Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(6):2605-2621. doi:10.7150/thno.92672 This issue Cite
Research Paper
IL4 receptor targeting enables nab-paclitaxel to enhance reprogramming of M2-type macrophages into M1-like phenotype via ROS-HMGB1-TLR4 axis and inhibition of tumor growth and metastasis
Sri Murugan Poongkavithai Vadevoo1,2,3,*, Yeoul Kang4,*, Gowri Rangaswamy Gunassekaran1,2,3, Seok-Min Lee1,2,3, Min-Sung Park1,2,3, Dong Gyun Jo1,2,3, Sang-Kyun Kim5, Ho Lee6, Won Jong Kim4, ![]() , Byungheon Lee1,2,3,
, Byungheon Lee1,2,3, ![]()
1. Department of Biochemistry and Cell Biology, School of Medicine, Kyungpook National University, 680 Gukchaebosang-ro, Jung-gu, Daegu 41944, Republic of Korea.
2. Department of Biomedical Science, School of Medicine, Kyungpook National University, 680 Gukchaebosang-ro, Jung-gu, Daegu 41944, Republic of Korea.
3. CMRI, School of Medicine, Kyungpook National University, 680 Gukchaebosang-ro, Jung-gu, Daegu 41944, Republic of Korea.
4. Department of Chemistry, POSTECH-CATHOLIC Biomedical Engineering Institute, Pohang University of Science and Technology (POSTECH), 77 Cheongam-ro, Nam-gu, Pohang, Gyeongbuk 37673, Republic of Korea.
5. Laboratory Animal Center, K-Medi Hub, 88 Dongnae-ro, Dong-gu, Daegu 41061, Republic of Korea.
6. Laboratory Animal Research Facility, National Cancer Center, 323 Ilsan-ro, Ilsandong-gu, Koyang, Kyunggi 10408, Republic of Korea.
* These authors equally contributed.
Received 2023-11-28; Accepted 2024-4-2; Published 2024-4-8
Citation:
Vadevoo SMP, Kang Y, Gunassekaran GR, Lee SM, Park MS, Jo DG, Kim SK, Lee H, Kim WJ, Lee B. IL4 receptor targeting enables nab-paclitaxel to enhance reprogramming of M2-type macrophages into M1-like phenotype via ROS-HMGB1-TLR4 axis and inhibition of tumor growth and metastasis. Theranostics 2024; 14(6):2605-2621. doi:10.7150/thno.92672. https://www.thno.org/v14p2605.htm
Other stylesAbstract

Rationale: Nab-paclitaxel (Abx) is widely employed in malignant tumor therapy. In tumor cells and pro-tumoral M2-type macrophages, the IL4 receptor (IL4R) is upregulated. This study aimed to elucidate the selective delivery of Abx to M2-type macrophages by targeting IL4R and reprogramming them into an anti-tumoral M1-type.
Methods: Abx was conjugated with the IL4R-binding IL4RPep-1 peptide using click chemistry (IL4R-Abx). Cellular internalization, macrophage reprogramming and signal pathways, and tumor growth and metastasis by IL4R-Abx were examined.
Results: IL4R-Abx was internalized into M2 macrophages more efficiently compared to the unmodified Abx and control peptide-conjugated Abx (Ctrl-Abx), which was primarily inhibited using an anti-IL4R antibody and a receptor-mediated endocytosis inhibitor compared with a macropinocytosis inhibitor. IL4R-Abx reprogrammed the M2-type macrophages into M1-like phenotype and increased reactive oxygen species (ROS) levels and extracellular release of high mobility group box 1 (HMGB1) in M2 macrophages at higher levels than Abx and Ctrl-Abx. The conditioned medium of IL4R-Abx-treated M2 macrophages skewed M2 macrophages into the M1-like phenotype, in which an anti-HMGB1 antibody and a toll-like receptor 4 (TLR4) inhibitor induced a blockade. IL4R-Abx accumulated at tumors, heightened immune-stimulatory cells while reducing immune-suppressing cells, and hampered tumor growth and metastasis in mice more efficiently than Abx and Ctrl-Abx.
Conclusions: These results indicate that IL4R-targeting allows enhancement of M2-macrophage shaping into M1-like phenotype by Abx through the ROS-HMGB1-TLR4 axis, improvement of antitumor immunity, and thereby inhibition of tumor growth and metastasis, presenting a new approach to cancer immunotherapy.
Keywords: IL4 receptor, M2-macrophage, nab-paclitaxel, reprogramming, immunotherapy
Introduction
There is an abundance of tumor-associated macrophages (TAMs) in the tumor microenvironment. Majority of TAMs feature a pro-tumoral M2 phenotype, secreting immune-suppressive cytokines, such as transforming growth factor-β and IL10, and recruiting immune-suppressive cells, including myeloid-derived suppressor cells (MDSCs) and regulatory Treg cells [1-3]. Furthermore, in tumor tissues, an increase in M2-type TAM population is a marker of poor cancer prognosis [4]. Thus, M2-type macrophages in tumor tissues represent a promising target for cancer therapy.
An albumin-bound paclitaxel nanoparticle called nab-paclitaxel (trade name Abraxane, hereafter Abx) has been used in treating metastatic breast tumor, lung tumor, and pancreatic tumor [5]. Paclitaxel is an antineoplastic agent that binds to microtubule tubulin and inhibits microtubule disassembly during mitosis, leading to cell cycle arrest and cancer cell apoptosis. In addition to tumor cell cytotoxicity, paclitaxel has been reported to activate macrophages into pro-inflammatory phenotype by binding to toll-like receptor 4 (TLR4) on the macrophage cell surface [6] or by binding to the TLR4 accessory protein, myeloid differentiation factor 2, in the cytoplasm [7]. Abx is internalized into macrophages via macropinocytosis and drives pro-inflammatory M1 macrophage polarization that demonstrates antitumor activity [8]. These findings indicate the potential of Abx as an immunotherapeutic agent that reprograms M2-type macrophages into the M1 phenotype in the tumor microenvironment and as a cytotoxic agent against tumor cells. However, the macrophage activation mechanism by Abx remains poorly explored.
In certain types of immune cells such as Th2 T-cells, the IL4 receptor (IL4R) comprising of IL4Rα and IL2Rγc subunits (named type 1 IL4R) is expressed [9, 10]. Conversely, IL4R consisting of IL4Rα and IL13Rα subunits (named type II IL4R) is highly upregulated in certain types of tumors such as breast, lung, and pancreatic cancers, which is associated with poor prognosis [11-13]. IL4 binds to the IL4Rα subunit of IL4R and eventually recruits the IL2Rγc or IL13Rα subunit [10]. To our interest, IL4R is expressed in M2 macrophages at higher levels than in M1 macrophages [14-17]. IL4 binding to IL4R induces M2 macrophage polarization [18]. Additionally, IL4 and IL4R interaction enhances the pro-tumorigenic M2-macrophage activity and cathepsin protease production by TAMs [19, 20], which contributes to tumor progression and metastasis. These findings indicate that IL4R is a promising cell surface receptor to target M2 macrophages in the tumor microenvironment.
Selective nanoparticle delivery to tumors using ligands such as antibodies and peptides specific to molecules overexpressed in the tumor cell surface empowers nanoparticles to differentiate tumor cells from normal cells. Peptides have a lower affinity and shorter circulating time and are more vulnerable to degradation compared to antibodies. In contrast, peptides have a deeper tissue penetration ability, more efficient internalization into cells, and lesser immune reaction possibility [21-24]. We have identified the IL4RPep-1 peptide consisting of CRKRLDRNC amino acid sequence using phage display, which is homologous to the KRLDRN sequence of IL4 and binds to IL4R [25]. IL4RPep-1 has been successfully used as a tumor-homing peptide for the selective delivery of nanoparticles and liposomes carrying chemotherapeutic drugs or siRNA to tumors [26-28]. This study aimed to enhance Abx delivery to IL4R-high M2-type macrophages via the surface modification of Abx with IL4RPep-1 and reprogramming of M2-type macrophages into M1-like phenotype for cancer immunotherapy. Furthermore, the pathways included in the reprogramming of M2 macrophages through the IL4R-targeted Abx were examined.
Materials and Methods
Cell cultures
4T1 mouse breast tumor cells were cultured in Roswell Park Memorial Institute media supplemented with 10% fetal bovine serum (FBS, Thermo Fisher, Waltham, MA). Lewis lung carcinoma (LLC) mouse lung tumor cells and Panc-1 human pancreatic tumor cells were cultured in Dulbecco's modified Eagle's medium (DMEM, HyClone, South Logan, UT) supplemented with 10% FBS. Cell cultures were maintained at 37 °C in a humidified 5% CO2 atmosphere.
Bone marrow-derived monocyte (BMDM) preparation and polarization to M2-type macrophages
To differentiate into BMDMs, bone marrow cells were isolated from tibias and femurs of BALB/c mice and incubated with DMEM supplemented with 10 ng/mL of macrophage-colony-stimulating factor (Gibco, Carlsbad, CA) and 10% FBS for 7 days. The culture medium was altered every 2 to 3 days. For M2-macrohage polarization, BMDMs were incubated with 20 ng/mL of recombinant mouse IL-4 for 24-48 h.
Peptide synthesis
IL4RPep-1 (sequence: CRKRLDRNC) and control peptide (sequence: CNSSSVDK) were synthesized and purified using high-performance liquid chromatography to >90% purity by Peptron Inc. (Daejeon, Korea). To prepare fluorescent peptides, peptides were conjugated at the N-terminus with fluorescein isothiocyanate (FITC) or Flamma 675 near-infrared fluorescence dye (BioActs, Incheon, Korea) during synthesis. The amino acid sequence of the control peptide came from the phage coat protein, in which random peptide sequences were inserted to construct a phage-displayed peptide library.
Preparation of peptide-conjugated Abx
A total of 30 mg of Abx (containing 27 mg (or 406 nmol) of Albumin) was dissolved in 3 mL phosphate buffer saline (PBS) and mixed with 2.534 mg (487 nmol; 1.2 equivalent to Abx) of 1,8-bismaleimido-diethyleneglycol (BM(PEG)2, Thermo Fisher) and stirred for 1 h. After incubation, to eliminate unbound BM(PEG)2, the Abx-BM(PEG)2 conjugate was subjected to ultracentrifugation via an Amicon Ultra 30 kDa-4 mL centrifugal filter (Merck, Burlington, MA). To reduce the peptide disulfide bonds, each peptide (487 nmol; 1.2 equivalent to Abx) was incubated with 5 mM Tris-(2-carboxyethyl)phosphine hydrochloride (Sigma-Aldrich, St. Louis, MO) in 0.1-M Tris-HCl buffer for 10 min. The Abx-BM(PEG)2 was added to the reduced peptide solution and stirred for 24 h. Using the Amicon Ultra 30 kDa-4 mL centrifugal filter, the Abx-peptide conjugate was ultracentrifuged followed by freeze-drying. Thereafter, using the PD-10 desalting column and Sephadex G-25 resin (GE healthcare, Chicago, IL), the Abx-peptide conjugate was purified and was obtained as a yellowish powder.
