Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Material and Methods
Results
Discussion
Acknowledgements
Supplementary Material
References
Introduction
Material and Methods
Results
Discussion
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(16):3920-3932. doi:10.7150/thno.21389 This issue Cite
Research Paper
Decrease in Lymphoid Specific Helicase and 5-hydroxymethylcytosine Is Associated with Metastasis and Genome Instability
Jiantao Jia1,2,3,4*, Ying Shi1,3*, Ling Chen1,3, Weiwei Lai1,3, Bin Yan1,3, Yiqun Jiang1,3, Desheng Xiao5, Sichuan Xi6, Ya Cao1,3, Shuang Liu4, Yan Cheng7, Yongguang Tao1,2,3 ![]()
1. Key Laboratory of Carcinogenesis and Cancer Invasion (Ministry of Education), Xiangya Hospital, Central South University, Changsha, Hunan, 410008 China;
2. Department of Pathophysiology, Changzhi Medical College, Changzhi, Shanxi, 046000 China;
3. Cancer Research Institute, Central South University, Changsha, Hunan, 410078 China;
4. Institute of Medical Sciences, Xiangya Hospital, Central South University, Changsha, Hunan, 410008 China;
5. Department of Pathology, Xiangya Hospital, Central South University, Changsha, Hunan 410078 China;
6. Thoracic Surgery Section, Thoracic and GI Oncology Branch, Center for Cancer Research, National Cancer Institute, Bethesda, MD 20892, USA;
7. Department of Pharmacology, School of Pharmaceutical Sciences, Central South University, Changsha, Hunan 410078 China.
* equal contribution
Received 2017-6-8; Accepted 2017-7-5; Published 2017-9-5
Citation:
Jia J, Shi Y, Chen L, Lai W, Yan B, Jiang Y, Xiao D, Xi S, Cao Y, Liu S, Cheng Y, Tao Y. Decrease in Lymphoid Specific Helicase and 5-hydroxymethylcytosine Is Associated with Metastasis and Genome Instability. Theranostics 2017; 7(16):3920-3932. doi:10.7150/thno.21389. https://www.thno.org/v07p3920.htm
Other stylesAbstract

DNA methylation is an important epigenetic modification as a hallmark in cancer. Conversion of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC) by ten-eleven translocation (TET) family enzymes plays an important biological role in embryonic stem cells, development, aging and disease. Lymphoid specific helicase (LSH), a chromatin remodeling factor, is regarded as a reader of 5-hmC. Recent reports show that the level of 5-hmC is altered in various types of cancers. However, the change in 5-hmC levels in cancer and associated metastasis is not well defined. We report that the level of 5-hmC was decreased in metastatic tissues of nasopharyngeal carcinoma, breast cancer, and colon cancer relative to that in non-metastasis tumor tissues. Furthermore, our data show that TET2, but not TET3, interacted with LSH, whereas LSH increased TET2 expression through silencing miR-26b-5p and miR-29c-5p. Finally, LSH promoted genome stability by silencing satellite expression by affecting 5-hmC levels in pericentromeric satellite repeats, and LSH was resistant to cisplatin-induced DNA damage. Our data indicate that 5-hmC might serve as a metastasis marker for cancer and that the decreased expression of LSH is likely one of the mechanisms of genome instability underlying 5-hmC loss in cancer.
Keywords: LSH, 5-hmC, TET2, satellites, genome instability.
Introduction
Heterochromatin is a specialized region of organized high-order chromosome structures that contains largely repetitive sequences derived from transposable elements. These repetitive nucleotide elements that contain numerous CpG dinucleotides include the centromeric satellite repeats satellite 2 (Sat2) and α-satellite (α-Sat) and pericentromeric satellite repeats, major satellite repeats (M-Sat) [1, 2]. Generally, Sat2, α-Sat and M-Sat are transcribed by RNA polymerase II at a low level, indicating high genome stability; however, when both the centromeric and pericentromeric regions are relaxed, the transcripts of Sat2 α-Sat, and M-Sat are increased, indicating genome instability [3]. DNA hypomethylation in cancer occurs in repeats at a genome-wide level, which leads to genomic instability links with cancer genome instability by increasing expression levels of transcribed repeats [1, 2]. Clearly, heterochromatin protects genome integrity and stability [4]. When cells encounter DNA damage from various stresses, the structure of heterochromatin is modulated by unique mechanisms from those used by euchromatin [5]. For example, the phosphorylation of histone H2A.x (H2AX) at Ser-139, which is a hallmark of DNA double-stranded breaks (DSBs), is refractory in heterochromatin but is enriched within euchromatin when cells encounter DNA damage stimuli [6].
DNA hypomethylation is one of the most important markers of tumor cell DNA. Alterations in DNA methylation, including the hypomethylation of oncogenes and the hypermethylation of tumor suppressor genes, indicate that DNA methylation plays an important role in tumorigenesis [7-9]. 5-Hydroxymethylcytosine (5-hmC) is an epigenetic marker that can be converted to 5-methylcytosine (5-mC) by the ten-eleven translocation (TET) gene family through a multistep process in which TETs and other proteins are involved. TET genes were initially identified as a chromosomal translocation partner in leukemia and represent a key enzyme for DNA demethylation [10, 11]. Interestingly, TET2 is a critical regulator for hematopoietic stem cell homeostasis, and functional impairment of this homeostasis leads to hematological malignancies [12]. Loss of 5-hmC in solid tumors does not have driver properties in cancer initiation but rather reinforces the molecular networks functioning in cancer cells [13]. However, the effect of 5-hmC and TETs on metastasis at a global level remains uncertain.
Lymphoid specific helicase (LSH), also called HELLS (helicase, lymphoid specific) or PASG (proliferation-associated SNF2-like), a protein belonging to the SNF2 family of chromatin-remodeling ATPases, is critical for the normal development of plants and mammals by establishing correct DNA methylation levels and patterns [14-17]. LSH maintains genome stability in mammalian somatic cells [18, 19]. LSH serves as a driver in several types of cancer under different mechanisms [20-25]. Interestingly, significant levels of 5-hmC have been detected in mouse embryonic stem cells, and 5-hmC levels decline during differentiation, which indicates the critical role of 5-hmC in the maintenance of stem cell status [26]. Several 5-hmC readers including LSH have been identified in mouse embryonic stem cells, neuronal progenitor cells and adult mouse brain [27]. Therefore, it will be very interesting to identify the potential factors such as LSH involved in carcinogenesis.
Epigenetic changes including DNA methylation are key events in the process of carcinogenesis. DNA methylation is a crucial factor of epigenetics and plays a critical part in the development of several malignancies [28, 29], but its role in the progress of metastasis remains unknown. In particular, it is not well known whether the level of 5-hmC is altered in metastasis and whether altered 5-hmC is associated with metastasis. Thus, in this study, we investigated the alteration of 5-hmC in metastasis in human patients. Our data show that 5-hmC might serve as a metastasis marker and that decreased expression of LSH is likely one of the mechanisms as an intact complex with TET2 underlying the loss of 5-hmC in metastasis.
Material and Methods
Ethics Statement
The written informed consent of patients to the handling of clinical data was obtained by the referring physicians at admission for surgery. Because the study posed no risk to patients or their privacy, the approval of the use of pathology specimens with a waiver of consent was granted by the Review Board of Xiangya Hospital and by the Ethics Committees of Xiangya Hospital. A coded database was prepared by clinical researchers unaware of molecular data, and deidentified coded samples were obtained from the pathology archives for molecular analysis.
Immunohistochemistry (IHC) analysis
Nasopharyngeal carcinoma, breast cancer and colon cancer biopsies were obtained from the Pathology Department of Xiangya Hospital. The nasopharyngeal tissue array was purchased from Pantomics (Richmond, CA, USA). IHC analysis of paraffin sections from nasopharyngeal, breast and colon tissues or xenograft samples was performed as previously described. The sections were incubated with antibodies as indicated. The images were surveyed and captured using a CX41 microscope (OLYMPUS, Tokyo, Japan) with a DP-72 Microscope Digital Camera System (OLYMPUS, Tokyo, Japan) and differentially quantified by two pathologists from Second Xiangya Hospital, Changsha, China.
