Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Integrin structure, ligand...
3. Approaches to integrin...
4. αIIbβ3 integrin
5. α4 integrins
6. αv integrins
7. α5β1 integrin...
8. Conclusion
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2011; 1:154-188. doi:10.7150/thno/v01p0154 This volume Cite
Review
Integrin Targeted Therapeutics
Melissa Millard, Srinivas Odde, Nouri Neamati ![]()
Department of Pharmacology and Pharmaceutical Sciences, University of Southern California, School of Pharmacy, PSC304 1985 Zonal Avenue, Los Angeles, CA 90089
Published 2011-2-17
Abstract
Integrins are heterodimeric, transmembrane receptors that function as mechanosensors, adhesion molecules and signal transduction platforms in a multitude of biological processes. As such, integrins are central to the etiology and pathology of many disease states. Therefore, pharmacological inhibition of integrins is of great interest for the treatment and prevention of disease. In the last two decades several integrin-targeted drugs have made their way into clinical use, many others are in clinical trials and still more are showing promise as they advance through preclinical development. Herein, this review examines and evaluates the various drugs and compounds targeting integrins and the disease states in which they are implicated.
Keywords: Integrin-targeted therapeutics, abciximab, tirofiban, eptifibatide, natalizumab, cilengitide, abegrin, acute coronary syndromes, Multiple Sclerosis, Crohn's Disease, cancer, angiogenesis, osteoporosis, age related macular degeneration, integrin-targeted small molecules, integrin-targeted peptides, integrin-targeted therapeutic antibodies
1. Introduction
Integrins are heterodimeric cell surface receptors found in nearly all metazoan cell types, composed of non-covalently linked α and β subunits. In mammals, eighteen α-subunits and eight β-subunits have been identified to date [1]. From this pool, 24 distinct heterodimer combinations have been observed in vivo that confer cell-to-cell and cell-to-ligand specificity relevant to the host cell and the environment in which it functions [2]. Integrin-mediated interactions with the extracellular matrix (ECM) are required for the attachment, cytoskeletal organization, mechanosensing, migration, proliferation, differentiation and survival of cells in the context of a multitude of biological processes including fertilization, implantation and embryonic development, immune response, bone resorption and platelet aggregation. Integrins also function in pathological processes such as inflammation, wound healing, angiogenesis, and tumor metastasis. In addition, integrin binding has been identified as a means of viral entry into cells [3].
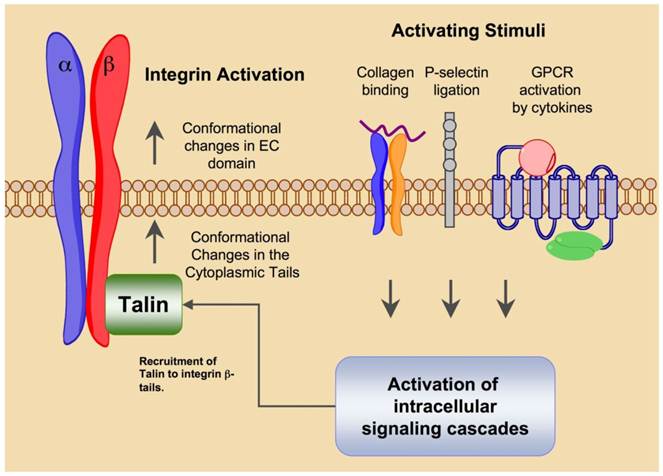
Integrin mediated cellular adhesion is dynamic process that is both indicative of and influenced by cell status. Integrins are not constitutively active but rather exist in multiple activation states wherein ligand binding stability is related to the activation status of the receptor. The biodirectional nature of integrin mediated signaling allows both extra- and intracellular cues to regulate activation [4] (Fig. 1). Extracellular factors that influence integrin activation are ligand binding, divalent cation concentration, chemokine signaling and mechanical stress. Intracellular signaling events that converge on the cytoplasmic domain of integrin tails are also capable of activating the high affinity state. Intracellular signals that result in integrin activation are referred to as 'inside-out' signaling events.
Integrin “Inside-out” signaling promotes integrin activation and heightens ligand affinity. External activating stimuli such as integrin or selectin ligation or cytokine binding to G-protein coupled receptors triggers intracellular signaling cascades that converge on integrin β-subunit tails. The activating signals induce conformational changes in the integrin tails that facilitate the docking of focal adhesion components such as Talin. The conformational changes in the tail region are transduced to the extracellular domain where structural rearrangments promote activation and increased affinity for ligand.
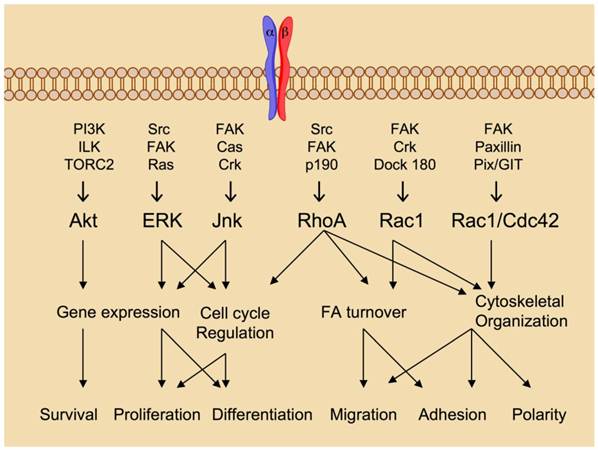
Integrin ligation via cell-to-cell and cell-to-matrix contacts transduces intracellular signaling events in a process called 'outside-in' signaling (Fig. 2). The integrin tails have no intrinsic kinase activity but rather serve as a site for the docking of various kinases and related adaptor proteins that comprise focal adhesions. The β-tail is the primary docking site in the formation of focal adhesions, but the alpha tail in some cases may also serve as a signaling scaffold for calcium dependent signaling events. Signals emanating from focal adhesions have been shown to promote survival, differentiation and proliferation [5]. In the absence of integrin ligation, these processes are abrogated therefore pharmacological inhibition of integrin ligation is of great interest for the therapy of numerous diseases resulting from abberant integrin mediated signaling.
Integrin “Outside-in” signaling governs cellular processes. Ligand binding serves as the initiation point for transduction of intracellular signaling cascades that regulate a multitude of biological processes. Figure adapted from reference 5 with permission.
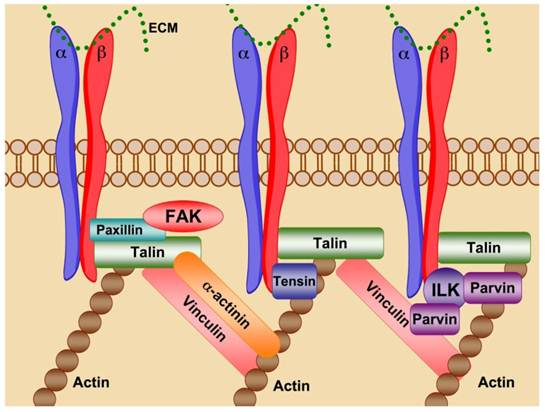
Through cell-cell and cell-ECM contacts, integrins behave as mechanosensors, bi-directionally transducing information both into and out of the cell, directing the strength, stability and growth of focal adhesions to promote cell adhesion, spreading and motility (Fig. 3). Talin binding to activated integrin β-tails promotes cytoskeletal organization via the crosslinking of both vinculin and actin. Association of talin with vinculin promotes focal adhesion growth, while crosslinking of viculin and actin through talin provides stable adhesion [5]. Disruption of focal adhesions prevents integrin mediated cell adhesion and impaired cell motility and migration. Prolonged integrin inhibition in adhesion dependent cells results in anoikis, apoptotic cell death due to ECM deprivation [6].
Integrins are the site of focal adhesions. Integrin ligation promotes receptor clustering and the formation of focal adhesions. Talin forms the initial contacts between integrin β-tails and the actin cytoskeleton. Vinculin crosslinks with Talin and actin to strengthen focal adhesions promoting focal adhesion growth. α-actinin can interact with integrin β-tails, vinculin, talin and actin to further reinforce crosslinking of the cytoskeleton. ILK associates with actin through its binding partner parvin creating a core scaffold for focal adhesion formation. Focal adhesion kinase (FAK) is involved in focal adhesion stabilization and turnover likely through post-translational modification of focal adhesion components.
By integrating and transducing information both into and out of the cell, integrins mediate cell localization, shape, spreading and motility and thus are critical determinants of both health and disease. Integrins have been implicated in the pathogenesis of inflammatory disease, platelet aggregation, tumor progression as well as osteoporosis and macular degeneration. The role of integrins in pathological conditions coupled with their 'druggability' by means of cell surface accessibility makes them attractive pharmacological targets.
2. Integrin structure, ligand binding and activation
The structural classification of integrins is based on the presence or absence of an extracellular A-type domain in the α- subunit. A-type domain integrins contain an ~180 amino acid metal ion coordinating insert that is required for ligand binding [7]. Within this domain a five amino acid motif termed the metal ion dependent adhesion site (MIDAS) acts to coordinate a divalent metal ion. Ligand binding places a glutamate residue within MIDAS to participate in metal coordination [8]. Integrins lacking the A-type domain contain a similar domain within the β-subunit, termed the βA domain. Ligand binding to βA integrins occurs through a divalent cation dependent site that requires glutamate or a similar acidic residue within the ligand to stabilize the metal coordination [9]. In contrast to the αA-type domain, in the absence of ligand a proximal glutamate can serve as an alternate coordinating residue.
Information regarding integrin structure has been obtained by NMR, electron microscopy and rotary shadowing techniques. High resolution crystal structures are the method of choice for structural determination for the development of therapeutics. High-resolution crystal structures of membrane spanning proteins are a challenge to obtain due to the fluid nature of most biological membranes. For this reason there are no entire integrin crystal structures. The most complete information regarding integrin atomic structure to date has been derived from the crystal structures of the ectodomains of αvβ3 and αIIbβ3 in both the presence and absence of ligand [10-12]. Due to the homology observed between β subunits and to a lesser degree, α subunits, and supporting NMR and crystal structure data it can be concluded that most integrins adopt a similar structural conformation to those seen in the αvβ3, αIIbβ3 crystal structures.
The α and β integrin subunits form a non-covalently associated dimer, each consisting of an NH2-terminal extracellular domain, a transmembrane domain (TM) and a cytoplasmic tail. The extracellular domain protrudes from the cell surface to interact with and bind, in a divalent cation-dependent manner, ligands of the extracellular matrix. The domains that comprise the extracellular portion of αvβ3 adopt a conformation that is often likened to a head and two tails. The transmembrane domain traverses the cell membrane linking the extracellular domain and cytoplasmic tail. The cytoplasmic tails act as adaptor proteins for intracellular signaling molecules as well as a scaffold for the formation of focal adhesions and interactions with the cytoskeleton.
Each individual integrin pair recognizes its own specific set of ECM proteins. There is to some extent overlap between integrins in their ligand recognition, as they generally recognize a single sequence motif common among matrix components. Based on the αvβ3 crystal stucture, the head-piece is the site of ligand binding and is formed by the β-propeller of the αv subunit and the hybrid and βA-domains of β3. The ECM ligand binding site appears to be formed more or less equally by both subunits (Fig. 4). Ligand recognition in the αvβ3 integrin is through the arginine-glycine-aspartate (RGD) sequence of ECM molecules such as vitronectin and fibronectin (Fig. 5). The arginine residue forms salt bridges with aspartate residues within the β-propeller while the carboxylate group within the aspartate residue of the RGD sequence forms a series of polar interactions in the βA domain, and the glycine residue appears to form hydrophobic contacts solely with the αv subunit.
CycloRGDFV bound to αvβ3 (RCSB code: 1L5G). Metal ions Mn2+ are depicted as spheres.
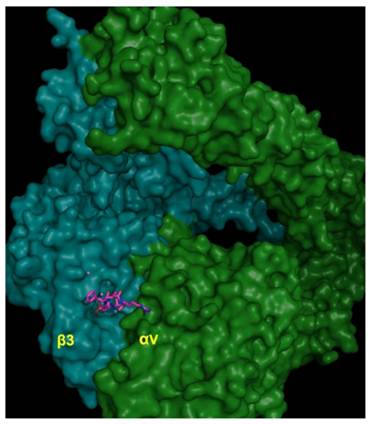
Ligand binding site of αvβ3 integrin (RCSB code: 1L5G) in complex with RGD peptide showing close contact residues of cycloRGDFV at the integrin dimer interface.
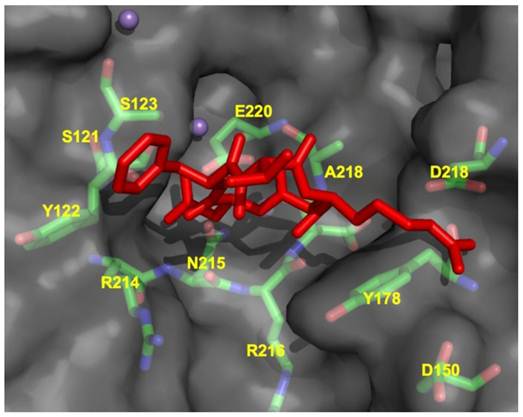
The seven-bladed, β-propeller is formed by the arrangement of 4-stranded anti-parallel β-sheets. The radial arrangement of the β-sheets forms a channel or cup-like structure composed mainly of aromatic residues in a consensus sequence termed the cage motif. Within the β-propeller are 4 Ca2+ coordination sites that may impart structural rigidity in the contacts between domains. The βA-domain is a six stranded beta sheet flanked by eight alpha helices. In these and other crystal structures, the integrin appears in a bent or folded over conformation that is believed to represent the low affinity state. In addition to the 4 Ca2+ binding sites, another metal ion dependent adhesion site exists is the βA domain which mediates a magnesium ion necessary for ligand binding.
Structural changes caused by ligand binding activate outside-in signaling. ECM binding causes displacement of residues within the ligand binding site that promote further structural rearrangements of the ectodomain that ultimately cause the separation of the intracellular tails [5]. This spatial separation facilitates the formation of focal adhesions and signaling complexes required for downstream effects. Two models of integrin activation have emerged based on NMR and crystal structure data, the switchblade and deadbolt models [13-14]. Both models propose that conformational rearrangements activate integrin binding and increase infinity. In the switchblade model, movements within the transmembrane regions of the dimer promote extension of the extracellular domain leading to increased ligand avidity. The deadbolt model proposes an activated state that retains its bent conformation but allows movement of the transmembrane domain in a manner that opens the ligand binding site to heighten receptor avidity.
3. Approaches to integrin targeting
The functionality of integrins is dependent on activation, ligand binding, focal adhesion formation and cytoskeletal contacts. Inhibition of any of these events abrogates integrin regulated functions and thus each comprise a valid pharmacological target. Based on a survey of current anti-integrin therapeutics and preclinical literature the most common approach to integrin antagonism involves targeting at or near the receptor binding site but knowledge of integrin activation and downstream signaling suggests that alternate approaches are possible.
