Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Experimental Section
Results and Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Experimental Section
Results and Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(20):5529-5547. doi:10.7150/thno.29039 This issue Cite
Research Paper
Targeting Mitochondrial Dysfunction and Oxidative Stress in Activated Microglia using Dendrimer-Based Therapeutics
Anjali Sharma1*, Kevin Liaw1,2*, Rishi Sharma1, Zhi Zhang3, Sujatha Kannan3,4,5, Rangaramanujam M. Kannan1,2,4,5 ![]()
1. Center for Nanomedicine, Department of Ophthalmology, Wilmer Eye Institute Johns Hopkins University School of Medicine, Baltimore, MD 21231, USA;
2. Department of Chemical and Biomolecular Engineering, Johns Hopkins University, Baltimore MD, 21218, USA.
3. Department of Anesthesiology and Critical Care Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA.
4. Hugo W. Moser Research Institute at Kennedy Krieger, Inc., Baltimore MD, 21205, USA.
5. Kennedy Krieger Institute - Johns Hopkins University for Cerebral Palsy Research Excellence, Baltimore, MD 21218, USA.
*These authors contributed equally
Received 2018-8-6; Accepted 2018-10-10; Published 2018-11-5
Citation:
Sharma A, Liaw K, Sharma R, Zhang Z, Kannan S, Kannan RM. Targeting Mitochondrial Dysfunction and Oxidative Stress in Activated Microglia using Dendrimer-Based Therapeutics. Theranostics 2018; 8(20):5529-5547. doi:10.7150/thno.29039. https://www.thno.org/v08p5529.htm
Other stylesAbstract

Mitochondrial oxidative stress is associated with many neurodegenerative diseases, such as traumatic brain injury (TBI). Targeted delivery of antioxidants to mitochondria has failed to translate into clinical success due to their nonspecific cellular localization, poor transport properties across multiple biological barriers, and associated side effects. These challenges, coupled with the complex function of the mitochondria, create the need for innovative delivery strategies.
Methods: Neutral hydroxyl-terminated polyamidoamine (PAMAM) dendrimers have shown significant potential as nanocarriers in multiple brain injury models. N-acetyl cysteine (NAC) is a clinically used antioxidant and anti-inflammatory agent which has shown significant potency when delivered in a targeted manner. Here we present a mitochondrial targeting hydroxyl PAMAM dendrimer-drug construct (TPP-D-NAC) with triphenyl-phosphonium (TPP) for mitochondrial targeting and NAC for targeted delivery to mitochondria in injured glia. Co-localization and mitochondrial content of mitochondria-targeted and unmodified dendrimer were assessed in microglia and macrophages in vitro via immunohistochemistry and fluorescence quantification. Therapeutic improvements of TPP-D-NAC over dendrimer-NAC conjugate (D-NAC) and free NAC were evaluated in vitro in microglia under oxidative stress challenge. In vivo neuroinflammation targeting was confirmed in a rabbit model of TBI.
Results: TPP-conjugated dendrimer co-localized significantly more with mitochondria than unmodified dendrimer without altering overall levels of cellular internalization. This targeting capability translated to significant improvements in the attenuation of oxidative stress by TPP-D-NAC compared to D-NAC and free NAC. Upon systemic administration in a rabbit TBI model, TPP-conjugated dendrimer co-localized specifically with mitochondria in activated microglia and macrophages in the white matter of the ipsilateral/injured hemisphere, confirming its BBB penetration and glial targeting capabilities.
Conclusion: D-NAC has shown promising efficacy in many animal models of neurodegeneration, and this work provides evidence that modification for mitochondrial targeting can further enhance its therapeutic efficacy, particularly in diseases where oxidative stress-induced glial cell death plays a significant role in disease progression.
Keywords: Hydroxyl PAMAM dendrimer, mitochondria, activated microglia, sub-cellular targeting, oxidative stress, N-acetyl cysteine
Introduction
Mitochondria are primarily responsible for energy production in the form of ATP through oxidetive phosphorylation, which is essential for all active cellular processes [1]. In addition to power generation, mitochondria play a pivotal role in performing other vital physiological functions such as calcium homeostasis, intracellular trafficking, cell cycle regulation, amino acids and nitrogen metabolism, and apoptosis [2]. There is a growing body of literature that indicates that under pathological conditions, mitochondria act as a double-edged sword and cause oxidative/nitrosative stress by over-production of reactive oxygen/nitrogen species (ROS/RNS), disturb calcium metabolism, and induce programmed cell death [3, 4]. Recovering mitochondrial function from pathological to physiological is crucial to avoid the activation of the cascade of events that cause irreversible mitochondrial and cellular damage [5].
Mitochondrial dysfunction is associated with several neurological diseases such as Alzheimer's disease [6], Parkinson's disease [7], amyotrophic lateral sclerosis [8], multiple sclerosis [9], as well as acute brain insults such as cerebral ischemia [10] and traumatic brain injury (TBI) [11, 12]. TBI is a leading health and socioeconomic problem which affects over 2.5 million Americans each year with no successful therapy to prevent long-term effects [13]. The initial insult after TBI results in microglial and macrophage (mi/ma) activation and inflammation leading to mitochondrial ROS and RNS generation [14]. One potential therapeutic approach is to target mitochondrial oxidative stress. The major limitation in this regard is the failure of antioxidants to preferentially accumulate in the mitochondria of the diseased tissue/cells in the brain. Even if the drug has the adequate physicochemical properties to overcome the blood-brain barrier (BBB) to reach the diseased tissue and cells, it still must cross several membranes to reach its targeted subcellular territory inside the mitochondrion. Moreover, most drugs undergoing preclinical and clinical trials for use in TBI have only focused on reversing or preventing neuronal cell death, neglecting the detrimental contributions of activated mi/ma to the disease progression [15, 16]. Amelioration of oxidative stress at the mitochondria level in activated mi/ma is a key potential target to mitigate TBI-induced secondary insult.
N-Acetyl cysteine (NAC) is an anti-oxidant and anti-inflammatory agent currently being used clinically for multiple indications including cystic fibrosis and acetaminophen poisoning. The antioxidant capacity of NAC is two-fold. First, NAC is able to directly scavenge for reactive oxygen species via its sulfhydryl active group [17]. Second, as a glutathione precursor, NAC stimulates the production of endogenous antioxidant glutathione (GSH), which prevents the formation of mitochondrial ROS and RNS, thereby ameliorating mitochondrial dysfunction and decreasing oxidative stress [18]. However, NAC has poor bioavailability due to protein interactions in serum via its thiol groups, which necessitates multiple administrations at high doses to observe neuroprotective effects [19]. Additionally, cellular internalization of NAC occurs through the cysteine-glutamate antiporter, which results in glutamate release and subsequent excitotoxicity and neuronal damage [20]. Nanoparticle-mediated targeted delivery of NAC to mitochondria of activated mi/ma at the site of injury in the brain can be a potential strategy to reduce oxidative stress and restore mitochondrial function associated with neurological diseases.
Several nanotechnology-based approaches are currently being developed for targeted delivery of drugs to mitochondria,[21-24] but endeavors to specifically target mitochondria of the diseased cells in the brain are rare [6]. Dendrimers are hyperbranched, monodisperse macromolecules which have shown significant promise for site-specific gene and drug delivery [25-33]. Introducing heterogeneity at nanoscale level is a formidable challenge in designing drug delivery systems. The highly precise molecular structure and the multivalent surface of dendrimers provide an ideal platform to incorporate different functional moieties such as therapeutic, imaging and targeting agents on a single nanocarrier in a well-defined manner. We have previously reported in several small and large animal models that generation 4 hydroxyl-terminated PAMAM dendrimers (D-OH), both unmodified and covalently conjugated to drugs, imaging agents, and targeting ligands, have the unique ability to cross the impaired BBB upon systemic administration and selectively target the activated mi/ma and astrocytes in the injured regions of the brain while exhibiting minimal accumulation in healthy brain tissue [34-39]. We have also shown that intravenous administration of a single dose of dendrimer-NAC conjugate (D-NAC) successfully delivered NAC to activated mi/ma and produced striking neuroprotective effects in a neonatal rabbit white matter injury model [40]. Here, we envisioned that the targeted subcellular delivery of NAC to the dysfunctional mitochondria of activated glia at the site of neuroinflammation could be a viable approach to treat oxidative stress at the mitochondrial level in the brain.
Triphenyl phosphinuim (TPP) is a well-known mitochondrial targeting ligand and has shown potential to target small molecule drugs and nanoparticles to mitochondria both in vitro and in vivo [41, 42]. The negative plasma membrane potential (30-60 mV, negative inside) and highly negative mitochondrial inner membrane potential (150-180 mV, negative inside) drives the accumulation of lipophilic TPP several folds more in mitochondria compared to the cytosol [43]. Here, we present a novel microglia specific systemic mitochondrial targeting NAC delivery platform by conjugating both NAC and TPP to the surface of D-OH dendrimer. In this report we describe the synthesis and evaluation of mitochondrial-targeted dendrimer and dendrimer-NAC in human macrophages and murine microglia and in a pediatric TBI model.
Experimental Section
Materials
1-[3-(Dimethylamino)propyl]-3-ethylcarbodiimide methiodide (EDC), 4(dimethylamino)pyridine (DMAP) (99%), BOC-GABA-OH, trifluoroacetic acid (TFA), N-hydroxy succinimide (NHS), N, N-diisopropyl ethyl amine (DIPEA), and N-acetyl cysteine (NAC) were purchased from Sigma Aldrich US, and used as received. Cy5 NHS ester was purchased from GE healthcare and used as received. Ethylenediamine-core PAMAM dendrimer (Biomedical grade generation 4 consisting 64 hydroxyl end-groups) (D-OH) was received from Dendritech as a solution in methanol. As received dendrimer methanol solution was evaporated and used as it is. Dialysis membrane (MWCO 1kDa) was purchased from Spectrum Laboratories Inc. All other solvents were used as received in their anhydrous forms. All reactions in the organic medium were performed in standard oven-dried glassware under an inert nitrogen atmosphere unless otherwise stated.
Dulbecco's Modified Eagle Medium (DMEM), fetal bovine serum (FBS), penicillin-streptomycin (P/S), 0.05% trypsin-EDTA, NucBlue fixed cell stain ReadyProbes, goat anti-rabbit Alexafluor 488 secondary antibody, and MTT reagent were obtained from Invitrogen (Carlsbad, CA, USA). Lysine-coated glass-bottom culture dishes were purchased from MatTek Inc. (Ashland, MA, USA). Lectin Dylight 594 was purchased from Vector Labs (Burlingame, CA, USA). Anti-AIF antibody for mitochondria marker and tetramethylrhodamine, ethyl ester (TMRE) were purchase from Abcam (Cambridge, UK). Analytical grade methanol and 10% formalin were purchased from Sigma-Aldrich. Mitochondria isolation kits were purchased from ThermoFisher. MitoSOX mitochondria superoxide indicator was purchased from Molecular Probes (Eugene, OR, USA). Trypan blue was obtained from Corning (Manassas, VA, USA).
Synthesis and characterization of intermediates and dendrimer conjugates
Characterization
Nuclear Magnetic Resonance (NMR)
NMR spectra were recorded on a Bruker 500MHz spectrometer at ambient temperatures. The chemical shifts in ppm are reported relative to tetramethylsilane as an internal standard for 1H NMR spectra. Residual protic solvent of CDCl3 (1H, δ 7.27 ppm; 13C, δ 77.0 ppm (central resonance of the triplet)), and DMSO-d6 (1H, δ2.50 ppm) were used for chemical shifts calibration.
Mass spectroscopy
Matrix assisted laser desorption ionization time of flight (MALDI-TOF) experiments were performed on Bruker Autoflex MALDI-TOF instrument. The conjugate was dissolved in ultra-purified water at 5 mg/mL and 2, 5-dihydroxybenzoic acid (DHB) matrix was dissolved in 50:50 (v/v) acetonitrile:water mixture at 10 mg/mL concentration. The samples were prepared by mixing 10 µL of conjugate solution with 10 µL of DHB solution after which 3 µL of the sample was spotted on a MALDI plate. Laser power used for this purpose was 55-100%.
