Impact Factor
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusions
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(10):2910-2923. doi:10.7150/thno.30879 This issue Cite
Research Paper
M2 microglia-derived exosomes protect the mouse brain from ischemia-reperfusion injury via exosomal miR-124
Yaying Song1,2, Zongwei Li2, Tingting He1, Meijie Qu1,2, Lu Jiang2, Wanlu Li2, Xiaojing Shi2, Jiaji Pan2, Linyuan Zhang1,2, Yongting Wang2, Zhijun Zhang2 ![]() , Yaohui Tang2
, Yaohui Tang2 ![]() , Guo-Yuan Yang1,2
, Guo-Yuan Yang1,2 ![]()
1. Department of Neurology, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China;
2. Neuroscience and Neuroengineering Research Center, Med-X Research Institute and School of Biomedical Engineering, Shanghai Jiao Tong University, Shanghai 200030, China.
Received 2018-10-22; Accepted 2019-4-2; Published 2019-5-4
Abstract

Rationale: Microglia play a critical role in modulating cell death and neurobehavioral recovery in response to brain injury either by direct cell-cell interaction or indirect secretion of trophic factors. Exosomes secreted from cells are well documented to deliver bioactive molecules to recipient cells to modulate cell function. Here, we aimed to identify whether M2 microglia exert neuroprotection after ischemic attack through an exosome-mediated cell-cell interaction.
Methods: M2 microglia-derived exosomes were intravenously injected into the mouse brain immediately after middle cerebral artery occlusion. Infarct volume, neurological score, and neuronal apoptosis were examined 3 days after ischemic attack. Exosome RNA and target protein expression levels in neurons and brain tissue were determined for the mechanistic study.
Results: Our results showed that the M2 microglia-derived exosomes were taken up by neurons in vitro and in vivo. M2 microglia-derived exosome treatment attenuated neuronal apoptosis after oxygen-glucose deprivation (p<0.05). In vivo results showed that M2 microglia-derived exosome treatment significantly reduced infarct volume and attenuated behavioral deficits 3 days after transient brain ischemia (p<0.05), whereas injection of miR-124 knockdown (miR-124k/d) M2 microglia-derived exosomes partly reversed the neuroprotective effect. Our mechanistic study further demonstrated that ubiquitin-specific protease 14 (USP14) was the direct downstream target of miR-124. Injection of miR-124k/d M2 exosomes plus the USP14 inhibitor, IU1, achieved comparable neuroprotective effect as injection of M2 exosomes alone.
Conclusions: We demonstrated that M2 microglia-derived exosomes attenuated ischemic brain injury and promoted neuronal survival via exosomal miR-124 and its downstream target USP14. M2 microglia-derived exosomes represent a promising avenue for treating ischemic stroke.
Keywords: brain ischemia, exosomes, miR-124, microglia, USP14
Introduction
Microglia are resident immune cells in the brain that play an important role in regulating immune response and brain function [1]. Stroke-induced brain injury activates microglia and polarizes microglia into either the “classically activated” M1 phenotype or “alternatively activated” M2 phenotype [2, 3]. Mounting evidence now suggesting that M1 microglia produce high levels of pro-inflammatory cytokines and exacerbate acute brain damage. In contrast, the M2 phenotype is consistently considered the “healing” phenotype, exhibiting neuroprotective effects on the brain and improving long-term neurological outcomes after stroke [4-6] through phagocytosis of neuron debris and secretion of multiple trophic factors such as brain-derived neurotrophic factor (BDNF), C-X-C motif chemokine ligand 12 (CXCL12), and galectin-3 [7, 8]. M2 microglia-conditioned medium has also been well documented to be neuroprotective [9]; however, the underlying mechanism of M2 microglia-mediated neuroprotection remains largely unknown.
Exosomes are membranous vesicles that are secreted by different cell types [10] and capable of crossing the blood-brain barrier (BBB) [11]. Increasing evidence indicates that exosomes play an important role in mediating cell-cell communication. Cell-derived exosome cargo contains a variety of microRNAs (miRNAs), proteins, mRNAs and DNA, which can work locally or be stably transferred to recipient cells. Among them, miRNA is the most investigated and has been shown to be involved in many physiological and pathological processes [12]. Over the past decade, exosomes have been employed as a therapeutic strategy for ischemic stroke. Mesenchymal stem cell (MSC)-derived exosomes enriched with the miR-17-92 cluster and miR-133b benefit neurite remodeling and functional recovery after ischemic stroke [13, 14]. Neuronal exosomal miR-132 has been reported as an intercellular signal mediating neurovascular communication [15]. Microglia-derived microvesicles have been reported to regulate neuronal excitability [16]. A recent study showed that increased miR-124 derived from microglial exosomes following traumatic brain injury inhibits neuronal inflammation and contributes to neurite outgrowth via the transfer of these exosomes into neurons [17]. However, the effect of M2 microglia on the function of neurons after stroke is still unclear.
In this study, we hypothesized that M2 BV2-derived exosomes (M2-EXO) exert neuroprotective effects in the ischemic mouse brain via transfer of miR-124 into neurons and regulation of its corresponding downstream genes. We injected M2-EXO and miR-124 knockdown (miR-124k/d) M2-EXO into ischemic mice to test our hypothesis and explore the downstream mechanisms.
Methods
BV2 cell activation and identification
BV2 was cultured under complete medium which was prepared using Dulbecco's modified Eagle medium (DMEM, HyClone, Logan, UT) with 10% Fetal Bovine Serum (FBS, Gibco) and 1% penicillin streptomycin antibiotic (HyClone). BV2 cells were treated with interleukin 4 (IL-4) at 20 ng/mL for 48 h to activate M2 BV2 cells. M2 BV2 cells were identified using immunostaining, PCR and western blotting assay.
Exosome isolation, identification and labeling
Exosomes were purified from the cell culture supernatant of M2 BV2 cells. Prior to culture medium collection, BV2 cells were washed twice with PBS, and the medium was switched to exosome-free medium (ultracentrifugation at 100000 X g for 16 h at 4℃) upon M2 stimulation. The cells were then cultured for 48 h. The supernatant was collected and went through sequential ultracentrifugation at 2000 X g for 30 min, 10000 X g for 30 min, and 100000 X g for 70 min at 4℃. The exosomes were washed once with PBS at 100000 X g for 70 min and suspended for further characterization.
A transmission electron microscope (TEM, Thermo Scientific, Waltham, MA) was used to identify the form of the exosomes. Nanoparticle tracking analysis (NTA, Brookhaven, New York) was used to measure exosome diameter and particle number. The protein content was measured using BCA protein assay (Thermo Scientific), and exosome markers CD9, CD63 and tumor susceptibility gene 101 (TSG101) were detected by western blot analysis.
Fluorescence labeling of exosomes was performed according to the protocol previously described [18]. The PKH26 kit was used according to the instruction manual (Sigma-Aldrich, San Louis, MO). The labeled exosomes were washed at 100000 g for 1 h, and the exosome pellet was diluted in PBS and used for the uptake experiment.
Primary neuron culture
Primary neurons were prepared from cerebral cortices of 16 to 18-day-old-ICR mouse embryos. Briefly, dissociated cortical cells were spread on 6-well plates coated with poly-D-lysine (Sigma-Aldrich) and cultured in DMEM at a density of 7 X 105 cells/well. After 4 h of seeding, the medium was changed to Neurobasal medium (Gibco, Carlsbad, NM) supplemented with B-27 (Gibco). Cells were cultured in a humidified incubator at 37℃ with 5% CO2. Cultures were used for experiments 7 to 10 days after seeding.
The oxygen-glucose deprivation (OGD) and reoxygenation model
Oxygen-glucose deprivation (OGD) experiments were performed using a specialized sealed chamber containing an anaerobic gas mixture (95% N2 and 5% CO2). Culture medium was also replaced with deoxygenated glucose-free DMEM (Gibco). After a 45-min challenge, cultures were removed from the chamber, and the OGD solution in the cultures was replaced with maintenance medium. Cells were then allowed to recover for 12 h in a regular incubator.