Targeting the ligand binding site has been accomplished through the use of antibodies, cyclic peptides, disintegrins, peptidomimetics and small molecules antagonists. These compounds are meant to bind the targeted integrin in a manner similar to the endogenous ligand thereby displacing it and preventing integrin ligation. This method of antagonism has been widely utilized for several reasons. The paucity of informative structural data regarding the relevant integrins allows the rational design of antagonists based on known ligands. In addition, extracellular regions are preferable protein targets as they circumvent the problems associated with intracellular drug delivery such as lysosomal degradation and activation of drug efflux pumps. In addition the extracellular domains are the only regions accessible using antibody-based therapeutics, which constitute a large portion of the antagonists in clinical use and under development. Finally, the majority of high throughput screening methods utilize solid phase receptor binding assays that screen for compounds that are able to inhibit ligand binding and thus by design favor the identification of compounds showing affinity to the ligand binding site. A similar method of antagonism involves the binding of molecules or antibodies in close proximity to the natural ligand binding site. This strategy provides steric hindrance of the ligand binding site such that endogenous binding site is obscured and ligand binding is prevented.
Proposed crystallographic and NMR-based models maintain integrin activation is dependent on global structural rearrangements. Targeting sites of allosteric control may represent an alternate means by which to antagonize integrin function. Our current understanding of integrin activation postulates that both extra- and intra-cellular cues can propagate signaling cascades that act on intracellular portions of integrins to activate inside-out signaling. The presence of metal ion dependent adhesion sites (MIDAS) and calcium coordination sites further supports the assertion that function is dependent on specific conformational arrangements. Therefore dependence of activation on conformational arrangements suggests that sites of allosteric control exist either intra- or extracellularly on integrins. This is significant for the design of anti-platelet integrin antagonists for which conformational changes induced by ligand mimetics have been proposed to lead to the adverse effects associated with paradoxical platelet activation.
Targeting the intracellular 'tails' of integrins represents another underutilized therapeutic approach to antagonism. Focal adhesion formation occurs at integrin tails and is the earliest intracellular event following ECM ligation. The formation of the signaling platform at this level impinges on cellular fate. Post-translational modification of integrin tails determines the composition of the signaling platform which ultimately regulates the nature of the signals transduced downstream. Pharmacological modification of integrin tails therefore has the potential of regulation the nature and extent of integrin signaling to mediate the potential outcome of ligand engagement. Integrin tails are also extensively crosslinked with the actin cytoskeleton via interactions with talin and vinculin. Pharmaceutical inhibition of cytoskeletal organization can inhibit cell proliferation and migration for the treatment of inflammatory disorders or inhibition of tumor growth.
Integrin targeting therapeutics come in many forms, each type having its own inherent advantages and limitations. In addition the approach to integrin specific drug design is dependent on multiple factors including the expected clinical application and the availability of reliable structural information. In this review we will primarily discuss three types of integrin therapeutics, targeted antibodies, peptide-based drugs and small molecule, peptidomimetics.
Based on their status in current clinical development, therapeutic antibodies (Fig. 6A) are the most abundant integrin targeting drugs. Therapeutic integrin-targeted antibodies are currently under preclinical development and clinical evaluation for the treatment of cancer, multiple sclerosis, Crohn's disease, psoriasis, rheumatoid arthritis and acute coronary syndromes. Antibodies are advantageous in that they exhibit very high target specificity and affinity, limiting toxicities relating to 'off-target' effects. From a drug design standpoint antibodies are more easily developed because high-resolution structural data is not necessary for therapeutic development. Once a function-blocking antibody is identified it can be modified to increase specificity and binding affinity and lower immunogenicity. Antibodies can have long circulation lifetimes compared to small molecule and peptide-based drugs thus increasing the duration of therapy, that depending on the intended application may or may not be advantageous. The disadvantages associated with antibody therapeutics are the high cost of production, the need for intravenous administration in a clinical setting, and the propensity for host immunogenicity and infusion reactions.
Examples of integrin antagonists (A) therapeutic antibody (B) cyclic peptide (C) small molecule antagonist.
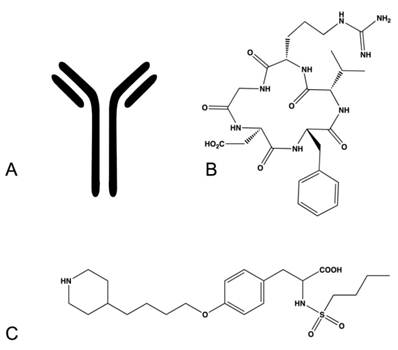
Peptide-based drugs (Fig. 6B) represent another class of drugs designed to target integrins. These drugs incorporate peptide sequences similar to ligand recognition sequences in endogenous integrin ligands and thus compete for the ligand binding site within integrins. This type of drug is advantageous for drug design as most of the endogenous ligands and their recognition sequences have been determined for each integrin pair. Peptide-based integrin targeting drugs have moderate to high target affinity but may lack specificity, as the same ligand recognition sequences are shared amongst many integrins. Varying the identity of specific peptides flanking the recognition sequence has been demonstrated to impart greater specificity between integrins by exploiting the subtle variations in topology amongst the dimer interfaces. Steric constraints imparted by peptide cyclization can also improve specificity and protect against proteolytic cleavage in vivo. The addition of D-amino acids is another means of prolonging circulation half-life and protecting against hydrolysis. Peptide-based drugs have additional disadvantages such as requiring injection or intravenous administration, high production cost, and limited stability.
Small molecule compounds (Fig. 6C) represent another class of integrin targeting drugs. Small molecule drugs are synthetic compounds with low molecular weight having favorable biopharmaceutical properties. They are readily synthesized, are less costly to manufacture than antibody and protein based drugs, have better stability profiles, and in many cases can be administered orally. Despite these advantages the development of small molecule antagonists that target integrins remains a challenge. Several methods have been applied for the development of small molecule integrin antagonists, including pharmacophore based modeling, bioisosteric substitution and high-throughput library screening. Most compounds in the clinic and under development retain structural features that preserve the molecular interactions observed between the integrin and its native ligand. These molecules tend to be zwitterionic in nature and generally encounter limitations in bioavailability, serum protein binding and integrin selectivity.
4. αIIbβ3 integrin
The αIIbβ3 integrin is comprised of a 136 kDa αIIb subunit non-covalently associated with the 92kDa β3 subunit. The αIIbβ3 integrin exists in various states of activation. Activated, ligand occupied and resting states have been demonstrated. Activating signals are required for ligand binding and lead to a conformational change in αIIbβ3 integrin that increase ligand affinity, receptor clustering leading to increased ligand avidity. αIIbβ3 integrin recognizes two separate consensus sequences, the arginine-glycine-aspartic acid (RGD) sequence [15] in fibrinogen, fibronectin, and Von Willebrand Factor and the lysine-glutamine-alanine-glycine-aspartate-valine sequence found in the gamma chain of fibrinogen [16-17]. As with αvβ3 integrin, coordination of Ca2+ ions within MIDAS and ADMIDAS are required for ligand binding.
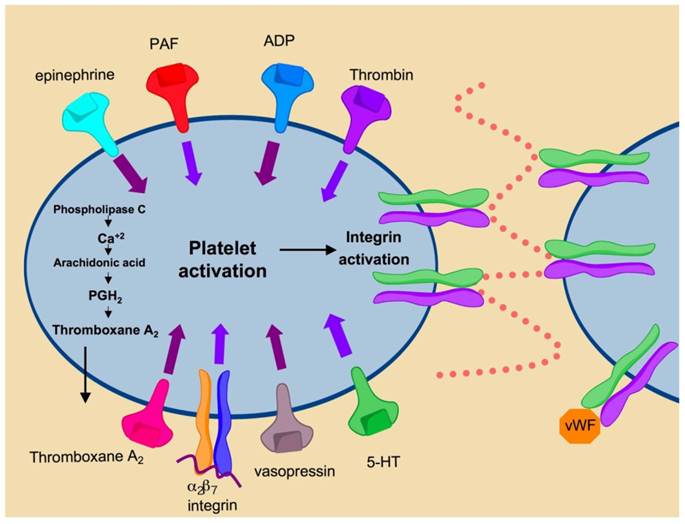
αIIbβ3 integrin (GPIIa/IIIb) is highly expressed on the surface of platelets and to a lesser degree, on megakaryocytes [18]. αIIbβ3 integrin comprises approximately 80% of the total surface proteins found on platelets. Engagement of this integrin is induced by platelet activation and represents the final common pathway in blood coagulation (Fig. 7). Under normal conditions integrin αIIbβ3 is maintained in the inactivated state. Soluble factors in the blood such as thrombin, ADP and thromboxane A2 bind their respective platelet receptors to activate inside-out signaling pathways that cause conformational changes in αIIbβ3 integrin [18]. Changes in conformation lead to modulation of ligand binding affinity and promote receptor clustering. The subsequent increases in receptor affinity and avidity promote platelet aggregation and clot formation through increased cell-to-cell contacts and cell-matrix contacts mediated by integrin αIIbβ3. Aberrant platelet aggregation or thrombosis is central to the pathophysiology of multiple Acute Coronary Syndromes (ACS), unstable angina, ischemic stroke and sickle cell anemia. Inhibition of αIIbβ3 prevents platelet aggregation and therefore has shown efficacy in the prevention of thrombosis for the treatment of ACS.
Mechanisms of platelet activation. Binding of secreted factors to cognate ligands promotes platelet activation leading to αIIbβ3 activation. Activation of αIIbβ3 promotes integrin clustering and binding of fibrinogen and von Willebrand Factor leading to platelet aggregation and thrombus formation.
αIIbβ3 integrin antagonists in Percutaneous Cardiac Intervention, Acute Coronary Syndromes and Myocardial Infarction. Currently, there are three FDA approved therapeutics targeting αIIbβ3 integrin, Abciximab, eptifibatide and tirofiban. These therapeutics are directed against αIIbβ3 integrin and act in the prevention of platelet aggregation. They are utilized during stent placement and other percutaneous coronary intervention (PCI) procedures for the treatment of both ST-elevated myocardial infarction (STEMI) and non ST-elevated myocardial infarction (nSTEMI). All three antagonists have been extensively studied in large randomized, placebo controlled clinical trials and demonstrated advantages over previous anti-platelet treatment modalities such as aspirin and the thienopyridines in preventing thrombosis and mortality. Although all three compounds act at the level of integrin αIIbβ3 to prevent thrombus formation, they represent separate classes of drug and thus differ in their pharmacokinetic and pharmacodynamic properties. The variations between each of these antagonists determine the extent of their utility in the treatment of various ACS. The clinical success of these agents in the prevention of platelet adhesion have prompted evaluation for the treatment of disorders and disease states in which abberant platelet aggregation is central to the pathology, such as ischemic stroke and sickle cell disease.
Abciximab. Abciximab (ReoPro, c7E3) was approved for use by the U.S. Food and Drug Administration for perioperative use in percutaneous coronary intervention in 1994 and is the first integrin antagonist to be approved for clinical use. Abciximab is the Fab fragment of a monoclonal, chimeric mouse-human antibody with specificity towards the αIIbβ3 integrin as well as the αvβ3 [19] and αMβ2 (MAC-1) integrins [20]. The platelet integrin (αIIbβ3) epitope for Abciximab has been identified as a region within the β-chain proximal to the RGD-sequence binding site and the secondary fibrinogen binding site [20]. Hence Abciximab inhibits platelet aggregation via steric hindrance of ligand binding.
For use in PCI, the treatment of MI and ACS, Abciximab is administered parenterally and the unbound fraction undergoes rapid proteolytic cleavage in the bloodstream. Due to short plasma half-life it is administered as a single bolus followed by continuous infusion over a period of 12-96 hours. The plasma concentrations drop rapidly upon discontinuation, the first phase half-life is close to 10 minutes and the second phase half-life is about 30 minutes due to rapid receptor occupancy. A slow decline in drug concentration results in an overall half-life of approximately 7h and continued low-grade receptor blockade has been noted for periods up to 10 days post infusion [21]. Abciximab is recommended for the treatment of both nSTEMI and STEMI [22].
Eptifibatide. Eptifibatide (Fig. 8) is a synthetic cyclic heptapeptide derived from barbourin, a snake venom disintigrin. Modifying the KGD sequence of barbourin, to homo-ArgGD and cyclization by introduction of a disulfide bridge produced a competitive inhibitor that shows high selectivity for αIIbβ3 at lower concentrations than the linear peptide [23]. Eptifibatide has low binding affinity and rapidly dissociates from αIIbβ3 in circulation, such that by 4 hours post administration, platelet binding ability is restored. Dosing is usually administered parenterally as a single or double bolus in combination with continuous infusion. The plasma half life is approximately 2.5 h and the drug is cleared mainly by the kidneys as epitifabitide, deamidated eptifabitde or other polar metabolites. Due to the extensive renal clearance, dosing modifications must be considered for renal impaired patients [24].
Structures of αIIbβ3 antagonists Eptifibatide and Tirofiban.
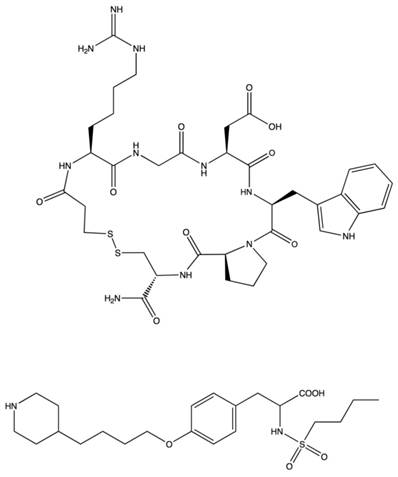
Tirofiban. Tirofiban (Fig. 8) is a non-peptide, small molecule RGD-mimetic antagonist specific for αIIbβ3 integrin. Tirofiban was developed through sequential bioisosteric substitutions of a candidate molecule pharmacophore chosen to approximate the bond distances observed between the carboxy and amino portions of the RGD-scaffold. This produced a small molecule, tyrosine-derivative with increased potency and selectivity for αIIbβ3. Tirofiban prevents fibrinogen binding and platelet aggregation in vitro by 50% at concentrations of 5 and 11 nM, respectively [25]. Tirofiban is administered intravenously. As with eptifibatide, this small molecule competes with fibrinogen and von Wilebrand Factor for the RGD-sequence binding pocket of platelet integrin αIIbβ3. It binds with higher affinity than eptifibatide but has a comparable volume of distribution and plasma half-life. Tirofiban is not highly bound to plasma in the bloodstream and the volume of distribution at steady state suggests that a large portion distributes within the extracellular space. Tirofiban has a plasma half-life of 1.5-2h and is excreted mostly as unchanged compound in the urine and feces. Renal insufficient patients require dose reductions and severe renal failure prohibits use. Hepatic failure does not require dose adjustment [26].