Cell lines and treatment conditions
The breast cancer cell lines (MCF7), cervical cell line (Hela), and embryonic kidney cell line (HEK-293) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). All cell lines were maintained under culture conditions as suggested by ATCC. CNE1, HNE3 and HK1 are nasopharyngeal squamous carcinoma cell lines. C666-1 is an NPC cell line consistently harboring the Epstein-Barr virus. Cells were cultured in RPMI-1640 (GIBCO, Life Technologies, Basel, Switzerland) medium with fetal bovine serum (FBS) to a final concentration of 10%. All cell lines were maintained at 37°C with 5% CO2.
Expression and shRNA constructs
The cDNA clone for full-length human LSH was obtained from Open Biosystems (Clone ID:4109340) and subcloned into lentivirus vector GV166 (Genchem, Shanghai, China) and transfected into HK1 and CNE1 cells. Stable expressing cells were selected with puromycin. For 'stable' knockdown of LSH, a shRNA lentivirus set of five clones (RHS4533-NM_018063 from Open Biosystems) was used, and stable expressing cells were selected with 1 μg/ml of puromycin. Cells that had been transfected with the empty control vector pLKO.1 served as control cells. The sequences of the five hairpins are available on the company's webpage http://www.openbiosystems.com.
MicroRNAs selection and detection, and reporter gene assay
The putative targets of miRNAs were analyzed using the following two databases: miRecords (http://mirecords.biolead.org/) and TargetScan (http://genes.mit.edu/targetscan). The selected miRs were further confirmed using the UCSC genome browser (hg19 assembly) (http://genome.ucsc.edu/).
Cellular miRNA extraction was performed using the mirVanaTM miRNA Isolation Kit (AM1560, Ambion) according to the manufacturer's instructions. Total RNA extraction was performed using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. The miRs quantitative real-time PCR assay was performed using TaqMan® MicroRNA Assays (RIBBIO, www.sima.cn). The relative expression level was determined as 2-ΔΔCt. Cells were transfected with miR-144, miR-26b-5p and miR-29c-3p Pre-miR miRNA Precursor Molecules, or Pre-miR miRNA Precursor Molecules Negative Control or anti-miR™ miRNA Inhibitor Negative Control (RIBBIO, www.sima.cn) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
The 3′ UTRs of TET2 and antise of 3′ UTRs of TET2 were PCR amplified from human MCF-10A cells, and inserted downstream of CMV-driven firefly luciferase cassette in the pMIR-REPORT vector (Ambion) between HindIII and SpeI sites. For miRNA target validation, approximately 2 × 104 293 cells per well in 24-well plates were transiently transfected with 25 to 50 ng of each of 3′ UTR-pMIR-REPORT constructs or those antisense constructs (Ambion), and miR-26b-5p and miR-29c-3p construct. Renilla luciferase vector was used to normalize transfection efficiency. Approximately 48 hours after transfection, firefly and Renilla luciferase activities were assayed. Normalized relative light units represent firefly luciferase activity/Renilla luciferase activity.
Quantitative real-time PCR
Cells were harvested with Trizol (Invitrogen), and cDNAs were synthesized with SuperScript II (Invitrogen) according to the manufacturer's protocol. Real-time PCR analysis was performed using the Applied Biosystems 7500 Real-Time PCR System according to the manufacturer's instructions. The reactions were performed in triplicate for three independent experiments; the results were normalized to β-actin. The following primer sequences were used: Satellite forward primer, 5'- CATCGAATGGAAATGAAAGGAGTC -3'; reverse primer, 5'- ACCATTGGATGATTGCAGTCAA -3'; α-Sat Forward primer, CTGCACTACCTGAAGAGGAC-3'; α-Sat reverse primer, GATGGTTCAACACTCTTACA; Major Sat forward primer, GACGACTTGAAAAATGACGAAATC; Major sat reverse primer, CATATTCCAGGTCCTTCAGTGTGC; TET1 forward primer, 5'-TCTGTTGTTGTGCCTCTGGA-3'; TET1 reverse primer, 5'-TTTTGTTCTTCCCCATGACC-3'; TET2 forward primer, 5'-ATTCTCAGGGGTCACTGCAT-3', TET2 reverse primer, 5'-AACGTGAAGCTGCTCATCCT-3'; TET2 reverse primer, 5'-AACGTGAAGCTGCTCATCCCT-3'; TET3 forward primer, 5'-TGCGTCGAACAAATAGTGGA-3', TET3 reverse primer, 5'-CTCCTTCCCCGTGTAGATGA-3'; LSH forward primer, 5'-AGAAGGCATGGAATGGCTTAGG-3'; LSH reverse primer, 5'-GCCACAGACAAGAAAAGGTCC-3'; β-actin, 5'-CATGTACGTTGCTATCCAGGC-3'; reverse primer, 5'-CTCCTTAATGTCACGCACGAT-3'. The mean ± SD of three independent experiments is shown.
Immunofluorescence analysis
Cells were cultured and fixed in 4% paraformaldehyde for 30 min. To identify the potential presence of LSH and TET2 or TET3 proteins or 5-hmC, cells were incubated with an anti-LSH antibody (Sigma) and an anti-TET2 or TET3 antibody (Abcam) or anit-5-hmC (Active motif) and then with fluorescein isothiocyanate (FITC)-conjugated anti-IgG (Santa Cruz) and Cy3-conjugated anti-IgG (Sigma). To visualize the nuclei, the cells were stained with Hoechst (1:1000). Fluorescent images were observed and analyzed with a laser scanning confocal microscope (Bio-Rad MRC-1024ES).
Western blot analysis and co-immunoprecipitation (Co-IP) assay
Cells were harvested after the treatment as indicated, washed twice with ice-cold phosphate-buffered saline (PBS), lysed in RIPA buffer and centrifuged at 15,000 × g for 10 min after sonication. The supernatants were collected as whole-cell lysates. A mass of 50 μg of total protein was used for Western blot analysis. The following antibodies were used for Western blotting: γ-H2AX (9718, Cell Signaling) and β-actin (A5441, Sigma).
Immunoprecipitation was performed with transfected HK1 and HK1-LSH cells. Cells were plated overnight in 100 mm2 dishes (1.5×106/dish). In the immunoprecipitation assay buffer (1× PBS, 0.5% Nonidet P-40, 0.5% sodium deoxycholate and 0.1% SDS), 1 mg of protein was mixed with 40 μl of Protein A-Sepharose beads (Sigma), incubated at 4°C for 2 h with gentle agitation and centrifuged for 10 min at 2000 rpm for preclearing. The recovered supernatant was incubated with 10 μl of anti-Flag M2 agarose (Sigma) in the presence of 1× protease inhibitors at 4°C overnight with mild shaking. The M2 agarose-precipitated protein complex was recovered by brief centrifugation followed by three washes with the immunoprecipitation assay buffer. The harvested beads were resuspended in 30 μl of 2× SDS PAGE sample buffer and boiled for 5 min to release the bound protein. A 20 μg aliquot of cell lysate was used as an input control. The samples were analyzed by Western blot. The antibodies used for Western blot detection were the LSH antibody, TET2 and TET3 antibodies, as indicated.
5-hmC hMeDIP at satellites
Before carrying out hMeDIP, 10 mg of genomic DNA was sonicated to yield a size range of 200 to 700 bp. Four micrograms of gel purified DNA was used for each hMeDIP assay described previously [9, 30]. Real-time PCR analysis was performed; for each primer, the amplification efficiency was calculated and the data expressed as enrichment related to input. The following primer sequences were used: Satellite forward primer, 5'- CATCGAATGGAAATGAAAGGAGTC-3'; reverse primer, 5'- ACCATTGGATGATTGCAGTCAA-3'; α-Sat Forward primer, CTGCACTACCTGAAGAGGAC-3'; α-Sat reverse primer, GATGGTTCAACACTCTTACA; Major Sat forward primer, GACGACTTGAAAAATGACGAAATC; Major sat reverse primer, CATATTCCAGGTCCTTCAGTGTGC.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were essentially performed as previously described [9, 31]. ChIP DNA was analyzed by qPCR with SYBR Green (Bio-Rad) in ABI-7500 (Applied Biosystems) using the primers specified in the following, site 1: miR-26b-5p forward primer: 5'- CCTGTGGAGATTGATGGGGT -3', reverse primer: 5'- TCTCTGGGCCTCTGACATTC -3'; site 2: miR-26b-5p forward primer: 5'- AGCCTTGCAGTTGATTGTGG -3', reverse primer: 5'- TAGGGAGGCACAAGTTGGAG -3'. site 1: miR-29c-3p forward primer: 5'- TCTGTTGACTCCTAGCAGCC -3', reverse primer: 5'- ACTGATGGTGTCGATGTGGA -3'; site 2: miR-29c-3p forward primer: 5'- GATTGCAGGTTCATGGGGTC -3', reverse primer: 5'- CACCCTATCTGCCTGTGGAA -3'. The LSH antibody was used from Sigma and normal mouse IgG was from Millipore.
