Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2023; 13(12):4288-4302. doi:10.7150/thno.82132 This issue Cite
Research Paper
Protein phosphatase 2A-B55β mediated mitochondrial p-GPX4 dephosphorylation promoted sorafenib-induced ferroptosis in hepatocellular carcinoma via regulating p53 retrograde signaling
Bo Qian1†, Lin Che1†, Ze-Bang Du1†, Ni-Jun Guo1†, Xin-Mou Wu1, Lei Yang1, Zhao-Xuan Zheng1, Yun-Lu Gao1, Ming-Zhu Wang1, Xiao-Xuan Chen1, Ling Xu1, Zi-Jian Zhou1,2 ![]() , Yu-Chun Lin1
, Yu-Chun Lin1 ![]() , Zhong-Ning Lin1
, Zhong-Ning Lin1 ![]()
1. State Key Laboratory of Vaccines for Infectious Diseases, Xiang An Biomedicine Laboratory; State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics; National Innovation Platform for Industry-Education Integration in Vaccine Research; School of Public Health, Xiamen University, Xiamen, China.
2. Center for Molecular Imaging and Translational Medicine, School of Public Health, Xiamen University, Xiamen, China.
† These authors contributed equally.
Received 2022-12-24; Accepted 2023-7-15; Published 2023-7-31
Abstract

Rationale: As a key endogenous negative regulator of ferroptosis, glutathione peroxidase 4 (GPX4) can regulate its antioxidant function through multiple post-translational modification pathways. However, the effects of the phosphorylation/dephosphorylation status of GPX4 on the regulation of inducible ferroptosis in hepatocellular carcinoma (HCC) remain unclear.
Methods: To investigate the effects and molecular mechanism of GPX4 phosphorylation/dephosphorylation modification on ferroptosis in HCC cells. Sorafenib (Sora) was used to establish the ferroptosis model in HCC cells in vitro. Using the site-directed mutagenesis method, we generated the mimic GPX4 phosphorylation or dephosphorylation HCC cell lines at specific serine sites of GPX4. The effects of GPX4 phosphorylation/dephosphorylation modification on ferroptosis in HCC cells were examined. The interrelationships among GPX4, p53, and protein phosphatase 2A-B55β subunit (PP2A-B55β) were also explored. To explore the synergistic anti-tumor effects of PP2A activation on Sora-administered HCC, we established PP2A-B55β overexpression xenograft tumors in a nude mice model in vivo.
Results: In the Sora-induced ferroptosis model of HCC in vitro, decreased levels of cytoplasmic and mitochondrial GPX4, mitochondrial dysfunction, and enhanced p53 retrograde signaling occurred under Sora treatment. Further, we found that mitochondrial p53 retrograded remarkably into the nucleus and aggravated Sora-induced ferroptosis. The phosphorylation status of GPX4 at the serine 2 site (GPX4Ser2) revealed that mitochondrial p-GPX4Ser2 dephosphorylation was positively associated with ferroptosis, and the mechanism might be related to mitochondrial p53 retrograding into the nucleus. In HCC cells overexpressing PP2A-B55β, it was found that PP2A-B55β directly interacted with mitochondrial GPX4 and promoted Sora-induced ferroptosis in HCC. Further, PP2A-B55β reduced the interaction between mitochondrial GPX4 and p53, leading to mitochondrial p53 retrograding into the nucleus. Moreover, it was confirmed that PP2A-B55β enhanced the ferroptosis-mediated tumor growth inhibition and mitochondrial p53 retrograde signaling in the Sora-treated HCC xenograft tumors.
Conclusion: Our data uncovered that the PP2A-B55β/p-GPX4Ser2/p53 axis was a novel regulatory pathway of Sora-induced ferroptosis. Mitochondrial p-GPX4Ser2 dephosphorylation triggered ferroptosis via inducing mitochondrial p53 retrograding into the nucleus, and PP2A-B55β was an upstream signal modulator responsible for mitochondrial p-GPX4Ser2 dephosphorylation. Our findings might serve as a potential theranostic strategy to enhance the efficacy of Sora in HCC treatment through the targeted intervention of p-GPX4 dephosphorylation via PP2A-B55β activation.
Keywords: Ferroptosis, Glutathione peroxidase 4, Retrograding p53 signal, Protein phosphatase 2A-B55β subunit, Hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) is one of the most common causes of cancer-related death globally [1]. Sorafenib (Sora) is one of the medically-approved systemic treatments that has demonstrated a survival benefit in patients with unresectable HCC [2]. However, some HCC patients are of limited effectiveness to Sora, and some patients acquire drug resistance [2]. Ferroptosis is a new approach for HCC treatment characterized by the accumulation of toxic lipid peroxidation (LPO) due to the loss of glutathione peroxidase 4 (GPX4) [3]. GPX4 is the only enzyme that can diminish lipid hydroperoxides of biological membranes and plays a primary role in regulating cells that undergo ferroptosis [4]. The anti-cancer effects of Sora may be partially explained by its ability to induce ferroptosis via interfering with the antioxidant function of GPX4 [5]. GPX4 seems to be particularly relevant in cancer development. The overall survival of glioma patients with low GPX4 expression was longer than that of GPX4 high expression group [3, 6].
Moreover, the inhibition of GPX4 induces growth suppression of tumors, especially for drug-resistance tumors [7, 8]. Combining androgen-deprivation therapy drugs with GPX4 inhibitors has achieved a remarkable synergistic effect on resistant prostate cancer treatment [9]. Therefore, targeting GPX4 inhibition might be a potential approach to improve the susceptibility of HCC to Sora treatment through enhancing ferroptosis. The current approach for GPX4 inhibition is achieved using a pharmacological method. The existing GPX4 inhibitors are covalently reacted with GPX4 through the reactive alkyl chloride, giving poor selectivity and pharmacokinetics [10]. It is thus imperative to find an effective intervention approach for targeting GPX4 to improve the anti-cancer effects via ferroptosis induction.
The different subcellular locations of GPX4 bring out its distinct roles. Among them, mitochondrial GPX4 was more efficient than non-mitochondrial GPX4 in protecting cells from mitochondria damage and extracellular oxidative stress (such as t-butyl-hydroperoxide, photosensitizers, H2O2, and 15-hydroperoxy-eicosatetraenoic acid) [11, 12]. Despite the role of non-mitochondrial GPX4 in ferroptosis being clearly defined [13], the underlying molecular mechanisms of mitochondrial GPX4 in ferroptosis remain unclear. p53 was involved in the GPX4 modulation via multiple pathways [14, 15], whereas the relationship and crosstalk between mitochondrial GPX4 and p53 are still missing. Mitochondrial p53 alterations might be the upstream signals for Sora-induced ferroptosis. Sora-induced mitochondrial translocation of p53 in the hepatic stellate cells triggered ferroptosis through direct interaction with the solute carrier family 25 member 28 [16]. Moreover, the translocation of p53 from the mitochondria to the nucleus, called the p53 retrograde signaling, is also an early signal of mitochondrial dysfunction [17]. Given the vital role of mitochondrial p53 in regulating mitochondria homeostasis and mitochondria-related ferroptosis, it is reasonable to speculate that mitochondrial p53 alterations might be involved in mitochondrial GPX4-regulated ferroptosis.
Evidence recently revealed that post-translational modification (PTM) of GPX4 might be a new strategy for ferroptosis modulation. Ding et al. pointed out that the ubiquitination of GPX4 triggered by DMOCPTL (a derivative of the natural product parthenolide) could induce triple-negative breast cancer cell ferroptosis [18]. Moreover, the mechanism of RSL3-induced ferroptosis was evidenced to be associated with the amino acid modification of GPX4 in the active region [19]. The online prediction software suggested that serine (Ser)/threonine (Thr) residues in GPX4 might be potential phosphorylation/dephosphorylation modification targets. Therefore, exploring the role of GPX4 phosphorylation/dephosphorylation modification in ferroptosis might provide an attractive intervention strategy for ferroptosis-associated cancer treatment.
The present study revealed that the dephosphorylation of mitochondrial p-GPX4Ser2 participated in Sora-induced ferroptosis in HCC by inducing mitochondrial p53 translocation into the nucleus and PP2A-B55β (encoded by the PPP2R2B gene) was involved in p-GPX4Ser2 dephosphorylation. As a result, the combination using of the PP2A-B55β activator and Sora is a novel theranostic approach for HCC treatment.