αIIbβ3 antagonists in clinical trials. All three drugs have undergone extensive clinical trials in the clinical setting for the treatment of various coronary syndromes including percutaneous coronary intervention, myocardial infarction and unstable angina and non-ST elevated myocardial infarction. Abciximab was the first αIIbβ3 targeted platelet antagonist to enter clinical trials. Phase I trials established dosing regimens and the effects of combination with common anticoagulants such as heparins. From this it was determined that optimal receptor occupancy was achieved with single bolus dosing followed with continuous infusion. Weight-adjusted heparin dosing reduced the propensity for bleeding events. Subsequent large-scale randomized trials examined the impact of abciximab on endpoints such as mortality, need for revascularization and occurrence of myocardial infarction [27]. Meta-analysis of the eleven major Phase III trials of abciximab showed significant overall decreases in death at 30 day endpoint, decreased need for revascularization and reduced occurance of myocardial infarction in patients receiving abciximab during percutaneous coronary intervention, as compared to fibrinolytic agent in myocardial infarction and during stent placement for the treatment of unstable angina [28]. Due to possible immunogencity related to the chimeric nature of abciximab, the safety of re-administration was examined in the ReoPro readministration trial. Abciximab was found to be safe for repeat administration [29]. Trials of eptifibatide were designed in a similar manner as the abciximab trials. Phase I studies examined various dosing levels alone and in combination with weight-adjusted heparin. Initially dosing was under estimated as the use of citrate anticoagulant chelated calcium ions necessary for receptor ligand binding and activation and produced falsely decreased readings of platelet aggregation that led to lower than anticipated performance in meeting protocol endpoints for survival, restenosis and myocardial infarction [30-31]. Subsequent trials utilized anti-coagulants that did not perturb measurements of platelet aggregation and dosing was increased from single bolus 135 mg/ kg to double bolus 180 mg/kg with 2 mg/kg/min infusion for up to 24 h [32]. This dosing resulted in significant reduction in mortality, restenosis and myocardial infarction when used in the ESPRIT trial [33]. The Randomized Efficacy Study of Tirofiban for Outcomes and Restenosis (RESTORE) trial evaluated tirofban versus placebo in patients at risk for arterial obstruction due to multiple acute coronary syndromes and those undergoing angioplasty for myocardial infarction. Significant reduction in primary endpoints were noted at day 2 but decreased by day 30 [34]. Overall meta-analysis of 12 trials of αIIbβ3 antagonists in over 20,000 patients demonstrated a significant reduction in mortality and myocardial infarction at 30 days [35].
Factors impacting the therapeutic efficacy of αIIbβ3 antagonists. The efficacy of αIIbβ3 antagonists is modulated by the total number of receptors available for drug binding. It is generally accepted that roughly 80% of the total platelet αIIbβ3 integrins should be bound to obtain therapeutic dosing [36]. Platelet count is the primary determinant of receptor availability as αIIbβ3 integrins are expressed almost solely on the surface of platelets, at levels of approximately 50-100,000 receptors per platelet. Although the in vivo significance of platelet count on drug efficacy has not been clearly established ex vivo studies suggest an inverse relationship between platelet count and integrin inhibition [37]. Additional factors that contribute, albeit less so, to receptor occupancy are the ratio of internalized receptors versus receptors at the platelet surface, the degree of receptor activation, and blood plasma fibrinogen levels.
Resting platelets typically maintain an internal receptor pool that upon activating stimuli such as ADP and Thromboxin A2 binding can be externalized to increase surface expression by 40-50%. Of this internalized pool, a portion may emerge at the platelet surface bound to soluble fibrinogen released from α-particles within the platelet. Abciximab is unable to completely inhibit this internal pool and the mimetic antagonists as competitive inhibitors may also lose efficacy, but likely to a lesser degree in light of their rapid dissociation kinetics [38].
Bleeding events associated with the use of αIIbβ3 antagonists. As potent antiplatelet drugs, administration of αIIbβ3 antagonists carry with them the risk of adverse bleeding events. Early on, the initial clinical trials involving αIIbβ3 antagonists demonstrated an increased risk of bleeding complications. Evaluation of Abciximab in clinical trials for the prevention of ischemic complications (EPIC trial) during angioplasty established the superiority of unfractionated heparin administration combined with abciximab bolus and continued infusion over UFH alone, but also revealed a two-fold increase in bleeding complications among the combined treatment group [39]. The propensity for bleeding complications was recapitulated in the CAPTURE trial, wherein patients with refractory unstable angina were treated with heparin and placebo or heparin and abciximab [30]. To address this significant concern, subsequent clinical trials of abciximab as well as eptifabitde and tirofiban employed weight-adjusted dosing for heparins. In doing so, the risks of serious bleeding complications fell, with little or no impact on drug efficacy. Despite alterations in dosing, bleeding events remain a concern for the use of αIIbβ3 antagonists. The therapeutic window, or the range of plasma concentrations at which these antiplatelet drugs act in both a safe and efficacious manner, is quite narrow and is dependent on the number of receptors available for drug binding. Drug plasma concentrations that promote receptor occupancy below 80% fail to achieve therapeutic inhibition, while levels that result in greater than 90% receptor occupancy are associated with higher risk of adverse bleeding events [40].
Drug-induced thrombocytopenia following αIIbβ3 antagonist treatment. A small percentage of patients receiving αIIbβ3 antagonists experience thrombocytopenia, a severe drop in platelet number (platelet count < 50,000) that is associated with increased incidence of ischemic events, bleeding related complications and requirement of plasma transfusions [41]. The primary cause of thrombocytopenia in patients treated with αIIbβ3 antagonists appears to be immune mediated [42], although non-immune mediated mechanisms have been reported for the RGD-mimetics, eptifibatide and tirofiban [43]. Bleeding events can range from minor occurrences such as leakage at the site of IV insertion, to systemic, life-threatening episodes requiring rapid intervention. As a humanized chimeric antibody, abciximab has demonstrated a propensity for the induction of human antichimeric antibody response upon first administration in approximately 1% of patients and in 4% upon repeat administration [29]. Thrombocytopenia caused by immune-mediated platelet destruction following Abciximab treatment may be of rapid or delayed onset [44]. Acute thrombocytopenia occurs within hours of infusion and may be accompanied by fever, drop in blood pressure, and anaphylaxis. Rapid, immune-mediated platelet destruction occurs through the binding of IgG and IgM antibodies that recognize murine sequences of the Fab' fragment or platelet conformational changes induced by abciximab binding. Delayed-type thrombocytopenia has also been noted to occur up to 8 days following cessation of treatment and may be caused by newly formed antibodies or through the stimulation of existent antibodies [45]. The rate of drug-induced thrombocytopenia is of lower frequency in patients receiving the ligand mimetics, Tirofiban and Eptifabitide compared to Abciximab treated patients [46]. The onset is usually acute, within hours of treatment and can be accompanied by symptoms similar to those accompanying abciximab immune thrombocytopenia. In contrast to Abciximab, the clearance rate of the RGD mimetics is much faster, thus limiting to the time period in which patients are at risk of bleeding events due to thrombocytopenia. The severity of complications varies from asymptomatic to minor bleeding to major hemorrhage. Immune mediated thrombocytopenia is believed to result from the clonal expansion of haptens, antibodies that recognize drug-bound receptors.[44] Monitoring platelet counts prior to and following administration of all three platelet inhibitors is warranted considering these issues. Depending on the severity of related bleeding events, platelet transfusion may be necessary to restore haemostasis.
Oral αIIbβ3 antagonists as preventive treatment for coronary syndromes. Prolonged inhibition of platelet aggregation using aspirin and thienopyredine has been demonstrated to significantly reduce the risk of ischemic events [47]. It was hypothesized that, as more potent inhibitors of platelet aggregation, the αIIbβ3 antagonists would show increased benefit in long-term ischemia risk reduction. Based on the success and clinical efficacy of the parenterally administered αIIbβ3 antagonists, an effort to develop orally available small molecule antagonists for the long-term prevention of cardiovascular disease was undertaken and led to the clinical evaluation of several antagonists having similar structural characteristics and pharmacological properties (Fig. 9). Most of the drugs from this class are prodrugs, requiring hepatic conversion to the active state and having limited bioavailability in the range of 7-38% [48]. The plasma elimination half-life for these compounds is generally in the range of 4-20 h with the exception of roxifiban that has an elimination half-life of up to 5 days. Renal excretion accounts for the primary means of drug elimination within this class. Pharmacokinetic studies established that efficacious dosing was in the range of 5-30 mg. Higher doses given once daily were associated with increased bleeding events, lower more frequent dosing provided similar effect and reduced the bleeding risks. Renal insufficient patients required lower dosages to prevent increased risk of bleeding presumably to decreased clearance rates [48]. Despite the dose adjustment efforts taken in risk reduction, the outcomes of dose ranging studies revealed overall increase in the risk of bleeding amongst all antagonists tested [48].
Structures of orally active, small molecule αIIbβ3 antagonists.
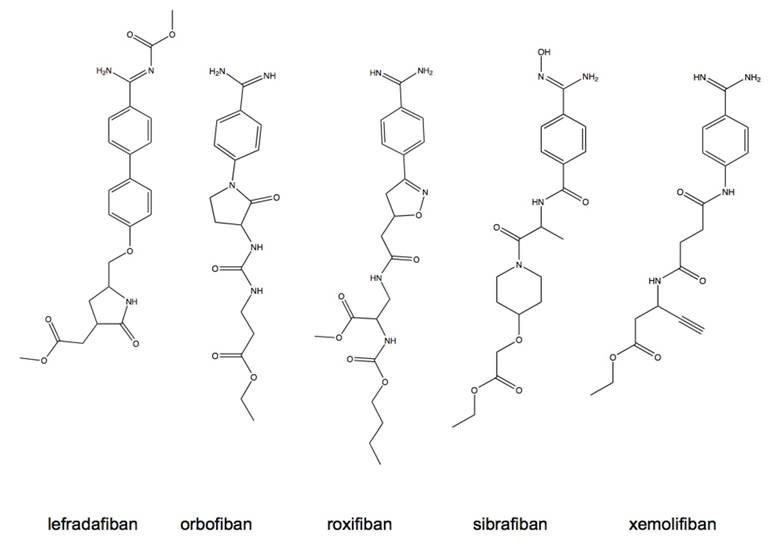
Following the dose-ranging studies, large scale randomized, double-blinded, placebo-controlled Phase III trials of xemilofiban, orbofiban, sibrafiban, and lotrafiban were undertaken. None of these studies were able to demonstrate decrease in the incidence of either short-term or long-term ischemic events. Trials of orbofiban, sibrafiban and lotrafiban were terminated early due to increased excess mortality risk. Meta-analysis of 4 Phase III trials carried out in over 33,000 patients confirmed a 31% increase in overall mortality associated with the oral platelet antagonists, halting the inception of further trials and development [49].
The mechanism leading to increased mortality is confounding based on evaluation of the meta-analysis of concurrently measured endpoints such as myocardial infarction, need for urgent revascularization and major bleeding events. The incidence of myocardial infarction was not significantly different between patients receiving study drug and those receiving aspirin alone. The incidence of urgent need for revascularization was significantly reduced in 2 of 4 trials analyzed, and not significantly different in the remaining two trials [49]. These results suggest thrombosis may not be the primary factor leading to increased mortality. A statistically significant three percent increase in major bleeding events due to platelet inhibition contradicts lack of antagonist efficacy as a contributing factor to increase in mortality.
Alternate mechanisms have been proposed to account for the increased mortality observed with oral antagonists including drug related toxicity, partial agonist activity, non-platelet mediated mechanisms and patient inter-variability. No one mechanism has been explicitly proven to account for the increased mortality and it is likely that the cause is multi-factorial.
αIIbβ3 antagonists for the treatment of ischemic stroke. Similar to acute coronary syndromes, inhibition of platelet aggregation and thrombus formation using aspirin has shown benefit in ischemic stroke by reducing recurrence and mortatlity, albeit with modest effects. As potent antagonists of platelet aggregation and thrombus formation, is was expected that αIIbβ3 inhibitors would demonstrate increased benefits over aspirin for the reduction of stroke related complications. Contrary to expected, meta-analysis of several large randomized clinical trials failed to demonstrate conclusive evidence supporting the use of αIIbβ3 antagonists for the treatment of ischemic stroke [50]. Furthermore, a few recent trials have terminated enrollment due to increased incidence of intracranial hemmorhage.
αIIbβ3 antagonists for the treatment of sickle cell disease. Sickle cell anemia is a hereditary disorder in which mutation in the β-globulin gene gives rise to abnormal hemoglobin that disrupts the structural integrity of red blood cell and leukocyte membranes leading to decreased pliability, perturbations in ion balance and increased adhesion and viscosity in blood vessels. Sickle cell anemia is characterized by episodes of ischemia-repurfusion [51], increased levels of inflammatory mediators [52] and abnormal activation of granulocytes and monocytes [53]. Activated platelets are thought to contribute to the pathology of Sickle cell anemia through multiple processes. Activation of platelets increases endothelial cell activation [54], P-selectin exposure, the exposure and release of CD40L [55] and it promotes vasoconstriction. Platelet released factors increase adhesion of RBC to vascular endothelium, promote coagulation [56], induce vasoconstriction [57]. Exposure and release of CD40L triggers immune and inflammatory responses, triggers B-cell proliferation, increases the expression of endothelial cell adhesion molecules and upregulates tissue factor. Platelet adhesivity and adhesion is mediated through αIIbβ3 integrin as well as granule secretion and procoagulant activity [56, 58]. Blockade of integrin αIIbβ3 with eptifibatide, abciximab or tirofiban prevents thrombus formation, prothrombin activation and the release of CD40L from activated platelets, events which are central to the pathology of sickle cell anemia and therefore, may offer benefit in the treatment of sickle cell anemia. Phase I trials evaluating the safety and pharmacokinetics of eptifibatide in sickle cell anemia patients in the non-crisis state have shown promising results. Eptifibatide administered as two 180 mg/kg boluses separated by 10 minute interval combined with 2 µg/kg/min infusion reduced levels of inflammatory cytokines and myoglobin, a marker of muscle injury. The vasodialotors MMP-2 and MMP-9 were increased with eptifibatide treatment, as was the adipokine leptin. Eptifibatide treatment had no effect on platelet reactivity or aggregate formation. The observed effects of eptifibatide were optimal by 6 h and fully reversible as early as 24h following infusion, suggesting that αIIbβ3 antagonist therapy may be beneficial for the treatment of the acute painful crises characteristic of sickle cell anemia [59].
5. α4 integrins
α4 integrin chains associate with β1 or β7 integrin family members, via non-covalent linkages, to form dimers. The α4β1 integrin, also known as VLA-4, Very Late Antigen-4, and CD49d/CD29, is constitutively expressed on the surface of lymphocytes and most leukocytes [3]. The primary ligands of α4β1 are endothelial VCAM1 and the extracellular matrix (ECM) glycoprotein, fibronectin. α4β1 interacts with VCAM1 through the QID(40)SPL site in domain 1, in which aspartate 40 appears to be essential for ligand binding. Interaction with the ECM occurs through the LDV sequence of the connecting segment-1 (CS-1) region of fibronectin. Other α4β1 ligands have been identified, including the acidic glycoprotein osteopontin, juntional adhesional molecule B and molecule B and mucosal addressin cellular adhesion molecule (Mad-CAM) [60].
Similarly, integrin α4β7 (LPAM, Lymphocyte Peyer's Patch Adhesion Molecule) is also expressed on the surface of lymphocytes and leukocytes. Interactions of this integrin with VCAM-1 and fibronectin occur through the same epitope as α4β1 integrin, although the α4β7 avidity for VCAM-1 is considerably lower than that of α4β1. In addition, α4β7 binds the mucosal addressin cellular adhesion molecule -1 (MAdCAM-1) expressed on high endothelial venules of gut mucosal lymphoid tissue such as Peyer's Patches and mesenteric lymph nodes, as well as lamina propria venules [61].