The comet assay, FACS and Cell viability assay
The comet assay was described previously [9]. Flow cytometry was used to quantify cells in each phase of the cell cycle described previously [31, 32]. Cells (2 × 105) were plated into 6-well plates and treated with cisplatin. Cells were harvested at 0, 24, and 48 h and washed twice with PBS. Pellets were resuspended in 0.5 mL PBS, fixed in 4.5 mL of 70% ethanol, and incubated overnight at 4°C. To detect the fluorescence intensity of certain proteins, cells were counterstained in the dark with 50 μg/mL of phosphatidyl inositol (PI) and 0.1% ribonuclease A (RNase A) in 400 μl of PBS at 25°C for 30 min. Stained cells were assayed and quantified using a FACSort Flow Cytometer (Becton Dickinson, U.S.A).
MTS assay were essentially performed as previously described [9, 31]. Cell viability was measured using a CellTiter-Glo Luminescent Cell Viability Assay Kit (MTS) purchased from Promega Corp (Madison, WI) and used according to the manufacturer's protocol. Measuring the absorbance at 450 nm using a microplate reader assessed the number of viable cells.
Nude mice and study approval
Xenograft tumor formation was performed as previously described [9]. Mice were purchased from the Hunan SJA Laboratory Animal Co.Ltd and were injected with cells as indicated (2 × 106 cells/mice) and their corresponding stable clones with knockdown of LSH expression via the mammary fat pad. Mice injected with cells were imaged along the dorsal and ventral planes once per week. Visible tumor nodules were examined macroscopically or detected in paraffin-embedded sections stained with H&E. Data were analyzed using Student's t-test; a p value less than 0.05 was considered significant.
All animal study procedures were approved by the Institutional Animal Care and Use Committee of the Central South University of Xiangya School of Medicine and conform to the legal mandates and federal guidelines for the care and maintenance of laboratory animals.
Statistics
The experiments were repeated at least three times. Results are expressed as mean ± SD or SEM as indicated. A two-tailed Student's t-test was used for intergroup comparisons. A p value less than 0.05 was considered statistically significant (*p<0.05, ** p <0.01, *** p <0.001).
Figure 1
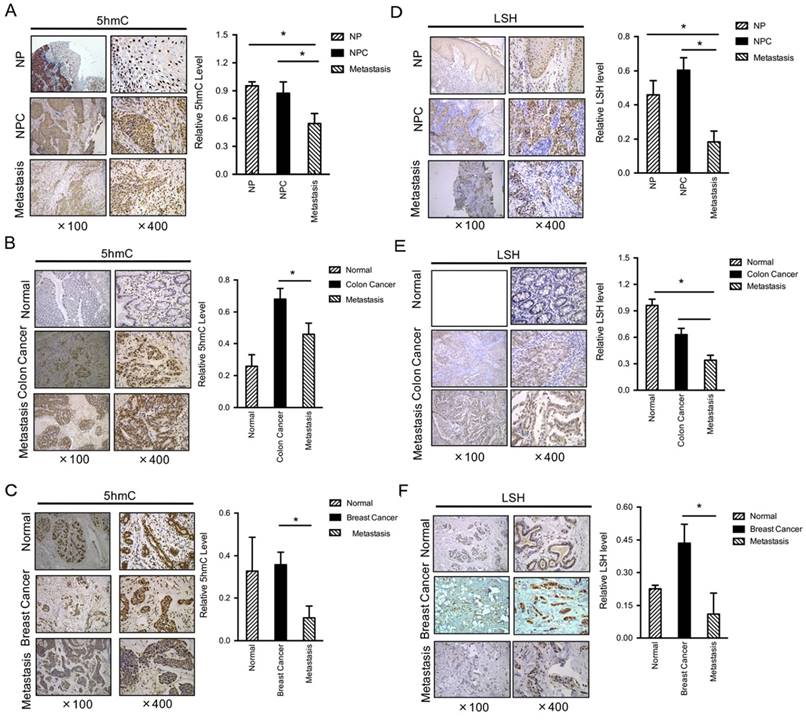
Both 5-hmC and LSH levels are decreased in metastasis tumors. IHC was performed using antibodies against 5-hmC in human normal, cancer without metastasis and cancer with metastasis tissues from the nasopharynx (A), colon (B) and breast (C). The mean values of the IHC quantification are shown in the right panel. IHC was performed using antibodies against LSH in human normal, cancer without metastasis and cancer with metastasis tissues from the nasopharynx (D), colon (E) and breast (F). The mean values of the IHC quantification are shown in the right panel. Data are presented as mean ± s.d. *P<0.05, **P<0.01, ***P<0.001.
Results
Both 5-hmC and LSH are decreased in human metastasis tissues
To determine the levels and distribution of 5-hmC in human normal and cancer tissues of nasopharyngeal carcinoma (NPC), a prevalent cancer in Southern China, we performed immunohistochemistry (IHC) analysis using a specific antibody that has been verified [33]. We assessed 5-hmC levels in a panel section of 52 NPC and 31 metastasis cancer and 12 normal nasopharyngeal tissues (left panel of Figure 1A). Interestingly, normal nasopharyngeal and NPC without metastasis displayed similar 5-hmC levels. However, a significant decrease in 5-hmC was observed in NPC with metastasis (right panel of Figure 1A). To further confirm this finding, we selected other types of human cancer tissues, including colon and breast cancer tissues; we found that the levels of 5-hmC were significantly reduced in 19 human colon cancers with metastasis when compared with the matched colon cancers, whereas the levels of 5-hmC were significantly increased in colon cancers compared to normal colon (P<0.01) (Figure 1B). In human breast, we evaluated a total of 23 samples and observed a reduction in 5-hmC in the tumor samples with metastasis relative to the levels measured in the matched breast cancer samples or normal breast tissues (P<0.01) (Figure 1C).
LSH is identified as a 5-hmC reader in mouse embryonic stem cells, neuronal progenitor cells and the adult mouse brain [27]. Based on our recent findings that LSH was overexpressed in NPC [21, 24], whereas we observed a reduction in LSH in tumor samples with metastasis when compared with the matched cancer (P<0.001) (Figure 1D). Furthermore, we found that the levels of LSH were significantly reduced in 19 human colon cancers with metastasis when compared with the matched colon cancers, however LSH was highly expressed in the normal colon tissues (P<0.01) (Figure 1E). In human breast, we evaluated a total of 23 samples and observed a reduction in LSH in the tumor samples with metastasis when compared with the matched breast cancers, whereas the levels of LSH were significantly increased in colon cancers compared to normal colon (P<0.01) (Figure 1F). Then, we analyzed the correlation of 5-hmC and LSH in cancer and metastasis, the evaluation of LSH and 5-hmC levels of all 125 biopsies confirms the positive correlation between LSH and 5-hmC in cancer and metastasis tissues (R=0.751, P <0.001).
LSH induces TET2 and TET3 gene expression in vitro and in vivo
Two mechanisms that would lead to decreased levels of 5-hmC in human tumors have been reported. One is loss-of-function mutations targeting the TET2 gene, and the other is inhibition of TET activity by the decrease in α-KG and accumulation of 2-HG resulting from IDH1/2 mutations. However, to date, neither the TET2 nor IDH1/2 gene has thus far been shown to be mutated in NPC cell lines (Data not shown). Because only TET2 and TET3 are detectable in NPC cells [9], we selected TET2 and TET3 for our study.