Characterization of peptide-conjugated Abx
The molecular weight of Abx-BM(PEG)2 and IL4R-Abx was determined using matrix-assisted laser desorption-ionization/time of flight mass spectrometry (MALDI-TOF-MS). MALDI-TOF-MS analyses were conducted using an Autoflex speed LRF, Bruker (smart beam-II laser (355 nm)) operating in linear mode/positive ion mode with a mass-to-charge (m/z) ratio range of 30-210 kDa. The laser fluence was set at 90%, with a laser frequency of 2000 Hz, and more than 10000 laser shots accumulated. The sample (4 mg/mL) was dissolved in an aqueous solution containing 50% acetonitrile (ACN) with 0.1% TFA (trifluoroacetic acid). Sinapic acid was served as the matrix. The hydrodynamic size of nanoparticle was measured by dynamic light scattering (DLS) with Zetasizer Nano S90 systems (Malvern, UK). All groups were dissolved at a concentration of 10 mg/mL in PBS buffer. To examine structural feature, transmission electron microscopy (TEM) images were obtained using JEM-1011 instrument (JEOL Ltd, Japan), and analyzed with Gatan digital microscope software. All groups are dissolved at a concentration of 0.01 mg/mL in deionized water.
Internalization assays
Abx was labeled with FITC using fluorescein-EX protein labeling kit (Thermo Fisher). A 0.5 mL solution of 2 mg/mL abraxane was mixed with 50 µl of 1 M bicarbonate and incubated with the reactive dye for 1 h at room temperature. The labeled Abx was purified from free dye using a spin column (MW > 20 kDa). Cells (1 × 105 cells/well of a four-well chamber) were incubated with 1% bovine serum albumin at 24°C for 30 min and then with 5 μg/mL of FITC-labeled Abx at 37 °C for 1 h. Cells were fixed with 4% paraformaldehyde followed by nuclear staining with 4', 6-diamidino-2-phenylindole (DAPI) after washing and observed under a confocal microscope (Nanoscope, Daejeon, Korea), or cells were analyzed using Attune NxT flow cytometer (Thermo Fisher). For the internalization inhibition, cells were incubated with 50 μM of 5-(n-ethyl-n-isopropyl) amiloride (EIPA, Thermo Fisher), 50 μM of chlorpromazine (CPZ, Sigma-Aldrich), and 100 μM of anti-IL4Rα blocking antibody or anti-IgG antibody (R&D Systems, Minneapolis, MN) for 30 min prior to incubation with FITC-labeled Abx.
Cytotoxicity assays
Cells (5 × 103 cells/well in a 96-well plate) were incubated with IL4R-targeted and unmodified Abx for 24 h at 37°C. Cells were incubated with a fresh culture medium containing 10% CCK-8 reagents (Dojindo, Kumamoto, Japan) for 1-4 h after treatments. Absorbance was measured at 450 nm. GraphPad Prism 7 software (GraphPad, New York, NY) was used to calculate the half maximal inhibitory concentration (IC50).
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Using a miRNeasy mini kit (Qiagen, Hilden, Germany), RNA was isolated from the M2 macrophages and subjected to qRT-PCR. Primers for arg1, il-10, il-4, tgf-β, il-12p40, ifn-γ, and β-actin were obtained from Bioneer Inc. (Daejeon, Korea). cDNA was synthesized using PrimerScript 1st strand cDNA Synthesis Kit (Takara, Shiga, Japan). qPCR using SYBRGreen (Qiagen) was conducted on a real time cycler (Bio-Rad, Hercules, CA). According to the Livak method [29], data for relative expression were analyzed.
Measurement of reactive oxygen species (ROS) levels and inhibition of ROS, high mobility group box 1 (HMGB1), and TLR4
Cells were incubated with IL4R-targeted and untargeted Abx for 6 h and then with 1 μM of 2',7'-dichlorodihydrofluorescein diacetate (H2DCFDA, Thermo Fisher) at 37 °C for 1 h, which is a cell-permeable ROS indicator and generates fluorescence through ROS. The fluorescence levels were observed under a fluorescence microscope (Nikon, Tokyo, Japan).Cells were incubated with 50 mM of N-acetyl-l-cysteine (NAC) for 30 min before treatment with IL4R-Abx and Abx to inhibit or block ROS activity. To inhibit or block HMGB1 and TLR4 activity, cells were incubated for 30 min with 10 μg/mL of anti-HMGB1 antibody (Arigo Biolaboratories, Hsinchu, Taiwan) and 500 nM of CLI095 (Thermo Fisher), respectively, before treatments with a conditioned medium (CM) of IL4R-Abx- and Abx-treated M2 macrophages.
Analysis of intracellular and extracellular HMGB1
To examine the location of intracellular HMGB1, M2 macrophage cells were seeded at a density of 1 × 105 cells in an 8-well chamber (Thermo Fisher) and allowed to attach overnight. Cells were incubated with Abx and IL4R-Abx (5 μg/mL) at 37 °C for 24 h. After incubation, cells were fixed with 4% PFA, permeabilized with 0.1% Triton X100, and incubated with an anti-HMGB1 antibody (Abcam, 1/400 dilution) at 4 °C overnight. Cells were then incubated with Alexa Fluor 647-labeled secondary antibody in dark at room temperature for 1 h. Cells were incubate with DAPI for nuclear staining and observed under a confocal scanning microscope (Nanoscope).
To examine the levels of extracellular HMGB1 released into the culture medium using western blotting, the culture medium of M2 macrophages treated for 24 h was collected and concentrated using a centrifugal concentrator (Vivaproducts, Littleton, MA). Samples were electrophoresed in 10% polyacrylamide gel and transferred to a polyvinylidene difluoride membrane. The membrane was blocked with 5% skimmed milk in Tris-buffered saline containing 0.1% Tween-20 and then incubated with antibodies against HMGB1 (Abcam, Cambridge, UK) and β-actin (Santa Cruz Biotechnology, Dallas, TX) at 4 οC overnight followed by incubation with a horse radish peroxidase-conjugated secondary antibody (Cell Signaling Technology, Danvers, MA) at room temperature for 1 h. A substrate (Thermo Fisher) was used to incubate the membrane, and a chemiluminescence detection reagent (FujiFilm, Tokyo, Japan) was used for visualization. The protein band intensity was quantified using ImageJ software (National Institute of Health (NIH), Bethesda, MD).
To measure the concentrations of HMGB1 in the culture medium using ELISA, 100 μL of the samples were added to each well of a 96-well plate coated with anti-mouse HMGB1 antibody and incubated at 37 °C for 2 h. After incubation, 100 μL of biotin-labeled anti-mouse HMGB1 antibody was added and incubated at 37 °C for 1 h followed by incubation with streptavidin-horseradish peroxidase conjugate at 37 °C for 1 h. The plate was incubated with tetramethylbenzidine substrate at room temperature in dark for 20 min, and the reaction was terminated by addition of 50 μL of stop solution. The absorbance at 450 nm was measured with a microplate reader (Thermo Fisher).
Paclitaxel concentrations in tissues
Approximately 3 μL of each of standard paclitaxel solutions in acetonitrile was mixed with 27 μL of plasma and added to 270 μL of acetonitrile containing an internal standard solution (gliclazide at 100 ng/mL concentration). Tumor and control organs were dissected at 6 h after injection of Abx, added with cold PBS at a weight ratio of 1:4, and homogenized using a high-speed homogenizer. The supernatants acquired after homogenate centrifugation were used as sample solutions. The 30-μL sample solutions were added to 270 μL of acetonitrile containing the internal standard solution. A 5-μL aliquot of the sample and paclitaxel standard solutions were injected into the ultraperformance liquid chromatography-mass spectrometry/mass spectrometry system (Agilent, Santa Clara, CA). The paclitaxel concentrations in samples were calculated using TargetLynx software (Waters, Milford, MA).
Immune cell population analysis in tissues
Tumors were excised and fragmented into several pieces to prepare the tumor-infiltrating leukocytes. The fragmented tissues were further minced into 2-3 mm3 pieces and incubated with collagenase D (Roche, Basel, Switzerland) at 37 °C for 40 min. The tissue samples were filtered through a 100-µm cell strainer. Dead cells and cellular debris were removed using Ficoll (GE Healthcare, Buckinghamshire, UK) gradient centrifugation. Collected cells were re-suspended in PBS containing 2% FBS. Utilizing a flow cytometer, cells were gated upon CD45 expression. Cells were incubated for 20 min in the dark with fluorescently labeled antibodies against CD11b, CD3, CD4, CD8, CD45, F4/80, MHCII, Foxp3, and Ly6C (BioLegend, San Diego, CA) for surface biomarker staining. Cells were analyzed using Attune NxT flow cytometer (Thermo Fisher).
Animal models
Six- to 8-week old mice were purchased from Orient Bio (Seongnam, Korea). Animal care was conducted in accordance with the Guidelines of the Institutional Animal Care and Use Committee of Kyungpook National University (permission No. 2015-0017). BALB/c mice were injected with 1×106 4T1 cells in the lower right mammary fat pad to establish 4T1 orthotopic breast tumor. To establish subcutaneous LLC lung tumor and Panc-1 pancreatic tumor models, C57BL/6 mice and BALB/c nude mice were injected with 5×105 LLC and Panc-1 cells in the lower right flank, respectively. K-rasLA2 mutant transgenic mice provided by the NIH (Bethesda, MD) were mated with wild-type (WT) C57BL/6N mice to establish a spontaneous lung tumor. For genotyping, complementary DNA was purified through QIAquick columns (Qiagen) and amplified using PCR with an annealing temperature of 60°C for 35 rounds using PCR primers as previously described [30]. KPC (LSL-KrasG12D/+LSL-p53R172H/+PDX-1 Cre) transgenic pancreatic tumor mice were obtained through cross breeding LSL-KrasG12D/+ PDX-1 Cre with LSL-p53R172H/+PDX-1 Cre. For genotyping, PCR primers used were as follows: PDX-1 Cre (F), CTG GAC TAC ATC TTG AGT TGC; PDX-1 Cre (R), TTC TTG CGA ACC TCA TCA CTC GTT G; K-ras WT (F), TGT CTT TCC CCA GCA CAG T; K-ras Com (R), CTG CAT AGT ACG CTA TAC CCT GT; K-ras Mut (F), GCA GGT CGA GGG ACC TAA TA; P53 KI WT, AGG TGT GGC TTC TGG CTT C; P53 KI Com, GAA ACT TTT CAC AAG AAC CAG ATC A; P53 KI Mut, CCA TGG CTT GAG TAA GTC TGC A.
Whole-body and ex vivo fluorescence imaging
Through the tail vein, mice were intravenously injected with 100 µL (at 5 μg/mL) of targeted and untargeted Abx conjugated with peptides labeled with Flamma 675 near-infra red fluorescent dye (BioActs) at the N-terminus during synthesis. After a 6-h circulation, mice were anesthetized, and in vivo images were taken using the IVIS imaging system (PerkinElmer, Waltham, MA). After in vivo imaging, the mice were sacrificed, and the tumors and other organs were isolated and subjected to ex vivo imaging using the IVIS imaging system, and the total flux (photon/second) was measured using Living Image 4.5 software (PerkinElmer).
Histological analysis
Frozen tissue sections (8 µm thickness) were stained at 37 oC for 1 h using anti-mouse IL4Rα (Santa Cruz Biotechnologies). Alexa594-conjugated antibody (Life technologies) was used as a secondary antibody. Tissue samples were counter-stained with DAPI, incubated with Prolong Gold® anti-fade mounting reagent (Life technologies), and observed under a confocal microscope (Nanoscope).