High Performance Liquid Chromatography (HPLC)
HPLC was utilized to analyse the purity of intermediates and final dendrimer conjugates. HPLC (Waters Corporation, Milford, MA) is equipped with a 1525 binary pump, an In-Line degasser AF and a 717 plus autosampler. It includes a 2998 photodiode array detector and a 2475 multi λ fluorescence detector interfaced with Waters Empower software. The specifications of the column used for HPLC analysis are symmetry 300 C18 column, 5µm, 4.6x250mm. The HPLC chromatograms of dendrimer-related intermediates and final conjugates were monitored at 210 nm. The fluorescently labelled dendrimer was monitored at both 650 and 210 nm using PDA and fluorescence detectors. A gradient flow was used in HPLC starting with 100:0 (Solvent A: 0.1% TFA in water; Solvent B: 0.1% TFA in ACN); gradually increasing to 10:90 (A:B) at 20 min, finally returning to 100:0 at 25 minutes maintaining a flow rate of 1 mL/min.
Dynamic light scattering (DLS) and Zeta potential (ζ)
The size distribution and ζ-potential distribution of dendrimer conjugates were determined by using a Zetasizer Nano ZS (Malvern Instrument Ltd. Worchester, U.K.) equipped with a 50mW He-Ne laser (633 nm). For size measurement, the conjugate was dissolved in deionized water (18.2 Ω) to make a solution with a final concentration of 0.5mg/mL. The sample solution was vortexed for 1 minute and then sonicated for 3 minutes followed by filtration through 0.2µm syringe filters (Pall Corporation, 0.2µm HT Tuffryn membrane) directly into the cell (UV transparent disposable cuvette, Dimensions: 12.5 x 12.5 x 45mm, SARSTEDT). Size measurements were performed in triplicate. For zeta potential measurement, the sample was prepared at a concentration of 0.2mg/mL in 10mM NaCl. The pH of both the solutions (D-OH and D-TPP; 0.2mg/mL in 10mM NaCl) was 5.5. The solution was filtered through 0.2µm syringe filters directly into the cell (Malvern Zetasizer Nanoseries disposable folded capillary cells) and three measurements were taken.
Synthesis
NAC-NHS linker (8) was synthesized using recently published literature procedure by our group [44]. D-Cy5 [40] (6) and D-NAC [44] (10) were synthesized with minor modifications of our published protocols.
General procedure A (for synthesis of BOC-protected bifunctional dendrimer)
BOC-GABA-OH and DMAP were added to a stirring solution of D-OH (compound 1) in anhydrous DMF. The solution was stirred at RT for 5 minutes to make a clear solution. EDC·HCl was added in portions to the reaction mixture. The reaction mixture was stirred for 48 h at room temperature. The reaction mixture was transferred to 1kDa MW cut-off cellulose dialysis membrane and was dialysed against water for 24 h, periodically changing water 3-4 times. The contents of dialysis tubing were transferred to pre-weighed 50 mL falcon tubes and lyophilized to get the desired product as white solid.
General procedure B (for BOC deprotection)
The BOC-protected dendrimer (2) was placed in an oven-dried round bottom flask and DCM was added under nitrogen atmosphere. The solution was sonicated for 15 minutes to make a cloudy suspension followed by the addition of TFA. The ratio of DCM to TFA is 4:1. The solution turned clear with the addition of TFA. The reaction mixture was stirred vigorously for 12 h at room temperature. The color of the reaction changed from colorless to light brown. Once completed, DCM was evaporated on a rotary evaporator. The reaction mixture was diluted with methanol and evaporated using a rotary evaporator. This procedure was repeated until excess of TFA was completely removed. The reaction mixture was left at high vacuum for 3 hours to remove any trace of solvents to afford bifunctional dendrimer as an off-white fluffy hygroscopic material, which was directly used for the next step without any further purification.
Synthesis of compound 2a
D-OH (1.1g, 0.077 mmol), EDC (265.7 mg, 1.386 mmol), DMAP (112.8 mg, 0.924) and GABA-BOC-OH (187.5, 0.924 mmol) were reacted in DMF (10 mL) using general procedure A to obtain compound 2a as white solid with an 80% yield.
1H NMR: (500 MHz, DMSO) δ 8.09 -7.77 (m, internal amide H, 124 H), 6.82 (s, GABA amide H), 4.70 (s, surface OH), 4.01 (t, ester linked -CH2, 15H), 3.39-3.32 (m, dendrimer -CH2), 3.27 (d, J = 5.3 Hz, dendrimer -CH2), 3.19 - 3.02 (m, dendrimer-CH2), 2.92 (d, J = 6.2 Hz, dendrimer-CH2), 2.70 - 2.55 (m, dendrimer-CH2 ), 2.45 - 2.39 (m, dendrimer-CH2), 2.23 - 2.12 (m, dendrimer-CH2), 1.67 - 1.56 (m, GABA linker-CH2, 16H), 1.37 (s, BOC H, 78H).
Synthesis of compound 3a
Compound 2a was reacted with a mixture of DCM (12 mL) and TFA (3 mL) using general procedure B to afford compound 3a as off-white product in quantitative yield.
1H NMR (500 MHz, DMSO) δ 8.15-7.82 (m, 124H), 4.72 (m, surface-OH), 4.10 (m, ester linked H), 3.40 - 3.12 (m, dendrimer -CH2), 2.66 (m, dendrimer -CH2), 2.44 (m, dendrimer -CH2), 2.21 (m, dendrimer -CH2), 1.80 (m, GABA linker-CH2).
Synthesis of compound 4
To a stirring solution of compound 3a (186 mg, 0.012 mmol) in DMF (5 mL), we added DIPEA (0.2 mL) followed by Cy5 NHS ester (10.2 mg, 0.015 mmol) dissolved in DMF (1 mL). The reaction mixture was stirred for 12 h. Upon completion, the contents of the reaction flask were transferred to 1kDa dialysis membrane and were dialyzed against DMF for 12 h followed by water dialysis for another 12 h to obtain the product as blue solid with a 78% yield.
1H NMR: (500 MHz, DMSO) δ 8.36 (m, Cy5 Ar H), 8.18 (m, Cy5 Ar H), (8.07-7.78 (m, internal amide H),7.33 (Cy5 H), 6.58 (Cy5 H), 6.30 (Cy5 H), 4.71 (m, surface-OH), 4.02 (m, ester linked H), 3.40 - 3.12 (m, dendrimer -CH2), 2.66 (m, dendrimer -CH2), 2.44 (m, dendrimer -CH2), 2.21 (m, dendrimer -CH2), 1.80 (m, GABA linker-CH2), 1.26 (Cy5 H) and 1.13 (Cy5 H).
Synthesis of compound 5
To a stirring solution of carboxy propyl triphenyl phosphinium bromide (43.62 mg, 0.102 mmol) in DMSO (5 mL), we added NHS (20 mg, 0.173 mmol) and EDC (32 mg, 0.167 mmol) to activate the acid. The stirring continued at RT for three hours followed by the addition of compound 4 (175 mg, 0.011 mmol) dissolved in DMSO (3 mL). Stirring continued for an additional 12 h. The contents of the reaction flask were then transferred to 1kDa dialysis membrane and were dialyzed against DMF for 12 h followed by water dialysis for another 12 h to obtain the product as blue solid with an 84% yield.
1H NMR: (500 MHz, DMSO) δ 8.36 (m, Cy5 Ar H), 8.18 (m, Cy5 Ar H), (7.98-7.38 (m, internal amide H and TPP Ar H),7.34 (Cy5 H), 6.59 (Cy5 H), 6.33 (Cy5 H), 4.72 (m, surface-OH), 3.99 (m, ester linked H), 3.54 (t, TPP inker -CH2), 3.50 - 3.12 (m, dendrimer -CH2), 2.71 (m, dendrimer -CH2), 2.25 (m, dendrimer -CH2), 1.71 (m, GABA linker-CH2 and Cy5 H) and 0.13 (Cy5 H).
Synthesis of compound 2b
D-OH (1.0 g, 0.070 mmol), EDC (67.23 mg, 0.350 mmols), DMAP (25.63 mg, 0.21 mmol) and GABA-BOC-OH (42.63 mg, 0.21 mmol) were reacted in DMF (10 mL) using general procedure A to obtain compound 2b as white solid with an 82% yield.
1H NMR: (500 MHz, DMSO) δ 8.09 -7.77 (m, internal amide H), 6.83 (s, GABA amide H), 4.71 (s, surface OH), 4.00 (t, ester linked -CH2, 15H), 3.39-3.32 (m, dendrimer -CH2), 3.11 - 3.02 (m, dendrimer-CH2), 2.70 - 2.55 (m, dendrimer-CH2 ), 2.45 - 2.39 (m, dendrimer-CH2), 2.23 - 2.12 (m, dendrimer-CH2), 1.67 - 1.56 (m, GABA linker-CH2), 1.37 (s, BOC H).
Synthesis of compound 3b
Compound 2b was reacted with a mixture of DCM (12 mL):TFA (3 mL) using general procedure B to afford compound 3b as an off-white product with a quantitative yield.
1H NMR (500 MHz, DMSO) δ 8.14-7.79 (m, dendrimer amide H), 4.72 (m, surface-OH), 4.03 (m, ester linked H), 3.40 - 3.12 (m, dendrimer -CH2), 2.66 (m, dendrimer -CH2), 2.44 (m, dendrimer -CH2), 2.21 (m, dendrimer -CH2), 1.79 (m, GABA linker-CH2).
Synthesis of compound 6
To a stirring solution of compound 3b (150 mg, 0.010 mmol) in DMF (5 mL) we added DIPEA (0.2 mL) followed by Cy5 NHS ester (8.5 mg, 0.012 mmol) dissolved in DMF (1 mL). The reaction mixture was stirred for 12 h. Upon completion, the contents of the reaction flask were transferred to 1 kDa dialysis membrane and were dialyzed against DMF for 12 h followed by water dialysis for another 12 h to obtain the product as blue solid with a 76% yield.
1H NMR: (500 MHz, DMSO) δ 8.37 (m, Cy5 Ar H), 8.06 (m, Cy5 Ar H), (8.07-7.78 (m, internal amide H),7.33 (Cy5 H), 6.60 (Cy5 H), 6.32 (Cy5 H), 4.72 (m, surface-OH), 4.12 (m, -CH2), 4.00 (m, ester linked H), 3.40 - 3.12 (m, dendrimer -CH2), 2.66 (m, dendrimer -CH2), 2.44 (m, dendrimer -CH2), 2.21 (m, dendrimer -CH2), 1.80 (m, GABA linker-CH2), 1.30-1.15 (Cy5 H).
Synthesis of compound 2c
D-OH (1g, 0.070 mmol), EDC (336 mg, 1.75 mmol), DMAP (188 mg, 1.54 mmol) and GABA-BOC-OH (312.62 mg, 1.54 mmol) were reacted in DMF using general procedure A to obtain compound 2c as white solid with an 80% yield.
1H NMR: (500 MHz, DMSO) δ 8.12 -7.76 (m, internal amide H), 6.81 (s, GABA amide H), 4.69 (s, surface OH), 4.01 (t, ester linked -CH2, 15H), 3.46-3.17 (m, dendrimer -CH2), 2.91 (m, dendrimer -CH2), 2.66 (m, dendrimer-CH2), 2.45 - 2.39 (m, dendrimer -CH2), 2.29-2.20 (m, dendrimer -CH2), 1.62 (m, GABA linker-CH2), 1.36 (s, BOC H).
Synthesis of compound 3c
Compound 2c was reacted with a mixture of DCM (12 mL)/TFA (3 mL) using general procedure B to afford compound 3c as an off-white product with a quantitative yield.
1H NMR (500 MHz, DMSO) δ 8.56-7.75 (m, dendrimer amide H), 4.04 (m, ester linked H), 3.40 - 3.15 (m, dendrimer -CH2), 2.84 ((m, dendrimer -CH2), 2.63 (m, dendrimer -CH2), 2.42 (m, dendrimer -CH2), 2.21 (m, dendrimer -CH2), 1.81 (m, GABA linker -CH2).