Results
Ferroptosis resistance and high GPX4 expression were associated with HCC
To explore the relationship between ferroptosis and HCC development. First, we analyzed the expression of ferroptosis-related genes in tumor tissues (n = 152) and peritumor tissues (n = 91) of HCC patients from the GEO dataset (GSE102079). As displayed in Figure 1A, compared to that in peritumor tissues, the expression of ferroptosis-inducing genes, including PTGS2, ALOX12, TFR2, and HMOX1, was decreased in tumor tissues, whereas ferroptosis-suppressor genes, including SLC7A11 and GPX4, were increased. Next, we analyzed the expression of ferroptosis-related genes in tumor tissues (n = 373) and peritumor tissues (n = 50) of HCC patients from the TCGA-LIHC dataset. Similarly, compared to that in peritumor tissues, the expression of ferroptosis-inducing genes, including PTGS2, HMOX1, TFR2, and NCOA4, was down-regulated in tumor tissues, whereas ferroptosis-suppressor genes SLC7A11 and FTH1 were upregulated (Figure 1B). KEGG analysis showed that the DEGs between tumor tissues and peritumor tissues of HCC patients from the TCGA-LIHC dataset were enriched in several ferroptosis-related pathways, such as p53 signaling pathway, fatty acid metabolism, fatty acid degradation, arachidonic acid metabolism, and alanine, aspartate, and glutamate metabolism (Figure 1C). Furthermore, GSEA analysis showed that the ferroptosis pathway was upregulated in peritumor tissues but not in tumor tissues of HCC patients from the TCGA-LIHC dataset (Figure 1D, normalized enrichment scores = 2.46, P < 0.05). The evidence above suggested that ferroptosis resistance was associated with the development of HCC.
Ferroptosis resistance and high GPX4 expression were associated with HCC. A. The expression of ferroptosis-related genes in tumor tissues (n = 152) and peritumor tissues (n = 91) of HCC patients from the GEO database (GSE102079). B. The expression of ferroptosis-related genes in tumor tissues (n = 373) and peritumor tissues (n = 50) of HCC patients from the TCGA-LIHC database. C. KEGG analysis of DEGs between tumor tissues and peritumor tissues in HCC patients from TCGA-LIHC database. D. GSEA analysis of the ferroptosis pathways in tumor tissues and peritumor tissues of HCC patients from TCGA-LIHC database. E. GPX4 expression in the paired tumor tissues (n = 40) and the paired peritumor tissues (n = 40) from the TCGA-LIHC database. F. GPX4 expression in two cohorts of HCC patients from GEPIA. G. Kaplan-Meier analysis showed the overall survival of HCC patients from GEPIA (TCGA-LIHC) with different levels of GPX4 expression. GPX4 expression was a binary variable divided into high or low expression according to the quartile. H. Protein levels of p53 and GPX4 in the paired peritumor tissues (P) and the paired tumor tissues (T) from the HCC patients, n = 6. I. Representative IHC images of p53 and GPX4 in the paired peritumor tissues and paired tumor tissues from the HCC patients. *, P < 0.05.
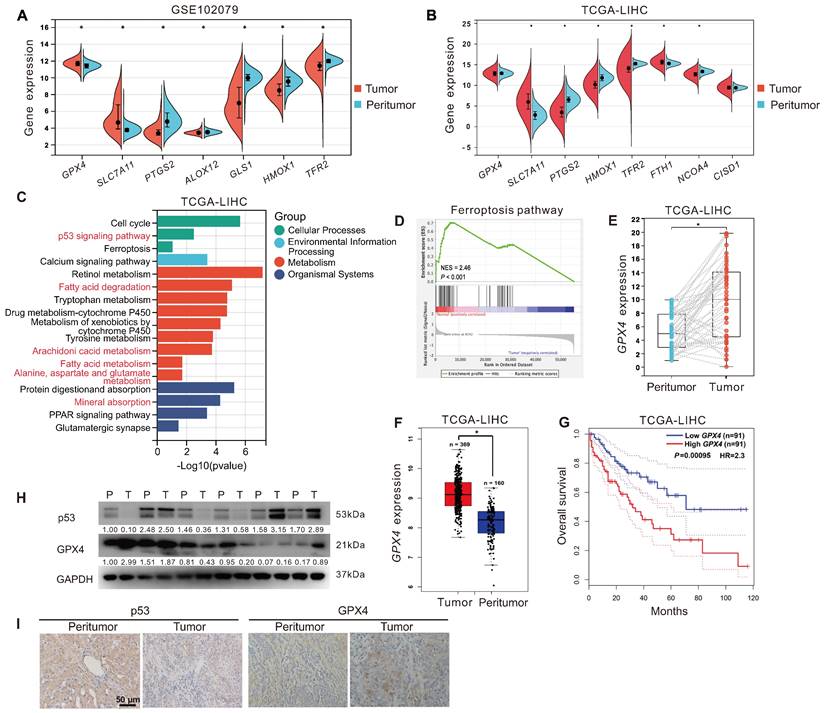
As GPX4 is a crucial endogenous negative regulator of ferroptosis, we analyzed the expression of GPX4 in 40 tumor tissues and 40 paired peritumor tissues of HCC patients from the TCGA-LIHC dataset. The results showed that the expression of GPX4 in tumor tissues was upregulated compared to in peritumor tissues (P < 0.05, Figure 1E). Besides, we further analyzed the correlation between GPX4 and HCC prognosis in HCC patients from the GEPIA database. Compared to that in peritumor tissues (n = 160), the expression of GPX4 gene was significantly increased in tumor tissues (n = 369) of HCC patients (Figure 1F). As shown in Figure 1G, compared to the HCC patients with higher GPX4 mRNA expression (n = 91), HCC patients with lower GPX4 mRNA expression (n = 91) had a higher overall survival rate (P < 0.05), indicating that GPX4 was negatively associated with HCC prognosis. Additionally, compared to that in peritumor tissues, the protein level of GPX4 was increased in the majority (2/3) of tumor tissues from HCC patients, whereas the protein level of p53 was decreased in half (1/2) of tumor tissues from HCC patients (Figure 1H-I). The above results indicated that resistance to ferroptosis and high expression of GPX4 were associated with the development of HCC.
Downregulation of GPX4 and mitochondrial dysfunction in Sora-induced ferroptosis of HCC cells
Sora was reported to exert anti-tumor effects on HCC via inducing ferroptosis [20]. We next explored the role of GPX4 in Sora-induced ferroptosis. As shown in Figure 2A, the cell viability of HCC cells decreased in a dose-dependent manner of Sora treatment, while the Sora-induced cell death could be rescued by ferroptosis inhibitor Fer-1. However, in contrast to that under the low-dose Sora (10 µM), Sora-induced cell death cannot be rescued to near-normal levels by Fer-1 under high-dose conditions (20 or 40 µM) (Figure 2A), suggesting that high-doses Sora may reduce the ferroptosis sensitivity of HCC cells. Low-dose Sora was evidenced to be more efficient and safer than high-dose Sora in NASH-induced HCC of mice [20]. Using of 10 μM Sora in the following experiments was based on the pharmacokinetic analysis of Sora in the plasma (9.7-12 µM) of patients with HCC [21]. As shown in Figure 2B and Figure S1A, Sora-induced cell death cannot be rescued by the apoptosis inhibitor (Zvad), necroptosis inhibitor (Nec-1), or autophagy inhibitor (chloroquine, CQ) (Figure 2B), but rescued by the co-treatment of Fer-1, indicating that Sora specifically induced ferroptosis in HCC. Next, the GPX4 level was measured in Sora-treated HCC cells. As expected, Sora treatment decreased the protein level of GPX4 in HCC cells, and the changes were alleviated with Fer-1 co-treatment (Figure 2C). Also, we examined the effects of Sora on system Xc- mediated GSH synthesis and iron metabolism in HCC cells. As shown in Figure S1B-D, the expression of the solute carrier family 7 member 11 (SLC7A11) subunit of system Xc- and the level of GSH were decreased in Sora-treated HCC cells, whereas there were no significant changes observed in the expression of transferrin receptor (TFRC), Fe3+ reductase six-transmembrane epithelial antigen of prostate 3 (STEAP3), ferritin heavy chain 1 (FTH1), and the iron-pumping protein ferroportin 1 (FPN1) in Sora-treated HCC cells. These findings demonstrated that Sora induced GPX4-related ferroptosis in HCC cells.
Downregulation of GPX4 and mitochondrial dysfunction in sorafenib-induced ferroptosis of HCC cells. A. HCC cells were treated with different doses of sorafenib (Sora, 5, 10, 20, and 40 µM, for 12 h and 24 h) alone or combined with ferroptosis inhibitor Fer-1 (1 µM) for 24 h. Cell viability was measured with the MTS assay. B. HCC cells were treated with Sora (10 µM) individually or combined with inhibitors of different cell death patterns, including Fer-1 (1 µM), Z-VAD (10 µM), Nec-1 (10 µM), or CQ (5 µM) for 24 h. Cell viability was measured with the MTS assay. C. WB detection of GPX4 protein expression in HCC cells after treatment with different doses of Sora (5, 10, 20 µM, 24 h) alone or combined with Fer-1 (1 µM, 24 h). D. TEM observation of mitochondrial morphological characteristics (red arrows) of ferroptosis in HCC cells treated with Sora (10 µM, 24 h). N, nucleus; M, mitochondria. Scale bar, 0.5 μm. E. FCM detection of LPO levels in HCC cells treated with Sora (10 μM, 24 h) individual or combined with Fer-1 (1 μM, 24 h). F. IF detection of mitochondrial LPO in HCC cells treated with Sora (10 µM, 24 h). Scale bars, 20 μm. G. FCM detection of mitochondrial ROS in HCC cells treated with Sora (10 µM, 24 h) individual or combined with Mito-Tempo (20 µM, pretreatment for 2 h). H. FCM detection of mitochondrial membrane potential in HCC cells treated with CCCP (10 µM, 6 h) and Sora (10 µM, 24 h) individual or combined with Fer-1 (1 μM, 24 h) in HCC cells. *, P < 0.05.