To investigate the possible role of LSH in TETs underlying the link between the decrease in 5-hmC and LSH level, we determined the mRNA expression of TET2 and TET3 in NPC cell lines. We generated stable overexpression of LSH in NPC cell lines HK1-LSH and HNE3-LSH using a lentivirus expressing plasmid; then, total RNA was later extracted and analyzed by real-time PCR. We found that stable overexpression of LSH significantly increased expression of TET2 and TET3 (Figure 2A and Figure 2B). To further validate the association of LSH with TET2 and TET3 mRNA levels, we generated stable LSH knockdown in C666-1 cancer cells. The knockdown approach successfully reduced LSH mRNA levels to less than 20% from two different target sequences to LSH (left column of Figure 2C). The knockdown of LSH resulted in the decrease in TET2 and TET3 mRNA expression levels in C666-1 cells. Notably, the decrease in TET2 and TET3 varied, with TET2 decreasing more significantly.
To further extend these observations, we examined TET2 in biopsies from xenograft tumors in mice. In the xenograft model, we found that LSH significantly increased tumor weight in HK1 cells (Figure 2D) and HNE3 cells (Figure 2E), whereas LSH depletion significantly impaired tumor weight in C666-1 cells (Figure 2F), indicating that LSH promoted tumor growth in vivo. Immunohistochemistry confirmed that the expression levels of TET2 were increased in LSH overexpression when compared with those measured for the HK1 (Figure 2G) and HNE1 groups (Figure 2H). Moreover, we also found that TET2 expression decreased in the depletion of LSH (Figure 2I). Therefore, these observations indicate a potential link between LSH and TET2.
Furthermore, we found that the levels of TET2 were significantly reduced in 19 human colon cancers with metastasis when compared with the matched colon cancers (Figure 2J). In human breast, we evaluated a total of 23 samples and observed a reduction in TET2 in the tumor samples with metastasis when compared with the matched breast cancers (Figure 2K). A significant decrease in TET2 was observed in colon cancer and breast cancer with metastasis (Figure 2L). The evaluation of LSH and TET2 levels of all 42 biopsies confirms the positive correlation between LSH and TET2 in cancer tissues (R=0.642, P <0.05). Lastly, a Kaplan-Meier plotter performed on a cohort of these breast cancers showed that the lower expression of TET2 and LSH was associated with longer overall survival in all breast cancer (supplementary Figure S1A-B).
Both miR-26b-5p and miR-29c-3p inhibit TET2 and TET3 expression and are downregulated by LSH
To address the mechanism through which LSH regulates TET2 and TET3 expression, we first detected whether LSH is directly recruited into the promoters of TET2 and TET3 as a chromatin modifier. A quantitative ChIP assay of LSH revealed that LSH was not recruited into the promoters of TET2 and TET3 in MCF-7 cells using two different primers that amplified the region of transcriptional start sites of TET2 and TET3 (Data not shown). Then we screened the unique microRNAs (miRs) that repress either TET2 or TET3 3'UTRs according to the UCSC genome browser. We chose four miRs, miR-139-5p, miR-144, miR-26b-5p and miR-29c-3p, for further study. We found that miR-139-5p and miR-144 did not affect TET2 or TET3 expression at the protein or mRNA level after the ectopic expression of miR-139-5p and miR-144 (supplementary Figure S2A-C), whereas both TET2 and TET3 mRNA levels decreased significantly after the introduction of miR-26b-5p and miR-29c-5p into MCF-7 cells (Figure 3A-C), which was further confirmed at the protein level in HNE3 and HK1 cells (Figure 3D-E). Additional experiments were performed using pMiR-report vectors with sense or antisense 3'UTRs of TET2, transiently transfected with miR-26b-5p or miR-29c-5p precursor constructs or control vectors into 293 cells. As shown in Figure 3F, luciferase activities increased 3.5 to 4.5 folds in cells overexpressing miR-26b-5p or miR-29c-5p relative to luciferase activities in cells transfected with control vectors. Antisense of TET2 3'UTR abolished increase mediated by miR-26b-5p or miR-29c-5p (Figure 3F). We found that total level of 5-hmC in MCF-7 cells decreased after the introduction of miR-26b-5p and miR-29c-3p into MCF-7 cells (Figure 3G).
Figure 2
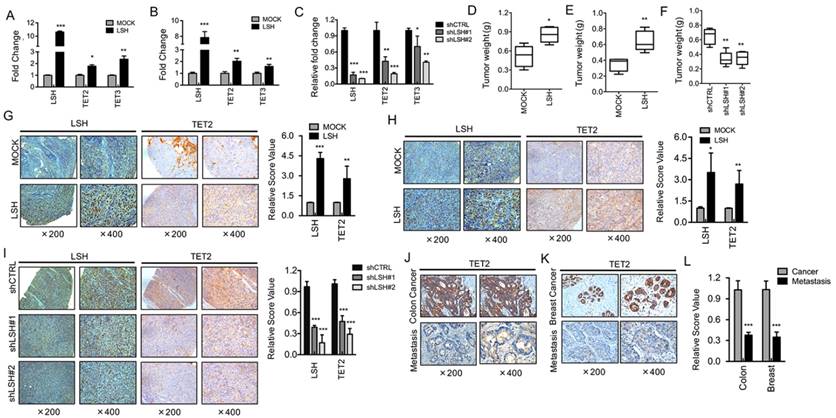
LSH induces TET2 expression in vitro and in vivo. RT-PCR analysis was conducted to detect TET2 and TET3 genes using total RNA derived from HK1 (A) and HNE3 (B) cells and matching LSH overexpressed cell lines. The level of gene expression was normalized against the housekeeping gene β-actin and is represented as fold change compared with HK1 and HNE2 cells. (C) Two stable LSH knockdown cell lines (shLSH#1 and shLSH#2) were established by transfecting shLSH sequences into C666-1 cells. RT-PCR analysis was used to detect TET2 and TET3 mRNA after knockdown of LSH. The means and s.d. values were derived from three to four independent experiments. A xenograft model of tumor weight was established in nude mice to evaluate the overexpression of LSH in HK1 (D) and HNE3 (E) cells and the control cells. (F) A xenograft model of tumor weight was established in nude mice to evaluate the knockdown of LSH in C666-1 cells. IHC was performed using antibodies against LSH and TET2 in xenograft tissues from HK1 cells (G) and HNE3 cells (H) together with matching LSH ectopic expression of LSH. (I) IHC was analyzed using antibodies against LSH and TET2 in xenograft tissues from C666-1 cells in the depletion of LSH. The mean values of the IHC quantification are shown in the right panel. IHC was performed using antibodies against TET2 in human cancer without metastasis and cancer with metastasis tissues from colon (J) and breast (K). (L) The mean values of the IHC quantification are shown. * p <0.05, ** p <0.01, ***P<0.001.
Furthermore, we found that both miR-26b-5p and miR-29c-3p levels decreased after ectopic expression of LSH in HK1 and HNE3 cells (Figure 3H-I). Lastly, we addressed whether LSH is directly recruited into the promoters of miR-26b-5p and miR-29c-3p as a chromatin modifier. A quantitative ChIP assay of LSH revealed that LSH was recruited into the promoters of 26b-5p and miR-29c-3p in MCF-7 cells using two different primers that amplified the region of transcriptional start sites (Figure 3J-M). Taken together, these results suggest that LSH might upregulate TET2 and TET3 expression by silencing miRs such as miR-26b-5p and miR-29c-3p that is silenced by LSH directly.
The intact complex of LSH and TET2 affects 5-hmC levels
TET enzymes have been shown to catalyze 5-hmC and to be involved in the regulation of gene expression [10, 11], while LSH might function as a 5-hmC reader [27], indicating that it is possible that TET2 or TET3 interacts directly with LSH protein. We found that TET3 did not show any colocalization of LSH (Data not shown), whereas TET2 colocalized with LSH in MCF-7 cells at endogenous level, as indicated by immunofluorescence analysis (Figure 4A). Next, we sought to further confirm the potential interaction of TET2 with LSH in an HK1 cell line stably expressing Flag-LSH. After immunoprecipitating LSH with Flag-M2 agarose, we detected the potential interaction of TET2 with LSH using anti-TET2 antibody (Figure 4B). Furthermore, we confirmed that the intact complex of LSH and TET2 existed in MCF-7 cells at the endogenous level using co-immunopreciptation assay (Figure 4C). Data indicated that LSH and TET2 could form an intact complex.
