Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results and discussion
Conclusion
Materials and methods
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Results and discussion
Conclusion
Materials and methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(8):1145-1159. doi:10.7150/thno.15257 This issue Cite
Research Paper
Synthesis and Preclinical Evaluation of Sulfonamido-based [11C-Carbonyl]-Carbamates and Ureas for Imaging Monoacylglycerol Lipase
Lu Wang1*, Wakana Mori2*, Ran Cheng1,3, Joji Yui2, Akiko Hatori2, Longle Ma1, Yiding Zhang2, Benjamin H. Rotstein1, Masayuki Fujinaga2, Yoko Shimoda2, Tomoteru Yamasaki2, Lin Xie2, Yuji Nagai2, Takafumi Minamimoto2, Makoto Higuchi2, Neil Vasdev1, Ming-Rong Zhang2 ![]() , Steven H. Liang1
, Steven H. Liang1 ![]()
1. Gordon Center of Medical Imaging, Division of Nuclear Medicine and Molecular Imaging, Massachusetts General Hospital & Department of Radiology, Harvard Medical School, Boston, MA, 02114, USA.
2. Molecular Imaging Center, National Institute of Radiological Sciences, Chiba, 263-8555, Japan.
3. School of Pharmaceutical Science and Technology, Tianjin University, Tianjin, 300072, China.
*These authors contributed equally to the work.
Received 2016-2-12; Accepted 2016-3-18; Published 2016-5-21
Citation:
Wang L, Mori W, Cheng R, Yui J, Hatori A, Ma L, Zhang Y, Rotstein BH, Fujinaga M, Shimoda Y, Yamasaki T, Xie L, Nagai Y, Minamimoto T, Higuchi M, Vasdev N, Zhang MR, Liang SH. Synthesis and Preclinical Evaluation of Sulfonamido-based [11C-Carbonyl]-Carbamates and Ureas for Imaging Monoacylglycerol Lipase. Theranostics 2016; 6(8):1145-1159. doi:10.7150/thno.15257. https://www.thno.org/v06p1145.htm
Other stylesAbstract

Monoacylglycerol lipase (MAGL) is a 33 kDa member of the serine hydrolase superfamily that preferentially degrades 2-arachidonoylglycerol (2-AG) to arachidonic acid in the endocannabinoid system. Inhibition of MAGL is not only of interest for probing the cannabinoid pathway but also as a therapeutic and diagnostic target for neuroinflammation. Limited attempts have been made to image MAGL in vivo and a suitable PET ligand for this target has yet to be identified and is urgently sought to guide small molecule drug development in this pathway. Herein we synthesized and evaluated the physiochemical properties of an array of eleven sulfonamido-based carbamates and ureas with a series of terminal aryl moieties, linkers and leaving groups. The most potent compounds were a novel MAGL inhibitor, N-((1-(1H-1,2,4-triazole-1-carbonyl)piperidin-4-yl) methyl)-4-chlorobenzenesulfonamide (TZPU; IC50 = 35.9 nM), and the known inhibitor 1,1,1,3,3,3-hexafluoropropan-2-yl 4-(((4-chlorophenyl)sulfonamido) methyl)piperidine-1-carboxylate (SAR127303; IC50 = 39.3 nM), which were also shown to be selective for MAGL over fatty acid amide hydrolase (FAAH), and cannabinoid receptors (CB1 & CB2). Both of these compounds were radiolabeled with carbon-11 via [11C]COCl2, followed by comprehensive ex vivo biodistribution and in vivo PET imaging studies in normal rats to determine their brain permeability, specificity, clearance and metabolism. Whereas TZPU did not show adequate specificity to warrant further evaluation, [11C]SAR127303 was advanced for preliminary PET neuroimaging studies in nonhuman primate. The tracer showed good brain permeability (ca. 1 SUV) and heterogeneous regional brain distribution which is consistent with the distribution of MAGL.
Keywords: positron emission tomography, monoacylglycerol lipase, MAGL, carbon-11, nonhuman primate, SAR127303.
Introduction
The endocannabinoid (eCB) system is a neurotransmission network in the central and peripheral nervous system, which functions primarily through two membrane-bound G protein-coupled receptors, namely, cannabinoid receptors CB1 and CB2 and several endogenous signaling lipids, for instance, 2-arachidonoylglycerol (2-AG) and anandamide (AEA).[1] The dysregulation of CB1/2 receptors and AEA / 2-AG levels in brain and other major organs has been identified in a wide spectrum of physiopathological conditions including nociception, cognitive function, appetite, emotion, motor control, memory and pain perception,[2-4] all of which have led to continued efforts in the search of eCB-targeting drugs to modulate these conditions.[5, 6] Since direct stimulation of CB1 receptors is generally accompanied by undesirable side effects,[7] drug discovery has focused on alteration of 2-AG and/or AEA metabolism, through inhibiting the functions of two serine hydrolases: monoacylglycerol lipase (MAGL; metabolizes 2-AG, Scheme 1) and fatty acid amide hydrolase (FAAH; metabolizes AEA).[1, 8, 9] Based on preclinical 2-AG mapping studies of the mouse brain, MAGL (84.2% homology to human) is responsible for ca. 85% of 2-AG catabolism and the remaining 15% was assigned to other serine hydrolases, including α/β hydrolase domain 6 (ABHD6) and ABHD12.[10] Blockade of MAGL has not only resulted in anti-nociceptive, anxiolytic, and anti-emetic responses through enhancing eCB signaling, but also simultaneously showed anti-inflammatory effects and provided protection against neuroinflammation via decrease of arachidonic acid (AA) in the brain (Scheme 1). In a mouse model of Parkinson's disease (PD), inhibition of MAGL significantly prevented dopaminegic neuronal loss in substania nigra and striatum, which was primarily caused by reduced AA and prostaglandin levels.[11] Genetic ablation of MAGL also showed an attenuation on neuroinflammation and a substantial reduction of amyloid plaques in MAGL-/-/PS1/APP+ mouse model of Alzheimer's disease (AD).[12] Inhibition of MAGL represents a possible therapeutic approach for the treatment of eCB disorders, such as drug addiction and anxiety, as well as neurodegenerative diseases, including PD, AD and multiple sclerosis.[13-18] Several pharmaceutical pipelines include MAGL as therapeutic target, and the first clinical trial with a drug (ABX-1431) targeting this enzyme was launched in 2015. [7]
Scheme 1
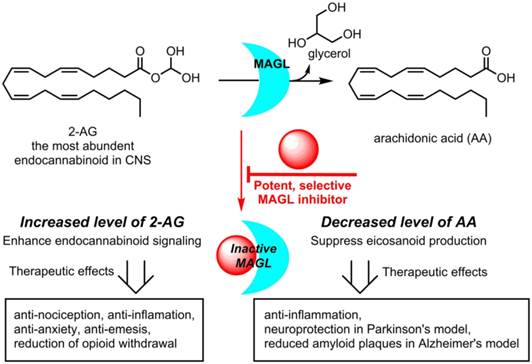
Therapeutic potential for MAGL Inhibitors.
Positron emission tomography (PET) is a non-invasive imaging technology that is capable of quantifying biochemical and pharmacological processes in vivo. Quantification of MAGL in the living brain by PET would enable investigations of the eCB system under normal and disease conditions, assessment of MAGL distribution in the brain and periphery, and target engagement for validation of promising drug candidates in clinical trials. Unlike several FAAH-targeting PET radiotracers in human use,[19, 20] there is an unmet need for probing MAGL in clinical research and drug development studies of this central enzyme in the eCB pathway. Figure 1 shows recent small molecule MAGL inhibitors that could be potentially utilized as lead compounds for PET tracer development for MAGL. For example, JZL184, a selective and carbamate based inhibitor, demonstrates 100-fold selectivity for MAGL over FAAH and most other serine hydrolases. MAGL blockade with JZL184 has been shown to exhibit a wide range of beneficial effects including alleviation of pain, inflammation, anxiety and cancer pathogenicity.[21] KML29, an O-hexafluoroisopropyl analog of JZL184, supersedes cross-reactivity that JZL184 displays for FAAH and carboxylesterase enzymes both in acute and chronic dosing, and is devoid of hypothermia and hypomotility effects induced by JZL184.[22, 23] Most recently, a structurally distinct carbamate, SAR127303, has been reported as a novel orally-active MAGL inhibitor with IC50 of 48 nM and displayed a 10-fold increase of potency compared with JZL184 (IC50 = 0.48 μM).[24] Based on these promising scaffolds, Hicks et al reported the first evaluation of a series of carbon-11 (11C; β+; t1/2 = 20.4 min) labeled MAGL ligands for PET in rat brain, including [11C]KML29, [11C]JJKK-0048, [11C]ML30 and [11C]JW642 via an efficient 11CO2 fixation library radiosynthesis protocol. [11C]JJKK-0048 showed moderate brain uptake (ca. 0.8 SUV) and specificity (ca. 50% blockade) to the target.[25] In addition to our ongoing efforts,[25-27] during the preparation of this manuscript, a preliminary evaluation of [11C]SAR127303 in rats was recently reported.[28] Herein we describe our medicinal chemistry efforts to identify new potent and selective MAGL inhibitors, radiolabeling and comprehensive in vivo evaluation of novel sulfonamido-based [11C-carbonyl]-labeled carbamate and urea ligands in rodent models and preliminary PET imaging studies in non-human primate (NHP).
Figure 1
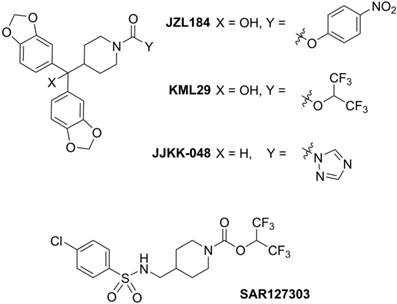
Representative small molecule MAGL inhibitors.