Animal Experimental design
Forty-eight mice were randomly divided into 6 groups including 1) Sham group: mice that underwent sham surgery; 2) PBS treated group: 200 μl PBS was injected through the tail vein for 3 consecutive days; 3) M2-EXO treated group: M2-BV2 cell-derived exosomes were injected through the tail vein for 3 consecutive days; 4) miR-124k/d EXO treated group: miR-124 were knocked down by using lentivirus (miR-124k/d) in M2-BV2 cells, then the exosomes were isolated and injected through the tail vein for 3 consecutive days; 5) miR-124k/d EXO plus IU1 treated group: miR-124k/d EXO was injected through the tail vein and given an intraperitoneal injection of 400 μg/kg IU1 for 3 consecutive days; and 6) miRNA vector treated group (miR-cn EXO): M2-type BV2 cells were stimulated to the M2 type and then treated with a control lentivirus vector, and the exosomes were isolated and injected through the tail vein for 3 days. Each group contained 8 mice, and all mice underwent neurobehavioral observation. Three days after tMCAO, 4 mice from each group were used for protein and RNA collection, and the remaining 4 mice were used for immunostaining.
Transient Middle cerebral artery occlusion (tMCAO)
Animal procedures were performed according to a protocol approved by the Institutional Animal Care and Use Committee of Shanghai Jiao Tong University, Shanghai, China. The experimental design is shown in Figure 2E. Sixty-four adult male ICR mice weighing 25 to 30 g were used. Mice were anesthetized with 1.5-2% isoflurane and 30%/70% oxygen/nitrous oxide. tMCAO was performed as previously described with minor modifications [19]. Briefly, the common carotid artery, internal carotid artery and external carotid artery were separated. A 6-0 nylon suture coated with silica gel was inserted from the external carotid artery, followed by the internal carotid artery and gently inserted into the middle cerebral artery. The success of occlusion was determined by monitoring the decrease in surface cerebral blood flow (CBF) to 10% of baseline CBF using a laser Doppler flowmetry (Moor Instruments, Devon, UK). Reperfusion was performed by withdrawing the suture 1 h after tMCAO. After the suture was withdrawn, exosomes were immediately injected through the tail vein, and this injection was repeated for a total of 3 days, 100 μg per day.
Neurobehavioral tests
Neurobehavioral tests were performed before and 3 days after tMCAO by an investigator blinded to the experimental design. Neurological evaluations used the modified neurological severity score (mNSS), which is a composite score of motor, reflex, and balance tests [20]. The severity score was graded at a scale from 0 to 14, in which 0 represents normal, and a higher score indicates a more severe injury [21].
Infarct volume assessment
Mouse brains were perfused with 0.9% saline and 4% paraformaldehyde (PFA, Sinopharm Chemical Reagent, Shanghai, China). After perfusion, brains were removed, placed in 4% PFA overnight and then fully dehydrated in 30% sucrose for 2 days. A series of 30-μm-thick brain sections were collected for a total thickness of nearly 3.6 mm. Infarct volume was determined by staining the brain sections with Cresyl violet solution and calculating the ratio of staining in the ipsilateral and contralateral hemispheres using ImageJ (National Institutes of Health, Bethesda, MD). The brain volume was estimated by the following formula: V = ∑h/3[∆Sn+(∆Sn*∆Sn+1)1/2+∆Sn+1]. In the formula, V represents volume, h represents the distance between the two adjacent brain sections, ∆Sn and ∆Sn+1 represent the different area between the two adjacent sections.
Immunohistochemistry
Immunohistochemistry was performed according to the protocol previously described [22]. Briefly, the brain tissue was removed from the anti-freezing solution, and the immunohistochemistry procedures were followed. The primary antibodies used were those targeting arginase (ARG; 1:50; Santa Cruz, CA), CD206 (1:200; Abcam, Cambridge, MA), Iba-1 (1:200 dilution, WAKO, Osaka, Japan), mitogen-activated protein 2 (MAP2; 1:200, 1:100; Millipore, Billerica, MA) and NeuN (1:200, Millipore). TdT-mediated dUTP nick end labeling (TUNEL) and NeuN double staining was performed following the manufacturer's instructions on double immunostaining (Roche Diagnostics, Basel, Switzerland). We performed statistical analysis of the immunohistochemistry data by counting the number of CD206 and IBA1, ARG and IBA1, CD16/32 and IBA1, TUNEL and DAPI double-positive cells and GFAP positive cells in 5 regions in the striatum of peri-infarct area of each brain slice, with 2 brain sections (600 μm apart) in each mouse, 4 mice per group.
TUNEL staining
To determine whether treatment with M2-EXO reduces apoptosis in cortical neurons, we performed TUNEL assays. Cells were fixed with 4% para-formaldehyde. After the cells were washed with PBS, the TUNEL assay was performed according to the manufacturer's protocol using the fluorescein-conjugated probe. Cells were counterstained with DAPI to assess the nuclear morphology. DAPI (1:1000, Beyotime, China) staining was performed by incubating the slides for 10 min at room temperature. The slides were washed and imaged with fluorescence microscopy. In order to determine whether M2-EXO treatment could reduce apoptosis in neuronal cells, we performed TUNEL assay. In peri-infarct area of brain section, we counted the number of NeuN/TUNEL double-positive cells and NeuN-positive cells in 5 regions of each brain section. For in vitro study, we counted the number of DAPI/TUNEL double-positive cells and DAPI positive cells in 5 region of each cell slide in each group for statistical analysis. To evaluate the proportion of injury, we calculated the ratio of DAPI/TUNEL double-positive cells to DAPI-positive cells.
Lactate dehydrogenase (LDH) and cell survival assay
The death of primary neurons was measured by LDH release in the culture medium. Levels of LDH release in the supernatants of cultured cells were measured using the LDH assay kit (Beyotime). Percent cell death was calculated using the formula: % cytotoxicity = LDH release (OD492)/maximum (OD492). Cell survival assays were performed by using Cell Counting Kit-8 solution (Dojindo, Kumamoto, Japan). The CCK-8 solution was added to each well 2 h prior to the sample collection time point. Absorbance was measured at 450 nm using a microplate reader.
Western blot analysis
Western blotting of exosomes was performed as previously described [23]. Tissue samples were collected from the ipsilateral hemisphere and quantified with the BCA protein assay (Pierce, Rockford, IL, USA).The primary antibodies were those targeting CD9 (1:1000), TSG101 (1:500), ARG (1:200), CD206 (1:1000), total caspase-3 (1:1000, Cell Signaling technology, Beverly, MA), cleaved caspase-3 (1:1000), USP14 (1:1000 dilution, Abcam), and β-actin (1:1000). For quantify western blot data, we analyzed the band gray values of 4 samples. After calculating the gray value of the band relative to the expression of the internal reference, we standardized the levels to those of the control group and then performed statistical analysis.
RNA extraction and real-time PCR
Total RNA from exosomes was extracted by TRIzol LS reagent (Invitrogen, Carlsbad, CA), and total RNA from tissues was extracted by TRIzol reagent (Invitrogen) 3 days after tMCAO. Total RNA was isolated according to the manufacturer's protocol. Single-strand cDNA was synthesized using a universal cDNA synthesis kit (Qiagen, Hilden, Germany) under the following conditions: 42°C for 1 h and then 95°C for 5 min. The expression of miRNA was tested by a fast real-time PCR system (7900 HT, ABI, Foster City, CA) using a SYBR Green master mix (Qiagen) with the following cycling conditions: 95°C for 10 min followed by 40 cycles of 95°C for 10 s and 60°C for 1 min. miR-16 was used as the exosomal miRNA endogenous control and GPADH as the control for tissues and cells. The relative expression was normalized to that in the control group.