LSH is identified as a 5-hmC reader in mouse embryonic stem cells, neuronal progenitor cells and the adult mouse brain [27]. We therefore addressed the potential intact complex by immunofluorescence analysis. We found that LSH colocalized with 5-hmC in HK1 cells after stable expression of LSH (Figure 4D). Furthermore, we quantified the 5-hmC content of nuclear DNA as the percentage of 5-hmC levels by ELISA, and we found that the 5-hmC level increased significantly in both HK1 and HNE3 cells in the presence of LSH (Figure 4E), indicating that LSH promotes 5-hmC through the interaction of TET2.
Figure 3
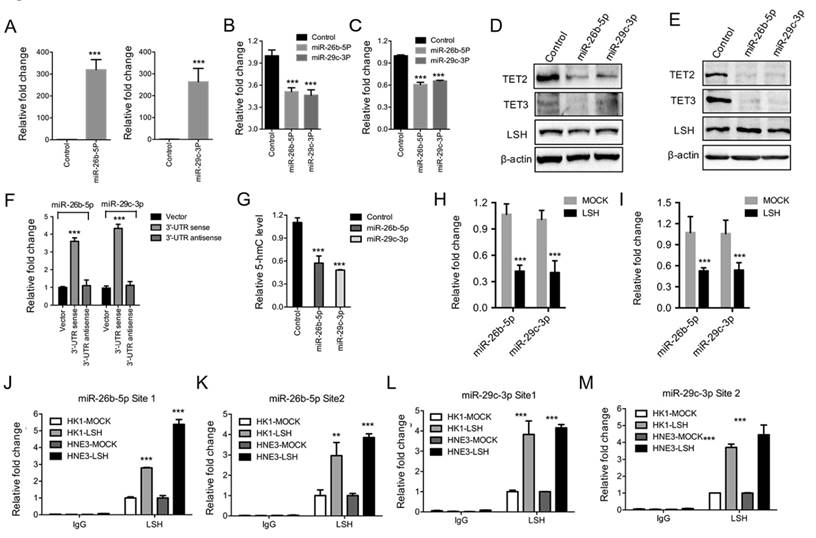
Both miR-26b-5p and miR-29c-3p inhibited TET2 and TET3 expression and were silenced by LSH. (A) Ectopic expression of miR-26b-5p (Left) and miR-29C-3p (Right) in MCF-7 cells. Relative expression levels of TET2 (B) and TET3 (C) were detected by RT-PCR after the transfection of miRs as indicated. (D, E) Expression levels of TET2 and TET3 were analyzed after the transfection of miRs as indicated in HK1 (F) and HNE3 (G) cells, whereas LSH protein level unchanged. (F) Luciferase reporter assay in 293 cells together with miRs as indicated, and with transfection of pMiR-Report-TET2 3'-UTR sense or pMiR-Report-TET2 3'-UTR antisense as indicted. (G) ELISA of 5-hmC was used to assess 5-hmC levels from genomic DNA derived from HNE3 cells after introduction of miRs as indicated. (H, I) miR levels of miR-26b-5p and miR-29c-3p in HK1 (H) and HNE3 (I) cells after overexpression of LSH. (J-M) The recruitment of LSH the promoter regions of miR-26b-5p (J and L) and miR-29c-3p (K and M) was analyzed in HK1 and HNE3 cells after overexpression of LSH. * p <0.05, ** p <0.01, ***P<0.001.
To further extend these observations, we examined 5-hmC in biopsies from xenograft tumors in mice. Immunohistochemistry confirmed that the expression levels of 5-hmC increased in overexpression of LSH tissues when compared with the levels measured for the HK1 and HNE1 groups (Figure 4F-G). A significant increase in 5-hmC was quantified (Figure 4H). Moreover, we found that 5-hmC levels decreased in the depletion of LSH (Figure 4I), and a significant decrease in 5-hmC was quantified (Figure 4J). These observations suggest a potential link between LSH and 5-hmC through miR-26b-5p and miR-29c-3p that silenced TET2 and TET3.
LSH expression increases pericentromeric heterochromatin relaxation and resistance in response to cisplatin
The relaxation of heterochromatin may result in genome instability. We selectively evaluated the transcripts of Sat2, α-Sat and M-Sat as markers for the alteration of heterochromatin structure [3]. The relative expression levels of Sat2 and M-Sat were significantly elevated, whereas α-Sat levels did not change in the presence of LSH in HK1 (Figure 5A) and HNE3 (Figure 5B) cells. Next, we found that after depletion of LSH in C666-1 cells, the expression levels of Sat2 and M-Sat decreased significantly (Figure 5C). Accordingly, a quantitative hMeDIP assay also revealed that 5-hmC was down-regulated in both M-Sat and Sat2 loci in the presence of LSH, whereas 5-hmC of α-Sat increased (Figure 5D). Notably, the decrease in 5-hmC in satellites varied, with M-Sat of 5-hmC decreasing most significantly, whereas while 5-hmC levels of Sat2 decreased only slightly. These data suggest that LSH mainly increases heterochromatin relaxation at pericentromeric regions, which correlates with the loss of 5-hmC in M-Sat of heterochromatic loci.
Finally, the ability of LSH to hydrolyze ATP is necessary for efficient phosphorylation of H2AX at DNA double-strand breaks and successful repair of DNA damage in response to irradiation in MEF cells [19]. We treated NPC cells with cisplatin over time. FACS indicated that LSH exited the cell cycle from the S stage and entered the G2/M stages after the treatment of cisplatin (supplementary Figure S3), suggesting that LSH is possibly linked to the resistance to DNA damage response. We first found that LSH reduced DNA damage using HNE3 and HK1 cells (Supplementary Figure S4A-B). We demonstrated that LSH reduced the increase in the phosphorylation of H2AX, a DNA damage marker that responds to cisplatin, compared with the absence of LSH in the HK1 (Figure 5E) and HNE3 (Supplementary Figure S4C). We found that LSH increased cell survival in both HK1 and HNE3 cells after the treatment of cisplatin (Figure 5F). Additional experiments indicated that LSH was resistant to apoptosis in both HK1 and HNE3 cells after the treatment of cisplatin cell lines (Figure 5G and supplementary Figure S4D). Finally, we showed that both LSH and TET2 could increase the global level of 5-hmC after introduction of LSH and TET2 into C666-1 cells that were stably depleted LSH expression (Figure 5I). These results suggest that LSH has the ability to exhibit resistance to DNA damage through a response involving cisplatin.
Discussion
Although recent studies have suggested that loss of 5-hmC is pervasive in several solid cancers, the putative association between 5-hmC in satellites and genome stability is not yet fully understood. In this study, we found that loss of 5-hmC was linked to cancer metastasis, with the interaction of LSH with TET2 contributing to 5-hmC and gene instability. To the best of our knowledge, this is the first study to show concurrent 5-hmC, satellite and genome instability in human nasopharyngeal carcinoma and other solid tumors.
