA-HNSCC cohort (Figure 1F, Figure S1D). However, we failed to reveal any significant associations between BRD2, BRD3 and BRD4 mRNA expression and patient overall survival in TCGA-HNSCC cohort, when the median values of their mRNAs were used as cutoff to stratify patients into low and high expression subgroups (Figure S1E). Noticeably, genetic alternations of BRD4 in TCGA-HNSCC dataset were approximately 10% and mostly of which were missense mutations (Figure S2), thus precluding the possibility that genetic aberrations of BRD4 might be not primarily responsible for its upregulation in HNSCC.
Figure 1
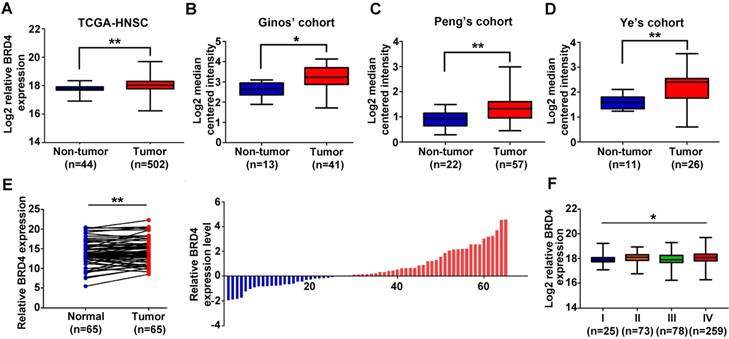
Overexpression of BRD4 mRNA associates with tumor aggressiveness in HNSCC. A-D. The expression level of BRD4 mRNA (log2-transformed) was compared between HNSCC samples and normal counterparts in multiple patient cohorts. The original data were retrieved from TCGA and Oncomine database and then plotted. Y-axis represents the median intensity, 25th and 75th percentile data. Student's t test, *P < 0.05; **P < 0.01. E. The relative expression of BRD4 mRNA was measured by qRT-PCR in freshly collected HNSCC samples and paired adjacent non-tumor mucosa. The height of column represents the fold change (log2-transformed) in BRD4 expression in these 65 patients (right panel). Paired t test, *P < 0.05; **P < 0.01. F. Relative expression of BRD4 mRNA (Log2-transformed) was compared among TCGA-HNSC subgroups stratified by pathological grade. Y-axis represents the median intensity, 25th, and 75th percentile data. ANOVA test, *P < 0.05; **P < 0.01.
Overexpression of BRD4 protein associates with tumor aggressiveness and prognosis in HNSCC
To determine the protein expression of BRD4 in HNSCC, its abundance in a panel of HNSCC cell lines, freshly collected and archived clinical samples was measured. As displayed in Figure S3A, increased BRD4 protein was observed in all cancerous cell lines relative to immortalized non-tumorigenic cells HOK. Additionally, the expression of BRD4 protein was also determined in 40 pairs of fresh HNSCC samples and adjacent non-tumor tissues. As shown in Figure S3B-C, markedly elevation of BRD4 was detected in HNSCC relative to corresponding non-tumor tissue (P < 0.01, Paired t test). Next, we measured BRD4 expression by immunohistochemistry in a retrospecttive cohort of 103 primary HNSCC samples. As shown in Figure 2A-C, positive nuclear staining of BRD4 was identified in cancerous cells, whereas weak or negative staining was detected in the normal counterparts. Based on our immunohistochemical staining scores, high BRD4 expression was identified in approximately 69.9% (72/103) in cancer samples and 29.1% (7/24) in normal counterparts, thus indicating aberrant BRD4 overexpression in a large fraction of HNSCC (P < 0.001, Chi-square test, Table S2).
To further understand the clinical significance of BRD4 overexpression in HNSCC, we sought to identify the potential associations between BRD4 expression and patients' clinicopathological parameters. As shown in Table S3, there were no significant correlations found between BRD4 and patient age, gender, smoking, alcohol use, cervical node metastasis and clinical stage. Noticeably, high BRD4 abundance significantly associated with large tumor size and advanced pathological grade (Fisher exact test, P < 0.05). Moreover, patients with high BRD4 had much lower overall and disease-free survival rates compared to those with low BRD4 as estimated by Kaplan-Meier analyses (Figure 2D-E, Log-rank test, P=0.0162 and 0.0432). Importantly, BRD4 abundance was further identified as an independent factor for patients' prognosis (P=0.021 and 0.026, Cox proportional hazards regression model, Table S4). Taken together, our data reveal that overexpressed BRD4 protein associates with aggressive clinicopathological features and unfavorable prognosis in primary HNSCC.
BRD4 is involved in chemical-induced HNSCC tumorigenesis
Having revealed high expression of BRD4 in human HNSCC samples, we next utilized a well-established chemical-induced animal model to further characterize the expression pattern of BRD4 during HNSCC initiation and progression (Figure 2F) [27]. 4NQO treatment resulted in various pathological lesions in tongue which included epithelial hyperplasia, dysplasia, carcinoma in situ and invasive SCC, thus largely recapitulating the multiple-staged tumorigenic process in human HNSCC. As displayed in Figure 2G-J, immunohistochemical staining of BRD4 in these samples indicated negative or low staining in normal tongue mucosa and epithelial with hyperplasia, while prominent nuclear staining in dysplasia/carcinoma in situ and invasive carcinoma. As detailed in Table S5, positive staining of BRD4 was observed in carcinoma (87.5%, 7/8), whereas much less were detected in healthy mucosa (33.3%, 2/6), samples with hyperplasia (16.7%, 1/6) or dysplasia/carcinoma in situ (66.7%, 4/6). Together, our finding from chemical-induced animal model provide support that BRD4 might be critically involved in HNSCC development by serving as a putative pro-oncogenic factor.
BRD4 promotes cell proliferation, migration/invasion, chemoresistance and cancer stem cell maintenance in HNSCC
To delineate the tumorigenic roles of BRD4 in HNSCC, two shRNA sequences targeting human BRD4 were designed and delivered into Cal27 and Fadu cells with relatively high endogenous BRD4. As shown in Figure 3A, the protein abundance of BRD4 was greatly diminished after shRNA transfection, accompanied by a marked decrease of c-Myc, a well-established downstream target of BRD4, thus in part verifying the efficiency and specificity of knockdown. Subsequently, the relevant phenotypic changes of cells after BRD4 knockdown were monitored in detail. Impaired cell proliferation and increased proportions of apoptotic cells were observed in cells with stable BRD4 knockdown as measured by MTT, colony formation and flow cytometry assays (Figure 3B-D). In addition, several markers of cell apoptosis including cleaved PARP, cleaved Caspase3 and Bax were upregulated with concomitant Bcl-2 downregulation following BRD4 depletion (Figure 3E). Moreover, BRD4 knockdown also significantly enhanced the therapeutic sensitivity of the chemotherapeutic agents 5-FU and cisplatin in vitro (Figure S4). Furthermore, BRD4 depletion resulted in impaired migration and invasion as detected via wound healing, transwell invasion assays and expression of EMT-relevant markers (Figure 3F-H). As shown in Figure 3I-K and Figure S5, BRD4 and c-Myc protein were markedly elevated in tumorsphere compared with cell cultured in monolayer. BRD4 depletion pronouncedly impaired the abilities of tumorsphere formation and reduced expression of cancer stem cell markers like CD44, Sox2 and Bmi1, suggesting BRD4 might be the critical regulator for stem-cell like traits in HNSCC. To further unravel the tumorigenic roles of BRD4 in HNSCC, we generated stable BRD4-overexpressing cells in HN6 (relatively low endogenous BRD4) by antibiotics selection (Figure S6A). Enforced BRD4 overexpression accelerated cell proliferation, migration and invasion as evidenced via results from MTT, colony formation, wound healing and transwell invasion assays (Figure S6B-D).