The α4 integrin family members, α4β1 and α4β7, function in leukocyte recruitment from the peripheral circulation to sites of inflammation within tissue [62-63] (Fig. 10A). The local release of chemokines at sites of inflammation causes upregulation of α4β1 and α4β7 on the circulating immune cells and adhesion molecules such as VCAM and/or MAdCAM, on the surface of proximal endothelial cells. Increased adhesion molecule density enhances the probability of forming contacts between circulating, immune competent leukocytes and the vascular endothelium. As blood flows, binding between leukocyte P-, L- or E-selectin molecules and endothelial cell carbohydrates or leukocyte α4 integrins and endothelial cell adhesion molecules (VCAM or MAdCAM) provide the initial contacts needed to capture leukocytes traveling through the circulation. The increased cell to cell contact between leukocytes and the endothelium slows the rate of leukocyte travel through the circulation, a process known as rolling adhesion. The decreased velocity imparted by the initial contact allows chemokines on the surface of vascular endothelium to bind G-protein coupled receptors on the leukocyte surface, transducing an inside-out signal that further activates integrins. The high avidity of activated α4 integrins for their cognate ligands results in firm adhesion to the endothelium. Firm adhesion is a requirement for diapedesis, extravasation of leukocytes through the endothelium into the ECM. Once leukocytes extravasate, α4 mediated interactions with fibronectin of the ECM facilitate migration through the chemokine gradient to the site of inflammation.
Mechanism of immune-cell mediated inflammation and its inhibition by α4 antagonists. Left Panel- α4 integrins are up-regulated on the surface of activated immune cells. Through contacts with endothelial cell adhesion molecules such as selectins and glycoproteins, these cells are captured from the circulation. The intial contacts are strengthened when α4 integrins make contact with MAdCAM and VCAM on endothelial cells. Extravasation and migration to sites of inflammation are mediated in part through α4 integrins. Right Panel- In the presence of α4 antagonists such as natalizumab, firm adhesion and subsequent migration are inhibited preventing immune cell infiltration within tissue.
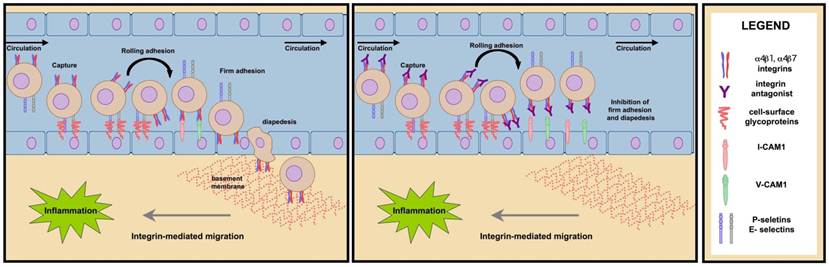
The infiltration of leukocytes and lymphocytes is central to the pathology of many inflammatory diseases. The α4 integrin family members, α4β1 and α4β7 act both in the formation of initial contacts and firm adhesion and therefore, are key constituents in the process of cellular influx to inflamed tissue. Suppression of α4 integrin binding inhibits cellular migration, thereby attenuating the inflammatory response (Fig. 10B). The therapeutic potential of α4 antagonists in the attenuation of inflammatory response has been investigated for the treatment of several chronic diseases.
Multiple Sclerosis (MS) is an autoimmune disease affecting the central nervous system (CNS). The pathology of MS is caused by increased entry of activated immune cells through the blood-brain barrier (BBB) into the CNS. Activated immune cells initiate processes of inflammation, edema and demyelination leading to the formation of acute CNS lesions [64]. The progressive accumulation of lesions in the CNS is responsible for the debilitating symptoms of MS, such as fatigue, imbalance, loss of mobility, sensory symptoms, bladder and bowel dysfunction, memory loss and concentration deficits, spasticity, visual problems, pain and sexual dysfunction [65].
Contacts between circulating immune cells and the BBB is an early and critical event in the pathogenesis of MS. Antibody directed inhibition of α4β1 mediated contacts between immunocompetent cells in the circulation and VCAM-1 on the vascular endothelium of the BBB has been demonstrated to prevent the development and even reverse disease progression by preventing the infiltration of inflammatory cells through the BBB. Targeting α4 integrin mediated cell binding to attenuate immune cell migration therefore represents a significant means by which to prevent disease progression.
Crohn's disease and ulcerative colitis are idiopathic inflammatory diseases of the intestinal tract characterized by focal or continuous regions of mucosal inflammation, respectively, which produce symptoms of abdominal pain, diarrhea and rectal bleeding and over time physical manifestations such as strictures and perforations of the intestinal wall. Dysregulated immune responses contribute to the pathogenesis of both types of IBD causing increased infiltration of immune cells into muscosal tissue of the gut. Interactions between immune cell α4β7 and VCAM and MAdCAM-1 on the gut mucosa facilitate transmigration and are believed to contribute to the pathogenesis of IBD [65].
Asthma is a chronic inflammatory disease of the lung characterized by increased infiltration of activated T-cells and eosinophils resulting in bronchoconstriction, airway hyperresponsiveness and airway remodeling. Inhibition of T-cell migration via blocking of α4 mediated cell interactions may reduce the severity of asthma symptoms.
In addition α4 antagnoists have been explored as a therapeutic means of attenuating the lymphocyte infiltration into joint synovium that is characteristic of the chronic autoimmune disease, rheumatoid arthritis. The rationale for targeting α4 integrin to modulate aberrant immune cell migration and activation is supported by in vitro as well as cell- and animal-based studies. Antibodies that block the function of α4 and β1 integrin chains decrease T cell and monocyte binding to VCAM-1 in frozen sections of inflamed blood vessels [66]. Inhibition of activated T-cell migration across the BBB using α4β1 blocking antibodies prevented and in some cases, reversed inflammation-induced neurological symptoms in mouse, rat and guinea pig models of experimental autoimmune encephalopathy. Similar results were obtained by blocking the α4 ligand VCAM-1 [67]. Inhibition of α4β7 reduces the development of spontaneous ulcerative colitis in tamaran monkeys [68] and alleviates inflammation in mouse models of colitis [69]. Inhibition of the α4β7 ligand, VCAM-1, produces similar results in mouse models of induced colitis [70]. Studies using monoclonal antibodies directed toward α4 integrins or VCAM in animal models of antigen-induced asthma suggest a critical role for this interaction in disease pathophysiology [71]. Various small molecule α4 antagonists have shown efficacy in allergen-induced asthma models.
α4 antagonists for the treatment of inflammatory diseases. To date, there is only one α4 antagonist approved by the U.S. Food and Drug Administration (FDA). Natalizumab is approved for treatment of relapsing, remitting multiple sclerosis and Crohn's disease. Natalizumab is a humanized mouse monoclonal antibody directed against the α4 integrin. Natalizumab binds α4β1 and 4β7 integrin on the surface of circulating T lymphocytes preventing interaction with cellular adhesion molecules that facilitate extravasation and migration from the circulation to tissue.
Following the administration of 300 mg I.V. bolus Cmax was 110 ± 52 µg/mL. Mean average steady state concentrations were between 23 and 29 µg/mL and the time to steady state was 24 weeks after 4 weeks dosing. The mean t1/2 is approximately 11 days, with a volume of distribution of 5.7 and clearance of 16 ml/hour. Clearance is influenced by bodyweight and the presence of neutralizing antibodies, increases in both results in a faster rate of clearance. Age and sex do not affect the rate of clearance [72-73].
Natalizumab underwent a series of clinical trials for the treatment of relapsing-remitting multiple sclerosis, and Crohn's disease. Natalizumab showed remarkable efficacy for the treatment of MS in Randomized clinical trials. α4-integrin blockade resulted in a decrease in the number of CD4+ and CD8+ T-cells, CD19+ B-lymphocytes and CD138+ plasma cells in CSF for up to 6 months following treatment cessation.[74] Based on measurements of 2-year outcomes, natalizumab decreased clinical measures of disease such as relapse rates and the risk of sustained disability progression by 68 and 54% respectively and reduced MRI based disease measures such as the appearance of new T2 lesions, new gadolinium positive lesions and lowered overall disease burden by 83, 92 and 18 % respectively [75]. The efficacy of natalizumab treatment appeared to improve with the duration of treatment. The difference in clinical and MRI measures of disease and progression between treated and placebo groups were significantly greater in the second year compared to the first. By the second year of treatment greater than 35% of natalizumab treated patients were free of all disease activity and more than 40% showed only one measure of disease activity [76]. In both highly active disease and non-highly active disease subgroups, the proportion of patients free of disease activity was significantly higher than members of the same group that received placebo only. Health related quality of life measures relating to patient perceptions regarding disease status were also improved in natalizumab treated patients [77]. Clinical measures of efficacy in Crohn's disease were initially less significant than those observed in the treatment of MS. Although treatment with natalizumab demonstrated efficacy in disease response and remission among a high percentage of treated patients in the ENACT 1 and ENACT 2 randomized phase III trials, both failed to meet their primary endpoints of statistically significant reduction in disease activity across the total population. However, the follow-up trial ENCORE was able to demonstrate disease response and remission in active Crohn's patients receiving 300 mg intravenous infusion every two weeks over a period of 12 weeks. In this randomized, placebo controlled, phase III trial, 51% of natalizumab treated patients showed response by week 4 after one infusion. The percent of patients in response at weeks 8 through 12 was similar to the initial results, 50% of natalizumab treated patients sustained response versus only 37% of patients receiving placebo [78].
The most prevalent side effects reported for natalizumab treatment are headache (38%), fatigue (27%) and infusion reaction (24%) [72]. By far the most serious side effect associated with natalizumab is the onset of progressive multifocal leukoencephalopathy (PML) [79]. PML is a rare, life-threatening, demyelinating infection caused by the JC virus. A large portion of the general population (~80%) is infected with latent JC virus. Active PML infections resulting from JC virus are often seen immunocompromised individuals such as HIV patients. The increased incidence of PML in patients receiving natalizumab is believed to be caused by impaired immune surveillance in the CNS as a direct result of α4 inhibition on circulating immune cells [80]. The risk of developing PML is believed to increase with duration of Natalizumab treatment and may be hastened in patients receiving prior or concurrent immunosuppressive therapies. The incidence of PML is calculated to be 1 case per 1000 patients treated with natalizumab for 17.9 months. To date, 11 cases of PML including 3 fatalities have been reported in patients receiving Natalizumab either as monotherapy or in combination [79, 81-83]. Additional opportunistic infections have been observed in patients receiving natalizumab, although the role of natalizumab in causation is as yet undetermined. Infections reported include viral meningitis and encephalitis (2), acute cytomegalovirus (2), pulmonary aspergillosis (2), cryptosporidial gastroenteritis (1), Pneumocystis carinii pneumonia (1), varicella pneumonia (1), mycobacterium avium intracellulare complex pneumonia (1), and Burkholderia cepacia pneumonia (1). Values in parentheses represent the number of reported cases. In an effort to decrease the inherent risks accompanying the obvious benefits to MS patients, FDA recommendations suggest the use of natalizumab in limited populations including those refractory to other therapies or those with particularly aggressive disease under strict clinical supervision.
Because the increased incidence of PML is believed to be caused as a direct result of α4 antagonism, improvement of the biopharmaceutical properties of compounds targeting α4 integrin would therefore have no bearing on the likelihood of developing PML. Though, presumably, a small molecule or peptidomimetic antagonist could present lower risk; the shorter plasma half-life of these molecules compared to antibodies would prevent the need for costly and invasive plasma exchange procedures currently utilized for the removal of natalizumab from the blood in the event PML symptoms are manifest [84]. Ultimately, due to the serious risks that increase with duration of administration, α4β1 integrin antagonist based therapies should be limited for the treatment of conditions that are life threatening or do not respond to first-line treatments.
Antagonism of VLA-4 may also have implications for fetal and embryonic development. Cell-to-cell interactions mediated by α4β1 integrin are critical to reproductive events such as fertilization, implantation, placental formation and cardiac development. α4β1 knockout mice are embryonic lethal and show disruption in placentation, defects in allantois-chorion fusion, cardiac abnormalities, malformation and hemorrhage. VCAM-1 knockout mice show similar defects further supporting the role of α4 integrins in embryonic development.
Based on animal studies of embryonic and post-natal development it has been deemed that prolonged, high dose natalizumab treatment caused no developmental defects [85-87]. These results cannot be extrapolated to other α4 antagnoists. In fact, basic teratogenicity assays of several small molecule α4β1 antagonists in preclinical development showed variability in propensity to cause defects (Fig. 11). The teratogenic potential of α4β1 antagonists appears to be related to the affinity state of the integrin to which the compound binds. Integrins exist in multiple activation states wherein ligand binding stability is proportional to the affinity or activation status of the receptor. Compounds that bind to VLA-4 at both low and high affinity states appear carry a greater risk of toxicity and embryonic defects compared to compounds selective for the activated integrin [88]. Therefore integrin activation state affinity is a major consideration for the development of successful clinically useful inhibitors.
Small molecule α4 antagonists vary in their propensity for teratogenic effects. IVL984 (potent teratogen), HMR1031 (mild teratogen) and IVL745 (non-teratogenic) are structurally similar yet have different teratogenicity profiles. The difference is attributed to their affinity for non-activated α4 integrin. Compounds that bind α4 in both activated and resting states appear to be more teratogenic.
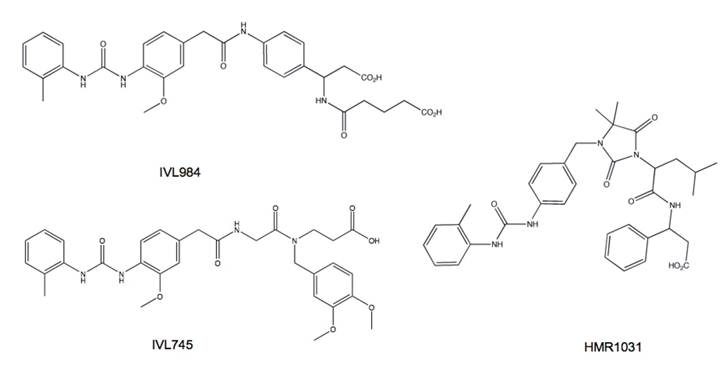
α4 antagonists under clinical evaluation. MLN-00002 MLN-0002 is a human antibody specific for the α4β7 integrin dimer. In phase II trials, MLN-00002 showed dose dependent efficacy as induction therapy for the treatment of active Crohn's disease and ulcerative colitis. Treatment consisted of two infusions, at day 1 and 29. Primary endpoint was a greater than 70 point decrease in the CDAI (Crohn's Disease Activity Index) score achieved in both studies. In the phase II trials patients were screened for the presence of latent JC virus prior to the administration of MLN-0002 and were monitored for signs of PML through out the course of treatment [89].
Firategrast. Firategrast is a small molecule α4 antagonist developed by Glaxo Smith Kline that has recently completed Phase I clinical trials for the treatment of Relapsing, Remitting Multiple Sclerosis. This trial evaluated pharmacokinetic parameters following oral administration of 900 mg once daily in men and women diagnosed with multiple sclerosis. Further Phase II studies have been initiated to evaluate the effect of firategrast on white blood cell counts in the cerebrospinal fluid.
IVL745. IVL745 (Fig. 11) is a small molecule antagonist of α4β1 antagonist that previously demonstrated efficacy in animal models of asthma and airway inflammation and allergen induced bronchoconstriction. IVL745 binds activated α4β1 and is non-teratogenic [90]. Orally administered IVL745 has low bioavailability and absorption properites, short plasma half-life. IVL745 is not highly metabolized and therefore is excreted mainly through the biliary route [91]. These limitations in biopharmaceutical properties typify the problems inherent with most small molecule α4 antagonists. Bioavailability is improved when administered by inhalation. Safety studies in both healthy and asthmatic volunteers demonstrated tolerability by inhalation at dosing up to 20 mg BID. Subsequent trials in human asthma patients failed to demonstrate efficacy greater than that observed with placebo [92-93] suggesting that the role of α4 integrins in asthma and allergen induced air inflammation may be more complicated or less influential than previously predicted [94].