Antitumor therapy
4T1, LLC, and Panc-1 tumor-bearing mice were subjected to randomization and grouping for initiation of therapy at 2 - 3 weeks after tumor inoculation when the tumor sizes reached approximately 100 mm3. K-rasLA2 mutant mice and KPC mice were used for treatments at 10 weeks of age. Targeted and untargeted Abx was injected intravenously via the tail vein of mice (5 or 10 mg/kg body weight, weekly for 4 weeks). In 4T1, LLC, and Panc-1 tumors, tumor size was measured using a caliper during treatments. At the treatment completion, mice were sacrificed and tumors and lungs were isolated, weighed, and assessed for metastatic tumor masses. Additionally, tumors were subjected to analysis of immune cell populations using flow cytometry or immunohistochemistry. At the end of treatments, mouse blood was collected and analyzed for liver function, such as aspartate transaminase (AST) and alanine transaminase (ALT), and kidney function, such as blood urea nitrogen (BUN) and creatinine (CRE), by K-Medi Hub (Daegu, Korea).
Statistical analysis
Data are presented as the mean ± standard deviation (SD). GraphPad Prism 7 software was used for all statistical analyses. Statistical significance was determined using one-way analysis of variance (ANOVA) followed by Tukey's multiple post hoc test or two-way ANOVA followed by Bonferroni multiple post hoc test. *, P < 0.05; ** P < 0.01; ***, P < 0.001.
Results
Preparation and characterization of IL4R-targeted Abx
To confer an IL4R-targeting activity to Abx, a sulfur-group-containing Abx on the albumin surface was conjugated with a bismaleimide linker and then conjugated to a sulfur group of cysteine residue of IL4RPep-1 (CRKRLDRNC, MW = 1,163 g/mol) using the same linker and called the IL4R-Abx (Figure 1A). The increase in molecular weight of albumin resulting from the connection of the free thiol group of albumin and the cysteine residue of the IL4RPep-1 using BM(PEG)2 was measured by MALDI-TOF-MS spectrometry. This increase was observed from 67,096 g/mol (theoretical MW = 66,348 g/mol [31]) to 67,287 g/mol, and subsequently to 68,273 g/mol (Figure 1B and S1A). Considering that the MW of albumin in Abx and BM(PEG)2 were 67,096 g/mol and 308.29 g/mol, respectively, the conjugation ratio of BM(PEG)2 was calculated to be 62%. Subsequently, the MW of IL4R-Abx was determined to be a mixture of 67,827 and 68,273 g/mol, indicating the presence of Abx-BM(PEG)2 and IL4R-Abx with one IL4RPep-1 conjugated. The modification of Abx with IL4RPep-1 preserved the physical properties of Abx, including its size of approximately 160 nm and a spherical structure through analysis using DLS (Figure 1C and S1B-C) and TEM (Figure 1D and S1D), respectively.
Additionally, using FITC-conjugated IL4RPep-1, the conjugation efficiency was determined by analyzing the absorbance of Abx and FITC at 280 and 495 nm, respectively, following the quantification method of dye-to-protein ratio. The extinction coefficient and correction factor were calculated from each absorbance spectra of Abx and FITC-IL4RPep-1 (Figure S2A-E). Subsequently, the conjugation efficiency was calculated from the absorbance of FITC-IL4R-Abx at 280 and 495 nm, applying constant values (Figure S2F and S2G). The resulting conjugation efficiency was calculated to be 59.69%, corresponding to approximately 0.6 molecules of IL4RPep-1 per molecule of albumin. These results indicated that IL4RPep-1 was successfully conjugated to Abx.
IL4R-targeted Abx was internalized into M2 macrophages through IL4R-mediated endocytosis
IL4R-Abx was internalized into M2 macrophages more efficiently than the unmodified Abx and control peptide-conjugated Abx (Ctrl-Abx) as determined by fluorescence microscopy (Figure 2A) and mean fluorescence intensity (MFI, Figure 2B) and percent internalization (Figure S3A and S3B) by flow cytometry. Ctrl-Abx was included to examine whether the conjugation of Abx with peptides affected the targeting activity of Abx regardless of the peptide sequence. Abx is known to be internalized into macrophages through macropinocytosis [8]. Macropinocytosis is a receptor-independent and actin-driven endocytic pathway [32]. To examine whether IL4R-Abx is internalized into macrophages via macropinocytosis, M2 macrophages were pre-treated with EIPA, a macropinocytosis inhibitor, before treatments with IL4R-Abx. EIPA efficiently inhibited Abx internalization while minimally reducing that of IL4R-Abx (Figure 2A-B and S3A-B). Conversely, pre-treatment with CPZ, a clathrin-mediated endocytosis inhibitor, efficiently reduced IL4R-Abx internalization while increasing that of Abx (Figure 2A-B and S3A-B). Cells were pre-treated with an anti-IL4R blocking antibody as the clathrin-mediated endocytosis is a receptor-dependent pathway. An anti-IL4R antibody significantly inhibited IL4R-Abx internalization while demonstrating a negligible effect on Abx (Figure 2A-B and S3A-B). Ctrl-Abx internalization was lower than Abx and IL4R-Abx and reduced by EIPA while being not inhibited by CPZ and anti-IL4R antibody (Figure 2A-B and S3A-B). In addition, EIPA and CPZ reduced the IL4R-Abx internalization but did not appear to hinder its cell surface binding, while the anti-IL4R antibody inhibited both the internalization and surface binding of IL4R-Abx (Figure 2A and 2B). These results indicate that IL4R-Abx internalization into M2 macrophages is a primarily IL4R targeted and the receptor-mediated endocytosis rather than macropinocytosis.
Figure 1
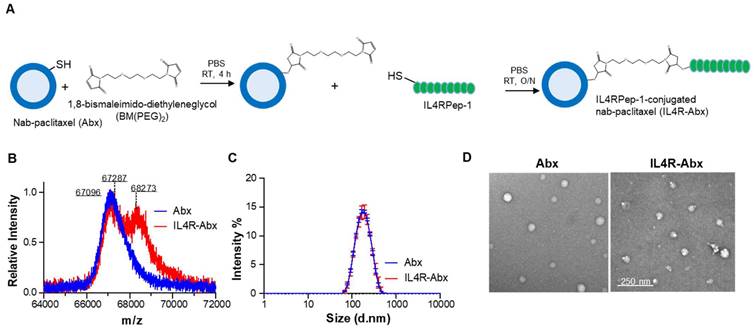
Preparation and characterization of IL4R-Abx. (A) Synthetic schemes of IL4RPep-1-conjugated Abx (IL4R-Abx). (B) MALDI-TOF-MS spectra of albumin part (64-72 kDa) in Abx (theoretical MWalbumin = 66348 g/mol) and IL4R-Abx. (C) Size distribution of Abx and IL4R-Abx by DLS. (D) TEM image of Abx and IL4R-Abx. Scale bars, 250 nm.
Figure 2
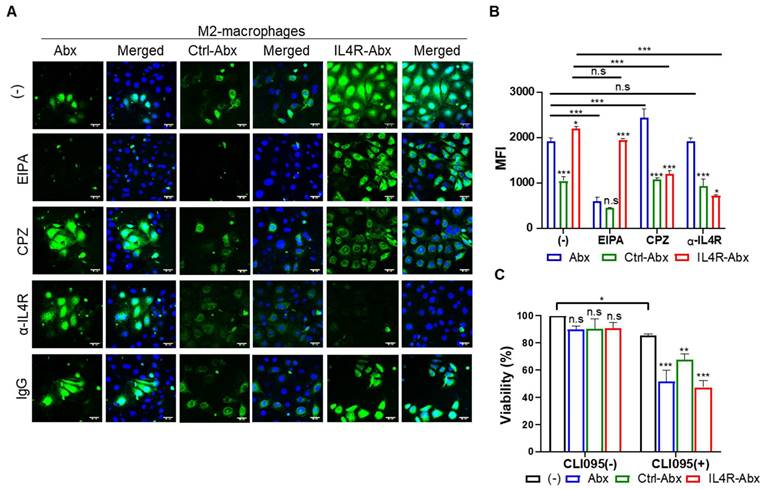
Internalization and cytotoxicity of IL4R-Abx in M2 macrophages. (A) Immunofluorescence analysis of the internalization. M2 macrophages were incubated using 2 μg/mL of fluorescein isothiocyanate (FITC) dye (green)-labeled Abx, Ctrl-Abx, and IL4R-Abx for 2 h with or without pre-treatment with 5-(n-ethyl-n-isopropyl) amiloride (EIPA) (50 μM), chlorpromazine (CPZ) (50 μM), anti-IL4R antibody (100 μM), and IgG (100 μM) for 30 min. Nucleus was stained with 4',6-diamidino-2-phenylindole (DAPI) (blue), and merging of images was performed. Scale bars, 20 μm. (B) Flow cytometry analysis of internalization. M2 macrophages were treated as described in (A), and the mean fluorescence intensity (MFI) was quantified using a flow cytometer. Data are expressed as mean ± standard deviation (SD) in the three separate experiments. P values of Ctrl-Abx and IL4R-Abx compared to Abx in each group are shown on the top of bars. *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant through one-way analysis of variance (ANOVA) followed by Tukey's multiple post hoc test. (C) Cytotoxicity assay. M2 macrophages were treated with 5 μg/mL of Abx, Ctrl-Abx, and IL4R-Abx for 24 h with or without pre-treatment with 500 nM of CLI095 for 30 min and subjected to cytotoxicity assays. Data are expressed as mean ± SD in the three separate experiments. P values compared to untreated control in each group are shown on the top of bars. *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant by two-way ANOVA followed by Bonferroni multiple post hoc test.
M2 macrophage treatments with Abx, Ctrl-Abx, and IL4R-Abx failed to significantly affect cell viability compared to untreated control (Figure 2C). However, M2 macrophage pre-treatment with CLI095, a TLR4 inhibitor, reduced cell viability through the above treatments compared to untreated control (Figure 2C), suggesting TLR4 involvement in the M2-macrophage viability against IL4R-targeted and untargeted Abx.
IL4R-Abx enhanced M2-macrophage reprogramming into M1-like phenotype
Abx and paclitaxel reprogram M2-type macrophages into the M1 phenotype [6, 8]. M2 macrophages were treated with untargeted and IL-4R-targeted Abx to examine the reprogramming of the macrophage phenotype. qRT-PCR analysis revealed that the relative mRNA levels of M1-macrophage markers, such as il12p40, il6, and ifn-γ, were significantly increased after IL4R-Abx treatment compared with untreated control, Abx, and Ctrl-Abx (Figure 3A-C). Conversely, the relative mRNA levels of M2-macrophage markers, such as fizz1, tgf-β, and il-10, were significantly decreased after IL4R-Abx treatment compared to untreated control and Ctrl-Abx, while the decreases in those of tgf-β and il-10 were not significant compared with Abx (Figure 3D-F). Compared to fizz1 and tgf-β, the decreases in the relative mRNA levels of il-10 by Abx and IL4R-Abx were weaker and showed lower responses (Figure 3F). In addition to the relative mRNA levels, the protein levels of M1-macrophage markers, such as IL12p70 (Figure 3G), CD86 (Figure 3H and S4A), and iNOS (Figure S4C), were increased after IL4R-Abx treatment compared with untreated control, Abx, and Ctrl-Abx. Conversely, the protein levels of M2-macrophage markers, such as TGF-β (Figure 3I), CD206 (Figure 3J and S4B), and Arg1 (Figure S4C), were decreased after IL4R-Abx treatment compared with untreated control, Abx, and Ctrl-Abx. Ctrl-Abx demonstrated weaker effects on the levels of M1- and M2-macrophage markers than unmodified Abx, indicating that Abx conjugation with non-targeting peptide interferes with naïve Abx activity (Figure 3A-J). These results suggest that IL4R-Abx reprograms the M2 macrophage into M1-like phenotype more efficiently than untargeted and control peptide-labeled Abx.