To verify the tumorigenic roles of BRD4 in vivo, we next developed a HNSCC xenograft model in which stable BRD4 knockdown cells were inoculated into left flanks of nude mice. As shown in Figure 4A,C, tumor growth was compromised in samples formed from BRD4-silencing cells as compared to those from control cells as evidenced by reduced tumor volume and final weight. Moreover, immunohistochemical staining of tumor samples revealed significantly reduced BRD4, c-Myc and Ki67 positive staining and increased cleaved caspase 3 in samples from BRD4 knockdown cells as compared to control (Figure 4B,D). Collectively, these findings reveal that BRD4 is critically involved in HNSCC growth and progression.
Pharmacological inhibition of BRD4 phenocopies BRD4 knockdown in vitro
Previous pioneering studies have identified several chemicals targeting BRD4 with potency and demonstrated their promising utility as single drug or in combination with others against human cancer, thus highlighting translational potential of BRD4 targeting as a novel anticancer strategy [15, 21, 37, 38]. We first determined whether two BRD4 inhibitors, JQ1 and OTX015, were able to inhibit its functions in vitro (Figure S7A). The values of IC50 of JQ1 and OTX015 were significantly lower in Cal27 and Fadu cells as compared to non-tumor cells HOK and bone mesenchymal stem cells (BMSCs), indicative of less toxicity of these inhibitors in non-tumor cells (Figure S7B). As expected, both JQ1 and OTX015 exposure remarkably reduced c-Myc mRNA and protein abundance in a dose-dependent manner in vitro (Figure 5A-B, Figure S7C-D). However, the mRNA expression of BRD4 was not affected by JQ1 or OTX015 treatment (Figure S7E). Similar findings were obtained when another housekeeping gene 18sRNA was utilized as loading control in these qRT-PCR assays (Figure S8). Next, we measured the phenotypic changes of HNSCC cells following these inhibitors' exposure. Not surprisingly, impaired cell proliferation and migration, tumorsphere formation as well as increased apoptosis were evident upon JQ1 or OTX015 treatment (Figure 5C-J and Figure S9A-G). Moreover, when cells were treated with JQ1 alone or together with 5-FU or cisplatin, more significantly anti-proliferative effects of combinational treatments were observed as compared to single agent treatment (Figure S9H), suggesting the potential synergic effects of JQ1 and these agents.
Figure 2
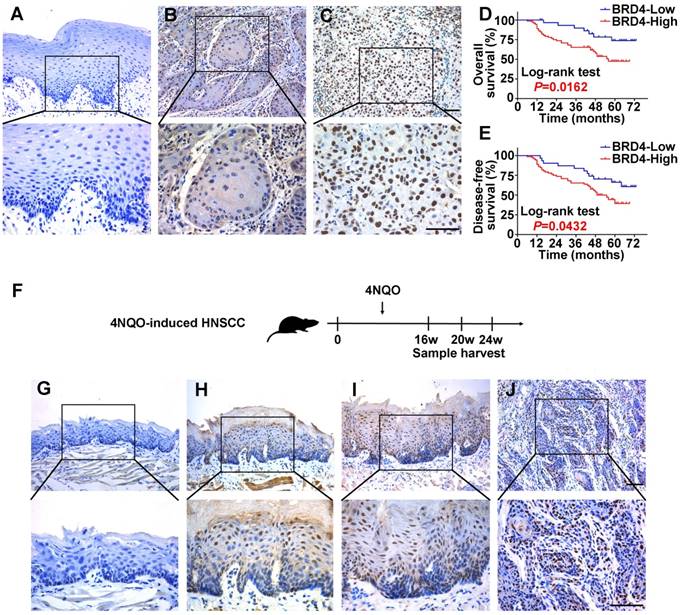
Expression patterns of BRD4 protein in HNSCC samples and 4NQO-induced HNSCC animal model and its overexpression positively associates with patients' survival. A-C. Representative BRD4 expression in human normal oral mucosa (A) and HNSCC specimens (B, C) was evaluated by immunohistochemical staining. Images (upper panel) marked by black-box were shown in larger magnification as images (lower panel), respectively. Scale bar: 50μm. D, E. Overall (D) and disease-free (E) survival analyses of patients with high or low expression of BRD4 based on IHC data were estimated by Kaplan-Meier method and compared with Log-rank test. F. Experimental design of 4NQO-induced HNSCC animal model. G-J. Immunohistochemical staining of BRD4 in samples from diverse stages in 4NQO-induced HNSCC animal model. Images in the upper panel were representative of BRD4 staining in normal (G), epithelial with hyperplasia (H), epithelial with severe dysplasia/carcinoma in situ (I) and squamous cell carcinoma (J), respectively. Images in the lower panel were magnified from the black box area in the images in the upper panel, respectively. Scale bar: 50μm.
Therapeutic potentials of JQ1 in HNSCC xenograft, chemical-induced primary HNSCC and HNSCC-PDXs
Having revealed anti-tumor roles of BRD4 inhibitors in vitro, we further developed three pre-clinical animal models including tumor xenograft, 4NQO-induced HNSCC and patient derived xenograft (PDX) to determine the therapeutic effects of JQ1 in vivo. Following xenograft establishment, tumor-bearing animals received JQ1 (50mg/kg) for successive 15 days (Figure S10A). The tumor volume and weight of xenograft treated with JQ1 were significantly lower than those treated with the vehicle (Figure S10B-C). In agreement, less c-Myc, Ki-67 staining and increased cleaved caspase 3 staining were observed in samples from JQ1 treated mice compared with control samples (Figure S10D-E). Next, we administered JQ1 into animals starting at 24th week for 3 consecutive weeks in 4NQO-induced HNSCC model (Figure 6A). Upon animal sacrifice, the tongue and cervical lymph nodes were harvested for histopathological analysis. As shown in Figure 6B, much less significant changes of tongue had been observed in JQ1-treated animals, while neoplastic or ulcerous lesions were frequently found in vehicle-treated mice. Histopathological examinations revealed that the numbers of dysplasia and SCC were significantly fewer following JQ1 administration compared with control (Figure 6C). In addition, much fewer metastatic lymph nodes were found in JQ1-treated mice relative to control (Figure 6D). Moreover, decreased Ki-67 staining and increased cleaved caspase 3 staining were detected in samples from JQ1-treated animals compared to control (Figure 6E).