Results and discussion
Medicinal Chemistry
With the goal to develop potent, selective and brain penetrant neuroimaging ligands for MAGL, we designed an array of small molecule inhibitors based on the irreversible inhibitor SAR127303. X-ray crystallography of a co-crystal between SAR127303 and MAGL showed that both the piperidine moiety and sulfonamide contributed key interaction with the enzyme.[24] Therefore we synthesized a series of eleven sulfonamido carbamates/ureas with alterations at the terminal aryl moieties, linkers and leaving groups (Scheme 2A). As summarized in Scheme 2B, a series of benzenesulfonyl chlorides 1 were condensed with primary amines 2 under basic conditions to afford benzenesulfonamides 3. Deprotection of 3 with trifluoroacetic acid gave secondary amines 4.
Several coupling strategies were used for introducing different carbonyl-R2 groups. For 5a-5e and 5i-5k, the combination of 4-nitrophenyl chloroformate and 1,1,1,3,3,3-hexa fluoro-2-propanol in the presence of pyridine and DMAP was found to be optimal and generated the corresponding carbamate moieties in 10%-35% yields. Triphosgene showed superior reactivity as an activating reagent to produce the corresponding triazolyl and trifluoroethyl carbonyls 5f and 5g in 30% and 10% yields, respectively. Compound 5h was synthesized from amine 4a and N,N'-disuccinimidyl carbonate directly in 55% yield.
Compounds 5a-5k were subsequently screened in vitro for their ability to inhibit MAGL activity using human recombinant MAGL and 4-nitrophenylacetate as the substrate. A primary single-point MAGL inhibition assay was performed with only one concentration (50 nM) of test compounds to assess preliminary structure activity relationships (Figure S1 in supporting information; SI). The two most potent compounds, namely the known MAGL inhibitor 1,1,1,3,3,3-hexafluoro propan-2-yl 4-(((4-chloro phenyl)sulfonamido) methyl) piperidine-1-carboxy late (5a or SAR127303[24]) and a novel N-((1-(1H-1,2,4-triazole-1-carbonyl)piperidin-4-yl)methyl)-4-chlorobenzenesulfonamide (5f or TZPU), possessed IC50 values of 35.9 nM and 39.3 nM in this assay, respectively (Figure 2). While carbamate SAR127303 (5a) showed at least 30-fold selectivity for MAGL over FAAH,[24] urea 5f exhibited reduced selectivity with 75% inhibition of FAAH enzyme at the concentration of 500 nM. No significant CB1 and CB2 receptors agonism or antagonism was observed from compound SAR127303 (5a) and TZPU (5f) at concentrations up to 30 µM (Figure S2 in SI).
Lipophilicity of candidate compounds can be used as a predictive parameter for assessing blood-brain barrier permeability.[29] The cLogD values were predicted using ADMET PredictorTM software, which is suitable for estimation of LogD values based on known MAGL radioligands (see Table S1 in SI). The cLogD values of compounds 5a-5k ranged from 1.28 to 4.54 (Table 1). The leaving groups in the molecule had the most pronounced affect on the lipophilicity (HFIP > TFE > Tz > NHS; cf. Scheme 2 for structures), while arene substituents and linkers slightly influenced lipophilicity (5b and 5c). Since these inhibitors were designed for irreversible acylation of a serine hydrolase, we also evaluated the stability of testing compounds under physiological conditions (PBS buffer, pH = 7.4). Most compounds showed reasonable stability (t½ > 60 min) with the exception of 5i (t½ = 23.6 min), 5j (t½ < 15 min) and 5k (t½ < 15 min), indicating that the length of the linker groups may be an important determination of stability. Brain permeability was estimated in vitro by parallel artificial membrane permeation assay for blood brain barrier (PAMPA-BBB assays).[31] Compounds SAR127303 (5a) and TZPU (5f) showed good potential for BBB penetration with in vitro effective permeability (Pe) values of ca. 2 x 10-6 cm/s and 5 x 10-6 cm/s with verapamil (13.6 x 10-6 cm/s) and hydrocortisone (0.8 x 10‑6 cm/s) serving as positive and negative control standards, respectively. Considering 1,2,4-triazole based [11C]JJKK-0048 demonstrated the highest brain uptake in the previous report,[25] together with in vitro binding data and physiochemical properties, SAR127303 (5a) and TZPU (5f) were selected to be the most promising ligands for radiolabeling with carbon-11 and in vivo evaluation in rodents.
Table 1
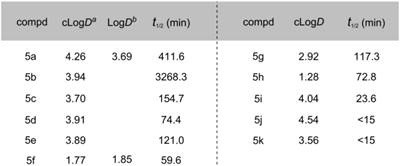
Physiochemical properties of compounds 5a-5k. a cLogD values were calculated with ADMET Predictor™ software (See Table S1 for details in SI). b LogD values were determined in the n-octanol/phosphate buffer (pH = 7.40) by the shaking flask method (n = 3, maximum range ± 2%). c t1/2 (min) values were measured in 1 mL phosphate buffer (10 mM, pH 7.4) and 0.5 mL DMSO and incubated at 37ºC.
Scheme 2
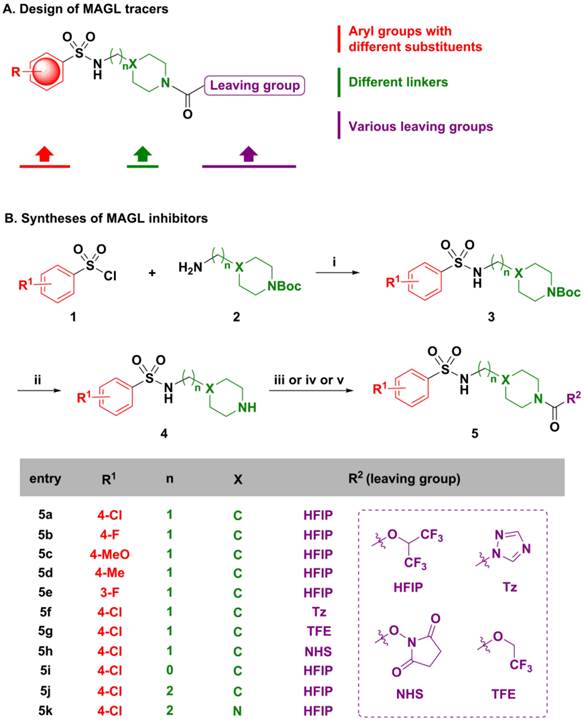
Design of medicinal chemistry and syntheses of MAGL inhibitors. aReagents and conditions: (i) triethylamine, CH2Cl2, rt, 6 h; (ii) trifluoroacetic acid, CH2Cl2, rt, overnight, then Na2CO3; (iii) 1,1,1,3,3,3-hexafluoro-2-propanol, 4-nitrophenyl chloroformate, DMAP, pyridine, triethylamine, CH2Cl2, 0 °C-rt; (iv) 1,2,4-triazole (for 5f) or 2,2,2-Trifluoroethanol (for 5g), triphosgene, DMAP, triethylamine, CH2Cl2, 0 °C-rt; (v) N,N'-disuccinimidyl carbonate, triethylamine, CH2Cl2, rt, overnight. DMAP = 4-dimethylaminopyridine; HFIP = 1,1,1,3,3,3-hexafluoropropan-2-ol; Tz = 1,2,4-triazole; TFE = trifluoroethan-1-ol; NHS = N-Hydroxysuccinimide.
Figure 2
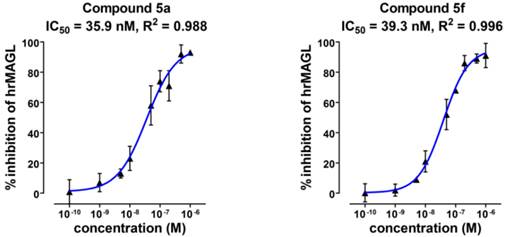
IC50 values of two most potent inhibitors 5a and 5f.
Scheme 3
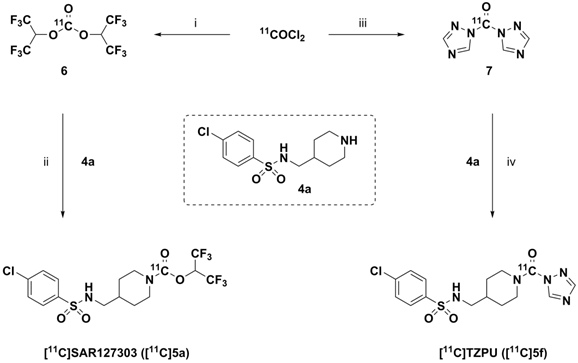
Radiosynthesis of [11C]SAR127303 ([11C]5a) and [11C]TZPU ([11C]5f). aReagents and conditions: (i) HFIP, PMP, THF, rt, 1 min; (ii) 4a, PMP, THF, 30 °C, 3 min; (iii) 1,2,4-triazole, THF, 30 °C, 3 min; (iv) 4a, THF, 30 °C, 3 min. HFIP = 1,1,1,3,3,3-hexafluoropropan-2-ol; PMP = 1,2,2,6,6-pentamethylpiperidine; THF = tetrahydrofuran.