Lentiviral vector transduction
BV2 cells were transduced with miR-124 inhibitor-expressing lentiviruses at a multiplicity of infection of 200 particles/cell in DMEM (HyClone), and incubated at 37°C in 5% CO2 in 24-well plates. Three days after transduction GFP signal was observed by fluorescence microscopy. No-load shRNA lentivirus was used as a control.
Luciferase reporter assay
293T cells are commonly used in luciferase reporter detection systems. miRNAs act primarily through the 3'UTR acting on target genes. The purpose of this experiment was to verify the relationship between miR-124 and USP14. Therefore, we inserted the 3'UTR region of USP14 into a vector and transfected the vector into 293T cells. Additionally, we performed site-directed mutagenesis to further determine the site of action of the miRNA and the 3' UTR of the target gene. Luciferase activity was assayed using the Dual-Luciferase® Reporter Assay System (Promega, Fitchburg, WI), and the ratio of Renilla (OBIO, Shanghai, China) luciferase to firefly luciferase activity was determined. The mouse USP14 3'UTR was amplified and cloned into a pGL4 vector containing the firefly luciferase reporter gene (ObiO Co., Ltd, Shanghai, China). For the luciferase assay, 293T cells were cotransfected with 100 ng firefly luciferase constructs, 10 ng pRL-TK Renilla luciferase plasmid, and 100 nmol/L synthetic miR-124 mimic and no-load vector. The results are expressed as relative luciferase activity (firefly luciferase/Renilla luciferase).
Statistical analysis
All data are expressed as the mean±standard deviation (SD). ANOVA with Student-Newman-Keuls multiple-comparisons posttest was used to compare gene expression. Comparisons between 2 groups were made by Student's t test. A p<0.05 was considered significantly different. Statistical analysis was carried out with Prism GraphPad 6.
Results
M2 BV2-conditioned medium protected neuronal survival
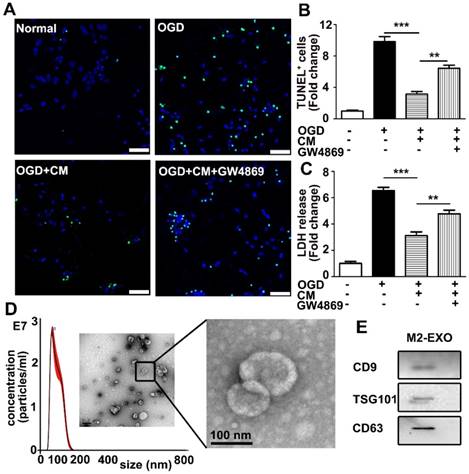
To investigate if M2 BV2-derived exosomes are involved in neuroprotection, BV2 cells were first polarized to the M2 type by the addition of 20 ng/ml IL-4, which significantly increased the expression of M2 BV2 markers ARG and CD206 (Figures S1A-C). As shown in Figures 1A and 1B, M2-conditioned medium significantly decreased apoptosis of neurons and increased viability of neurons subjected to OGD, while the addition of GW4869 (a small-molecule inhibitor that reduces exosomal secretion) in conditioned medium partly reversed the protective effect, suggesting that exosomes derived from M2 BV2 cells are involved in neuroprotection after OGD. To quantify the protective effect of M2 BV2-conditioned medium on neurons that underwent OGD, an LDH assay was performed for rapid quantification of cell viability. Cellular injury was quantified by the amount of LDH released [24]. To quantify the protective effect of M2 microglia conditioned medium on neurons that underwent OGD, Lactate dehydrogenase (LDH) assay was performed. LDH assay allows rapid evaluation of cellular injury by measuring the amount of LDH released [24]. The level of LDH was normalized to that in the control group. OGD treatment increased cell injury by 6.5-fold and the addition of conditioned medium decreased cell death to 3-fold that in the control group. Notable, GW4869 reversed the protective effect of M2 microglia-conditioned medium (Figure 1C).
Exosomes were then isolated from M2 BV2 cells by ultracentrifugation and identified using a TEM, nanoparticle tracking analysis (NTA) and western blot analysis. As shown in Figure 1D, the typical cup-shaped membrane vesicle morphology was observed, and the diameter of particles was in the range of 30-120 nm. Western blot analysis showed that exosome markers including CD9, TSG101, and CD63 were expressed in exosomes (Figure 1E).
M2 microglia-conditioned medium protected neuronal survival and identification of M2-EXO. A. TUNEL staining showed apoptosis of: 1) neurons under normal culture conditions (Normal), 2) neurons underwent OGD treatment (OGD), 3) neurons pretreated with M2-BV2-conditioned medium (CM) and exposed to OGD injury (OGD+CM), 4) neurons pretreated with M2-BV2 CM plus GW4869 and exposed to OGD injury (OGD+CM+GW4869). Scale bar=50 μm. B. Quantification of A shows the fold change of TUNEL-positive cells relative to control. C. LDH release of neurons treated with M2 microglia CM and CM plus GW4869 after OGD injury. D. Nanoparticle tracking analysis and transmission electron microscopy imaging of M2 microglia-derived exosomes. Scale bar =200 nm. The enlarged picture shows the structure of the exosomes. Scale bar =100 nm. E. Expression of exosomal markers CD9, TSG101 and CD63 in M2-EXO. 1B and 1C showed the fold change to the control group. All data are presented as the mean±SD. *, p<0.05; **, p<0.01; ***, p<0.001.
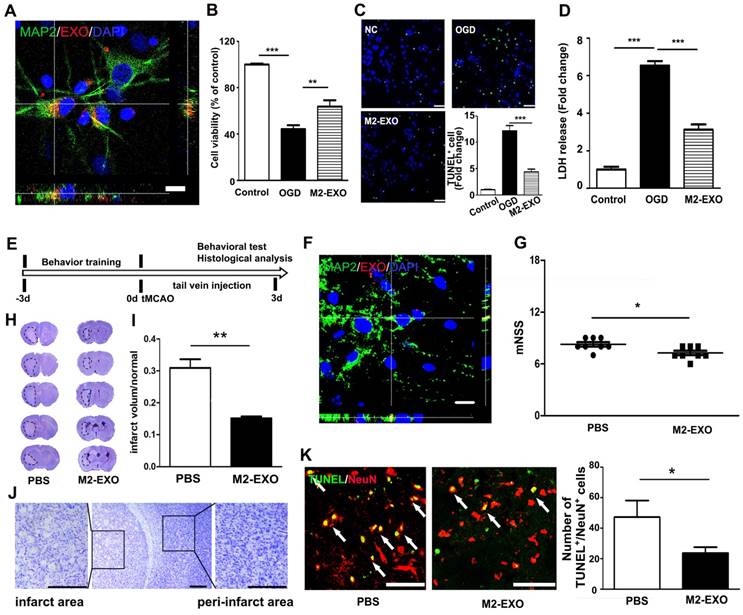
M2-EXO attenuated neuronal apoptosis and improved neurobehavioral recovery after stroke. A. Confocal imaging showing that PKH-26-labeled exosomes (Red) were taken up by neurons (Green) in vitro. Scale bar=50 μm. B. Cell viability of neurons treated with M2-EXO after OGD. C. TUNEL staining of cultured neuron and bar graph showing the number of TUNEL+ neurons. Scale bar=50 μm. D. Cell viability of neurons treated with M2-EXO after OGD. E. In vivo experimental scheme. F. Confocal imaging of brain sections showing that PKH-labeled exosomes (Red) were taken up by MAP-2+ neurons after tail vein injection. Scale bar=10 μm. G. Neurological score showing neurobehavioral recovery of mice treated with PBS and M2-EXO. H. Cresyl violet staining of brain sections treated with PBS and M2-EXO for 3 days after tMCAO. Dashed line shows the infarcts. Zoomed-in images of the ischemic area are shown in order to closely observe the ischemic area. I. Bar graph showing the percentage of infarct volume normalized to the normal side. Scale bar=50 μm. J. Zoomed-in image of brain cresyl violet staining and partial enlargement of the infarct area and peri-infarct area. Scale bar=50 μm. K. Costaining and statistical analysis of TUNEL and NeuN double-positive cells in mice treated with PBS and M2-EXO. Scale bar=50 μm. M2-EXO: neurons pretreated with exosomes derived from M2-BV2 cells. NC: negative control. *, p<0.05; **, p<0.01.
