Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(9):2424-2438. doi:10.7150/thno.30941 This issue Cite
Research Paper
EBV(LMP1)-induced metabolic reprogramming inhibits necroptosis through the hypermethylation of the RIP3 promoter
Feng Shi1,2,3, Min Zhou1,2,3, Li Shang4, Qianqian Du1,2,3, Yueshuo Li1,2,3, Longlong Xie1,2,3, Xiaolan Liu1,2,3, Min Tang1,2,3, Xiangjian Luo1,2,3, Jia Fan5, Jian Zhou5, Qiang Gao5, ShuangJian Qiu5, Weizhong Wu5, Xin Zhang6, Ann M. Bode7, Ya Cao1,2,3,8,9 ![]()
1. Key Laboratory of Carcinogenesis and Invasion, Chinese Ministry of Education, Xiangya Hospital, Central South University, Changsha 410078, China
2. Cancer Research Institute and School of Basic Medical Science, Xiangya School of Medicine, Central South University, Changsha 410078, China
3. Key Laboratory of Carcinogenesis, Chinese Ministry of Health, Changsha 410078, China
4. Department of Pathology, Xiangya Hospital, Central South University, Changsha 410078, China
5. Key Laboratory for Carcinogenesis and Cancer Invasion, Chinese Ministry of Education, Zhongshan Hospital, Shanghai Medical School, Fudan University, Shanghai 200000, China
6. Department of Otolaryngology Head and Neck Surgery, Xiangya Hospital, Central South University, Changsha 410078, China
7. The Hormel Institute, University of Minnesota, Austin, MN 55912, USA
8. Research Center for Technologies of Nucleic Acid-Based Diagnostics and Therapeutics Hunan Province, Changsha 410078, China
9. National Joint Engineering Research Center for Genetic Diagnostics of Infectious Diseases and Cancer, Changsha 410078, China
Received 2018-10-24; Accepted 2019-3-12; Published 2019-4-13
Citation:
Shi F, Zhou M, Shang L, Du Q, Li Y, Xie L, Liu X, Tang M, Luo X, Fan J, Zhou J, Gao Q, Qiu S, Wu W, Zhang X, Bode AM, Cao Y. EBV(LMP1)-induced metabolic reprogramming inhibits necroptosis through the hypermethylation of the RIP3 promoter. Theranostics 2019; 9(9):2424-2438. doi:10.7150/thno.30941. https://www.thno.org/v09p2424.htm
Other stylesAbstract

EBV infection is a recognized epigenetic driver of carcinogenesis. We previously showed that EBV could protect cancer cells from TNF-induced necroptosis. This study aims to explore the epigenetic mechanisms allowing cancer cells with EBV infection to escape from RIP3-dependent necroptosis.
Methods: Data from the TCGA database were used to evaluate the prognostic value of RIP3 promoter methylation and its expression. Western blotting, real-time PCR, and immunochemistry were conducted to investigate the relationship between LMP1 and RIP3 in cell lines and NPC tissues. BSP, MSP and hMeDIP assays were used to examine the methylation level. Induction of necroptosis was detected by cell viability assay, p-MLKL, and Sytox Green staining.
Results: RIP3 promoter hypermethylation is an independent prognostic factor of poorer disease-free and overall survival in HNSCC patients, respectively. RIP3 is down-regulated in NPC (a subtype of HNSCC). EBV(LMP1) suppresses RIP3 expression by hypermethylation of the RIP3 promoter. RIP3 protein expression was inversely correlated with LMP1 expression in NPC tissues. Restoring RIP3 expression in EBV(LMP1)-positive cells inhibits xenograft tumor growth. The accumulation of fumarate and reduction of α-KG in EBV(LMP1)-positive cells led to RIP3 silencing due to the inactivation of TETs. Decreased FH activity caused fumarate accumulation, which might be associated with its acetylation. Incubating cells with fumarate protected NPC cells from TNF-induced necroptosis.
Conclusion: These results demonstrate a pathway by which EBV(LMP1)-associated metabolite changes inhibited necroptosis signaling by DNA methylation, and shed light on the mechanism underlying EBV-related carcinogenesis, which may provide new options for cancer diagnosis and therapy.
Keywords: Epstein-Barr virus, Nasopharyngeal carcinoma, Necroptosis, Receptor-interacting protein 3, Fumarate.
Introduction
Similar to apoptosis, necroptosis is a novel form of programmed cell death that is considered to be a barrier to tumorigenesis. Tumor necrosis factor (TNF)-induced necroptosis is a canonical model that depends on the TNF receptor, receptor- interacting protein 1/3 (RIP1/3), and mixed lineage kinase-domain like protein (MLKL). When the necroptosis signaling activates, RIP1, RIP3 and MLKL are recruited and phosphorylated in the necrosome successively, and phosphorylation of MLKL is involved in executing necroptosis. RIP3 is a key regulator of TNF-induced necroptosis [1, 2]. Lack of RIP3 expression is observed in a majority of cancer cell lines due to DNA hypermethylation of the RIP3 promoter near its transcriptional start site (TSS) [3]. Several viral species, such as the Epstein-Barr virus (EBV), herpes simplex virus (HSV), and cytomegalovirus (CMV), are reported by our and other labs to protect cells from necroptosis [4, 5]. Further research is still needed to shed light on the mechanism by which cancer cells can resist necroptosis induced by virus infection, especially EBV.
EBV is the first characterized oncogenic virus and is implicated in the etiology of nasopharyngeal carcinoma (NPC), a subtype of head and neck squamous cell cancer) [6, 7]. EBV infection is a recognized epigenetic driver of tumorigenesis. Epigenetic alterations, especially aberrant DNA methylation driven by EBV, are significant in the pathogenesis of EBV-related cancers [8]. DNA methylation is the best known epigenetic modification and plays an important role in cancer development. Hypermethylation in the promoter of tumor suppressor genes (TSGs) is a major mechanism of the silencing of TSGs [9]. The balance of DNA methylation- demethylation dynamics is maintained by DNA methyltransferases (DNMTs) and ten-eleven translocation methylcytosine dioxygenases (TETs). Any disequilibrium would result in aberrant DNA methylation. The EBV-encoded latent membrane protein 1 (EBV-LMP1, a well-documented onco-protein of EBV) , is reported to induce DNA methylation by up-regulating DNMT1 expression [10, 11]. Positive LMP1 expression was significantly associated with poorer overall survival in nasopharyngeal carcinoma and non-Hodgkin lymphoma patients [12]. We have previously shown that EBV(LMP1) enhances both DNMT1 expression and its DNA methyltransferase activity [13]. Thus far, few studies have examined the association between EBV(LMP1) and TETs.
Genetic and epigenetic alterations of enzymes in the tricarboxylic acid (TCA) cycle lead to accumulation of oncometabolites, which in turn participate in the pathogenesis and progression of cancer [14]. 2-Hydroxyglutarate (2-HG), succinate, and fumarate are known oncometabolites in the TCA cycle, and their accumulation mainly results from mutations in isocitrate dehydrogenase (IDH), succinate dehydrogenase (SDH), and fumarate hydratase (FH), respectively. With a similar structure to α-ketoglutarate (α-KG), they competitively inhibit α-KG-dependent dioxygenases, including TETs and histone lysine demethylases (KDMs), which subsequently cause abnormal epigenetic modifications through DNA or histone methylation [15]. Our previous study demonstrated that EBV(LMP1) changes the cellular metabolic profile and plays an important part in cancer cell metabolic reprogramming [13, 16, 17]. Therefore, an interesting question is raised as to whether EBV(LMP1)-associated metabolites are involved in epigenetic modifications.
In this study, we will further examine the pathway of necroptosis resistance induced by EBV infection and the connection between EBV-associated oncometabolites and DNA methylation, in order to better understand the mechanism underlying EBV-related carcinogenesis.
Methods
Cell culture
The human NPC cell lines, HK1, HK1-EBV, C666-1, and C666-1 shLMP1, and the immortalized human nasopharyngeal epithelial cell lines, NP460hTERT and NP460hTERT-EBV, have been described previously [13, 18-20]. NPC cell lines were cultured in RPMI-1640 medium (Cat: 11875500, Gibco, Grand Island, NY, USA) supplemented with 10% FBS (Cat: 04-001-1, BI, Kibbutz Beit-Haemek, Israel). NP460hTERT and NP460hTERT-EBV cells were cultured in a 1:1 mixture of Defined Keratinocyte-SFM and EpiLife medium (Cat: 10744019 and MEPI500CA, Gibco). All cells were cultured at 37°C in 5% CO2.