Radiochemistry. The importance of the labeling site at the 11C-carbonyl position was shown by Wilson et. al. to be mechanistically critical for the design of FAAH radiotracers, including the 11C-carbamate [11C]CURB[32] and the 11C-unsymetrrical urea [11C]PF-04457845.[33] Targeting an analogous irreversible acylation mechanism for MAGL, we utilized [11C]COCl2 to label the 11C-carbamate and 11C-urea of SAR127303 (5a) and TZPU (5f), respectively. (Scheme 3). Reaction of [11C]COCl2 [34] with HFIP gave 11C-carbonate intermediate 6 in the presence of 1,2,2,6,6-pentamethylpiperidine (PMP). Addition of piperidine 4a then furnished [11C]SAR127303 ([11C]5a, 32.9 ± 10.6 mCi, n = 7) in ca. 20% decay-corrected radiochemical yield, relative to starting 11CO2. The specific activity was high (typically >1 Ci/µmol for baseline studies, see Table S2 for detailed values for each PET imaging studies) and radiochemical purity was higher than 99% (Figure S3 in SI for analytical and semi-prep HPLC chromatograms). The overall synthesis time was ca. 35 min and no radiolysis was observed up to 90 min after formulation in a saline solution (3 mL) containing 100 µL of 25% ascorbic acid in sterile water and 100 µL of 20% Tween® 80 in ethanol. [11C]TZPU ([11C]5f, 25.7 ± 7.3 mCi, n = 5) was synthesized in a similar fashion from 11C-urea 7. The radiochemical yield was 19% (decay-corrected, based on starting 11CO2) with > 99% radiochemical purity and high specific activity (typically >1 Ci/µmol for baseline studies, see Table S2 for detailed values for each PET imaging studies). The synthesis time was 38 min and no evidence of radiolysis was observed within 90 min after formulation (vide supra). Using liquid-liquid partition between n-octanol and PBS (“shake flask method”), LogD7.4 values of [11C]SAR127303 ([11C]5a) and [11C]TZPU ([11C]5f) were determined to be 3.69 ± 0.06 and 1.85 ± 0.003, respectively (Table 1). The efficient radiosyntheses, radiochemical purities and specific activities of [11C]5a and [11C]5f enabled the subsequent ex vivo biodistribution and preclinical in vivo PET imaging studies.
Whole body biodistribution studies. The uptake, distribution and clearance of [11C]SAR127303 ([11C]5a) and [11C]TZPU ([11C]5f) were studied in mice at five time points (1, 5, 15, 30 and 60 min) post injection. The results are expressed as the percentage of injected dose per gram of wet tissue (%ID/g) in Figure 3 and standardized uptake value (SUV) in Table S3 - S6 (SI). High uptake (>3% ID/g) was observed in the blood, heart, lungs, adrenal glands and kidneys at 1 min post injection of [11C]5f, after which it declined slowly in most tissues over 60 min. Peak brain uptake was low-to-moderate at ca. 1% ID/g. A different whole body distribution was observed for [11C]SAR127303 ([11C]5a). At 1 min post injection of [11C]5a, high uptake (>3% ID/g) was observed in the heart, lungs, liver, pancreas, adrenal glands, kidneys, small intestine, brain and brown adipose tissue. Washout from most tissues was rapid, other than activity levels in the liver, adrenal glands, brain and brown adipose tissue, which gradually increased to a plateau at 5 min with limited washout. High uptake of SAR127303 (5a) and TZPU (5f) was observed in the liver, kidneys and small intestine, indicating urinary and hepatobiliary excretion, as well as possible intestinal re-uptake. The distribution of [11C]5a in the present study was consistent with the distribution of MAGL in mice, as high expression in the brown adipose tissue, adrenal glands, liver, brain and kidneys has been previously observed.[35] In particular, [11C]5a showed characteristic high uptake (> 10 %ID/g) in brown adipose tissue, which has the most abundant MAGL expression in rodents.[36]
Figure 3
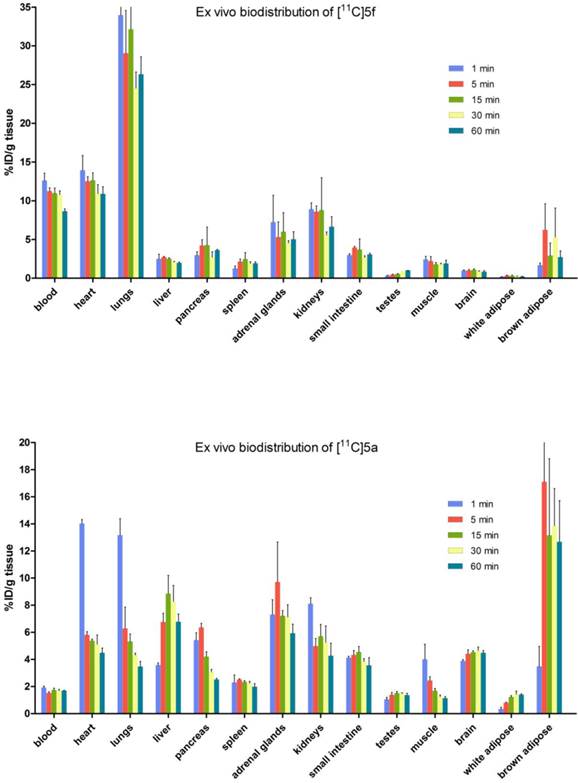
Ex vivo biodistribution of [11C]5f (top) and [11C]5a (bottom) in mice. Data are expressed as %ID/g (mean ± SD, n = 3).
Figure 4
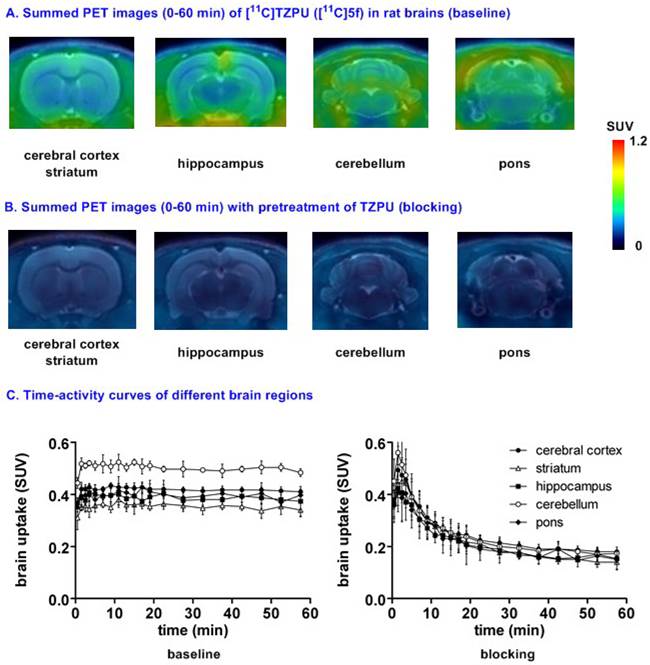
Representative PET images and time-activity curves of [11C]TZPU in rat brains (baseline and blocking) aBlocking conditions: TZPU (1 mg/kg, 30 min i.v. before injection); bData are expressed as SUV (mean ± SD, n = 3).
Small animal PET studies of [11C]TZPU ([11C]5f) in rat brain. Dynamic PET acquisitions were performed first with [11C]TZPU ([11C]5f) in Sprague-Dawley rats for 60 min. Representative PET images (summed images from 0-60 min) in whole brain and detailed brain regions, as well as time-activity curves of [11C]5f in normal rat are shown in Figure S4 (SI) and Figure 4, respectively. [11C]5f showed limited whole brain uptake (ca. 0.4 SUV) in rat and no obvious washout (ratio of SUV5 min / SUV60 min = 1) was observed during 60 min dynamic scans (Figure S4 in SI). The distribution of [11C]5f was heterogeneous (Figure 4A and 4C) with the highest radioactivity observed in the cerebellum (0.43 SUV), followed by cerebral cortex, pons, hippocampus and the lowest uptake in striatum (0.34 SUV). Pretreatment with non-radiolabeled TZPU (1 mg/kg, 30 min i.v. before injection) reduced whole brain uptake by 48% (Figure 4B and 4C) and abolished heterogeneity in various regions of interest, including cerebral cortex (42% reduced uptake), striatum, hippocampus, cerebellum (53% reduced uptake) and pons (Figure S5 in SI for detailed brain regions). Although the distribution pattern of [11C]5f was aligned with MAGL distribution,[37] the blocking studies indicated certain modest specificity of [11C]5f in rat brain.
PET imaging studies of [11C]TZPU in PgP/Bcrp knockout mice. The insufficient brain penetration of [11C]TZPU may be caused by ATP-binding cassette (ABC) efflux transporters at the blood-brain barrier, [38, 39] particularly, P-glycoprotein (PgP) and breast cancer resistance protein (Bcrp). To test this hypothesis, we carried out PET imaging studies of [11C]TZPU on wild-type and PgP/Bcrp knockout (ABCB1a/1b-/-ABCG2-/-) mice, and compared pharmacokinetic profiles, including brain uptake and clearance. As shown in Figure 5 and Figure S6 in SI (detailed regional brain analysis), peak uptake in whole brain was 0.6 SUV in PgP/Bcrp knockout mice, indicating no significant difference compared with brain uptake (0.66 SUV) in wild type controls. Overall brain uptake changed merely ca. 10% in PgP/Bcrp knockout mice (Figure 5 and Figure S7 in SI for changes in different brain regions); therefore, these results indicate [11C]TZPU lacks intensive interactions with efflux pumps on the murine brain and is not likely a Pgp/Bcrp substrate in mice.
Figure 5
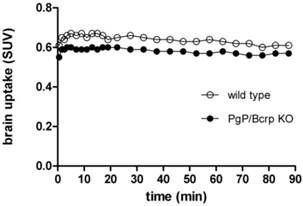
Time-activity curves of [11C]TZPU in wild-type and PgP/Bcrp Knockout mice.