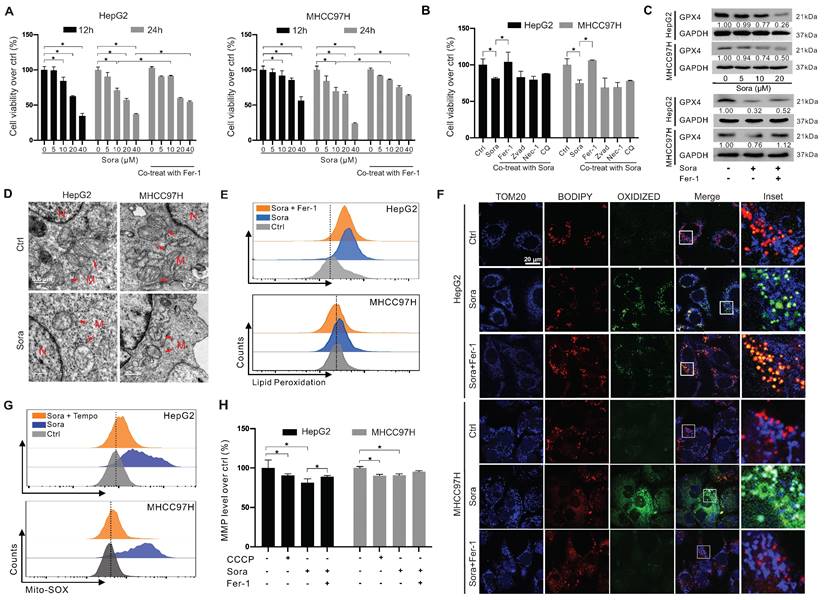
It was reported that GPX4 regulated ferroptosis by controlling mitochondrial function [22]. We explored the mitochondrial function in Sora-treated HCC. As shown in Figure 2D, Sora-treated HCC cells presented a decrease in the mitochondrial cristae (one of the ferroptosis markers) and an increase in the electron density of the outer mitochondrial membrane (OMM) using the TEM technique. Ferroptosis is accompanied by the massive generation of LPO [22]. FCM analysis showed that Sora induced obvious LPO accumulation in HCC cells (Figure 2E). With IF staining, the oxidized lipid (green dots) and its overlay (cyan dots) with the OMM protein TOM20 (blue dots) were increased in Sora-treated HCC cells (Figure 2F), suggesting that Sora induced LPO accumulation in mitochondria of HCC cells. Moreover, cellular ROS (Figure S1E) and mitochondrial ROS (Figure 2G) were increased in HCC cells under Sora treatment. Additionally, consistent with the change in CCCP (a positive inducer of mitochondrial membrane damage) treated cells, Sora decreased the mitochondrial membrane potential (MMP) level of HCC cells (Figure 2H). These Sora-driven mitochondrial changes were alleviated by Fer-1 co-treatment. These results indicated that mitochondrial dysfunction was associated with Sora-induced ferroptosis in HCC cells.
Mitochondrial p53 retrograde into the nucleus was involved in Sora-induced ferroptosis
The retrograde signal of p53 was reported as an early event of mitochondrial dysfunction [17]. We investigated whether mitochondrial p53 translocation was involved in Sora-induced mitochondrial dysfunction and ferroptosis. Along with the decline in mitochondrial GPX4, diminished mitochondrial p53 and elevated nucleus p53 were observed in Sora-treated HCC cells (Figure 3A-B). Consistently, Sora treatment attenuated p53 (green) localization in mitochondria (red), whereas it enhanced the p53 (green) signal in the nucleus (blue) (Figure 3C), further revealing that mitochondrial p53 retrograding into the nucleus occurred under Sora treatment.
Mitochondrial p53 retrograde into the nucleus was involved in Sora-induced ferroptosis. A. Protein levels of p53 in cytosolic (Cyto) or mitochondrial (Mito) fraction of HCC cells treated with Sora (10 µM, 24 h). B. Protein levels of p53 in cytosolic (Cyto) or nucleus (Nu) fraction of HCC cells treated with Sora (10 µM, 24 h). C. IF assay of the p53 distribution in mitochondria and nucleus of HCC cells treated with Sora individual (10 µM, 24 h) or combined with Fer-1 (1 µM, 24 h). Cells were subjected to IF staining of p53 (green), Mitotracker (red), and DAPI (blue). Scale bars, 20 μm. D. FCM detection of LPO in Hep3B cells. Staining cells with C11 BODIPY probe. E. FCM detection of LPO in Hep3B cells transfected with a p53-expressing plasmid or p53 nuclear localization sequence deleted (ΔNLS) plasmid. Staining cells with C11 BODIPY probe. F. Hep3B cells transfected with a p53-expressing plasmid or p53 ΔNLS plasmid were treated with Sora (10 µM, 24 h) individually or combined with Fer-1 (1 µM, 24 h). Cell viability was measured with the MTS assay. G-H. HepG2 cells transfected with a p53-expressing plasmid or p53 ΔNLS plasmid were treated individually with Sora (10 µM, 24 h) or combined with Fer-1 (1 µM, 24 h). Staining cells with a C11 BODIPY probe and LPO levels were measured with the FCM assay (G). Cell viability was measured with the MTS assay (H). *, P < 0.05.
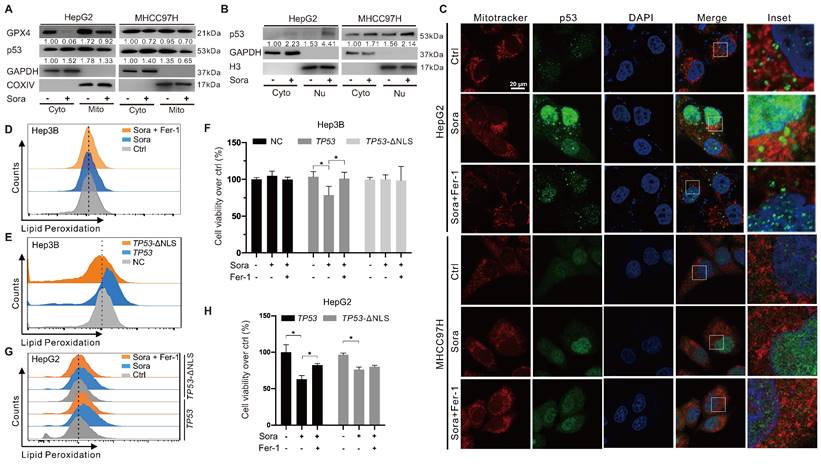
Hep3B cells with a null-p53 background were selected for the subsequent studies to determine whether p53 retrograding into the nucleus affected the occurrence of ferroptosis. As shown in Figure 3D, Sora treatment did not alter LPO levels in Hep3B cells. Next, we transfected Hep3B cells with a p53-expressing plasmid (TP53), and p53 nuclear sequence deleted plasmid (TP53-ΔNLS). Compared to the empty vector-transfected control cells, the LPO level was significantly increased in TP53-transfected Hep3B cells but not in TP53-ΔNLS-transfected Hep3B cells (Figure 3E). Moreover, Sora-induced cell death was observed in TP53 plasmid-transfected Hep3B cells but not in control or TP53-ΔNLS-transfected Hep3B cells (Figure 3F). We also transfected HepG2 cells with TP53 and TP53-ΔNLS plasmids and found that Sora-induced LPO accumulation was only observed in TP53 plasmid-transfected HepG2 cells but not in TP53-ΔNLS-transfected HepG2 cells (Figure 3G). Coincidentally, Sora-induced cell death was more obvious in TP53 plasmids-transfected HepG2 cells (Figure 3H). The above evidence suggested that mitochondrial p53 retrograding into the nucleus initiated the LPO phenomenon and promoted Sora-induced ferroptosis, but the upstream mechanism remained unclear.
Mitochondrial p-GPX4Ser2 dephosphorylation triggered ferroptosis and induced mitochondrial p53 translocation
GPX4 PTM plays a vital role in ferroptosis regulation. To explore whether the phosphorylation status of mitochondrial GPX4 was involved in Sora-induced ferroptosis, we examined the level of phosphorylated GPX4 (p-GPX4) in mitochondria of HCC cells upon Sora treatment. The results showed that the Sora treatment decreased the mitochondrial p-GPX4 level of HCC cells (Figure 4A). Next, we screened the potential kinases responsible for the phosphorylation of mitochondrial GPX4. As shown in Figure S2A and Figure 4B, among several common kinases, we found that Sora treatment decreased the protein level of PINK 1 and its interaction with mitochondrial GPX4 in HCC cells. Meanwhile, PINK1 knockdown decreased the level of p-GPX4 in HepG2 cells (Figure S2B), indicating that PINK1 might be one of the kinases responsible for mitochondrial GPX4 phosphorylation. Further, using 5 online phosphorylation prediction tools, we identified that the Ser2, Ser40, Ser45, and Ser112 sites were the unique and potential phosphorylation sites of GPX4 (Figure 4C), and only the Ser2 site mutation decreased the mitochondrial p-GPX4 level in HCC cells (Figure 4D), suggesting that the Ser2 site was one of the functional regulatory sites associated with GPX4 phosphorylation.