Figure 3
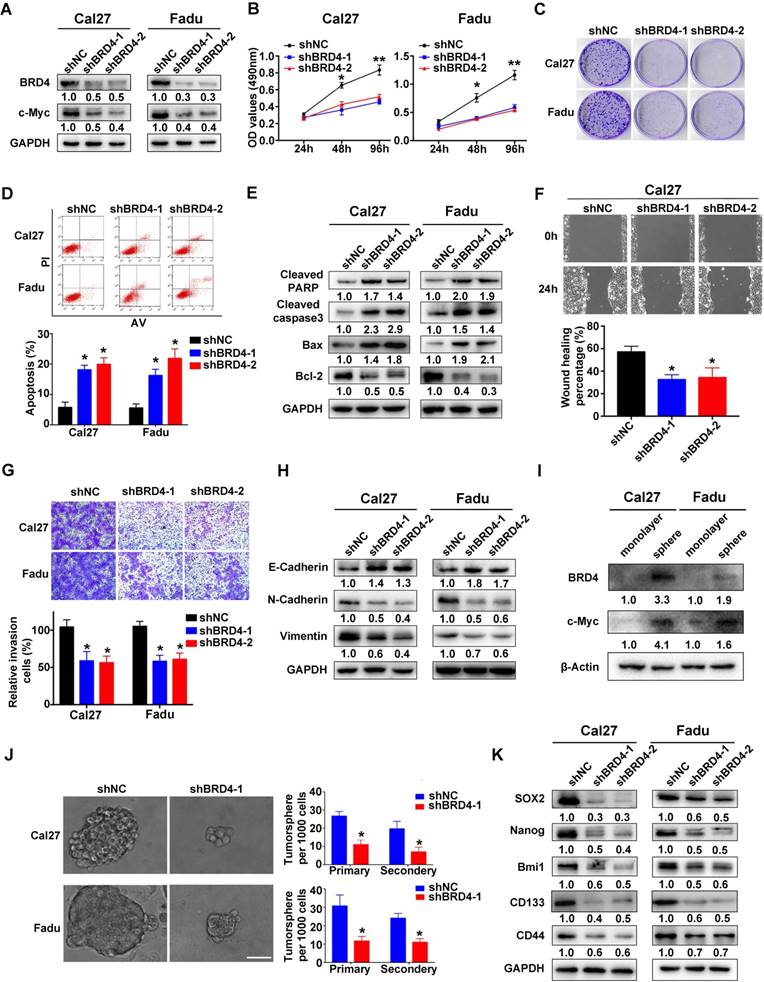
BRD4 knockdown inhibits cell proliferation, migration and invasion and tumorsphere formation and triggers apoptosis in HNSCC cells. A. Endogenous BRD4 was efficiently reduced by shRNA targeting BRD4 in Cal27 and Fadu cells. Representative images of western blot were shown. B-C. Cell proliferation was remarkably suppressed upon BRD4 knockdown as measured by MTT (B) and colony formation (C) assays. D-E. Increased percentages of apoptotic cells were observed following BRD4 knockdown as assayed by Annexin V-PI staining and expression changes of relevant markers. Representative images were shown. F-G. The migration and invasion abilities were significantly reduced in shBRD4-transfected cells in wound healing and transwell assays, respectively. H. The abundance of E-cadherin, N-cadherin and Vimentin was measured by western blot following BRD4 knockdown. Representative images were shown. I. The protein expression of BRD4 and c-Myc cultured in monolayer and tumorsphere were detected by western blot. Representative images were shown. J. The ability of tumorsphere formation was significantly reduced in shBRD4-transfected cells relative to cells with control shRNA. Scale bar: 50μm. K. The abundance of cancer stem cells markers was probed in Cal27 and Fadu cells with stable infected BRD4 knockdown. Representative images of western blot were shown. Student's t test, *P < 0.05; **P < 0.01.
Figure 4
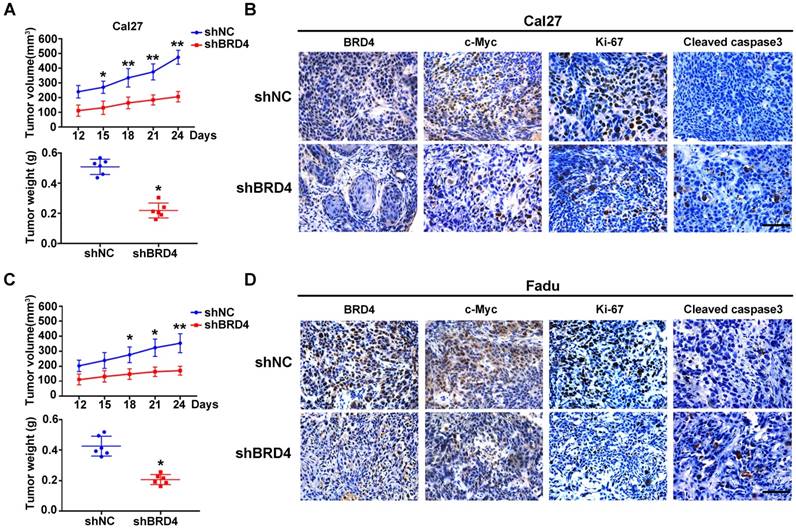
BRD4 knockdown results in impaired xenograft tumor growth in vivo. A, C. Tumor volume (upper panel) and final weight (lower panel) were monitored in xenograft samples derived from Cal27 (A) or Fadu (C) cells with stable BRD4 knockdown or controls. Student's t test, *P < 0.05; **P < 0.01. B, D. Immunohistochemical staining of BRD4, c-Myc, Ki67 and Cleaved caspase3 in xenograft samples derived from Cal27 (B) or Fadu (D) cells with stable BRD4 knock-down or controls. Representative images of IHC were shown. Scale bar: 50μm.
Given the inherent heterogeneity of HNSCC, to more accurately mimic HNSCC in human, we developed PDX model from patients' samples to further assess the therapeutic efficacy of JQ1. A series of PDX mice were successfully established and then randomly divided into subgroups receiving vehicle or JQ1 when PDX volume reach approximately 100 mm3 (Figure 6F). As displayed in Figure 6G-J, JQ1 administration remarkably inhibited tumor growth probably via repressing cell proliferation and inducing cell apoptosis as evidenced by quantification of Ki-67 and cleaved caspase 3 staining in samples. Furthermore, we plotted Kaplan-Meier survival curves and compared the progression-free survival in animals receiving treatment. Tumor progression was delayed in JQ1-treated animals with a median volume doubling of 9 days as compared to 6 days for vehicle-treated mice (P=0.0022, Log-rank test, Figure 6K).
JQ1-regulated gene risk score correlates patients' survival in HNSCC
To explore the transcriptional targets which might be responsible for therapeutic effects of JQ1 in HNSCC, we subjected total RNA samples from Cal27 and Fadu cells treated with JQ1 (0.5μM, 24h) to genomic-wide transcriptional profiling (Figure 7A). A total number of 478 upregulated genes and 614 downregulated genes by 2-fold or greater was identified in both cells (Figure 7B, Figure S11A). Genes with more than 5-fold changes (22 genes upregulated, 52 genes downregulated) were clustered in Figure 7C. Expression of ten candidates with downregulation were selected and independently validated by qRT-PCR. Our results revealed that expression levels of most candidates were reduced upon JQ1 exposure, although difference of FOFX2 mRNA in Cal27 and GPR68 mRNA in Fadu failed to reach statistical significance (Figure 7D, Figure S12). Moreover, Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses indicated that those JQ1-downregulated genes were significantly enriched in multiple human cancers such as thyroid cancer, melanoma, bladder cancer and basal cell carcinoma, etc. Gene Ontology (GO) and GSEA analyses further revealed that these JQ1-regulated genes were significantly enriched in multiple cancer-related categories including Pathways in cancers and regulation of cell proliferation and growth (Figure 7E, Figure S11B, Figure S13-14). Furthermore, our data mining and interrogation of TCGA-HNSCC dataset found that among 52 genes downregulated by JQ1, 14 genes were significantly overexpressed in HNSCC samples and 4 genes were significantly associated with patient survival in patients from TCGA-HNSCC cohort (Figure S11C-D).
