Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results and Discussion
Conclusions
Acknowledgements
Supplementary Material
References
Introduction
Materials and Methods
Results and Discussion
Conclusions
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(15):4450-4460. doi:10.7150/thno.35393 This issue Cite
Research Paper
Controllable Autocatalytic Cleavage-Mediated Fluorescence Recovery for Homogeneous Sensing of Alkyladenine DNA Glycosylase from Human Cancer Cells
Li-Juan Wang1*, Ming-Li Luo1*, Xiao-Yun Yang2*, Xiao-Fang Li2*, Yanxia Wu2, Chun-Yang Zhang1 ![]()
1. College of Chemistry, Chemical Engineering and Materials Science, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Key Laboratory of Molecular and Nano Probes, Ministry of Education, Shandong Provincial Key Laboratory of Clean Production of Fine Chemicals, Shandong Normal University, Jinan 250014, PR China
2. Department of Pathology, Affiliated Hospital of Guangdong Medical University, Zhanjiang 524001, China
*These authors contributed equally to this work.
Received 2019-4-1; Accepted 2019-5-17; Published 2019-6-9
Citation:
Wang LJ, Luo ML, Yang XY, Li XF, Wu Y, Zhang CY. Controllable Autocatalytic Cleavage-Mediated Fluorescence Recovery for Homogeneous Sensing of Alkyladenine DNA Glycosylase from Human Cancer Cells. Theranostics 2019; 9(15):4450-4460. doi:10.7150/thno.35393. https://www.thno.org/v09p4450.htm
Other stylesAbstract

DNA alkylation and oxidation are two most common forms of cytotoxic damage with the characteristics of mutagenic and carcinogenic. Human alkyladenine DNA glycosylase (hAAG) is the only glycosylase known to repair a wide variety of alkylative and oxidative DNA lesions. However, few approaches are capable of real-time monitoring hAAG activity.
Methods: Herein, we develop a facile fluorescent strategy for homogeneous and sensitive sensing of hAAG activity based on the controllable autocatalytic cleavage-mediated fluorescence recovery. The presence of hAAG enables the cleavage of hairpin probe 1 (HP1) at the damaged 2′-deoxyinosine site by AP endonuclease 1 (APE1), forming a DNA duplex. The trigger 1 built in the resultant DNA duplex may hybridize with hairpin probe 2 (HP2) to induce the T7 exonuclease (T7 exo)-catalyzed recycling cleavage of HP2 (Cycle I) to release trigger 2. The trigger 2 can further hybridize with the signal probe (a fluorophore (FAM) and a quencher (BHQ1) modified at its 5′ and 3′ ends) to induce the subsequent recycling cleavage of signal probes (Cycle II) to liberate FAM molecules. Through two-recycling autocatalytic cleavage processes, large amounts of fluorophore molecules (i.e., FAM) are liberated from the FAM-BHQ1 fluorescence resonance energy transfer (FRET) pair, leading to the amplified fluorescence recovery.
Results: Taking advantage of the high accuracy of in vivo DNA repair mechanism, the high specificity of T7 exo-catalyzed mononucleotides hydrolysis, and the high efficiency of autocatalytic recycling amplification, this strategy exhibits high sensitivity with a detection limit of 4.9 × 10-6 U/μL and a large dynamic range of 4 orders of magnitude from 1 × 10-5 to 0.1 U/μL, and it can further accurately evaluate the enzyme kinetic parameters, screen the potential inhibitors, and even quantify the hAAG activity from 1 cancer cell.
Conclusion: The proposed strategy can provide a facile and universal platform for the monitoring of DNA damage-related repair enzymes, holding great potential for DNA repair-related biochemical research, clinical diagnosis, drug discovery, and cancer therapy.
Keywords: DNA alkylation and oxidation, alkyladenine DNA glycosylase, autocatalytic recycling amplification, fluorescence recovery, clinical diagnosis
Introduction
The Watson-Crick-pairing bases in DNA double helix constitute the genetic codes that are critically important for all organisms [1]. However, the heterocyclic bases (i.e., A, T, G, and C) contain an large number of nucleophilic / redox-active sites, which are susceptible to the reactions with the extracellularly and endogenously produced electrophiles / reactive oxygens to yield a vast array of DNA alkylative and oxidative lesions (e.g., 3-alkyladenine, 7-alkylguanine, 1-N2-ethenoguanine, 1-N6-ethenoadenine, and hypoxanthine) [2-4]. Recent researches indicate that the alkylative and oxidative lesions may either block DNA replication by impeding polymerases or cause mutations by creating abasic sites and eventually induce carcinogenesis progressions [3, 5-7]. Human alkyladenine DNA glycosylase (hAAG) is the only DNA glycosylase identified to date in human cells that can specially recognize and structurally excise a wide variety of DNA alkylative and oxidative lesions [8], and its aberrant activity may cause the malfunction of alkylated DNA damage repair, resulting in multiple human diseases (e.g., chronic inflammation, Crohn's and Wilson's diseases) [9-11] and cancers (e.g., colon, liver, lung, cervix, glioblastoma, lymphomas, melanoma and leukemia cancers) [10-14]. Therefore, hAAG has been proposed as a biomarker in the alkylation- and oxidation-mediated diseases. The importance of hAAG has provided the impetus for developing accurate quantification methods.
Despite the important roles of hAAG in biomedical and clinical researches, few approaches are capable of real-time monitoring the hAAG activity due to the limited knowledge about its catalytic pathway. The reported methods for hAAG assay are usually based on gel electrophoresis using the labeled (γ-32P) DNA substrates [15, 16], immunoblot analysis with the horseradish peroxidase-conjugated immunoglobulins [17], single-molecule counting by DNA repair-responsive molecular beacons [12], and fluorescence monitoring through target-directed hyperbranched amplifcation / base-excision repair-mediated triple amplification [18, 19]. However, the gel-based radiometric assays [15, 16] usually suffer from the hazardous radiation, complicated manipulation, and time-consuming procedures; The enzyme-linked immunoassay [17] generally requires the costly protein antibodies, stringent reaction conditions, and labor-consuming operations. Notably, these assays are only capable of semi-quantitatively detecting hAAG activity. Even though some reported fluorescence assays [12, 18, 19] can quantitatively measure hAAG activity with improved sensitivity, the single-molecule counting-based fluorescence assay [12] involves the complicated molecular beacon designs, expensive instruments, and sophisticated manipulation, and the hyperbranched amplifcation- / triple amplification-based fluorescence assays [18, 19] require multiple tool enzymes, complicated probe / template preparations, and cumbrous experimental procedures. Thus, the development of facile, accurate and sensitive methods for hAAG assay is highly desirable.