Reagents and antibodies
Human TNFα was purchased from Peprotech (Cat: 300-01A, Rocky Hill, NJ, USA). Dimethyl fumarate (DMF), DMSO, and z-VAD-fmk were obtained from Sigma-Aldrich (Cat: V900731, D2650, and C2105, St. Louis, MO, USA). Octyl-α-ketoglutarate (octyl-α-KG) was from Cayman (Cat: 11970, Ann Arbor, MI, USA). Trichostatin A (TSA) and nicotinamide (NAM) were purchased from MCE (Cat: HY-15144 and HY-B0150, Monmouth Juncton, NJ, USA). Smac mimetic and 5-aza-dC were from Selleck (Cat: S7010 and S1200, Houston, TX, USA). TNF-α (T, 100 ng/ml), Smac mimetic (S, 5 μM), and z-VAD-fmk (Z, 20 μM) were used as necroptosis inducers (T/S/Z).
Antibodies used were mouse anti-acetylation (Cat: 66289-1-Ig, Proteintech, Rosemont, IL, USA), mouse anti-β-actin (Cat: A5441, Sigma), rabbit anti-FH (Cat: 11375-1-AP, Proteintech), rabbit anti-LMP1 (Cat: ab78113, Abcam, Cambridge, MA, USA), rabbit anti-MLKL (Cat: M6697, Sigma), rabbit anti-phosphorylated MLKL (Cat: ab187091, Abcam), rabbit anti-RIP3 (Cat: ab56164, Abcam), mouse anti-TET1 (Cat: 61444, Active Motif, Carlsbad, CA, USA).
Immunohistochemistry
Nasopharyngitis and NPC tissues were collected from the Department of Pathology at Xiangya Hospital, Central South University, Changsha, China. All patients signed informed consent forms for sample collection. The study was approved by the Medical Ethics Committee of Xiangya Hospital, Central South University (No. 201803134). Immunohistochemical analysis was conducted as described previously [21].
Tumorigenicity assay
The study was approved by the Medical Ethics Committee (for experimental animals) of Xiangya Hospital, Central South University (No. 201803135). The analysis was performed as described previously [22]. 3×106 cells per animal were injected subcutaneously into the flank regions of the 6-week-old female BALB/c-nu mice. The tumors were measured twice a week. The tumor volume was calculated by the following formula: V = 1/6π×length×width2. The mice were sacrificed after five weeks, and the weight of xenograft tumors was obtained at the same time.
Western blotting
Western blot analysis was conducted as previously described [13]. Because of the rapid degradation of the LMP1 protein, cells were incubated with 10 mM MG132 (Cat: HY-13259, MCE) for 6 h before harvest to accumulate LMP1 for detection [23].
RNA isolation and real-time PCR
Total mRNA was isolated using the NucleoZOL reagent (Cat: 740404, MACHEREY- NAGEL GmbH & Co. KG, Düren, Germany). The RevertAid First Strand cDNA Synthesis Kit (Cat: K1622, Invitrogen, Carlsbad, CA, USA) was used for reverse transcription. Real-time PCR analysis was performed in triplicate using the SYBR™ Green Master Mix (Cat: A25742, Invitrogen) on the ABI7500 Real-Time System (Applied Biosystems).
Cell viability assay
Cells were seeded on 96-well plates in replicates of three. A CellTiter 96® AQueous One Solution Cell Proliferation Assay (Cat: G5430, Promega, Madison, WI, USA) was used to evaluate cell viability. Absorbance was measured at a wavelength of 570 nm using the BioTek Elx800 microplate reader.
Cell permeability assay
The cell permeability assay was performed using Sytox Green (Cat: S34860, Invitrogen). Cells were incubated with 30 nM Sytox Green for 20 min in the dark at room temperature, and then visualized with the Leica DMI3000 fluorescence microscope.
DNA methylation
The genomic sequence near the transcription start site of RIP3 was retrieved from UCSC Genome Browser (GRCh37/hg19). The CpG island in the RIP3 promoter was predicted by MethPrimer [24]. Methylation-specific PCR (MSP) primers for RIP3 designed by MethPrimer were as follows: MF: 5'-GATTGTAGTGAGAACGTCGAG-3'; MR: 5'-AAATATCGCCCACTAACCGA-3'; UF: 5'-GGATTGTAGTGAGAATGTTGAGT-3'; UR: 5'-AAAAATATCACCCACTAACCAACC-3'. MSP analysis was conducted as previously reported [13].
Bisulfite-sequencing PCR (BSP) analysis was performed by TsingKe Biological Technology (Beijing, China). Briefly, 2 μg of genomic DNA was modified by the bisulfate reaction. Bisulfate-treated DNA was amplified with the following primers: F: 5'-TTATGGTGAGTAGGGAGTGGTATG-3' and R: 5'- CATCRTAACCCCACTTCCTATATTAC -3'. The PCR product was cloned into pUC18, and clones were randomly picked for sequencing. The methylation level of each site is shown as the mean percentage of the total methylation according to sequencing data obtained from 8-10 clones.
DNMT, TET, and FH activity measurement
Nuclear proteins were extracted using nuclear and cytoplasmic extraction reagents (Cat: 78835, Thermo Scientific, Waltham, MA, USA). DNMT and TET activities from nuclear extracts were quantified using the EpiQuik™ DNMT activity assay kit and Epigenase™ 5-mC hydroxylase TET activity assay kit (Cat: P3009 and P3086, EpigenTek, Farmingdale, NY, USA), respectively. FH activity was measured by a fumarase activity colorimetric assay kit (Cat: K596, Biovision, Milpitas, CA, USA).
Hydroxymethylated DNA immunoprecipitation (hMeDIP) assay
The hMeDIP assay was performed using the EpiQuik™ Hydroxymethylated DNA immunoprecipitation assay kit (Cat: P1038, EpigenTek).
Fumarate, α-KG, succinate, and 2-HG assays
α-KG and 2-HG were measured using the alpha-ketoglutarate colorimetric fluorometric assay kit and D-2-hydroxyglutarate colorimetric assay kit (Cat: K677 and K213, Biovision), respectively. Fumarate and succinate were measured using the respective fumarate assay kit and succinate colorimetric assay kits (Cat: MAK060 and MAK184, Sigma).
Database analysis
The cBioPortal for the Cancer Genomics Database can be acquired online (http://www.cbioportal.org/index.do). The TCGA dataset (N = 530) was used to analyze the prognostic value of RIP3 in head and neck squamous cell carcinoma. The parameters provided in the database for survival analysis were as follows: sex, age, overall survival months, disease-free survival months, survival status, histology grade, pathologic TNM stage, clinical stage, gene expression, and methylation status.
Statistical analysis
All statistical analyses were performed using SPSS 17.0 software. The experimental results were statistically evaluated using the Student t test, the Pearson chi-square test, the ANOVA test, COX regression analysis, and Kaplan-Meier analysis. A value of p < 0.05 was considered statistically significant.
Results
RIP3 promoter hypermethylation indicates a poor prognosis for head and neck squamous cell cancer (HNSCC) patients
Kaplan-Meier curves showed that patients with RIP3 mRNA negative-expression had a shorter disease-free survival (N=392; median survival time: 53.1 months vs. 71.2 months) and overall survival (N=518; median survival time: 48.2 months vs 60.4 months) compared to those with positive expression in HNSCC patients from the TCGA database (Figure 1A). Hypermethylation of the RIP3 promoter is considered to cause RIP3-negative expression [3]. We analyzed the relationship between the RIP3 promoter methylation and mRNA expression, and a significantly negative correlation was observed between the two (Figure S1). The survival analysis revealed that patients with RIP3 promoter methylation had a shorter disease-free survival (N=392; median survival time: 50.0 months vs. a cumulative probability of survival > 50%) and overall survival (N=518; median survival time: 48.2 months vs. 71.2 months) compared to those with no methylation (Figure 1B). The combination of RIP3 promoter methylation and RIP3 mRNA expression could stratify patients more accurately (Figure 1C). No significant relationship was found between RIP3 promoter methylation and clinico-pathological parameters (Table S1).