Next, we compared the transcriptional profiling data with others wherein diverse cancer cells were exposed to JQ1 and found that they were not significantly overlapped, thus suggesting that transcriptional targets modulated by JQ1 might be cancer-type specific (Figure S15). Finally, to determine whether these JQ1-regulated genes have prognostic significance in HNSCC, we developed a JQ1-related prognostic score comprising 4 genes (FRMD5, MXD4, PITPNM3 and TRIB3) by sequential univariate regression analysis, Robust likelihood-based modelling and multivariate regression analysis using TCGA-HNSCC dataset as training cohort (as schematic illustrated in Figure S16). As shown in Figure S17, the expression changes of these 4 genes upon BRD4 knockdown or JQ1 exposure were consistent with our microarray results. This risk score was calculated by the following formula: risk score = FRMD5×0.11890+MXD4×(-0.27234)+PITPNM3×(-0.20711)+TRIB3×0.20080. Distributions of this risk score for each patient among these three cohorts were shown in Figure S18A-C. The optimal cutoff for risk this score was 1.005 derived from ROC curve using TCGA-HNSCC dataset (Figure S18D). Then Kaplan-Meier analyses indicated that patients in high risk subgroup had markedly reduced survival as compared to those with low risk in TCGA-HNSCC dataset (P<0.0001, Figure 7F). To reinforce the prognostic value of this risk score and rule out other confounding factors, we performed univariate and multivariate cox regression analyses and found that this risk score was an independent prognostic predictor for overall survival in the training cohort (Table S6). Furthermore, data from another two independent cohorts of HNSCC samples (GSE41613 and GSE42743) were further utilized as testing and validation cohorts to verify the prognostic utility of this risk score. Consistent with the findings from training set, patients with high risk score had significantly lower OS ratios as compared to those with low risk score in both testing and validation cohorts with sensitivity and specificity (P=0.00083, 0.0028; Log-rank test; Figure 7G-H, Figure S18E-F).
Discussion
Dysregulation of chromatin modifiers has been increasingly appreciated as a hallmark of cancer [39]. Therapeutic strategies that selectively modulate the recruitment and/or activities of these modifiers like BET proteins at chromatin hold great promise as targeted anticancer therapies [7, 18]. Here, our data revealed that aberrant BRD4 overexpression significantly associated with cancer aggressiveness and unfavorable prognosis in HNSCC via serving as a putative pro-oncogenic factor driving cell proliferation, migration and invasion. Pharmacologic targeting of BRD4 by JQ1 potently inhibited tumor growth and progression in preclinical animal models. These findings provide ample evidence for the therapeutic utility of BRD4 inhibitors in HNSCC.
BRD4 overexpression as a novel biomarker with diagnostic and prognostic significance in HNSCC
Growing evidence has indicated that BRD4 is usually upregulated in a broad spectrum of human malignancies and significantly associated with aggressive clinicopathological features and poor survival, thus indicative of BRD4 as a novel diagnostic and prognostic biomarker with translational potentials [11, 33, 40]. For example, BRD4 was aberrantly overexpressed in gastric cancer, renal cell carcinoma and melanoma, and its upregulation significantly correlated with advanced tumor stage, lymph node metastasis and inferior patient survival [11, 33, 40]. Consistent with these reports, our data mining from publically available databases and results from cell lines and clinical samples indicated that both mRNA and protein levels of BRD4 were aberrantly upregulated in a large fraction of HNSCC samples. In addition, its overexpression significantly correlated with malignant features such as advanced pathological grade, large tumor size and cervical lymph node metastasis as well as reduced survival. Importantly, BRD4 expression status was identified as an independent prognostic factor to predict patient survival by multivariate Cox regression analyses. However, we failed to identify positive association between BRD4 mRNA and prognosis in TCGA- HNSCC dataset and reasoned that this discrepancy might be due to patient heterogeneity, genetic background, etiological factors, cutoff values for patient stratification and possible inconsistency between mRNA and protein et al. Collectively, our data together with others indicate that BRD4 is a novel diagnostic and prognostic biomarker for HNSCC. Pre-treatment measurement of BRD4 abundance in HNSCC samples might provide information regarding patient stratification, prognostic prediction and indication for follow-up management. Of course, large number of prospectively enrolled patients from multiple centers is still needed to confirm the prognostic utility of BRD4 in HNSCC before its translation into the clinic.
Figure 5
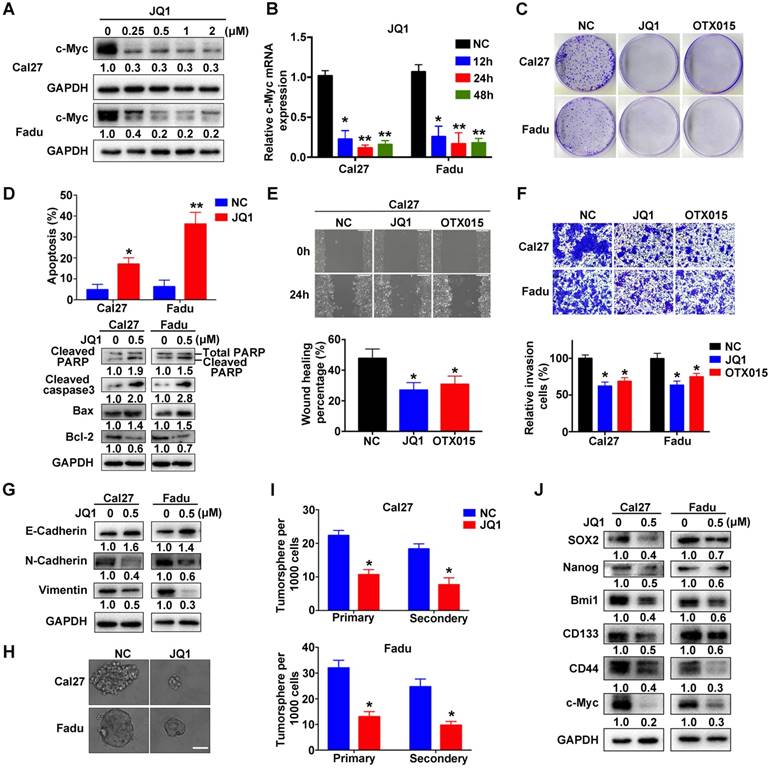
JQ1 treatment results in impaired proliferation, migration and tumorsphere formation and induces cell apoptosis in HNSCC cells. A. Endogenous c-Myc protein was efficiently inhibited by JQ1 in a dosage-dependent manner in Cal27 and Fadu cells for 48h. Representative images of western blot were shown. B. The mRNA levels of c-Myc were significantly decreased following JQ1 treatment (0.5μM) for indicated time. ANOVA, *P < 0.05, **P < 0.01. C. The colony formation potential was inhibited following JQ1/OTX015 treatment (0.5μM) as compared to vehicle treatment. D. Increased percentages of apoptotic cells were evident following JQ1 exposure (0.5μM, 48h) as assayed by Annexin V-PI staining (upper panel). The expression of multiple markers involved in cell apoptosis was measured by western blot (lower panel). Student's t test, *P < 0.05, **P < 0.01. E-F. The migration and invasion abilities were significantly reduced following JQ1/OTX015 treatment (0.5μM) in wound healing and transwell assays, respectively. Student's t test, *P < 0.05, **P < 0.01. G. The abundances of E-cadherin, N-cadherin and Vimentin were measured by western blot following JQ1 treatment. Representative images were shown. H-I. The ability of tumorsphere formation was significantly reduced in JQ1-treated cells relative to cells treated with vehicle. Representative images were shown. Scale bar: 50μm. Student's t test, *P < 0.05, **P < 0.01. J. The abundance of cancer stem cells markers was probed in Cal27 and Fadu cells after JQ1 treatment. Representative images of western blot were shown.