T7 exonuclease (T7 exo) is a unique sequence-independent nuclease, and it can selectively catalyze the stepwise hydrolysis of mononucleotides from 5′-phosphoryl (PO4) / 5′-hydroxyl (OH) termini / at gaps and nicks of the double-stranded DNA (dsDNA), but it does not act on the single-stranded DNA (ssDNA) [20]. Moreover, T7 exo can degrade RNA and DNA from the RNA/DNA hybrids in the 5' to 3' direction, but it is not active on the double-stranded and single-stranded RNA [21]. Importantly, T7 exo exhibits high resolution in discriminating single-base mismatch [22]. Taking advantage of its unique characteristics (e.g., sequence independence, substrate diversity, high catalytic activity, and excellent single-base discrimination), T7 exo is ideal for constructing the cyclic enzymatic signal amplification (CESA) systems [23]. The CESA systems are generally based on nucleases, in which one input target may lead to multiple cycles of target-dependent nuclease cleavage of reporter probes for the amplified output signal, enabling simple, rapid and sensitive detection of nucleic acids [24-26]. Recently, nucleases such as exonuclease III (Exo III) [27, 28], deoxyribonuclease I (DNase I) [29], duplex-specific nuclease (DSN) [30, 31] and endonucleases (e.g., Nb.BbvCI [32], Nt.AiwI [33], and Fok I [34] have been recruited to fabricate different CESA systems for the detection of various biomolecules including nucleotides [28-31, 34], enzymes [27, 33], proteins [32], and small molecules [32]. Due to the intrinsic properties of nucleases, these CESA systems have some unavoidable limitations in practical applications. For example, Exo III as the most frequently used tool nuclease can remove mononucleotides from the blunt /recessed 3′-OH termini of dsDNA, but it requires 4 bases / longer 3′-protruding sequence to prevent the cleavage, complicating the probes design [35]. In addition, Exo III activity may be disturbed by many factors including sequence structure, salt concentration and ratio of enzyme to DNA substrates [36]. DNase I not only nonspecifically digests the dsDNA into oligodeoxyribonucleotides, but also degrades the ssDNA, and thus it is more suitable for purifying the contaminant DNA from the non-RNA preparations [29]. DSN can specifically degrade DNA in both dsDNA and DNA-RNA hybrids, with little activity on single-stranded nucleic acids and double-stranded RNA, but it exhibits cleavage activity only towards the sequences of dsDNA and DNA-RNA hybrids longer than 10 bp and 15 bp, respectively [37]. For endonucleases (e.g., Nb.BbvCI [32], Nt.AiwI [33] and Fok I [38], they are a special family of restriction endonucleases that can recognize a specific sequence known as a restriction site along a dsDNA and they only cut one strand to leave a nick in dsDNA, but they all have strict requirements of the sequence and length in DNA probe design. To overcome the limitations of above nucleases, we alternatively utilize T7 exo to construct a new CESA system for facile, homogeneous and sensitive detection of human alkyladenine DNA glycosylase (hAAG) based on the controllable autocatalytic cleavage-mediated fluorescence recovery. Upon the specific cleavage of hairpin substrate (i.e., hairpin probe 1 (HP1)) at the damaged 2′-deoxyinosine site by hAAG and APE1, the hairpin structure of HP1 is unfolded to generate a DNA duplex. Trigger 1 built in the resultant DNA duplex may partially hybridize with HP2 through the toehold-mediated strand displacement reaction (TMSDR) to induce the T7 exo-catalyzed recycling cleavage of HP2 to release trigger 2. Trigger 2 is partially complementary with the signal probe (a 10-nt DNA fragment modified by a fluorophore (FAM) and a quencher (BHQ1) at its 5′ and 3′ ends), and it can induce the subsequent recycling cleavage of signal probes. Through two-recycling autocatalytic cleavage processes, large amounts of fluorophore molecules (i.e., FAM) are liberated from the FAM-BHQ1 fluorescence resonance energy transfer (FRET) pair, leading to the amplified fluorescence recovery. Taking advantage of the ubiquitous DNA repair mechanisms in vivo, the excellent features of T7 exo and the intrinsic superiorities of fluorescence strategy, the proposed method can provide a facile and robust biosensing platform for homogeneous detection of hAAG activity with high sensitivity and good specificity.
Materials and Methods
Chemicals and materials
Human alkyladenine DNA glycosylase (hAAG), 10× ThermoPol reaction buffer (200 mM trizma hydrochloride (Tris-HCl), 100 mM ammonium sulfate ((NH4)2SO4), 100 mM potassium chloride (KCl), 20 mM magnesium sulfate (MgSO4), 1% Triton X-100, pH 8.8), human apurinic/apyrimidinic endonuclease (APE1), 10× NEBuffer 4 (500 mM potassium acetate (KAc), 200 mM tris-acetate (Tris-Ac), 100 mM magnesium acetate (Mg(Ac)2), 10 mM DL-Dithiothreitol (DTT), pH 7.9), T7 exonuclease (T7 exo), and uracil-DNA glycosylase (UDG) were purchased from New England BioLabs (Beverly, MA, USA). Thymine DNA glycosylase (TDG) was bought from R&D System (Minneapolis, Minnesota, USA). Tris-HCl (PH 8.0), ammonium sulfate ((NH4)2SO4), MgSO4, sodium chloride (NaCl), magnesium chloride (MgCl2), chromium(II) chloride (CdCl2), and bovine serum albumin (BSA) were obtained from Sigma Aldrich Company (St. Louis, MO, USA). SYBR Gold was bought from Life Technologies (Carlsbad, CA, USA). The rabbit anti-hAAG polyclonal antibody was obtained from Shenzhen ZIKER Biotech Co., Ltd. (Shenzhen, China). The actin and histone H3 were bought from Wuhan Service Biotech Co., Ltd. (Wuhan, China). All other chemicals and solvents with analytical grade were used as received without further purification. Solutions were prepared using high-purity water obtained from a Millipore filtration system (Millipore, Milford, MA, USA). Human cervical carcinoma cell line (HeLa cells) and lung adenocarcinoma cell line (A549 cells) were bought from Cell Bank, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China). The oligonucleotides (Table 1) were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China).
Table 1
Sequences of the Oligonucleotidesα
| note | sequences (5'-3') |
|---|---|
| hairpin probe 1 | GTA GTG AGG TAG GTT GTA TIG TTG GGT TGA ACT ATA CAA CCT ACC |
| hairpin probe 2 | TGT ATA GTT CAA CCC GGG ACC TAA GAG CAT TCT ACA CCT CTT AGG TCC CTG C |
| signal probe | FAM-AAG AGG TGT A-BHQ1 |
| cleavage product | GTA GTG AGG TAG GTT GTA T |
| trigger 1 | GTT GGG TTG A AC TAT ACA ACC TAC C |
| trigger 2 | CAT TCT ACA CCT CTT AGG TCC CTG C |
αIn hairpin probe 1 (HP1), the underlined base “I” indicates a 2′-deoxyinosine, and the italicized regions indicate the complementary sequences. In hairpin probe 2 (HP2), the italicized regions indicate the complementary sequences. In signal probe, a 6-carboxy-fluorescein (FAM) and a black hole quencher 1 (BHQ1) are modified at the 5' and 3' ends, respectively.
DNA repair-controlled T7 exo-assisted autocatalytic recycling signal amplification
Before performing the amplification reaction, all synthesized oligonucleotides were dissolved in 1× Tris-EDTA as the stock solutions, stored at -20 °C. The hairpin probes (i.e., HP1 and HP2) were diluted to 1 μM with hybridization buffer (10 mM Tris-HCl, 1.5 mM MgCl2, pH 8.0), and incubated at 95 °C for 5 min, followed by slowly cooling to room temperature for folding into a perfect hairpin structure. The hAAG assay involves two consecutive reaction steps: (1) 2 µL of HP1 (1 μM) were added into the excision reaction system (20 µL) including variable-concentration hAAG, 2 µL of 10× ThermoPol reaction buffer, 0.3 U/µL APE1, 2 µL of 10× NEBuffer 4, incubated at 37 °C for 60 min; (2) 5 μL of above excision products was added into the amplification reaction system (20 µL) including 250 nM HP2, 700 nM signal probes, 15 U of T7 exo, 2 µL of 10× NEBuffer 4, followed by incubation at 25 °C for 50 min in the dark for T7 exo-assisted autocatalytic recycling amplification reaction.