Mitochondrial p-GPX4Ser2 dephosphorylation triggered ferroptosis and induced mitochondrial p53 translocation. A. Mitochondrial-IP (Mito-IP) detection of the mitochondrial p-GPX4 level in HCC cells upon Sora treatment (10 µM, 24 h). B. Mito-IP detection of the interaction between GPX4 and PINK1 in mitochondria of HCC cells treated with Sora (10 µM, 24 h). C. Venn diagram showed the shared and unique phosphorylation sites of GPX4 predicated by DISPHOS 1.3, PhosphoSVM, MusiteDeep, Gps 6.0, and NetPhon3.1. D. IP detection of the phosphorylation levels of GPX4 in the constructed S2A, S40A, S45A, and S112A cells of HepG2. E. Sequences alignment of the conserved serine residues on GPX4. F. Mito-IP detection of the mitochondrial p-GPX4 levels in WT cells and the constructed S2D and S2A cells of HepG2. G. FCM detection of the LPO levels in WT, S2D, S112D, S2A, and S112A cells of HepG2. Staining cells with C11 BODIPY probe. H. WT, S2D, S112D, S2A, and S112A cells of HepG2 were treated with Sora (10 µM) individually or combined with Fer-1 (1 µM, 24 h). Cell viability was measured with the MTS assay. I. PLA detection of the interaction between p53 and GPX4 in HCC cells upon Sora treatment (10 µM, 24 h). J. PLA detection of the interaction between p53 and GPX4 in S2A and S112A cells of HepG2. K. Mito-IP detection of the interaction between GPX4 and p53 in S2D and S2A cells of HepG2 treated with Sora (10 µM, 24 h). L. Protein levels of p53 in cytosolic (Cyto), mitochondrial (Mito), and nucleus (Nu) fraction of WT, S2D, and S2A cells of HepG2. M. IF assay of the distribution of p53 in mitochondria and nucleus of S2D and S2A cells of HepG2 upon Sora treatment (10 µM, 24 h). Cells were subjected to IF staining of p53 (green), mitotracker (red), and DAPI (blue). Scale bars, 20 μm. *, P < 0.05.
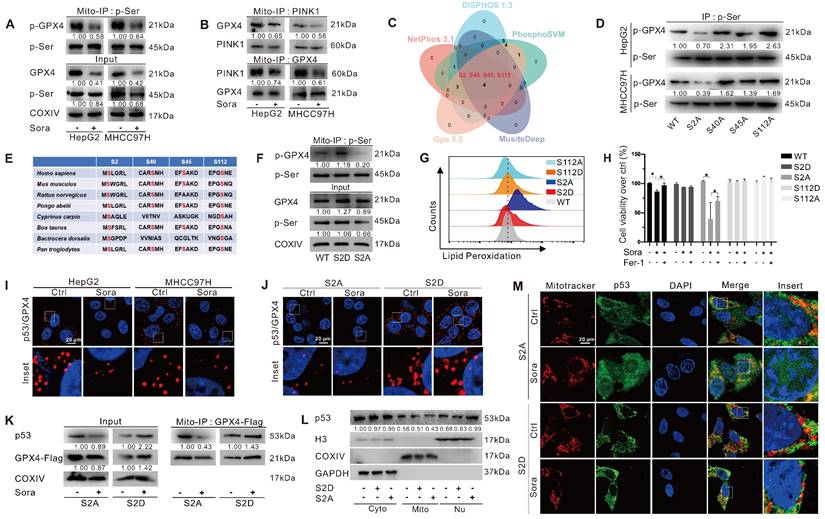
As Ser2 and Ser112 sites were highly conserved (Figure 4E), we explored the functions of the regulated Ser2 site phosphorylation in ferroptosis, and the Ser112 site was selected as a control. We mutated Ser2 and Ser112 sites of GPX4 to alanine (A) or aspartate (D) to generate GPX4 mimic dephosphorylation (S2A, S112A) or phosphorylation (S2D, S112D) cell lines of HepG2. As expected, the p-GPX4 level in mitochondria was increased in S2D cells, whereas it was reduced in S2A cells compared to that in WT cells (Figure 4F). Distinct from the ferroptosis resistance effects observed in HepG2 cells overexpressing WT and the other mutations of GPX4, Sora-induced LPO accumulation (Figure 4G) and cell death (Figure 4H) were only observed in S2A cells, suggesting that p-GPX4Ser2 dephosphorylation accelerated ferroptosis in HCC.
We next explored whether p-GPX4Ser2 dephosphorylation affected mitochondrial p53 translocation. As shown in Figure 4I, Sora treatment decreased the interaction between mitochondrial p53 and GPX4. Moreover, upon Sora treatment, a decreased interaction between mitochondrial p53 and GPX4 was only observed in S2A cells but not in S2D cells (Figure 4J-K). Further, upon Sora treatment, decreased mitochondrial p53 and increased nuclear p53 were displayed in S2A cells but not in S2D cells (Figure 4L-M and Figure S2C), implying that mitochondrial p-GPX4 dephosphorylation at the Ser2 site promoted the p53 translocation from mitochondria to the nucleus.
PP2A-B55β regulated p-GPX4Ser2 dephosphorylation and promoted Sora-induced ferroptosis in HCC
Protein phosphatase 2A (PP2A), a serine/threonine phosphatase, regulates the activity of numerous enzymes and proteins via dephosphorylation modification [23-27]. To investigate whether PP2A participated in regulating p-GPX4Ser2 dephosphorylation, we first analyzed the expression of the substrate recognition subunit of PP2A in HCC patients. As shown in Figure S3A, the levels of PPP2R2A, PPP2R2B, and PPP2R2D genes expression in tumor tissues of HCC patients from the GEO (GSE76427) and TCGA-LIHC dataset were lower than those in peritumor tissues. Further, we explored the relationship between the expression of PP2A subunits and GPX4 in HCC patients. The analysis revealed a negative correlation between GPX4 and PPP2R2A, PPP2R2B, or PPP2R5E expression in HCC patients from the TCGA-LIHC dataset (Figure S3B). HCC patients with high PPP2R2B gene expression had a higher overall survival rate (Figure S3C). Moreover, upon Sora treatment, the protein level of PP2A-B55β (coded by PPP2R2B) was dose-dependent increased in HCC cells (Figure 5A). These results suggested that PPP2R2B might be against the development of HCC via the negative modulating of GPX4.
PP2A-B55β regulated p-GPX4Ser2 dephosphorylation and promoted Sora-induced ferroptosis in HCC cells. A. Protein levels of B55α (coded by PPP2R2A), B55β (coded by PPP2R2B), and B55ε (coded by PPP2R5E) in HCC cells treated with Sora (5, 10, 20 µM, 24 h). B. Mito-IP detection of the interaction between GPX4 and B55β in HCC cells treated with Sora (10 µM, 24 h). C. PLA detection of the interaction between B55β and GPX4 in HCC cells treated with Sora (10 µM, 24 h). Interaction events were shown as red dots. Scale bars, 20 μm. D. IP detection of the interaction between B55β and GPX4 in WT cells and the constructed S2A, S40A, S45A, and S112A cells. E. PLA detection of the interaction between B55β and GPX4 in WT, S2A, S40A, S45A, and S112A cells. Interaction events were shown as red dots. Scale bars, 20 μm. F. HepG2 cells were treated with si2R2B and/or OA, while the whole cell lysates were isolated and subjected to PhosTag™ gel electrophoresis. The protein levels of the phosphorylated GPX4 (p-GPX4) and non-phosphorylated GPX4 (N-GPX4) were tested. G. Mito-IP detection of the interaction between GPX4 and p53 in HCC cells transfected with the PPP2R2B-overexpression plasmid. H. Protein levels of B55β and GPX4 in HepG2 cells treated with PPP2R2B-overexpression plasmid and/or Sora (10 µM, 24 h). I. FCM detection of LPO levels in HepG2 cells transfected with the PPP2R2B-overexpression plasmid. Staining cells with C11 BODIPY probe. J. Cell viability of HCC cells treated with Sora (10 µM) alone or combined with PPP2R2B-overexpression plasmid or PP2A agonists DT-061 (20 µM) and iHAP1 (10 µM) for 24, 48, 72, and 96 h. K. EdU staining and colony formation assay detection of the cell proliferation ability of HCC cells treated with Sora (10 µM) alone or combined with PPP2R2B-overexpression plasmid or PP2A agonists DT-061 (20 µM) and iHAP1 (10 µM) for 24 h. *, P < 0.05.