6. αv integrins
Integrins αvβ3 and αvβ5 are highly expressed on the surface of osteoclasts, angiogenic endothelial cells, and some solid tumors. Their antagonism has been explored for the purpose of preventing/reversing osteoporosis, inhibition of angiogenesis and induction of tumor regression. Currently there are no FDA-approved antagonists of αvβ3 and/or αvβ5 integrins on the market, but several compounds have shown efficacy in preclinical evaluation and are currently under evaluation in clinical trials for the treatment of malignancies, rheumatoid-arthritis and osteoporosis.
αv integrin antagonists as anti-angiogenic agents. Angiogenesis is the process of neovascularization in which new blood vessels sprout from existing ones. The 'angiogenic switch' is an early event required for tumor progression. Initially tumor growth is supported by nutrients and oxygen derived from proximal vasculature. As tumor mass increases, local blood supply becomes inadequate, resulting in a hypoxic, nutrient-deprived tumor environment. The inducible transcription factor, hypoxia-ducible factor-1 (HIF-1) is activated in response to the local hypoxic conditions, leading to the release of trophic factors such as vascular endothelial growth factor (VEGF). Tumor associated macrophages have recently been identified as another means by which the angiogenic switch can be tripped. Tumor-released cytokines stimulate the migration of macrophage precursors, that once differentiated in the tumor mass can secrete pro-angiogenic growth factors and tissue proteases. In an autocrine fashion, proliferating tumors cells release VEGF, basic fibroblast growth factor (bFGF) and tumor necrosis factor alpha (TNF-α). VEGF-receptor 2 (VEGFR2) acting in concert with αvβ5 and FGF-receptor (FGFR) co-signaling with αvβ3 stimulate distinct signaling pathways that can be integrated to heighten the angiogenic response. Growth factor release stimulates the proliferation of quiescent endothelial cells and signals the migration of angiogenic endothelial progenitor cells from the bone marrow stroma to the tumor bed. αvβ3 and αvβ5 integrins are highly expressed on the surface of angiogenic endothelial cells as compared to resting endothelial cells. In the process of differentiation from bone marrow-derived endothelial cell progentior to terminal, committed endothelial cell, αvβ3 integrin expression increases. Integrin αvβ3 and αvβ5 recognize the RGD sequence in ECM components such as fibronectin, fibrinogen, and vitronectin. Through interactions between αvβ3 and αvβ5 integrins and the ECM, migrating endothelial cells are captured from the circulation and participate in the formation of vasculature to feed the growing tumor. The association of integrin αvβ3 with matrix proteases including matrixmetalloprotease -2 (MMP-2), matrixmetalloprotease-9 (MMP-9) and urokinase plasminogen activator (uPA) facilitates ECM degredation allowing tissue extravasation. Integrins on the leading edge of lamelapodia use ECM components to generate the traction necessary for cell migration. Adhesion and spreading are mediated through integrin binding to ECM. Cell-to-cell contacts are mediated through integrins during tube formation and vessel maturation. During angiogenesis integrins stimulate the processes of endothelial cell migration, proliferation, differentiation through FAK/Src/p130Cas and Raf/MEK/Erk signaling pathways [95].
Endothelial survival in both nascent and established tumor vasculature is supported by integrin mediated signaling events that are dependent on ligation with the ECM. Integrin ligation promotes cell survival primarily through activation of the PI3K/Akt pathway. Cross talk between integrins and growth factor receptors, possibly through trimeric interaction between growth factors, growth factor receptors and integrins [96], inhibits the activation of intrinsic and extrinsic pathways of apoptosis. In addition, integrin ligation promotes the increased expression of the pro-survival molecules Bcl-2 and FLIP, increases NFkB signaling and decreases p53 activation. Hyper-stimulation of pro-survival signaling mechanisms aids endothelial cells as they form vasculature in the hypoxic tumor microenvironment. Conversely, unligated integrins can promote apoptosis through the interruption of pro-survival signaling and the initiation of pro-apoptotic signaling cascades [6, 97].
The significance of αvβ3 and αvβ5 integrins in the processes of angiogenesis is supported by a wealth of experimental data obtained using in vitro, cell culture and in vivo methods. Angiogenic factors and tumor-secreted cytokines increase the expression of αvβ3 integrin in animal and human models of angiogenesis. Inhibition of αvβ3 and αvβ5 integrins prevents endothelial cell migration, adhesion, differentiation, tube formation and survival in cell culture models and has been demonstrated to inhibit angiogenesis and vascular tube formation in animal models. Mice null of αv integrin expression show a high rate of embryonic lethality due to bleeding and placental defects, as do β3 knockout mice. Surviving αv-knockout mice die prematurely due to complications of vessel malformation in the brain and gut. In contrast, surviving β3 knockout mice exhibit normal vasculature and increased angiogenic potential, presumably through up-regulation of VEGFR2 mediated compensatory mechanisms. The reasons for the pro-angiogenic effect of β3 knockdown and the implications for the role of αvβ3 integrins in angiogenesis is a source of continued debate. It is important to keep in mind that the observed effects of β3-knockdown during development may differ from those seen post-natally [97-98].
The primary goal of anti-angiogenic therapy is to prevent tumor growth by inhibiting the formation of new vasculature and destroying established tumor blood vessels that feed oxygen and nutrients to the growing neoplasm. Additional evidence suggests that anti-angiogenic therapies may act to transiently stabilize tumor vasculature increasing oxygenation and perfusion. The resulting increase in tumor blood flow may explain the seemingly paradoxical potentiation in efficacy observed when anti-angiogenic agents are combined with standard chemotherapies [99]. Anti-angiogenic agents are generally well tolerated and can be administered at high doses with few dose-limiting toxicities. With low levels of toxicity, anti-angiogenic agents can easily be combined with standard drug regimens with little or no additional adverse effects. The anti-angiogenic effects of radiation therapy can synergize with angiogenic inhibitors to increase cell kill [100]. Additionally, as non-transformed cells, the genetic stability of endothelial cells precludes the development of drug resistance, making them an attractive target.
αv integrin antagonists as anti-tumor agents. Inhibition of integrins has demonstrated growth inhibitory effect independent of angiogenesis. As in endothelial cells, αvβb integrins support tumor cell adhesion, proliferation, survival migration and invasion. Integrin αvβ3 is expressed on a variety of cancer cells including melanoma, glioma, breast, prostate, cervical, ovarian and pancreatic cancers. Patients whose tumors express high levels of αvβ3 tend toward a poorer prognosis than patients with low tumor expression levels. Expression of αvβ3 integrins in tumors is associated with an aggressive phenotype. The increased expression of αvβ3 imparts metastatic potential and invasive traits. In some instances, αvβ expression increases during transition to a metastatic phenotype supporting a role for this integrin in metastases and invasion. Cell to platelet interactions between integrins αvβ3 on tumor cells and αIIbβ3 on activated platelets facilitates circulating tumor cell capture from the blood, a critical initial step in the metastatic cascade. Extravasation, migration, adhesion and proliferation are supported by integrin ligation with the extracellular matrix. Association of αvβ3 integrin with Src promotes anchorage independent cell growth. αvβ3 mediated signaling through focal adhesion kinase (FAK) supports the survival of cancer stem cells. Inhibtion of αvβ3 is associated with decreased tumor cell growth in breast and prostate cancers, melanomas and gliomas. Supression of tumor αvβ3 integrin expression in animal models decreases the ability of tumor to migrate and metastasize [6].
αv integrin antagonists for the treatment of osteoporosis. Osteoporosis is a disease characterized by the progressive loss of bone density due to imbalances in the rate of bone resportion by osteoclasts and bone formation by osteoblasts. The disease is common in post-menopausal women, as age-related declines in estrogen levels increase osteoclast activity. Osteoporosis imparts increased risk of fractures leading to augmented rates of morbidity and mortality among osteoporotic women [65]. The development of antiresorptive agents that prevent osteoclast function to successfully reverse bone loss and increase bone mineral density (BMD) are of great interest for the treatment of osteoporosis. αvβ3 integrins are highly expressed on the bone surface and have been implicated in cell spreading, migration and signaling processes central to osteoclast function. Inhibition of αvβ3 has been demonstrated to prevent bone resorption both in vitro and in vivo. The RGD-disintegrin, echistatin prevents bone loss in hypercalcemic mice on a low calcium diet [101]. It also inhibits serum calcium level increases following parathryroid hormone treatment in parathyroid/thyroidectomized mice and prevents bone resporption in ovarectomized mice [102-103]. Similarly, β3 function blocking antibody, mAb F11 blocks the effect of parathyroid hormone on blood calcium levels [104]. At the genetic level, disruption of β3 induces late onset osteoporosis in knockout mice. Osteoclasts derived from these mice are unable to spread, lack cytoskeletal organization and show decreased capacity to resorp bone [105]. The exact mechanisms by which αvβ3 integrin inhibition prevent bone resorption are not clear as the role of αvβ3 in osteoclast function are not completely delineated. In vitro studies suggest that αvβ3 antagonism prevents bone resorption through inhibition of osteoclast migration [106-107]. A few orally available, small molecule αvβ3 antagonists have been developed for the treatment of osteoporosis and are in the early stages of clinical development.
Therapeutic antibodies. A series of therapeutic antibodies targeting αv integrins are currently under development and in clinical trials as antiangiogenic and anti-tumor agents. These antibodies are summarized in Table 1.
αvβ3 targeted therapeutic antibodies.
| Antibody | Source | Reactivity | Specificity | Activity | Clinical Development |
|---|---|---|---|---|---|
| LM609 | mouse monoclonal | human, rabbit, chicken | αvβ3, αIIbβ3, αvβ5 dimers | Blockade of TNFα and bFGF induced angiogenesis. Induces apoptosis. | Immunogenicity and human anti-mouse antibody responses preclude its use in clinical applications. Further modifcations produced vitaxin. |
| Vitaxin | humanized mouse monoclonal derived from LM609 | human, mouse | recognizes αvβ3 dimer | Blockade of TNFα and bFGF induced angiogenesis. Induces apoptosis | Deemed safe in Phase I trials; Ineffective for advanced cancers. |
| Etaracizumab (Abegrin) | humanized mouse monoclonal derived from LM609 | human, mouse | recognizes αvβ3 dimer | Blockade of TNFα and bFGF induced angiogenesis. Induces apoptosis | In Phase I trials, long term safety established; failed to demonstrate efficacy. |
| CNTO 95 | humanized IgG1 | human, does not recognize murine | αv integrin specific αvβ3, αvβ5 | Inhibits melanoma cell adhesion, migration and invasion in vitro. Inhibits tumor growth in mouse melanoma xenografts | Phase I trials established safety, 1 prolonged response observed. Phase II trials are underway. |
| c7E3 | chimeric mouse human | human, murine | αIIbβ3, αvβ3, MAC-1 | Potenet antiplatelet drug. Shows anti-cancer activity in preclinical studies. | FDA approved for treatment/ prevention of thrombosis during angioplasty and in Acute Coronary Syndromes. Increased risk of bleeding events may preclude use in cancer drug regimens. |
| 17E6 | mouse monoclonal | human, does not recognize murine | αv integrin specific αvβ3, αvβ5, αvβ1 | Inhibits tumor growth in αvβ3 positive tumor xenografts. Activity attributed to blockade of tumor integrins. | Immunogenicity and human anti-mouse antibody responses preclude its use in clinical applications. |
LM609 LM609 was developed in the laboratory of David Cheresh and was subsequently used in the discovery and characterization of αvβ3 integrin as adhesion molecules in endothelial and melanoma cell lines. As such it is a mouse monoclonal antibody that recognizes human αvβ3 integrin dimers. It displays cross reactivity with cognate rabbit and chicken but not murine αvβ3 integrins [108]. LM609 binds αvβ3 integrin and prevents the adhesion and spreading of endothelial cells to ECM proteins fibrinogen, vitronectin, von Wilebrand Factor mulitmers, and RGD-peptide coated surfaces but not collagen or laminin as these interactions are mediated through alternate adhesion receptors [98]. LM609 is unable to lift attached endothelial cells from the former mentioned ECM components presumably due to epitope masking as a result of intergrin interactions with matrix proteins. In cell based models of bone resorption LM609 supresses osteopontin-induced cytosolic calcium reduction and subsequent osteoclast function through the inhibition of αvβ3 signaling [109]. In quail embryo models of embryonic neovasculogenesis, LM609 effectively disrupts the process of vessel formation [110]. LM609 also inhibits angiongenesis in mouse-human skin graft tumor models [111]. The anti-angiogenic and anti-tumor properties observed with LM609 treatment in experimental models occurs primarily via the inhibition of αvβ3 mediated TNFα and bFGF signaling pathways. The murine origin of LM-609 limits its clinical utility, raising the concern of antigenic responses when administered to humans. Humanization of LM-609 using phage display strategy produced the second generation αvβ3 antibody, Vitaxin.
Vitaxin (MEDI-523, Applied Molecular Evolution, San Diego, CA) Vitaxin is the humanized version of LM609 containing the mouse CDR region grafted to human IgG1 Kappa. Humanization using phage display libraries produced greater affinity while retaining epitope specificity and reduced antigenicity [112]. In pre-clinical evaluation Vitaxin inhibited growth-factor mediated endothelial cell proliferation and prevented angiogenesis. Vitaxin has been evaluated in two Phase I clinical trials for the treatment of advanced, treatment refractory solid tumors. The initial Phase I trial evaluated dosing in range of 0.1 to 4 mg/kg administered weekly as a 90 minute intravenous infusion over a period of six weeks among 17 patients. Adverse effects were observed in 88% of the patients but were mild and of low grade in the initial studies. The most common adverse event was infusion reaction including fever, nausea and chills but preadministration of aspirin and diphenhydramine abrogated this effect and no further reactions were noted with pre-treatment. No immunogenicty, anti-vitaxin antibodies or propensities for prolonged bleeding or delayed wound healing were noted among any of the patients. The volumes of distribution were consistent with that of plasma distribution. The low dose regimen of 0.1 mg/kg resulted in plasma half-life of 14.6 hours, while the highest dose tested, 4.0 mg/kg produced much longer plasma half-lives between 63-138 hours. The AUC was increased with dose but not proportionally. Vitaxin treatment resulted in 7 instances of stable disease and one partial response. Continued treatment up to 93 weeks in 3/17 patients was well tolerated and delayed disease progression but did not result in tumor regression [113]. The second Phase I trial evaluated Vitaxin administered as two doses of either 10, 50 or 200 mg seperated by a three week interval, with a focus on radioimaging and gathering of pharmacokinetic data for Phase II studies. Vitaxin was administered as two doses of 10, 50 or 200 mg administered at day 0 and 21 as a 90 minute intravenous infusion. No toxicity was noted for the lowest dose cohort, the most common adverse event in the other cohorts was infusion related response such as fever and nausea. Grade 3 pain and hepatic toxicity and grade 1 headache were also reported among patients in these cohorts. No anti-vitaxin antibodies or immunogenicty was noted in this trial. Pharmacokinetic parameters for the 10 mg dose were not obtained due to low plasma recovery following the 90 minute infusion. Higher doses of vitaxin demonstrated greater plasma recovery. Doses of 200 mg produce plasma half-life (t1/2) of ~7 days and circulating plasma levels of ≥ 1mg/ mL, a level consistent with saturation of αvβ3 receptors in vitro. No tumor response was noted in any of the patients in any of the treatment cohorts. Two patients in the 50 mg cohort and 1 patient in the 200 mg cohort were assessed at day 43 as having achieved stable disease, received 4 additional doses of vitaxin and were taken off study at day 85 with stable disease. Two patients in the 10 mg cohort and 1 patient each from the 50 and 200 mg cohorts were taken off study at day 43 due to disease progression. Two patients, one each from the 10 and 200 mg cohorts were removed from study after 1 dose due to disease progression. Radio-imaging was not successful due to label instability.[114] In general, vitaxin was well tolerated in patients even with prolonged administration. Pharmacokinetic data obtained suggests that dosing within ranges of 200 to 800 mg is likely to produce plasma concentrations consistent with complete receptor occupation.