Fluorescence measurement and gel electrophoresis
The 20 µL of amplification products were diluted to 60 µL with ultrapure water. At the excitation wavelength of 491 nm, the fluorescence spectra were scanned in the range from 505 to 620 nm by a HitachiF-7000 fluorescence spectrophotometer (Tokyo, Japan). The fluorescence intensity at 520 nm was recorded for data analysis. To analyze the excision products and the amplification products, 12% nondenaturating polyacrylamide gel electrophoresis (PAGE) was performed in 1× TBE buffer containing 9 mM Tris-HCl, 0.2 mM EDTA, 9 mM boric acid, pH 7.9, with SYBR Gold as the fluorescent indicator. After electrophoresis at 110 V constant voltages for 45 min at room temperature, the gels were imaged with a ChemiDoc MP Imaging System (Hercules, California, USA).
Kinetic assay
The hAAG-catalyzed 2′-deoxyinosine-excision reaction was carried out at 37 °C in 20 μL of excision reaction system (0.1 U/µL hAAG, 2 µL of 10× ThermoPol reaction buffer, 0.3 U/µL APE1, 2 µL of 10× NEBuffer 4 and variable-concentration HP1) for 5 min. After excision reaction, the excision products were added into the amplification reaction system (250 nM HP2, 700 nM signal probes, 15 U of T7 exo, 2 µL of 10× NEBuffer 4), and the hAAG activity was measured according to the procedures described above. The obtained experimental data were fitted to the equation 1 to obtain the enzyme kinetic parameters:
V = Vmax[S] / (Km + [S]) (1)
where Vmax is the maximum initial velocity, [S] is the concentration of DNA substrate, and Km is the Michaelis-Menten constant corresponding to the concentration at half-maximal velocity.
Inhibition assay
Variable-concentration CdCl2 was incubated with 0.1 U/µL hAAG at 37 °C for 15 min. Then 2 µL of 10× ThermoPol reaction buffer, 0.3 U/µL APE1, 2 µL of 10× NEBuffer 4, and 100 nM HP1 were added into the reaction mixture, followed by incubation at 37 °C for 60 min. After the excision reaction, 5 μL of excision products was added into the amplification reaction system (250 nM HP2, 700 nM signal probes, 15 U of T7 exo, and 2 µL of 10× NEBuffer 4) for hAAG assay according to the procedures described above. The relative activity (RA) of hAAG was determined based on the equation 2:
RA = (Fi - Fo) / (Ft - Fo) (2)
where F0 is the fluorescence intensity in the absence of hAAG, and Ft is the fluorescence intensity in the presence of 0.1 U/µL hAAG , and Fi is the fluorescence intensity in the presence of both 0.1 U/µL hAAG and variable-concentration CdCl2. The half maximal inhibition (IC50) was determined from the fitted curve of RA versus the CdCl2 concentration.
Cell culture and preparation of cellular extracts
Human cervical carcinoma cell line (HeLa cells) and lung adenocarcinoma cell line (A549 cells) were cultured with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Gibco, USA) in Dulbecco's modified Eagle's medium (DMEM) at 37 °C in a humidified chamber with 5% CO2. In the exponential phase of growth, HeLa and A549 cells were collected and washed with ice-cold PBS (pH 7.4, Gibco, USA), followed by centrifugation at 800 rpm for 5 min. The hAAG enzyme was extracted by a nuclear extract kit (40010) (Active Motif, Carlsbad, CA, USA). The obtained supernatant was immediately subjected to the hAAG activity assay.
Western blotting and ELISA analyses
For western blotting analysis, the rabbit anti-hAAG polyclonal antibody (ZIKER-2412R, ZIKER Bio, Shenzhen, China) was used against hAAG expressed in HeLa cells. HeLa cells were collected, and the hAAG enzyme was extracted from the cytoplasm, nucleus, and whole cell, respectively, with the nuclear extract kit (40010) according to the procedures described above. The obtained supernatants from different parts of HeLa cells were analyzed by western blotting. With actin (GB12001, Servicebio, Wuhan, China) and histone H3 (RLM3038, RuiYing Bio, Wuhan, China) as the internal reference proteins, the levels of hAAG protein from cytoplasm, nucleus and whole cell extracts were evaluated with a western blot detection kit (E-IR-R304A) (Elabscience, Wuhan, China). The immune complexes were detected by an excellent chemiluminescent substrate detection kit (E-BC-R347) (Elabscience, Wuhan, China), and the protein strips can be clearly displayed on the X-ray film. The intensities of strips were determined by densitometic scanning on Epson V300 scanner (Epson, Suwa, Japan) and quantified by Alpha Ease FC software (Alpha Innotech, San. Leandro, CA, USA). For ELISA analysis, the supernatants from different parts of 1000 HeLa cells were obtained according to the same procedures described above, and analyzed by using an ELISA kit (ZK-H2553) (ZIKER Bio, Shenzhen, China). The corresponding optical densities (O.D.) were quantified by using a SpectraMax i3× multi-mode microplate reader (Molecular Devices, San. Jose, CA, USA) at a wavelength of 450 nm.
Results and Discussion
HAAG-catalyzed damaged base-excision repair
HAAG is a type of monofunctional DNA glycosylates with only glycosylase activity [39]. As shown in Figure 1, upon the exposure of genomic DNA to alkylative or oxidative damages, hAAG can specifically recognize and excise lesions by flipping the damaged nucleotides 180° out of the double helix and hydrolyzing the C1-N glycosidic bond, leaving an apurinic / apyrimidinic (AP) site [42]. The AP site can be recognized and excised by human AP endonuclease 1 (APE1) through cleaving the phosphodiester bond 5′ to the damaged site, leaving 5′-deoxyribose phosphate (5′-dRP) and 3′-OH termini [39]. The subsequent repair process will be completed by DNA polymerase and DNA ligase.
Figure 1
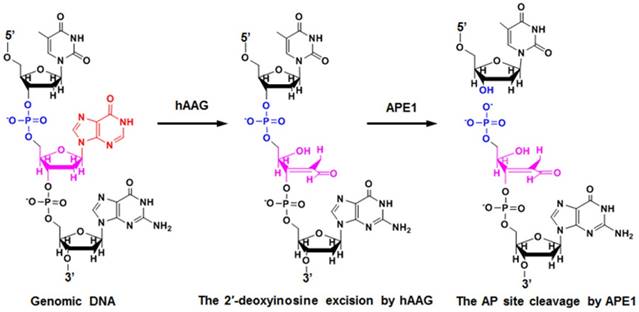
Mechanism of hAAG-catalyzed base-excision repair. The hAAG can remove the damaged 2′-deoxyinosine (red color) to produce an AP site. The AP site will subsequently be cleaved by APE1 to generate the 5′-dRP and 3′-OH termini.