M2-EXO attenuated neuronal apoptosis both in vitro and in vivo
To examine if M2-EXO could be taken up by neurons, M2-EXO were incubated with neurons for 24 h. 3D confocal images showed that PKH26-labeled exosomes (red) were localized in the cytoplasm of MAP2+ neurons (green), suggesting exosomes were taken up by neurons (Figure 2A). To investigate the effects of M2-EXO in neuroprotection, we treated primary neurons with M2-EXO for 24 h to allow sufficient exosome uptake and then performed OGD for 45 min. After reoxygenation for 12 h, treatment with M2-EXO significantly increased cell survival and decreased neuronal apoptosis, as demonstrated by neuronal survival, TUNEL staining and the LDH assay (Figures 2B-2D).
We next examined the effect of M2-EXO in ischemic mice. PKH26-labeled exosomes were detected in the ischemic mouse brain after 3 days of injection, and exosomes were localized to the cytoplasm of neurons (Figure 2F). The mNSS further showed that treatment with M2-EXO greatly attenuated neurobehavioral deficits 3 days after stroke compared to PBS treatment (Figure 2G). Compared to PBS treatment, treatment with M2-EXO significantly reduced infarct volume 3 days after tMCAO (Figure 2H and 2I). A zoomed-in image of the ischemic area and peri-infarct area are also shown (Figure 2J). Furthermore, the M2-EXO group had fewer apoptotic neurons than the PBS group (Figures 2K).
Exosomal miR-124 was involved in neuroprotection
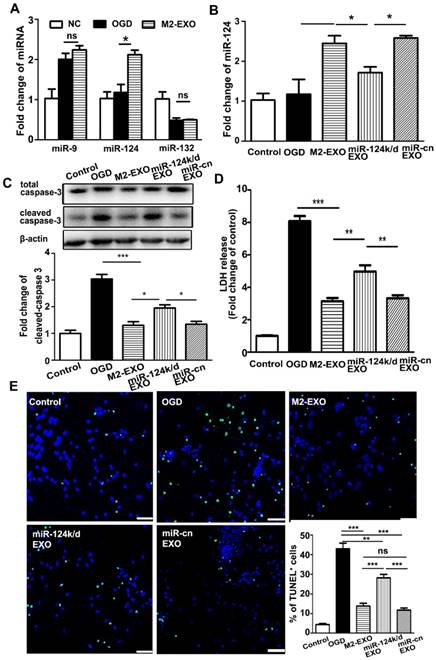
Exosomes are well documented to be enriched with miRNAs and able to deliver miRNA from host cells to target cells to further regulate the function of recipient cells. To explore the underlying mechanism of M2-EXO-mediated neuroprotection, we examined several miRNAs related to neuroprotection including miR-9, miR-124, and miR-132. Our PCR results showed that after treatment with M2-EXO, only miR-124 was significantly upregulated, while the expression of miR-9 and miR-132 were comparable among all groups in vitro (Figure 3A).
Next, we verified the underlying mechanism by knocking down miR-124 in M2-EXO. Transfection of lentivirus carrying miR-124-shRNA downregulated the expression of miR-124 not only in M2-EXO (miR-124k/d-EXO, Figure S2) but also in neurons treated with miR-124k/d-EXO (Figure 3B). Cleaved-caspase 3 expression reduced in the M2-EXO treated group. miR-124k/d M2-EXO could also partly rescue its expression (Figure 3C). As shown in Figure 3D, LDH was higher in the group treated with miR-124k/d EXO than in the group treated with M2-EXO. Treatment with miR-124k/d-EXO partly reversed the neuroprotective effect of M2-EXO; however, treatment with either M2-EXO or miR-124k/d-EXO had no effect on neuronal death under normal conditions (Figure S3). The number of TUNE-positive cells was reduced in M2-EXO treated group, and the number of apoptotic cells rescued by miR-124k/d M2-EXO was decreased than M2-EXO group, while miR-cn EXO showed the similar result to M2-EXO (Figure 3E). Therefore, M2-EXO could reduce neuronal apoptosis caused by OGD, and miR-124 played a role in neuroprotection.
Treatment with M2-EXO protected neuronal survival via miR-124. A. Expression of miR-132, miR-9 and miR-124 in neurons treated with M2-EXO after OGD. B. Expression of miR-124 in neurons treated with M2-EXO, miR-124 knockdown exosomes (miR-124k/d EXO) and control shRNA after OGD. C. Expression of total caspase-3 and cleaved caspase-3 in neurons treated with M2-EXO, miR-124k/d EXO and control shRNA after OGD. D. LDH assay of neurons treated with M2-EXO, miR-124k/d EXO and control shRNA after OGD. E. Number of TUNEL+ neurons treated with M2-EXO, miR-124k/d EXO and control shRNA after OGD. Scale bar=50 μm. Data are presented as the mean±SD. *, p<0.05; **, p<0.01; ***, p<0.001.
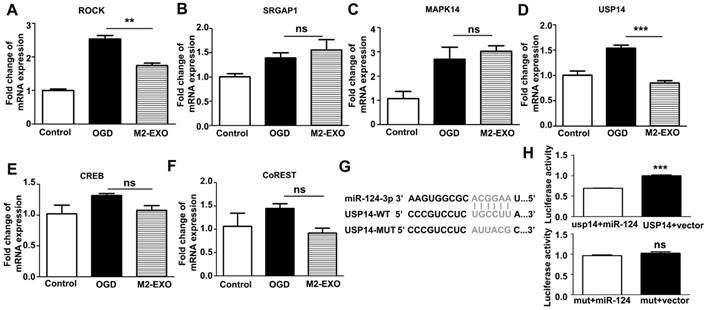
Expression of putative miR-124 targets. A-F. Expression of putative miR-124 targets ROCK, SRGAP1, USP14, CREB, Co-REST, and MAPK14 in neurons treated with M2-EXO after OGD. G-H. Dual luciferase assays of miR-124 targeting effects on USP14 3'-UTR. Data are presented as the mean±SD. ***, p<0.001.
miR-124 derived from M2-EXO increased neuronal survival by regulating its downstream target USP14
To investigate the mechanism of the miR-124-mediated neuroprotective effects, we scanned the miRBase and determined the expression of several miR-124 targets that are related to neuroprotection. The expression of ROCK, SRGAP1, MAPK14, USP14, CREB, and Co-REST was detected in cultured neurons treated with M2-EXO (Figures 4A-4F). Treatment with M2-EXO significantly reduced the expression of ROCK and USP14. A dual-luciferase reporter system further demonstrated that USP14 is a direct target of miR-124. miR-124 mimic significantly inhibited luciferase activity in 293T cells transfected with the USP14 3'-UTR. However, the USP14 mutant did not change the luciferase activity level compared to the blank vector, suggesting that USP14 was a direct target of miR-124. Our result demonstrated USP14 was the direct target of miR-124 (Figure 4G, 4H). Furthermore, treatment with M2-EXO decreased USP14, while treatment with miR-124k/d-EXO increased USP14, suggesting that M2-EXO regulates USP14 expression through miR-124 (Figure S4).