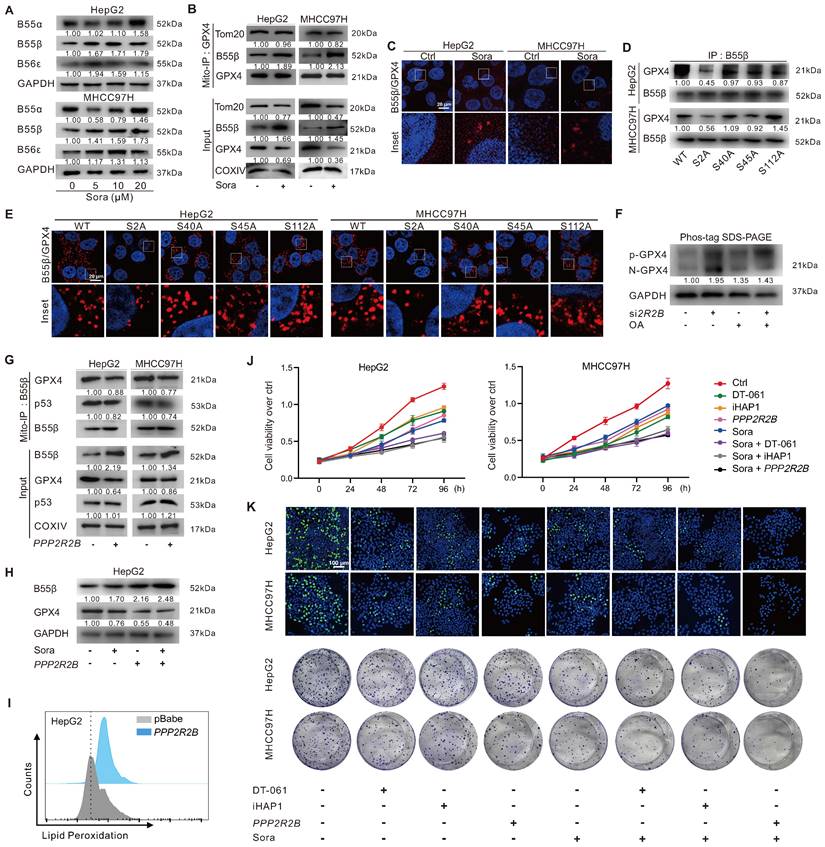
Next, the interaction between PP2A-B55β and GPX4 was explored. Molecular docking showed potential binding ability (Etotal = -591.4 kcal/mol, Eshape = -724.6 kcal/mol, Eforce = 133.2 kcal/mol) between PP2A-B55β and GPX4 (Figure S4A). The Mito-IP assay revealed that PP2A-B55β could interact with GPX4 on mitochondria, and the interaction was further enhanced upon treatment of Sora (Figure 5B). The IF method and proximity ligation assay (PLA) showed the increased PP2A-B55β and GPX4 interaction in HCC cells upon Sora treatment (Figure S4B and Figure 5C). Moreover, decreased PP2A-B55β and GPX4 interaction was only observed in S2A cells but not in S40A, S45A, or S112A cells (Figure 5D-E and Figure S4C), indicating that the Ser2 site of GPX4 played a crucial role in the regulation of PP2A-B55β-targeted GPX4 dephosphorylation. Additionally, PP2A-B55β knockdown (si2R2B) or PP2A inhibition (okadaic acid, OA, an inhibitor of PP2A) increased the protein level of p-GPX4 (Figure 5F), suggesting that PP2A-B55β was the upstream modulator responsible for the dephosphorylation regulation of p-GPX4.
Further, we explored whether PP2A-B55β could affect the p53 retrograde signaling and ferroptosis in HCC cells. As shown in Figure 5G, genetic (PPP2R2B) overexpression of B55β reduced the interaction between GPX4 and p53 on mitochondria. Meanwhile, B55β overexpression accelerated the GPX4 reduction (Figure 5H) and LPO accumulation (Figure 5I) in HepG2 cells. Moreover, using MTS assay, EdU assay, and colony formation assay, we found that the cell proliferation ability of HCC cells was decreased under Sora treatment, and the effects were further aggravated by the co-treatment of PPP2R2B overexpression plasmid or PP2A agonists DT-061 and iHAP1 (Figure 5J-K), indicating that PP2A-B55β activation enhanced the inhibitory effect of Sora on the proliferation ability of HCC cells. Our data revealed that PP2A-B55β might regulate the dephosphorylation of p-GPX4Ser2 and promote Sora-induced ferroptosis in HCC cells.
PP2A-B55β promoted the anti-tumor effect of Sora via aggravation of ferroptosis
Using the xenograft tumor assay, we explored the synergistic anti-tumor effects of genetic PP2A-B55β upregulation and Sora administration (Figure 6A). Xenograft tumor results of 24-day growth curves (Figure 6B), sizes (Figure 6C), and weight (Figure 6D) revealed that tumor growth of the B55β-overexpressing HCC cells was significantly suppressed while compared to the xenograft tumors with control cells. Moreover, compared to the obvious tumor-like mass lesions observed in the control xenograft tumors, B55β-overexpressing tumors were loosely arranged, and necrosis occurred (Figure 6E). These changes were aggravated under treatment with Sora, suggesting that PP2A-B55β expression in HCC enhanced the tumor-suppressive effect of Sora.
PP2A-B55β promoted the anti-tumor effect of Sora via aggravation of ferroptosis. A. The diagram depicted the experimental design of the in vivo study. BALB/c nude mice were subcutaneously inoculated with the constructed HepG2-PPP2R2B cells with B55β-overexpression, while HepG2-pBabe cells were used as control cells. Mice harbored with HCC-xenograft tumors were randomly divided into 4 groups (n = 4 for each group): pBabe-Ctrl, pBabe-Sora, PPP2R2B-Ctrl, PPP2R2B-Sora. The Sora (10 mg/kg) was injected via the tail vein every two days. B. The growth curves of xenograft tumors in nude mice. Tumor volume = (W2 × L)/2, where L is the longer dimension and W is the shorter one. C-D. Representative images (C) and weight (D) of xenograft tumors. E. Representative H&E staining images of xenograft tumor tissues. F-G. Levels of GSH (F) and MDA (G) in xenograft tumor tissues. H. TEM observation of mitochondrial morphological characteristics (red arrows) of HCC cells in xenograft tumor tissues. N, nucleus; M, mitochondria. Scale bar, 0.5 μm. I. Representative IHC staining images of GPX4 and B55β expression in xenograft tumor tissues. J. Protein levels of p53 in the nucleus and in the mitochondrial or cytoplasmic fractions of xenograft tumor tissues. *, P < 0.05.
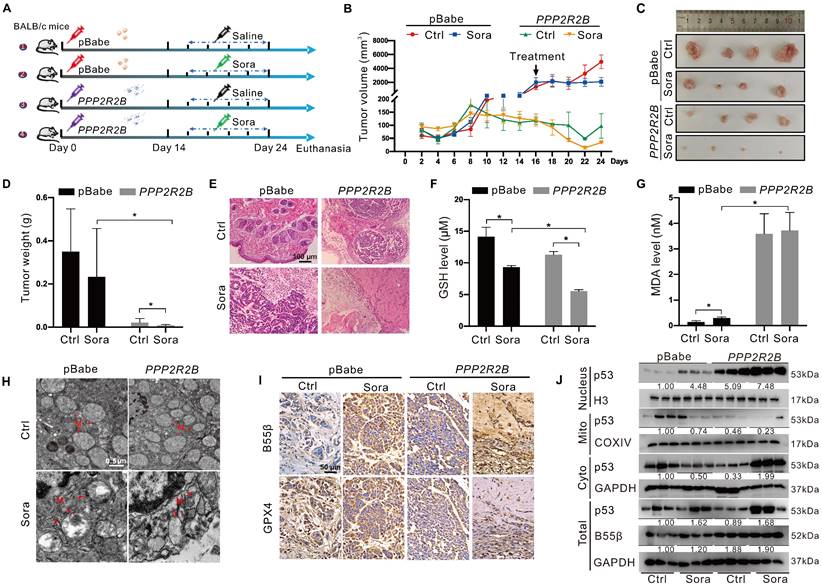
Further, we detected the effect of PP2A-B55β on the occurrence of ferroptosis in the xenograft tumors. Compared to the control tumors, reduced GSH and increased MDA were observed in the PPP2R2B overexpression tumors (Figure 6F-G). Meanwhile, TEM showed that the mitochondria became smaller in the PPP2R2B overexpression tumors, while the mitochondrial cristae disappeared, a characteristic change of ferroptosis (Figure 6H). Moreover, immunohistochemistry (IHC) analysis of the dissected xenograft tissues showed that GPX4 expression level was decreased in PPP2R2B overexpression tumors (Figure 6I). These changes were aggravated under the treatment of Sora, indicating that PP2A-B55β promoted Sora-induced ferroptosis in the HCC xenograft tumors. Then, we explored the effects of PP2A-B55β on the retrograde signal of mitochondrial p53 in the xenograft tumors. Consistent with the results in vitro, genetic PPP2R2B overexpression increased the protein level of nucleus p53 but decreased the protein level of mitochondrial p53 (Figure 6J). Altogether, these results obtained from the xenograft animal model suggested that PP2A-B55β enhanced Sora-induced ferroptosis, thereby inhibiting tumor growth.
Discussion
HCC is the most common type of liver cancer with high morbidity and lethality, and its incidence continues to increase worldwide [28]. Though Sora has shown significant effects in mitigating the development of HCC, drug resistance and poor prognosis to Sora therapy remain formidable challenges in human HCC.