Principle of hAAG assay
The principle of the controllable autocatalytic cleavage-induced fluorescence recovery for hAAG assay is illustrated in Scheme 1. We designed two hairpin probes (i.e., HP1 and HP2) with a protruding 5′-OH terminus for the prevention of the cleavage by T7 exo [20]. In HP1, one 2′-deoxyinosine (I) lesion is modified on the stem at the site 3 bases away from the loop, which functions as the recognition site for hAAG [12]. The signal probe is a 10-nt DNA sequence with a 6-carboxy-fluorescein (FAM) and a black hole quencher 1 (BHQ1) modified at the 5′ and 3′ termini, respectively, with the fluorescence of FAM being quenched by BHQ1 through the fluorescence energy resonance transfer (FRET) effect [40]. T7 exo is the “heart” of this CESA system, and it is in charge of the running of autocatalytic recycling signal amplification. The proposed strategy mainly involves two consecutive reaction steps: (1) the specific cleavage of HP1 at 2′-deoxyinosine site by hAAG and APE1, and (2) T7 exo-assisted autocatalytic recycling signal amplification. In the presence of hAAG, it will specifically recognize and cleave the HP1 at the 2′-deoxyinosine site (I, Scheme 1, green color), unfolding the hairpin structure, and simultaneously generating a dsDNA duplex (І) with the protruding 5′-OH and 5′-dRP) termini. Because the red region of resultant dsDNA (І) is complementary with the yellow region of hairpin probe 2 (HP2), one strand with the protruding 5′-dRP termini (i.e., trigger 1) in dsDNA (І) will partially hybridize with the stem of HP2 through the TMSDR, forming a dsDNA duplex (ІI) with a recessed 5′-OH terminus. The resultant dsDNA duplex (ІI) can function as the substrate of T7 exo and is specifically hydrolyzed from the recessed 5′-OH terminus in HP2, releasing trigger 1 and trigger 2 which is originally built in HP2. Because the signal probe (Scheme 1, purple color) is complementary with the pink region of trigger 2, the released trigger 2 will hybridize with the signal probe to form a dsDNA duplex (III) with a recessed 5′-OH terminus, which can act as the substrate of T7 exo for the initiation of hydrolyzation from the recessed 5′-OH terminus of signal probe. As a result, the signal probe in dsDNA (III) is selectively hydrolyzed to liberate the FAM molecule, and the trigger 2 remains intact to be released from the dsDNA (III) simultaneously. Benefiting from strategically keeping the 5′-OH termini in the protruding states during the autocatalytic hydrolysis process, trigger 1 and trigger 2 in dsDNA duplexes are protected from the cleavage by T7 exo. Consequently, the released trigger 1 can partially hybridize with the excess HP2 to induce T7 exo-catalyzed recycling cleavage of HP2 (Cycle I) to release multiple trigger 2, and the released trigger 2 can subsequently hybridize with free signal probes to induce T7 exo-catalyzed recycling cleavage of signal probes (Cycle II) to liberate numerous FAM molecules. Through two-recycling autocatalytic cleavage processes, large amounts of fluorophore molecules (i.e., FAM) are liberated from the FAM-BHQ1 FRET pair, leading to a significantly amplified fluorescence signal. In the absence of hAAG, the 2′-deoxyinosine lesion cannot be recognized and the HP1 cannot be cleaved by APE1. The HP1 and HP2 with the protruding 5′-OH termini may hinder T7 exo-assisted autocatalytic recycling cleavage process without the generation of an amplified fluorescence signal. Taking advantage of the high accuracy of in vivo DNA repair mechanism, the high specificity of T7 exo-catalyzed mononucleotides hydrolysis in DNA duplex, and the high efficiency of autocatalytic recycling amplification reaction, the proposed strategy provides a facile and robust platform for the detection of hAAG activity with high specificity and sensitivity.
Scheme 1
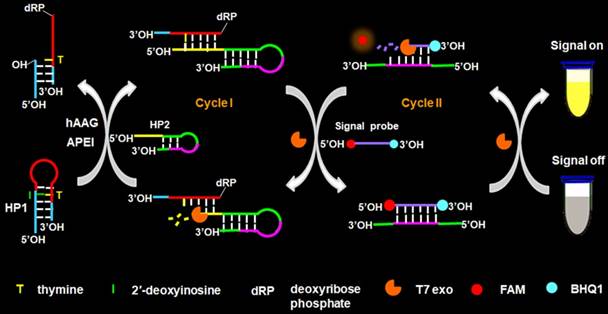
Schematic illustration of the controllable autocatalytic cleavage-induced fluorescence recovery for hAAG assay. This strategy involves two consecutive steps: (1) the specific cleavage of HP1 at 2′-deoxyinosine site by hAAG and APE1, and (2) T7 exo-assisted autocatalytic recycling signal amplification.
Validation of the hAAG assay
We used nondenaturating gel electrophoresis to investigate whether hAAG can catalyze the cleavage of HP1 at the 2′-deoxyinosine site with the coexistence of APE1 to induce the subsequent T7 exo-assisted autocatalytic recycling cleavage reaction. As shown in Figure 2A, in the presence of APE1 + HP1, only one band from the original HP1 (45 nt) (Figure 2A, lane 3) is observed (Figure 2A, lane 4), indicating that the absence of hAAG cannot induce the cleavage of HP1 at the 2′-deoxyinosine site. While in the presence of hAAG + APE1 + HP1, the characteristic bands of 19 nt and 25 nt are detected (Figure 2A, lane 5), which are exactly the sizes of the 19-nt cleavage product (Figure 2A, lane 1) and the 25-nt trigger 1 (Figure 2A, lane 2), demonstrating that hAAG can specifically excise the 2′-deoxyinosine and induce the subsequent cleavage of HP1 by APE1 to generate two short DNA fragments (i.e., the 19-nt cleavage product and the 25-nt trigger 1). To investigate the subsequent T7 exo-directed autocatalytic recycling cleavage process, 5 μL of the above cleavage products (Figure 2A, lanes 4 and 5) were added into 20 µL of amplification reaction system containing HP2, signal probe and T7 exo for the autocatalytic recycling cleavage reaction. Upon the termination of reaction, nondenaturating gel electrophoresis was performed to analyze the reaction products. As shown in Figure 2B, in the absence of hAAG, only the band of HP2 (52 nt) is detected (Figure 2B, lane 1), suggesting that no cleavage reaction happens. While in the presence of hAAG, another characteristic band (25 nt) is observed (Figure 2B, lane 3), with the same size of the synthesized trigger 2 (25 nt) (Figure 2B, lane 2), indicating that the 25-nt trigger 1 (i.e., hAAG- and APE1-catalyzed the cleavage product of HP1) can partially hybridize with HP2 through TMSDR to initiate the T7 exo-catalyzed recycling cleavage of HP2 to release large amounts of trigger 2 (25 nt, Figure 2B, lane 3). These results (Figures 2A and 2B) clearly demonstrate that hAAG and APE1 can catalyze the cleavage of HP1 at the 2′-deoxyinosine site to generate a 25-nt trigger 1 for the initiation of subsequent T7 exo-directed autocatalytic recycling cleavage of HP2 to release trigger 2. Notably, no extra band is observed in the presence of APE1 + HP1 (Figure 2A, lane 4), hAAG + APE1 + HP1 (Figure 2A, lane 5), and hAAG + APE1 + T7 exo + HP1 + HP2 (Figure 2B, lane 3), suggesting the high specificity of the DNA repair-controlled T7 exo-assisted autocatalytic recycling amplification without the generation of nonspecific DNA fragments. The extremely high specificity may be attributed to three factors: (1) the high accuracy of in vivo DNA repair mechanism, (2) the high specificity of hAAG-catalyzed 2′-deoxyinosine excision, and (3) the high resolution of T7 exo-mediated single-base mismatch discrimination.