miR-124 derived from M2-EXO attenuated neural deficits and neuronal apoptosis in mice after stroke by downregulating USP14
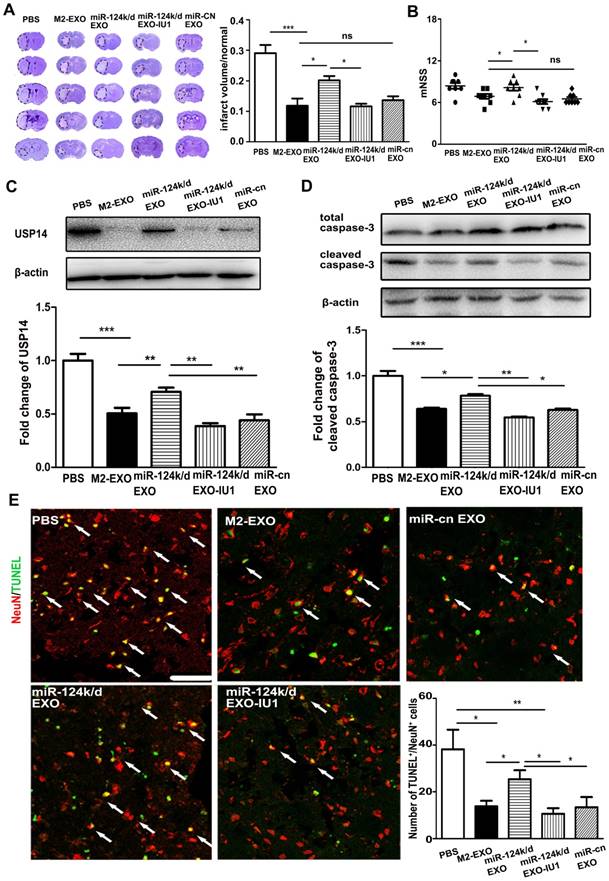
To investigate whether M2-EXO exerted neuroprotective effects in the ischemic mouse brain by miR-124 and its downstream target USP14, miR-124k/d-EXO and the inhibitor of USP14, IU1, were administered in a mouse model of stroke. IU1 is the abbreviation of 1-[1-(4-Fluorophenyl)- 2,5-dimethyl-1H-pyrrol-3-yl]-2-(1-pyrrolidinyl) ethanone. It is the specific reversible inhibitor of the catalytic site of USP14 [25]. IU1 treatment significantly downregulated the expression of USP14 in neurons (p<0.01) (Figure S4). As shown in Figure 5A, treatment with M2-EXO significantly reduced the infarct volume 3 days post ischemia. Injection of miR-124 k/d EXO partly increased the infarct volume, while the addition of IU1 substantially decreased the infarct volume to a size comparable to that in the group treated with M2-EXO. An assessment of neurological score was performed to investigate the effect of M2-EXO on the neural deficit after stroke (Figure 5B). Treatment with M2-EXO greatly attenuated the neurobehavioral deficit at day 3 after stroke, and the addition of IU1 substantially improved neurological outcomes. The USP14 level was examined in the groups treated with PBS, M2-EXO, miR-124k/d EXO, miR-124k/d EXO plus IU and miR-cn EXO. The PBS group showed a high level of USP14, and treatment with M2-EXO reduced USP14 expression. Treatment with miR-124k/d-EXO plus IU1 was shown to substantially decrease the expression of USP14 that resulted from miR-124 downregulation (Figure 5C). TUNEL staining and western blot analysis were performed to test the neuroprotective effects of M2-EXO in vivo. As shown in Figure 5D, western blot analysis showed that both treatment with M2-EXO and treatment with miR-124k/d-EXO plus IU1 substantially decreased the expression of cleaved caspase-3. Treatment with M2-EXO resulted in significantly fewer apoptotic neurons than did treatment with PBS or miR-124k/d-EXO (p<0.05), and miR-124k/d-EXO plus IU1 treatment also significantly decreased the number of apoptotic neurons (Figure 5E).
Treatment with M2-EXO promoted neuronal survival by downregulation of USP14 expression. A. Cresyl violet-staining of brain sections treated with PBS, M2-EXO, miR-124k/d EXO and miR-124k/d EXO+IU1 for 3 days after tMCAO. Dashed line shows the infarcts. Bar graph shows the percentage of infarct volume normalized to the contralateral side. B. Neurological score shows the neurobehavioral recovery of mice treated with PBS, M2-EXO, miR-124k/d EXO, miR-124k/d EXO plus IU1 and miR-cn EXO. C. Expression of USP14 in mice treated with PBS, M2-EXO, miR-124k/d EXO, miR-124k/d EXO plus IU1 and miR-cn EXO. D. Expression of total caspase-3 and cleaved caspase-3 in mice treated with PBS, M2-EXO, miR-124k/d EXO, miR-124k/d EXO plus IU1 and miR-cn EXO. E. TUNEL and NeuN costaining showed apoptosis of neurons in mice treated with PBS, M2-EXO, miR-124k/d EXO, miR-124k/d EXO+IU1 and miR-cn EXO. Arrows indicate apoptotic neurons. Scale bar=50 μm. Bar graph shows the number of apoptotic neurons in mice treated with PBS, M2-EXO, miR-124k/d EXO, miR-124k/d EXO+IU1 and miR-cn EXO. miR-cn EXO: control shRNA EXO. Data are presented as the mean±SD. *, p<0.05; **, p<0.01; ***, p<0.001.
Discussion
Previous studies have shown that the neuroprotective effect of M2 microglia was mainly mediated by the phagocytosis and paracrine secretion of neurotrophic factors [26]. The molecular signal that mediates interactions between microglia and neurons to further promote neuronal survival is unclear. Unveiling the molecular mechanism by which M2-BV2 exert protective effects on neurons would help develop new strategies for the treatment of ischemic stroke. Our present study demonstrated that exosomes derived from M2-BV2 cells are taken up by neurons and further promote their survival, and this process is mediated by exosomal miR-124 and its target USP14.
Recent studies have revealed that microglia play an important role in regulating the function of neurons, including cell survival [27], neurogenesis [28], and synaptogenesis [29]. The communication between microglia and neurons is essential to synchronize diverse functions with brain activity. Accumulating evidence has shown that extracellular vesicles secreted from microglia are key players in mediating microglia-neuron interactions and neuro-inflammation by mRNA, miRNA and proteins [30]. For example, microglia-derived exosomes contribute to the neurologic prognosis of Alzheimer's disease by affecting the propagation of Tau [31]. Chang et al reported that α-synuclein induced an increase in exosomal secretion by microglia, and these activated exosomes expressed a high level of MHC class II molecules and tumor necrosis factor (TNF)-α, thereby enhancing the progression of Parkinson's disease [32]. Thus, microglial exosomes may play a significant role in regulating the neurologic functional recovery in acute neurologic diseases such as ischemic stroke. Indeed, in our study we found that M2-BV2-derived medium significantly decreased neuronal apoptosis and death after OGD, while the exosome secretion inhibitor GW4869 partly reversed such protective effects, suggesting that exosomes derived from M2-BV2-conditioned medium participated in the neuroprotection. Notably, the addition of GW4869 only partly reversed the neuroprotective effects provided by M2-BV2-conditioned medium (20%), suggesting that other factors such as fibroblast growth factor (FGF), nerve growth factor (NGF) and BDNF secreted by M2-microglia could also potentiate neuronal survival [27].
In the central nervous system, many miRNAs were closely related to neuroprotection after ischemic stroke. miR-9 plays an important role in neuronal survival and regeneration after ischemia. miR-9 mimic transfection enhanced post-ischemic neuronal cell viability, and increased the proliferation and neurite elongation of neurons, which contributes to ischemic stroke recovery [33]. miR-132 is a neuron-specific miRNA that plays a key role in synapse development, synaptic plasticity, and structural remodeling and is important in the growth and remodeling of dendrites in the development of neural circuits [34-36]. Previous studies demonstrated that the dysregulation of miR-132 and its target protein is associated with epilepsy, Alzheimer's disease, and the pathophysiological process of Parkinson's disease [37-39]. In ischemic injury, inhibition of miR-132 increased neuronal damage after cerebral ischemia while overexpression of miR-132 in hippocampal neurons provided strong protection against ischemia-induced neuronal death [40]. Downregulation of miR-124 can aggravate hypoxic injury of the neuronal cell line PC12 via intercellular adhesion molecule-1 (ICAM-1) [41]. Overexpression of miR-124 attenuates MPP+-induced neuronal damage by targeting signal transducer and activator of transcription-3 (STAT3) [42]. In addition, overexpression of miR-124 can reduce M1 macrophage activation and promote the M2 regulatory phenotype, and inhibition of miR-124 can increase M1 levels [43, 44]. miR-9 and miR-124 play important roles in microglial regulation and activation in neuroinflammation and neurodegenerative diseases [45]. Therefore, we selected miR-9, miR-124 and miR-132 in the study. Our exosomal miRNA array data validated miR-124 was up-regulated in M2-BV2 cell-derived exosomes. In the present study, neuronal cells were subjected to 45 min of OGD, and miR-124 was measured 12 h after re-oxygenation, a previous study showed that miR-124 was upregulated 24 h after re-oxygenation [46]. The upregulation of miR-124 expression may be time dependent. Our in vivo study demonstrated miR-124 was up-regulated in the ischemic penumbra region at 72 h after tMCAO (Figure S5), which is consistent with previous studies [46-48].