Univariate Cox proportional hazards regression analysis showed that advanced T stage, lymph node metastasis, advanced clinical stage, and RIP3 promoter methylation were significantly correlated with poorer overall survival (Table 1). Multiple Cox proportional hazards regression analysis indicated that RIP3 promoter methylation was an independent prognostic factor for HNSCC patients (Table 1). In addition, the multivariate Cox regression analysis revealed that lymph node metastasis and RIP3 promoter methylation were independent prognostic factors of poorer disease-free survival for HNSCC patients (Table 2).
Table 1
Univariate and multivariate COX regression analysis of disease-free survival of HNSCC patients
| Variable | Univariate analysis | Multivariate analysis | |||||
|---|---|---|---|---|---|---|---|
| HR | 95% CI | P value | HR | 95% CI | P value | ||
| Age (≥61 vs <61) | 1.088 | 0.785-1.507 | 0.613 | ||||
| Sex (Male vs Female) | 1.461 | 0.976-2.186 | 0.0656 | ||||
| Grade (G3/G4 vs G1/G2) | 0.722 | 0.478-1.089 | 0.120 | ||||
| pT (T3/T4 vs T1/T2) | 1.511 | 1.046-2.183 | 0.0279* | 1.351 | 0.851-2.144 | 0.202 | |
| pN (N1/N2/N3 vs N0) | 1.668 | 1.149-2.421 | 0.00717* | 1.488 | 0.996-2.222 | 0.0523* | |
| Stage (IV vs I/II/III) | 1.531 | 1.077-2.176 | 0.0177* | 1.294 | 0.828-2.022 | 0.258 | |
| RIP3 promoter (M vs U) | 1.497 | 1.072-2.092 | 0.0179* | 1.566 | 1.075-2.282 | 0.0193* | |
HR, hazard ratio; CI, confidence interval; G1, well differentiated; G2, moderately differentiated; G3, poorly differentiated; G4, Undifferentiated; pT, pathologic T stage; pN, lymph node metastases; M, methylated; U, unmethylated; *, significant difference.
Table 2
Univariate and multivariate COX regression analysis of overall survival of HNSCC patients
| Variable | Univariate analysis | Multivariate analysis | |||||
|---|---|---|---|---|---|---|---|
| HR | 95% CI | P value | HR | 95% CI | P value | ||
| Age (≥61 vs <61) | 1.259 | 0.964-1.644 | 0.0914 | ||||
| Sex (Male vs Female) | 1.156 | 0.851-1.570 | 0.353 | ||||
| Grade (G3/G4 vs G1/G2) | 0.775 | 0.555-1.083 | 0.136 | ||||
| pT (T3/T4 vs T1/T2) | 1.252 | 0.942-1.666 | 0.122 | ||||
| pN (N1/N2/N3 vs N0) | 1.789 | 1.297-2.468 | 3.96×10-4* | 1.882 | 1.355-2.612 | 1.59×10-4* | |
| Stage (IV vs I/II/III) | 1.103 | 0.842-1.445 | 0.475 | ||||
| RIP3 promoter (M vs U) | 1.354 | 1.035-1.772 | 0.0271* | 1.384 | 1.018-1.882 | 0.0381* | |
HR, hazard ratio; CI, confidence interval; G1, well differentiated; G2, moderately differentiated; G3, poorly differentiated; G4, Undifferentiated; pT, pathologic T stage; pN, lymph node metastases; M, methylated; U, unmethylated; *, significant difference.
EBV(LMP1) is responsible for RIP3 down-expression in NPC
NPC is a subtype of HNSCC. From the Oncomine database, we found that RIP3 mRNA is significantly down-regulated in NPC tissues (p < 0.001, Figure S2). Lack of RIP3 protein expression was detected in NPC tissues, whereas high expression was present in nasopharyngitis tissues (Figure 2A).
Figure 1
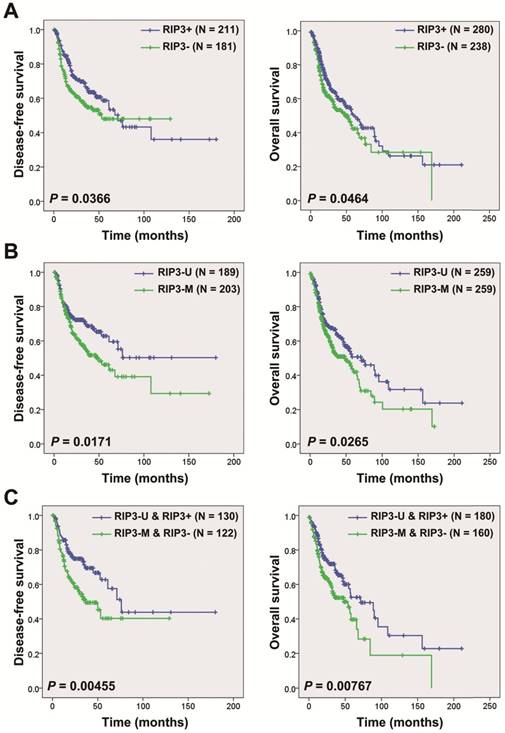
Kaplan-Meier analysis according to RIP3 promoter methylation and mRNA expression status in HNSCC patients. A, Disease-free survival (left) and overall survival (right) analysis according to RIP3 mRNA expression. HNSCC patients were divided into two groups: good prognosis (positive expression of RIP3 mRNA) and poor prognosis (negative expression of RIP3 mRNA; “-”, negative; “+”, positive). B, Disease-free survival (left) and overall survival (right) analysis according to RIP3 promoter methylation. HNSCC patients were divided into two groups: good prognosis (unmethylated RIP3 promoter) and poor prognosis (methylated RIP3 promoter). U, unmethylated; M, methylated. C, Disease-free survival (left) and overall survival (right) analysis according to the combination of RIP3 promoter methylation and mRNA expression. The combination could stratify HNSCC patients more accurately than just one factor.
EBV latent infection plays a crucial role in NPC, thus we determined whether EBV is involved in RIP3 down-regulation. RIP3 expression was examined in EBV- uninfected/infected cells cell lines. Results indicated that RIP3 mRNA and protein expression were significantly reduced in the EBV-infected immortalized nasopharyngeal epithelial cell line, NP460hTERT-EBV, and the nasopharyngeal carcinoma cell line, HK1-EBV (Figure 2B-C). LMP1 is a well-documented onco-protein in the EBV latent infection stage and can be detected in NP460hTERT-EBV and HK1-EBV (Figure 2B and S3) cells. Then we examined whether LMP1 is involved in EBV-mediated RIP3 down-regulation. C666-1 cells consistently harbor EBV and express LMP1, while C666-1 cells stably transfected with LMP1-shRNA have been established as described previously [13, 23]. The level of RIP3 mRNA and protein expression in C666-1 shLMP1 cells was higher than that in C666-1 cells (Figure 2B-C and S3). Ectopic expression of LMP1 in HK1 cells decreased the expression of RIP3 (Figure 2B-C and S3). This suggested that EBV down-regulated RIP3 mRNA and protein expression through LMP1 and the regulation was at the transcriptional level.
Next the relationship of LMP1 and RIP3 was analyzed in NPC tissues. The expression was detected by IHC with consecutive sections from the same NPC tissues. Representative photos are shown in Figure 2D. Among the 42 NPC specimens, the percentage of LMP1- and RIP3-positive staining was 64.3% (27/42) and 45.2% (19/42), respectively (Figure 2E). A significantly negative correlation by the Pearson χ2 test was observed between the two proteins (p < 0.05, Table 3).