Figure 4
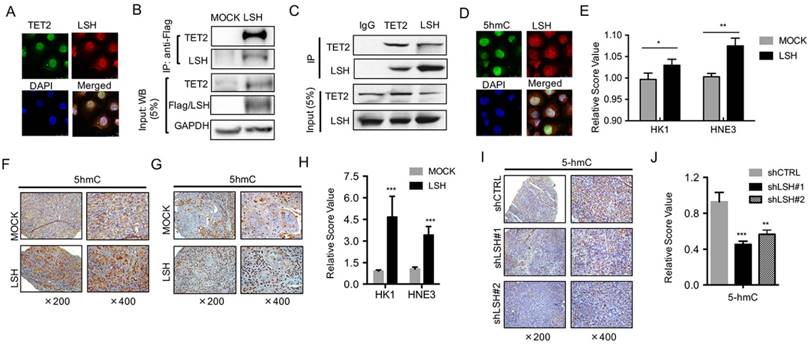
LSH interacts with TET2 and affects 5-hmC levels. (A) LSH colocalized with TET2. C666-1 cells were stained with the indicated antibodies. Colocalization of LSH and TET2 is shown in yellow in the merged image. (B) Equal amounts of protein from HK1 and HK1-LSH were immunoprecipitated (IP) with an anti-Flag M2 agarose and were immunoblotted to detect LSH or TET2. (C) Equal amounts of protein from MCF-7 cells were immunoprecipitated (IP) with TET2 or LSH and were immunoblotted to detect LSH or TET2 as indicated. (D) LSH colocalized with 5hmC. C666-1 cells were stained with the indicated antibodies. Colocalization of LSH and TET2 is shown in yellow in the merged image. (E) ELISA of 5-hmC was used to assess 5-hmC levels from genomic DNA derived from HK1 and HNE3 cells and matching LSH overexpressed cell lines. IHC was performed using antibodies against 5-hmC in xenograft tissues from HK1 cells (F) and HNE3 (G) cells. (H) The mean values of the IHC quantification were shown. (I) IHC was analyzed using antibodies against 5-hmC in xenograft tissues from C666-1 cells in the depletion of LSH. (J) The mean values of the IHC quantification of 5-hmC are shown. (K) ELISA of 5-hmC was used to assess 5-hmC levels from genomic DNA derived from MCF-7 cells after introduction of shLSHs as indicated. * p <0.05, ** p <0.01, ***P<0.001.
Figure 5
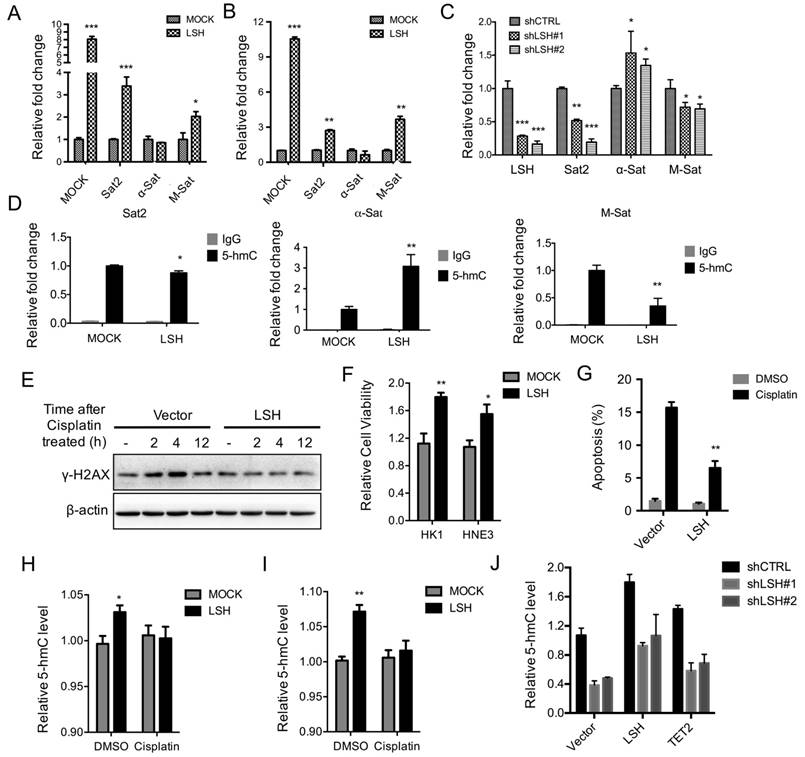
LSH decreases heterochromatin relaxation at pericentromeric repeats and resistance in response to cisplatin. RT-PCR analysis was conducted to detect Sat2, α-Sat, and M-Sat using total RNA derived from HK1 (A) and HNE3 (B) cells and matching LSH overexpressed cell lines. The level of gene expression was normalized against the housekeeping gene β-actin and is represented as a fold change compared with HK1 and HNE2 cells. (C) RT-PCR analysis was used to detect Sat2, α-Sat, and M-Sat mRNA after knockdown of LSH. (D) hMeDIP was analyzed to detect Sat2, α-Sat, and M-Sat using 5-hmC antibody in HK1 and matching LSH overexpressed cells. The means and s.d. values were derived from three to four independent experiments. (E) Western blot assay was conducted to detect γ-H2Ax using total protein derived from HK1 cells and matching LSH overexpressed cell lines after the treatment of cisplatin at the indicated time. The level of gene expression was normalized against the housekeeping gene β-actin. (F) The MTT assay was performed to assess cell viability in HK1 and HNE3 cells and matching LSH overexpressed cell lines after the treatment of cisplatin for 72 hrs. (G) FACS assay was performed to assess apoptosis in HNE3 cells and matching LSH overexpressed cell lines after the treatment of cisplatin for 72 hrs. (H, I) ELISA of 5-hmC was used to assess 5-hmC levels from genomic DNA derived from HK1 (H) and HNE3 (I) cells and matching LSH overexpressed cell lines after the treatment of cisplatin for 72 hrs. (J) ELISA of 5-hmC was used to assess 5-hmC levels from genomic DNA derived from C666-1cells in the depletion of LSH after introduction of LSH and TET2 as indicated. *p<0.05, **p<0.01, ***p<0.001.
DNA methylation was the first well-characterized epigenetic modification and has been demonstrated to play an important role in carcinogenesis [2, 34]. Recently, the balance of DNA methylation and demethylation in epigenetic modification has become a popular topic in cancer research. Studies have demonstrated that aberrant DNA methylation in cancer is not only associated with the repression of chromatin related to specific genes but also with the repression of large chromosomal regions [1, 2]. The recent discovery of 5-hmC as a novel DNA modification marker in mammalian genomes has raised many questions regarding the role of DNA demethylation in epigenetic regulation [35]. TETs are associated with malignancy and tumorigenesis, and research in this field has mainly focused on TET2. In myeloma, myelodysplastic syndrome and lymphoma, 5-hmC decreases in patients due to TET2 depletion or mutation-induced enzymatic dysfunction, even though the whole-genome distribution of methylation does not change in an appreciable manner [36-40]. The somatic conditional depletion of TET2 leads to increased hematopoietic stem cell self-renewal and myeloproliferation, suggesting that TET2 contributes to normal hematopoietic and lymphatic development or malignant progression [39, 41]. This behavior may be influenced by the TET2-targeted gene itself, indicating that the loss of 5-hmC is an epigenetic hallmark in cancer [40]. Our data showed no mutations of TET2 involved in NPC tissues or cells, which is consistent with the results reported by other groups.
MicroRNA (miR) deregulations including circulating miRs are also known to contribute to several malignancies [34, 42-44]. A combined approach involving assessment of miR expression (using microarrays) and bioinformatic prediction of miR targets has revealed that distinct miR signatures fine-tune each step of malignancies; in turn, miRs that target critical suppressors could act as powerful proto-oncogenes. Several miRs, including miR-22, miR-29b and miR-26a, exert control by negatively regulating TET protein levels in mouse and human [45-47]. In this study, we demonstrated that both miR-26b-5p and miR-29c-5p silenced TET2 expression, whereas LSH was not recruited to the promoters of TET2 and TET3, providing a novel finding that LSH upregulated TET2 expression by silencing the expression levels of miRs.
The level of 5-hmC has been observed in different types of cancers and might play an important role in the pathogenesis of cancers [33, 48, 49]. However, it is not well known whether 5-hmC levels are changed in NPC and whether altered 5-hmC levels are associated with metastasis in patients with NPC. Thus, in this study we investigated the alteration of 5-hmC levels in NPC in human patients and metastasis. Our data show that low levels of 5-hmC in NPC were associated with metastasis patients with NPC, it is a little inconsistent with other reported observations of reduced levels of 5-hmC in other types of cancers [33, 50-53]. Therefore, our observations suggest a potential different molecular mechanism for the observed loss of 5-hmC and metastasis in NPC, a prevalent cancer in China. Therefore, exploring the correlation between malignant progression-associated genes including repeats and 5-hmC in NPC will be an important area of research in the near future. Furthermore, we demonstrated that low levels of 5-hmC in NPC were correlated with NPC.