BV2-derived exosomes were taken up by neurons both in vitro and in vivo. Through time-lapse recording, we found that neurons began taking up BV2-derived exosomes as early as 45 s after the addition of exosomes to neuronal culture (Figure S6, video 1, 2). Our in vivo studies showed that exosomes were able to cross the BBB. Only a small number of exosomes were taken up by neurons after 3 continuous days of tail vein injection, and very few exosomes were detected in the brain at day 3 if only one injection was applied immediately after tMCAO (data not shown), which is consistent with the results observed in another study [49], as most of the exosomes may be cleared out due to metabolism. Thus, there is a high demand to promote target delivery of exosomes to neurons. In the figure 2F, the uptake of M2-EXO by neurons was showed in the ischemic penumbra region. There was a high demand to promote the targeted delivery of exosomes into neurons. The exosomes were able to enter the body through circulating blood and were engulfed by cells in the ischemic core, ischemic penumbra, and the sham group mouse brain, but the numbers of exosomes in each area differed (Figure S7). Previous methods such as fusing the neuron-specific rabies virus glycoprotein (RVG) peptide to MSC-derived exosomes have shown increased target delivery of exosomes to neurons, further improving neurobehavioral recovery than unmodified exosomes [17]. The use of this strategy to promote target delivery of M2-EXO to neurons should be examined in the future. However, this is a proof-of-concept study, and the main purpose of this study was to investigate whether treatment with M2-EXO was neuroprotective tMCAO. Neuronal death is known to occur from minutes to hours after tMCAO; therefore, we hypothesized that injection of M2-EXO immediately after tMCAO would maximize its neuroprotective effect. Our results demonstrated that the delivery of M2-EXO immediately after tMCAO reduced ischemic brain injury. The ideal time window of exosome delivery is an important issue, and whether a prolonged latency to injection of M2-EXO is still able to reduce brain injury needs to be studied in the future.
In addition to neuron cell survival, we also examined microglia and astrocyte activity. We examined 2 brain slices from each of 4 mice in each group and counted the number of glial fibrillary acidic protein (GFAP)- and ionized calcium binding adaptor molecule (IBA1)-positive cells in 3 regions from each brain slice. The number of astrocytes was decreased in the M2-EXO group than in the control group. We used IBA1 and CD16/32 double staining and IBA1 and ARG double staining to identify microglia and astrocytes, respectively and found that activation of M1 microglia cells decreased and M2 microglia cells increased. The expression of inflammatory factors such as IL-1 and TNF-α was lower in groups treated with EXO than in the control group, and the expression of IL-10 and transforming growth factor (TGF)-β was higher in the group treated with M2-EXO than in the control group (Figure S8). The results suggested that M2-EXO treatment decreased the inflammation and immune responses after tMCAO.
Several labeling strategies were used for in vivo exosome tracking such as luciferase reporters [50, 51], fluorescent proteins [52, 53], and fluorescent lipophilic dyes [52, 54-57]. Among them, fluorescent lipophilic dyes including Dil, DiR, PKH26 and PKH67 were mostly widely used owing to important chemical features such as 1) no significant effect on the viability and other physiological properties of the target cell; 2) fast diffusion into and uniform labeling of the plasma membrane; and 3) cost-efficient and easy to operate and observe, thereby making them ideal for in vivo applications. Other molecular probe-based approaches such as superparamagnetic iron labeling, radionuclides labeling have their own limitations, which included requirement of large instruments, complicated probe synthesis, cost expensive and potential toxicity. Therefore, we chose PKH26 staining for exosome labeling in our study. We further examined the distribution of exosomes in other organs post injection by histology. We found that PKH26 labeled exosomes were mostly present in spleen and liver after intravenous injection (Figure S9). After exosome delivery, we did not find swelling and inflammation in the spleen and liver. Exosome treatment did not cause animal weight loss and death, suggesting exosomes are not toxic to organs. In addition, previous studies have revealed effects of extracellular vesicle in various organs of the mouse including the thymus, heart, lung, liver, spleen, kidney, adrenal gland, ovary, uterus and brain. Studies have also demonstrated that repeated administration of extracellular vesicles in mice does not cause visible toxicity, assessed by changes in body weight, immune response and tissue pathological changes up to 3 weeks [58]. One study suggested that naturally occurring secreted membrane vesicles are less toxic and better tolerated in the body than exogenous vesicles, as evidenced by their ubiquitous presence in biological fluids, and have an intrinsic homing ability. miRNA is abundant in the contents of exosomes. Exosomes mainly exert their function by transferring exosomal miRNA to recipient cells, thereby modulating downstream gene expression to further regulate the function of recipient cells. We evaluated the expression of miR-9, miR-132 and miR-124, all of which are abundant in microglial exosomes and have been reported to play important roles in regulating the function of neurons [59]. However, only the expression of miR-124 was greatly increased in M2-EXO compared to that in M0-EXO. MiR-124 has been shown to promote M2 polarization in microglia and exert an anti-inflammatory effect on injured neurons via microglial exosome transfer after traumatic brain injury [60, 61]. Downregulation of miR-124 expression is an indicator of neuro-inflammation in various diseases such as experimental autoimmune encephalomyelitis [44] and intracerebral hemorrhage [62]. Previous studies have also reported that injection of miR-124 promotes neuroprotection and function recovery upon stroke onset [17, 60]. In this study, we demonstrated that exosomes can be transmitted horizontally to neurons, thereby exerting neuroprotective effects. Through coculture with exosomes, neuronal miR-124 expression was upregulated, and survival increased. M2 microglia exosome treatment increased miR-124 expression in neurons, and treatment of neurons with M2 microglial exosomes with miR-124 expression knocked down decreased miR-124 expression and reversed the neuroprotective effects of the exosomes, suggesting that M2 microglial exosomes exert their protective effect mainly via transfer of miR-124 to neurons, rather than by stimulating endogenous miR-124 expression in neurons. Importantly, other miRNAs in addition to miR-124 may also mediate the neuroprotective effects of M2-EXO. Exosomes contain many other miRNAs in addition to miR-124. Therefore, we used an exosomal miRNA array to explore more possible functional miRNAs in exosomes (Figure S10). miR-124 was shown to be highly expressed. Other neuroprotection-related miRNAs such as miR-219, miR-218 and miR-186 were upregulated, and miR-145, miR-125 and miR-207 were downregulated. Further studies are warranted to determine whether other miRNAs are involved in this process. In addition, we envision that injection of miR-124 hyper-expressing M2-EXO could be a more effective treatment for stroke.