Table 3
RIP3 expression negatively correlates with LMP1 expression in NPC
| RIP3 | LMP1 | Total | |
|---|---|---|---|
| Negative | Positive | ||
| Negative | 5 | 18 | 23 |
| Positive | 10 | 9 | 19 |
| Total | 15 | 27 | 42 |
Pearson Chi-Square Test, P = 0.038
Figure 2
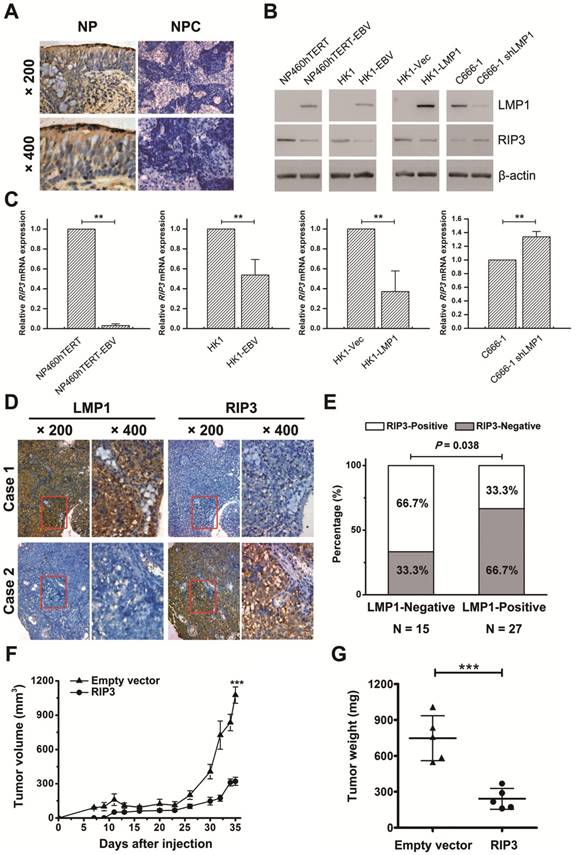
RIP3 expression is negatively associated with EBV(LMP1), and restoring RIP3 expression inhibits EBV(LMP1)-mediated tumorigenesis. A, Representative IHC photos for the expression of RIP3. Compared with NP tissues, RIP3 is expressed at low levels in NPC tissues. NP: nasopharyngitis; NPC: nasopharyngeal carcinoma. B, RIP3 protein expression is down-regulated in EBV(LMP1)-positive cells detected by Western blot analysis. EBV- uninfected/infected cells: NP460hTERT/ NP460hTERT-EBV (immortalized human nasopharyngeal epithelial cell), HK1/ HK1-EBV (differentiated NPC cells), C666-1 (undifferentiated NPC cells harboring EBV). LMP1-associated cells: HK1-Vec/ HK1-LMP1 (HK1 cells were transfected with LMP1- overexpression vector), C666-1/ C666-1 shLMP1 (C666-1 cells stably transfected with shLMP1). C, RIP3 mRNA expression is down-regulated in EBV(LMP1)-positive cells detected by RT-PCR (columns = mean; bars = S.D.; n = 3; **, p < 0.01). D, Representative IHC photos for the expression of LMP1 and RIP3 in consecutive sections of NPC tissues. E, RIP3 expression is calculated according to LMP1 expression in NPC tissues. F, Tumor growth curves of C666-1 xenografts in groups indicated. Data are expressed as mean values ± S.D. (n = 5; ***p < 0.001). G, Tumor weight was measured at the end of the experiment. Data are expressed as mean values ± S.D. (n = 5; ***p < 0.001).
Restoring RIP3 expression in EBV(LMP1)-positive cells inhibits xenograft tumor growth in nude mice
To investigate the effect of RIP3 on EBV(LMP1)-mediated tumorigenesis in vivo, we performed a xenograft tumor formation assay. C666-1 stably transfected RIP3 (C666-RIP3) cells or empty vector-transfected cells (C666-EV) were injected subcutaneously into nude mice. The average volume and weight of xenograft tumors in mice injected with C666-RIP3 cells were significantly lower compared with C666-EV cells (Figure 2F-G and S4A). LMP1 and RIP3 expression in xenograft tumors was examined by IHC (Figure S4B).
RIP3 is silenced by EBV(LMP1) due to hypermethylation of its promoter
We considered whether DNA hypermethylation was responsible for EBV(LMP1)-mediated RIP3 down-regulation. BSP analysis showed heavily methylated alleles in EBV(LMP1)-positive cell lines (NP460hTERT-EBV and HK1-EBV), but was barely detectable in EBV(LMP1)-negative cell lines (Figure 3A-B). RIP3 promoter methylation was also examined by MSP analysis. DNA hypermethylation occurred in the RIP3 promoter near its TSS in EBV-infected cell lines (NP460hTERT-EBV, HK1-EBV, and C666-1) and LMP1-positive cells (HK1-LMP1), while the methlylation level was reduced in LMP1-knockdown cells (C666-1 shLMP1, Figure 3C).
Then we investigated whether RIP3 promoter hypermethylation contributed to its low-expression. A hypomethylating agent, 5-aza-dC, was used to treat EBV-infected cell lines. The methylation level of the RIP3 promoter and RIP3 mRNA expression were responsive to 5-aza-dC (Figure 3D and S5). Moreover, the RIP3 protein was re-expressed in a time-dependent manner with 5-aza-dC treatment in EBV-infected cell lines (Figure 3E). The 5-aza-dC treatment also restored RIP3 expression in LMP1-positive cells (Figure 3F). These results indicate that EBV(LMP1) induces RIP3 promoter hypermethylation, which in turn results in the silencing of RIP3.
TETs are involved in EBV(LMP1)-mediated RIP3 promoter hypermethylation
The dynamic balance of DNA methylation is maintained by methyltransferases (DNMTs) and demethylases (TETs). We previously showed that LMP1 promoted both DNMT1 expression and its enzymatic activity [13]. In this study DNMT1 was altered in the same direction in C666-1 cells, but not in NP460hTERT/NP460hTERT-EBV and HK1/HK1-EBV cells (Figure S6). Next we examined TETs expression and enzymatic activity. The enzymatic activity of TETs was impaired in EBV(LMP1)-positive cell lines (NP460hTERT-EBV, HK1-EBV, and C666-1), and was restored in LMP1-knockdown cells (C666-1 shLMP1) (Figure 4A), while TETs expression was not significantly changed (Figure S7). TET enzymes catalyze the oxidation of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC), which is the first step for active DNA demethylation. Then we used the hMeDIP assay to detect the 5-hmC level in the RIP3 promoter. As shown in Figure 4B, RIP3 promoter was poorly hydroxymethylated in EBV(LMP1)-positive cell lines, whereas high levels of 5-hmC were observed in LMP1-knockdown cells. These findings suggest that the reduction of TETs enzymatic activity probably results in RIP3 promoter hypermethylation.
EBV(LMP1) promotes the accumulation of fumarate and reduction of α-KG
As members of the α-KG-dependent dioxygenase family, TET enzymes require α-KG as a co-substrate to catalyze the conversion of 5-mC to 5-hmC. A few intermediates in the TCA cycle, such as fumarate, succinate, and 2-HG, can competitively inhibit the enzymatic activity of TETs [25]. We previously investigated EBV(LMP1)-modulated metabolic changes using a metabolomic approach [17]. Fumarate, succinate, 2-HG, and α-KG levels were confirmed in this study. No significant changes of succinate concentration were found between EBV(LMP1)-positive and -negative cell lines (Figure S8A). Levels of 2-HG changed less than one fold (significantly) in EBV(LMP1)-positive cell lines (Figure S8B). Fumarate levels were appreciably increased in EBV(LMP1)-positive cell lines, and knockdown of LMP1 lessened the level (Figure 4C). Moreover, the concentration of α-KG decreased in EBV(LMP1)-positive cell lines (Figure 4D). This suggested that EBV(LMP1) significantly promotes the accumulation of fumarate and reduction of α-KG.
EBV(LMP1)-associated metabolite changes regulate TET activity and RIP3 expression
We determined whether fumarate accumulation could affect RIP3 expression. Incubating NP460hTERT or HK1 cells with DMF (a cell-permeable derivative of fumarate) decreased the protein expression of RIP3 in a time-dependent manner (Figure 5A). Promoter methylation and mRNA expression levels of RIP3 were also depressed with DMF treatment (Figure 5B-D). Fumatate is a competitive inhibitor of TETs, so 5mC-hydroxylase activity was examined. As shown in Figure 5C-D, DMF treatment inhibited TET enzymatic activity and the 5-hmC levels in the RIP3 promoter were also lower in DMF-treated cells (Figure 5C-D).