Abegrin (etaracizumab, MEDI-522, MedImmune) The absence of objective disease responses seen in the vitaxin phase I and II trials was attributed to limitations of affinity and stability in vivo. It was believed that improvements in these parameters would result in a better therapeutic outcome. Therefore, the epitope affinity of Vitaxin was improved using step-wise affinity maturation producing the humanized monoclonal antibody, Abegrin [115]. This antibody maintained specificity for αvβ3, yet inhibited binding to vitronectin and fibrinogen with 7.2 fold greater potency than its predecessors LM-609 and Vitaxin. Abegrin was highly active in preclinical evaluations, preventing cell adhesion, migration, proliferation and integrin mediated signaling events in a dose dependent manner at clinically achievable concentrations [115]. Phase I studies evaluated pharmacokinetic parameters and angiogenic response in patients with advanced stage, treatment refractory neoplasms representing a wide spectrum of cancer types with particular focus on late-stage colorectal cancers, melanomas and lymphomas. Reports of Phase I trials involving 25 lymphoma and solid tumor patients showed dosing in the range of 2-10 mg/kg/ weekly to be well tolerated. Pharmacokinetic parameters such as peak and trough plasma concentrations, Cmax and AUC were increased in a dose dependent manner and accumulated with continued dosing such that values recorded at week 4 were higher than those observed in week 1. Plasma concentration half-life was longer at week 4 compared to week 1. There were no significant differences in accumulation between doses. No major or serious adverse events were recorded and as such MTD was not achieved in this study. Antigenicity was not observed in any of the treated patients. In total 8 of 25 patients achieved stable disease at the 8 week evaluation. Stable disease was recorded in 4 patients with metastatic renal cell carcinoma following 16-95+ months on treatment. Although no significant changes were observed in vascular area, endothelial cell proliferation and apoptosis or β3 integrin expression levels, surrogate measures of angiogenic potential were altered in response to Abegrin treatment. Tumor blood mean transit time was increased in one renal cell carcinoma patient following treatment, perhaps due to decreased tumor vascularization resulting from αvβ3 integrin inhibition. Levels of phosphorylated Focal adhesion kinase (pFAK) were significantly decreased in skin blood vessels following one round of treatment, suggesting that Abegrin may act through disruption of focal adhesions [116].
Abegrin has completed Phase II clinical trials for the treatment of multiple cancers, rheumatoid arthritis and plaque psoriasis either as a single agent or in combination with clinically relevant drugs. Based on results of the Phase II trial of Abegrin in combination with the DNA alkylating agent, dacarbazine in metastatic melanomas, in which treatment alone or in combination did not significantly impact overall survival, further oncology trials have been suspended pending further investigation [117].
CNTO95 CNTO95 is a fully humanized antibody produced by immunization of transgenic mice carrying the human repetoire of immunoglobulins. CNTO95 binds αv integrins including αvβ1, αvβ3, αvβ5 and αvβ6. CNTO95 binds purified αvβ3 and αvβ5 with dissociation constants (Kd) of 2.1 and 2.5 nM respectively and cell-surface αvβ3 and αvβ5 Kd in the range of 5- 24 nM [118]. In preclinical evaluation CNTO95 showed significant anti-angiogenic and anti-tumor activity. CNTO95 prevented adhesion and migration of human umbilical vein endothelial cells (HUVECs) and human melonoma cells plated on the αvβ3 and αvβ5 ECM ligands vitronectin, fibrinogen, and fibrin [118]. In a dose dependent fashion, CNTO95 inhibited HUVEC and human melanoma cell proliferation and inhibited neovascularization in both in vitro and in vivo models of angiogenesis [118]. In xenograft studies, CNTO95 significantly inhibited tumor growth independent of its anti-angiogenic effect and potentiated the effects of fractionated radiation on tumor growth [119]. CNTO95 distributes systemically and binds αv integrin on the surface of both normal and neoplastic tissue. Despite systemic tissue distribution, no adverse effects were noted after prolonged administration up to 6 months in macaque monkeys [120]. CNTO95 is currently under investigation in Phase I and II clinical trials as a single agent for the treatment of advanced melanomas or in combination with dacarbazine or avastin for treatment of solid tumor or prednisone and docetaxel for treatment of metastatic hormone refractory prostate cancer. Results of the Phase I, dose-escalating trials in patients harboring solid tumors CNTO demonstrated a favorable safety profile in which infusion reactions controlled with acetominophen appeared to be the most common adverse event. Pharmacokinetic analysis of dosing between 0.1 and 10 mg/kg showed rapid clearance at lower dosage and non-linear decrease in the rate of clearance indicative of tissue binding. Accumulation upon repeated administration was noted only in the two highest dose cohorts (3, 10 mg/kg). Analysis of a post-treatment biopsy confirmed the presence of CNTO 95 within the tumor cytoplasm and decreased expression of bcl-2 following treatment. A partial response and stable disease were documented in 1 and 6 of 24 patients respectively, although of the six with stable disease, five entered the trial with this status. Following treatment, stable disease was recorded for up to seven months [121]. Based on the favorable safety profile, tissue distribution and incidence of partial response, Phase II trials will be pursued further.
c7E3 (Abcixmab) c7E3 is currently approved by the FDA for treatment of multiple acute coronary syndromes, for use in percutaneous coronary intervention and myocardial infarction and has been reviewed for these purposes in a previous section. Abciximab recognizes αvβ3 integrin with nearly the same affinity as αIIbβ3 and for this reason may also have anti-angiogenic and antitumor activities. In addition through the inhibition of αIIbβ3 on platelets, Abciximab may have anti-metastatic activity by preventing the adhesion of αvβ3 adhesion competent tumors to platelet thrombi.
RGD-based peptides Integrins αvβ3 and αvβ5 bind cellular adhesion molecules through the interaction with RGD-containing recognition motifs found within endogenous ligands such as vitronectin, fibrinogen and fibronectin. The first peptide-based pharmacological antagonists of the αvβ3 integrin were protein fragments based on the RGD-sequence. These flexible linear peptides lacked integrin specificity and were susceptible to proteolytic degradation in vivo making them unsuitable for drug development. Structure activity relationships based on thoughtful chemical modifications proved to be quite informative in the development of more potent, selective and biologically stable peptide drugs. Cyclization of the RGD peptide and addition of D-amino acids created steric constraints to better mimic the spatial orientation of the natural ligand while simultaneously protecting against enzymatic hydrolysis thereby increasing binding affinity and biological stability. Variations in peptide length (i.e. penta- versus hexa- peptides) were found to alter integrin selectivity. Introduction of D-isoforms and N-methylation of RGD- flanking amino acids was found to increase binding affinity. Cilengitide, a cyclic RGD peptide clearly exemplifies the effects cyclization and amino acid modification impart on receptor specificity and peptide stability. Cilengitide binds integrin αvβ3 4 times more potently than cRGDfV, a cyclic pentapeptide containing D-phenylalanine and 362 times more potently than the straight chain peptide GRGDSPK [122].
Cilengitide (EMD 121974, Merck) Cilengitide (Fig. 12) is a cyclic pentapeptide containing the RGD sequence, D-phenylalanine and N-methyl valine, denoted c-[Arg-Gly-Asp-DPhe-(NMeVal)] (Fig. 12). It is a potent, selective inhibitor of integrins αvβ3 and αvβ5. In solid phase receptor binding assays the IC50 for inhibition of integrin αvβ3 and αvβ5 binding to vitronectin are 0.58 nM and 37 nM respectively. Cilengitide shows considerably lower affinity for other integrins, with IC50 concentrations 10- to 100-fold higher in receptor binding assays. These differences in IC50 concentration among the various integrins are indicative of the selectivity of cilengitide for αvβ3 and αvβ5. Cilengitide binds the RGD-binding cleft of integrins αvβ3 and αvβ5 obstructing adhesion to the extracellular matrix and inhibiting integrin mediated cell-cell interactions. The EC50 for cell attachment varies with cell type but is generally in the high nanomolar to low micromolar range for both αvβ3 and αvβ5. By inhibiting integrin binding, cilengitide attenuates integral cellular functions that are mediated through adhesion, cytoskeletal organization and intracellular signaling pathways [123].
Structures of αvβ3 antagonists in clinical development.
Cilengitide has shown promising potential for therapeutic development based on preclinical studies. In contrast to the therapeutic antibodies targeting αv integrins currently in clinical evaluation, cilengitide is able to lift matrix-adherent cells, and does so at concentrations similar to that inhibiting cell attachment. Thus, by competitive inhibition, cilengitide may offer increased benefit by targeting both nascent and established tumor vasculature. Additionally, cilengitide has shown anti-cancer activity in cell lines and animal models that express αvβ3 and αvβ5 integrins. The interactions between tumor cells and angiogenic endothelium are complex and thus experimental evidence has not clearly delineated the interplay between these two cell types.
In cell culture models, cilengitide attenuates growth, proliferation and survival in endothelial cells and tumor cells expressing αvβ3 and αvβ5 integrins by preventing interactions between integrins and the extracellular matrix. Cilengitide inhibits endothelial cell migration, attachment and spreading on vitronectin at low micromolar concentrations. Similar effects have been noted following cilengitide treatment in αvβ3, αvβ5 positive glioma [124], breast [125], melanoma [126] and non-small cell lung cancer cell lines [127]. Following cilengitide treatment, endothelial and glioma cells show disruption of actin organization and tight junction formation consistent with inhibition of cytoskeletal organization [128]. Cilengitide inhibits cell signaling through FAK-Src-Akt and Erk mediated pathways in endothelial and tumor cells and attenuates the effect of VEGF stimulation on growth factor signaling [129]. At sublethal concentrations, cilengitide inhibits the NFkB component, p65Rel-A in U251 glioblastoma cells [100]. Cilengitide has shown significant anti-angiogenic and anti-tumor activity in vivo. In several cases, the tumor sensitivities and drug combinations that have been determined in preclinical models have been recapitulated in clinical trials.
In chick chorioallantoic membrane (CAM) assays, injection of cilengitide into CAM vasculature prevented the growth of implanted U87MG glioma and DAOY medulloblastoma tumor fragments compared to the inactive peptide EMD135981. The inhibition of tumor growth was attributed to the anti-angiogenic activity of cilengitide as membrane vascularity surrounding the tumor graft was greatly decreased by cilengitide treatment as compared to the inactive control peptide [130]. In animal models, cilengitide, as a single agent effectively inhibited the growth and proliferation of orthotopically implanted malignant glioma and medulloblastoma cell lines in nude mice. Daily treatment of established xenografts with 100-200 µg cilengitide significantly delayed tumor growth and prolonged survival compared to controls. The delayed tumor growth was accompanied by increases in CD31 and tumor doubling times, elevated apoptotic index and decreases in proliferative index and tumor vessel number and density [130]. Interestingly, the efficacy of cilengitide as a single agent appears to be limited to brain tumors, as results have reported a modest effect of cilengitide on melanoma, breast and lung cancer xenografts [100, 125, 127]. The disparity in the anti-angiogenic effects of cilengitide may be dependent on the microenvironment in which a tumor resides as parallel heterotopic and orthotopic implantation of identical glioma cell xenografts exhibit differential sensitivities to cilengitide treatment [130].
Radiation therapy has been demonstrated to have effects on tumor vasculature in addition to its direct cytotoxic effects on tumor cells. Interestingly, studies have shown that radiation can increase the expression of integrin αvβ3 up to two fold in endothelial cells as well as breast and lung cancer cells. In vitro and in vivo studies have shown benefit from combination of radiation therapy and cilengitide treatment. In cell based assays combination of radiation and cilengitide treatment enhances cell detachment, inhibits endothelial cell tube formation, decreases cell proliferation and increases apoptosis. In a human breast cancer xenograft tumor model, combination of radioimmunotherapy (RIT) with cilengitide resulted in radiation dose dependent increase of cure rate from 15% (RIT alone) to 53% (RIT plus cilengitide) at 260 µCi [125]. A more recent study using U251 human glioma cells showed that cilengitide blocked tumor cell adhesion in vitro with no effect on responses of these cells to radiotherapy. In contrast, cilengitide strongly amplified radiation effects on endothelial cell survival. These effects were translated to prolonged survival (50 days from control, 110 days radiation only, and >200 days combination of cilengitide and radiation) along with enhanced apoptosis and suppressed tumor growth. The synergism was determined to be highly schedule dependent, as a single dose of cilengitide (4 mg/kg) given between 4 and 8 hour prior to radiation was required to produce this effect. Dosing at shorter and longer time periods failed to prolong survival [100].
Based on preclinical animal models, cilengitide has been demonstrated to synergize with the DNA alkylating agent, temozolomide in malignant melanoma. Combination of 100 mg/kg/day temozolomide with 40 mg/kg/day cilengitide significantly inhibits B16 melanoma growth compared to control or cilengitide treated mice. This effect was accompanied by low overall toxicity suggesting potential clinical application [126-127].
Phase I trials of cilengitide in solid tumors and malignant gliomas. Cilengitide has been evaluated in six open-label, Phase I trials with the intent to establish safety and pharmacokinetic profiles, determine maximal tolerated dose and optimal biological dose and correlate responses with potential angiogenic and genomic biomarkers. The initial two trials enrolled adult patients with advanced stage solid tumors refractory to conventional treatment or lacking standardized therapy [131-132]. Two subsequent trials evaluated cilengitide in adults with various types of malignant glioma [133] and in children with refractory brain tumors [134]. Phase I trials in patients with locally advanced or metastatic cancers and Kaposi's sarcoma have been completed, but results have not been released. Phase I studies in patients with advanced solid tumors or lymphomas are currently underway.
In the first phase I study, cilengitide was administered parenterally at doses between 30-1600mg/m2 twice weekly in 37 patients with solid tumors [132]. In the subsequent Phase I trial of cilengitide in patients with advanced solid tumors, 20 enrollees received similar infusions at doses of 600 and 1200 mg/m2 for up to 50 courses [131]. The Phase I trials in adults with recurrent gliomas enrolled 51 patients and followed similar treatment schedules as previous trials. Dose escalation was performed in the range of 120-2400 mg/m2 [133]. The pediatric study followed similar dose escalation protocols, with 35 patients receiving cilengitide infusions between 120-2400 mg/m2 [134].