Potential downstream target of miR-124 related to neuroprotection, such as ROCK, SRGAP1, MAPK14, USP14, CREB and Co-REST, were selected from miRBase and TargetScan. However, only the expression of ROCK and USP14 was found to be significantly downregulated after treatment with M2-EXO. USP14 was a deubiquitinated protein that banded back to the proteasome to trim the K48 Ub chain and negatively regulate proteasome activity [63]. Inhibition of USP14 activity by IU1 enhanced proteasome activity [64], thus reduced oxidative stress-induced cell death in vitro [65]. It was reported that miR-124 played a neuroprotective role by directly targeting USP14 after cerebral ischemia [48]. ROCK was a key regulator of the actin cytoskeleton, and was proven to play an important role in neuroprotection after stroke [66]. Studies showed that miR-124 modulates the function of neural regeneration by targeting Rho/ROCK pathway [67]. However, whether miR-124 exerted its neuroprotection through modulating ROCK was unclear. We purposed to investigate the target of exosomal miR-124 mediated neuroprotection after stroke, therefore, we chose USP14 as one potential target. USP14 could not be the only target for miR-124, whether M2-exosomal miR-124 protected neuronal survival via targeting ROCK needs to be further investigated. In this study, we examined whether USP14 is involved in the brain damage caused by ischemic stroke and inhibits it by miR-124 to promote neuroprotection. USP14 is a major regulator of the proteasome that has been thought to inhibit the degradation of ubiquitin-protein conjugates and be very important in regulating synaptic activity in mammals [68]. The inhibitor of USP14, IU1, is associated with reduced protein aggregates and considered a therapeutic target in the stroke model [69]. Both the dual luciferase assay and loss-of-function study strongly indicated that USP14 is the direct downstream target of miR-124 and that M2-EXO exerted their neuroprotective effects through modulation of miR-124 and USP14.
Conclusions
In conclusion, our results indicate that M2-EXO can improve the outcome of tMCAO, and the associated mechanism may be partly related to miR-124 and IU1. We hypothesize that M2-EXO may be a potential therapeutic target for treating ischemic stroke and that may be partly related to miR-124 and IU-1.
Supplementary Material
Supplementary figures and tables.
Video 1.
Video 2.
Abbreviations
BBB: blood-brain barrier; M2-EXO: M2 BV2 cell-derived exosomes; miRNA: microRNA; MSCs: mesenchymal stem cells; OGD: oxygen-glucose deprivation; tMCAO: transient middle cerebral artery occlusion; USP14: ubiquitin-specific protease 14; TUNEL: TdT-mediated dUTP nick end labeling.
Acknowledgements
This work was supported by the Science and Technology Commission of Shanghai Municipality (17ZR1413600, ZZ), the National Natural Science Foundation of China (NSFC) projects 81771251 (GYY), 81771244(ZZ), 81801170 (YT), 81870921 (YW), 81471178 (GYY), 81522015 (YW), National Key R&D Program of China #2016YFC1300602 (GYY), and K. C. Wong Education Foundation (GYY).
Contributions
Y.S. designed and performed the experiments, analyzed the data and drafted the manuscript and figures; G.Y. and Y.T. conceived the project designed the experiments and edited the paper; Z.L. participated in the design of the study and contributed to the isolation of exosomes; T.H. and L.J. contributed to the animal model. M.Q. contributed to the mRNA analysis. W.L. and L.Z. contributed to the behavior test; Z.Z. and Y.W. helped to design the experiment and interpret the data.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ma Y, Wang J, Wang Y, Yang GY. The biphasic function of microglia in ischemic stroke. Prog Neurobiol. 2017;157:247-72
2. Hu X, Li P, Guo Y. et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063-70
3. Zhang M, Wu X, Xu Y. et al. The cystathionine beta-synthase/hydrogen sulfide pathway contributes to microglia-mediated neuroinflammation following cerebral ischemia. Brain Behav Immun. 2017;66:332-46
4. Hu X, Leak RK, Shi Y. et al. Microglial and macrophage polarization-new prospects for brain repair. Nat Rev Neurol. 2015;11:56-64
5. Liu X, Liu J, Zhao S. et al. Interleukin-4 Is Essential for Microglia/Macrophage M2 Polarization and Long-Term Recovery After Cerebral Ischemia. Stroke. 2016;47:498-504
6. Nikolakopoulou AM, Dutta R, Chen Z, Miller RH, Trapp BD. Activated microglia enhance neurogenesis via trypsinogen secretion. Proc Natl Acad Sci U S A. 2013;110:8714-9
7. Yan YP, Lang BT, Vemuganti R, Dempsey RJ. Galectin-3 mediates post-ischemic tissue remodeling. Brain Res. 2009;1288:116-24
8. Bouhy D, Malgrange B, Multon S. et al. Delayed GM-CSF treatment stimulates axonal regeneration and functional recovery in paraplegic rats via an increased BDNF expression by endogenous macrophages. FASEB J. 2006;20:1239-41
9. Kigerl KA, Gensel JC, Ankeny DP. et al. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435-44
10. Sica A, Melillo G, Varesio L. Hypoxia: a double-edged sword of immunity. J Mol Med (Berl). 2011;89:657-65
11. Zhuang X, Xiang X, Grizzle W. et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol Ther. 2011;19:1769-79
12. Ratajczak MZ, Ratajczak J. Horizontal transfer of RNA and proteins between cells by extracellular microvesicles: 14 years later. Clin Transl Med. 2016;5:7
13. Zhang H, Li Y, Yu J. et al. Rho kinase inhibitor fasudil regulates microglia polarization and function. Neuroimmunomodulation. 2013;20:313-22
14. Xin H, Li Y, Liu Z. et al. MiR-133b promotes neural plasticity and functional recovery after treatment of stroke with multipotent mesenchymal stromal cells in rats via transfer of exosome-enriched extracellular particles. Stem Cells. 2013;31:2737-46
15. Xu B, Zhang Y, Du XF. et al. Neurons secrete miR-132-containing exosomes to regulate brain vascular integrity. Cell Res. 2017;27:882-97
16. Antonucci F, Turola E, Riganti L. et al. Microvesicles released from microglia stimulate synaptic activity via enhanced sphingolipid metabolism. EMBO J. 2012;31:1231-40
17. Yang J, Zhang X, Chen X, Wang L, Yang G. Exosome Mediated Delivery of miR-124 Promotes Neurogenesis after Ischemia. Mol Ther Nucleic Acids. 2017;7:278-87
18. Bang C, Batkai S, Dangwal S. et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124:2136-46
19. Yang G, Chan PH, Chen J. et al. Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke. 1994;25:165-70
20. Li Y, Chen J, Chen XG. et al. Human marrow stromal cell therapy for stroke in rat: neurotrophins and functional recovery. Neurology. 2002;59:514-23
21. Tang Y, Cai B, Yuan F. et al. Melatonin Pretreatment Improves the Survival and Function of Transplanted Mesenchymal Stem Cells after Focal Cerebral Ischemia. Cell Transplant. 2014;23:1279-91
22. Tang Y, Wang J, Lin X. et al. Neural stem cell protects aged rat brain from ischemia-reperfusion injury through neurogenesis and angiogenesis. J Cereb Blood Flow Metab. 2014;34:1138-47
23. Chen Y, Song Y, Huang J. et al. Increased Circulating Exosomal miRNA-223 Is Associated with Acute Ischemic Stroke. Front Neurol. 2017;8:57
24. Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83-90
25. Sareen-Khanna K, Papillon J, Wing SS, Cybulsky AV. Role of the deubiquitinating enzyme ubiquitin-specific protease-14 in proteostasis in renal cells. Am J Physiol Renal Physiol. 2016;311:F1035-F46