Reports show that LSH contributes to the malignant progression of prostate cancer, melanoma, head and neck cancer, nasopharyngeal carcinoma and gliomas etc. [20, 22, 32, 54, 55]. LSH is an ATP-dependent chromatin remodeler, and the ATP binding site is critical for chromatin function and establishment of DNA methylation [15, 16, 56, 57]. Previous studies have demonstrated that decreased levels of 5-hmC in tumors are due to the reduced expression of TET1/2/3 and IDH2 genes or tumor-derived IDH1 and IDH2 mutations [58, 59]. We did not find that TET2/3 mRNA levels changed in NPC tumors compared with those measured in non-tumor tissues. However, an intact complex of TET2, not TET3, with LSH was observed. These findings suggest that LSH might play an important role in the conversion of 5-mC to 5-hmC in NPC through TET2.
LSH is crucial for the control of heterochromatin at pericentromeric regions consisting of satellite repeats under the direct interaction of LSH with repetitive sites in the genome [60, 61], indicating that LSH is a guardian of heterochromatin. Depletion of LSH increases DNA hypomethylation in 5-mC in repeats, leading to transcribed satellite repeat expression, indicating LSH control genome stability. Interestingly, 5-hmC levels are significantly associated with long interspersed nucleotide element -1 (LINE-1) methylation, which is regarded as a surrogate marker for global DNA methylation levels. However, TET mRNA expression is not associated with LINE-1 methylation, indicating that another factor is involved in the process [62]. Our data show that LSH might increase 5-hmC levels at major satellites, not Sat 2, via interaction with TET2, suggesting that LSH also contributes pericentromeric regions by DNA demethylation. This contribution is attributed to the fact that LSH directly localizes in pericentromeric regions. Moreover, LSH is essential for the efficient repair of DNA double-strand breaks, and this function of LSH is neither related to alterations in DNA methylation nor caused by mis-expression of known genes involved in DNA repair [19, 63]. In this study, our data show that LSH is resistant to cisplatin-induced DNA damage repair.
Taken together, our data demonstrate that 5-hmC might serve as a metastasis marker for NPC and genome-wide DNA demethylation and that 5-hmC, as measured in satellites, is associated with LSH. The decreased expression of LSH is likely one of the mechanisms underlying 5-hmC loss in NPC metastasis. These findings might hold considerable clinical implications.
Acknowledgements
We would like to thank all laboratory members for their critical discussion of this manuscript.
This work was supported by the National Basic Research Program of China [2015CB553903(Y.T.)].
This work was supported by the National Natural Science Foundation of China [81372427 and 81672787(Y.Tao), 81271763 and 81672991(S.Liu), 81302354(Y.S.), 81422051, 81472593, and 31401208 (Y.Cheng)].
Contributions
Conception and design: Y.T., and S.L.
Development of methodology: J.J., L.C., Y.J., Y.S., W.L., D.X. S.L., S.X. and Y.T.
Acquisition of data (provided animals, provided facilities, etc.): J.J., L.C., S.X., Y.Cheng. Y.J., Y.S., S.L. and Y.T.
Analysis and interpretation of data (e.g. statistical analysis, biostatistics, computational analysis): Y.T., J.J., Y.S. Y.Cao and S.L.
Writing, review, and/or revision of the manuscript: Y.T., J.J., and S.L.
Administrative, technical, or material support (i.e. reporting or organizing data, constructing databases): Y.T., S. L., and J.J.
All authors have given final approval of the version to be published.
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists. This manuscript has been read and approved by all the authors, and not submitted or under consideration for publication elsewhere.
References
1. Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta. 2007;1775:138-62
2. Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148-59
3. Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM. et al. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature. 2011;477:179-84
4. Hennig W. Heterochromatin. Chromosoma. 1999;108:1-9
5. Cann KL, Dellaire G. Heterochromatin and the DNA damage response: the need to relax. Biochem Cell Biol. 2011;89:45-60
6. Falk M, Lukasova E, Kozubek S. Chromatin structure influences the sensitivity of DNA to gamma-radiation. Biochim Biophys Acta. 2008;1783:2398-414
7. Jiang Y, Liu S, Chen X, Cao Y, Tao Y. Genome-wide distribution of DNA methylation and DNA demethylation and related chromatin regulators in cancer. Biochimica et biophysica acta. 2013;1835:155-63
8. Liu S, Tao Y. Interplay between chromatin modifications and paused RNA polymerase II in dynamic transition between stalled and activated genes. Biological reviews of the Cambridge Philosophical Society. 2013;88:40-8
9. Jiang Y, Yan B, Lai W, Shi Y, Xiao D, Jia J. et al. Repression of Hox genes by LMP1 in nasopharyngeal carcinoma and modulation of glycolytic pathway genes by HoxC8. Oncogene. 2015;34:6079-91
10. Delatte B, Deplus R, Fuks F. Playing TETris with DNA modifications. EMBO J. 2014;33:1198-211
11. Shen L, Zhang Y. 5-Hydroxymethylcytosine: generation, fate, and genomic distribution. Curr Opin Cell Biol. 2013;25:289-96
12. Solary E, Bernard OA, Tefferi A, Fuks F, Vainchenker W. The Ten-Eleven Translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia. 2014;28:485-96
13. Ficz G, Gribben JG. Loss of 5-hydroxymethylcytosine in cancer: cause or consequence? Genomics. 2014;104:352-7
14. Zemach A, Kim MY, Hsieh PH, Coleman-Derr D, Eshed-Williams L, Thao K. et al. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell. 2013;153:193-205
15. Yu W, McIntosh C, Lister R, Zhu I, Han Y, Ren J. et al. Genome-wide DNA methylation patterns in LSH mutant reveals de-repression of repeat elements and redundant epigenetic silencing pathways. Genome Res. 2014;24:1613-23
16. Tao Y, Xi S, Shan J, Maunakea A, Che A, Briones V. et al. Lsh, chromatin remodeling family member, modulates genome-wide cytosine methylation patterns at nonrepeat sequences. Proc Natl Acad Sci U S A. 2011;108:5626-31
17. Myant K, Termanis A, Sundaram AY, Boe T, Li C, Merusi C. et al. LSH and G9a/GLP complex are required for developmentally programmed DNA methylation. Genome Res. 2011;21:83-94
18. Fan T, Yan Q, Huang J, Austin S, Cho E, Ferris D. et al. Lsh-deficient murine embryonal fibroblasts show reduced proliferation with signs of abnormal mitosis. Cancer Res. 2003;63:4677-83
19. Burrage J, Termanis A, Geissner A, Myant K, Gordon K, Stancheva I. The SNF2 family ATPase LSH promotes phosphorylation of H2AX and efficient repair of DNA double-strand breaks in mammalian cells. J Cell Sci. 2012;125:5524-34
20. von Eyss B, Maaskola J, Memczak S, Mollmann K, Schuetz A, Loddenkemper C. et al. The SNF2-like helicase HELLS mediates E2F3-dependent transcription and cellular transformation. EMBO J. 2012;31:972-85
21. Liu S, Tao YG. Chromatin remodeling factor LSH affects fumarate hydratase as a cancer driver. Chin J Cancer. 2016;35:72
22. Xiao D, Huang J, Pan Y, Li H, Fu C, Mao C. et al. Chromatin Remodeling Factor LSH is Upregulated by the LRP6-GSK3beta-E2F1 Axis Linking Reversely with Survival in Gliomas. Theranostics. 2017;7:132-43
23. Keyes WM, Pecoraro M, Aranda V, Vernersson-Lindahl E, Li W, Vogel H. et al. DeltaNp63alpha is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell. 2011;8:164-76
24. He X, Yan B, Liu S, Jia J, Lai W, Xin X. et al. Chromatin Remodeling Factor LSH Drives Cancer Progression by Suppressing the Activity of Fumarate Hydratase. Cancer research. 2016;76:5743-55
25. Jiang Y MC, Yang R, Yan B, Shi Y, Liu X, Lai W, Liu Y, Wang X, Xiao D, Zhou H, Cheng Y, Yu F, Cao Y, Liu S, Yan Q, Tao Y. EGLN1/c-Myc Induced Lymphoid-Specific Helicase Inhibits Ferroptosis through Lipid Metabolic Gene Expression Changes. Theranostics. 2017;7:3293-305
26. Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J. et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200-13
27. Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C. et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146-59
28. Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339:1567-70
29. Chen T, Dent SY. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet. 2014;15:93-106
30. Tao Y, Xi S, Briones V, Muegge K. Lsh mediated RNA polymerase II stalling at HoxC6 and HoxC8 involves DNA methylation. PLoS One. 2010;5:e9163
31. Shi Y, Tao Y, Jiang Y, Xu Y, Yan B, Chen X. et al. Nuclear epidermal growth factor receptor interacts with transcriptional intermediary factor 2 to activate cyclin D1 gene expression triggered by the oncoprotein latent membrane protein 1. Carcinogenesis. 2012;33:1468-78
32. Xu Y, Shi Y, Yuan Q, Liu X, Yan B, Chen L. et al. Epstein-Barr Virus encoded LMP1 regulates cyclin D1 promoter activity by nuclear EGFR and STAT3 in CNE1 cells. Journal of experimental & clinical cancer research: CR. 2013;32:90
33. Yang H, Liu Y, Bai F, Zhang JY, Ma SH, Liu J. et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene. 2013;32:663-9
34. Poh WJ, Wee CP, Gao Z. DNA Methyltransferase Activity Assays: Advances and Challenges. Theranostics. 2016;6:369-91
35. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930-5
36. Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A. et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289-301
37. Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M. et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41:838-42
38. Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS. et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839-43
39. Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O. et al. TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and Is a Recurrent Event during Human Lymphomagenesis. Cancer Cell. 2011;20:25-38
40. Lian CG, Xu Y, Ceol C, Wu F, Larson A, Dresser K. et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell. 2012;150:1135-46
41. Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C. et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer cell. 2011;20:11-24
42. Bertoli G, Cava C, Castiglioni I. MicroRNAs: New Biomarkers for Diagnosis, Prognosis, Therapy Prediction and Therapeutic Tools for Breast Cancer. Theranostics. 2015;5:1122-43
43. James AM, Baker MB, Bao G, Searles CD. MicroRNA Detection Using a Double Molecular Beacon Approach: Distinguishing Between miRNA and Pre-miRNA. Theranostics. 2017;7:634-46
44. Wang J, Ye H, Zhang D, Cheng K, Hu Y, Yu X. et al. Cancer-derived Circulating MicroRNAs Promote Tumor Angiogenesis by Entering Dendritic Cells to Degrade Highly Complementary MicroRNAs. Theranostics. 2017;7:1407-21
45. Tu J, Ng SH, Luk AC, Liao J, Jiang X, Feng B. et al. MicroRNA-29b/Tet1 regulatory axis epigenetically modulates mesendoderm differentiation in mouse embryonic stem cells. Nucleic Acids Res. 2015;43:7805-22
46. Song SJ, Ito K, Ala U, Kats L, Webster K, Sun SM. et al. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell. 2013;13:87-101
47. Fu X, Jin L, Wang X, Luo A, Hu J, Zheng X. et al. MicroRNA-26a targets ten eleven translocation enzymes and is regulated during pancreatic cell differentiation. Proc Natl Acad Sci U S A. 2013;110:17892-7
48. Haffner MC, Chaux A, Meeker AK, Esopi DM, Gerber J, Pellakuru LG. et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget. 2011;2:627-37
49. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH. et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer cell. 2011;19:17-30
50. Kraus TF, Globisch D, Wagner M, Eigenbrod S, Widmann D, Munzel M. et al. Low values of 5-hydroxymethylcytosine (5hmC), the "sixth base," are associated with anaplasia in human brain tumors. Int J Cancer. 2012;131:1577-90
51. Zhang Y, Wu K, Shao Y, Sui F, Yang Q, Shi B. et al. Decreased 5-Hydroxymethylcytosine (5-hmC) predicts poor prognosis in early-stage laryngeal squamous cell carcinoma. Am J Cancer Res. 2016;6:1089-98
52. Gao F, Xia Y, Wang J, Lin Z, Ou Y, Liu X. et al. Integrated analyses of DNA methylation and hydroxymethylation reveal tumor suppressive roles of ECM1, ATF5, and EOMES in human hepatocellular carcinoma. Genome Biol. 2014;15:533
53. Larson AR, Dresser KA, Zhan Q, Lezcano C, Woda BA, Yosufi B. et al. Loss of 5-hydroxymethylcytosine correlates with increasing morphologic dysplasia in melanocytic tumors. Mod Pathol. 2014;27:936-44
54. Ryu B, Kim DS, Deluca AM, Alani RM. Comprehensive expression profiling of tumor cell lines identifies molecular signatures of melanoma progression. PLoS One. 2007;2:e594
55. Waseem A, Ali M, Odell EW, Fortune F, Teh MT. Downstream targets of FOXM1: CEP55 and HELLS are cancer progression markers of head and neck squamous cell carcinoma. Oral Oncol. 2010;46:536-42
56. Yu W, Briones V, Lister R, McIntosh C, Han Y, Lee EY. et al. CG hypomethylation in Lsh-/- mouse embryonic fibroblasts is associated with de novo H3K4me1 formation and altered cellular plasticity. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:5890-5
57. Ren J, Briones V, Barbour S, Yu W, Han Y, Terashima M. et al. The ATP binding site of the chromatin remodeling homolog Lsh is required for nucleosome density and de novo DNA methylation at repeat sequences. Nucleic Acids Res. 2015;43:1444-55
58. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A. et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer cell. 2010;18:553-67
59. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP. et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer cell. 2010;17:510-22
60. Huang J, Fan T, Yan Q, Zhu H, Fox S, Issaq HJ. et al. Lsh, an epigenetic guardian of repetitive elements. Nucleic Acids Res. 2004;32:5019-28
61. Yan Q, Cho E, Lockett S, Muegge K. Association of Lsh, a regulator of DNA methylation, with pericentromeric heterochromatin is dependent on intact heterochromatin. Mol Cell Biol. 2003;23:8416-28
62. Murata A, Baba Y, Ishimoto T, Miyake K, Kosumi K, Harada K. et al. TET family proteins and 5-hydroxymethylcytosine in esophageal squamous cell carcinoma. Oncotarget. 2015;6:23372-82
63. Lai W, Li H, Liu S, Tao Y. Connecting chromatin modifying factors to DNA damage response. International journal of molecular sciences. 2013;14:2355-69
Author contact
![]() Corresponding author: Email: taoyongedu.cn; Tel. +(86) 731-84805448; Fax. +(86) 731-84470589.
Corresponding author: Email: taoyongedu.cn; Tel. +(86) 731-84805448; Fax. +(86) 731-84470589.
Citation styles
APA
Jia, J., Shi, Y., Chen, L., Lai, W., Yan, B., Jiang, Y., Xiao, D., Xi, S., Cao, Y., Liu, S., Cheng, Y., Tao, Y. (2017). Decrease in Lymphoid Specific Helicase and 5-hydroxymethylcytosine Is Associated with Metastasis and Genome Instability. Theranostics, 7(16), 3920-3932. https://doi.org/10.7150/thno.21389.
ACS
Jia, J.; Shi, Y.; Chen, L.; Lai, W.; Yan, B.; Jiang, Y.; Xiao, D.; Xi, S.; Cao, Y.; Liu, S.; Cheng, Y.; Tao, Y. Decrease in Lymphoid Specific Helicase and 5-hydroxymethylcytosine Is Associated with Metastasis and Genome Instability. Theranostics 2017, 7 (16), 3920-3932. DOI: 10.7150/thno.21389.
NLM
Jia J, Shi Y, Chen L, Lai W, Yan B, Jiang Y, Xiao D, Xi S, Cao Y, Liu S, Cheng Y, Tao Y. Decrease in Lymphoid Specific Helicase and 5-hydroxymethylcytosine Is Associated with Metastasis and Genome Instability. Theranostics 2017; 7(16):3920-3932. doi:10.7150/thno.21389. https://www.thno.org/v07p3920.htm
CSE
Jia J, Shi Y, Chen L, Lai W, Yan B, Jiang Y, Xiao D, Xi S, Cao Y, Liu S, Cheng Y, Tao Y. 2017. Decrease in Lymphoid Specific Helicase and 5-hydroxymethylcytosine Is Associated with Metastasis and Genome Instability. Theranostics. 7(16):3920-3932.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.