Small animal PET studies of [11C]SAR127303 ([11C]5a) in rat brains. Representative PET images (summed images 0-60 min) of whole brain and detailed brain regions after the injection of [11C]SAR127303([11C]5a) are shown in Figure S8 (SI) and Figure 6, respectively. [11C]SAR127303, which rapidly crossed the blood brain barrier and increased to 1.7 SUV at 2 min, demonstrated ca. 4 times higher peak brain uptake compared with that of [11C]TZPU. The brain uptake of [11C]SAR127303 was comparable with the previous work.[28] Radioactivity in brain tissues reached plateau (1.5 SUV) after 10 min, and remained this level within 60 min scan time without significant clearance (Figure S8). The distribution of [11C]5a was heterogeneous (Figure 6A and 6B) with the highest radioactivity observed in the cerebral cortex and striatum (1.75 SUV both), followed by cerebellum (1.58 SUV), hippocampus (1.55 SUV), and the lowest uptake in pons (1.25 SUV). The distribution pattern of [11C]SAR127303([11C]5a) is consistent with the distribution of MAGL in the rat brain and the previous report.[28,37] As shown in Figure 6C, pretreatment with KML29, a potent MAGL inhibitor (cf. Figure 1 for structure, IC50 (mouse MAGL) = 15 nM, 3 mg/kg, 30 min i.v. before injection). Blocking studies with SAR127303 (1 mg/kg, 30 min i.v. before injection) also decreased uptake in whole brain (50% reduction) and various brain regions (51% cerebellum, 56% cerebral cortex; Figure S9 in SI), and abolished the difference of uptakes in regions of interest, including cerebral cortex, striatum, hippocampus, cerebellum and pons (Figure 6D). It is worthy of note that blocking studies with another MAGL inhibitor, JZL184 (IC50 (mouse MAGL) = 8 nM, 3 mg/kg, 5 min i.v. before injection) did not show significant blockade, which is consistent with the prior report.[28] (Figure 6E). Blocking studies with a potent FAAH inhibitor, URB597 (3 mg/kg, 30 min i.v. before injection), showed no effect on the uptake and distribution in rat brain, as predicted for this selective MAGL radiotracer (Figure 6E). The percentage of unchanged [11C]5a in rat brain homogenate at 30 min post injection was determined to be 53% with 47% associated with polar metabolites (see Figure S10 in SI). As affinities of PET radiotracers targeting MAGL are known to differ amongst species,[13] we focused our efforts on preliminary translation to the higher species (nonhuman primates) which have slower metabolism, which more closely reflect that of humans.
Preliminary PET imaging studies in nonhuman primate. Towards development of a suitable PET tracer for MAGL for clinical use, we transitioned [11C]SAR127303 ([11C]5a) into NHP imaging studies and studied brain permeability, in vivo distribution and clearance profiles in the living brain. [11C]SAR127303([11C]5a) readily crossed the blood-brain barrier (ca. 1 SUV) and was retained for the duration of the 90 min acquisition time (Figure 7A). Radioactivity in the living brain reached plateau after 20 min, and remained this level within 90 min scan time without a significant clearance (Figure 7B). The distribution of [11C]5a was heterogeneous with the highest uptake observed in the occipital cortex (0.93 SUV), followed by striatum (0.81 SUV), cerebellum (0.75 SUV), and the lowest uptake in pons (0.55 SUV), which is consistent binding with the distribution of MAGL.[37]
Conclusion
We have efficiently synthesized an array of sulfonamido-based MAGL inhibitors and radiolabeled the two most promising ligands, namely [11C]TZPU and [11C]SAR127303 with carbon-11 in good radiochemical yields and high radiochemical purities. The lipophilicity, pKa, whole body distribution, brain penetration, clearance and metabolism studies were evaluated to determine the suitability of these radiotracers to quantitate MAGL in the brain. Comprehensive PET imaging studies with [11C]SAR127303 in rats showed a high level of brain permeability and specificity, followed by a proof-of-concept PET imaging study in NHP. Imaging validation including detailed radiometabolite analysis is underway to assess the suitability of [11C]SAR127303 as a potential lead for clinical translation.
Figure 6
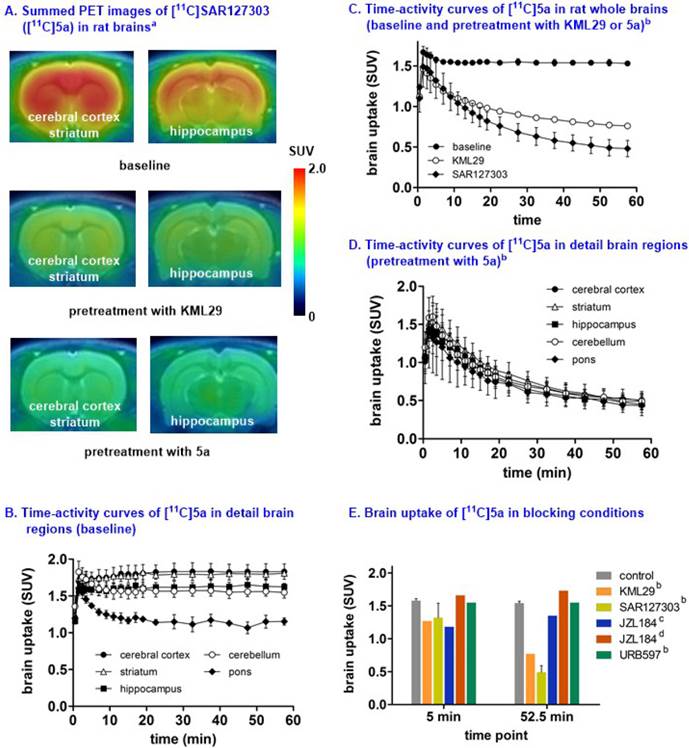
Representative PET images and time-activity curves of [11C]SAR127303 in rat brains (baseline and blocking) aSummed PET images from 0 - 60 min; bBlocking conditions: KML29 (3 mg/kg), SAR127303 (1 mg/kg), or URB 597 (3 mg/kg), 30 min i.v. before injection; cBlocking conditions: JZL184 (1 mg/kg), 5 min i.v. before injection; dBlocking conditions: JZL184 (40 mg/kg), 60 min i.p. before injection; eData are expressed as SUV (mean ± SD, n = 3).
Figure 7
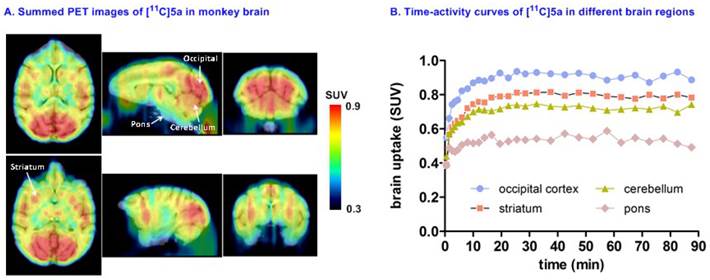
Representative PET images and time-activity curves of [11C]SAR127303 in NHP brain.
Materials and methods
General considerations. All the starting materials used in the syntheses were purchased from commercial vendors and used without further purification. Thin-layer chromatography (TLC) was conducted with 0.25 mm silica gel plates (60F254) and visualized by exposure to UV light (254 nm) or stained with potassium permanganate. Flash column chromatography was performed using silica gel (particle size 0.040-0.063 mm). H-Nuclear magnetic resonance (NMR) spectra were obtained on a 300 MHz on Bruker spectrometers. 13C NMR spectra were obtained at 75 MHz. Chemical shifts (δ) are reported in ppm and coupling constants are reported in Hertz. The multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sext = sextet, sept = setpet, m = multiplet, br = broad signal, dd = doublet of doublets. For all the HRMS measurements, the ionization method is ESI and the mass analyzer type is TOF. Lipophilicity was calculated by ADMET Predictor™ (Simulations Plus, Inc., USA). URB597 and JZL184, and KML29 were purchased from Cayman Chemical, Inc. Melting point was measured by Thomas Hoover capillary melting point apparatus (PA, USA). Carbon-11 (11C) was produced by 14N (p, α)11C nuclear reactions using a GE PETtrace cyclotron (16.5 meV) or a sumitomo CYPRIS HM-18 cyclotron The animal experiments were approved by the Institutional Animal Care and Use Committee of Massachusetts General Hospital or the Animal Ethics Committee at the National Institute of Radiological Sciences. DdY mice (male; 7 weeks, 34-36 g) and Sprague-Dawley rats (male; 7 weeks; 210-230 g) were kept on a 12 h light/12 h dark cycle and were allowed food and water ad libitum.
General procedure for synthesis of amide 3. Sulfonyl chloride 1 (5 mmol) was added to a solution of primary amine 2 (5 mmol) and triethylamine (5 mmol) in CH2Cl2 (15 mL). The mixture was stirred at ambient temperature for 6 hours. Upon the completion of reactions, H2O (15 mL) was added into the mixture and aqueous phase was extracted with CH2Cl2 (15 mL x 3). The combined organic layers were washed with brine, dried over MgSO4, and concentrated under reduced pressure. The product was used in the next step without further purification.
General procedure for syntheses of amide 4. Trifluoroacetic acid (100 mmol) was added to a solution sulfonyl amide 3 in CH2Cl2 (15 mL). The mixture was stirred at ambient temperature overnight. Upon the completion of reactions, the mixture was neutralized with saturated aqueous Na2CO3, and extracted with ethyl acetate (25 mL x 3). The combined organic layers were washed with brine, and dried over MgSO4, and concentrated under reduced pressure. The residue could be used in the next steps without extra purification.