Synthesis of compound 7
To a stirring solution of carboxy propyl triphenyl phosphinium bromide (330 mg, 0.769 mmol) in DMSO (5mL) we added NHS (166 mg, 1.44 mmol) and EDC (276 mg, 1.44 mmol) to activate the acid. The stirring continued at RT for three hours followed by the addition of compound 3c (1.58 g, 0.096 mmol) dissolved in DMSO (7mL). The stirring continued for an additional 12 h. The contents of the reaction flask were then transferred to 1kDa dialysis membrane and were dialyzed against DMF for 12 h followed by water dialysis for another 12 h to obtain compound 7 as white solid with a 76% yield.
1H NMR: (500 MHz, DMSO) δ 8.22-7.70 (m, internal amide H and TPP Ar H), 4.19 (m, -CH2), 4.00 (m, ester linked H), 3.54 (t, TPP inker -CH2), 3.56 - 2.29 (m, dendrimer -CH2), 1.79-1.63 (m, linker -CH2).
Synthesis of compound 8
We added a drop-wise addition of a solution of N-acetyl cysteine (NAC, 5.74 g, 35.24 mmol, 1.1 eq) dissolved in THF (30 mL) to a stirring solution of N-succinimidyl 3-(2-pyridyldithio)-propionate (SPDP) (10 g, 32.04 mmol) in anhydrous tetrahydrofuran (THF, 30 mL) under inert atmosphere. The reaction mixture turned yellow within a few minutes. The reaction mixture was stirred at RT for 2 h. Upon completion, the crude product was purified using pre-packaged high performance redisep gold Rf™ 80-gram silica cartridge on CombiFlash system from Teledyne (Lincoln, NE) keeping the flow at 60 mL/minute. The pure desired product was collected in 4% MeOH in dichloromethane as a white powder with a 75.4% yield (8.8 g).
1H NMR (500 MHz, CDCl3): δ 6.67 (d, J = 6.9 Hz, 1H), 4.87 (d, J = 5.5 Hz, 1H), 3.31 (ddd, J = 48.8, 14.2, 4.7 Hz, 2H), 3.13 - 2.99 (m, 4H), 2.88 (s, 4H), 2.09 (s, 3H).13C NMR (126 MHz, CDCl3) δ 171.9, 171.2, 167.0, 52.0, 40.0, 32.8, 31.1, 25.5, 22.9.
Synthesis of compound 9
A round bottom flask was charged with compound 7 (600 mg, 0.028 mmol) dissolved in anhydrous DMF (7 mL) with constant stirring under inert atmosphere. The pH of the reaction mixture was adjusted to 7.0 - 7.5 by addition of DIPEA. The reaction mixture was stirred for 30 minutes and once the pH was stable the slow addition of compound 8 (276 mg, 0.758 mmol) dissolved in DMF (5 mL) was performed. The stirring continued at room temperature for 12 h. The reaction mixture was transferred to 1kDa cut-off dialysis bag and dialyzed against DMF for 6 h followed by water for 24 hours, periodically changing solvent every 2-3 hours. The contents of dialysis tubing were transferred to pre-weighed 50 mL falcon tubes and were lyophilized to get compound 9 as white, fluffy solid with 82% yield.
1H NMR (500 MHz, DMSO) δ 8.25 - 7.76 ((m, amide H and TPP Ar H), 7.36-7.26 (amide H) 4.4511qq (s, NAC-CH-), 3.99 (s, ester linked -CH2), 3.53 - 2.20 (m, dendrimer -CH2), 1.85 (s, N-Acetyl -CH3), 1.79-1.63 (m, linker-CH2).
Synthesis of compound 2d
D-OH (1g, 0.07 mmol), EDC (268 mg, 1.4 mmol), DMAP (128.1 mg, 1.05 mmol) and GABA-BOC-OH (213 mg, 1.05 mmol) were reacted in DMF (10 mL) using general procedure A to obtain compound 2d as a white solid with an 84% yield.
1H NMR: (500 MHz, DMSO) δ 8.09 -7.77 (m, internal amide H), 6.82 (s, GABA amide H), 4.70 (s, surface OH), 4.01 (t, ester linked -CH2), 3.39-3.32 (m, dendrimer -CH2), 3.27 (d, dendrimer -CH2), 3.19 - 3.02 (m, dendrimer -CH2), 2.92 (d, dendrimer -CH2), 2.70 - 2.55 (m, dendrimer -CH2), 2.45 - 2.39 (m, dendrimer -CH2), 2.23 - 2.12 (m, dendrimer-CH2), 1.67 - 1.56 (m, GABA linker-CH2, 16H), 1.37 (s, BOC H).
Synthesis of compound 3d
Compound 2d was reacted with a mixture of DCM (12 mL)/TFA (3 mL) using general procedure B to afford compound 3d as an off-white product with a quantitative yield.
1H NMR (500 MHz, DMSO) δ 8.15-7.82 (m, internal amide H), 4.72 (m, surface-OH), 4.10 (m, ester -CH2), 3.40 - 3.12 (m, dendrimer -CH2), 2.66 (m, dendrimer -CH2), 2.44 (m, dendrimer -CH2), 2.21 (m, dendrimer -CH2), and 1.80 (m, GABA linker -CH2).
Synthesis of compound 10
A round bottom flask was charged with compound 3d (400 mg, 0.026 mmol) dissolved in anhydrous DMF (8 mL) with constant stirring under inert atmosphere. The pH of the reaction mixture was adjusted to 7.0 -7.5 by addition of DIPEA. The reaction mixture was stirred for 30 minutes and once the pH was stable the slow addition of compound 8 (142 mg, 0.39 mmol) dissolved in DMF (5 mL) was performed. The stirring continued at room temperature for 12 h. The reaction mixture was then transferred to 1kDa cut-off dialysis bag and dialyzed against DMF for 6 hours followed by water for 24 hours, periodically changing solvent every 2-3 hours. The contents of dialysis tubing were transferred to pre-weighed 50 mL falcon tubes and were lyophilized to get compound 10 as a white, fluffy solid with an 86% yield.
1H NMR (500 MHz, DMSO) δ 8.20 - 7.75 (m, amide H), 4.46 (s, NAC -CH), 4.01 (s, ester linked H), 3.43 - 3.34 (m, , dendrimer -CH2), 3.18 - 3.03 (m, , dendrimer -CH2), 2.94 - 2.82 (m, dendrimer -CH2), 2.75 - 2.65 (m, , dendrimer -CH2), 2.35 - 2.13 (m, , dendrimer -CH2), 1.85 (s, N-Acetyl H), 1.66 (s, GABA linker -CH2).
In vitro mitochondria colocalization, cellular uptake, and anti-oxidant evaluation
Cell culture
HMC3 human macrophages were acquired from ATCC (Manassas, VA). BV2 murine microglia cell line was obtained from Children's Hospital of Michigan's cell culture facility. Both cell lines were cultured in DMEM supplemented with 10% heat inactivated FBS and 1% P/S (full serum media) in a sterile incubator at 37 °C and 5% CO2. Cells were collected for passing or seeding by incubating for 2 minutes in 0.05% trypsin-EDTA. Cell lines were maintained by passaging every 2 days, and cells were seeded on well plates for experiments when 80-90% confluent.
Confocal microscopy and mitochondrial colocalization
HMC3 human macrophages were seeded in glass-bottom culture dishes in full serum media. HMC3 cells were used due to their large cell bodies for optimal visualization of mitochondria targeting. Cells were treated with Cy5 fluorescently labelled dendrimer (D-Cy5) and TPP-conjugated Cy5-labelled dendrimer (TPP-D-Cy5) for 48 hours in DMEM with 5% heat inactivated FBS and 1% P/S (half-serum media). The cells were then rinsed three times in sterile PBS and fixed in 5% formalin solution. Cells were then stained with rabbit anti-mouse AIF primary antibody (1:200) and goat anti-rabbit Alexafluor 488 (1:200) to label mitochondria, Lectin Dylight 594 (1:1000) to label microglia/macrophage membranes, and NucBlue to label cell nuclei. Cells were imaged using an LSM710 confocal microscope (Zeiss) under a 63x oil objective. Image capture settings, such as laser power, pinhole, gain, and averaging, were kept constant across samples.
Image processing was performed in Zen 2011 software (Zeiss). For colocalization analyses, untreated cells were used to set background fluorescence thresholds to calculate colocalization coefficients between anti-AIF mitochondria marker and Cy5 dendrimer signal, signifying the percentage of overlapped pixels between dendrimer and mitochondria out of total mitochondrial pixels. Representative fluorescence line profiles between mitochondria and dendrimer signals were obtained in the Zen 2011 software.
Mitochondrial isolation and dendrimer quantification
Dendrimer quantification from cell extracts were performed as previously described with modifications for mitochondrial isolation [45]. For quantification of internalized dendrimer and cell fractionation, BV2 murine microglia were seeded on 60mm culture dishes in full serum media. BV2 cells were used for quantification rather than HMC3 macrophages because BV2 cells yielded greater isolated mitochondria pellets. Once ~80% confluent, cells were treated with 50 µg/ml D-Cy5 or TPP-D-Cy5 in serum-free media for 24 hours. Cells were collected by trypsinization and centrifugation. Cells were freshly fractionated to isolate mitochondria fractions to quantify dendrimer content targeted to the mitochondria using the mitochondria isolation from cultured cells kit (Thermofisher). Isolated mitochondria pellets were resuspended in isolation buffer, and dendrimer were extracted via three cycles freeze/ thaw in liquid nitrogen. Dendrimer fluorescence was measured using a microcuvette (Starna Cell, CA, USA) on a Shimadzu RF-3501PC spectrofluorophotometer (Shimadzu Corporation, Columbia, MD). Fluorescence calibration curves were constructed in mitochondrial isolation buffer from known concentrations of each type of dendrimer. Untreated cells were used to subtract fluorescence background signal, and fluorescence intensity readings were converted to a mass basis. Both the mitochondrial and non-mitochondrial fractions were measured, and total cellular dendrimer content was calculated as the sum of these two fractions. Mitochondrial partitioning was calculated by dividing the dendrimer content in mitochondria by the total cellular content.
Mitochondrial superoxide and transmembrane potential detection
In acute oxidative stress injury, BV2 microglia were seeded on 96-well plates at a density of 10,000 cells per well in full serum media. Once near confluence, cells were stimulated with 50µM hydrogen peroxide (H2O2) in half-serum medium for 2 hours followed by treatment with dendrimer-conjugated NAC (D-NAC) and TPP-conjugated D-NAC (TPP-D-NAC) in half-serum medium at 0.5, 1.0, and 10 µg/ml on a NAC drug basis. 1% DMSO in medium was used to solubilize the treatment compounds. Mitochondrial superoxide formation was assessed using Molecular Probes MitoSOX superoxide indicator and read on a Synergy MX plate reader (Biotek, VT, USA). To test transmembrane potential, BV2 cells were likewise seeded in 96-well plates and stimulated with 50µM H2O2. Cells were then treated in half-serum medium with free NAC, D-NAC, or TPP-D-NAC for 6 hours. Mitochondrial transmembrane potentials were assessed with tetramethylrhodamine, ethyl ester (TMRE), which localizes to the mitochondrial membrane in a membrane polarization dependent manner. Cells were treated with 500nM TMRE for 30 minutes following drug treatment and analyzed on the plate reader.
In chronic oxidative stress injury, BV2 cells were seeded in 96-well plates at a density of 10,000 cells per well in full-serum medium. Cells were stimulated with 50µM H2O2 for 2 hours, followed by cotreatment of free NAC, D-NAC, and TPP-D-NAC with 5µM H2O2 for 24 hours. Mitochondria superoxide was measured on a plate reader after treatment with MitoSOX superoxide indicator.
Cell viability for cytotoxicity and oxidative stress induced cell death
Cell viability was assessed via MTT assay in healthy BV2 murine microglia to confirm the cytotoxicity profile of TPP and TPP-conjugated dendrimer (D-TPP). Cells were seeded in 96-well plates in full-serum medium and treated for 24 hours in half-serum medium. Cells were treated with 1, 10, 100, and 1000 µg/ml D-TPP and 1, 10, and 100 µg/ml free TPP. 1% DMSO was used to solubilize the treatment compounds, and control cells were treated with equivalent DMSO content. For the MTT assay, 100 µL fresh media was applied to wells along with 10 µL of 12mM MTT solution for 4 hours following treatment. The resulting converted formazan was dissolved in 150µL DMSO and analyzed on a plate reader. Cell viability was normalized to control cells.