Figure 3
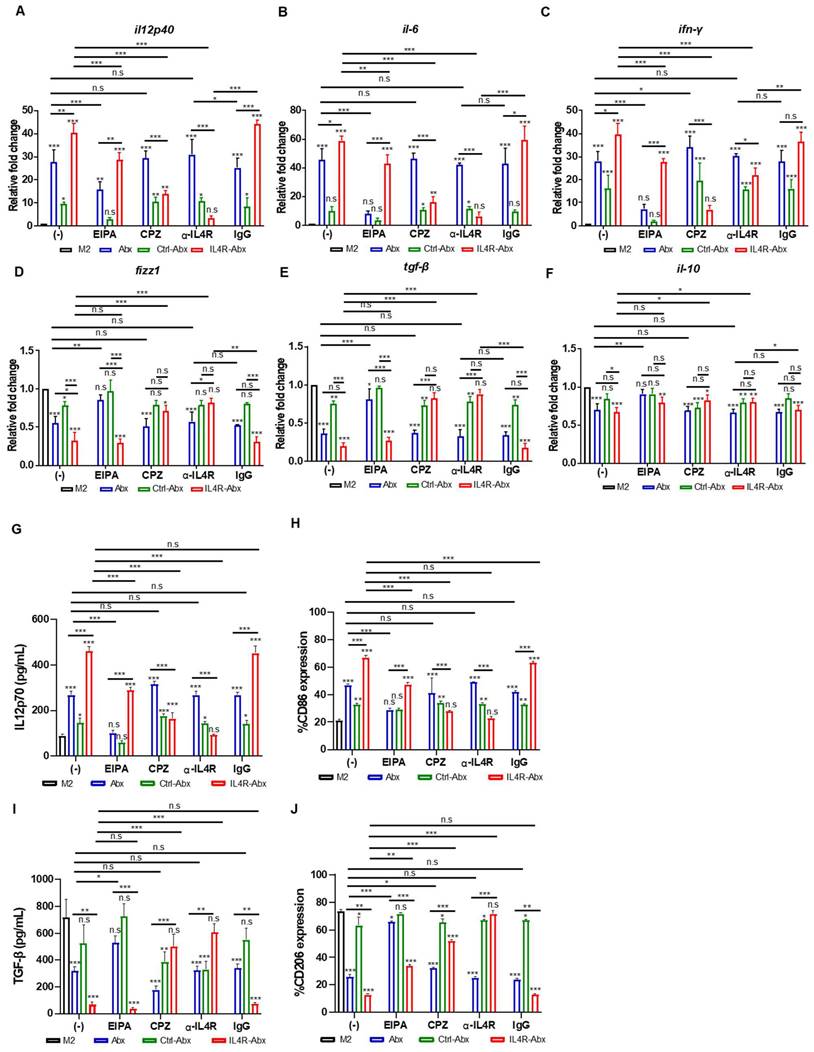
Reprogramming of M2 macrophages into M1-like phenotype by IL4R-Abx. (A-F) M2 macrophages were incubated with 5 μg/mL of Abx, Ctrl-Abx, and IL4R-Abx for 24 h without (-) or with pre-treatment with EIPA (50 μM), CPZ (50 μM), anti-IL4Rα antibody (100 μM), and IgG (100 μM) for 30 min. Relative mRNA levels of il12p40 (A), il6 (B), ifn-γ (C), fizz1 (D), tgf-β (E), and il10 (F) were analyzed using quantitative reverse transcription-polymerase chain reaction (qRT-PCR) using a real time cycler. (G, I) ELISA of the concentrations of IL12p70 (G) and TGF-β (I) secreted into the culture medium. (H, J) Flow cytometry analysis of CD86 (H) and CD206 (J) expression. Data are expressed as mean ± SD in the three separate experiments. P values compared to untreated M2-macrophage control (M2, black bar) are shown on the top of bars. *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant by one-way ANOVA followed by Tukey's multiple post hoc test.
Figure 4
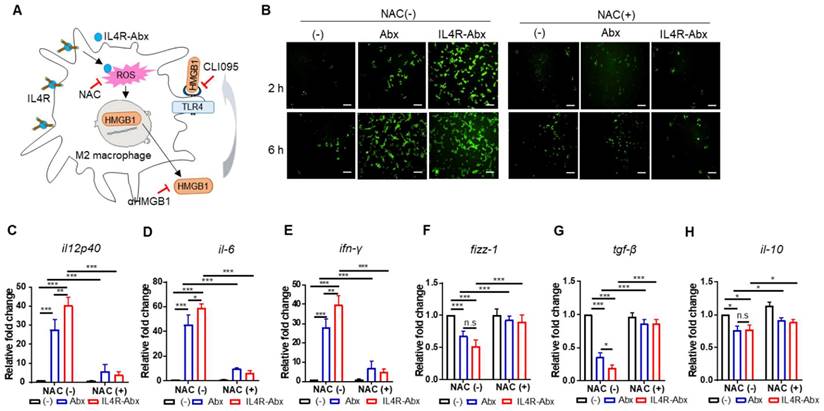
IL4R-Abx reprograms M2 macrophages into M1-like phenotype via increase in ROS. (A) Suggested pathways and inhibitors of IL4R-Abx-induced M2-macrophage reprogramming. (B) ROS analysis. M2 macrophages were incubated with 5 μg/mL of Abx and IL4R-Abx for 2 and 6 h with or without pre-treatment with 50 mM of N-acetyl-l-cysteine (NAC) for 30 min. A fluorescent microscope was used to image the fluorescence of H2DCFDA generated by ROS. (C-H) M2 macrophages were incubated with 5 μg/mL of Abx and IL4R-Abx for 24 h with or without pre-treatment with 50 mM of NAC for 30 min. Relative mRNA levels of M1-macrophage markers, such as il12p40 (C), il-6 (D), and ifn-γ (E), and M2-macrophage markers, such as fizz-1 (F), tgf-β (G), and il-10 (H), were measured via qRT-PCR analysis. (-), untreated M2-macrophage control (black bar).
Pre-treatment of M2 macrophages with EIPA hampered the increase of M1-macrophage markers, including relative mRNA levels of il12p40, il6, and ifn-γ (Figure 3A-C) and protein levels of IL12p70 (Figure 3G) and CD86 (Figure 3H and S4A), by Abx and IL4R-Abx. EIPA failed to inhibit the decrease of M2-macrophage markers, including relative mRNA levels of fizz1, tgf-β, and il-10 (Figure 3D-F), protein levels of TGF-β (Figure 3I), and CD206 (Figure 3J and S4B), by IL4R-Abx while still efficiently inhibiting that of Abx. Contrarily, M2-macrophage pre-treatment with CPZ and anti-IL4R blocking antibody remarkably inhibited the increase of M1-macrophage related mRNA and protein levels and the decrease of M2-macrophage related mRNA and protein levels by IL4R-Abx, while demonstrating negligible effects by Abx (Figure 3A-J and S4A-B).
IL4R-Abx reprograms M2 macrophages to M1-like phenotype via the ROS-HMGB1-TLR4 axis
Paclitaxel increases ROS accumulation and triggers the extracellular release of HMGB1 from macrophages, which contributes to paclitaxel-induced peripheral neuropathy [33]. Extracellular HMGB1 binds to TLR4 and activates pro-inflammatory cytokine secretion and skews M2 macrophage to the M1 phenotype [34-37]. According to these previous findings, we explored whether Abx and IL4R-Abx shape M2 macrophages toward the M1 phenotype via ROS-HMGB1-TLR4 axis stimulation (Figure 4A). IL4R-Abx increased the ROS levels in M2 macrophages at higher levels at 2 h after treatment compared to untreated cells and Abx, while the increases in ROS levels by Abx and IL4R-Abx were similar at 6 h after treatments, which were blocked by pre-treatment of cells with NAC, an ROS scavenger (Figure 4B). Moreover, NAC significantly impeded the increase of the relative mRNA levels of M1-macrophage markers (il12p40, il-6, and ifn-γ; Figure 4C-E), including the decrease of M2-macrophage markers (fizz-1, tgf-β, and il-10; Figure 4F-H) by Abx and IL4R-Abx. These results indicate that IL4R-Abx more rapidly and efficiently increases ROS levels in M2 macrophages and induces the skew of the macrophage polarity compared to Abx.
Next, the extracellular release and nuclear to cytoplasmic translocation of HMGB1 and subsequent activation of TLR4 after treatments with Abx and IR4R-Abx were examined. IL4R-Abx increased the extracellular release of HMGB1 into the CM of M2 macrophages as determined by ELISA (Figure 5A) and western blotting (Figure 5B and S5) more efficiently than Abx. In the same context, the nuclear to cytoplasmic translocation of HMGB1 was more remarkable after treatment with IL4R-Abx compared to Abx (Figure 5C). The extracellular release and nuclear to cytoplasmic translocation of HMGB1 were blocked by pre-treatment with NAC (Figure 5B, 5C, and S5). The CM of M2 macrophages treated with IL4R-Abx increased the relative mRNA levels of M1-macrophage markers (Figure 5D-F) and decreased those of the M2-macrophage markers (Figure 5G-I) in recipient M2 macrophages cultured on separate plates more efficiently than the CM of M2 macrophages incubated with Abx. Pre-treatment of recipient M2 macrophages with an anti-HMGB1 blocking antibody inhibited the alterations in the macrophage polarity markers induced by CM treatment (Figure 5D-I). Moreover, pre-treatment of recipient M2 macrophages with CLI095 also impeded the changes in the macrophage polarity markers induced by CM treatment (Figure 5D-I). These results indicate that after being internalized into M2 macrophages, Abx and IL4R-Abx skew them into M1-like phenotype through the ROS-HMGB1-TLR4 axis.