Figure 3
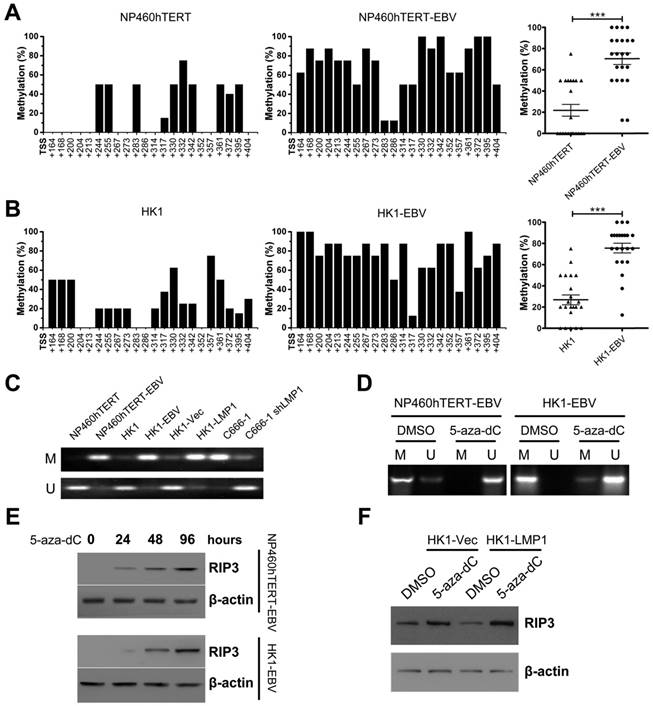
RIP3 is silenced by methylation in EBV(LMP1)-positive cell lines. A, Methylation levels of CpG sites in the RIP3 promoter near its TSS (+164 to +404) in NP460hTERT-EBV cells are higher than those in NP460hTERT cells as detected by BSP (left and middle). The results are statistically significant (right). Dot, the mean methylation level of a single CpG island (***, p < 0.001). B, Methylation levels of RIP3 promoter are significantly higher in HK1-EBV cells compared to HK1 cells (the same as above). C, Promoter methylation of RIP3 in EBV(LMP1)-positive/negative cell lines validated by MSP (M, methylated; U, unmethylated.). D, With 5-aza-dC (10 μM, 4 days) treatment, methylation level of RIP3 promoter is down-regulated as detected by MSP (M, methylated; U, unmethylated). E, RIP3 protein is restored in a time-dependent manner in EBV-infected cells treated with 5-aza-dC (10 μM) as detected by Western blotting. F, RIP3 protein is restored in LMP1-positive cells treated with 5-aza-dC (10 μM, 4 days) as detected by Western blotting.
Next, we determined whether raising the α-KG concentration could restore EBV(LMP1)-induced RIP3 silencing. Cell-permeable octyl-α-KG was used a supplement of α-KG [26]. With octyl-α-KG treatment, RIP3 expression, TET activity, and 5-hmC levels in the RIP3 promoter were increased in EBV(LMP1)-positive cells (Figure 5E-F).
These observations indicate that EBV(LMP1)-associated fumarate accumulation and α-KG reduction cause RIP3 silencing due to TET activity-dependent hypermethylation.
EBV(LMP1) suppresses FH activity
Loss-of-function mutation of FH results in accumulation of its substrate fumarate [14]. As shown in Figure 6A, FH activity decreased in EBV(LMP1)-positive cell lines, and increased in LMP1-knockdown cells. However, no mutations were found in 56 NPC tissues by searching the cBioPortal for Cancer Genomics Database for the mutation status of FH and other frequently mutated genes involved in the TCA cycle (Figure S9). Western blot analysis revealed no significant alteration in the FH protein expression (Figure 6B).This demonstrated that the reduction of FH activity is responsible for fumarate accumulation, instead of FH mutations or its abnormal expression.
Most of the metabolic enzymes in the TCA cycle, including FH, are acetylated, and their enzyme activity or protein stability relies on lysine acetylation [27, 28]. We determined whether the acetylation of FH was affected by EBV(LMP1). Figure 6B shows that acetylation of endogenous FH decreased in EBV(LMP1)-positive cell lines and increased in LMP1-knockdown cells. To determine the effect of acetylation on FH enzymatic activity, we treated 460hTERT-EBV/ HK1-EBV cells with TSA (a class I, II, IV HDAC inhibitor) and NAM (a NAD+-dependent class III SIRT inhibitor), respectively. TSA treatment increased endogenous FH acetylation and activity (Figure 6C-D). This suggested that acetylation may play a role in EBV(LMP1)-mediated reduction of FH activity.
Figure 4
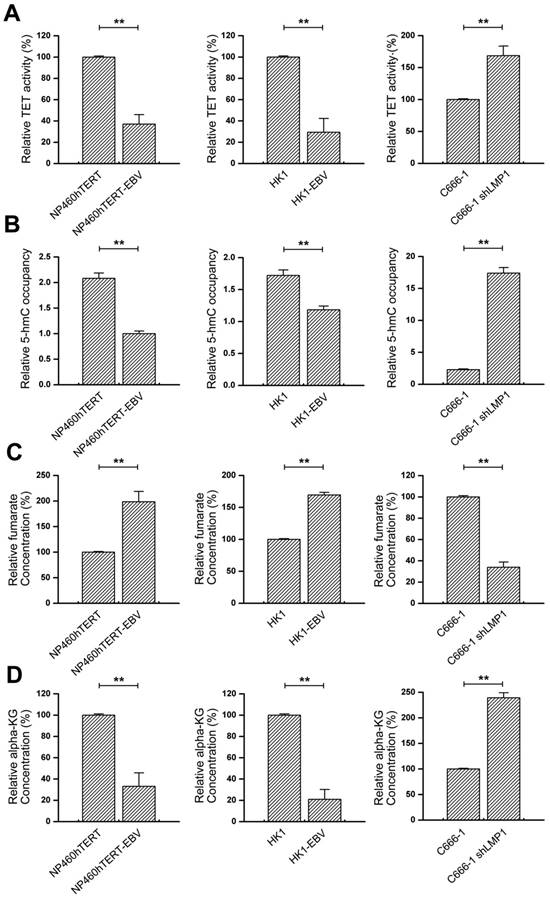
EBV(LMP1) regulates TET enzymatic activity and metabolic changes. A, TET 5mC-hydroxylase activity is down-regulated in EBV-infected cells (left and middle). Knockdown of LMP1 expression in C666-1 restored TET activity (right; columns = mean; bars = S.D.; n = 3; **, p < 0.01). B, Hydroxymethylated DNA immunoprecipitation (hMeDIP) assays to determine 5-hmC levels in the RIP3 promoter. The 5-hmC level is reduced in EBV-infected cells (left and middle) and knockdown of LMP1 expression in C666-1 elevates the 5-hmC level (right; columns = mean; bars = S.D.; n = 3; **, p < 0.01). C, Fumarate accumulates in EBV-infected cells (left and middle). The level of fumarate is decreased in LMP1-knockdown cells (right; columns = mean; bars = S.D.; n = 3; **, p < 0.01). D, The level of α-KG is decreased in EBV-infected cells (left and middle) and the level is increased in LMP1-knockdown cells (right; columns = mean; bars = S.D.; n = 3; **, p < 0.01).
Figure 5
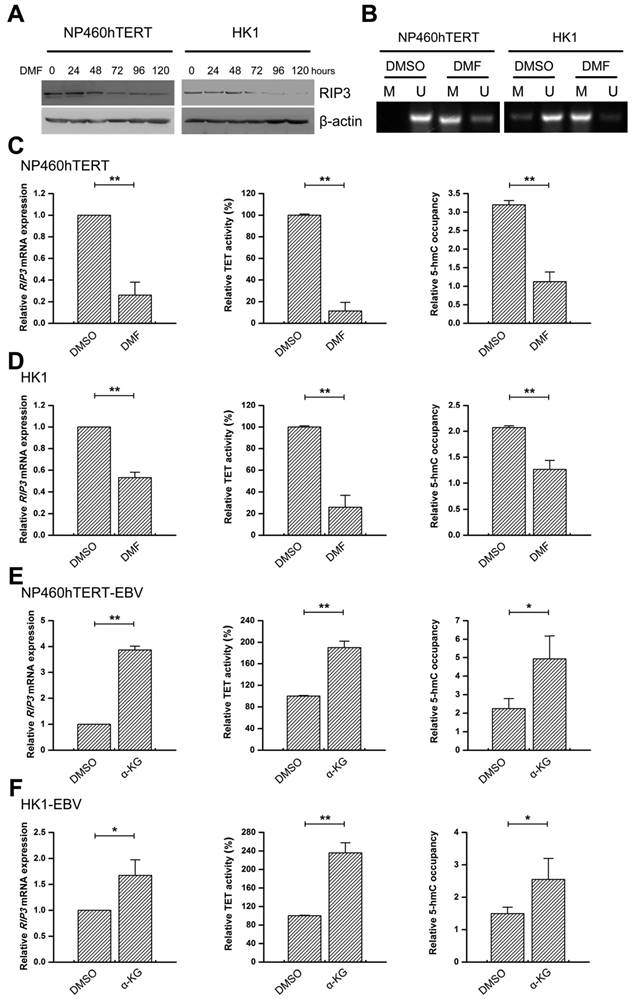
EBV(LMP1)-associated metabolite changes regulate TET activity and RIP3 expression. A, Western blot analysis of RIP3 expression in NP460hTERT (left) and HK1 (right) cells treated with DMF (50 μM) for the indicated times. Treatment wtih DMF decreases the protein expression of RIP3 in a time-dependent manner. B, DMF (50 μM, 4 days) treatment increases methylation levels of the RIP3 promoter as detected by MSP (M, methylated; U, unmethylated). C-D, NP460hTERT (C) and HK1 (D) cells were treated with DMSO or DMF (50 μM, 4 days). The mRNA expression of RIP3 (left), TET 5mC-hydroxylase activity levels (middle), and 5-hmC levels of RIP3 promoter (right) are decreased in the DMF-treated group (columns = mean; bars = S.D.; n = 3; **, p < 0.01). E-F, NP460hTERT-EBV (E) and HK1-EBV (F) cells were treated with DMSO or octyl-α-KG (1 mM, 24 hours). The mRNA expression of RIP3 (left), TET 5mC-hydroxylase activity levels (middle), and 5-hmC levels of RIP3 promoter (right) are increased in the α-KG -treated group (columns = means; bars = S.D.; n = 3; *, p < 0.05; **, p < 0.01).