Oxidative stress-induced cell death was obtained from modified previously reported protocols.[46, 47] BV2 cells were pretreated for 24 hours with free NAC, D-NAC, or TPP-D-NAC. 1% DMSO was used to dissolve the treatments, and control cells were treated with equivalent DMSO content. Cells were then stimulated with H2O2 for 3 hours at 500 µM. For cell viability experiments via MTT assay, cells were seeded in 96-well plates for treatment and assessed with MTT as described above. For cell viability experiments via trypan blue exclusion, cells were seeded in 12-well plates. Cells were collected by trypsinization and resuspended in fresh medium. Nonviable cells were labeled by mixing cell solutions 1:1 with trypan blue solution. Living and dead cells were counted with a hemocytometer (Hausser Scientific, PA, USA) under a light microscope.
Statistical Analysis
Error bars presented in figures denote mean ± standard error of the mean (SEM). Comparisons between groups were performed with Student's t-tests. Two-way Analysis of Variance (ANOVA) tests were performed to compare treatment groups across multiple time points or doses. For colocalization analysis of confocal images, 4-5 images containing 2-4 cells each were averaged and analyzed for statistics. For quantification studies and efficacy studies, n = 3 independent trials were run with n = 2 internal replicates each. Statistical analyses were performed in IBM SPSS v21 software (IBM Corporation, NY, USA). Graphs were formulated with Graphpad Prism v5.0 (Graphpad Prism, CA, USA).
In vivo evaluation in rabbit model of TBI
Animals
New Zealand white rabbits were purchased from Robinson Services Inc. (Mocksville, NC) and arrived at the facility one week before in-house breeding. All of the kits were delivered naturally and remained with their mother. All animals were housed under ambient conditions (22°C, 50% relative humidity, and a 12-h light/dark cycle), and necessary precautions were taken throughout the study to minimize pain and stress. Experimental procedures were approved by the Johns Hopkins University Animal Care and Use Committee (ACUC).
TBI surgical procedures
TBI surgery was performed as previously described [48]. In brief, rabbits were anesthetized on postnatal day 5 (PND5) with dexmedetomidine hydrochloride (30 μg/kg, subcutaneously) and anesthesia was maintained with 2% inhaled isoflurane. A computer-operated thermal blanket pad and a rectal thermometer allowed maintenance of body temperature within normal limits (∼37°C). Respirations were monitored, and no apnea or hypoventilation was noted. An 8-mm craniotomy was made on the left hemisphere lateral to the sagittal suture and centered between bregma and lambda. The skull at the craniotomy site was removed without disrupting the underlying dura. A controlled cortical impact (CCI) device was used to injure the exposed cortex (6-mm flat impactor tip, velocity of 5.5 m/sec, duration of 50-60 msec, and a depth of 2 mm). After the injury, the skull was replaced over the injury site and secured with dental cement. The skin was then sutured together, and the animals were placed in an animal incubator to recover.
TPP-D-Cy5 injection and immunohistochemistry
TPP-D-Cy5 (55mg/kg) was administered intravenously at 6 hours post-TBI injury. Rabbit kits were anesthetized and transcardially perfused with saline at 24 hours post TPP-D-Cy5 injection. The brains were removed, post-fixed in 10% formalin for 48 hours and cryoprotected in 30% sucrose. Coronal brain sections (40 μm, 1:6 series) were sectioned using a cryostat (Leica) and mounted on gelatin-coated glass slides. Sections were incubated overnight at 4°C with goat-anti-IBA1 antibody (1:500, Abcam, Cambridge, MA) and mouse anti-mitochondria [MTC02] antibody (1:250, Abcam, Cambridge, MA). Sections were subsequently washed and incubated with fluorescent secondary antibody (1:250; Thermo Scientific, Waltham, MA) for 2 h at room temperature. All sections were incubated with DAPI (Thermo Fisher Scientific, MA, USA) (1:1000) for 15 min. Images were acquired using LSM710 confocal microscope (Zeiss, Thornwood, NY, USA) and processed with Zen 2011 software (Zeiss, Thornwood, NY, USA).
Results and Discussion
Synthesis and characterization of TPP-D-Cy5 and D-Cy5
One of the key limitations of existing mitochondrial-targeting nanocarriers is that these cannot differentiate between healthy and diseased cells. In contrast, the hydroxyl PAMAM dendrimer nanoplatform presented here has the inherent ability to accumulate selectively in the activated glia at the site of inflammation in the brain upon systemic administration [20, 34, 49, 50]. Moreover, PAMAM dendrimers have outstanding cellular internalization potential which makes them excellent carriers for cellular and organelle delivery [51-53]. To incorporate both mitochondrial targeting and imaging functionalities in a single dendrimer, we developed a multifunctional dendrimer conjugate (TPP-D-Cy5 (5), Figure 1A) by the covalent attachment of mitochondrial targeting moiety TPP and near infra-red imaging dye cyanine 5 (Cy5) with amide linkages. We envisioned the attachment of only 6-7 TPP molecules (~10% of the 64 OH surface groups) on D-OH to maintain the dendrimer's transport properties and its inherent targeting potential to accumulate in activated mi/ma [37, 53]. Moreover, TPP is a highly hydrophobic moiety and attachment of more groups can compromise dendrimer's aqueous solubility. Recent studies have also shown around 5 TPP moieties as the optimum number to conjugate on cationic G4 PAMAM dendrimer to achieve significant mitochondrial targeting [42]. However, the potential translation of cationic dendrimers is hampered by their toxicity [54]. Therefore, we started the synthesis from neutral D-OH (1, Figure 1A), which exhibits a superior safety profile and mi/ma targeting compared to previously studied cationic PAMAM dendrimer [54, 55]. D-OH was reacted with BOC-GABA-OH in the presence of coupling reagents EDC and DMAP to get BOC protected bifunctional dendrimer (2a). Since all the reagents and side products were water soluble, the conjugate was purified through extensive water dialysis using 2kDa cut-off dialysis membrane. The successful attachment of GABA-BOC linkers was confirmed by 1H NMR analysis which showed the presence of protons at δ 1.37 ppm corresponding to the tert-butyl groups of BOC and methylene protons of dendrimer next to the ester group at δ 4.02 ppm (Figure 2A, bottom Red NMR). By comparing the integration of internal amide protons of dendrimer (δ 8.14-7.71 ppm) to BOC protons of linker and ester methylene protons of the dendrimer, we confirmed the attachment of approximately 8 GABA-BOC molecules on the dendrimer surface.
Figure 1
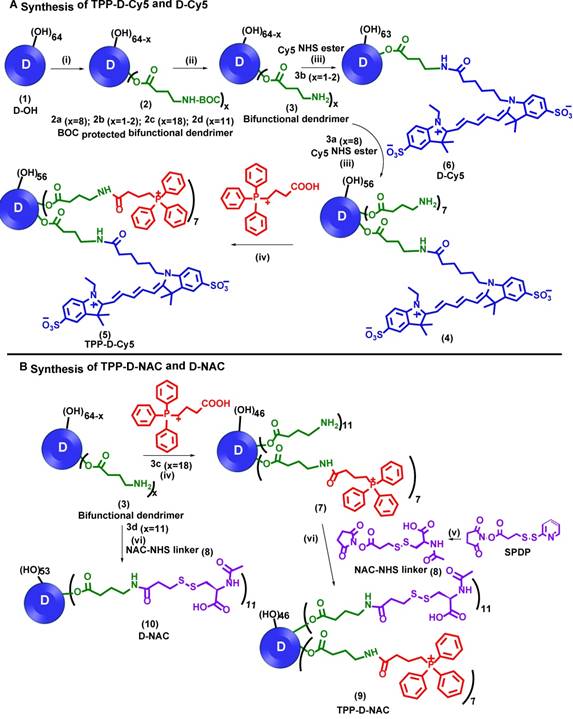
Synthesis of non-targeting and mitochondria targeting dendrimer conjugates. A Synthesis of TPP-D-Cy5 and D-Cy5; and B synthesis of TPP-D-NAC and D-NAC: Reagents and conditions: i) GABA-BOC-OH, EDC, DMAP, DMF, 48 h, RT; ii) DCM:TFA (4:1), 12 h, RT; iii) DIPEA, DMF, 12 h, RT; iv) NHS, EDC, DMSO, 12 h, RT; v) N-Acetyl-L-cysteine, anhydrous THF, 2h vi) DIPEA, DMF, 12 h, RT.
Figure 2
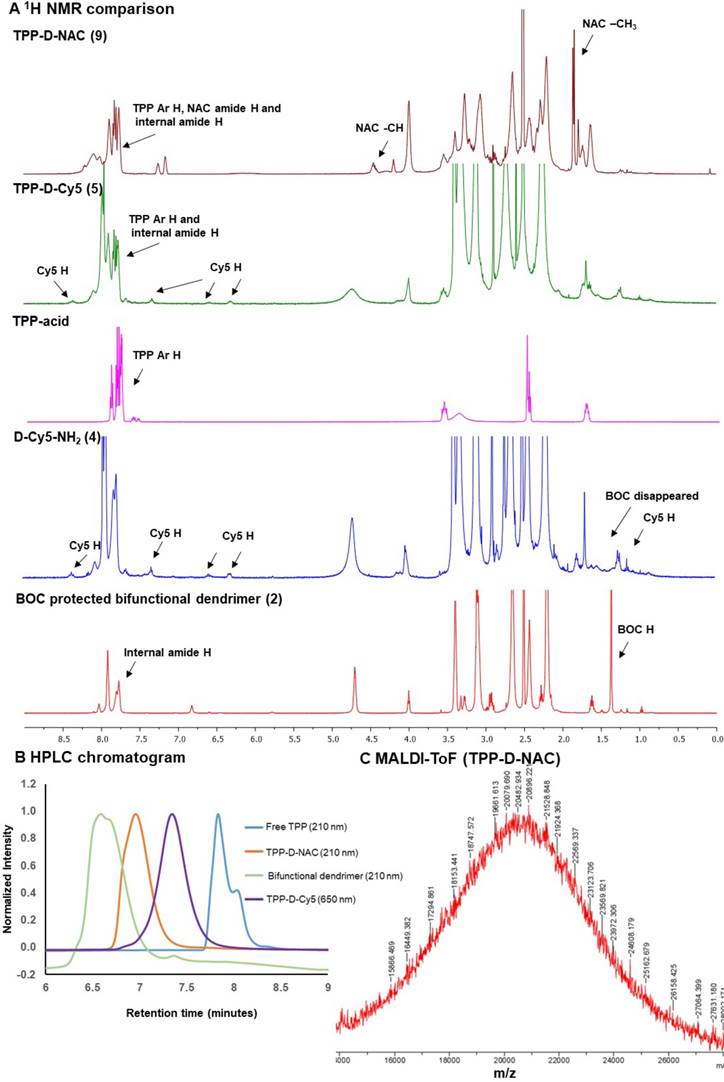
Characterization of intermediates and dendrimer conjugates. A 1H NMR comparison of intermediates and final dendrimer conjugates showing the appearance/disappearance of characteristic proton signals; B HPLC traces of intermediates and final conjugates showing shifts in the retention time (6.6 minutes-bifunctional dendrimer; 7.8 minutes-free TPP; 7.3 minutes-TPP-D-Cy5 and 7.0 minutes-TPP-D-NAC); and C MALDI-ToF spectrum of TPP-D-NAC.