Syntheses of MAGL inhibitor 5
1,1,1,3,3,3-hexafluoropropan-2-yl 4-(((4-chlorophenyl)sulfonamido)methyl)piperidine-1-carboxylate (SAR127303; 5a). A solution of 4-nitrophenyl chloroformate (4 mmol) in CH2Cl2 (10 mL) was added dropwise to a solution of 1,1,1,3,3,3-hexafluoro-2-propanol (10 mmol), pyridine (10 mmol) and 4-dimethylamino- pyridine (0.24 mmol) in CH2Cl2 (10 mL) at 0°C under argon. The mixture was stirred at ambient temperature overnight, followed by the addition of 4-chloro-N-(piperidin-4-ylmethyl)benzenesulphonamide 4a obtained in previous step and triethylamine (12 mmol) in CH2Cl2 (16 mL). The mixture was stirred at ambient temperature for 5 hours, then evaporated to dryness and re-dissolved in ethyl acetate (100 mL). The organic phase was washed with H2O (100 mL x 4), 1 M aqueous potassium carbonate solution (100 mL) and brine (100 mL). The combined organic layers were dried over MgSO4 and evaporated to dryness. The residue was purified by chromatography on silica gel, elution being carried out with a 25 : 75 mixture of ethyl acetate and hexanes. The product was obtained as white solid as previously reported.[24] Yield for three steps: 35%. Melting point (°C): 135-137. 1H NMR (300 MHz, CDCl3) δ 7.79 (d, J = 8.4 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 5.73 (m, 1H), 4.50 (t, J = 6.6 Hz, 1H), 4.16 (t, J = 12.1 Hz, 1H), 2.78-2.95 (m, 4H), 1.60-1.80 (m, 3H), 1.05-1.17 (m, 2H).
1,1,1,3,3,3-hexafluoropropan-2-yl 4-(((4-fluo-rophenyl)sulfonamido)methyl)piperidine-1-carboxylate (5b). Compound 5b was prepared in a manner similar to that described for 5a in 28% yield for three steps as a white solid. Melting point (°C): 109-111. 1H NMR (300 MHz, CDCl3) δ 7.80-7.91 (m, 2H), 7.16-7.25 (m, 2H), 5.73 (m, 1H), 4.58 (t, J = 6.3 Hz, 1H), 4.16 (t, J = 12.2 Hz, 1H), 2.78-2.95 (m, 4H), 1.65-1.80 (m, 3H), 1.05-1.22 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 165.1(d, JC-F = 253.6 Hz), 151.3, 135.8 (d, J C-F = 3.2 Hz), 129.6 (d, J C-F = 9.2 Hz), 120.6 (q, J C-F = 281.1 Hz), 116.4 (d, J C-F = 22.5 Hz), 68.0 (sept, J C-C-F = 34.2 Hz), 48.1, 44.6, 44.0, 36.0, 29.3, 29.0; 19F NMR (CDCl3, 282 MHz) δ -73.7 (d, J = 6.2 Hz), -104.8 (sept). HRMS (m/z): [M + H]+ calcd. for C16H18F7N2O4S, 467.0876; found 467.0881.
1,1,1,3,3,3-hexafluoropropan-2-yl 4-(((4-meth-oxyphenyl)sulfonamido)methyl)piperidine-1-carboxylate (5c). Compound 5c was prepared in a manner similar to that described for 5a in 21% yield for three steps as a white solid. Melting point (°C): 127-128. 1H NMR (300 MHz, CDCl3) δ 7.79 (dt, J = 9.0 Hz, 2.6 Hz, 2H), 6.99 (dt, J = 9.0 Hz, 2.6 Hz, 2H), 5.73 (sept, 1H), 4.61 (t, J = 6.6 Hz, 1H), 4.14 (t, J = 11.7 Hz, 1H), 3.88 (s, 3H), 2.75-2.95 (m, 4H), 1.60-1.80 (m, 3H), 1.00-1.20 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 162.9, 151.3, 131.2, 129.1, 120.7 (q, J C-F = 281.3 Hz), 114.3, 68.0 (sept, J C-C-F = 34.2 Hz), 55.5, 48.1, 44.6, 44.0, 35.9, 29.4, 29.0;. 19F NMR (CDCl3, 282 MHz) δ -73.7 (d, J = 6.2 Hz). HRMS (m/z): [M + H]+ calcd. for C17H21F6N2O5S, 479.1075; found 479.1072.
1,1,1,3,3,3-hexafluoropropan-2-yl 4-(((4-meth-ylphenyl)sulfonamido)methyl)piperidine-1-carboxylate (5d). Compound 5d was prepared in a manner similar to that described for 5a in 53% yield for three steps as a white solid. Melting point (°C): 118-119. 1H NMR (300 MHz, CDCl3) δ 7.74 (d, J = 8.1 Hz, 2H), 7.32 (d, J = 8.1 Hz, 2H), 5.73 (sept, 1H), 4.60 (t, J = 6.5 Hz, 1H), 4.14 (t, J = 12.0 Hz, 1H), 2.75-2.95 (m, 4H), 2.44 (s, 3H), 1.60-1.80 (m, 3H), 1.10-1.20 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 151.3, 143.6, 136.7, 129.7, 127.0, 120.7 (q, J C-F = 280.4 Hz), 68.0 (sept, J C-C-F = 34.2 Hz), 48.1, 44.6, 44.0, 36.0, 29.4, 29.0, 21.4;. 19F NMR (CDCl3, 282 MHz) δ -73.7 (d, J = 6.2 Hz). HRMS (m/z): [M + H]+ calcd. for C17H21F6N2O4S, 463.1126; found 463.1129.
1,1,1,3,3,3-hexafluoropropan-2-yl 4-(((3-fluo-rophenyl)sulfonamido)methyl)piperidine-1-carboxylate (5e). Compound 5e was prepared in a manner similar to that described for 5a in 19% yield for three steps as a white solid. Melting point (°C): 71-73. 1H NMR (300 MHz, CDCl3) δ 7.63-7.70 (m, 1H), 7.47-7.59 (m, 2H), 7.26-7.35 (m, 1H), 5.73 (sept, 1H), 4.63 (t, J = 6.3 Hz, 1H), 4.16 (t, J = 11.4 Hz, 1H), 2.72-2.95 (m, 4H), 1.63-1.80 (m, 3H), 1.05-1.25 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 162.5 (d, JC-F = 250.3 Hz), 151.3, 141.8 (d, J C-F = 6.5 Hz), 131.0 (d, J C-F = 7.7 Hz), 122.6 (d, J C-F = 3.3 Hz), 120.7 (q, J C-F = 280.0 Hz), 120.0 (d, J C-F = 21.1 Hz), 114.3 (d, J C-F = 24.2 Hz), 68.0 (sept, J C-C-F = 34.2 Hz), 48.2, 44.6, 44.0, 36.1, 29.3, 29.0; 19F NMR (CDCl3, 282 MHz) δ -73.7 (d, J = 6.2 Hz), -109.3 (sext). HRMS (m/z): [M + H]+ calcd. for C16H18F7N2O4S, 467.0876; found 467.0880.
1,1,1,3,3,3-hexafluoropropan-2-yl 4-((4-chlo-rophenyl)sulfonamido)piperidine-1-carboxylate (5i). Compound 5i was prepared in a manner similar to that described for 5a in 22% yield for three steps as a white solid. Melting point (°C): 150-152. 1H NMR (300 MHz, CDCl3) δ 7.82 (dt, J = 8.7 Hz, 2.3 Hz, 2H), 7.51 (dt, J = 8.7 Hz, 2.3 Hz, 2H), 5.70 (sept, 1H), 4.69 (d, J = 7.2 Hz, 1H), 3.87-4.05 (m, 2H), 3.30-3.45 (m, 1H), 3.00 (q, J = 12.3 Hz, 2H), 1.81-1.88 (m, 2H), 1.30-1.50 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 151.2, 139.4, 139.3, 129.6, 128.3, 120.6 (q, J C-F = 280.7 Hz), 68.1 (sept, J C-C-F = 34.5 Hz), 50.1, 43.2, 42.6, 32.5, 32.1;. 19F NMR (CDCl3, 282 MHz) δ -73.7 (d, J = 6.5 Hz). HRMS (m/z): [M + H]+ calcd. for C15H16ClF6N2O4S, 469.0423; found 469.0425.
1,1,1,3,3,3-hexafluoropropan-2-yl 4-(2-((4-ch-lorophenyl)sulfonamido)ethyl)piperidine-1-carboxylate (5j). Compound 5j was prepared in a manner similar to that described for 5a in 16% yield for three steps as a semi-solid. 1H NMR (300 MHz, CDCl3) δ 7.80 (dt, J = 8.7 Hz, 2.3 Hz, 2H), 7.50 (dt, J = 8.7 Hz, 2.3 Hz, 2H), 5.73 (sept, 1H), 4.40 (t, J = 6.2 Hz, 1H), 4.13 (t, J = 12.2 Hz, 2H), 3.00 (q, J = 6.8 Hz, 2H), 2.84 (q, J = 13.7 Hz, 2H), 1.50-1.75 (m, 3H), 1.45 (q, J = 6.9 Hz, 2H), 1.05-1.23 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 151.3, 139.3, 138.2, 129.5, 128.4, 120.7 (q, J C-F = 280.6 Hz), 68.0 (sept, J C-C-F = 34.3 Hz), 45.0, 44.4, 40.4, 35.8, 32.6, 31.6, 31.2;. 19F NMR (CDCl3, 282 MHz) δ -73.7 (quint, J = 1.6 Hz). [M + H]+ calcd. for C17H20ClF6N2O4S, 497.0736; found 497.0733.