Figure 6
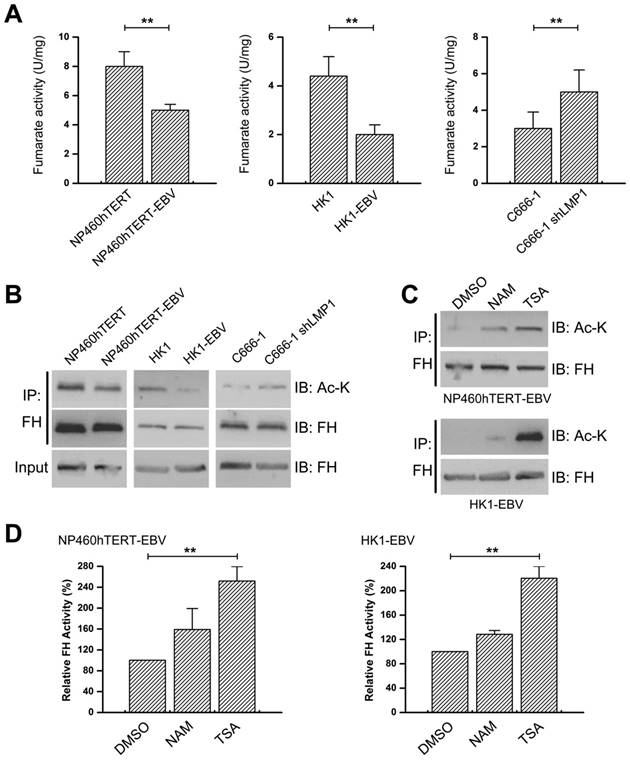
EBV(LMP1) inhibits FH activity. A, FH activity declines in EBV-infected cells (left and middle) and the activity is increased in LMP1-knockdown cells (right; columns = mean; bars = S.D.; n = 3; **, p < 0.01). B, Endogenous FH was immunoprecipitated (IP) and probed with FH and acetyl-Lys (Ac-K) antibodies. The whole cell extract was used as input. C-D, Cells were treated with 5 μM TSA or 5 mM NAM for 16 hours. The endogenous FH was immunoprecipitated and analyzed by Western blotting (C). FH activity were also detected (D; columns = means; bars = S.D.; n = 3; **, p < 0.01).
Fumarate inhibits TNF-induced necroptosis
RIP3 is a key regulator of TNF-induced necroptosis [1, 2]. We next investigated whether fumarate accumulation might play a role in TNF-induced necroptosis. Cell viability assay results showed that the necroptosis inducer (T/S/Z) led to massive cell death and DMF pre-treatment restored cell viability (Figure 7A). MLKL is a key RIP3 downstream component and its phosphorylation is indispensable for TNF-induced necroptosis [29]. Western blot analysis revealed that T/S/Z treatment induced the phosphorylation of MLKL, while the phosphorylation was hardly observed in the DMF pre-treated group (Figure 7B). The rupture of the plasma membrane is a remarkable feature of necroptosis [30], and therefore we examined cell permeability using Sytox Green staining. Our results showed that T/S/Z treatment increased the Sytox Green fluorescence signal, while the staining was suppressed by DMF pre-treatment (Figure 7C). Overall, changes in EBV(LMP1)-associated metabolites inhibit TNF-induced necroptosis (Figure 8).
Figure 7
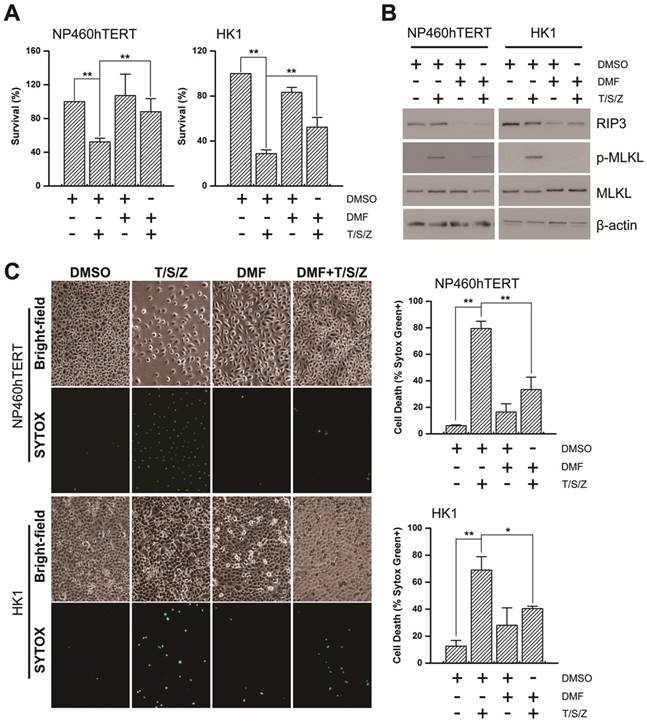
Fumarate inhibits T/S/Z-induced necroptosis. NP460hTERT and HK1 cells were untreated or pretreated with DMF (50 μM, 4 days) followed by treatment with TNF-α (T, 100ng/ml)/ Smac mimetic (S, 5 μM)/ z-VAD-fmk (Z, 20 μM) for 24 (NP460hTERT) or 48 h (HK1). A, Cell viability was determined by MTS (columns = means; bars = S.D.; n = 3; **, p < 0.01. B, Western blot analysis of RIP3, p-MLKL and MLKL expression. C, The integrity of cellular membrane was determined by Sytox Green fluorescence staining (left). The results are statistically significant (right; columns = means; bars = S.D.; n = 3; *, p < 0.05; **, p < 0.01).
Figure 8
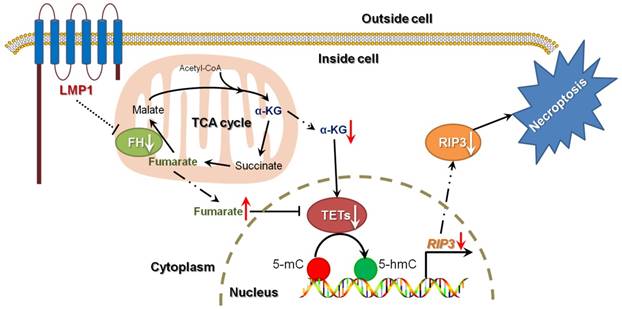
A schematic for EBV-induced epigenetic reprogramming of RIP3. The metabolic reprogramming caused by EBV(LMP1) expression inhibits necroptosis signaling through the hypermethylation of the RIP3 promoter, which sheds light on the mechanism underlying EBV-related carcinogenesis.
Discussion
Necroptosis is considered to be a defense against viral infection. Several viruses can escape from necroptosis by encoding inhibitors, such as vIRA/M45 (MCMV), ICP6 (HSV-1), and ICP10 (HSV-2) [4]. We have shown that EBV and EBV-encoded LMP1 could inhibit necroptosis through post-translational modification of RIP1/3 [5, 31]. In this study, we revealed a novel mechanism of necroptosis resistance by silencing RIP3 expression in a methylation-dependent manner in cells with EBV infection (Figure 8).