During the next step, we performed the BOC deprotection under mildly acidic conditions using DCM:TFA mixture in the ratio 4:1 to get bifunctional dendrimer 3a with amine groups as a TFA salt. The 1H NMR confirmed the absence of BOC protons showing the successful deprotection. The amine groups were then available to attach both targeting and imaging agents. We used one to two amine groups to attach Cy5 and the rest were utilized for the conjugation of TPP moieties. Bifunctional dendrimer 3a was reacted with the Cy5-N-hydroxysuccinimidyl (Cy5-NHS) ester in the presence of N, N diisopropyl ethyl amine (DIPEA) to afford fluorescent dendrimer 4. The proton NMR analysis showed the presence of protons corresponding to Cy5 (Figure 2A, blue). The remaining amine groups on dendrimer 4 were then reacted with (3-carboxypropyl) triphenylphosphonium bromide via activated acid-amine coupling reaction to obtain final mitochondrial targeting fluorescent conjugate 5 (TPP-D-Cy5). The proton NMR spectra in figure 2A (magenta is Free TPP, green is TPP-D-Cy5) depicts the presence of aromatic protons of phenyl rings of TPP confirming the product formation. We further analyzed the purity of intermediates and the final product using HPLC (Figure 2B). The HPLC chromatogram showed shifts in retention time with the formation of each successive intermediate and final conjugate. The retention time of bifunctional dendrimer in the HPLC is 6.6 minutes, whereas the hydrophobic free TPP elutes later at 7.9 minutes at 210 nm. Upon conjugation of TPP and Cy5 (TPP-D-Cy5), the conjugate elutes at 7.4 minutes. After conjugation of the fluorescent tag, the dendrimer starts to show strong absorbance at 650 nm. HPLC traces further confirmed the absence of any impurities in the final product. We further analyzed the size and zeta potential distribution of D-TPP using dynamic light scattering. We synthesized D-TPP without Cy5 (Supplementary Scheme S1) for size and zeta measurements using DLS since fluorescent dyes interfere with these measurements. After the addition of TPP on dendrimer surface, its size increased from 4.4 nm to 5.2 nm (Table 1) and zeta potential changed from neutral (+4.5mV, Table 1) to cationic (+17.8 mV, Table 1). For comparison purposes we also synthesized non-mitochondrial targeting fluorescent dendrimer conjugate (D-Cy5, 6, Figure 1A) using our previously published protocol with minor modifications.[40] Briefly, we synthesized BOC-protected dendrimer 2b which was converted to bifunctional dendrimer 3b using TFA, and further reacted with Cy5-NHS ester. The final conjugate D-Cy5 (6) was characterized using NMR and HPLC (Supplementary Figures S7 and S16). The stability of Cy5 conjugated dendrimer (D-Cy5) at physiological conditions has been extensively evaluated previously by our group [40].
Table 1
Dynamic light scattering (DLS) and Zeta potential (ζ) measurements of D-OH, D-TPP and TPP-D-NAC:
| Dendrimer | Size(nm) | Zeta potential (ζ, mV) |
|---|---|---|
| D-OH | 4.4±0.2 | 4.5 ± 0.6 |
| D-TPP | 5.2±0.1 | 17.8±0.5 |
| TPP-D-NAC | 7.5±0.2 | 16.9±0.4 |
Synthesis and characterization of TPP-D-NAC and D-NAC
One of the current common strategies to develop mitochondrial targeting therapeutics is to directly conjugate potent anti-oxidant drugs to TPP to enhance their cellular or organelle delivery, for example, mitoNAC and mitoQ etc. [56, 57]. However, the poor pharmacokinetics, undesirable biodistribution profiles, and nonspecific cellular targeting associated with certain drugs cannot be further improved using this approach. Free NAC is known for non-specific binding to plasma proteins due to the presence of its thiol group and is thus given in high doses to observe a therapeutic effect [19]. The dendrimer-based platform presented here can not only improve the cellular trafficking of NAC specifically to the mitochondria of diseased cells but also enhance its stability and pharmacokinetic profile. Moreover, the dendrimer-based platform allows the delivery of high drug payloads to the desired target mitochondrial location compared to the direct one-to-one TPP conjugation to antioxidant.
To evaluate the impact of mitochondrial targeting on the efficacy of the dendrimer-NAC conjugate, we synthesized TPP-D-NAC and D-NAC (Figure 1B). The synthesis of TPP-D-NAC started with the modification of D-OH (1, Figure 1A) to get BOC-protected bifunctional dendrimer (2c) which was further converted to bifunctional dendrimer 3c by deprotecting BOC groups. The bifunctional dendrimer (3c) was synthesized to have around 18 GABA arms with amine termini; 7 of which were utilized to attach TPP and the rest were consumed by NAC attachment. The dendrimer (3c) was first reacted with carboxy propyl TPP using an activated acid-amine coupling to obtain dendrimer 7. Separately, NAC-NHS linker (8) was synthesized by reacting NAC with SPDP in tetrahydrofuran followed by the reaction with dendrimer (7) at pH 7.0-7.5 to obtain TPP-D-NAC (9). The confirmation of intermediates and the final conjugate was achieved by proton NMR and HPLC. 1H NMR of TPP-D-NAC (Figure 2A, dark red) showed the presence of aromatic TPP protons, NAC amide protons and internal amide protons of dendrimer (δ 8.26-7.68 ppm) along with NAC protons corresponding to -CH (δ 4.46 ppm) and -CH3 groups (δ 1.86 ppm). HPLC analysis (Figure 2B) showed a retention time of 7.1 minutes for TPP-D-NAC. MALDI-ToF spectrum depicted a peak around 20,482 Da (Figure 2C) and the theoretical molecular weight of TPP-D-NAC is 20,430 Da. After the addition of TPP and NAC on dendrimer surface, its size increased from 4.4 nm to 7.5 nm (Table 1) and zeta potential changed from neutral (+4.5 mV, Table 1) to cationic (+16.9 mV, Table 1). The delocalized nature of the cationic charge with TPP prevents the significant cytotoxic effects displayed by amine-terminated dendrimers despite similar zeta potential (~+18 mV). A small reduction in zeta potential for TPP-D-NAC (+16.9 mV) as compared to D-TPP (+17.8 mV) might be due to the presence of carboxylic acid groups of NAC. We further synthesized non-mitochondrial targeting D-NAC (10) conjugate for comparison purpose. D-NAC was synthesized with minor modifications using recently published procedure by our group (Figure 1B).[44] Briefly, D-OH was modified to get dendrimers 2d and 3d with 11 GABA arms attached. Bifunctional dendrimer (3d) was finally reacted with the NAC-NHS linker (8) at pH 7.0-7.5 to obtain D-NAC (10). D-NAC was characterized by NMR and HPLC (Supplementary Figures S14 and S18). The proton NMR spectra for all the intermediates and final conjugates are available in the supplementary information (Supplementary Figures S1 to S14).
TPP conjugation enhances dendrimer co-localization with mitochondria in vitro
We next looked in vitro to further explore the mitochondrial targeting capabilities of TPP-conjugated dendrimer. To assess the targeting capabilities of the TPP-conjugated dendrimer compared to unmodified dendrimer, human macrophage cell line HMC3 cells were incubated with Cy5-labeled dendrimers for 48 hours. Macrophages were chosen as the cell type of interest based on our previous in vivo work with dendrimers, where we have shown that dendrimer uptake is predominantly localized to activated microglia and macrophages in multiple models of neurodegenerative disorders, as opposed to other cells like neurons [34, 36, 37, 40]. TPP-D-Cy5 exhibits highly punctated signal corresponding to the mitochondria marker, indicating a high degree of colocalization, as denoted by the yellow color signifying the overlapping of the red dendrimer signal and green mitochondrial signal (Figure 3A). In contrast, D-Cy5 exhibits the expected diffuse cytosolic signal previously shown in both in vitro and in vivo macrophages [45, 50, 55]. D-Cy5 treated cells do exhibit some mitochondria-dendrimer signal overlap, but this overlap appears to arise from the broadness of cytosolic dendrimer signal rather than specific interactions with mitochondria. Semi-quantitative fluorescence colocalization analysis of treated cells confirms that TPP-D-Cy5 colocalizes with mitochondria to a significantly greater degree than D-Cy5 (Figure 3B). Approximately 75% of mitochondria pixels are colocalized with dendrimer signal with TPP-D-Cy5 treatment, compared to only approximately 45% exhibited by D-Cy5 treatment (p < 0.001). Similarly, fluorescence line profiles through cells illustrate the close correspondence between TPP-D-Cy5 and mitochondria signals, while D-Cy5 profiles show regions of mitochondrial signal absent dendrimer signal and vice versa (Figure 3C). Notably, the mitochondrial colocalization exhibited by D-Cy5 of approximately 45% is much greater than has been previously reported for non-targeting nanoparticles, which typically exhibit mitochondrial colocalization levels of 10-20%, and is similar to the colocalization levels exhibited by mitochondria-targeting modified versions of these nanoparticles of 40-60%.[6, 23] This observed effect may partially arise from the relatively high cytosolic content of the dendrimer compared to other nanoparticles of similar size, which broadly overlaps with mitochondrial signal nonspecifically [45, 58, 59]. This suggests that hydroxyl-terminated PAMAM dendrimers are superior vehicles for intracellular targeting, and that their inherent intracellular targeting capabilities can be significantly enhanced through modification with targeting moieties.
TPP-conjugated dendrimer partitions preferentially to mitochondria without altering overall cellular internalization
To further evaluate the mitochondrial targeting capabilities of the modified dendrimer, mitochondrial isolation and fluorescence quantification were performed. BV2 murine microglia were treated with TPP-D-Cy5 or D-Cy5 for 48 hours, followed by mitochondrial isolation. Dendrimer was extracted from mitochondria and cytosolic fractions and measured for fluorescence intensities, which were then converted to mass quantities through the use of appropriate calibration curves. Surface conjugation with 6-7 TPP molecules did not influence cellular internalization of the dendrimer (p > 0.2) despite the additional mass and cationic charge provided by the TPP molecules (Figure 4A). This indicates that the overall transport properties of the dendrimer were retained with approximately 13 wt% loading of the TPP targeting moiety, consistent with previous studies where we have shown that up to 20 wt% loading of conjugated drug molecules does not affect biological interactions between dendrimers and cells [37]. However, TPP-D-Cy5 exhibited significantly greater partitioning to the isolated mitochondrial fraction compared to D-Cy5 (p < 0.01), and this effect increased with longer incubation times (Figure 4B). After 24 hours, TPP-D-Cy5 exhibits approximately 1.5-fold greater accumulation in the mitochondrial fraction, while after 48 hours this effect increases to a 2-fold difference.
Figure 3
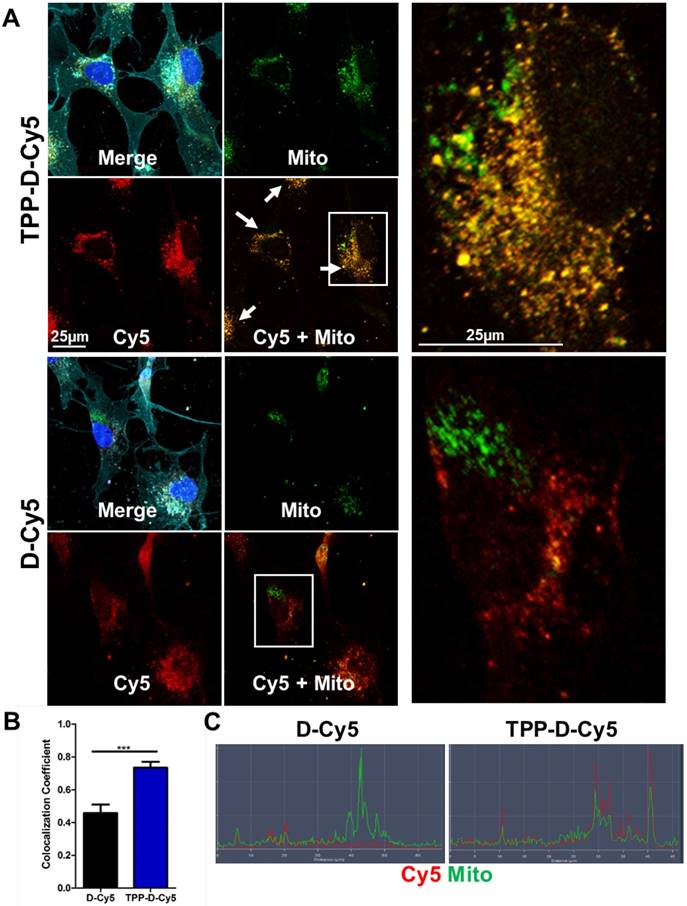
TPP enhances dendrimer colocalization with mitochondria. HMG3 human macrophages were treated with 50 µg/mL DCy5 or TPPDCy5 (red) for 48 hours, fixed, and stained with anti-AIF to mark mitochondria (green), DAPI to label nuclei (blue), and lectin to label cell membranes (cyan). A TPPDCy5 colocalizes strongly with mitochondrial signal, as illustrated by the yellow signal of overlapping red and green, while DCy5 exhibits diffuse signal with regions of dendrimer-free mitochondria and vice versa. B Semi-quantitative analysis of colocalization demonstrates that TPPDCy5 exhibits significantly greater colocalization with mitochondria than DCy5. *** p < 0.001 in Student's t-test. C Fluorescence line profile through representative cells show that TPPDCy5 signal aligns closely with mitochondria signal while DCy5 signal does not.