1,1,1,3,3,3-hexafluoropropan-2-yl 4-(2-((4-ch-lorophenyl)sulfonamido)ethyl)piperazine-1-carboxylate (5k). Compound 5k was prepared in a manner similar to that described for 5a in 10% yield for three steps as light yellow solid. Melting point (°C): 90-92. 1H NMR (300 MHz, CDCl3) δ 7.82 (dt, J = 8.7 Hz, 2.3 Hz, 2H), 7.51 (dt, J = 8.7 Hz, 2.3 Hz, 2H), 5.73 (sept, 1H), 5.10 (t, J = 5.0 Hz, 1H), 3.50 (d, J = 4.5 Hz, 4H), 3.04 (q, J = 5.5 Hz, 2H), 2.45-2.50 (m, 2H), 2.35 (s, 4H); 13C NMR (75 MHz, CDCl3) δ 151.2, 139.3, 138.2, 129.4, 128.5, 120.6 (q, J C-F = 281.2 Hz), 68.0 (sept, J C-C-F = 34.3 Hz), 55.9, 52.1, 51.8, 44.4, 44.1, 39.2;. 19F NMR (CDCl3, 282 MHz) δ -73.6 (d, J = 6.2 Hz). HRMS (m/z): [M + H]+ calcd. for C16H19ClF6N3O4S, 498.0689; found 498.0690.
N-((1-(1H-1,2,4-triazole-1-carbonyl)piperidin-4-yl)methyl)-4-chlorobenzenesulfonamide (5f). A solution of triphosgene (2 mmol) in CH2Cl2 (2 mL) was added dropwise to a solution of 1,2,4-triazole (6 mmol) and triethylamine (6 mmol) in CH2Cl2 (6 mL) at 0 oC under argon. The mixture was left at ambient temperature for 5 hours, followed by the addition of crude 4-chloro-N-(pipe-ridin-4-ylmethyl)benzenesulphonamide 4a obtained in previous step and triethylamine (20 mmol) in CH2Cl2 (20 mL). The mixture was stirred at ambient temperature for 5 hours, then evaporated to dryness and re-dissolved in ethyl acetate (100 mL). The organic phase was washed with H2O (100 mL) and then extracted with ethyl acetate (100 mL x 3). The combined organic layers were washed with brine (100 mL), dried over MgSO4, and the solvent was evaporated under reduced pressure. The residue was purified by chromatography on silica gel, elution being carried out with a 50 : 50 mixture of ethyl acetate and hexane. The product was obtained as white solid. Yield for three steps: 30%. Melting point (°C): 109-110. 1H NMR (300 MHz, CDCl3) δ 8.75 (s, 1H), 8.00 (s, 1H), 7.79 (dt, J = 8.7 Hz, 2.3 Hz, 2H), 7.50 (dt, J = 8.7 Hz, 2.3 Hz, 2H), 4.81 (t, J = 6.3 Hz, 1H), 4.54 (s, 2H), 2.95 (t, J = 12.6 Hz, 2H), 2.88 (t, J = 6.5 Hz, 2H), 1.70-1.90 (m, 3H), 1.20-1.40 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 151.8, 148.5, 146.5, 139.2, 138.3, 129.5, 128.4, 48.1, 46.2, 36.2, 29.4. HRMS (m/z): [M + H]+ calcd. for C15H19ClN5O3S, 384.0897; found 384.0896.
2,2,2-trifluoroethyl 4-(((4-chlorophenyl)sulfon-amido)methyl)piperidine-1-carboxylate (5g). 5g was prepared in a manner similar to that described for 5f in 10% yield for three steps as light yellow solid. Melting point (°C): 152-153. 1H NMR (300 MHz, CDCl3) δ 7.79 (d, J = 8.6 Hz, 2H), 7.50 (d, J = 8.7 Hz, 2H), 4.58 (t, J = 6.5 Hz, 1H), 4.46 (q, J = 8.5 Hz, 2H), 4.15 (t, J = 15.2 Hz, 2H), 2.65-2.90 (m, 4H), 1.60-1.80 (m, 3H), 1.03-1.18 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 153.2, 139.3, 138.3, 129.5, 128.4, 123.1 (q, J C-F = 275.7 Hz), 61.3 (q, J C-C-F = 36.1 Hz), 48.3, 43.9, 43.8, 36.2, 29.4, 29.1; 19F NMR (CDCl3, 282 MHz) δ -74.2 (d, J = 8.5 Hz). HRMS (m/z): [M + H]+ calcd. for C15H19ClF3N2O4S, 415.0706; found 415.0702.
2,5-dioxopyrrolidin-1-yl 4-(((4-chlorophenyl)-sulfonamido)methyl)piperidine-1-carboxylate (5h). N,N'-disuccinimidyl carbonate (5 mmol) was added to a solution crude 4-chloro-N-(piperidin-4-ylmeth-yl)benzenesulphonamide 4a obtained in previous step in CH2Cl2 (20 mL). The mixture was stirred at ambient temperature for 5 hours, then evaporated to dryness and re-dissolved in ethyl acetate (100 mL). The organic phase was washed with H2O (100 mL) and then extracted with ethyl acetate (100 mL x 3). The combined organic layers were washed with brine, and dried over MgSO4, and the solvent was evaporated under reduced pressure. The residue was purified by chromatography on silica gel, elution being carried out with a 40 : 60 mixture of ethyl acetate and hexane. The product was obtained as white solid. Yield for three steps: 55%. Melting point (°C): 168-170. 1H NMR (300 MHz, CDCl3) δ 7.79 (dt, J = 8.7 Hz, 2.3 Hz, 2H), 7.50 (dt, J = 8.7 Hz, 2.1 Hz, 2H), 4.80 (t, J = 6.5 Hz, 1H), 4.05-4.23 (m, 2H), 2.73-3.02 (m, 8H), 1.63-1.80 (m, 3H), 1.18-1.38 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 169.8, 150.3, 139.3, 138.4, 129.5, 128.4, 48.2, 45.1, 44.3, 36.0, 29.2, 28.9, 25.5; 19F NMR (CDCl3, 282 MHz) δ -73.7 (d, J = 6.5 Hz). HRMS (m/z): [M + H]+ calcd. for C17H21ClN3O6S, 430.0840; found 430.0845.
The melting points of compounds 5a-5i and 5k are summarized in Table S7 in the supplementary information.
Measurement of physiochemical properties
Measurement of lipophilicity (“shake flask method”). The measurement of LogD value was commenced by mixing a 11C-labeled compound (radiochemical purity ~100%) with n-octanol (3.0 g) and PBS (0.1 M, 3.0 g) in a test tube. The tube was first vortexed for 5 min, followed by centrifuge (~3500-4000 rpm) for additional 5 min. PBS and n-octanol (0.60 mL each) were aliquoted, weighted and the radioactivity in each component was measured using a 1480 Wizard autogramma counter. (Perkin Elmer, Inc.) The LogD was determined by Log [ratio of radioactivity between the n-octanol and PBS solutions] (n = 3). The procedure was applied for the determination of LogD of two 11C-labeled compounds 5a and 5f, and the values were shown in the Radiochemistry section and Table 1.
Measurement of hydrolysis rate. The stability of compounds in buffer solutions was measured using a HPLC method adapted from a literature method.[25] Briefly, compounds 5a-5k (0.25 µmol) were dissolved in 1 mL phosphate buffer (10 mM, pH 7.4) and 0.5 mL DMSO and incubated for different time at 37 ºC. The rate of hydrolysis was monitored by HPLC (Luna analytical column, 4.6 x 250 mm, 5 µm, CH3CN / H2O + 0.1% formic acid). T1/2 values for compounds 5a-5k were measured and shown in Table 1.
In vitro binding assays
In vitro MAGL/FAAH inhibition enzyme assays. % Inhibition on a single point MAGL assay for compounds 5a-5k ([C] = 50 nM) and IC50 values of testing compounds 5a and 5f were determined by a literature procedure[24] and manufacturer's instructions from commercially available MAGL / FAAH inhibitor screening kits (Cayman Chemical, Inc.). Dose-response simulation function in GraphPad Prism was used for data processing.
Binding affinities to CB1 and CB2 receptors. CB1 and CB2 binding profiles of 5a and 5f were determined according to published literatures [40, 41] and supported by the National Institute of Mental Health's Psychoactive Drug Screening Program. The detailed procedures “assay protocol book” are listed on the website (https://pdspdb.unc.edu/pdspWeb/).