With regard to the mechanisms responsible for aberrant BRD4 overexpression in cancer, several mechanistic models have been proposed at diverse regulatory levels. First, BRD4 amplification (19p13.1) and overexpression was found in 19/33 (57.5%) of epithelial ovarian cancer without BRCA mutations [41]. However, we failed to identify genetic amplification of BRD4 in TCGA-HNSCC dataset as evidenced by the facts that approximately 10% samples harbored genetic alternations and quite few amplification, thus precluding the possibility that genetic amplification of BRD4 was responsible for its overexpression in HNSCC. Second, several tumor suppressive microRNAs including miR-204 and miR-608 have been identified to directly modulate BRD4 mRNA expression by post-transcriptional silencing way in a few cancers [42, 43]. Moreover, recent studies have documented several post- translational modifications like phosphorylation, ubiquitination or deubiquitination induced by CDK9, SPOP or DUB3 as key mechanisms underlying BRD4 upregulation in specific types of cancer [44-46]. However, our initial results from bioinformatics analyses using TCGA-HNSCC datasets failed to support these abovementioned candidates as mediators responsible for BRD4 upregulation in HNSCC with consistent and ample evidence (data not shown). Therefore, the precise cellular and molecular mechanisms underlying BRD4 overexpression in HNSCC remain an open interesting question which is warranted in future in-depth investigations.
Figure 6
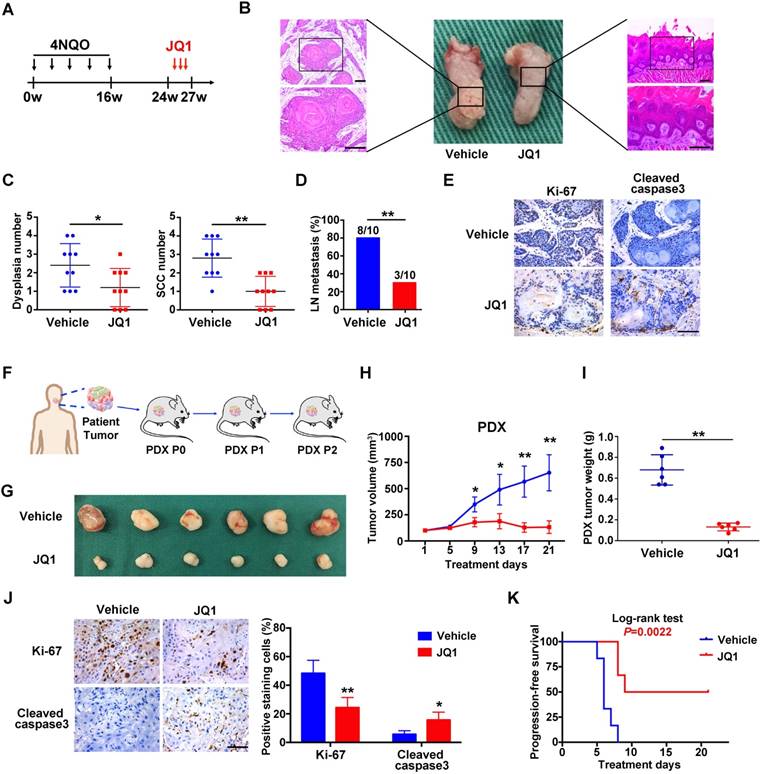
The BRD4 inhibitor JQ1 inhibits growth and metastasis in 4NQO-HNSCC and PDXs models. A. Experimental schedule for therapeutic interference of JQ1 in 4NQO-induced HNSCC model. Mice were randomly divided into two groups (n=10 for each group), and treated by either vehicle or JQ1 (50mg/kg/day) for consecutive 3 weeks. B. Representative image of tongue lesions and H&E staining. Images in the left or right panel were magnified from the black box area in tongue specimen. Scale bar: 50μm C. Quantification of dysplasia and SCC in mice treated with JQ1 and vehicle. Values are mean ± SD from the pool of two independent experiments. Student's t test, *P < 0.05, **P < 0.01. D. The ratio of mice having lymph node metastasis after treatments with JQ1 and vehicle. Student's t test, *P < 0.05, **P < 0.01. E. Representative immunohistochemical staining of Ki-67 and Cleaved caspase 3 in samples from mice treated with vehicle and JQ1. Scale bar: 50μm. F. Schematic description of experimental procedure for HNSCC PDX generation and passaging. Here, the third generation (P2) mice were randomly divided into two group (n=6 per group) when the volume of PDX tumor reached about 100 mm3 and treated with vehicle or JQ1 (50mg/kg/day) for consecutive 3 weeks. G. Image of HNSCC-PDX samples harvested from vehicle- and JQ1-treated animals. H-I. Tumor volume changes of PDX samples were monitored from treatments initiated (H). Final weight of tumor masses harvested from JQ1-treated or control animals was compared (I). Student's t test, *P < 0.05, **P < 0.01. J. Representative immunohistochemical staining of Ki-67, Cleaved caspase 3 in samples from PDX mice treated with vehicle and JQ1 (left panel). Quantification data of positive staining cells were shown (right panel). Student's t test, *P < 0.05, **P < 0.01. Scale bar: 50μm. K. Cumulative incidence plot depicting the percentage of tumors in each treatment group that have doubled in volume as a function of time. Log-rank test.
Figure 7
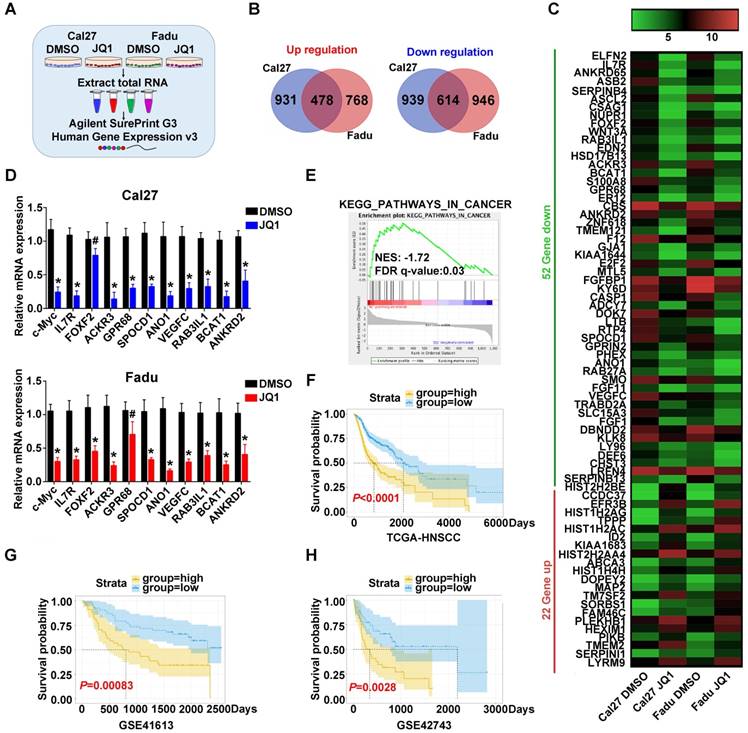
Prognostic risk score generated from JQ1-regulated genes correlates with survival of HNSCC patients. A. The experimental design of genomic transcriptional profiling of HNSCC cells treated with JQ1. Cal27 or Fadu were treated with JQ1 (0.5μM) or vehicle for 24h and RNA were analyzed by Agilent SurePrint G3 Human Expression v3 platform. B. Overlapped genes upregulated (left panel)or downregulated (right panel) in Cal27 and Fadu cells following JQ1 exposure. C. The top upregulated (red) and downregulated (green) genes with more than 5-fold change upon JQ1 treated in Cal27 and Fadu cells were clustered and shown in heatmap. D. The mRNA levels of ten gene candidates were determined by RT-PCR and the c-Myc was selected as a positive control. GAPDH was utilized as loading control. Student's t test, # not significant, *P < 0.05, **P < 0.01. E. GSEA analyses revealed that JQ1-regulated gene in Fadu cells was significantly enriched in KEGG category_Pathway in Cancer. F-H. A JQ-1 regulated gene signature was developed by univariate regression assay using HNSCC dataset and Robust likelihood-based modelling with Rbsurv package and multivariate Cox regression analysis from those overlapped and differentially expressed genes upon JQ1 exposure in Cal27 and Fadu cells (as detailed in Materials and methods part and Figure S16). The Kaplan-Meier analyses of overall survival in patients from TCGA-HNSCC (F) and GEO datasets (GSE41613 (G), GSE42743 (H)) who were stratified by a prognostic risk score derived from JQ1-regulated gene signature (Log-rank test).