The toxic effect of Sora on cancer cells partly relies on inducing ferroptosis. GPX4 is the only enzyme that can diminish lipid hydroperoxides of biological membranes, which is vital in protecting cells from oxidative damage in ferroptosis [4]. Tumor cells may develop resistance to ferroptosis-dependent cancer treatment by augmenting GPX4 expression, and GPX4-targeted attenuation of its activity has been proposed as a potential approach to sensitize HCC to the treatment of Sora [29]. In the present study, we found that a novel route enhanced the anti-tumor effect of Sora by affecting the mitochondrial GPX4 PTM. GPX4 (S2 site) Ser-to-Ala mutant (S2A), which mimics GPX4 dephosphorylation, failed to reverse the LPO generation in HCC cells and sensitized HCC to Sora-induced ferroptosis. Similar to us, the stable expression of GPX4 Sec-to-Ser mutant in the GPX4 deficiency cell model did not reverse cell death from lack of GPX4 [30]. A previous study reported that, compared to the Ala-to-Cys mutant of GPX4, the Ala-to-Sec mutant contributed to GPX4 binding of RSL3, a selective ferroptosis activator for tumor cells carrying oncogenic RAS [19]. Furthermore, Michael et al. found that covalently modifying the Cys93 site of GPX4 by fumarates that accumulated in fumarate hydratase inactivation conditions promoted ferroptosis in renal carcinoma cells, highlighted a crucial role of GPX4 PTM in ferroptosis regulation [31]. The current results and those of the previous studies suggested that mitochondrial GPX4 PTM might be an available strategy for regulatory ferroptosis in HCC. Our study firstly revealed that the dephosphorylation of mitochondrial GPX4 positively modulated ferroptosis in HCC.
Given the positive role of p-GPX4Ser2 dephosphorylation in Sora-induced ferroptosis, combining protein phosphatase activation and Sora may exert more effective anti-cancer effects in HCC treatment. PP2A is a major member of serine/threonine protein phosphatase family and is vital in mitochondrial quality control and cancer development [23-27]. In response to endogenous and exogenous stimulation, the multiple regulatory B subunits of PP2A can be selectively translocated to mitochondria and are involved in various mitochondria-mediated physiopathological processes [24-27]. Herein, we verified that PP2A-B55β not only directly interacted with mitochondrial GPX4 but might affect the stability of GPX4 by regulating the phosphorylation status of GPX4. Furthermore, PP2A-B55β overexpression facilitated Sora-induced GPX4 reduction and LPO accumulation in HCC cells and xenograft tumors in vitro and in vivo. The above evidences suggested that PP2A-B55β-mediated dephosphorylation of the p-GPX4 might result in the degradation of GPX4 and promote the occurrence of ferroptosis. The related research on PP2A and ferroptosis was very limited. FTY720, an FDA-approved immunosuppressant, showed obvious anti-tumor properties in many cancer models by inducing PP2A activation [32]. Studies recently further verified that PP2A activation was essential for FTY720-induced ferroptosis in multiple myeloma cells, highlighting the role of PP2A in ferroptosis-associated cancer development [33]. Although some pharmacological inhibitors (such as statins) could sensitize cancer cells to ferroptosis via targeted inhibition of GPX4, oxidative damage to normal cells and newly developed drug resistance restrict its clinical application [8, 34]. The combined use of PP2A-B55β activation and Sora might be a promising strategy for enhancing ferroptosis-based HCC therapy.
Another novel finding of the present study was that GPX4 could interact with p53 in mitochondria under hyperphosphorylation status. Moreover, dephosphorylation of p-GPX4Ser2 decreased the GPX4 and p53 interaction and caused an obvious reduction of mitochondrial p53. p53 plays a vital role in the nucleus to mitochondrial communication. p53 translocation from the nucleus to mitochondria was proven to maintain mitochondrial homeostasis via interaction with multiple mitochondrial proteins [35]. Furthermore, inconsistent with the dynamics of p53 in mitochondria, p-GPX4Ser2 dephosphorylation increased the p53 level in the nucleus, indicating that p-GPX4Ser2 dephosphorylation enhanced the p53 translocation from mitochondria to the nucleus (known as the retrograde signal of p53). The retrograde signal of p53 is an essential feedback approach for mitochondria-to-nucleus interorganellar communication. However, an excessive retrograde signal can lead to malignant cell fate [36]. Herein, we found that the increased nucleus signal of p53 aggravated the occurrence of ferroptosis in HCC, whereas the phenomenon was not obvious in p53 nucleus deletion cells. Consistent with us, rotenone exposure triggered the retrograde signal of p53, and the signals preceded mitochondrion dysfunction and cell death [17]. p53 has been verified to be a positive regulator in promoting ferroptosis, and multiple ferroptosis-related genes and proteins were proved to directly interact with p53 or be the transcriptional targets of p53 [37]. Therefore, the retrograde signal of p53 might be an early event of ferroptosis, which was one of underlying mechanisms for elevating ferroptosis via the regulation of p-GPX4Ser2 dephosphorylation.
We also noted that there were some limitations of the present study. First, since the phosphorylation modification of the Ser2 site of GPX4 was not confirmed by LC/MS/MS analysis in HEK293T cells, the functional screening and regulation of p-GPX4Ser2 in Sora-treated HCC cells should be concerned with caution and remained for further validation. Moreover, the present study suggested that PINK1 might be the potential kinase responsible for mitochondrial GPX4 phosphorylation, which would be another research direction that remains verified by the in vitro phosphorylation and IP analysis, as well as for the targeted intervention of GPX4 phosphorylation. Nevertheless, the present study identified that the B55β/p-GPX4Ser2/p53 axis was a novel regulatory pathway of ferroptosis. Mitochondrial p-GPX4Ser2 dephosphorylation initiated ferroptosis in HCC via promoting the retrograde signaling of p53. The targeting regulation of PP2A-B55β on the dephosphorylation of p-GPX4Ser2 could serve as a novel targeted intervention strategy for enhancing ferroptosis-dependent tumor theranostics.
Methods
Bioinformatics analysis
Public datasets, including the GEO database (GSE102079) and the Cancer Genome Atlas (TCGA) liver hepatocellular carcinoma (LIHC) dataset, were used to extract RNA expression data from HCC patients. We utilized LIMMA to conduct the differentially expressed genes (DEGs) analysis. The Log2 (fold change) and P-value were 1.0 and 0.05, respectively. The DEGs analysis and Gene Ontology (GO) enrichment were performed on R studio software (version 4.1.1). Gene set enrichment analysis (GSEA) was performed using the GSEA software (version 4.1.0) to analyze the enrichment of the ferroptosis pathway in tumors. Expression and survival analysis of GPX4 in LIHC were conducted using the GEPIA database (http://gepia.cancer-pku.cn/index.html).
HCC tissue specimens
HCC tumor tissues and the corresponding peritumor tissues were obtained from six HCC patients at Xiang'an Hospital of Xiamen University (Xiamen, China). HCC tissue specimens were immediately frozen in liquid nitrogen and then stored at -80 °C for the follow-up experiments. Part of the HCC tissue specimens were fixed and paraffin-embedded before the pathological examination. Western blotting (WB) and immunohistochemistry (IHC) staining were used to detect the protein levels of p53 and GPX4. All patients signed the informed consent. The present study was approved by the Ethics Committee of Xiamen University and performed following the Helsinki Declaration.
In vivo xenograft tumor study
Six-week-old BALB/c nude mice were purchased from SLAC Laboratory Animal Co. Ltd (Shanghai, China). HepG2-PPP2R2B cells with B55β-overexpression were constructed, while HepG2-pBabe cells were used as control cells. 3 × 106 cells (HepG2-pBabe or HepG2-PPP2R2B) in 50 μL PBS combined with 50 μL of Matrigel (BD, CA, USA) were subcutaneously inoculated into the right flank of each mouse. When xenograft tumors reached palpable size (at day 14 after injection), inoculated mice were randomly divided into 4 groups (n = 4): pBabe-Ctrl, pBabe-Sora, PPP2R2B-Ctrl, PPP2R2B-Sora. The saline or Sora (10 mg/kg) was injected via the tail vein every two days. The dimensions of xenograft tumors were measured using a digital caliper. The formula (W2 × L)/2 was used to estimate tumor volume, where L is the longer dimension and W is the shorter one. After five 2-day cycles of treatment, the mice were executed and xenograft tumors were excised and weighed. Xenograft tumor tissues were collected for subsequent Western blotting (WB) or tissue sectioning and IHC analysis. All experiments were approved by the Experimental Animal Ethics Committee of Xiamen University (Ethic protocol code: XMULAC20180094).
IHC analysis and hematoxylin-eosin (HE) staining
The dissected xenograft tumor tissues were fixed in a 4% paraformaldehyde (PFA) fix solution, followed by paraffin embedding and serially sectioning at 4 μm thickness. Sections were deparaffinized with xylene and dehydrated with an ethanol series of increasing concentrations. IHC detection was accomplished with the UltraSensitiveTM SP IHC Kit (MXB, Fuzhou, China) as previously described [38]. Information on the antibodies, including anti-B55β, anti-GPX4, and anti-p53, were listed in Table S1. Sections were subsequently stained with HE for 10 min. Then, the sections were washed with xylene after dehydration in gradient ethanol solutions, sealed with neutral resin, and observed by an inverted microscope.