26. Franco R, Fernandez-Suarez D. Alternatively activated microglia and macrophages in the central nervous system. Prog Neurobiol. 2015;131:65-86
27. Zhang SC, Fedoroff S. Neuron-microglia interactions in vitro. Acta Neuropathol. 1996;91:385-95
28. Arno B, Grassivaro F, Rossi C. et al. Neural progenitor cells orchestrate microglia migration and positioning into the developing cortex. Nat Commun. 2014;5:5611
29. Mosser CA, Baptista S, Arnoux I, Audinat E. Microglia in CNS development: Shaping the brain for the future. Prog Neurobiol. 2017;149-150:1-20
30. Budnik V, Ruiz-Canada C, Wendler F. Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci. 2016;17:160-72
31. Asai H, Ikezu S, Tsunoda S. et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18:1584-93
32. Chang C, Lang H, Geng N. et al. Exosomes of BV-2 cells induced by alpha-synuclein: important mediator of neurodegeneration in PD. Neurosci Lett. 2013;548:190-5
33. Nampoothiri SS, Rajanikant GK. miR-9 Upregulation Integrates Post-ischemic Neuronal Survival and Regeneration In Vitro. Cell Mol Neurobiol. 2019;39:223-40
34. Siegel G, Saba R, Schratt G. microRNAs in neurons: manifold regulatory roles at the synapse. Curr Opin Genet Dev. 2011;21:491-7
35. Schratt G. microRNAs at the synapse. Nat Rev Neurosci. 2009;10:842-9
36. Wayman GA, Davare M, Ando H. et al. An activity-regulated microRNA controls dendritic plasticity by down-regulating p250GAP. Proc Natl Acad Sci U S A. 2008;105:9093-8
37. Lau P, Bossers K, Janky R. et al. Alteration of the microRNA network during the progression of Alzheimer's disease. EMBO Mol Med. 2013;5:1613-34
38. Lungu G, Stoica G, Ambrus A. MicroRNA profiling and the role of microRNA-132 in neurodegeneration using a rat model. Neurosci Lett. 2013;553:153-8
39. Wong HK, Veremeyko T, Patel N. et al. De-repression of FOXO3a death axis by microRNA-132 and -212 causes neuronal apoptosis in Alzheimer's disease. Hum Mol Genet. 2013;22:3077-92
40. Hwang JY, Kaneko N, Noh KM, Pontarelli F, Zukin RS. The gene silencing transcription factor REST represses miR-132 expression in hippocampal neurons destined to die. J Mol Biol. 2014;426:3454-66
41. Hu X, Liu J, Zhao G, Zheng J, Qin X. Long non-coding RNA GAS5 aggravates hypoxia injury in PC-12 cells via down-regulating miR-124. J Cell Biochem. 2018;119:6765-74
42. Geng L, Liu W, Chen Y. miR-124-3p attenuates MPP(+)-induced neuronal injury by targeting STAT3 in SH-SY5Y cells. Exp Biol Med (Maywood). 2017;242:1757-64
43. Veremeyko T, Siddiqui S, Sotnikov I, Yung A, Ponomarev ED. IL-4/IL-13-dependent and independent expression of miR-124 and its contribution to M2 phenotype of monocytic cells in normal conditions and during allergic inflammation. PLoS One. 2013;8:e81774
44. Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat Med. 2011;17:64-70
45. Veremeyko T, Kuznetsova IS, Dukhinova M. et al. Neuronal extracellular microRNAs miR-124 and miR-9 mediate cell-cell communication between neurons and microglia. J Neurosci Res. 2019;97:162-84
46. Doeppner TR, Kaltwasser B, Sanchez-Mendoza EH. et al. Lithium-induced neuroprotection in stroke involves increased miR-124 expression, reduced RE1-silencing transcription factor abundance and decreased protein deubiquitination by GSK3beta inhibition-independent pathways. J Cereb Blood Flow Metab. 2017;37:914-26
47. Sun Y, Gui H, Li Q. et al. MicroRNA-124 protects neurons against apoptosis in cerebral ischemic stroke. CNS Neurosci Ther. 2013;19:813-9
48. Doeppner TR, Doehring M, Bretschneider E. et al. MicroRNA-124 protects against focal cerebral ischemia via mechanisms involving Usp14-dependent REST degradation. Acta Neuropathol. 2013;126:251-65
49. Hwang DW, Choi H, Jang SC. et al. Noninvasive imaging of radiolabeled exosome-mimetic nanovesicle using (99m)Tc-HMPAO. Sci Rep. 2015;5:15636
50. Lai CP, Tannous BA, Breakefield XO. Noninvasive in vivo monitoring of extracellular vesicles. Methods Mol Biol. 2014;1098:249-58
51. Takahashi Y, Nishikawa M, Shinotsuka H. et al. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. J Biotechnol. 2013;165:77-84
52. Wiklander OP, Nordin JZ, O'Loughlin A. et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles. 2015;4:26316
53. Suetsugu A, Honma K, Saji S. et al. Imaging exosome transfer from breast cancer cells to stroma at metastatic sites in orthotopic nude-mouse models. Adv Drug Deliv Rev. 2013;65:383-90
54. Hoshino A, Costa-Silva B, Shen TL. et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329-35
55. Alvarez-Erviti L, Seow Y, Yin H. et al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341-5
56. Wen Z, Shimojima Y, Shirai T. et al. NADPH oxidase deficiency underlies dysfunction of aged CD8+ Tregs. J Clin Invest. 2016;126:1953-67
57. Skog J, Wurdinger T, van Rijn S. et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470-6
58. Zhu X, Badawi M, Pomeroy S. et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J Extracell Vesicles. 2017;6:1324730
59. Saraiva C, Esteves M, Bernardino L. MicroRNA: Basic concepts and implications for regeneration and repair of neurodegenerative diseases. Biochem Pharmacol. 2017;141:118-31
60. Hamzei Taj S, Kho W, Riou A, Wiedermann D, Hoehn M. MiRNA-124 induces neuroprotection and functional improvement after focal cerebral ischemia. Biomaterials. 2016;91:151-65
61. Huang S, Ge X, Yu J. et al. Increased miR-124-3p in microglial exosomes following traumatic brain injury inhibits neuronal inflammation and contributes to neurite outgrowth via their transfer into neurons. FASEB J. 2018;32:512-28
62. Yu A, Zhang T, Duan H. et al. MiR-124 contributes to M2 polarization of microglia and confers brain inflammatory protection via the C/EBP-alpha pathway in intracerebral hemorrhage. Immunol Lett. 2017;182:1-11
63. Lee MJ, Lee BH, Hanna J, King RW, Finley D. Trimming of ubiquitin chains by proteasome-associated deubiquitinating enzymes. Mol Cell Proteomics. 2011;10:R110 003871
64. Xu D, Shan B, Sun H. et al. USP14 regulates autophagy by suppressing K63 ubiquitination of Beclin 1. Genes Dev. 2016;30:1718-30
65. Lee BH, Lee MJ, Park S. et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179-84
66. Lapchak PA, Han MK. Simvastatin improves clinical scores in a rabbit multiple infarct ischemic stroke model: synergism with a ROCK inhibitor but not the thrombolytic tissue plasminogen activator. Brain Res. 2010;1344:217-25
67. Temchura VV, Tenbusch M, Nchinda G. et al. Enhancement of immunostimulatory properties of exosomal vaccines by incorporation of fusion-competent G protein of vesicular stomatitis virus. Vaccine. 2008;26:3662-72
68. Wilson SM, Bhattacharyya B, Rachel RA. et al. Synaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin-specific protease. Nat Genet. 2002;32:420-5
69. Min JW, Lu L, Freeling JL, Martin DS, Wang H. USP14 inhibitor attenuates cerebral ischemia/reperfusion-induced neuronal injury in mice. J Neurochem. 2017;140:826-33
Author contact
![]() Corresponding author: Zhijun Zhang, Yaohui Tang, and Guo-Yuan Yang, Med-X Research Institute and School of Biomedical Engineering, Shanghai Jiao Tong University, Shanghai 200030, China. Tel: +86-21-62933186 Fax: +86-21-62932302 E-mail: gyyangedu.cn, yaohuitangedu.cn and zhangzjedu.cn
Corresponding author: Zhijun Zhang, Yaohui Tang, and Guo-Yuan Yang, Med-X Research Institute and School of Biomedical Engineering, Shanghai Jiao Tong University, Shanghai 200030, China. Tel: +86-21-62933186 Fax: +86-21-62932302 E-mail: gyyangedu.cn, yaohuitangedu.cn and zhangzjedu.cn