Key tumorigenic roles of BRD4 in promoting HNSCC initiation and progression
The clinical significance of BRD4 overexpression and/or hyperactivation in human cancer strongly implies its essential tumorigenic roles. Several lines of evidence have shown that BRD4 is required for cell transformation and cancer maintenance in select contexts. For example, both BRD4-NUT oncogenic in-frame fusion and BRD4 hyperphosphorylation drive neoplastic transformation by specifically activate Sox2 and c-MYC in NUT midline carcinoma [45, 47]. BRD4 was critically required for maintenance of acute myeloid leukemia as evidenced that BRD4 depletion led to robust anti-leukemic in vitro and in vivo, accompanied with cell cycle arrest, apoptosis, terminal myeloid differentiation and eradication of leukemia stem cells [48]. In line with these findings, our results indicated that increased BRD4 expression was observed along with disease progression from hyperplasia to invasive SCC in 4NQO-induced HNSCC animal model. Moreover, significantly impaired tumorigenicity and tumor overgrowth were observed upon BRD4 knockdown in the xenograft model. We reasoned that this might be in part attributed to the regulatory roles of BRD4 in the cancer stem cell, the unique subpopulation responsible for cancer initiation and tumorigenicity in HNSCC, since we found BRD4 enrichments in CSC subpopulation isolated from cell lines and cells cultured as tumorsphere, reduced self-renewal properties upon BRD4 depletion. This is also consistent with previous findings that BRD4 has been increasingly appreciated as a key regulator underlying CSC traits in multiple cancers [17, 49, 50].
A growing body of evidence has revealed that BRD4 facilitates cell proliferation, cell cycle progression, invasion and chemotherapeutic resistance in diverse cancer contexts [21, 45]. BRD4 inhibition by genetic or pharmacologic approaches resulted in marked induced of apoptosis, G0/G1 arrest by transcriptional inhibition at key genes like Bcl2, c-Myc and CDK6 [17, 40]. Additionally, BRD4 physically interacted with diacetylated Twist to form an activated Twist/BRD4/P-TEFb/RNA-Pol II complex at the WNT5A promoter and enhancer and in turn activated its transcription, thus enhancing invasion, CSC-like properties and tumorigenicity of basal-like breast cancer cells [51]. In accordance with these reports, our findings derived from shRNA-mediated BRD4 knockdown and pharmacological inhibition by JQ1 and OTX-015 indicated that impaired cell proliferation and migration/invasion were observed with concomitantly increased cell apoptosis and chemosensitivity in HNSCC cells upon BRD4 depletion. Noticeably, these vitro findings were further substantiated by the in vivo results wherein impaired tumorigenicity, reduced tumor growth and metastasis were observed in xenograft model and chemical-induced model. Complementary, these positive associations between overexpression of BRD4 and tumor size, cervical node metastasis in clinical samples provided additional support for these oncogenic roles of BRD4 in driving HNSCC progression. Taken together, our data offer compelling evidence to establish BRD4 as a bone fide pro-oncogenic mediator facilitating HNSCC initiation and progression.
Several lines of evidence have revealed that BRD4 is preferentially enriched at super-enhancers that drive the expression of key oncogene like c-Myc, Runx1 and Bcl2 and cooperates with lineage-specific transcriptional factors at these sites [52, 53]. Given the well-known oncogenic roles of c-Myc in HNSCC and c-Myc as a well-established downstream target of BRD4 [54, 55], we reasoned that c-Myc might be at least in part account for pro-oncogenic functions of BRD4 in HNSCC and anti-cancer effects induced by BET inhibitors like JQ1. Of course, we believe that other key and unknown mediators beyond c-Myc exist, which warrants further investigations of the accurate downstream targets of BRD4 in HNSCC to better pinpoint its mechanistic insights and develop potent therapeutic strategies.
Pharmacologic inhibition of BRD4 inhibits tumor growth and progression in HNSCC
Pioneering works have established that multiple small-molecule inhibitors targeting BET proteins have promising therapeutic utilities in several preclinical cancer models, highlighting the translational potential of BET targeting in cancer [15, 21, 22, 56]. For example, BRD4 inhibition by JQ1 was more efficacious than direct androgen receptor antagonism to block tumor growth in castration-resistant prostate cancer xenograft model [15]. BRD4 depletion by shRNA significantly impaired tumor growth and lung metastasis in melanoma xenograft models [11]. More importantly, encouraging results from clinical trials about pharmacological BRD4 inhibition by OTX-015 in patients with lymphoma and multiple myeloma have revealed promising anti-cancer effects with acceptable toxic effects [37]. To explore the therapeutic potential of BRD4 targeting in HNSCC, we employed three types of HNSCC preclinical models including xenograft, chemical-induced as well as PDX model. At first, we compared IC50 of JQ1 and OTX015 in HNSCC, HOK and BMSCs and found that HOK and BMSCs were more resistant to these inhibitors as compared to HNSCC cells. These were also supported by following animal experiments wherein these chemicals were well-tolerated in animals. Moreover, our results from these models revealed that significantly reduced tumor growth and cervical node metastasis were observed upon JQ1 administration in vivo. This was collaborated by histopathological findings from chemical-treated samples wherein markedly reduced proliferation and increased apoptosis were detected. Thus, in agreement with effects of BETi in other hematological and solid malignancies, these findings indicated that therapeutic targeting BRD4 in HNSCC was feasible and efficacious. Furthermore, transcriptional profiling of HNSCC upon JQ1 exposure revealed that hundreds of genes were significantly affected, which were enriched in multiple cancer-related pathways. Intriguingly, comparisons of transcriptional profiling data between HNSCC cells and other types of cancer cells following JQ1 treatment revealed relatively small number of overlapped genes which was consistent with previous findings that specific changes predominated in diverse cellular contexts in which BET inhibitors treatment resulted in loss of BRD4 at super-enhancers and preferentially modulated key tissue- or cancer-specific genes, mainly lineage- specific transcriptional factors [19, 52, 53]. Given the well-established effects of BRD4 during transcription, we reasoned that therapeutic targeting of BRD4 by JQ1 was mediated by its profound effects on gene transcription. Recent reports suggested that BRD4 preferentially modulated target gene expression by enriching in their super-enhancer regions and genes regulated by super-enhancers were particularly sensitive to BET inhibition [17, 53]. However, considering the fact that both JQ1 and OTX015 are BET inhibitors but not strictly specific to BRD4 inhibition, we believe that these anti-cancer effects of JQ1 observed in HNSCC are probably dependent on BRD4 as evidenced by consistent results between BRD4 knockdown and JQ1 treatment. Of course, we here can't rule out the possibility that other uncharacterized targets of JQ1 were also involved. Thus, the precise targets responsible for these anti-cancer effects of JQ1 in HNSCC remain an open and interesting question. Furthermore, therapeutic resistance following BRD4 inhibition alone in selected cancer contexts has attracted attentions and several combinational therapeutic strategies like JQ1 and HDACi in preclinical models have shown synthetic effects with more sensitivity and potency [49, 57, 58]. Quite recently, Leonard B and his colleagues have reported that BRD4 targeting potently circumvented toxicities and acquired resistance of receptor tyrosine kinase inhibitor, and combination of cetuximab and JQ1 may be a promising therapeutic strategy for patients with HNSCC [59]. However, optimized therapeutic regime regarding BRD4 inhibition alone or in combination with other drugs has yet to be developed for HNSCC.