Figure 5
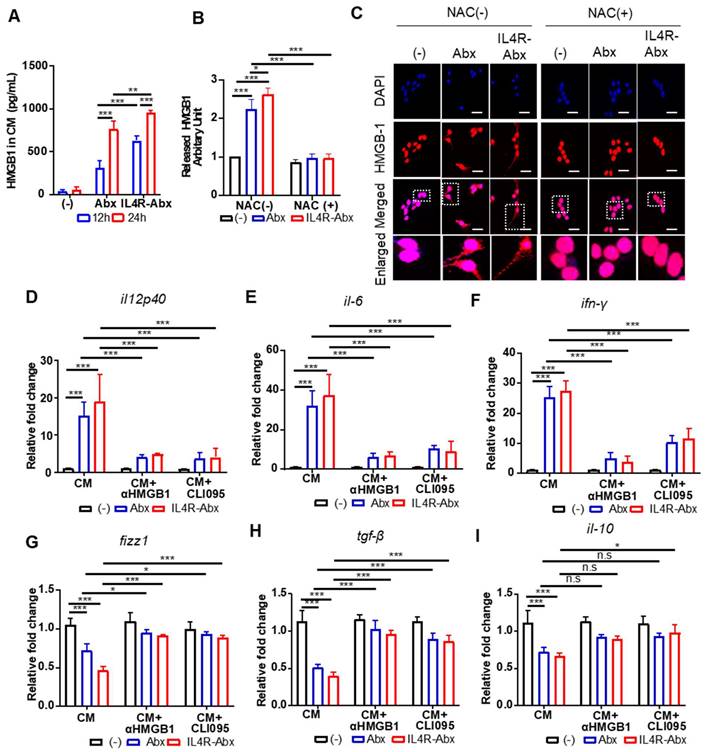
IL4R-Abx reprograms M2 macrophages into M1-like phenotype via the ROS-HMGB1-TLR4 axis. (A) M2 macrophages were incubated with 5 μg/mL of Abx and IL4R-Abx for 12 and 24 h. Concentrations of HMGB1 released into the conditioned medium were determined by ELISA. (B) M2 macrophages were incubated with 5 μg/mL of Abx and IL4R-Abx for 24 h with or without pre-treatment with 50 mM of NAC for 30 min. The conditioned medium was collected, concentrated, and subjected to western blotting using an anti-HMGB1 antibody. HMGB1 protein band intensity was quantified using Image J software. (C) M2 macrophages were incubated with Abx and IL4R-Abx for 24 h with or without pre-treatment with NAC. Cells were incubated with anti-HMGB1 antibody (red). Nucleus was stained with DAPI (blue), and images were merged. Boxes represent the enlarged area. Scale bars, 40 µm. (D-I) M2 macrophages were incubated using Abx or IL4R-Abx for 12 h with or without pre-treatment of anti-HMGB1 antibody (10 μg/mL, CM+αHMGB1) and CLI095 (500 nM, CM+CLI095) for 30 min. The CM of M2 macrophages was collected and treated to other sets of M2 macrophages for 24 h. Relative mRNA levels of M1-macrophage markers, such as il12p40 (D), il-6 (E), and ifn-γ (F), and M2-macrophage markers, such as fizz-1 (G), tgf-β (H), and il-10 (I), were measured via qRT-PCR analysis. Data are expressed as mean ± SD in the three separate experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant by one-way ANOVA followed by Tukey's multiple post hoc test. (-), untreated M2-macrophage control (black bar).
Figure 6
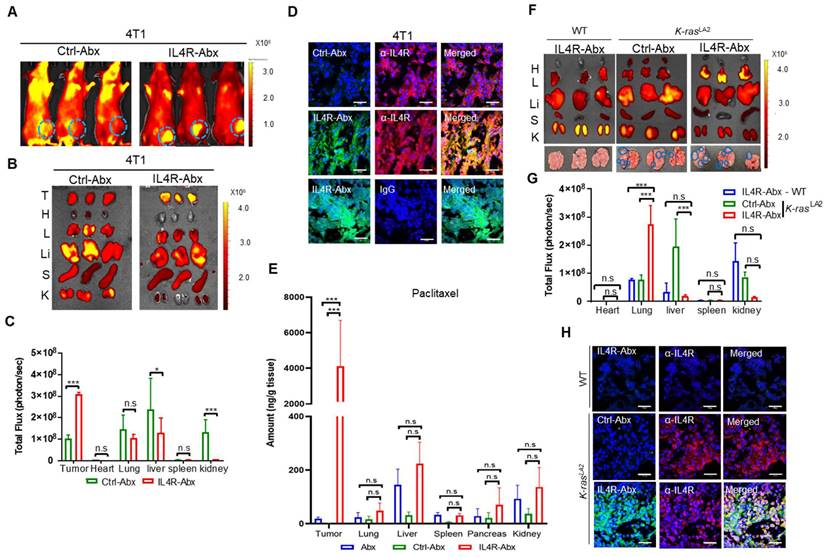
Tumor homing of IL4R-Abx in mice bearing 4T1 breast tumor and K-rasLA2 lung tumor. (A) Whole-body fluorescence imaging of mice bearing 4T1 tumor at 6 h after injection of near-infrared fluorescence dye-labeled Ctrl-Abx and IL4R-Abx (5 mg/kg body weight). Circles in cyan blue represent the region of interest (tumor site). (B, C) Ex vivo imaging of tumor and organs isolated after whole-body imaging (B) and total flux quantification (photon/s) of each organ (C). T, tumor; H, heart; L, lung; Li, liver; S, spleen; K, kidney. (D) Histochemical analysis. Tumor tissues isolated at 6 h after injection of FITC dye (green)-labeled Ctrl-Abx and IL4R-Abx underwent frozen section. Tissues were incubated using an anti-IL4R antibody or IgG (red) and with DAPI for nucleus staining (blue), and images were merged. Scale bars, 20 μm. (E) Amount of paclitaxel (ng/g tissue) in tumor and control organs were determined at 6 h after Abx, Ctrl-Abx, and IL4R-Abx injection (5 mg/kg body weight) in mice bearing 4T1 tumor. (F, G) Ex vivo imaging of tumor and organs isolated at 6 h after Ctrl-Abx and IL4R-Abx injection (5 mg/kg body weight) into K-rasLA2 mutant and wild-type (WT) littermate mice (F) and quantification of the total flux (photon/sec) of each organ (G). H, heart; L, lung; Li, liver; S, spleen; K, kidney. Circles (cyan blue) reflect tumor nodules in the lungs. (H) Histochemical analysis. Tumor tissues isolated at 6 h after injection of FITC dye (green)-labeled Ctrl-Abx and IL4R-Abx into K-rasLA2 mutant and WT littermate mice and then were subjected to frozen section. An anti-IL4R antibody (red) was used for tissue staining, with DAPI for nucleus staining (blue), and images were merged. Scale bars, 20 μm. Data are expressed as mean ± SD *, P < 0.05; ***, P < 0.001; n.s., not significant by two-way ANOVA followed by Bonferroni post hoc test (n = 3 per group).
IL4R-Abx selectively homes to tumors in mice
For in vivo fluorescence imaging, IL4R-Abx and Ctrl-Abx were conjugated with near-infrared fluorescence dye-labeled IL4RPep-1 and control peptide, respectively, and injected into mice bearing 4T1 breast tumor at the mammary fat pad via the tail vein. Whole-body fluorescence imaging acquired at 6 h after IL4R-Abx injection revealed higher levels of fluorescence signals at the tumor site and lower levels of background signals compared to Ctrl-Abx (Figure 6A). Ex vivo imaging and fluorescence intensity quantification of tumors and control organs isolated after the in vivo imaging also demonstrated higher levels of uptake at tumors and lower levels of accumulation at control organs, particularly at the liver, by IL4R-Abx than Ctrl-Abx (Figure 6B and 6C). Histological analysis further revealed higher levels of accumulation by IL4R-Abx than Ctrl-Abx in 4T1 tumor tissues where IL4R was abundantly expressed (Figure 6D). To examine paclitaxel amounts accumulated at tissues, mice bearing 4T1 breast tumor at the mammary fat pad were injected via the tail vein with targeted and untargeted Abx. Tumor and control organs such as the lung and liver were isolated, and the amounts of paclitaxel in tissues (ng/g tissue) were determined through mass spectrometry. In tumor tissues, significantly higher amounts of paclitaxel were detected with minimal accumulation at control organs in mice injected with IL4R-Abx compared to Abx and Ctrl-Abx (Figure 6E).
In addition to 4T1 breast tumor, we investigated tumor homing of IL4R-Abx in two lung tumor models: K-rasLA2 mutant transgenic mouse tumor and LLC syngeneic mouse tumor. Ex vivo imaging of lungs and control organs at 6 h after injection demonstrated that IL4R-Abx accumulated at lungs of K-rasLA2 mutant mice bearing tumor nodules at higher levels than at control organs such as the liver, whereas Ctrl-Abx accumulated mostly in the liver (Figure 6F and 6G). IL4R-Abx accumulation at lungs of WT mice was as low as Ctrl-Abx (Figure 6F and 6G). Histological analysis revealed higher levels of IL4R-Abx accumulation and IL4R expression in the lung tumor nodules of K-rasLA2 mutant mice compared with lung tissues of WT mice (Figure 6H). A similar pattern of tumor homing by IL4R-Abx was noted in mice bearing subcutaneous LLC lung tumor (Figure S6A-C). Moreover, higher levels of tumor accumulation of IL4R-Abx compared to Ctrl-Abx were observed up to 24 h after injection in Panc1 subcutaneous human pancreatic xenograft tumor (Figure S7A-D). These results suggested that IL4R-Abx more selectively homed to IL4R-high tumors with minimal accumulation at control organs than Abx and Ctrl-Abx.
IL4R-Abx inhibits tumor growth and metastasis and improves antitumor immunity
Mice bearing 4T1 tumor were injected intravenously (weekly for 4 weeks) with IL4R-targeted and untargeted Abx. IL4R-Abx decreased the tumor volumes (Figure 7A), tumor weights (Figure 7B), spleen weights or splenomegaly (Figure 7C and S8A), and the number of metastatic tumor nodules in the lungs (Figure 7D and S8B) more efficiently than Abx and Ctrl-Abx given at a similar dose (5 mg/kg body weight) and Abx given at a higher dose (10 mg/kg body weight). The decrease in the number of lung metastatic tumor nodules through Abx treatment was attained only at a 10 mg/kg body weight dose (Figure 7D). Next, immune cell population in tumor tissue and spleen were analyzed after treatments using flow cytometry (Figure S9A). Compared to Abx and Ctrl-Abx, IL4R-Abx more efficiently increased the population of M1-macrophage (Figure 7E and S9B) and CD8+ T-cells (Figure S9B and S9C) and CD8+ T-cell/Treg ratio (Figure 7G), while reducing the population of M2 macrophages (Figure 7F and S9B), MDSCs (Figure 7H and S9B), and Tregs (Figure S9B and S9D) in the tumor microenvironment. Conversely, untargeted Abx did not significantly change CD8+ T-cell/Treg ratio (Figure 7G) and the population of MDSC, CD8+ T-cells, and Tregs in tumor tissues (Figure 7H and S9B-D) even at a higher dose (10 mg/kg body weight). Similar to tumor tissues, IL4R-Abx changed immune cell population in the spleen of tumor-bearing mice and improved antitumor immunity more efficiently than Abx and Ctrl-Abx (Figure 7I-L).
Next, the therapeutic efficacy of IL4R-Abx in mouse lung tumor models was investigated. Ten-week old K-rasLA2 mutant mice and mice bearing LLC lung tumors were injected with IL4R-targeted and untargeted Abx (5 or 10 mg/kg body weight, weekly for 4 weeks). IL4R-Abx reduced the lung weights (Figure 8A), the total number of tumor lesions (Figure 8B and S10), and the number of tumor lesions above 3 mm in diameter (Figure 8C) and increased survival rates (Figure 8D) in K-rasLA2 mutant mice more efficiently than Abx and Ctrl-Abx, while no statistical difference was observed for the number of large lesions (above 3 mm in diameter) compared to Abx. In LLC lung tumor mice, IL4R-Abx decreased the tumor volumes (Figure 8E), tumor weights (Figure 8F), and number of metastatic tumor nodules in the lungs (Figure 8G) more efficiently than Abx and Ctrl-Abx. Additionally, IL4R-Abx increased the M1-macrophage population (Figure 8H) and CD8+ T-cell/Treg ratio (Figure 8J), while decreasing the M2-macrophage (Figure 8I) and MDSC population (Figure 8K) in LLC tumor tissues at higher levels than Abx and Ctrl-Abx. The reduction in the tumor volumes and increases in the M1-macrophage population and CD8/Treg ratio by IL4R-Abx were higher than those by Abx given at a higher dose (10 mg/kg body weight), while the decline in tumor weights, the number of metastatic nodules, and the M2-macrophage and MDSC population by IL4R-Abx was equivalent to those of Abx given at a higher dose (Figure 8E-K).