Detection of malondialdehyde (MDA) and glutathione (GSH) levels
Xenograft tumor tissues (about 5 mg) were washed with pre-cooled PBS. 200 μL of PBS was added to prepare tissue homogenate, followed by the dilution with saline and subsequent determination. According to the manufacturer's instructions, MDA and GSH levels were tested using the corresponding reagent kits (Nanjing Jiancheng, China). Optical density values were detected by the microplate reader, while the excitation wavelength was 530 nm for MDA and 405 nm for GSH. Protein concentrations were tested using bicinchoninic acid (BCA) protein assay to calculate MDA and GSH levels per mg of protein.
Cell culture and reagents
HepG2, MHCC97H, Hep3B, and HEK293T cell lines were stocked in our laboratory. Cells are maintained in Dulbecco's Modified Eagle Medium (DMEM, Gibco, NY, USA) with 10% fetal bovine serum (FBS, Gibco, CA, USA) and 1% penicillin-streptomycin (Gibco, CA, USA) at 37 °C in a 5% CO2 humidified incubator (Thermo, CO, USA). Dimethyl sulfoxide (DMSO) was purchased from Sigma (MO, USA). Sorafenib (Sora) and Ferrostatin-1 (Fer-1) were purchased from Selleck (TX, USA).
Establishment of GPX4-overexpressing and its phosphorylated site-directed mutagenesis cells
The construction of stable GPX4-overexpressing HepG2-GPX4 cells was the same as our previously described [38]. In brief, full-length GPX4 coding sequence was obtained and incorporated into the retroviral vector pBabe-puro to construct the pBabe-GPX4 recombinant plasmid. HEK293T cells were co-transfected with retroviral plasmid and pCL-Ampho vector in a ratio of 1:1. HepG2 cells were transfected with retroviruses produced by HEK293T cells. The established cell lines were screened with 0.6 μg/mL puromycin. The phosphorylation sites of GPX4 were predicated by DISPHOS 1.3 (http://www.dabi.temple.edu/disphos/), PhosphoSVM (http://sysbio.unl.edu/PhosphoSVM/), MusiteDeep (https://www.musite.net/), Gps 6.0 (http://gps.biocuckoo.cn/), and NetPhon3.1 (https://services.healthtech.dtu.dk/services/NetPhos-3.1/). The serine 2 (S2), serine 40 (S40), serine 45 (S45), and serine 112 (S112) sites of GPX4 were mutated to alanine (A) or aspartate (D) according to the instructions of the KOD-Plus Mutagenesis Kit (Toyobo, Osaka, Japan). Information of mutation primers was shown in Table S2.
Cell viability assay
Cell viability assay was performed as previously described [39]. In brief, 1 × 104 cells of each cell line were seeded on 96-well plates and treated with different administration. MTS solution was added and the cells were incubated for 3 h at 37 °C. The absorbance at 490 nm was quantified using a microplate spectrophotometer system (Multiskan, Thermo, USA). Fer-1 and inhibitors of different cell death patterns were used in combination with Sora to identify whether cell viability was affected by ferroptosis.
Transmission electron microscope (TEM) analysis
After treatment with 10 µM of Sora for 24 h, the cells were collected, fixed with 0.1 M glutaraldehyde buffer, post-fixed with 4% PFA, dehydrated with ethanol and propylene oxide, and finally embedded in epoxy resin. About 70-80 nm thickness sections were sliced and stained with uranyl acetate and lead citrate. Mitochondrial and nuclear ultrastructure was evaluated using a TEM (Tecnai 20, FEI, USA). Similarly, for animal study, xenograft tumor tissues were collected and followed the above methods.
Immunofluorescence (IF) analysis
Cells were mounted on coverslips for different experimental conditions and stained with 100 nM MitoTracker Red CMXRos (Life, CA, USA) in a culture medium at 37 °C for 30 min. Cells were fixed in 4% PFA and permeabilized with 0.5% Triton X-100 in PBS for 5 min. After blocked in PBS containing 1% BSA, cells were incubated overnight with primary antibodies (anti-B55β, anti-GPX4, or anti-p53), followed by the appropriate fluorescent secondary antibodies for 1 h in the dark. Detail information on the antibodies was listed in Table S1. Experiments for detecting protein changes in the nucleus need to be stained with DAPI for 5 min before viewing and shooting. Cells were viewed using a laser-scanning confocal microscope (Leica SP8, Wetzlar, Germany).
Proximity ligation assay (PLA)
The protein-protein interaction studies were performed with PLA. According to the manufacturer's instructions, a Duolink® In Situ Detection Reagent (Sigma, MO, USA) was used. Briefly, cells were mounted on sterile coverslips in 24-well plates at a density of 5 × 104 cells per well overnight. After being fixed with 4% PFA and permeabilized using 0.5% Triton X-100, cells were blocked in Duolink II solution for 1 h. The coverslips were incubated with anti-p53, anti-B55β, or anti-GPX4 at 4 °C overnight (the detailed information on the antibodies was listed in Table S1), followed by Duolink PLA anti-Rabbit PLUS and PLA anti-Mouse PLUS proximity probes. After washing the coverslips three times, the ligation reaction was conducted for 30 min, and the amplification was run for 100 min at 37 °C. Then the coverslips were visualized using a confocal microscope (Leica SP8, Wetzlar, Germany). Images were analyzed with IPP 6.0 software.
Lipid peroxidation (LPO) assay
Cells were seeded into 6-well plates with 3×105 per well. After the indicated treatment, the cells were digested and stained with 2 μM BODIPY-C11 (Invitrogen, CA, USA) for 40 min at 37 °C, followed by flow cytometry analysis (Beckman, CA, USA). At least 1×104 cells were analyzed for each condition, each experiment was independently performed at least 3 times, and representative experimental results were shown. Data analysis was performed using the FlowJo 10.4 software.
Isolation of mitochondrial, nuclear, and cytosolic fractions
Mitochondrial and cytosolic fractions were prepared using a mitochondrial isolation kit (Enzo Life, PA, USA). Briefly, 5×107 cells of each group were harvested and centrifuged at 600 × g at 4 °C for 5 min. The pellets were resuspended in mitochondria isolation buffer and centrifuged at 600 × g for 10 min. The supernatants were collected and centrifuged at 12,000 × g for 10 min. The pellet was mitochondria, and the supernatant was the cytosolic fraction. Resuspended the pellet in mitochondria lysate buffer and centrifuged at 12,000 × g at 4 °C for 10 min to achieve mitochondrial fractions. Proteins were quantified with the BCA protein assay and were used for Western blotting (WB) analysis.
As for the nuclear fraction, cells were harvested and centrifuged at 600 × g for 4 min at 4 °C to remove the supernatant. Cells were resuspended with PMSF-added cytoplasmic protein extraction reagent A followed by the ice bath for 15 min. Then cytoplasmic protein extraction reagent B was added and centrifuged at 16,000 × g at 4 °C for 5 min. The supernatant was a cytoplasmic fraction. After sucking up the supernatant, the nuclear protein extraction reagent containing PMSF was added and centrifuged at 16,000 × g for 10 min at 4 °C. The supernatant is the nuclear fraction.
Western blotting (WB) analysis and immunoprecipitation (IP) assay
Cells were washed with ice-cold PBS, and whole-cell lysates were prepared in an SDS/β-mercaptoethanol sample buffer containing protease inhibitors. Proteins were separated by 10-12% SDS-PAGE gels, transferred to PVDF membranes (Millipore, MA, USA), blocked in 5% skimmed milk for 1 h at room temperature then incubated with the primary antibodies at 4 °C overnight. After incubation with goat Anti-Rabbit-IgG-HRP or goat Anti-Mouse-IgG-HRP for 1 h at room temperature, proteins were visualized by enhanced chemiluminescence. IP assay was performed as described previously [40]. The detailed information on the primary antibodies including the manufacturers, code number, the dilution used in WB, IP, and IHC, molecule weight, and species were listed in Table S1.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from cells using TRIzol reagent (Ambion, TX, USA). cDNA was reversed from total RNA using PrimeScriptTM RT reagent kit (TaKaRa, Otsu, Japan) and qRT-PCR was subsequently performed the same in our previous study [38]. The relative mRNA transcription levels of genes were displayed using the 2-ΔΔCt method. The information of primers used in this study was listed in Table S2.
Statistics
Statistical analyses were performed using the Statistical Package for Social Sciences (SPSS) version 16.0 (SPSS, Chicago, IL, USA). Unless indicated, all data are shown as mean ± standard deviation (SD) of at least three independent experiments. Statistical comparison of mean values in two groups was assessed using the Student's t-test. Multiple groups were compared by one-way analysis of variance (ANOVA) with Dunnett's post-test. Pearson's correlation analysis was performed for the correlation between variables. A P-value of < 0.05 was considered to be statistically significant. All the experiments were repeated at least three times.