Figure 4
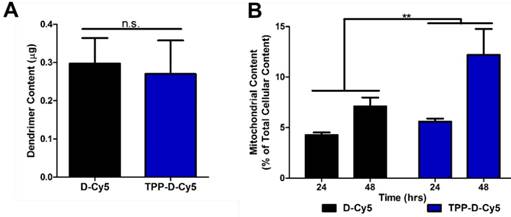
Quantitative assessment of mitochondrial dendrimer content. Mitochondrial isolation was performed on cells treated with dendrimer at 50 µg/mL. A Conjugation of TPP ligand to dendrimer does not affect overall cellular internalization after 24 hour treatment. n.s. p > 0.1 in Student's t-test. B TPPDCy5 exhibits significantly greater levels in mitochondria compared to DCy5 as a percentage of total cellular dendrimer content in a time-dependent manner. ** p < 0.01, F = 9.32 with treatment in two-way ANOVA.
Figure 5
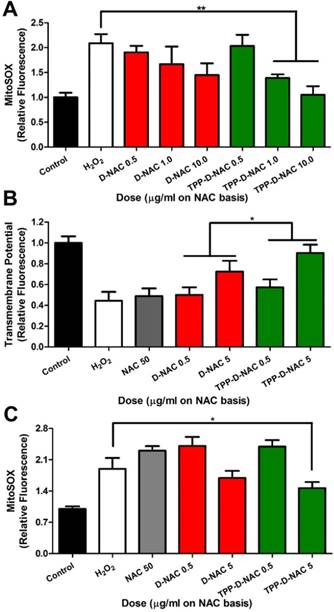
TPP conjugation enhances therapeutic efficacy of D-NAC in mitochondrial oxidative stress. A For acute oxidative stress injury, BV2 murine microglia were stimulated with 50 µM H2O2 for 2 hours, then treated with dendrimer for 6 hours, and analyzed with MitoSOX to measure mitochondrial superoxide levels. When compared to the H2O2 group, TPP-D-NAC exhibits a significant dose-dependent reduction in mitochondria superoxide while D-NAC did not under conditions of oxidative stress. ** p < 0.01 in Student's t-test compared to H2O2 group. B TPP-D-NAC ameliorates oxidative stress-induced membrane depolarization. * p < 0.05, F = 5.632 with treatment in two-way ANOVA. C For long term oxidative stress insult, cells were stimulated with 50 µM H2O2 for 2 hours, followed by cotreatment of free NAC, D-NAC, or TPP-D-NAC with 5 µM H2O2 for 24 hours. TPP-D-NAC exhibits greater reduction in mitochondrial superoxide than DNAC and free NAC. * p < 0.05 in Student's t-test compared to H2O2 group.
We next sought to leverage the mitochondria targeting capabilities of the modified dendrimer to deliver anti-oxidant compound N-acetyl cysteine (NAC) for improved anti-oxidant effects because of the central role of mitochondria in producing reactive oxygen species. We have previously reported the enhanced efficacy of dendrimer conjugated NAC (D-NAC) compared to freely administered NAC in a variety of in vitro and in vivo models [20, 34, 35]. In this work, we developed a new compound, with the dendrimer conjugated to TPP for mitochondrial targeting and NAC as an anti-oxidant (TPP-D-NAC) to further enhance the therapeutic efficacy. To ensure that results were not confounded by cytotoxic effects, MTT cell viability assays were performed to establish the in vitro toxicity of TPP and TPP-conjugated dendrimer at doses in the experimental range. Neither TPP-conjugated dendrimer nor free TPP exhibited any reduction in cell viability at the doses tested (Supplementary Figure S19). The safety profiles of unmodified hydroxyl PAMAM dendrimers and D-NAC have been extensively explored and found to be nontoxic far above the dosing range used in this study [60-62].
In vitro efficacy of TPP-D-NAC against mitochondrial oxidative stress
Dendrimers were evaluated under in vitro conditions of oxidative stress induced by treatment with hydrogen peroxide (H2O2). BV2 mouse microglial cells were stimulated with 50 µM H2O2 for 2 hours, followed by dendrimer treatment for 6 hours. Both D-NAC and TPP-D-NAC exhibited a dose-dependent reduction in mitochondrial superoxide production (Figure 5A). However, TPP-D-NAC exhibited slightly greater efficacy compared to D-NAC, with only TPP-D-NAC group (at 1 and 10 µg/ml on a NAC basis) exhibiting significantly reduced superoxide compared to untreated H2O2 stimulated cells (p < 0.01). At 10 µg/ml, TPP-D-NAC was able to return the mitochondrial superoxide levels back to within 10% of healthy control cells. We also evaluated the recovery of mitochondria transmembrane potential, which is decreased under oxidative stress due to membrane depolarization [63]. Similarly, both D-NAC and TPP-D-NAC exhibited a dose-dependent recovery in mitochondrial membrane polarization, while a 10-fold greater dose of freely administered NAC showed no efficacy (Figure 5B). TPP-D-NAC exhibited a significantly greater effect compared to D-NAC (p < 0.05), with the high dose of 5 µg/ml TPP-D-NAC restoring mitochondrial membrane potential back to approximately 90% of healthy cell levels. We also looked at continuous exposure to oxidative stress stimulus with a co-treatment of 5 µM H2O2 with dendrimer for 24 hours following the initial 2-hour stimulation. Free NAC exhibited no effect, while D-NAC exhibited a modest decrease in superoxide levels (Figure 5C). TPP-D-NAC exhibited slightly greater effect compared to D-NAC, with the high dose significantly lowering superoxide levels compared to H2O2 stimulated cells (p < 0.05).
Given the significantly greater mitochondrial targeting capabilities of TPP-conjugated dendrimers compared to unmodified dendrimers, we expected greater improvements in mitochondrial recovery. Studies with mitochondrial targeting properties generally report drastic improvements in efficacy when delivering drugs with mitochondrial targeting nanoparticles while unmodified nanoparticles exhibit mild or inconsistent effects [21, 23]. We attribute the modest improvements of TPP-D-NAC compared with D-NAC to the over-performance of D-NAC at addressing mitochondrial oxidative stress, as well as NAC conversion to glutathione in the cytosol. As discussed previously, unmodified dendrimer exhibits mitochondrial targeting capabilities similar to other nanoparticles modified with mitochondrial targeting ligands [6, 23]. Additionally, oxidative stress can compromise the integrity of the mitochondrial membrane to increase permeability, leading to an efflux of compounds that are typically localized to mitochondria [64]. Reactive oxygen species are therefore able to be released into the cytosol, where they can be equally accessed by D-NAC and TPP-D-NAC in the cytosol. Additionally, we have reported previously that D-NAC treatment significantly increases glutathione levels in vivo in models of Rett syndrome and cerebral palsy, indicating the contribution of delivered NAC in glutathione production [20, 34]. Therefore, the NAC from both TPP-D-NAC and D-NAC act as precursors for glutathione production and ROS scavengers in the cytosol, while the modestly enhanced efficacy of TPP-D-NAC is due to the greater mitochondrial targeting leading to more ROS scavenging in the mitochondrial compartment compared to D-NAC. Further exploration is necessary to understand the precise mechanisms and respective contributions to differences between D-NAC and TPP-D-NAC.
Next, we created a more severe oxidative stress injury to determine differences between D-NAC and TPP-D-NAC. BV2 cells were preincubated with the dendrimers for 24 hours, followed by stimulation with 500 µM H2O2 for 3 hours to create oxidative stress-induced cell death. At that point, cells begin to exhibit significant membrane blebbing, an effect which is mediated by the release of caspases from the mitochondrial intermembrane space [65]. In the MTT assay, TPP-D-NAC significantly preserved the metabolic activity of H2O2 stimulated cells (p < 0.01) (Figure 6A). In the context of oxidative stress and mitochondrial damage, the MTT assay serves as a measure of mitochondrial metabolism rather than overall cell viability. This is due to the role of NADPH (produced by the mitochondria) in both converting MTT to formazan and inactivating reactive oxygen species [66]. Therefore, superior scavenging of mitochondrial reactive oxygen species by TPP-D-NAC in the mitochondrial compartment results in minimal depletion of NADPH, which allows NADPH to instead act on the MTT to formazan conversion process. Due to the observed membrane blebbing, we also assessed cell viability via trypan blue exclusion for membrane integrity. In trypan blue exclusion, free NAC and D-NAC failed to exhibit any protective efficacy, while TPP-D-NAC exhibited a dose-dependent protection of cell viability (p < 0.001), with the high dose elevating cell viability up to approximately 75% of healthy cells (Figure 6B). There are two major mitochondrial-mediated mechanisms by which oxidative stress induces cell death: necrosis caused by ATP depletion and activation of the caspase-3 pathway by cytochrome C leakage into the cytosol [4, 67]. We theorize that the caspase-dependent pathway is being attenuated by our mitochondrial targeting intervention due to the presence of membrane blebbing, which is mediated by caspase release from the mitochondrial intermembrane space, although the precise mechanism of action remains to be explored in future studies [65]. Further understanding of this mechanism can inform the design of mitochondrial targeting dendrimer constructs targeted to specific pathways. The significantly improved efficacy of TPP-D-NAC compared to D-NAC and free NAC, along with improved amelioration of mitochondrial superoxide and transmembrane potential, demonstrate that this targeted construct could be highly beneficial for enhancing therapeutic outcomes in brain injuries where oxidative stress-induced cell death plays a significant role in disease progression, such as in neurodegenerative diseases including Parkinson's disease and Alzheimer's disease, as well as in and TBI.
In vivo uptake of TPP-D-Cy5 in a pediatric TBI model
Impaired mitochondrial function following TBI has been widely addressed as a central phenomenon of the post-traumatic neurometabolic cascade [68]. Studies show that injury induces prominent depolarization of the plasma membrane and massive Ca2+ influx into the cell, which results in mitochondrial dysfunction [69], and subsequent initiation of cell death pathways [70]. Drugs that target mitochondrial dysfunction specifically in the diseased cells and rescue cell death may provide therapeutic effects following TBI. In this study, we used a pediatric rabbit model of traumatic brain injury (PND 5) to focus on the long-lasting developmental and functional impacts of childhood brain trauma [48]. Even though we used a pediatric model, by PND5 the BBB is well-developed and intact in healthy animals, similar to what would be expected in adult animals. We have shown previously that the brains in neonatal rabbits show non-detectable Evans blue extravasation and high expression of occludin by PND1, confirming a well-developed BBB even at birth [34]. Moreover, we have shown that the dendrimer-cy5 uptake is barely detectable in the brain of healthy PND1 kits after systemic administration, which further indicates the high integrity and low permeability of BBB in PND1 kits [34, 40]. The PND5 kits have more mature BBB than PND1 kits. Therefore, the lack of BBB maturity is not the reason for dendrimer uptake, it is the BBB impairment due to injury. The results from this should be applicable to many models where the BBB is impaired due to injury, both in young and adult brain injury models. To assess the localization of TPP-D-Cy5, it was systemically administered in PND 5 rabbit kits at 6 hours post-injury, and the kits were sacrificed at 24 hours post TPP-D-Cy5 injection. We found that TPP-D-Cy5 exhibits high signal in the perinuclear cytosolic region of mi/ma at the site of injury, consistent with unmodified dendrimer localization we have previously reported [37, 39, 40, 71]. Moreover, this dominantly mi/ma uptake profile is consistent with our previous studies where we have shown that the dendrimers show minimal uptake in neurons and a modest uptake in astrocytes in other models of neurological disorders [37, 40]. Here, TPP-D-Cy5 exhibits overlap with the mitochondrial signal, indicating mitochondrial subcellular targeting, but the high cellular uptake of dendrimer corresponding to high mi/ma activation in the site of injury obfuscates its specificity for mitochondria (Figure 7A). In the corpus callosum where the white matter is outside the direct site of injury and mi/ma activation, the specificity of TPP-D-Cy5 for the mitochondria is clear, with the bulk of dendrimer signal overlapping with mitochondrial signal and minimal cytosolic dendrimer signal seen (Figure 7B). Moreover, there was no significant TPP-D-Cy5 uptake in the mi/ma at the corpus callosum of the contralateral site of injury where the microglia were not activated (Supplemental Figure S20). These results indicate that dendrimer conjugated TPP can target mitochondria in activated mi/ma following TBI in a pediatric model with well-developed BBB, similar to what would be expected in mature, well-developed BBB.