We used two pancreatic tumor models to further examine the antitumor effects of IL4R-Abx: KPC transgenic mice and Panc-1 human pancreatic tumor xenografts. KPC mice spontaneously developed pancreatic intraepithelial neoplasia at 8-10 weeks of age and invasive pancreatic ductal adenocarcinoma by the 16th week [38]. IL4R expression increased during 9-18 weeks of age in KPC mice (Figure S11A). IL4R-Abx decreased the population of Ki67+ proliferating cells and IL4R-expressing cells in tumor tissues more efficiently than Abx when systemically administered at 10 weeks of age (5 mg/kg body weight, weekly for 4 weeks) (Figure S11B). In the pancreatic tumor tissues of KPC mice, IL4R-Abx increased the M1-macrophage population (Figure S11C) and decreased the MDSC population (Figure S11F) at higher levels than Abx, while the M2-macrophage population (Figure S11D) and CD8+ T-cell/Treg ratio (Figure S11E) were equivalent to those of Abx. In Panc-1 human pancreatic tumor xenografts, IL4R-Abx inhibited the tumor volumes (Figure S12A), tumor weights (Figure S12B), number of metastatic nodules in the lungs (Figure S12C), and TUNEL+ apoptosis levels (Figure S12D) more effectively than Abx.
Serum levels of liver enzymes, such as AST and ALT, and kidney function indicators, such as BUN and creatinine, were measured at the culmination of the treatments to explore the systemic side effects of IL4R-Abx. IL4R-Abx did not induce a noticeable toxicity to the liver compared to saline; however, Abx increased the serum levels of AST in 4T1 (Figure S13A-D) and AST and ALT in K-rasLA2 mutant mice (Figure S13E-H) as well as Panc-1 tumor xenograft-bearing mice (Figure S13I-L), suggesting liver toxicity. The serum levels of BUN and creatinine after Abx and IL4R-Abx treatments were within normal ranges, suggesting no kidney toxicity. In addition, analysis of hematological parameters showed that white blood cell and neutrophil counts were above normal ranges (Figure S14A and S14B), while lymphocyte, monocyte, red blood cell, and platelet counts were close to or within normal ranges (Figure S14C-F), in 4T1 tumor-bearing mice treated with Abx and IL4R-Abx as well as saline. Furthermore, hemolysis tests showed that IL4R-Abx as well as Abx, Ctrl-Abx, and IL4RPep-1 peptide did not induce hemolysis (Figure S15A and S15B), indicating the biocompatibility of IL4R-Abx in the blood.
While the IL4R-Abx induced 4T1, LLC, and Panc-1 tumor cell cytotoxicity at higher levels than Ctrl-Abx, it revealed similar cytotoxicity levels and IC50 values in tumor cells compared to Abx (Figure S16A-C). IL4R-Abx induced lower levels of cytotoxicity in HEK293 normal cells than Abx and Ctrl-Abx (Figure S16D), suggesting preferential Abx delivery to tumor cells over normal cells. These results indicated that a more efficient inhibition of tumor growth and metastasis by IL4R-targeted Abx over untargeted Abx was primarily attributed to its effects on the M2-macrophage polarity rather than tumor cell viability.
Figure 7
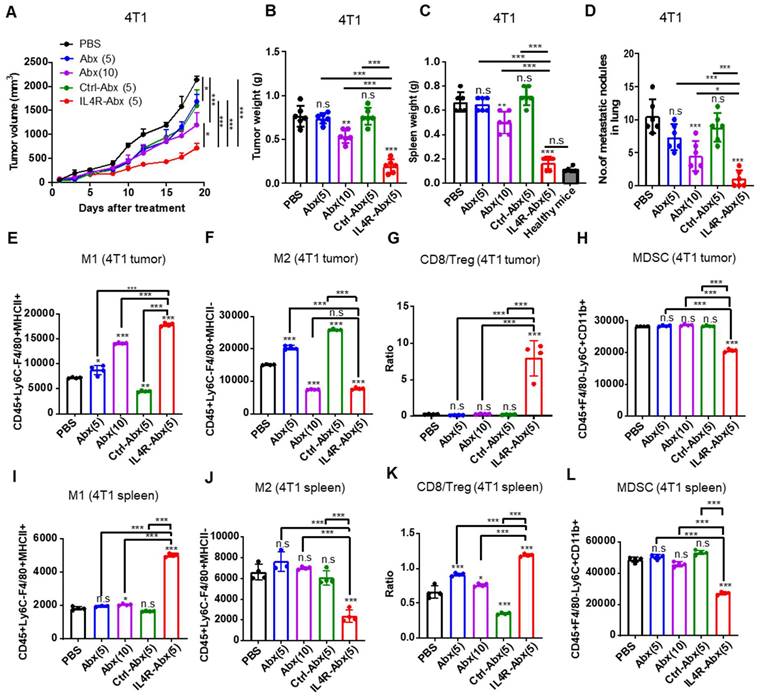
Inhibition of tumor growth and metastasis and improvement of antitumor immunity by IL4R-Abx in mice bearing 4T1 breast tumor. (A-D) Mice bearing 4T1 tumor were injected with Abx, Ctrl-Abx, and IL4R-Abx (5 or 10 mg/kg body weight, weekly for 4 weeks). The tumor volume (A), tumor weight (B), spleen weight (C), and number of metastatic nodules in the lungs (D) after treatments were measured. (E-L) At the culmination of the treatments, tumor and spleen were isolated for immune cell analysis using flow cytometry. The population of CD45+F4/80+Ly6C-MHC II+ M1 macrophages (E), CD45+F4/80+Ly6C-MHC II M2 macrophages (F), CD8/Treg ratio (G), and CD45+F4/80-Ly6C+CD11b+ MDSCs (H) in 0.1 g of tumor tissues and the population of M1 macrophages (I), M2 macrophages (J), CD8/Treg ratio (K), and MDSCs (L) in 105 cells of spleen were determined. Data are expressed as mean ± SD. P values compared to saline-treated group are shown on the top of bars. *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant by one-way ANOVA followed by Tukey's multiple post hoc test. (A, n = 10 per group; B-D, n = 6 per group; E-L, n = 4 per group).
M2 macrophages (F), CD8/Treg ratio (G), and CD45+F4/80-Ly6C+CD11b+ MDSCs (H) in 0.1 g of tumor tissues and the population of M1 macrophages (I), M2 macrophages (J), CD8/Treg ratio (K), and MDSCs (L) in 105 cells of spleen were determined. Data are expressed as mean ± SD. P values compared to saline-treated group are shown on the top of bars. *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant by one-way ANOVA followed by Tukey's multiple post hoc test. (A, n = 10 per group; B-D, n = 6 per group; E-L, n = 4 per group).
Figure 8
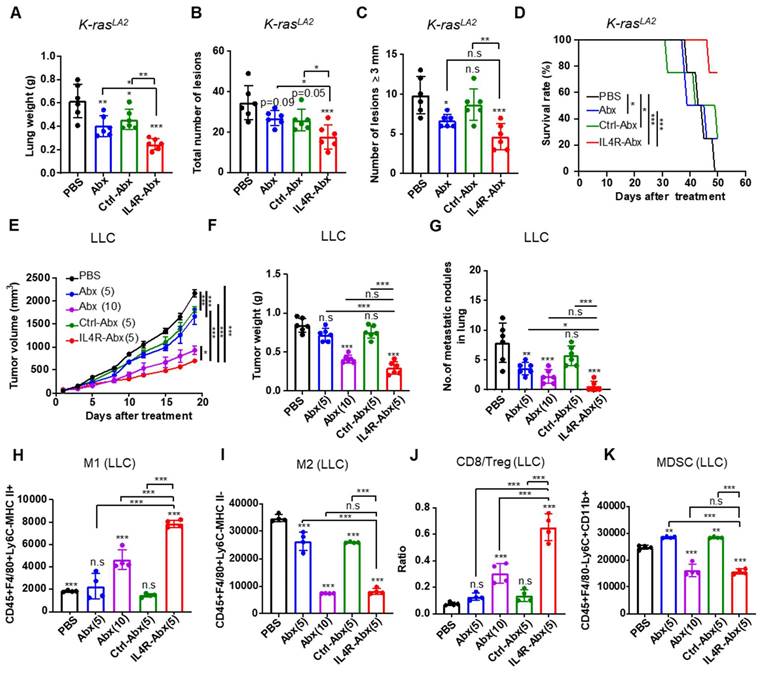
Therapeutic efficacy of IL4R-Abx in K-rasLA2 mutant and LLC lung tumor-bearing mice. (A-D) K-rasLA2 mutant mice bearing spontaneous lung tumors were intravenously injected with Abx, Ctrl-Abx, and IL4R-Abx (5 mg/kg body weight, weekly for 4 weeks). The lung weight (A), the total number of tumor lesions in the lungs (B), the number of tumor lesions above 3 mm in diameter (C), and survival rate (D) at the culmination of the treatments were determined. (E-K) Mice bearing subcutaneous LLC lung tumor were injected intravenously with Abx, Ctrl-Abx, and IL4R-Abx (5 or 10 mg/kg body weight, weekly for 4 weeks). The tumor volume (E), tumor weight (F), and the number of metastatic nodules in the lungs (G) were determined. The population of CD45+F4/80+Ly6C-MHC II+ M1 macrophages (H), CD45+F4/80+Ly6C-MHC II-M2 macrophages (I), CD8/Treg ratio (J), and CD45+F4/80-Ly6C+CD11b+ MDSCs (K) in 0.1 g of tumor tissues were determined via flow cytometry. Data are expressed as mean ± SD. P values compared to the saline-treated group are shown on the top of bars. *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant by one-way ANOVA followed by Tukey's multiple post hoc test. (A-D, n = 6 per group; E, n = 10 per group; F-G, n = 6 per group; H-K, n = 4 per group).
Discussion
The study findings demonstrated that IL4R targeting allowed Abx to improve M2-macrophage reprogramming into an M1-like phenotype through the ROS-HMGB1-TLR4 axis and enhance the antitumor immunity via increasing immune-stimulatory cells while decreasing immune-suppressive cells in the tumor microenvironment, contributing to the inhibition of tumor growth and metastasis in tumor-bearing mouse models (Figure S17). Treatment with IL4R-Abx at a lower dose (5 mg/kg body weight) exerted higher levels of therapeutic efficacy compared to Abx (10 mg/kg body weight), suggesting IL4R targeting can reduce the dose of Abx. Moreover, IL4R-Abx enhanced survival rates and was accompanied by negligible systemic side effects such as liver toxicity compared to Abx. The IL4R-targeting activity was attained through the conjugation of the IL4R-targeting IL4RPep-1 peptide to a sulfur group exposed on the albumin surface of Abx. Abx has a core-shell structure with paclitaxel comprising the core through hydrophobic interactions, while albumin coats the paclitaxel, forming a stabilizing layer around the payload [39, 40]. Given the layered nature of albumin, the conjugation reaction with albumin molecules at inner layers seems to be hindered by steric hindrance, resulting in the conjugation ratio of 60% in this study.