Detection sensitivity of hAAG assay
To achieve the best assay performance, we optimized the experimental conditions including the amplification reaction time, HP2 concentration and signal probe concentration, and the amount of T7 exo (Figures S1-S4, see Supporting Information). Under the optimally experimental conditions, we measured the variance of fluorescence spectra with different concentrations of hAAG (Figure 3A). With the hAAG concentration increasing from 1 × 10-5 to 0.1 U/μL, the fluorescence intensity enhances correspondingly (Figure 3B). Notably, a good linear relationship between the fluorescence intensity and the logarithm of hAAG concentration is obtained over a large dynamic range of 4 orders of magnitude from 1 × 10-5 to 0.1 U/μL (inset of Figure 3). The linear regression equation is F = 5137.9 + 901.8 log10 C with a correlation coefficient of 0.9942, where F represents the fluorescence intensity and C represents the hAAG concentration (U/μL). The detection limit is calculated to be as low as 4.9 × 10-6 U/μL by evaluating the average value of the negative control plus three times standard deviation. The sensitivity of this strategy is superior to those of the gel-based radiometric assays [15, 16], the immunoassay [17], and is improved by 18.4-fold compared with that of target-directed hyperbranched amplifcation-based fluorescence assay (9.0 × 10-5 U/μL) [18], 5.3-fold compared with that of base-excision repair-mediated triple amplification-based fluorescence assay (2.6 × 10-5 U/μL) [19], and is even comparable to that of single-molecule counting-based fluorescence assay (8.7 × 10-7 U/μL) [12]. The high sensitivity can be ascribed to the following factors: (1) the high accuracy of in vivo DNA repair mechanism, (2) the high specificity of T7 exo-catalyzed mononucleotides hydrolysis in DNA duplex, and (3) the high efficiency of autocatalytic recycling amplification. In addition, we evaluated the reproducibility of this strategy through 5 successive assays of 0.1 U/µL hAAG. The relative standard deviation (RSD) is determined to be 2.63%.
Selectivity of hAAG assay
To investigate the selectivity of the proposed assay, we used the nonspecific proteins (e.g., bovine serum albumin (BSA) [41], immunoglobulin G (IgG) [42], DNA glycosylases (thymine DNA glycosylase (TDG) [40], uracil-DNA glycosylase (UDG) [41], and 8-oxoguanine-DNA glycosylase (hOGG1)) [42] as the interferences. As shown in Figure 4, only an extremely low fluorescence signal is observed in the presence of BSA (Figure 4, pink column), IgG (Figure 4, green column), TDG (Figure 4, purple column), UDG (Figure 4, yellow column), hOGG1 (Figure 4, blue column), and the control with only reaction buffer (Figure 4, black column), respectively. While in the presence of hAAG, a significantly enhanced fluorescence signal is detected (Figure 4, red column), which is 11.6, 13.1, 12.8, 13.0, 11.7, and 15.3-fold higher than that in response to BSA, IgG, TDG, UDG, hOGG1, and the control, respectively, suggesting that only hAAG can specifically recognize and excise hairpin substrate (i.e., HP1) at 2′-deoxyinosine site to initiate the subsequent T7 exo-directed autocatalytic recycling cleavage-induced fluorescence signal amplification. These results demonstrate that the proposed strategy can discriminate hAAG against interference proteins with high specificity, holding great potential in biomedical applications.
Figure 2
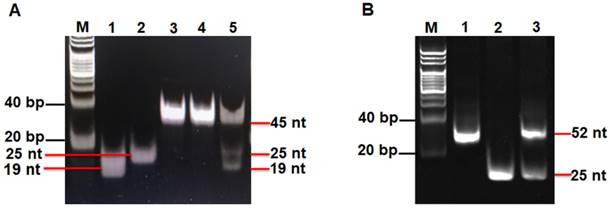
(A) Nondenaturing PAGE analysis of the products of hAAG-catalyzed cleavage reaction under different reaction conditions. Lane M, the DNA ladder marker; lane 1, the synthesized cleavage product; lane 2, the synthesized trigger 1; lane 3, the synthesized HP1; lane 4, in the presence of APE1 + HP1; lane 5, in the presence of hAAG + APE1 + HP1. (B) Nondenaturing PAGE analysis of the products of T7 exo-assisted autocatalytic recycling cleavage reaction. Lane M, the DNA ladder marker; lane 1, in the absence of hAAG; lane 2, the synthesized trigger 2; lane 3, in the presence of hAAG. SYBR Gold is used as the fluorescent indicator. The 0.1 U/µL hAAG and 0.3 U/µL APE1 were used in this experiment.
Figure 3
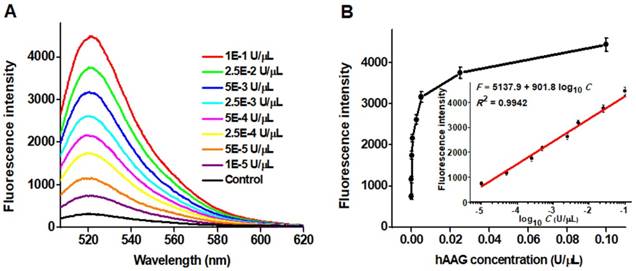
(A) Dependence of fluorescence spectra on different concentrations of hAAG. (B) Dependence of fluorescence intensity on hAAG concentration in the range from 1 × 10-5 to 0.1 U/μL. The inset shows the linear relationship between the fluorescence intensity and the logarithm of hAAG concentration in the range from 1 × 10-5 to 0.1 U/μL. Error bars represent the standard deviations of three experiments.
Figure 4
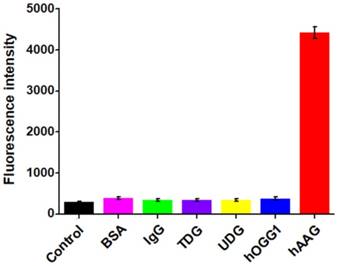
Measurement of fluorescence intensity in response to the control with only reaction buffer (black column), 0.1 g/L BSA (pink column), 0.1 g/L IgG (green column), 0.1 U/μL TDG (purple column), 0.1 U/μL UDG (yellow column), 0.1 U/μL hOGG1 (blue column), and 0.1 U/μL hAAG (red column), respectively. Error bars represent standard deviations from three replicates.