Membrane permeability assay for blood-brain barrier. The blood-brain barrier penetration was measured in vitro using a modified version of the parallel artificial membrane permeation assay (PAMPA-BBB) described previously.[42] Briefly, a testing compound (5a or 5f) was dissolved in DMSO at the concentration of 5 mg/mL, then diluted to a final concentration of 25 µg/mL. The filter membrane coated with porcine polar brain lipid in dodecane (ca. 4 µL volume of 20 mg/mL). A solution of testing compound (300 µL) was added to a donor well while phosphate buffer (pH 7.4, 200 µL) was added to the corresponding acceptor well. The acceptor well was carefully put on the donor plate and left for 18 hours. The concentration of testing compounds in the acceptor and donor wells was measured using a UV plate reader (SpectraMax M Series Multi-Mode Microplate Readers). Verapamil and hydrocortisone were used as positive and negative control compounds, respectively. All the experiments were conducted in triplicate. In vitro effective permeability (Pe) was calculated by using the established method.[42]
Radiochemistry
Radiosynthesis of [11C]SAR127303 (11C-carbonyl-labeled 1,1,1,3,3,3-hexafluoropropan-2-yl 4-(((4chlorophenyl)sulfonamido)methyl)piperi-dine-1-carboxylate, [11C]5a). 11CO2 was produced by 14N(p, α)11C nuclear reactions in cyclotron, and transferred into a pre-heated methanizer packed with nickel catalyst at 400 ºC to produce 11CH4, which was subsequently reacted with chlorine gas at 560 ºC to generate 11CCl4. 11COCl2 was produced via the reactions between 11CCl4 and iodine oxide and sulfuric acid,[34] and trapped in a solution of hexafluoroisopropanol (5.02 mg) and 1,2,2,6,6-penta methylpiperidine (PMP; 5.5 µL) in THF (200 µL) at 0 ºC. A solution of piperidine 4a (0.52 mg) and PMP (0.5 µL) in THF (200 µL) was added into the mixture and heated at 30 ºC for 3 min before cooling to ambient temperature. The reaction mixtures were concentrated to remove THF, then diluted with HPLC mobile phase (500 µL), followed by the injection on HPLC column. HPLC purification was performed on a Capcell Pak C18 column (10 x 250 mm, 10 µm) using a mobile phase of CH3CN / H2O + 0.1% TFA (70/30, v/v) at a flowrate of 5.0 mL/min. The retention time of [11C]5a was 8.7 min. The product solution was concentrated by evaporation, and reformulated in a saline solution (3 mL) containing 100 µL of 25% ascorbic acid in sterile water and 100 µL of 20% Tween® 80 in ethanol. The radiochemical and chemical purity were measured by an analytical HPLC (Capcell Pak C18, 4.6 x 250 mm, 5µm). The identity of [11C]5a was confirmed by the co-injection with unlabeled SAR127303. The radiochemical yield was 20% decay-corrected based on [11C]CO2 with > 99% radiochemical purity and high specific activity (typically >1 Ci/µmol for baseline studies, see Table S2 for detailed values for each PET imaging studies).
Radiosynthesis of [11C]TZPU (11C-carbonyl-labeled N-((1-(1H-1,2,4-triazole-1-carbonyl)piperidin-4-yl)methyl)-4-chlorobenzenesulfonamide, [11C]5f). Similar the radiosynthesis of [11C]SAR127303, 11COCl2 was produced via the reactions between 11CCl4 and iodine oxide and sulfuric acid, and trapped in a solution of 1,2,4-triazole (1.13 mg) in THF (200 µL) at -15 ºC. The reaction mixture was heated at 30 ºC for 3 min and evaporated to dryness. A solution of piperidine 4f (1.23) in THF (200 µL) was added into the mixture and heated at 30 ºC for 3 min before cooling to ambient temperature. The reaction mixtures were concentrated to remove THF, then diluted with HPLC mobile phase (500 µL), followed by the injection on HPLC column. HPLC purification was performed on a Capcell Pak C18 column (10 x 250 mm, 10 µm) using a mobile phase of CH3CN / H2O + 0.1% TFA (45/55, v/v) at a flowrate of 5.0 mL/min. The retention time of [11C]5f was 8.0 min. The product solution was concentrated by evaporation, and reformulated in a saline solution (3 mL) containing 100 µL of 25% ascorbic acid in sterile water and 100 µL of 20% Tween® 80 in ethanol. The radiochemical and chemical purity were measured by an analytical HPLC (Capcell Pak C18, 4.6 x 250 mm, 5 µm). The identity of [11C]5f was confirmed by the co-injection with unlabeled TZPU. The radiochemical yield was 19% decay-corrected based on [11C]CO2 with > 99% radiochemical purity and high specific activity (typically >1 Ci/µmol for baseline studies, see Table S2 for detailed values for each PET imaging studies).
Ex vivo biodistribution in mice
A saline solution of [11C]SAR127303 ([11C]5a) or [11C]TZPU ([11C]5f) (54 μCi / 200 µL) was injected into DdY mice via the tail vein. Three mice were sacrificed at 1, 5, 15, 30 and 60 min post injection. Major organs, including whole brain, heart, liver, lung, spleen, kidneys, small intestine (including contents), muscle, testes, and blood samples were quickly harvested and weighted. The radioactivity in these organs was measured by a 1480 Wizard autogramma counter. The results are expressed as the percentage of injected dose per gram of wet tissue (% ID/g) or standardized uptake value (SUV). All radioactivity measurements were decay-corrected.
Small-animal PET study in rats
PET scans were conducted by an Inveon PET scanner (Siemens Medical Solutions, Knoxville, TN, USA). Sprague-Dawley rats were kept under anesthesia with 1-2% (v/v) isoflurane during the scan. The radiotracer (1.08-1.35 mCi / 200 µL) was injected via a preinstalled catheter via tail vein. A dynamic scan in 3D list mode was acquired for 90 min. For pretreatment studies, URB597 (3 mg/kg), SAR127303 (1 mg/kg), JZL184 (3 mg/kg) or KML29 (3 mg/kg) dissolved in 300 µL saline containing 10% ethanol and 5% Tween® 80 was injected at 30 min via the tail vein catheter before the injection of [11C]SAR127303 ([11C]5a) or [11C]TZPU ([11C]5f). As we previously reported,[43] the PET dynamic images were reconstructed using ASIPro VW software (Analysis Tools and System Setup/Diagnostics Tool, Siemens Medical Solutions). Volumes of interest, including the whole brain, cerebral cortex, cerebellum, striatum, thalamus and pons were placed using ASIPro software. The radioactivity was decay-corrected and expressed as the standardized uptake value. SUV = (radioactivity per mL tissue / injected radioactivity) x body weight.
PET study in monkey
A male rhesus monkey (weighting 5.72 kg) underwent PET scan while awake. A solution of [11C]SAR127303 ([11C]5a; 3.71 mCi) in saline was injected into the monkey via a flexible percutaneous venous catheter, followed by a 90 min dynamic PET scan with the head centered in the field of view. The co-registration of PET image to individual MR image, which was transformed into brain template MR images, was based on a literature method.[44] The same parameter was used for the transformation of co-registered PET image into the brain template MR and for the individual MR image. Each ROI was delineated on the brain template MR image. Time-activity curves were extracted from the corresponding ROIs and brain uptake of radioactivity was decay-corrected to the time of injection and expressed as SUV.
Abbreviations
PET: positron emission tomography; MAGL: monoacylglycerol lipase; FAAH: fatty acid amide hydrolase; AEA: anandamide; 2-AG: 2-arochidonoylglycerol; [11C]TZPU: [11C-carbonyl] N-((1-(1H-1,2,4-triazole-1-carbonyl)piperidin-4-yl)methyl)-4-chlorobenzenesulfonamide; [11C]SAR127303: [11C-carbonyl] 1,1,1,3,3,3-hexa fluoropropan-2-yl 4-(((4-chlorophenyl)sulfonamido)methyl)piperidine-1-carboxylate; %ID/g: percentage of the injected dose per gram of wet tissue; SUV: standardized uptake value; HFIP: 1,1,1,3,3,3-hexafluoropropan-2-ol; TFE: trifluoro ethan-1-ol; NHS: N-Hydroxysuccinimide; Tz: 1,2,4- triazole; PMP: 1,2,2,6,6-pentamethylpiperidine; DMF: dimethylformamide; THF: tetrahydrofuran.
Supplementary Material
Supplementary tables and figures.