Importantly, we developed a novel prognostic risk score derived from JQ1-regulated genes in HNSCC cells by bioinformatics and statistical approaches. This risk score potently stratified patients into subgroups with favorable or inferior survival in three independent publically available HNSCC datasets. Of course, at present, optimal biomarkers, gene signature and risk score are urgently needed to identify patients who might be suitable or benefited from BRD4 inhibition.
Conclusions
Our data reveal that BRD4 serves as a novel pro-tumorigenic factor driving HNSCC progression and a robust biomarker for HNSCC prognostic prediction. Our findings from preclinical animal models highlight the feasibility, potency and values of therapeutic targeting of BRD4 against HNSCC.
Abbreviations
HNSCC: head neck squamous cell carcinoma; BET: bromodomain and extraterminal domain; HOK: Human Oral Keratinocytes; GSEA: gene set enrichment analysis; ROC: receiver operating characteristics; OS: overall survival; DFS: disease-free survival; KEGG: Kyoto Encyclopedia of Genes Genomes; GO: Gene Ontology.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work is financially supported, in whole or in part, by National Natural Science Foundation of China (81572669, 81602386, 81602378), Natural Science Foundation of Jiangsu Province (BK20151561, BK20161564, BK20161024), A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (2018-87), The Natural Scientific Foundation of the Jiangsu Higher Education Institutions of China (16KJB320002), Research grant from Nanjing Medical University and Southeast University (2017DN20) and Project from Nanjing Municipal Committee of Science and Technology (201803044).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7-30
2. Chi AC, Day TA, Neville BW. Oral cavity and oropharyngeal squamous cell carcinoma-an update. CA Cancer J Clin. 2015;65:401-21
3. Chinn SB, Myers JN. Oral Cavity Carcinoma: Current Management, Controversies, and Future Directions. J Clin Oncol. 2015;33:3269-76
4. Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576-82
5. Carrillo JF, Carrillo LC, Cano A, Ramirez-Ortega MC, Chanona JG, Aviles A. et al. Retrospective cohort study of prognostic factors in patients with oral cavity and oropharyngeal squamous cell carcinoma. Head Neck. 2016;38:536-41
6. Popovic R, Licht JD. Emerging epigenetic targets and therapies in cancer medicine. Cancer Discov. 2012;2:405-13
7. Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17:330-9
8. Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491-6
9. Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K. et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19:535-45
10. Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc Natl Acad Sci U S A. 2003;100:8758-63
11. Segura MF, Fontanals-Cirera B, Gaziel-Sovran A, Guijarro MV, Hanniford D, Zhang G. et al. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 2013;73:6264-76
12. Nagarajan S, Hossan T, Alawi M, Najafova Z, Indenbirken D, Bedi U. et al. Bromodomain Protein BRD4 Is Required for Estrogen Receptor-Dependent Enhancer Activation and Gene Transcription. Cell Rep. 2014;8:459-68
13. Andrieu G, Tran AH, Strissel KJ, Denis GV. BRD4 Regulates Breast Cancer Dissemination through Jagged1/Notch1 Signaling. Cancer Res. 2016;76:6555-67
14. Muhar M, Ebert A, Neumann T, Umkehrer C, Jude J, Wieshofer C. et al. SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science. 2018;360:800-5
15. Asangani IA, Dommeti VL, Wang XJ, Malik R, Cieslik M, Yang RD. et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278-82
16. Welti J, Sharp A, Yuan W, Dolling D, Rodrigues DN, Figueiredo I. et al. Targeting Bromodomain and Extra-Terminal (BET) Family Proteins in Castration-Resistant Prostate Cancer (CRPC). Clin Cancer Res. 2018;24:3149-62
17. Yokoyama Y, Zhu HR, Lee JH, Kossenkov AV, Wu SY, Wickramasinghe JM. et al. BET Inhibitors Suppress ALDH Activity by Targeting ALDH1A1 Super-Enhancer in Ovarian Cancer. Cancer Res. 2016;76:6320-30
18. Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13:339-58
19. Stathis A, Bertoni F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov. 2018;8:24-36
20. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI. et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529-33
21. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi JW, Jacobs HM. et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell. 2011;146:903-16
22. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O. et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067-73
23. Li Z, Wang Y, Zhu Y, Yuan C, Wang D, Zhang W. et al. The Hippo transducer TAZ promotes epithelial to mesenchymal transition and cancer stem cell maintenance in oral cancer. Mol Oncol. 2015;9:1091-105
24. Wang Y, Zhu Y, Wang Q, Hu H, Li Z, Wang D. et al. The histone demethylase LSD1 is a novel oncogene and therapeutic target in oral cancer. Cancer Lett. 2016;374:12-21
25. Liu LK, Jiang XY, Zhou XX, Wang DM, Song XL, Jiang HB. Upregulation of vimentin and aberrant expression of E-cadherin/beta-catenin complex in oral squamous cell carcinomas: correlation with the clinicopathological features and patient outcome. Modern Pathol. 2010;23:213-24
26. Li ZW, Wang YL, Qiu J, Li Q, Yuan CP, Zhang W. et al. The polycomb group protein EZH2 is a novel therapeutic target in tongue cancer. Oncotarget. 2013;4:2532-49
27. Vitale-Cross L, Molinolo AA, Martin D, Younis RH, Maruyama T, Patel V. et al. Metformin Prevents the Development of Oral Squamous Cell Carcinomas from Carcinogen-Induced Premalignant Lesions. Cancer Prev Res. 2012;5:562-73
28. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545-50
29. Renaud G, Stenzel U, Maricic T, Wiebe V, Kelso J. deML: robust demultiplexing of Illumina sequences using a likelihood-based approach. Bioinformatics. 2015;31:770-2
30. Kendall WL, Pollock KH, Brownie C. A likelihood-based approach to capture-recapture estimation of demographic parameters under the robust design. Biometrics. 1995;51:293-308
31. Lohavanichbutr P, Mendez E, Holsinger FC, Rue TC, Zhang YZ, Houck J. et al. A 13-Gene Signature Prognostic of HPV-Negative OSCC: Discovery and External Validation. Clin Cancer Res. 2013;19:1197-203