RIP3 is a critical regulator of necroptosis, which is considered to be a TSG. Lack of RIP3 expression could reduce the sensitivity of cells to necroptosis stimuli, and then promote cell viability and tumorigenesis. RIP3 is reported to be down-regulated in colorectal cancer, breast cancer, and esophageal squamous cell carcinoma, and the low expression of RIP3 is associated with a poor prognosis [3, 32, 33]. In this study, we showed that RIP3 s expressed at low levels in NPC tissues. Not only negative RIP3 protein expression, but RIP3 promoter methylation was correlated with a shorter disease-free and overall survival in HNSCC. Furthermore, RIP3 promoter hypermethylation was an independent prognostic factor for poor clinical outcome. Therefore, RIP3 promoter methylation seems to be a valuable factor in cancer diagnosis and treatment.
DNA methylation is considered to be a reversible progression with the balance maintained by DNMTs and TETs. DNMT1 is the most abundant DNMT in mammalian cells and it maintains methylation status during cell replication [34]. Over-expression of DNMT1 disrupts the DNA methylation-demethylation balance, which results in promoter hypermethylation and silencing of the expression of many TSGs [35]. Lack of RIP3 expression has been observed in a variety of cancer cell lines and breast cancer tissues, mainly because of DNMT1-dependent hypermethylation [3, 36, 37]. Unlike DNMTs and DNA methylation, TETs and DNA demethylation have not been well studied. As another regulator of the balance, TETs (including TET1, TET2, and TET3) oxidize 5-mC to 5-hmC and other oxidation products, thereby mediating active DNA demethylation. Decreased expression/ or activity of TETs and low levels of 5-hmC are hallmarks of various cancers [38, 39]. EBV infection is an epigenetic driver. EBV(LMP1) is known to upset the balance by increasing DNMT1 expression and activity [10, 11, 13]. Surprisingly, data regarding the relationship between EBV infection and TETs are limited. Here we demonstrated that EBV(LMP1) reduced the activity of TETs and consequently led to RIP3 promoter hypermethylation. Altogether, LMP1 could regulate DNA methylation by two axis: LMP1-DNMT1 and LMP1-TETs in various cell lines, because of tumor heterogeneity of nasopharyngeal carcinoma.
Fumarate, regarded as an oncometabolite, is reported to mediate DNA and histone demethylation by preventing α-KG-dependent dioxygenase, TETs and KDMs, activities [40, 41]. In multiple cancers, fumarate accumulation is caused by mutation of FH [14, 42]. Loss-of-function mutation of FH results in loss of 5-hmC [43, 44]. Few studies have focused on fumarate hydratase activity independent of mutation. Our data showed that FH protein acetylation, instead of FH mutation or its abnormal expression, may lead to its reduction in activity and fumarate accumulation, which might be a reason for weakened TETs enzymatic activity and RIP3 silencing. Interestingly, fumarate hydratase has been identified in the RIP3 complex [2]. Therefore, future studies need to explore the crosstalk between RIP3 and FH.
Metabolic reprogramming is a hallmark of viral oncogenesis [45]. We previously reported that EBV(LMP1) expression and activity resulted in various metabolite changes and high levels of glycolysis [13, 17]. EBV(LMP1) expression resulted in the accumulation of fumarate and reduction of α-KG, which silenced RIP3 expression in a methylation-dependent manner. In this study, EBV(LMP1) linked metabolic changes with epigenetic modifications. Our previous study revealed the mechanism as to how the EBV(LMP1)-mediated signaling axes led to metabolism reprogramming [13, 17, 46]. Here we showed that EBV(LMP1)-associated metabolite changes inhibited necroptosis signaling and promoted oncogenesis, which would enrich the EBV(LMP1) oncogenic signaling network.
In conclusion, our results demonstrate a novel pathway in which EBV(LMP1)-associated oncometabolites by DNA methylation led to necroptosis resistance, and also shed light on the mechanism underlying EBV-related carcinogenesis, which may provide new options for cancer diagnosis and therapy.
Abbreviations
2-HG: 2-hydroxyglutarate; 5-hmC: 5-hydroxymethylcytosine; 5-mC: 5-methylcytosine; α-KG: alpha ketoglutarate; BSP: bisulfite-sequencing PCR; CMV: cytomegalovirus; DNMT: DNA methyltransferase; EBV: Epstein-Barr virus; EBV-LMP1: EBV-encoded latent membrane protein 1; FH: fumarate hydratase; hMeDIP: hydroxymethylated DNA immunoprecipitation; HNSCC: head and neck squamous cell cancer; HSV: herpes simplex virus; IDH: isocitrate dehydrogenase; KDM: histone lysine demethylase; MSP: methlyation-specific PCR; MLKL: mixed lineage kinase-domain like protein; NP: nasopharyngitis; NPC: nasopharyngeal carcinoma; RIP1/3: receptor-interacting protein 1/3; SDH: succinate dehydrogenase; TET: ten-eleven translocation methylcytosine dioxygenase; TNF: tumor necrosis factor; TSG: tumor suppressor gene; TSS: transcriptional start site.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Prof. Sai Wah Tsao (University of Hong Kong, Hong Kong SAR, China) for providing the human NPC cell lines and the immortalized human nasopharyngeal epithelial cell lines; Prof. Yongguang Tao (Central South University, Changsha, China) for suggestions and discussions. This study was supported by National Natural Science Foundation of China (81430064, 81602402, 81874172), China Postdoctoral Science Foundation funded project (2017M612595), Hunan Provincial Natural Science Foundation of China (2018JJ3700), the Fundamental Research Funds for the Central Universities (502042004) and the Open-End Fund for the Valuable and Precision Instruments of Central South University (CSUZC201744).
Author contributions
Supervision: Ya Cao. Study concept and design: Feng Shi and Ya Cao. Drafting of the manuscript: Feng Shi, Li Shang and Ya Cao. Acquisition, analysis, or interpretation of data: Min Zhou, Li Shang, Qianqian Du, Yueshuo Li, Longlong Xie, Xiaolan Liu, Min Tang, Xiangjian Luo, Ann M. Bode. Technical or material support: Jia Fan, Jian Zhou, Qiang Gao, ShuangJian Qiu, Weizhong Wu, Xin Zhang.
Competing Interests
The authors have declared that no competing interest exists.