Abbreviations
Ala: alanine; Asp: aspartate; BCA: bicinchoninic acid; IP: immunoprecipitation; Cyto: cytosolic; DEGs: differentially expressed genes; FCM: flow cytometry; GEO: gene expression omnibus; GO: gene ontology; GPX4: glutathione peroxidase 4; GPX4ser2: GPX4 serine 2; GSEA: Gene set enrichment analysis; GSH: glutathione; HCC: hepatocellular carcinoma; H3: Histone 3; IHC: immunohistochemistry; IF: immunofluorescence; LIHC: liver hepatocellular carcinoma; LPO: lipid peroxidation; MDA: malondialdehyde; Mito: mitochondrial; MMP: mitochondrial membrane potential; OMM: outer mitochondrial membrane; PLA: proximity ligation assay; PP2A: phosphatase protein phosphatase 2A; PTM: post-translational modification; qRT-PCR: quantitative real-time polymerase chain reaction; ROS: reactive oxygen species; Ser: serine; Sora: sorafenib; System Xc-: cystine-glutamate antiporter; TCGA: the cancer genome atlas; TEM: transmission electron microscope; WT: wild type; WB: western blotting.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We are thankful for the grants from the National Natural Science Foundation of China (Nos. 81973082, 81874272, 82073588, 82273667), the Marine Science Programs of the Ocean and Fisheries Department of Fujian (No. FJHJF-L-2022-5), The Industry-University-Research Cooperation Project of Fujian Science and Technology Plan (No. 2022Y4009), XMU Undergraduate Innovation and Entrepreneurship Training Programs (Nos. S202010384747, 2019Y0810, 2019X0797), and Innovative Practice Platform for Undergraduate Students, School of Public Health Xiamen University (No. 202005).
Availability of data and materials
Please contact the corresponding author for all data requests.
Author contributions
Bo Qian, Lin Che, Ze-Bang Du, and Ni-Jun Guo: provision of study materials, reagents, and materials, specifically performing the experiments. Xin-Mou Wu, Lei Yang, Zhao-Xuan Zheng, Yun-Lu Gao, and Ming-Zhu Wang: data curation, data analysis, and original draft writing. Xiao-Xuan Chen and Ling Xu: providing necessary writing guidance. Zi-Jian Zhou, Yu-Chun Lin, and Zhong-Ning Lin: formulation of research goals and methodology design.
Ethical approval and consent to participate
The research followed the Principles and Guidelines for the Use of Animals in Research, Testing, and Education issued by the New York Academy of Sciences Adhoc Committee on Animal Research. All experiments were approved by the Experimental Animal Ethics Committee of Xiamen University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M. et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018
2. Chen J, Jin R, Zhao J, Liu J, Ying H, Yan H. et al. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2015;367:1-11
3. Louandre C, Marcq I, Bouhlal H, Lachaier E, Godin C, Saidak Z. et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015;356:971-7
4. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W. et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16:302-9
5. Wang Q, Bin C, Xue Q, Gao Q, Huang A, Wang K. et al. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021;12:426
6. Zhao HY, Ji B, Chen JG, Huang QF, Lu XG. Gpx 4 is involved in the proliferation, migration and apoptosis of glioma cells. Pathol Res Pract. 2017;213:626-33
7. Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA. et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol. 2018;14:507-15
8. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A. et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247-50
9. Ghoochani A, Hsu EC, Aslan M, Rice MA, Nguyen HM, Brooks JD. et al. Ferroptosis inducers are a novel therapeutic approach for advanced prostate cancer. Cancer Res. 2021;81:1583-94
10. Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hilpmann A, Ryan MJ. et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol. 2020;16:497-506
11. Arai M, Imai H, Koumura T, Yoshida M, Emoto K, Umeda M. et al. Mitochondrial phospholipid hydroperoxide glutathione peroxidase plays a major role in preventing oxidative injury to cells. J Biol Chem. 1999;274:4924-33
12. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H. et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586-90
13. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K. et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. 2018;172:409-22.e21
14. Zhang Y, Xia M, Zhou Z, Hu X, Wang J, Zhang M. et al. p53 promoted ferroptosis in ovarian cancer cells treated with human serum incubated-superparamagnetic iron oxides. Int J Nanomedicine. 2021;16:283-96
15. Chen C, Huang Y, Xia P, Zhang F, Li L, Wang E. et al. Long noncoding RNA Meg3 mediates ferroptosis induced by oxygen and glucose deprivation combined with hyperglycemia in rat brain microvascular endothelial cells, through modulating the p53/GPX4 axis. Eur J Histochem. 2021;65:3224
16. Zhang Z, Guo M, Shen M, Kong D, Zhang F, Shao J. et al. The BRD7-P53-SLC25A28 axis regulates ferroptosis in hepatic stellate cells. Redox Biol. 2020;36:101619
17. Green ML, Pisano MM, Prough RA, Knudsen TB. Release of targeted p53 from the mitochondrion as an early signal during mitochondrial dysfunction. Cell Signal. 2013;25:2383-90
18. Ding Y, Chen X, Liu C, Ge W, Wang Q, Hao X. et al. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J Hematol Oncol. 2021;14:19
19. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113:E4966-75
20. Jian C, Fu J, Cheng X, Shen LJ, Ji YX, Wang X. et al. Low-dose sorafenib acts as a mitochondrial uncoupler and ameliorates nonalcoholic steatohepatitis. Cell Metab. 2020;31:892-908.e11
21. Díaz-González Á, Sapena V, Boix L, Brunet M, Torres F, N LL. et al. Pharmacokinetics and pharmacogenetics of sorafenib in patients with hepatocellular carcinoma: Implications for combination trials. Liver Int. 2020;40:2476-88
22. Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K. et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. 2020;5:e132747
23. Sangodkar J, Farrington CC, McClinch K, Galsky MD, Kastrinsky DB, Narla G. All roads lead to PP2A: exploiting the therapeutic potential of this phosphatase. FEBS J. 2016;283:1004-24
24. Wlodarchak N, Xing Y. PP2A as a master regulator of the cell cycle. Crit Rev Biochem Mol Biol. 2016;51:162-84
25. Liu WH, Chou WM, Chang LS. p38 MAPK/PP2Acα/TTP pathway on the connection of TNF-α and caspases activation on hydroquinone-induced apoptosis. Carcinogenesis. 2013;34:818-27
26. Tomasella A, Blangy A, Brancolini C. A receptor-interacting protein 1 (RIP1)-independent necrotic death under the control of protein phosphatase PP2A that involves the reorganization of actin cytoskeleton and the action of cofilin-1. J Biol Chem. 2014;289:25699-710
27. Teixeira V, Medeiros TC, Vilaça R, Pereira AT, Chaves SR, Côrte-Real M. et al. Ceramide signalling impinges on Sit4p and Hog1p to promote mitochondrial fission and mitophagy in Isc1p-deficient cells. Cell Signal. 2015;27:1840-9
28. Jafri W, Kamran M. Hepatocellular carcinoma in Asia: A challenging situation. Euroasian J Hepatogastroenterol. 2019;9:27-33
29. Guerriero E, Capone F, Accardo M, Sorice A, Costantini M, Colonna G. et al. GPX4 and GPX7 over-expression in human hepatocellular carcinoma tissues. Eur J Histochem. 2015;59:2540
30. Ingold I, Aichler M, Yefremova E, Roveri A, Buday K, Doll S. et al. Expression of a catalytically inactive mutant form of glutathione peroxidase 4 (Gpx4) confers a dominant-negative effect in male fertility. J Biol Chem. 2015;290:14668-78
31. Kerins MJ, Milligan J, Wohlschlegel JA, Ooi A. Fumarate hydratase inactivation in hereditary leiomyomatosis and renal cell cancer is synthetic lethal with ferroptosis induction. Cancer Sci. 2018;109:2757-66
32. Young MM, Bui V, Chen C, Wang HG. FTY720 induces non-canonical phosphatidylserine externalization and cell death in acute myeloid leukemia. Cell Death Dis. 2019;10:847
33. Zhong Y, Tian F, Ma H, Wang H, Yang W, Liu Z. et al. FTY720 induces ferroptosis and autophagy via PP2A/AMPK pathway in multiple myeloma cells. Life Sci. 2020;260:118077
34. Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2019;133:144-52
35. Galluzzi L, Morselli E, Kepp O, Vitale I, Pinti M, Kroemer G. Mitochondrial liaisons of p53. Antioxid Redox Signal. 2011;15:1691-714
36. Vizioli MG, Liu T, Miller KN, Robertson NA, Gilroy K, Lagnado AB. et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020;34:428-45
37. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57-62
38. Zhuang Q, Zhou T, He C, Zhang S, Qiu Y, Luo B. et al. Protein phosphatase 2A-B55delta enhances chemotherapy sensitivity of human hepatocellular carcinoma under the regulation of microRNA-133b. J Exp Clin Cancer Res. 2016;35:67
39. He CY, Jiang SW, Jin HJ, Chen SZ, Lin G, Yao H. et al. Mitochondrial electron transport chain identified as a novel molecular target of SPIO nanoparticles mediated cancer-specific cytotoxicity. Biomaterials. 2016;83:102-14
40. Che L, Wu JS, Du ZB, He YQ, Yang L, Lin JX. et al. Targeting mitochondrial COX-2 enhances chemosensitivity via Drp1-dependent remodeling of mitochondrial dynamics in hepatocellular carcinoma. Cancers (Basel). 2022;14:821
Author contact
![]() Corresponding authors: zhouzedu.cn (Z.-J. Zhou), linychedu.cn (Y.-C. Lin), and linzhnedu.cn (Z.-N. Lin).
Corresponding authors: zhouzedu.cn (Z.-J. Zhou), linychedu.cn (Y.-C. Lin), and linzhnedu.cn (Z.-N. Lin).