These results indicate that dendrimer conjugated TPP can target mitochondria in activated microglia following TBI. Therefore, TPP-D-NAC conjugates should also be able to target microglial mitochondria and provide therapeutic effects, which will be future explored in our future study.
Figure 6
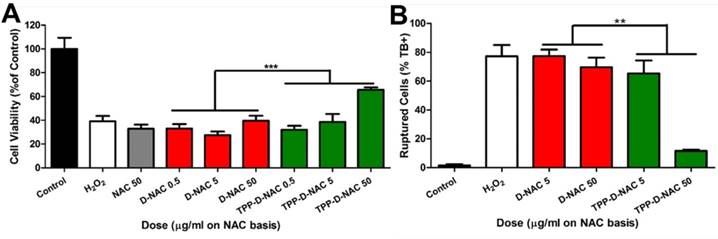
TPP-D-NAC enhances protection from oxidative stress induced cell death. Cells were pretreated for 24 hours with free NAC, D-NAC, or TPP-D-NAC, followed by oxidative stress insult of 500 µM H2O2 for 3 hours. A Cell viability was assessed with MTT assay. TPP-D-NAC exhibits significantly greater improvement in cell viability under oxidative stress compared to both DNAC and free NAC. *** p < 0.001, F = 33.706 with variable interaction in two-way ANOVA. B Cells were collected, treated with trypan blue, and counted in membrane exclusion assay. TPP-D-NAC exhibits significantly decreased cell death compared to D-NAC. ** p < 0.01, F = 9.569 with variable interaction in two-way ANOVA.
Figure 7
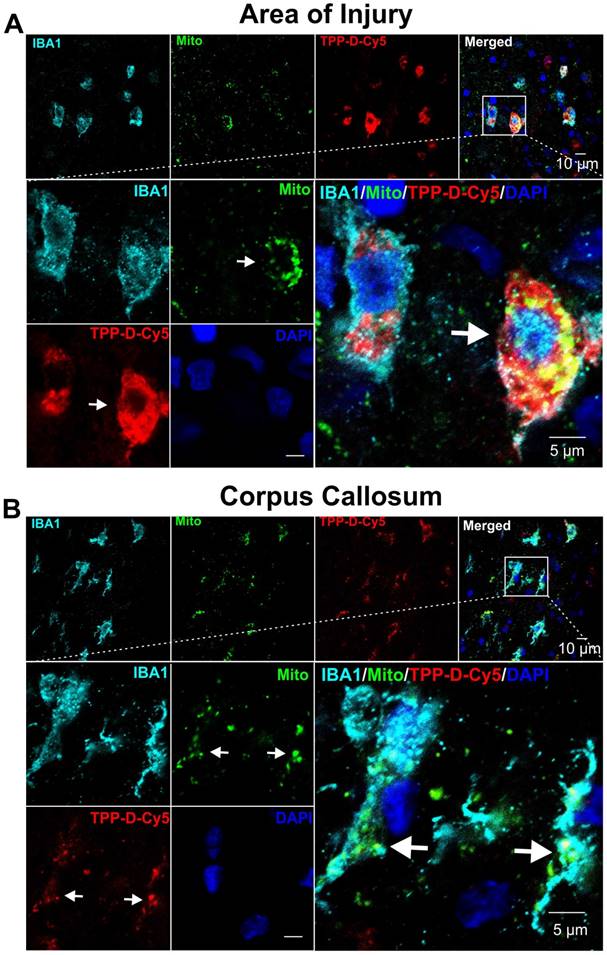
Confocal images showing that TPP-D-Cy5 exhibits co-localization with mitochondria in pediatric TBI rabbit kits. A At the area of injury, TPP-D-Cy5 distributed in the cytosol and mitochondria of activated microglial cells (IBA1 positive). B At the corpus callosum of the ipsilateral injury brain, TPP-D-Cy5 co-localizes mainly with mitochondrial signals.
Conclusions
We designed and developed multifunctional hydroxyl-terminated PAMAM dendrimer conjugates for in vivo targeting of mitochondrial function of activated mi/ma. The dendrimer surface is partially modified with TPP mitochondrial targeting moiety to confer intracellular targeting without affecting the favorable in vivo microglia targeting potential and cell internalization properties of the unmodified dendrimers. We have conjugated anti-oxidant NAC to TPP-modified dendrimer to enhance its efficacy against mitochondrial oxidative stress. We demonstrated that TPP-conjugated dendrimer exhibits significantly greater localization to the mitochondria and this greater localization was in addition to the inherent properties of unmodified dendrimers, which already exhibit mitochondrial localization superior to several other nanoparticles. It should be noted that while these dendrimers exhibit mitochondrial targeting, they are not selectively targeted only to mitochondria. The mitochondrial targeting capability of these dendrimers translated to enhanced efficacy in improvements of markers of oxidative stress, including mitochondrial superoxide production and transmembrane potential. In terms of oxidative stress-induced cytotoxicity, TPP-D-NAC performs significantly better in recovering cell viability compared to both D-NAC and freely administered NAC. Upon systemic administration TPP-D-Cy5 co-localized with mitochondria in activated mi/ma cells at the white matter area of the ipsilateral brain injury in a pediatric TBI model indicating that this conjugate has the potential to treat of oxidative stress in the area of injury in vivo. These results suggest that TPP-D-NAC may significantly improve the efficacy of D-NAC in CNS disease pathologies with significant oxidative stress-induced cell death, such as traumatic brain injury, Alzheimer's disease, and hypoxic ischemia.
Abbreviations
TBI: traumatic brain injury; PAMAM: polyamidoamine; NAC: N-acetyl cysteine; TPP: triphenyl-phosphonium; ROS/RNS: reactive oxygen/nitrogen species; BBB: blood-brain barrier; GSH: glutathione; EDC: 1-[3-(Dimethylamino)propyl]-3-ethylcarbodiimide methiodide; DMAP: 4(dimethylamino)pyridine; TFA: trifluoroacetic acid; NHS: N-hydroxy succinicmide; DIPEA: N, N-diisopropyl ethyl amine; DMEM: Dulbecco's Modified Eagle Medium; FBS: fetal bovine serum; NMR: Nuclear Magnetic Resonance; DLS: Dynamic light scattering.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was funded by the NIBIB R01EB018306 (RM Kannan). We thank the Wilmer Core Grant for Vision Research, Microscopy and Imaging Core Module (Grant #EY001865) for access to the Zen LSM710 confocal microscope.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Green DR, Reed JC. Mitochondria and Apoptosis. Science. 1998;281:1309-12
2. Heller A, Brockhoff G, Goepferich A. Targeting drugs to mitochondria. Eur J Pharm Biopharm. 2012;82:1-18
3. Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E. Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci. 2012;322:254-62
4. Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787
5. Sorrentino V, Menzies KJ, Auwerx J. Repairing Mitochondrial Dysfunction in Disease. Annu Rev Pharmacol Toxicol. 2018;58:353-89
6. Kwon HJ, Cha M-Y, Kim D, Kim DK, Soh M, Shin K. et al. Mitochondria-Targeting Ceria Nanoparticles as Antioxidants for Alzheimer's Disease. ACS Nano. 2016;10:2860-70
7. Hu Q, Wang G. Mitochondrial dysfunction in Parkinson's disease. Transl Neurodegener. 2016;5:14
8. Dupuis L, Gonzalez de Aguilar JL, Oudart H, de Tapia M, Barbeito L, Loeffler JP. Mitochondria in Amyotrophic Lateral Sclerosis: A Trigger and a Target. Neurodegener Dis. 2004;1:245-54
9. Mao P, Reddy PH. Is multiple sclerosis a mitochondrial disease? Biochim Biophys Acta. 2010;1802:66-79
10. Morin C, Simon N. Mitochondria: A Target for Neuroprotective Interventions in Cerebral Ischemia-Reperfusion. Curr Pharm Des. 2006;12:739-57
11. Di Pietro V, Lazzarino G, Amorini AM, Signoretti S, Hill LJ, Porto E. et al. Fusion or Fission: The Destiny of Mitochondria In Traumatic Brain Injury of Different Severities. Sci Rep. 2017;7:9189
12. Fischer TD, Hylin MJ, Zhao J, Moore AN, Waxham MN, Dash PK. Altered Mitochondrial Dynamics and TBI Pathophysiology. Front Syst Neurosci. 2016:10
13. A Hyder A, A Wunderlich C, Puvanachandra P, Gururaj G, Kobusingye O. The impact of traumatic brain injuries: A global perspective. NeuroRehabiliation. 2007;22(5):241-353
14. Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED. Time Course of Post-Traumatic Mitochondrial Oxidative Damage and Dysfunction in a Mouse Model of Focal Traumatic Brain Injury: Implications for Neuroprotective Therapy. J Cereb Blood Flow Metab. 2006;26:1407-18
15. Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31:596-604
16. Hernandez-Ontiveros DG, Tajiri N, Acosta S, Giunta B, Tan J, Borlongan CV. Microglia Activation as a Biomarker for Traumatic Brain Injury. Front Neurol. 2013;4:30
17. Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. 2003;60:6-20
18. Battin EE, Brumaghim JL. Antioxidant Activity of Sulfur and Selenium: A Review of Reactive Oxygen Species Scavenging, Glutathione Peroxidase, and Metal-Binding Antioxidant Mechanisms. Cell Biochem Biophys. 2009;55:1-23
19. Olsson B, Johansson M, Gabrielsson J, Bolme P. Pharmacokinetics and bioavailability of reduced and oxidized N-acetylcysteine. Eur J Clin Pharmacol. 1988;34:77-82
20. Nance E, Kambhampati SP, Smith ES, Zhang Z, Zhang F, Singh S. et al. Dendrimer-mediated delivery of N-acetyl cysteine to microglia in a mouse model of Rett syndrome. J Neuroinflammation. 2017;14:252
21. Sharma A, Soliman GM, Al-Hajaj N, Sharma R, Maysinger D, Kakkar A. Design and Evaluation of Multifunctional Nanocarriers for Selective Delivery of Coenzyme Q10 to Mitochondria. Biomacromolecules. 2012;13:239-52
22. Marrache S, Pathak RK, Dhar S. Detouring of cisplatin to access mitochondrial genome for overcoming resistance. Proc Natl Acad Sci. 2014;111:10444-9
23. Yang G, Xu L, Xu J, Zhang R, Song G, Chao Y. et al. Smart Nanoreactors for pH-Responsive Tumor Homing, Mitochondria-Targeting, and Enhanced Photodynamic-Immunotherapy of Cancer. Nano Lett. 2018
24. He H, Wang J, Wang H, Zhou N, Yang D, Green DR. et al. Enzymatic Cleavage of Branched Peptides for Targeting Mitochondria. J Am Chem Soc. 2018;140:1215-8
25. Sharma A, Neibert K, Sharma R, Hourani R, Maysinger D, Kakkar A. Facile Construction of Multifunctional Nanocarriers Using Sequential Click Chemistry for Applications in Biology. Macromolecules. 2011;44:521-9
26. Sharma R, Kottari N, Chabre YM, Abbassi L, Shiao TC, Roy R. A highly versatile convergent/divergent "onion peel" synthetic strategy toward potent multivalent glycodendrimers. Chem Commun. 2014;50:13300-3
27. Sharma R, Zhang I, Abbassi L, Rej R, Maysinger D, Roy R. A fast track strategy toward highly functionalized dendrimers with different structural layers: an "onion peel approach". Polym Chem. 2015;6:1436-44
28. Sharma R, Zhang I, Shiao TC, Pavan GM, Maysinger D, Roy R. Low generation polyamine dendrimers bearing flexible tetraethylene glycol as nanocarriers for plasmids and siRNA. Nanoscale. 2016;8:5106-19
29. Mignani S, El Kazzouli S, Bousmina M, Majoral J-P. Expand classical drug administration ways by emerging routes using dendrimer drug delivery systems: A concise overview. Adv Drug Deliv Rev. 2013;65:1316-30
30. Mignani S, Bryszewska M, Zablocka M, Klajnert-Maculewicz B, Cladera J, Shcharbin D. et al. Can dendrimer based nanoparticles fight neurodegenerative diseases? Current situation versus other established approaches. Prog Polym Sci. 2017;64:23-51