References
1. He S, Wang L, Miao L, Wang T, Du F, Zhao L. et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100-11
2. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC. et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332-6
3. Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS. et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015;25:707-25
4. Danthi P. Viruses and the Diversity of Cell Death. Annu Rev Virol. 2016;3:533-53
5. Liu X, Li Y, Peng S, Yu X, Li W, Shi F. et al. Epstein-Barr virus encoded latent membrane protein 1 suppresses necroptosis through targeting RIPK1/3 ubiquitination. Cell Death Dis. 2018;9:53
6. Lieberman PM. Virology. Epstein-Barr virus turns 50. Science. 2014;343:1323-5
7. Cao Y. EBV based cancer prevention and therapy in nasopharyngeal carcinoma. NPJ Precis Oncol. 2017;1:10
8. Kaneda A, Matsusaka K, Aburatani H, Fukayama M. Epstein-Barr virus infection as an epigenetic driver of tumorigenesis. Cancer Res. 2012;72:3445-50
9. Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148-59
10. Tsai CN, Tsai CL, Tse KP, Chang HY, Chang YS. The Epstein-Barr virus oncogene product, latent membrane protein 1, induces the downregulation of E-cadherin gene expression via activation of DNA methyltransferases. Proc Natl Acad Sci U S A. 2002;99:10084-9
11. Tsai CL, Li HP, Lu YJ, Hsueh C, Liang Y, Chen CL. et al. Activation of DNA methyltransferase 1 by EBV LMP1 Involves c-Jun NH(2)-terminal kinase signaling. Cancer Res. 2006;66:11668-76
12. Chen YP, Zhang WN, Chen L, Tang LL, Mao YP, Li WF. et al. Effect of latent membrane protein 1 expression on overall survival in Epstein-Barr virus-associated cancers: a literature-based meta-analysis. Oncotarget. 2015;6:29311-23
13. Luo X, Hong L, Cheng C, Li N, Zhao X, Shi F. et al. DNMT1 mediates metabolic reprogramming induced by Epstein-Barr virus latent membrane protein 1 and reversed by grifolin in nasopharyngeal carcinoma. Cell Death Dis. 2018;9:619
14. Sajnani K, Islam F, Smith RA, Gopalan V, Lam AK. Genetic alterations in Krebs cycle and its impact on cancer pathogenesis. Biochimie. 2017;135:164-72
15. Nowicki S, Gottlieb E. Oncometabolites: tailoring our genes. FEBS J. 2015;282:2796-805
16. Jiang Y, Yan B, Lai W, Shi Y, Xiao D, Jia J. et al. Repression of Hox genes by LMP1 in nasopharyngeal carcinoma and modulation of glycolytic pathway genes by HoxC8. Oncogene. 2015;34:6079-91
17. Xiao L, Hu ZY, Dong X, Tan Z, Li W, Tang M. et al. Targeting Epstein-Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene. 2014;33:4568-78
18. Cheung ST, Huang DP, Hui AB, Lo KW, Ko CW, Tsang YS. et al. Nasopharyngeal carcinoma cell line (C666-1) consistently harbouring Epstein-Barr virus. Int J Cancer. 1999;83:121-6
19. Hui KF, Chiang AK. Suberoylanilide hydroxamic acid induces viral lytic cycle in Epstein-Barr virus-positive epithelial malignancies and mediates enhanced cell death. Int J Cancer. 2010;126:2479-89
20. Tsang CM, Zhang G, Seto E, Takada K, Deng W, Yip YL. et al. Epstein-Barr virus infection in immortalized nasopharyngeal epithelial cells: regulation of infection and phenotypic characterization. Int J Cancer. 2010;127:1570-83
21. Shi F, Shang L, Pan BQ, Wang XM, Jiang YY, Hao JJ. et al. Calreticulin promotes migration and invasion of esophageal cancer cells by upregulating neuropilin-1 expression via STAT5A. Clin Cancer Res. 2014;20:6153-62
22. Shi F, Shang L, Yang LY, Jiang YY, Wang XM, Hao JJ. et al. Neuropilin-1 contributes to esophageal squamous cancer progression via promoting P65-dependent cell proliferation. Oncogene. 2018;37:935-43
23. Hau PM, Tsang CM, Yip YL, Huen MS, Tsao SW. Id1 interacts and stabilizes the Epstein-Barr virus latent membrane protein 1 (LMP1) in nasopharyngeal epithelial cells. PLoS One. 2011;6:e21176
24. Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427-31
25. Koivunen P, Laukka T. The TET enzymes. Cell Mol Life Sci. 2018;75:1339-48
26. Li J, He Y, Tan Z, Lu J, Li L, Song X. et al. Wild-type IDH2 promotes the Warburg effect and tumor growth through HIF1alpha in lung cancer. Theranostics. 2018;8:4050-61
27. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T. et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000-4
28. Weinert BT, Scholz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA. et al. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 2013;4:842-51
29. Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S. et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443-53
30. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700-14
31. Liu X, Shi F, Li Y, Yu X, Peng S, Li W. et al. Post-translational modifications as key regulators of TNF-induced necroptosis. Cell Death Dis. 2016;7:e2293
32. Feng X, Song Q, Yu A, Tang H, Peng Z, Wang X. Receptor-interacting protein kinase 3 is a predictor of survival and plays a tumor suppressive role in colorectal cancer. Neoplasma. 2015;62:592-601
33. Sun Y, Zhai L, Ma S, Zhang C, Zhao L, Li N. et al. Down-regulation of RIP3 potentiates cisplatin chemoresistance by triggering HSP90-ERK pathway mediated DNA repair in esophageal squamous cell carcinoma. Cancer Lett. 2018;418:97-108
34. Dhe-Paganon S, Syeda F, Park L. DNA methyl transferase 1: regulatory mechanisms and implications in health and disease. Int J Biochem Mol Biol. 2011;2:58-66
35. Zhang W, Xu J. DNA methyltransferases and their roles in tumorigenesis. Biomark Res. 2017;5:1
36. Morgan MJ, Kim YS. The serine threonine kinase RIP3: lost and found. BMB Rep. 2015;48:303-12
37. Yang Z, Jiang B, Wang Y, Ni H, Zhang J, Xia J. et al. 2-HG Inhibits Necroptosis by Stimulating DNMT1-Dependent Hypermethylation of the RIP3 Promoter. Cell Rep. 2017;19:1846-57
38. Huang Y, Rao A. Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet. 2014;30:464-74
39. Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016;30:733-50
40. Yang M, Soga T, Pollard PJ, Adam J. The emerging role of fumarate as an oncometabolite. Front Oncol. 2012;2:85
41. Sciacovelli M, Goncalves E, Johnson TI, Zecchini VR, da Costa AS, Gaude E. et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature. 2016;537:544-7
42. Morin A, Letouze E, Gimenez-Roqueplo AP, Favier J. Oncometabolites-driven tumorigenesis: From genetics to targeted therapy. Int J Cancer. 2014;135:2237-48
43. Hoekstra AS, de Graaff MA, Briaire-de Bruijn IH, Ras C, Seifar RM, van Minderhout I. et al. Inactivation of SDH and FH cause loss of 5hmC and increased H3K9me3 in paraganglioma/pheochromocytoma and smooth muscle tumors. Oncotarget. 2015;6:38777-88
44. Castro-Vega LJ, Buffet A, De Cubas AA, Cascon A, Menara M, Khalifa E. et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet. 2014;23:2440-6
45. Levy P, Bartosch B. Metabolic reprogramming: a hallmark of viral oncogenesis. Oncogene. 2016;35:4155-64
46. Lu J, Tang M, Li H, Xu Z, Weng X, Li J. et al. EBV-LMP1 suppresses the DNA damage response through DNA-PK/AMPK signaling to promote radioresistance in nasopharyngeal carcinoma. Cancer Lett. 2016;380:191-200
Author contact
![]() Corresponding author: Prof. Ya Cao, Key Laboratory of Carcinogenesis and Invasion, Chinese Ministry of Education, Xiangya Hospital, Central South University, Changsha 410078, China. Tel: +86-731-84805448. E-mail: ycao98sina.com.
Corresponding author: Prof. Ya Cao, Key Laboratory of Carcinogenesis and Invasion, Chinese Ministry of Education, Xiangya Hospital, Central South University, Changsha 410078, China. Tel: +86-731-84805448. E-mail: ycao98sina.com.
Citation styles
APA
Shi, F., Zhou, M., Shang, L., Du, Q., Li, Y., Xie, L., Liu, X., Tang, M., Luo, X., Fan, J., Zhou, J., Gao, Q., Qiu, S., Wu, W., Zhang, X., Bode, A.M., Cao, Y. (2019). EBV(LMP1)-induced metabolic reprogramming inhibits necroptosis through the hypermethylation of the RIP3 promoter. Theranostics, 9(9), 2424-2438. https://doi.org/10.7150/thno.30941.
ACS
Shi, F.; Zhou, M.; Shang, L.; Du, Q.; Li, Y.; Xie, L.; Liu, X.; Tang, M.; Luo, X.; Fan, J.; Zhou, J.; Gao, Q.; Qiu, S.; Wu, W.; Zhang, X.; Bode, A.M.; Cao, Y. EBV(LMP1)-induced metabolic reprogramming inhibits necroptosis through the hypermethylation of the RIP3 promoter. Theranostics 2019, 9 (9), 2424-2438. DOI: 10.7150/thno.30941.
NLM
Shi F, Zhou M, Shang L, Du Q, Li Y, Xie L, Liu X, Tang M, Luo X, Fan J, Zhou J, Gao Q, Qiu S, Wu W, Zhang X, Bode AM, Cao Y. EBV(LMP1)-induced metabolic reprogramming inhibits necroptosis through the hypermethylation of the RIP3 promoter. Theranostics 2019; 9(9):2424-2438. doi:10.7150/thno.30941. https://www.thno.org/v09p2424.htm
CSE
Shi F, Zhou M, Shang L, Du Q, Li Y, Xie L, Liu X, Tang M, Luo X, Fan J, Zhou J, Gao Q, Qiu S, Wu W, Zhang X, Bode AM, Cao Y. 2019. EBV(LMP1)-induced metabolic reprogramming inhibits necroptosis through the hypermethylation of the RIP3 promoter. Theranostics. 9(9):2424-2438.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.