32. Lee M, Tayyari F, Pinnaduwage D, Bayani J, Bartlett JMS, Mulligan AM. et al. Tumoral BRD4 expression in lymph node-negative breast cancer: association with T-bet plus tumor-infiltrating lymphocytes and disease-free survival. BMC Cancer. 2018;18:750
33. Zhu YX, Yang WJ, Ji GN, Lin N, Wu WH, Xiong P. et al. Bromodomain protein 4 is a novel predictor of survival for gastric carcinoma. Oncotarget. 2017;8:31092-100
34. Ginos MA, Page GP, Michalowicz BS, Patel KJ, Volker SE, Pambuccian SE. et al. Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res. 2004;64:55-63
35. Peng CH, Liao CT, Peng SC, Chen YJ, Cheng AJ, Juang JL. et al. A Novel Molecular Signature Identified by Systems Genetics Approach Predicts Prognosis in Oral Squamous Cell Carcinoma. Plos One. 2011;6:e23452
36. Ye H, Yu TW, Temam S, Ziober BL, Wang JG, Schwartz JL. et al. Transcriptomic dissection of tongue squamous cell carcinoma. BMC Genomics. 2008;9:69
37. Amorim S, Stathis A, Gleeson M, Iyengar S, Magarotto V, Leleu X. et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016;3:e196-e204
38. Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA. et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108:16669-74
39. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
40. Wu XC, Liu D, Gao XM, Xie F, Tao D, Xiao XY. et al. Inhibition of BRD4 Suppresses Cell Proliferation and Induces Apoptosis in Renal Cell Carcinoma. Cell Physiol Biochem. 2017;41:1947-56
41. Goundiam O, Gestraud P, Popova T, Rouge TD, Fourchotte V, Gentien D. et al. Histo-genomic stratification reveals the frequent amplification/overexpression of CCNE1 and BRD4 genes in non-BRCAness high grade ovarian carcinoma. Int J Cancer. 2015;137:1890-900
42. Li D, Cui C, Chen J, Hu Z, Wang Y, Hu D. Long noncoding RNA UCA1 promotes papillary thyroid cancer cell proliferation via miR204mediated BRD4 activation. Mol Med Rep. 2018;18:3059-67
43. He L, Meng D, Zhang SH, Zhang Y, Deng Z, Kong LB. microRNA-608 inhibits human hepatocellular carcinoma cell proliferation via targeting the BET family protein BRD4. Biochem Biophys Res Commun. 2018;501:1060-7
44. Jin X, Yan YQ, Wang DJ, Ding DL, Ma T, Ye ZQ. et al. DUB3 Promotes BET Inhibitor Resistance and Cancer Progression by Deubiquitinating BRD4. Mol Cell. 2018;71:592-605
45. Wang RR, Cao XJ, Kulej K, Liu W, Ma TC, MacDonald M. et al. Uncovering BRD4 hyperphosphorylation associated with cellular transformation in NUT midline carcinoma. Proc Natl Acad Sci U S A. 2017;114:E5352-E61
46. Dai X, Gan W, Li X, Wang S, Zhang W, Huang L. et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med. 2017;23:1063-71
47. Wang RR, Liu W, Helfer CM, Bradner JE, Hornick JL, Janicki SM. et al. Activation of SOX2 Expression by BRD4-NUT Oncogenic Fusion Drives Neoplastic Transformation in NUT Midline Carcinoma. Cancer Res. 2014;74:3332-43
48. Zuber J, Shi JW, Wang E, Rappaport AR, Herrmann H, Sison EA. et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524-8
49. Fong CY, Gilan O, Lam EYN, Rubin AF, Ftouni S, Tyler D. et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015;525:538-42
50. Alsarraj J, Walker RC, Webster JD, Geiger TR, Crawford NPS, Simpson RM. et al. Deletion of the Proline-Rich Region of the Murine Metastasis Susceptibility Gene Brd4 Promotes Epithelial-to-Mesenchymal Transition- and Stem Cell-Like Conversion. Cancer Res. 2011;71:3121-31
51. Shi J, Wang YF, Zeng L, Wu YD, Deng J, Zhang Q. et al. Disrupting the Interaction of BRD4 with Diacetylated Twist Suppresses Tumorigenesis in Basal-like Breast Cancer. Cancer Cell. 2014;25:210-25
52. Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J. et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24:777-90
53. Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR. et al. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell. 2013;153:320-34
54. Georgy SR, Cangkrama M, Srivastava S, Partridge D, Auden A, Dworkin S. et al. Identification of a Novel Proto-oncogenic Network in Head and Neck Squamous Cell Carcinoma. J Natl Cancer Inst. 2015;107:djv152
55. Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54:728-36
56. Zhang ZF, Ma PF, Jing Y, Yan Y, Cai MC, Zhang MY. et al. BET Bromodomain Inhibition as a Therapeutic Strategy in Ovarian Cancer by Downregulating FoxM1. Theranostics. 2016;6:219-30
57. Mazur PK, Herner A, Mello SS, Wirth M, Hausmann S, Sanchez-Rivera FJ. et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015;21:1163-71
58. Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM. et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529:413-7
59. Leonard B, Brand TM, O'Keefe RA, Lee ED, Zeng Y, Kemmer JD. et al. BET Inhibition Overcomes Receptor Tyrosine Kinase-Mediated Cetuximab Resistance in HNSCC. Cancer Res. 2018;78:4331-43
Author contact
![]() Corresponding author: Jie Cheng, D.D.S., Ph.D. Associate Professor. Department of Oral and Maxillofacial Surgery, Affiliated Stomatological Hospital, Nanjing Medical University. Tel:86-25-85031880. E-mail: jiecheng_dentaledu.cn.
Corresponding author: Jie Cheng, D.D.S., Ph.D. Associate Professor. Department of Oral and Maxillofacial Surgery, Affiliated Stomatological Hospital, Nanjing Medical University. Tel:86-25-85031880. E-mail: jiecheng_dentaledu.cn.
Citation styles
APA
Wu, Y., Wang, Y., Diao, P., Zhang, W., Li, J., Ge, H., Song, Y., Li, Z., Wang, D., Liu, L., Jiang, H., Cheng, J. (2019). Therapeutic Targeting of BRD4 in Head Neck Squamous Cell Carcinoma. Theranostics, 9(6), 1777-1793. https://doi.org/10.7150/thno.31581.
ACS
Wu, Y.; Wang, Y.; Diao, P.; Zhang, W.; Li, J.; Ge, H.; Song, Y.; Li, Z.; Wang, D.; Liu, L.; Jiang, H.; Cheng, J. Therapeutic Targeting of BRD4 in Head Neck Squamous Cell Carcinoma. Theranostics 2019, 9 (6), 1777-1793. DOI: 10.7150/thno.31581.
NLM
Wu Y, Wang Y, Diao P, Zhang W, Li J, Ge H, Song Y, Li Z, Wang D, Liu L, Jiang H, Cheng J. Therapeutic Targeting of BRD4 in Head Neck Squamous Cell Carcinoma. Theranostics 2019; 9(6):1777-1793. doi:10.7150/thno.31581. https://www.thno.org/v09p1777.htm
CSE
Wu Y, Wang Y, Diao P, Zhang W, Li J, Ge H, Song Y, Li Z, Wang D, Liu L, Jiang H, Cheng J. 2019. Therapeutic Targeting of BRD4 in Head Neck Squamous Cell Carcinoma. Theranostics. 9(6):1777-1793.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.