31. Sharma A, Khatchadourian A, Khanna K, Sharma R, Kakkar A, Maysinger D. Multivalent niacin nanoconjugates for delivery to cytoplasmic lipid droplets. Biomaterials. 2011;32:1419-29
32. Sharma A, Mejia D, Maysinger D, Kakkar A. Design and synthesis of multifunctional traceable dendrimers for visualizing drug delivery. RSC Adv. 2014;4:19242-5
33. Sharma A, Mejía D, Regnaud A, Uhlig N, Li C-J, Maysinger D. et al. Combined A3 Coupling and Click Chemistry Approach for the Synthesis of Dendrimer-Based Biological Tools. ACS Macro Lett. 2014;3:1079-83
34. Kannan S, Dai H, Navath RS, Balakrishnan B, Jyoti A, Janisse J. et al. Dendrimer-Based Postnatal Therapy for Neuroinflammation and Cerebral Palsy in a Rabbit Model. Sci Transl Med. 2012;4:130ra46-ra46
35. Nance E, Porambo M, Zhang F, Mishra MK, Buelow M, Getzenberg R. et al. Systemic dendrimer-drug treatment of ischemia-induced neonatal white matter injury. J Control Release. 2015;214:112-20
36. Mishra MK, Beaty CA, Lesniak WG, Kambhampati SP, Zhang F, Wilson MA. et al. Dendrimer Brain Uptake and Targeted Therapy for Brain Injury in a Large Animal Model of Hypothermic Circulatory Arrest. ACS Nano. 2014;8:2134-47
37. Nemeth CL, Drummond GT, Mishra MK, Zhang F, Carr P, Garcia MS. et al. Uptake of dendrimer-drug by different cell types in the hippocampus after hypoxic-ischemic insult in neonatal mice: Effects of injury, microglial activation and hypothermia. Nanomedicine: NBM. 2017;13:2359-69
38. Dai H, Navath RS, Balakrishnan B, Guru BR, Mishra MK, Romero R. et al. Intrinsic targeting of inflammatory cells in the brain by polyamidoamine dendrimers upon subarachnoid administration. Nanomedicine (London, England). 2010;5:1317-29
39. Zhang F, Mastorakos P, Mishra MK, Mangraviti A, Hwang L, Zhou J. Uniform brain tumor distribution and tumor associated macrophage targeting of systemically administered dendrimers. Biomaterials. 2015:52
40. Lesniak WG, Mishra MK, Jyoti A, Balakrishnan B, Zhang F, Nance E. Biodistribution of fluorescently labeled PAMAM dendrimers in neonatal rabbits: effect of neuroinflammation. Mol Pharm. 2013:10
41. Biswas S, Dodwadkar NS, Piroyan A, Torchilin VP. Surface conjugation of triphenylphosphonium to target poly(amidoamine) dendrimers to mitochondria. Biomaterials. 2012;33:4773-82
42. Bielski ER, Zhong Q, Brown M, da Rocha SRP. Effect of the Conjugation Density of Triphenylphosphonium Cation on the Mitochondrial Targeting of Poly(amidoamine) Dendrimers. Mol Pharm. 2015;12:3043-53
43. Murphy MP. Selective targeting of bioactive compounds to mitochondria. Trends Biotechnol. 1997;15:326-30
44. Sharma R, Sharma A. et al. Scalable Synthesis and Validation of PAMAM Dendrimer-N-acetyl cysteine Conjugate for Potential Translation. Bioeng Transl Med. 2018;3(2):87-101
45. Liaw K, Gök O, DeRidder LB, Kannan S, Kannan RM. Quantitative assessment of surface functionality effects on microglial uptake and retention of PAMAM dendrimers. J Nanopart Res. 2018;20:111
46. Yan-Kun Y, Bin M, Jun Z, Ya-Jie Y, Lu L, Chang-Li Y. et al. Neuroprotective Effect of Curcumin Against Oxidative Damage in BV-2 Microglia and High Intraocular Pressure Animal Model. J Ocul Pharmacol Ther. 2014;30:657-64
47. Hou RCW, Wu CC, Huang JR, Chen YS, Jeng KCG. Oxidative Toxicity in BV-2 Microglia Cells: Sesamolin Neuroprotection of H2O2 Injury Involving Activation of p38 Mitogen-Activated Protein Kinase. Ann N Y Acad Sci. 2005;1042:279-85
48. Zhi Z, Manda S. et al. A New Rabbit Model of Pediatric Traumatic Brain Injury. J Neurotrauma. 2015;32:1369-79
49. Sharma A, Porterfield JE, Smith E, Sharma R, Kannan S, Kannan RM. Effect of mannose targeting of hydroxyl PAMAM dendrimers on cellular and organ biodistribution in a neonatal brain injury model. J Control Release. 2018;283:175-89
50. Sharma R, Kim S-Y, Sharma A, Zhang Z, Kambhampati SP, Kannan S. et al. Activated Microglia Targeting Dendrimer-Minocycline Conjugate as Therapeutics for Neuroinflammation. Bioconjugate Chem. 2017;28:2874-86
51. Saovapakhiran A, D'Emanuele A, Attwood D, Penny J. Surface Modification of PAMAM Dendrimers Modulates the Mechanism of Cellular Internalization. Bioconjugate Chem. 2009;20:693-701
52. Li Y, He H, Lu W, Jia X. A poly(amidoamine) dendrimer-based drug carrier for delivering DOX to gliomas cells. RSC Adv. 2017;7:15475-81
53. Kambhampati SP, Mishra MK, Mastorakos P, Oh Y, Lutty GA, Kannan RM. Intracellular delivery of dendrimer triamcinolone acetonide conjugates into microglial and human retinal pigment epithelial cells. Eur J Pharm Biopharm. 2015;95:239-49
54. Marina AD. Dendrimers Effects on the Immune System: Insights into Toxicity and Therapeutic Utility. Curr Pharm Des. 2017;23:3134-41
55. Nance E, Zhang F, Mishra MK, Zhang Z, Kambhampati SP, Kannan RM. et al. Nanoscale effects in dendrimer-mediated targeting of neuroinflammation. Biomaterials. 2016;101:96-107
56. Jauslin ML, Meier T, Smith RAJ, Murphy MP. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003;17:1972-4
57. Sheu S-S, Nauduri D, Anders MW. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochim Biophys Acta. 2006;1762:256-65
58. Liu X, Huang N, Li H, Jin Q, Ji J. Surface and Size Effects on Cell Interaction of Gold Nanoparticles with Both Phagocytic and Nonphagocytic Cells. Langmuir. 2013;29:9138-48
59. Shi J, Choi JL, Chou B, Johnson RN, Schellinger JG, Pun SH. Effect of Polyplex Morphology on Cellular Uptake, Intracellular Trafficking, and Transgene Expression. ACS Nano. 2013;7:10612-20
60. Kurtoglu YE, Navath RS, Wang B, Kannan S, Romero R, Kannan RM. Poly(amidoamine) dendrimer-drug conjugates with disulfide linkages for intracellular drug delivery. Biomaterials. 2009;30:2112-21
61. Thiagarajan G, Greish K, Ghandehari H. Charge affects the oral toxicity of poly(amidoamine) dendrimers. Eur J Pharm Biopharm. 2013;84:330-4
62. Dobrovolskaia MA, Patri AK, Simak J, Hall JB, Semberova J, De Paoli Lacerda SH. et al. Nanoparticle size and surface charge determine effects of PAMAM dendrimers on human platelets in vitro. Mol Pharm. 2012;9:382-93
63. Lieven CJ, Vrabec JP, Levin LA. The Effects of Oxidative Stress on Mitochondrial Transmembrane Potential in Retinal Ganglion Cells. Antioxid Redox Signal. 2003;5:641-6
64. Guo C, Sun L, Chen X, Zhang D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res. 2013;8:2003-14
65. Wang C, Youle RJ. The Role of Mitochondria in Apoptosis. Annu Rev Genet. 2009;43:95-118
66. Śliwka L, Wiktorska K, Suchocki P, Milczarek M, Mielczarek S, Lubelska K. et al. The Comparison of MTT and CVS Assays for the Assessment of Anticancer Agent Interactions. PLOS ONE. 2016;11:e0155772
67. Smith RAJ, Hartley RC, Cochemé HM, Murphy MP. Mitochondrial pharmacology. Trends Pharmacol Sci. 2012;33:341-52
68. Stefano Signoretti, Roberto Vagnozzi, Barbara Tavazzi, Giuseppe Lazzarino. Biochemical and neurochemical sequelae following mild traumatic brain injury: summary of experimental data and clinical implications. Neurosurg Focus. 2010;29:E1
69. Vekaria HJ, Talley Watts L, Lin A-L, Sullivan PG. Targeting mitochondrial dysfunction in CNS injury using Methylene Blue; still a magic bullet? Neurochem Int. 2017;109:117-25
70. P.G. S, A.G. R, P.C. W, J.E. S. Mitochondrial permeability transition in CNS trauma: Cause or effect of neuronal cell death? J Neurol Sci. 2005;79:231-9
71. Grimm JC, Magruder JT, Wilson MA, Blue ME, Crawford TC, Troncoso JC. et al. Nanotechnology Approaches to Targeting Inflammation and Excitotoxicity in a Canine Model of Hypothermic Circulatory Arrest Induced Brain Injury. Ann Thorac Surg. 2016;102:743-50
Author contact
![]() Corresponding author: Rangaramanujam M. Kannan, Arnall Patz Distinguished Professor of Ophthalmology, Center for Nanomedicine at the Wilmer Eye Institute, 400 North Broadway, Baltimore, Maryland 21231, USA. Tel.: +1 443-287-8634; Fax: +1 443-287-8635; e-mail: krangar1edu
Corresponding author: Rangaramanujam M. Kannan, Arnall Patz Distinguished Professor of Ophthalmology, Center for Nanomedicine at the Wilmer Eye Institute, 400 North Broadway, Baltimore, Maryland 21231, USA. Tel.: +1 443-287-8634; Fax: +1 443-287-8635; e-mail: krangar1edu
Citation styles
APA
Sharma, A., Liaw, K., Sharma, R., Zhang, Z., Kannan, S., Kannan, R.M. (2018). Targeting Mitochondrial Dysfunction and Oxidative Stress in Activated Microglia using Dendrimer-Based Therapeutics. Theranostics, 8(20), 5529-5547. https://doi.org/10.7150/thno.29039.
ACS
Sharma, A.; Liaw, K.; Sharma, R.; Zhang, Z.; Kannan, S.; Kannan, R.M. Targeting Mitochondrial Dysfunction and Oxidative Stress in Activated Microglia using Dendrimer-Based Therapeutics. Theranostics 2018, 8 (20), 5529-5547. DOI: 10.7150/thno.29039.
NLM
Sharma A, Liaw K, Sharma R, Zhang Z, Kannan S, Kannan RM. Targeting Mitochondrial Dysfunction and Oxidative Stress in Activated Microglia using Dendrimer-Based Therapeutics. Theranostics 2018; 8(20):5529-5547. doi:10.7150/thno.29039. https://www.thno.org/v08p5529.htm
CSE
Sharma A, Liaw K, Sharma R, Zhang Z, Kannan S, Kannan RM. 2018. Targeting Mitochondrial Dysfunction and Oxidative Stress in Activated Microglia using Dendrimer-Based Therapeutics. Theranostics. 8(20):5529-5547.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.