Acknowledgements
We would like to thank the staff at the radiochemistry program, Gordon Center for Medical Imaging, Nuclear Medicine and Molecular Imaging, Massachusetts General Hospital, MA, USA and National Institute of Radiological Sciences, Chiba, Japan for their support with cyclotron operation, radioisotope production, radiosynthesis and animal experiments. We thank the National Institute of Mental Health's Psychoactive Drug Screening Program (NIMH PDSP) for the compound screening. The NIMH PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA. We also thank Drs. Thomas J. Brady and Lee Collier for helpful discussion. R.C. is supported by China Scholarship Council Fellowship (201506250036). S.H.L is a recipient of NIH career development award from the National Institute on Drug Abuse (NIDA 1K01DA038000).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ahn K, McKinney MK, Cravatt BF. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev. 2008;108:1687-707
2. Hohmann AG, Suplita RL. Endocannabinoid mechanisms of pain modulation. AAPS J. 2006;8:E693-708
3. Jhaveri MD, Richardson D, Chapman V. Endocannabinoid metabolism and uptake: novel targets for neuropathic and inflammatory pain. Br J Pharmacol. 2007;152:624-32
4. Bisogno T, Di Marzo V. Short- and long-term plasticity of the endocannabinoid system in neuropsychiatric and neurological disorders. Pharmacol Res. 2007;56:428-42
5. Di Marzo V, Bifulco M, De Petrocellis L. The endocannabinoid system and its therapeutic exploitation. Nat Rev Drug Discov. 2004;3:771-84
6. Di Marzo V. Targeting the endocannabinoid system: to enhance or reduce? Nat Rev Drug Discov. 2008;7:438-55
7. Owens B. Drug development: The treasure chest. Nature. 2015:525
8. Blankman JL, Cravatt BF. Chemical probes of endocannabinoid metabolism. Pharmacol Rev. 2013;65:849-71
9. Fowler CJ. The Potential of Inhibitors of Endocannabinoid Metabolism for Drug Development: A Critical Review. Handb Exp Pharmacol. 2015;231:95-128
10. Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347-56
11. Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC. et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334:809-13
12. Piro JR, Benjamin DI, Duerr JM, Pi Y, Gonzales C, Wood KM. et al. A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer's disease. Cell Rep. 2012;1:617-23
13. Labar G, Wouters J, Lambert DM. A review on the monoacylglycerol lipase: at the interface between fat and endocannabinoid signalling. Curr Med Chem. 2010;17:2588-607
14. Fowler CJ. Monoacylglycerol lipase - a target for drug development? Br J Pharmacol. 2012;166:1568-85
15. Mulvihill MM, Nomura DK. Therapeutic potential of monoacylglycerol lipase inhibitors. Life Sci. 2013;92:492-7
16. Kohnz RA, Nomura DK. Chemical approaches to therapeutically target the metabolism and signaling of the endocannabinoid 2-AG and eicosanoids. Chem Soc Rev. 2014;43:6859-69
17. Hernandez-Torres G, Cipriano M, Heden E, Bjorklund E, Canales A, Zian D. et al. A reversible and selective inhibitor of monoacylglycerol lipase ameliorates multiple sclerosis. Angew Chem Int Ed. 2014;53:13765-70
18. Chen R, Zhang J, Wu Y, Wang D, Feng G, Tang YP. et al. Monoacylglycerol lipase is a therapeutic target for Alzheimer's disease. Cell Rep. 2012;2:1329-39
19. Rusjan PM, Wilson AA, Mizrahi R, Boileau I, Chavez SE, Lobaugh NJ. et al. Mapping human brain fatty acid amide hydrolase activity with PET. J Cereb Blood Flow Metab. 2013;33:407-14
20. Joshi A, Li W, Sanabria S, Holahan M, Purcell M, Declercq R. et al. Translational studies with [11C]MK-3168, a PET tracer for fatty acid amide hydrolase (FAAH). J Nucl Med Meeting Abstracts. 2012;53:397
21. Nomura DK, Long JZ, Niessen S, Hoover HS, Ng S-W, Cravatt BF. Monoacylglycerol Lipase Regulates a Fatty Acid Network that Promotes Cancer Pathogenesis. Cell. 2010;140:49-61
22. Chang JW, Niphakis MJ, Lum KM, Cognetta AB 3rd, Wang C, Matthews ML. et al. Highly selective inhibitors of monoacylglycerol lipase bearing a reactive group that is bioisosteric with endocannabinoid substrates. Chem Biol. 2012;19:579-88
23. Ignatowska-Jankowska BM, Ghosh S, Crowe MS, Kinsey SG, Niphakis MJ, Abdullah RA. et al. In vivo characterization of the highly selective monoacylglycerol lipase inhibitor KML29: antinociceptive activity without cannabimimetic side effects. Br J Pharmacol. 2014;171:1392-407
24. Griebel G, Pichat P, Beeske S, Leroy T, Redon N, Jacquet A. et al. Selective blockade of the hydrolysis of the endocannabinoid 2-arachidonoylglycerol impairs learning and memory performance while producing antinociceptive activity in rodents. Sci Rep. 2015:5
25. Hicks JW, Parkes J, Tong J, Houle S, Vasdev N, Wilson AA. Radiosynthesis and ex vivo evaluation of [11C-carbonyl]carbamate- and urea-based monoacylglycerol lipase inhibitors. Nucl Med Biol. 2014;41:688-94
26. Wang L, Mori W, Cheng R, Yui J, Rotstein BH, Fujinaga M. et al. A Novel Class of Sulfonamido [11C-Carbonyl]-labeled Carbamates and Ureas as Radiotracers for Monoacylglycerol Lipase. J Nucl Med Meeting Abstracts. 2016;57:abstract1232
27. Wang L, Fujinaga M, Cheng R, Yui J, Shimoda Y, Rotstein BH. et al. Synthesis and preliminary evaluation of a 11C-labeled piperidin-4-yl azetidine diamide for imaging monoacylglycerol lipase. J Nucl Med Meeting Abstracts. 2016;57:abstract1183
28. Wang C, Placzek MS, Van de Bittner GC, Schroeder FA, Hooker JM. A Novel Radiotracer for Imaging Monoacylglycerol Lipase in the Brain Using Positron Emission Tomography. ACS Chemical Neuroscience. ASAP. DOI: 10.1021/acschemneuro.5b00293
29. Waterhouse RN. Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol Imaging Biol. 2003;5:376-89
30. Zhang L, Villalobos A, Beck EM, Bocan T, Chappie TA, Chen L. et al. Design and selection parameters to accelerate the discovery of novel central nervous system positron emission tomography (PET) ligands and their application in the development of a novel phosphodiesterase 2A PET ligand. J Med Chem. 2013;56:4568-79
31. Di L, Kerns EH, Carter GT. Strategies to assess blood-brain barrier penetration. Expert Opin Drug Discov. 2008;3:677-87
32. Wilson AA, Garcia A, Parkes J, Houle S, Tong J, Vasdev N. [11C]CURB: Evaluation of a novel radiotracer for imaging fatty acid amide hydrolase by positron emission tomography. Nucl Med Biol. 2011;38:247-53
33. Hicks JW, Parkes J, Sadovski O, Tong J, Houle S, Vasdev N. et al. Synthesis and preclinical evaluation of [11C-carbonyl]PF-04457845 for neuroimaging of fatty acid amide hydrolase. Nucl Med Biol. 2013;40:740-6
34. Kumata K, Yui J, Hatori A, Maeda J, Xie L, Ogawa M. et al. Development of [11C]MFTC for PET imaging of fatty acid amide hydrolase in rat and monkey brains. ACS Chem Neurosci. 2015;6:339-46
35. Karlsson M, Contreras JA, Hellman U, Tornqvist H, Holm C. cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase. Evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. J Biol Chem. 1997;272:27218-23
36. Tornqvist H, Belfrage P. Purification and some properties of a monoacylglycerol-hydrolyzing enzyme of rat adipose tissue. J Biol Chem. 1976;251:813-9
37. Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL. et al. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci. 2002;99:10819-24
38. Tatsuta T, Naito M, Oh-hara T, Sugawara I, Tsuruo T. Functional involvement of P-glycoprotein in blood-brain barrier. J Biol Chem. 1992;267:20383-91
39. Pike VW. PET Radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol Sci. 2009;30:431-40
40. Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang XP. et al. Automated design of ligands to polypharmacological profiles. Nature. 2012;492:215-20
41. Kroeze WK, Sassano MF, Huang XP, Lansu K, McCorvy JD, Giguere PM. et al. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat Struct Mol Biol. 2015;22:362-9
42. Di L, Kerns EH, Fan K, McConnell OJ, Carter GT. High throughput artificial membrane permeability assay for blood-brain barrier. Eur J Med Chem. 2003;38:223-32
43. Wang L, Yui J, Wang Q, Zhang Y, Mori W, Shimoda Y. et al. Synthesis and Preliminary PET Imaging Studies of a FAAH Radiotracer ([11C]MPPO) Based on alpha-Ketoheterocyclic Scaffold. ACS Chem Neurosci. 2015;17:17
44. McLaren DG, Kosmatka KJ, Oakes TR, Kroenke CD, Kohama SG, Matochik JA. et al. A population-average MRI-based atlas collection of the rhesus macaque. Neuroimage. 2009;45:52-9
Author contact
![]() Corresponding authors: M.-R. Zhang Tel: +81 433 823 709, Email: zhanggo.jp; S. H. Liang Tel: +1 617 726 6107, Email: liang.stevenharvard.edu.
Corresponding authors: M.-R. Zhang Tel: +81 433 823 709, Email: zhanggo.jp; S. H. Liang Tel: +1 617 726 6107, Email: liang.stevenharvard.edu.
Citation styles
APA
Wang, L., Mori, W., Cheng, R., Yui, J., Hatori, A., Ma, L., Zhang, Y., Rotstein, B.H., Fujinaga, M., Shimoda, Y., Yamasaki, T., Xie, L., Nagai, Y., Minamimoto, T., Higuchi, M., Vasdev, N., Zhang, M.R., Liang, S.H. (2016). Synthesis and Preclinical Evaluation of Sulfonamido-based [11C-Carbonyl]-Carbamates and Ureas for Imaging Monoacylglycerol Lipase. Theranostics, 6(8), 1145-1159. https://doi.org/10.7150/thno.15257.
ACS
Wang, L.; Mori, W.; Cheng, R.; Yui, J.; Hatori, A.; Ma, L.; Zhang, Y.; Rotstein, B.H.; Fujinaga, M.; Shimoda, Y.; Yamasaki, T.; Xie, L.; Nagai, Y.; Minamimoto, T.; Higuchi, M.; Vasdev, N.; Zhang, M.R.; Liang, S.H. Synthesis and Preclinical Evaluation of Sulfonamido-based [11C-Carbonyl]-Carbamates and Ureas for Imaging Monoacylglycerol Lipase. Theranostics 2016, 6 (8), 1145-1159. DOI: 10.7150/thno.15257.
NLM
Wang L, Mori W, Cheng R, Yui J, Hatori A, Ma L, Zhang Y, Rotstein BH, Fujinaga M, Shimoda Y, Yamasaki T, Xie L, Nagai Y, Minamimoto T, Higuchi M, Vasdev N, Zhang MR, Liang SH. Synthesis and Preclinical Evaluation of Sulfonamido-based [11C-Carbonyl]-Carbamates and Ureas for Imaging Monoacylglycerol Lipase. Theranostics 2016; 6(8):1145-1159. doi:10.7150/thno.15257. https://www.thno.org/v06p1145.htm
CSE
Wang L, Mori W, Cheng R, Yui J, Hatori A, Ma L, Zhang Y, Rotstein BH, Fujinaga M, Shimoda Y, Yamasaki T, Xie L, Nagai Y, Minamimoto T, Higuchi M, Vasdev N, Zhang MR, Liang SH. 2016. Synthesis and Preclinical Evaluation of Sulfonamido-based [11C-Carbonyl]-Carbamates and Ureas for Imaging Monoacylglycerol Lipase. Theranostics. 6(8):1145-1159.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.