Kinetic analysis of hAAG assay
To investigate the capability of the the proposed strategy for kinetic analysis, we measured the initial velocities (V) in the presence of 0.1 U/µL hAAG, 0.3 U/µL APE1, and different concentrations of hairpin substrate in 5 min of hAAG-induced 2′-deoxyinosine excision reaction at 37 °C to ensure ∼80% of the hairpin substrate unconsumed (i.e., in the initial-rate regime). As shown in Figure 5, the initial velocity (V) enhances with the increasing concentration of hairpin substrate from 0 to 16 nM. According to the Michaelis-Menten equation V = Vmax[S] / (Km + [S]) [43], where Vmax is the maximum initial velocity, [S] is the concentration of substrate, and Km is the Michaelis-Menten constant corresponding to the concentration at half-maximal velocity, the Vmax and Km values of hAAG are determined to be 56.6 min-1 and 22.1 nM, respectively. The obtained Km value is consistent with those obtained by the gel-based radioactive assay (13-25 nM) [44, 45] and single-molecule counting-based fluorescent assay (20.7 nM) [12]. These results demonstrate that the proposed strategy can be used to accurately determine the Michaelis-Menten kinetic parameters.
Figure 5
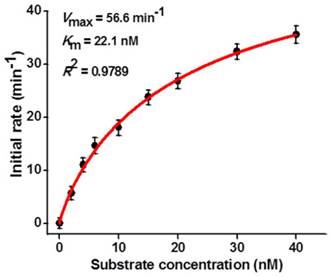
Dependence of initial velocity (V) on different concentrations of hairpin substrate. The 0.1 U/µL hAAG and 0.3 U/µL APE1 were used in this experiment. The time for hAAG-induced 2′-deoxyinosine excision reaction was 5 min. Error bars show the standard deviation of three experiments.
Screening of hAAG inhibitor
The feasibility of the proposed strategy to screen the potential hAAG inhibitors was evaluated by using chromium (II) chloride (CdCl2) as the model [12]. CdCl2 is a classic inhibitor of DNA glycosylases [12, 42], and it can inhibit hAAG activity through directly occupying the Zn2+ binding site (i.e., the hAAG active site) to inactivate the hAAG catalytic activity [46]. As shown in Figure 6, when the CdCl2 concentration increases from 0 to 200 µM, the relative activity of hAAG decreases in a concentration-dependent manner. According to the fitted calibration curve, the half maximal inhibition (IC50) of hAAG in the presence of 0.3 U/µL APE1 is measured to be 17.5 μM, much lower than the value (~100 µM) of hAAG alone measured by the radioactive assay [46] and the value (66.6 µM) in the presence both 0.1 U/µL hAAG and 0.1 U/µL APE1 measured by single-molecule counting-based fluorescent assay [12]. The lower IC50 value of hAAG measured in the presence of 0.3 U/µL APE1 may be attributed to that the Cd2+ can competitively occupy the Mg2+ binding sites within the APE1 active site to induce the inactivation of APE1 activity, which may block the repair of AP sites by cleaving the DNA backbone via a Mg2+-dependent reaction [47]. These results clearly demonstrate that the proposed strategy can be used to screen the hAAG inhibitors for the development of anticancer drugs.
Figure 6
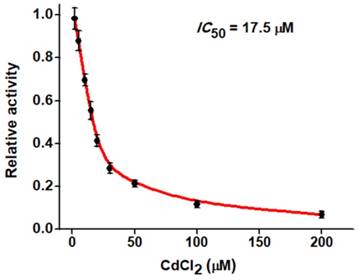
Dependence of the relative activity of hAAG on different concentrations of CdCl2. The 0.1 U/µL hAAG and 0.3 U/µL APE1 were used in this experiment. The error bars represent the standard deviations of the three experiments.
Real sample analysis
The capability of the proposed strategy for real sample analysis is of great importance to early clinical diagnosis and therapy of cancers. We used the human cervical carcinoma cell line (HeLa cells) and lung adenocarcinoma cell line (A549 cells) as the models for the detection of cellular hAAG activity [12, 13]. Considering the hAAG enzyme mainly located in nucleus [48], cellular extracts from the nucleus and whole cell of cancer cells were subjected to hAAG assay (Figures S5-S6, see Supporting Information). As shown in Figures 7A and B, the hAAG activity in nucleus extract and whole cell extract from 1000 HeLa cells and 1000 A549 cells were measured, respectively. Significant fluorescence signals are detected in both nucleus extract group (Figure 7A, red column and Figure 7B, pink column) and whole cell extract group (Figure 7A, green column and Figure 7B, blue column), respectively. In contrast, no distinct fluorescence signals are observed in control groups (Figure 7A, black column and Figure 7B, black column). These results indicate that this strategy exhibits good tolerance to the cellular interferences. Moreover, we further measured the hAAG activity from different number of HeLa and A549 cells. As shown in Figure 7C and D, the fluorescence intensity enhances significantly from 1 to 5000 cells and gradually levels off above 5000 cells. Notably, the fluorescence intensity exhibits a linear correlation with the logarithm of the number of cancer cells (insets of Figures 7C and D) in the range from 1 to 10000 cells. The regression equations are F = 295.5 + 684.7 log10 N (R2 = 0.9980) for HeLa cells and F = 432.3 + 979.1 log10 N (R2 = 0.9894) for A549 cells, where F represents the fluorescence intensity and N represents the number of cells. The directly measured detection limits are down to 1 HeLa cell and 1 A549 cell, respectively, much lower than that obtained by single-molecule counting-based fluorescent assay (9 cells) [12]. In addition, we measured the recovery of hAAG in the spiked normal human serum (Table S1, see Supporting Information). The recovery ratio is calculated to be 99.6%-101.0% with a relative standard deviation (RSD) of 0.98%-2.34%, consistent with the values (recovery ratio of 96.6-106.0% and RSD of 1.53-5.38%) obtained by base-excision repair-mediated triple amplification-based fluorescent assay [19]. These results demonstrate that the proposed strategy can be applied for the quantification of hAAG activity in real samples with high sensitivity even at the single-cell level, holding great potential for further applications in early clinical diagnosis and targeted cancer therapy.
Conclusions
In summary, we have developed a facile fluorescence strategy for homogeneous and sensitive detection of hAAG activity based on controllable autocatalytic cleavage-mediated fluorescence recovery. By taking advantage of the high accuracy of in vivo DNA repair mechanism, the high specificity of T7 exo-catalyzed mononucleotides hydrolysis in DNA duplex, and the high efficiency of autocatalytic recycling amplification, the proposed strategy exhibits high sensitivity with a detection limit of as low as 4.9 × 10-6 U/μL and it can even quantify hAAG activity from 1 HeLa cell and 1 A549 cell, superior to / comparable to those of the reported methods [12, 15-19]. Importantly, this strategy can be further applied for the evaluation of kinetic parameters and the screening of hAAG inhibitors. In addition to the high sensitivity, the proposed strategy exhibits distinct advantages: (1) the high specificity of hAAG-catalyzed excision and the high resolution of T7 exo on single-base mismatch endow this method with the excellent selectivity, greatly decreasing the background signal; (2) only one tool enzyme (i.e., T7 exo) is required in the proposed CESA system, greatly simplifying the operation procedure; (3) this strategy is carried out in a homogeneous format, without the involvement of any separation and washing steps, granting the high accuracy and good reproducibility; (4) the output fluorescent signal is measured by an accessible fluorescence spectrophotometer, without the involvement of sophisticated instrument and complicated manipulations. We believe that the proposed strategy can provide a robust and universal platform for the monitoring of DNA damage-related repair enzymes, holding great potential for DNA repair-related fundamental biochemical research, early clinical diagnosis, anticancer drug discovery, and targeted cancer therapy.