In each of the four Phase I trials reported, cilengitide was administered as a 1-hour intravenous infusion at doses between 30 and 2400 mg/m2 twice weekly, in 4- week cycles up to 50 cycles. Pharmacokinetics were similar amongst all trials including the pediatric glioma trial. Serum pharmacokinetics established a linear relationship between dose and AUC and Cmax. The terminal half life (t1/2) was between 2.5 - 4 hours. Kinetics is dose independent and time invariable. Systemic clearance, volume of distribution at steady state (Vss) and t1/2 were not significantly different between dose levels or following prolonged administration (day 1 compatred to day 15). The Vss is in line with distribution within the interstitial space as it correlates with the extracellular fluid volume. Systemic clearance is low compared to measurement of the rate of renal and heptic blood flow. Dose per body surface area did not affect clearance suggesting flat dosing was possible. Pharmacokinetic parameters are not affected by concurrent use of enzyme inducing anticonvulsants. Peak plasma concentrations at 120mg/m2 infusion of cilengitide reached the optimal concentration that inhibited tumor growth in preclinical models, but the short plasma half-life favors the adjustment of dose to obtain continuous exposure.
In the initial Phase I studies of cilengitide administered to patients with solid tumors as single treatment at dosing up to 2400 mg/m2 intravenously, twice a week for at least 4 consecutive weeks, dose-dependent toxicity and dose limiting toxicity (DLT) were not observed [131-132]. However, in the Phase I trial of cilengitide in adults with recurrent malignant glioma several grade 3 and 4 DLTs, possibly related to treatment were observed [133]. These DLTs included thrombosis, joint and bone pain, thrombocytopenia, anorexia, hypoglycemia, and hyponatremia, but were limited to one occurence per cohort. Grade 3 and 4 toxicities possibly related to study drug were noted in the pediatric glioma trials as well [134]. At doses of 2400 mg/m2, 3 of 13 pediatric patients developed intratumoral hemorrhage, two of which were asymptomatic and would under current standard be classified as Grade 1 or 2. These events were not considered dose limiting since their occurrence at days 44-157 post inception, was outside the timeframe of DLT observance. Other observed toxic effects of cilengitide treatment were mild to moderate in severity and similar amongst all studies. These effects included grade 1 to 2 fatigue, nausea, and headache, vomiting and anorexia. The toxicities were not dose-dependent and did not require dose reduction. In general, treatment was well tolerated, with mild to moderate toxicity (Grade 1 and 2 only) reported at all dose levels in three of four trials and a limited number of DLTs in the adult glioma trial. For this reason, definition of maximum-tolerated dose (MTD) was not achieved in any of the four trials.
Although patient response to treatment is not a primary endpoint in Phase I trials, Cilengitide as a signle agent did exhibit promising results particularly in the glioma patients. In trials of Cilengitide in adults with recurrent gliomas 2 complete responses were noted at doses of 360 and 2400 mg/m2 following 12 and 24 months of treatment. Remission was sustained up to 29 and 15 months post-treatment in these patients. In addition partial responses were recorded for three patients with a mean duration of 9.3 months. Stable disease was achieved in 16 patients for a mean period of 5.4 months. Of these patients, 4 sustained progression free survival for up to 29 months following Cilengitide treatment. The responses seen with Cilengitide in pediatric patients with refractory brain tumors appeared hopeful as well. One complete response was achieved at 1200 mg/m2 dosing following one year of treatment, with improvement in clinical symptoms sustained up to one year. In total six instances of stable disease were also associated with Cilengitide treatment in this trial. Two patients completing one year of treatment sustained stable disease up to 22 weeks. Three patients were evaluated as having stable disease following the second 4-week treatment cycle based on MRI results but failed to meet all protocol criteria for stable disease. There were no complete responses recorded for any patient in the trials of cilengitide for the treatment of refractory or advanced solid tumors. Taken together there were seven instances of stable disease ranging in duration from 4-15 months in duration associated with Cilengitide treatment [133].
Evaluation of endothelial cell proliferation markers, tumor and endothelial cell apoptosis and adhesion molecule expression failed to demonstrate correlates between patient response and the anti-angiogenic activity of cilengitide [131-133]. Correlation studies for serum angiogenesis factors, including P-selectin, I-CAM, V-CAM-1, VEGF, bFGF, systemic angiogenesis factors, endothelial cell apoptosis, gene expression profiles and tumor tissue mean vessel density, all showed no correlation or no reliable correlation with response or predicted anti-tumor activity except for a modest increase of P-selectin over time. Use of MRI to measure tumor perfusion detected slight differences between responders and non-responders but could not be fit with pharmacodynamic modeling. Decreased tumor perfusion was associated with higher cilengitide dose. The failure to establish correlates between biological markers and tumor response is not surprising, as the lack of experimentally validated biomarkers for the measurement of anti-angiogenic activity is a major impediment for the development and evaluation of agents of this type.
Interactions with cytochrome P450 and P-glycoprotein family members can alter drug exposure levels and duration, significantly impacting drug efficacy and toxicities. Concommitant administration of enzyme inducing or inhibitory drugs and population-wide differences in genetic constitution are two common factors affecting enzyme activity. Combination of cilengitide with cytochrome P450 enzyme inducing anticonvulsants had no effect on pharmacokinetic parameters suggesting that this family of drug metabolizing enzymes may not have a great impact on treatment [133]. Pharmacogenetic analysis of single nucleotide polymorphisms (SNPs) in the P-glycoprotein isoform ABCB1 in 15 pediatric patients receiving cilengitide suggests that SNPs in exon 26 may alter renal and overall clearance [134]. Further study in larger populations is warranted.
Overall assessment of Phase I trials demonstrated that Cilengitide treatment is well tolerated across a wide range of doses, with few serious toxicities. Dosing in the range of 120-360 mg/m2 appears sufficient to achieve plasma concentrations similar to those determined experimentally to be sufficient for anti-tumor activity, with higher dosing associated with response to treatment. The lack of serious toxicity suggests that cilengitide could easily be combined with conventional chemotherapies and targeted anti-cancer agents for improved results. In these preliminary evaluations, Cilengitide showed promise for the treatment of gliomas, a subset of cancers with poor prognoses that are notoriously difficult to treat. Taken together, the positive results obtained in Phase I trials have led to the design and implementation of numerous Phase II trials of cilengitide as both a single agent and in combination with cytotoxic and targeted therapies for the treatment of brain tumors, prostate cancer, melanomas, pancreatic cancer and lung cancer.
Phase II studies of cilengitide as a single agent and in combination. At least five Phase II trials of cilengitide as a single agent have been completed and several others are ongoing. Trials currently underway are evaluating cilengitide as a single agent for the treatment of metastatic melanoma, progressive and recurrent glioblastoma multiforme, recurrent glioma and localized and metastatic prostate cancers. Results of trials in glioblastoma multiforme and asymptomatic, androgen-independent prostate cancer have been reported and show modest response rates.
The results of a multi-center, randomized Phase II trial examined response to flat dosing in glioblastoma patients having undergone no greater than two previous cycles of standard treatment. In this trial, 81 patients were randomized to receive dosing of either 500 or 2000 mg infusions of cilengitide twice weekly for up to 48 weeks [135]. In addition to evaluation of safety and efficacy profiles, this trial compared plasma and cerebrospinal fuid (CSF) pharmacokinetic parameters to evaluate the efficiency of drug delivery to the tumor site. Plasma pharmacokinetic parameters were similar to those observed in Phase I trials. Cmax and AUC were linear and dose dependent with no accumulation over prolonged administration. Drug accumulation was confirmed in the cerebrospinal fluid. CSF drug concentrations were dose dependent and maximal concentrations were found to be approximately 100 fold less than those observed in the plasma. The CSF time to Cmax was delayed 2 h compared to plasma Cmax with elimination half-life up to two fold longer. No complete responses were observed in this trial, although 7 patients achieved a partial response. Progression free survival was mainted for 10-36 months amongst all responders. Grade 3-4 toxicities were infrequent and limited to hematological origin. None of the treatment groups achieved statistical significance in the defined endpoints of 6 months progression free survival, overall survival, radiologic response or quality of life measures. Patients in the 2000 mg treatment cohort did tend to have higher response rates as measured by radiologic response, media overall survival and progression free survival. The endpoint of 6 month progression free survival was met in a median 15% of patients in the 2000 mg cohort compared to 10% from the 500mg group. The overall survival (OS) was 9.9 months in the 2000 mg cohort versus 6.5 months in the 500 mg cohort. A higher percentage of patients in the 2000 mg cohort completed 12 or more treatment cycles, but the percentage of patients completing 24 cycles was the same. In the 2000 mg treatment group 13% of the patients completed >12 treatment cycles as compared to 7% in 500 mg treatment group. The number of patients receiving ≥ 24 cycles was 5% in both treatment groups. The overall response rates were deemed to be similar to those observed with temozolomide treatment suggesting that for the treatment of glioblastoma multiforme, cilengitide may act more potently when combined with standard therapies.
Preliminary results of a randomized Phase II trial of cilengitide as a single agent treatment for asymptomatic, androgen-independent prostate cancer showed modest effects, with a failure to meet the 6 month primary endpoint, a predicted rate of 75% delay in progression based on historical controls [136]. Patients randomized to receive flat dosing of 500 or 2000 mg infusions twice weekly in 6-week cycles showed 6 month progression free survival rates of 14 and 27%, respectively. Stable disease was observed in 27% and 36% of patients in the 500 and 2000 mg cohorts. Analysis of biological markers failed to establish correlates between treatment response and marker expression. Bone markers did not demonstrate significant correlation with response and the number of circulating tumor cells was increased in both treatment groups as disease progressed. As in past trials, treatment was well tolerated. Toxicities were limited to Grade 3 and lower in both treatment groups. Based on the lower than hypothesized impact on outcomes, further enrollment was halted.
In Phase II trials as a single agent, cilengitide was well tolerated and displayed uniform pharmacokinetic parameters amongst studies. Cilengitide is able to penetrate the blood brain barrier and accumulate in the CSF following treatment, a critical determinant for therapeutic delivery. Response rates, progression free survival and overall survival were moderate when adminstered as a single agent in both glioblastoma multiforme and androgen-independent prostate cancer patients. The utility of cilengitide appears more evident for the treatment of glioblastoma multiforme as current treatment modalities fail to produce significant remission rates or even delays in progression and no standard therapy currently exists in the likely event of recurrance. Under these circumstances, treatments that offer even modest response warrant further development.
Phase II trials of cilengitide in combination with standard therapies Angiogenesis is a complex process and current evidence suggests that combination of anti-angiogenic agents with cytotoxic and/or targeted drugs may be necessary for complete growth inhibition. Furthermore, preclinical studies demonstrate that Cilengitide sensitize tumor cells to radiation treatment which on its own has also shown anti-angiogenic effects. Cilengitide has a favorable safety profile over a wide range of doses and modest effects as a single agent suggesting the combination with cytotoxic and targeted drugs and/or radiotherapy would be a feasible approach to improve treatment modalities. To this end, numerous studies have investigated the effects of Cilengitide when combined with treatment regimens for pancreatic and non-small cell lung cancer, squamous cell carcinoma of the head and neck and glioblastoma multiforme. Results of trials in glioblastoma multiforme and pancreatic cancer have recently been reported.
Combination of cilengitide and gemcitabine. Survival rates for pancreatic cancer are historically low. In patients presenting with localized disease, median survival times are 6-10 months. Advanced disease survival times are roughly half that of early stage patients. Gemcitibine has replaced 5-fluoruracil as the standard of care for advanced disease, but improved treatment and regimens are still needed to increase overall survival rates. In a recent multi-national, open-label, controlled, randomized Phase II pilot study with advanced stage pancreatic cancer patients, combination of cilengitide with gemcitibine failed to demonstrate a difference in outcome compared to treatment with gemcitibine alone despite the significant role of angiogenesis in pancreatic tumor growth [137]. A total of 89 patients were enrolled and randomized to receive 600 mg/m2 cilengitide as intravenous infusion twice weekly in 4 week cycles combined with 1000 mg/m2 gemcitibine administered as intravenous infusion weekly for 3 weeks followed by one week rest or a similar schedule of gemcitibine alone. Study protocol defined the primary endpoint as overall survival and secondary endpoints of progression free survival, response rate and quality of life. CA19.9 served as a biomarker of disease progression and plasma VEFG and bFGF levels were evaluated as markers of angiogenic response. The combination of gemcitibine and Cilengitide was well tolerated and no influence of gemcitibine on Cilengitide pharmacokinetic parameters was detected. The number of cycles of treatment received and the duration of treatment was similar amongst both groups. Quality of life measures were not significantly different between groups. The combination of gemcitibine and Cilengitide did not change the overall rate of survival. With the single agent mean survival was 7.7 months compared to 6.7 months when combined with cilengitide. The progression free survival times were similar with median PFS 3.8 with gemcitibine and 3.6 months in the combination group. CA19.9 plasma levels increased with disease progression as expected and no relationship between VEGF and bFGF and disease response could be established. Combined treatment with cilengitide and gemcitibine could be safely administered to patients with no need for dose adjustment but failed to demonstrate benefit over gemcitibine alone. Based on these results, it is unlikely that cilengitide would benefit pancreatic cancer patients either alone or in combination.
In constrast to the less than significant results of combined gemcitibine and cilengitide treatment in pancreatic cancer patients, a case report details the partial remission of an advanced, refractory squamous cell carcinoma of the head and neck following combined cilengitide and gemcitibine treatment in a single patient [138]. This patient, in the fourth relapse with a 15cm diameter squamous cell carcinoma originating from the upper left jaw, was treated with 600 mg/m2 cilengitide twice weekly in combination with 1000 mg/m2 gemcitabine at day 1 and 8 every three weeks for 5 months and achieved partial response with improved functionality and quality of life measures. The patient maintained stable disease for 12 months on cilengitide maintenance therapy with no bleeding side effects. The squamous cell carcinoma in this case report was highly vascularized making it a good candidate for antiangiogenic therapies. Prior to combined treatment with cilengitide and gemcitibine, treatment with gemcitibine alone failed to stabilize disease in this patient. Therefore, unlike trials in pancreatic cancer, some benefit was seen with combination of gemcitibine and cilengitide in this iindividual case study.
Combination of cilengitide and radiation therapy and temozolomide. The addition of cilengitide to radiotherapy and temozolomide based treatment regimens has shown promising preliminary results in ongoing Phase II trials in both newly diagnosed and progressive glioblastoma multiforme [139-140]. In addition to the Phase II objectives sought, these trials are significant in that they represent progress that has made in determining tumor drug uptake and in identifying a subset of patients that may benefit from treatment. In a Phase II trial enrolling 52 patients with newly diagnosed glioblastoma multiforme receiving 500 mg cilengitide twice weekly during radiotherapy and in combination with temozolomide for 6 monthly cycles following radiotherapy, 69% achieved 6 months progression free survival compared to 54 % of patients receiving radiotherapy followed by temozolomide alone. The one-year overall survival was 67 and 62 % of patients for the cilengitide combination group and the radiotherapy and temozolomide group, respectively. Non-hematological grade 3-4 toxcities were limited, and included symptoms of fatigue, asthenia, anorexia, elevated liver function tests, deep vein thrombosis and pulmonary embolism in across a total of 5.7% of the patients. Grade 3-4 hematological malignancies were more common and included lymphopenia (53.8%), thrombocytopenia (13.4%) and neutropenia (9.6%). This trial is significant in the fact that is has provided the first evidence correlating a molecular biomarker with response to treatment. Decreased methylguanine methyltransferase (MGMT) expression was associated with favorable outcome. Patients harboring increased MGMT promoter methylation appeared to benefit more from combined treatment with cilengitide than did patients lacking promoter methylation. The significance of the MGMT promoter methylation in predicting response is likely due to inclusion of temozolomide in the treatment combination.