Compared to Abx, IL4R-Abx was more efficiently internalized into the M2 macrophages. The internalization of IL4R-Abx was primarily via the receptor-mediated endocytosis and was inhibited by an anti-IL4R antibody and a CPZ endocytosis inhibitor. Conversely, Abx was internalized primarily through the macropinocytosis as previously known [8]. The increase in Abx internalization by CPZ observed in this study might be explained by previous findings that the inhibition of the clathrin-mediated endocytosis can increase the clathrin-independent endocytosis including macropinocytosis [41, 42]. After internalization into macrophages, IL4R-Abx and Abx induced M2-macrophage reprogramming into M1-like phenotype through the ROS-HMGB1-TLR4 axis. IL4R-Abx and Abx increased intracellular ROS levels in M2 macrophages and extracellular release of HMGB1. NAC, a well-known ROS scavenger, inhibited intracellular ROS elevation and extracellular release of HMGB1 and subsequently inhibited the increase of M1-type cytokines and the decrease of M2-type cytokines induced by IL4R-Abx and Abx. Moreover, the CM of M2 macrophages pre-treated with IL4R-Abx and Abx elevated the M1-type cytokines and decreased the M2-type cytokines, indicating that an extracellular factor(s) is involved in this event. In the same context, paclitaxel has been shown to induce the extracellular release of HMGB1 in macrophages via the ROS/p38MAP kinase/NF-κB/histone acetyltransferase pathway [33]. Furthermore, either an anti-HMGB1 antibody or a TLR4 inhibitor CLI095 efficiently hampered the alterations in the cytokine levels induced by the CM of M2 macrophages treated with Abx and IL4R-Abx. Collectively, our results indicate that IL4R-Abx and Abx enhanced ROS level upregulation, and the release of HMGB1 and extracellular HMGB1 bound to TLR4 and activated TLR4, resulting in pro-inflammatory activation of M2 macrophages. However, macrophages have an increased p-glycoprotein expression as an efflux pump of paclitaxel [43]. Paclitaxel activates macrophages via a direct binding to TLR4 on the macrophage cell surface [6]. These previous findings suggest that paclitaxel may be released from M2 macrophages treated with IL4R-Abx and Abx into the CM and contribute to the CM-induced TLR4 activation in collaboration with HMGB1. IL4R-targeted Abx and the unmodified Abx did not trigger cytotoxicity in the M2 macrophages, which corroborates with previous findings [6, 8, 43], and rather induced M2-macrophage repolarization. TLR4-mediated NF-κB activation promotes a resistance to Abx-mediated cell death [44], supporting the increase in cell death after Abx and IL4R-Abx treatments when TLR4 was inhibited by CLI095.
Whole-body imaging, histologic analysis, and measurement of paclitaxel concentrations in tissues showed that IL4R targeting allowed Abx to selectively accumulate at tumors avoiding control organs and thereby delivering more paclitaxel to tumors. In vivo and ex vivo fluorescence signals by the homing of nanoparticles tend to be dependent on the size and depth in the body of each organ. To accurately determine the amount of paclitaxel accumulated at organs by IL4R-Abx, the mass spectrometry quantification of paclitaxel in tissues was included. Organs such as the liver were homogenized, and the total amount of paclitaxel in the organ was measured and represented as an amount per tissue weight (ng/g tissue). Because the liver was larger than tumor, the relative amount of paclitaxel in the liver after IL4R-Abx treatment was lower compared to tumor, while the ex vivo fluorescence signals of the liver were apparently high. This may explain the discrepancy between the fluorescence images and paclitaxel amounts in the liver and tumor.
Targeted nanoparticle delivery, such as Abx, to tumors depends on the enhanced permeability and retention effect of nanoparticles through leaky tumor blood vessels while avoiding mature and tightly sealed blood vessels of normal tissues [45]. An additional merit of albumin-bound nanoparticle formulation of paclitaxel is that it can eliminate the requirement for Cremophor, a toxic agent for solubility, and increase the circulation time of paclitaxel [45]. Affinity-based targeting or targeted delivery of Abx via albumin modification with a tumor-homing peptide has been first exploited by Ruoslahti et al. [46]. They conjugated Abx using CREKA peptide that binds to clotted plasma proteins in tumor blood vessels or LyP-1 peptide (CGNKRTRGC) that binds to p32 overexpressed on tumor cells; the CREKA- or LyP-1-guided Abx led to a significant inhibition of tumor growth compared to untargeted Abx [46]. Furthermore, the combined treatment of Abx and CEND-1 (or iRGD) peptide, a cyclic peptide that targets alpha V-integrin and neurophilin-1 and improves internalization of co-administered drugs into tumor cells, demonstrated an increased activity and acceptable safety profile in a clinical trial [47]. These results indicate that the direct Abx modification with a tumor-homing peptide or combined treatment of Abx and the iRGD internalizing peptide is a promising tool to improve tumor cell cytotoxicity of Abx.
Cancer therapy targeting M2 macrophages has been exploited in numerous methods [48, 49]. For example, TLR3 and TLR7 agonists activated NF-κB and triggered secretion of immune-stimulatory cytokines in macrophages and have been in clinical trials for cancer therapy [49]. An antibody against colony-stimulating factor-1 receptor hindered macrophage recruitment into tumors [50]. An antibody against MARCO, a pattern recognition receptor, reprogrammed M2 macrophages into pro-inflammatory M1 macrophages [51]. An agonistic anti-CD40 antibody triggered nitric oxide production in macrophages and inhibited tumor growth [52]. In addition to antibodies, trabectedin, a chemotherapeutic drug, exerted antitumor growth activity via monocyte and macrophage cytotoxicity [53]. Epigenetic therapies using DNA methylation and histone deacetylation inhibitors skewed M2 macrophages into the M1-like phenotype [54] and stimulated MDSC differentiation into a more-interstitial macrophage-like phenotype [55], leading to tumor progression and metastasis inhibition. Furthermore, M1-macrophage-derived exosomes engineered to target IL4R and stimulate pro-inflammatory polarization of macrophages skewed M2 macrophages toward M1-like macrophages [56].
Abx promotes multiple steps in the cancer-immunity cycle: antigen presentation; T-cell activation and infiltration; and tumor cell killing. Antigen peptides from tumor cells are generally presented to CD4+ T cells via MHC II on dendritic cells (DCs). Paclitaxel induces the expression of MHC II on DCs [57] and upregulates the expression of antigen processing machinery and costimulatory molecules such as CD86 in DCs [58]. In addition, Abx promotes activation of CD8+ T cells by inhibiting the function of immune-suppressive cells including MDSCs and Tregs [59]. Abx inhibits cancer-associated fibroblast proliferation and extracellular matrix synthesis, which promotes T-cell infiltration into tumor tissues [59]. Moreover, Abx inhibits microtubule of tumor cells and coopts with CD8+ T cells to kill tumor cells. To our interest, MDSCs in the tumor microenvironment also express high levels of IL4R, and the IL4R expression is critical for their immune-suppressive activity on CD8+ T cells [60]. These findings suggest that IL4R-Abx may enable an increased delivery of nab-paclitaxel to MDSCs as well as M2-macrophages, leading to more efficient inhibition of immune-suppressive myeloid cells compared to Abx. In addition to breast, lung, and pancreatic tumors exploited in this study, IL4R is upregulated in other types of tumors such as glioblastoma [61] and Hodgkin lymphoma [62]. Collectively, these findings suggest that IL4R-targeted Abx has a broad spectrum of applications in the management of patients with cancer.
Conclusions
Altogether, the current study suggests that IL4R-targeted Abx presents as a promising cancer immunotherapeutic agent targeting M2 macrophages.
Supplementary Material
Supplementary methods and figures.