Figure 7
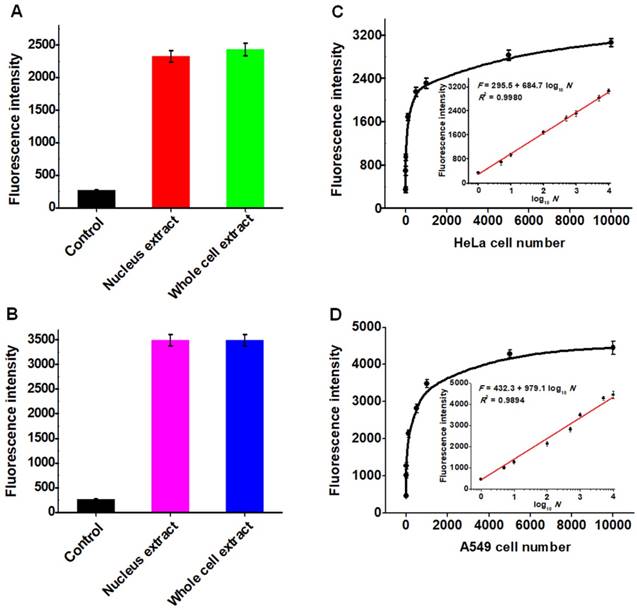
(A) Measurement of fluorescence intensity in the presence of control (only lysis buffer), nucleus extract (1000 HeLa cells), whole cell extract (1000 HeLa cells), respectively. (B) Measurement of fluorescence intensity in the presence of control (only lysis buffer), nucleus extract (1000 A549 cells), whole cell extract (1000 A549 HeLa cells), respectively. (C) Dependence of fluorescence intensity on different number of HeLa cells. The inset shows the linear relationship between the fluorescence intensity and the logarithm of the number of HeLa cells from 1 to 10000 cells. (D) Dependence of fluorescence intensity on different number of A549 cells. The inset shows the linear relationship between the fluorescence intensity and the logarithm of the number of A549 cells from 1 to 10000 cells. Error bars show the standard deviations of three independent experiments.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos. 21735003, 21527811, and 21705097), and the Award for Team Leader Program of Taishan Scholars of Shandong Province, China.
Supplementary Material
Supplementary information, figures and table.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schärer OD. Chemistry and Biology of DNA Repair. Angew Chem Int Ed. 2003;42:2946-74
2. Drabløs F, Feyzi E, Aas PA, Vaagbø CB, Kavli B, Bratlie MS. et al. Alkylation damage in DNA and RNA-repair mechanisms and medical significance. DNA Repair. 2004;3:1389-407
3. Sedgwick B. Repairing DNA-methylation damage. Nat Rev Mol Cell Bio. 2004;5:148-57
4. Shrivastav N, Li D, Essigmann JM. Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis. 2010;31:59-70
5. Fronza G, Gold B. The biological effects of N3-methyladenine. J Cell Biochem. 2004;91:250-7
6. Barbin A. Etheno-adduct-forming chemicals: from mutagenicity testing to tumor mutation spectra. Mutat Res/Rev Mutat. 2000;462:55-69
7. Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis. 2000;21:361-70
8. Leitner-Dagan Y, Sevilya Z, Pinchev M, Kramer R, Elinger D, Roisman LC. et al. N-Methylpurine DNA Glycosylase and OGG1 DNA Repair Activities: Opposite Associations With Lung Cancer Risk. J Natl Cancer I. 2012;104:1765-9
9. Bartsch H, Nair J. Potential role of lipid peroxidation derived DNA damage in human colon carcinogenesis: studies on exocyclic base adducts as stable oxidative stress markers. Cancer Detect Prev. 2002;26:308-12
10. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860-7
11. Nair U, Bartsch H, Nair J. Lipid peroxidation-induced DNA damage in cancer-prone inflammatory diseases: A review of published adduct types and levels in humans. Free Radical Bio Med. 2007;43:1109-20
12. Hu J, Liu M-h, Li Y, Tang B, Zhang C-y. Simultaneous sensitive detection of multiple DNA glycosylases from lung cancer cells at the single-molecule level. Chem Sci. 2018;9:712-20
13. Gines G, Saint-Pierre C, Gasparutto D. On-bead fluorescent DNA nanoprobes to analyze base excision repair activities. Anal Chim Acta. 2014;812:168-75
14. Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer. 2012;12:104-20
15. Abner CW, Lau AY, Ellenberger T, Bloom LB. Base Excision and DNA Binding Activities of Human Alkyladenine DNA Glycosylase Are Sensitive to the Base Paired with a Lesion. J Biol Chem. 2001;276:13379-87
16. Lee C-YI, Delaney JC, Kartalou M, Lingaraju GM, Maor-Shoshani A, Essigmann JM. et al. Recognition and Processing of a New Repertoire of DNA Substrates by Human 3-Methyladenine DNA Glycosylase (AAG). Biochemistry. 2009;48:1850-61
17. Paik J, Duncan T, Lindahl T, Sedgwick B. Sensitization of Human Carcinoma Cells to Alkylating Agents by Small Interfering RNA Suppression of 3-Alkyladenine-DNA Glycosylase. Cancer Res. 2005;65:10472-7
18. Wang L, Zhang H, Xie Y, Chen H, Ren C, Chen X. Target-mediated hyperbranched amplification for sensitive detection of human alkyladenine DNA glycosylase from HeLa cells. Talanta. 2019;194:846-51
19. Zhang H, Wang L, Xie Y, Zuo X, Chen H, Chen X. Base excision repair mediated cascading triple-signal amplification for the sensitive detection of human alkyladenine DNA glycosylase. Analyst. 2019;144:3064-71
20. Kerr C, Sadowski PD. Gene 6 Exonuclease of Bacteriophage T7: I. Purification and Properties of the Enzyme. J Biol Chem. 1972;247:305-10
21. Shinozaki K, Tuneko O. T7 gene 6 exonuclease has an RNase H activity. Nucleic Acids Res. 1978;5:4245-62
22. Wu Z-K, Zhou D-M, Wu Z, Chu X, Yu R-Q, Jiang J-H. Single-base mismatch discrimination by T7 exonuclease with target cyclic amplification detection. Chem Commun. 2015;51:2954-6
23. Liu X, Li W, Hou T, Dong S, Yu G, Li F. Homogeneous Electrochemical Strategy for Human Telomerase Activity Assay at Single-Cell Level Based on T7 Exonuclease-Aided Target Recycling Amplification. Anal Chem. 2015;87:4030-6
24. Li JJ, Chu Y, Lee BY-H, Xie XS. Enzymatic signal amplification of molecular beacons for sensitive DNA detection. Nucleic Acids Res. 2008;36:e36-e52
25. Zuo X, Xia F, Xiao Y, Plaxco KW. Sensitive and Selective Amplified Fluorescence DNA Detection Based on Exonuclease III-Aided Target Recycling. J Am Chem Soc. 2010;132:1816-8
26. Xuan F, Luo X, Hsing IM. Ultrasensitive Solution-Phase Electrochemical Molecular Beacon-Based DNA Detection with Signal Amplification by Exonuclease III-Assisted Target Recycling. Anal Chem. 2012;84:5216-20
27. Wang X, Hou T, Lu T, Li F. Autonomous Exonuclease III-Assisted Isothermal Cycling Signal Amplification: A Facile and Highly Sensitive Fluorescence DNA Glycosylase Activity Assay. Anal Chem. 2014;86:9626-31
28. Xu Q, Cao A, Zhang L-f, Zhang C-y. Rapid and label-free monitoring of exonuclease III-assisted target recycling amplification. Anal Chem. 2012;84:10845-51
29. Cui L, Lin X, Lin N, Song Y, Zhu Z, Chen X. et al. Graphene oxide-protected DNA probes for multiplex microRNA analysis in complex biological samples based on a cyclic enzymatic amplification method. Chem Commun. 2012;48:194-6
30. Xi Q, Zhou D-M, Kan Y-Y, Ge J, Wu Z-K, Yu R-Q. et al. Highly Sensitive and Selective Strategy for MicroRNA Detection Based on WS2 Nanosheet Mediated Fluorescence Quenching and Duplex-Specific Nuclease Signal Amplification. Anal Chem. 2014;86:1361-5
31. Hao N, Li X-L, Zhang H-R, Xu J-J, Chen H-Y. A highly sensitive ratiometric electrochemiluminescent biosensor for microRNA detection based on cyclic enzyme amplification and resonance energy transfer. Chem Commun. 2014;50:14828-30
32. Li J, Fu H-E, Wu L-J, Zheng A-X, Chen G-N, Yang H-H. General Colorimetric Detection of Proteins and Small Molecules Based on Cyclic Enzymatic Signal Amplification and Hairpin Aptamer Probe. Anal Chem. 2012;84:5309-15
33. Zhao Y, Chen F, Lin M, Fan C. A methylation-blocked cascade amplification strategy for label-free colorimetric detection of DNA methyltransferase activity. Biosens Bioelectron. 2014;54:565-70
34. Weizmann Y, Cheglakov Z, Willner I. A Fok I/DNA Machine that Duplicates its Analyte Gene Sequence. J Am Chem Soc. 2008;130:17224-5
35. Shevelev IV, Hübscher U. The 3′-5′ exonucleases. Nat Rev Mol Cell Bio. 2002;3:364-76
36. Richardson CC, Lehman IR, Kornberg A. A Deoxyribonucleic Acid Phosphatase-Exonuclease from Escherichia coli: II. Characterization of the Exonuclease Activity. J Biol Chem. 1964;239:251-8
37. Shagin DA, Rebrikov DV, Kozhemyako VB, Altshuler IM, Shcheglov AS, Zhulidov PA. et al. A Novel Method for SNP Detection Using a New Duplex-Specific Nuclease From Crab Hepatopancreas. Genome Res. 2002;12:1935-42
38. Wang Y, Qiu Y, Huang Z, Yisheng Z. Modelling on the Kinetics Mechanism of the FokI Restriction Endonuclease. 2007 1st International Conference on Bioinformatics and Biomedical Engineering. 2007 p. 9-12
39. Hendershot JM, Wolfe AE, O'Brien PJ. Substitution of Active Site Tyrosines with Tryptophan Alters the Free Energy for Nucleotide Flipping by Human Alkyladenine DNA Glycosylase. Biochemistry. 2011;50:1864-74
40. Wang L-j, Wang Z-y, Zhang Q, Tang B, Zhang C-y. Cyclic Enzymatic Repairing-Mediated Dual-Signal Amplification for Real-Time Monitoring Thymine DNA Glycosylase. Chem Commun. 2017;53:3878-81
41. Wang L-j, Ren M, Zhang Q, Tang B, Zhang C-y. Excision Repair-Initiated Enzyme-Assisted Bicyclic Cascade Signal Amplification for Ultrasensitive Detection of Uracil-DNA Glycosylase. Anal Chem. 2017;89:4488-94
42. Wang L-j, Ma F, Tang B, Zhang C-y. Base-Excision-Repair-Induced Construction of a Single Quantum-Dot-Based Sensor for Sensitive Detection of DNA Glycosylase Activity. Anal Chem. 2016;88:7523-9
43. Boeneman K, Mei BC, Dennis AM, Bao G, Deschamps JR, Mattoussi H. et al. Sensing Caspase 3 Activity with Quantum Dot-Fluorescent Protein Assemblies. J Am Chem Soc. 2009;131:3828-9
44. Xia L, Zheng L, Lee H-W, Bates SE, Federico L, Shen B. et al. Human 3-Methyladenine-DNA Glycosylase: Effect of Sequence Context on Excision, Association with PCNA, and Stimulation by AP Endonuclease. J Mol Biol. 2005;346:1259-74
45. O'Brien PJ, Ellenberger T. Dissecting the Broad Substrate Specificity of Human 3-Methyladenine-DNA Glycosylase. J Biol Chem. 2004;279:9750-7
46. Wang P, Guliaev AB, Hang B. Metal inhibition of human N-methylpurine-DNA glycosylase activity in base excision repair. Toxicol Lett. 2006;166:237-47
47. McNeill DR, Narayana A, Wong H-K, Wilson DM 3rd. Inhibition of Ape1 nuclease activity by lead, iron, and cadmium. Environ Health Persp. 2004;112:799-804
48. Saki M, Prakash A. DNA damage related crosstalk between the nucleus and mitochondria. Free Radical Bio Med. 2017;107:216-27
Author contact
![]() Corresponding author: Prof. Chun-yang Zhang. E-mail addresses: cyzhangedu.cn.
Corresponding author: Prof. Chun-yang Zhang. E-mail addresses: cyzhangedu.cn.
Citation styles
APA
Wang, L.J., Luo, M.L., Yang, X.Y., Li, X.F., Wu, Y., Zhang, C.Y. (2019). Controllable Autocatalytic Cleavage-Mediated Fluorescence Recovery for Homogeneous Sensing of Alkyladenine DNA Glycosylase from Human Cancer Cells. Theranostics, 9(15), 4450-4460. https://doi.org/10.7150/thno.35393.
ACS
Wang, L.J.; Luo, M.L.; Yang, X.Y.; Li, X.F.; Wu, Y.; Zhang, C.Y. Controllable Autocatalytic Cleavage-Mediated Fluorescence Recovery for Homogeneous Sensing of Alkyladenine DNA Glycosylase from Human Cancer Cells. Theranostics 2019, 9 (15), 4450-4460. DOI: 10.7150/thno.35393.
NLM
Wang LJ, Luo ML, Yang XY, Li XF, Wu Y, Zhang CY. Controllable Autocatalytic Cleavage-Mediated Fluorescence Recovery for Homogeneous Sensing of Alkyladenine DNA Glycosylase from Human Cancer Cells. Theranostics 2019; 9(15):4450-4460. doi:10.7150/thno.35393. https://www.thno.org/v09p4450.htm
CSE
Wang LJ, Luo ML, Yang XY, Li XF, Wu Y, Zhang CY. 2019. Controllable Autocatalytic Cleavage-Mediated Fluorescence Recovery for Homogeneous Sensing of Alkyladenine DNA Glycosylase from Human Cancer Cells. Theranostics. 9(15):4450-4460.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.