Acknowledgements
This work was supported by National Research Foundation of Korea (NRF) grants [2021R1A5A2021614 and 2022M3A9G8095334 to B. Lee] and [2022R1A3B1077354 to W.J. Kim] funded by the Ministry of Science and ICT of Korea.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39-51
2. Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22:231-7
3. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49-61
4. Komohara Y, Jinushi M, Takeya M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014;105:1-8
5. Aapro MS, Von Minckwitz G. Molecular basis for the development of novel taxanes in the treatment of metastatic breast cancer. Eur J Cancer Suppl. 2008;6:3-11
6. Wanderley CW, Colon DF, Luiz JPM, Oliveira FF, Viacava PR, Leite CA. et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1 profile in a TLR4-dependent manner. Cancer Res. 2018;78:5891-900
7. Zimmer SM, Liu J, Clayton JL, Stephens DS, Snyder JP. Paclitaxel binding to human and murine MD-2. J Biol Chem. 2008;283:27916-26
8. Cullis J, Siolas D, Avanzi A, Barui S, Maitra A, Bar Sagi D. Macropinocytosis of nab-paclitaxel drives macrophage activation in pancreatic cancer. Cancer Immunol Res. 2017;5:182-90
9. Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol. 1999;17:701-38
10. LaPorte SL, Juo ZS, Vaclavikova J, Colf LA, Qi X, Heller NM. et al. Molecular and structural basis of cytokine receptor pleiotropy in the interleukin-4/13 system. Cell. 2008;132:259-72
11. Leland P, Taguchi J, Husain SR, Kreitman RJ, Pastan I, Puri RK. Human breast carcinoma cells express type II IL-4 receptors and are sensitive to antitumor activity of a chimeric IL-4-Pseudomonas exotoxin fusion protein in vitro and in vivo. Mol Med. 2000;6:165-78
12. Kawakami M, Kawakami K, Stepensky VA, Maki RA, Robin H, Muller W. et al. Interleukin 4 receptor on human lung cancer: a molecular target for cytotoxin therapy. Clin Cancer Res. 2002;8:3503-11
13. Park MH, Kwon HJ, Kim JR, Lee B, Lee SJ, Bae YK. Elevated interleukin-13 receptor alpha 1 expression in tumor cells is associated with poor prognosis in patients with invasive breast cancer. Ann Surg Oncol. 2017;24:3780-7
14. Movahedi K, Laoui D, Gysemans C, Baeten M, Stangé G, Van den Bossche J. et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C (high) monocytes. Cancer Res. 2010;70:5728-39
15. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253-68
16. Shiao SL, Ruffell B, DeNardo DG, Faddegon BA, Park CC, Coussens LM. TH2-polarized CD4+ T cells and macrophages limit efficacy of radiotherapy. Cancer Immunol Res. 2015;3:518-25
17. Vadevoo SMP, Kim JE, Gunassekaran GR, Jung HK, Chi L, Kim DE. et al. IL4 Receptor-Targeted Proapoptotic Peptide Blocks Tumor Growth and Metastasis by Enhancing Antitumor Immunity. Mol Cancer Ther. 2017;16:2803-16
18. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176:287-92
19. DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N. et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16:91-102
20. Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL. et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010;24:241-55
21. Ladner RC, Sato AK, Gorzelany J, de Souza M. Phage display-derived peptides as therapeutic alternatives to antibodies. Drug Discov Today. 2004;9:525-9
22. Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today. 2015;20:122-8
23. Wang L, Wang N, Zhang W, Cheng X, Yan Z, Shao G. et al. Therapeutic peptides: Current applications and future directions. Signal Transduct Target Ther. 2022;7:48
24. Vadevoo SMP, Gurung S, Lee HS, Gunassekaran GR, Lee SM, Yoon JW. et al. Peptides as multifunctional players in cancer therapy. Exp Mol Med. 2023:1-11
25. Hong HY, Lee HY, Kwak W, Yoo J, Na MH, So IS. et al. Phage display selection of peptides that home to atherosclerotic plaques: IL-4 receptor as a candidate target in atherosclerosis. J Cell Mol Med. 2008;12:2003-14
26. Namgung R, Lee YM, Kim J, Jang Y, Lee BH, Kim IS. et al. Poly-cyclodextrin and poly-paclitaxel nano-assembly for anticancer therapy. Nat Commun. 2014;5:1-12
27. Chi L, Na MH, Jung HK, Vadevoo SMP, Kim CW, Padmanaban G. et al. Enhanced delivery of liposomes to lung tumor through targeting interleukin-4 receptor on both tumor cells and tumor endothelial cells. J Control Rel. 2015;209:327-36
28. Guruprasath P, Kim J, Gunassekaran GR, Chi L, Kim S, Park RW. et al. Interleukin-4 receptor-targeted delivery of Bcl-xL siRNA sensitizes tumors to chemotherapy and inhibits tumor growth. Biomater. 2017;142:101-11
29. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2- ΔΔCT method. Methods. 2001;25:402-8
30. Jung H, Park S, Gunassekaran GR, Jeon M, Cho YE, Baek MC. et al. A peptide probe enables photoacoustic-guided imaging and drug delivery to lung tumors in K-rasLA2 mutant mice. Cancer Res. 2019;79:4271-82
31. Mishra A, Vats A. Supervised machine learning classification algorithms for detection of fracture location in dissimilar friction stir welded joints. Frat Integrità Strutt. 2021;15:242-53
32. Kerr MC, Teasdale RD. Defining macropinocytosis. Traffic. 2009;10:364-71
33. Sekiguchi F, Domoto R, Nakashima K, Yamasoba D, Yamanishi H, Tsubota M. et al. Paclitaxel-induced HMGB1 release from macrophages and its implication for peripheral neuropathy in mice: Evidence for a neuroimmune crosstalk. Neuropharmacol. 2018;141:201-13
34. Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J. et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA. 2010;107:11942-7
35. Yang H, Wang H, Andersson U. Targeting inflammation driven by HMGB1. Front Immunol. 2020;11:484
36. Paudel YN, Angelopoulou E, Piperi C, Othman I, Aamir K, Shaikh MF. Impact of HMGB1, RAGE, and TLR4 in Alzheimer's disease (AD): from risk factors to therapeutic targeting. Cells. 2020;9:383
37. Schaper F, de Leeuw K, Horst G, Bootsma H, Limburg PC, Heeringa P. et al. High mobility group box 1 skews macrophage polarization and negatively influences phagocytosis of apoptotic cells. Rheumatol. 2016;55:2260-70
38. Lee JW, Komar CA, Bengsch F, Graham K, Beatty GL. Genetically engineered mouse models of pancreatic cancer: the KPC model (LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1-Cre), its variants, and their application in immuno-oncology drug discovery. Curr Protoc Pharmacol. 2016;73:14.39 1-14.39. 20
39. Desai N. Albumin-drug nanoparticles. Drug Delivery in Oncology: From Basic Research to Cancer Therapy. 2011:1133-61
40. Desai N. Nanoparticle albumin-bound paclitaxel (Abraxane®). In: Otagiri M and Chuang V.T.G. Ed. Albumin in medicine: Pathological and clinical applications.1st ed. Berlin: Springer. 2016:101-19
41. Sandvig K, Kavaliauskiene S, Skotland T. Clathrin-independent endocytosis: an increasing degree of complexity. Histochem Cell Biol. 2018;150:107-18
42. Mayor S, Parton RG, Donaldson JG. Clathrin-independent pathways of endocytosis. Cold Spring Harb Perspect Biol. 2014;6:a016758
43. Tanei T, Leonard F, Liu X, Alexander JF, Saito Y, Ferrari M. et al. Redirecting transport of nanoparticle albumin-bound paclitaxel to macrophages enhances therapeutic efficacy against liver metastases. Cancer Res. 2016;76:429-39
44. Ma Y, Temkin V, Liu H, Pope RM. NF-κB protects macrophages from lipopolysaccharide-induced cell death: the role of caspase 8 and receptor-interacting protein. J Biol Chem. 2005;280:41827-34
45. Davis ME, Chen Z, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771-82
46. Karmali PP, Kotamraju VR, Kastantin M, Black M, Missirlis D, Tirrell M. et al. Targeting of albumin-embedded paclitaxel nanoparticles to tumors. Nanomed. 2017;5:647-66
47. Dean A, Gill S, McGregor M, Broadbridge V, Järveläinen HA, Price T. Dual αV-integrin and neuropilin-1 targeting peptide CEND-1 plus nab-paclitaxel and gemcitabine for the treatment of metastatic pancreatic ductal adenocarcinoma: A first-in-human, open-label, multicentre, phase 1 study. Lancet Gastroenterol Hepatol. 2022;7:943-51
48. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399-416
49. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov. 2022;21:799-820
50. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V. et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer cell. 2014;25:846-59
51. Georgoudaki A-M, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Östling J. et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 2016;15:2000-11
52. Lum HD, Buhtoiarov IN, Schmidt BE, Berke G, Paulnock DM, Sondel PM. et al. Tumoristatic effects of anti-CD40 mAb-activated macrophages involve nitric oxide and tumour necrosis factor-α. Immunol. 2006;118:261-70
53. Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M. et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23:249-62
54. Vadevoo SMP, Gunassekaran GR, Yoo JD, Kwon TH, Hur K, Chae S. et al. Epigenetic therapy reprograms M2-type tumor-associated macrophages into an M1-like phenotype by upregulating miR-7083-5p. Front Immunol. 2022;13:976196
55. Lu Z, Zou J, Li S, Topper MJ, Tao Y, Zhang H. et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature. 2020;579:284-90
56. Gunassekaran GR, Vadevoo SMP, Baek MC, Lee B. M1 macrophage exosomes engineered to foster M1 polarization and target the IL-4 receptor inhibit tumor growth by reprogramming tumor-associated macrophages into M1-like macrophages. Biomater. 2021;278:121137
57. John J, Ismail M, Riley C, Askham J, Morgan R, Melcher A. et al. Differential effects of Paclitaxel on dendritic cell function. BMC Immunol. 2010;11:1-11
58. Shurin GV, Tourkova IL, Kaneno R, Shurin MR. Chemotherapeutic agents in noncytotoxic concentrations increase antigen presentation by dendritic cells via an IL-12-dependent mechanism. J Immunol. 2009;183:137-44
59. Chen Y, Liu R, Li C, Song Y, Liu G, Huang Q. et al. Nab-paclitaxel promotes the cancer-immunity cycle as a potential immunomodulator. Am J Cancer Res. 2021;11:3445
60. Mandruzzato S, Solito S, Falisi E, Francescato S, Chiarion-Sileni V, Mocellin S. et al. IL4Rα+ myeloid-derived suppressor cell expansion in cancer patients. J Immunol. 2009;182:6562-8
61. Sampson JH, Singh Achrol A, Aghi MK, Bankiewicz K, Bexon M, Brem S. et al. Targeting the IL4 receptor with MDNA55 in patients with recurrent glioblastoma: Results of a phase IIb trial. Neuro-oncol. 2023;25:1085-97
62. Natoli A, Lüpertz R, Merz C, Müller WW, Köhler R, Krammer PH. et al. Targeting the IL-4/IL-13 signaling pathway sensitizes Hodgkin lymphoma cells to chemotherapeutic drugs. Int J Cancer. 2013;133:1945-54
Author contact
![]() Corresponding authors: Byungheon Lee, Department of Biochemistry and Cell Biology, School of Medicine, Kyungpook National University, 680 Gukchaebosang-ro, Jung-gu, Daegu 41944, Korea. Phone: +82-53-420-4824, Fax: +82-53-422-1466, E-mail: leebhac.kr. Won Jong Kim, Department of Chemistry, POSTECH-CATHOLIC Biomedical Engineering Institute, Pohang University of Science and Technology (POSTECH), 77 Cheongam-ro, Nam-gu, Pohang, Gyeongbuk 37673, Korea. Phone: +82-10-9426-2104, E-mail: wjkimac.kr.
Corresponding authors: Byungheon Lee, Department of Biochemistry and Cell Biology, School of Medicine, Kyungpook National University, 680 Gukchaebosang-ro, Jung-gu, Daegu 41944, Korea. Phone: +82-53-420-4824, Fax: +82-53-422-1466, E-mail: leebhac.kr. Won Jong Kim, Department of Chemistry, POSTECH-CATHOLIC Biomedical Engineering Institute, Pohang University of Science and Technology (POSTECH), 77 Cheongam-ro, Nam-gu, Pohang, Gyeongbuk 37673, Korea. Phone: +82-10-9426-2104, E-mail: wjkimac.kr.
Citation styles
APA
Vadevoo, S.M.P., Kang, Y., Gunassekaran, G.R., Lee, S.M., Park, M.S., Jo, D.G., Kim, S.K., Lee, H., Kim, W.J., Lee, B. (2024). IL4 receptor targeting enables nab-paclitaxel to enhance reprogramming of M2-type macrophages into M1-like phenotype via ROS-HMGB1-TLR4 axis and inhibition of tumor growth and metastasis. Theranostics, 14(6), 2605-2621. https://doi.org/10.7150/thno.92672.
ACS
Vadevoo, S.M.P.; Kang, Y.; Gunassekaran, G.R.; Lee, S.M.; Park, M.S.; Jo, D.G.; Kim, S.K.; Lee, H.; Kim, W.J.; Lee, B. IL4 receptor targeting enables nab-paclitaxel to enhance reprogramming of M2-type macrophages into M1-like phenotype via ROS-HMGB1-TLR4 axis and inhibition of tumor growth and metastasis. Theranostics 2024, 14 (6), 2605-2621. DOI: 10.7150/thno.92672.
NLM
Vadevoo SMP, Kang Y, Gunassekaran GR, Lee SM, Park MS, Jo DG, Kim SK, Lee H, Kim WJ, Lee B. IL4 receptor targeting enables nab-paclitaxel to enhance reprogramming of M2-type macrophages into M1-like phenotype via ROS-HMGB1-TLR4 axis and inhibition of tumor growth and metastasis. Theranostics 2024; 14(6):2605-2621. doi:10.7150/thno.92672. https://www.thno.org/v14p2605.htm
CSE
Vadevoo SMP, Kang Y, Gunassekaran GR, Lee SM, Park MS, Jo DG, Kim SK, Lee H, Kim WJ, Lee B. 2024. IL4 receptor targeting enables nab-paclitaxel to enhance reprogramming of M2-type macrophages into M1-like phenotype via ROS-HMGB1-TLR4 axis and inhibition of tumor growth and metastasis. Theranostics. 14(6):2605-2621.
This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See http://ivyspring.com/terms for full terms and conditions.