A similar Phase II study evaluating safety and differences in overall survival among newly diagnosed glioblastoma multiforme patients receiving radiation therapy combined with temozolomide and varying doses of cilengitide is nearing completion. Preliminary reports specify that initial safety run-in studies in 18 patients receiving doses 500, 1000 and 2000 mg cilengitide found no dose limiting toxicities. Subsequently 94 patients were randomized to receive standard therapy plus 500 or 2000 mg cilengitide. Median survival time in both cohorts was 18.9 months. At 12 months the overall survival was 79.5 % (89/112 patients).
Delivery of ciengitide across the blood brain barrier. Passage across the blood brain barrier is necessary for effective drug delivery to brain neoplasms and successful treatment requires the accumulation of adequate amounts of drug at the tumor site. A Phase II trial of cilengitide in combination with surgical debulking demonstrated intratumoral drug levels following presurgical administration of drug. In this trial carried out by the North American Brain Tumor Consortium, 30 glioblastoma multiforme patients with up to two previous progressions were randomized to receive 3 doses of 500 or 2000 mg cilengitide in the 8 days prior to surgical debulking [140]. Following surgery both groups received 2000 mg cilengitide on a twice weekly dosing schedule. Mean intratumoral cilengitide concentrations 24 hours after the last dose were 919 ng/g and 2561 ng/g of tissue for 500 or 2000 mg doses, respectively. Based on the intratumoral cilengitide concentration tissue to plasma ratios were calculated to be 1.83 for the 500-mg dose and 4.17 for the 2000-mg dose. This study effectively demonstrates that cilengitide can be delivered across the blood-brain barrier and remain at the tumor site for at least 24 hours.
Based on these results, numerous additional trials of cilengitide as a single agent and in combination with standard therapies are currently underway, including one Phase III trial of cilengitide in combination with radiotherapy and temozolomide in glioblastoma multiforme patients with methylated MGMT gene promoter status. Further Phase II trials of cilengitide in combination with standard therapy for glioblastoma multiforme are underway to evaluate the significance of promoter methylation in treatment response. Additional trials are evaluating the safety and efficacy of cilengitide when combined with standard treatments for non-small cell lung cancers and squamous cell carcinomas of the head and neck.
RGD-containing disintegrins. The disintegrins are low molecular weight peptides derived from snake venoms, more specifically pit viper venoms. The disintegrins are classified into three classes, the RGD-, KTS and MLD- based disintegrins. The classification is based on ligand mimetic motifs distributed within their disulfide-rich primary sequences. The RGD-based members are the most studied disintegrins and have been investigated for their ability to inhibit angiogenesis and tumor growth and osteoporotic bone loss via blockade of RGD- liganded integrins. The RGD disintegrins bind with high affinity and specificity through an RGD-sequence to RGD-dependent integrins and thereby block their function. At least six separate disintegrins have been isolated and characterized for their RGD binding affinity. Although the disintegrins are highly effective in binding and inhibiting integrin function, their utility in clinical applications is limited due to the problems of immunogenicity and lability common to peptide-based drugs and the limited supplies typical of natural products. Efforts have been undertaken to reduce the immunogenicity of disintegrins using approaches such as targeted drug delivery. Recombinant DNA technology has been employed to produce large quantities of disintegrin. Liposomal preparations of recombinant contortrostatin have been shown to reduce the growth of MDA-MB-435 breast cancer xenografts in nude mice.
L000845704 L000845704 (Merck) (Fig. 12) is the first small molecule αvβ3 antagonist to progress through clinical trials for the treatment of osteoporosis. Incorporation of RGD-like pharmacophoric features on a chain shortened, imidazolidine core scaffold produdced L000845704, a selective, potent inhibitor of αvβ3 with favorable pharmacokinetic properties, lowered plasma protein binding affinity and good oral bioavailability. In scinitillation proximity assay the IC50 of αvβ3 binding is 0.08 nM. L000845704 shows at least 100 fold lower affinity for integrins αvβ5 and αIIbβ3. In preclinical animal models L000845704 was efficacious in modulating clinically relevant measures of osteoporosis. Ovarectimized female rats, administered L000845704 at 10 and 30 mg/kg/day over a period 28 days exhibited respective 8.9 and 12.8% (p values < 0.0002) increases in measures of bone mineral density compared to vehicle treated counterparts. L000845704 increased bone mineral density above baseline as effectively as, the antiresorptive, alendronate in growing young male rats (18.4 versus 20.5%, p values < 0.0002). The urinary marker of bone degredation crosslinked n-telopeptide was decreased by 39% in ovarectimized female rhesus monkeys following two weeks 15 mg/kg/ day oral dosing of L000845704 [141].
The favorable results in preclinical animal models led to the advancement of L000845704 into Phase I clinical trials. Dosing at 400 mg daily over a two-week period in post menopausal women was well tolerated and produced significant decreases in urinary n-telopeptide crosslinks suggesting antiresorptive activity. Short plasma half-life prompted the evaluation of varied doses and schedules to optimize evaluation of efficacy and safety parameter in the subsequent study. In this large scale, multicenter, randomized, double blind, placebo controlled 12 month study, 227 post-menopausal women received 2 week open label calcium carbonate, hydroxy vitamin D run-in therapy followed by randomization to either 100 mg qd, 400 mg qd, 200 mg bid or placebo treatment groups. 100mg was deemed the minimum effect dose and 400 mg was chosen on the premise that it would provide serum levels similar to those found to be necessary for efficacy in animal models. The 200 mg bid was chosen to evaluate its ability to provide higher serum trough concentrations. All active treatment groups met the primary endpoint of decrease from baseline in serum and urine n-telopeptide crosslink/creatine levels. No dose response was seen, and maximal decrease was achieved following two weeks treatment and maintained for 22 more weeks at similar levels in all treatment groups. A slight increase in levels was noted at 24 weeks but an overall decrease up to 42% was maintained through 52 weeks treatment. Serum collagen I n-telopeptide crosslinks (cTx) decreased in a manner similar to urinary telopeptides with the initial decrease after two weeks treatment with sustained suppression. At the end of 52 weeks treatment cTx levels were decreased 42.2, 46.6 and 48.4% over baseline. Markers of bone formation including osteocalcin and serum bone specific alkaline phosphatase steadily decreased following treatment, stabilizing after 24 weeks. This effect was in-line with other antiresorptive agents tested clinically. The secondary endpoint of increased lumbar spine bone mineral density was met in all three treatment groups. Increases of 2.1, 3.1 and 3.5% were noted in the 100 mg qd, 400 mg qd and 200 mg bid treatment groups respectively (p values < 0.001) In the remaining measures of total hip and femoral neck bone mineral density the 200 mg bid treatment appeared superior to other treatment levels. Total bone mineral density increased in all treatment groups and in the placebo group as well. In safety evaluations, L000845704 was well tolerated. No drug-related serious adverse events were recorded. Minor adverse events were headache, decreased appetite and hot flashes. Overall, L000845704 met the endpoints set forth in clinical protocol and exhibited a favorable safety profile. 200 mg bid dosing appears superior to 400 mg administered once daily [142].
SB273005 SB273005 (Fig. 12) is an orally active small molecule antagonist of αvβ3 that has shown efficacy in preclinical animal models. SB273005 binds strongly to αvβ3 and lesser degree to αvβ5. It is selective against αIIbβ3 and α5β1. Inhibtion of αvβ3 by SB273005 prevented in vivo bone loss in rat models of estrogen deprivation mediated bone loss [143]. As prophylactic or therapeutic agent, it also prevented and reduced edema and inflammation associated with arthritis in rat models of rheumatoid arthiritis.
7. α5β1 integrin antagonists
α5β1 integrin antagonists are currently under development for the treatment of cancers, retinal neovascularization in Age-related Macular Degeneration (AMD). α5β1 integrin expression is up-regulated in endothelial cells during angiogenesis and is expressed on tumor cells' surface. Ligation of α5β1 integrins promotes cell growth and migration through the activation of Akt and MAPK mediated signaling pathways. Inhibition of α5β1 integrin binding to fibronectin prevents angiogenesis and induces endothelial cell apoptosis.
Volocixmab Volociximab is the first α5b1 antagonist to progress to clinical development. It is a chimeric human-mouse monoclonal antibody with high affinity for the α5β1 integrin. Volociximab binds human α5β1 integrin with a Kd of 0.32 nM and the homologous rabbit integrin with a Kd of 7.24 nM [144]. It is not reactive with mouse α5β1 integrin [145]. The IC50 for inhibition of human α5β1 binding to fibronectin is 0.15 µg/mL. Through inhibition of α5β1, volociximab induces cell death and prevents capillary tube formation in cell based assays. The lack of additive anti-proliferative effect in endothelial cells when combined with VEGF blocking antibody led to the belief that volociximab inhibition of α5β1 is downstream, and therefore independent of growth factor stimulation. Volociximab has shown both anti-tumor and anti-angiongenic activity in animal models. Angiogenesis and tumor growth were diminished in CAM models following volociximab administration. In cynomolgus monkey models of choroid vascularization, volociximab prevented angiogenesis. In rabbit VX2 carcinoma xenograft models, 10 mg/kg volociximab prevented the growth of newly implanted as well as established tumors over a three week period. In the rabbit xenograft studies response to treatment was associated with higher serum levels. The differences in serum levels were attributed to the presence of rabbit anti-chimeric responses. Increased levels of serum anti-volociximab antibodies were noted in animals less responsive to treatment [145]. Volociximab was well tolerated in Phase I trials, patients with refractory solid tumors dosed with 0.15-15 mg /kg for up to 104 infusions showed no dose limiting toxicities. The most common toxicities noted included nausea, fatigue, headache and athraligia and myaligias; all were low grade. The incidence of anti-chimeric antibodies and neutralizing antibodies was low, occurring in 14.3% of patients. One minor response was noted in a renal cell carcinoma patient. Stable disease was confirmed in five patients for periods lasting 4-14 months [146].
In subsequent Phase II trials, volociximab showed a similar safety and tolerability profile and demonstrated promising potential efficacy in prolonging disease progression and overall survival. In a Phase II trial involving patients with metastatic clear cell renal cell carcinoma, 10 mg/kg IV infusion of volociximab delayed disease progression up to 22 months. Overall survival was 68% at 22 months and stable disease was achieved in 80% of patients. Another Phase II trial of evaluated volociximab 10 mg/ kg every two weeks in combination with 1000 mg/m2 gemcitibine on days 1, 8 and 15 in 20 male patients with metastatic pancreatic cancers. Toxicities included nausea, vomiting and constipation. Overall survival was 5.4 months and median time to progression was 4.3 months. Partial response was noted in one patient and stable disease was confirmed in 10 patients. Combination of 10 mg/kg volociximab with dacarbazine 1 g/m2 every four weeks resulted in stable disease in 22/40 patients with metastatic melanoma. One partial response was also observed [147].
Additional Phase II and III trials involving volociximab as a single agent and in combination regimens are currently underway for the treatment of metastatic melanoma, non-small cell lung cancer and peritoneal cancer. Phase I trials in non-small cell lung cancer in combination with carboplatin and paclitaxel have been terminated as have Phase II trials in metatstatic melanomas and renal cell carcinomas and platinum resistant ovarian cancer. Phase I trials of volociximab for the treatment of macular degeneration have also been initiated.
JSM6427 JSM6427 is a small molecule antagonist selective for α5β1 integrin. It is currently under evaluation in a Phase I clinical trial in patients with neovascular age-related macular degeneration. This study will establish pharmacokinetic, safety and tolerability profiles for JSM6427 following single and repeated intravitreal injection of dosing in the range of 1.5 to 15 mg/mL. The basis for clinical development is supported by preclinical evidence demonstrating a role for α5β1 antagonists as a therapeutic intervention to inhibit neovascularization. JMS6427 induces HUVEC apoptosis and inhibits fibronectin-induced Erk phosphorylation in ARPE cells both in a dose dependent fashion. In a mouse model of choroidal neovascularization induced by laser damage to the retina, continous infusion of JSM6427 induced endothelial cell apoptosis and prevented choroidal neovascularization in established lesions at dosing between 1.5 and 10 µg/h. Intravitreal injection failed to produce similar results in newly acquired lesions suggesting that inhibition of neovascularization by JSM6427 requires sustained plasma drug concentrations [148].
8. Conclusion
In the last two decades great progress has been made in the discovery and development of integrin targeted therapeutics. Years of intense research into integrin function has provided an understanding of the potential applications for the treatment of disease. Advances in structural characterization of integrin-ligand interactions has proved beneficial in the design and development of potent, selective inhibitors for a number of integrins involved in platelet aggregation, inflammatory responses, angiongenesis, neovascularization and tumor growth.
The αIIbβ3 integrin antagonists were the first inhibitors to make their way into clinical use and have proven to be effective and safe drugs, contributing to the reduction of mortality and morbidity associated with acute coronary syndromes. Interestingly, the prolonged administration of small molecules targeting this integrin for long-term prevention of thrombosis related complications have not been successful, for reasons that are not yet fully understood. This suggests that modulating the intensity, duration and temporal aspects of integrin function may be more effective than simply shutting off integrin signaling in some instances. Further research into the dynamics of platelet activation and thrombosis formation may elucidate the mechanisms by which integrin activation is modulated.
The introduction of α4 targeted therapies held great promise for the treatment of inflammatory diseases. The development of Natalizumab greatly improved the quality of life for multiple sclerosis patients and those suffering with Crohn's Disease compared to previous treatments, but the role in asthma related inflammation could not be validated. Unfortunately for MS and Crohn's patients, immune surveillance in the central nervous system was also compromised as a direct effect α4β7 antagonism, with potentially lethal effects. Thus Natalizumab and related α4β7 targeting drugs are now limited to patients refractory to standard therapies. The design and development of α4β1 antagonists for the treatment of Crohn's Disease may offer benefit with decreased risks. The involvement of these integrins in fetal development also raises concerns for widespread clinical use.
Integrin antagonists that target angiogenesis are progressing through clinical trials. Cilengitide has shown promising results for the treatment of glioblastomas and recurrent gliomas, cancers with notoriously low survival and cure rates. The greatest challenge facing the development of anti-angiogenic integrin targeted therapies is the overall lack of biomarkers by which to measure treatment efficacy.
Overall, the road to clinical application has been marked with both success and failure and sometimes unexpected results as we find that integrin function may be more complex and intricate than once believed.
Acknowledgements
This study was supported through funds provided by the National Cancer Institutes, National Institutes of Health (RO1CA120188 grant).
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Shimaoka M, Springer T. Therapeutic antagonists and conformational regulation of integrin function. Nat Rev Drug Discov. 2003;2:703-16
2. Takada Y, Ye X, Simon S. The integrins. Genome Biol. 2007;8:215
3. Takada Y, Strominger J, Hemler M. The very late antigen family of heterodimers is part of a superfamily of molecules involved in adhesion and embryogenesis. Proc Natl Acad Sci U S A. 1987;84:3239-43
4. Hynes R. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673-87
5. Legate K, Wickström S, Fässler R. Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev. 2009;23:397-418
6. Desgrosellier J, Cheresh D. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9-22
7. Michishita M, Videm V, Arnaout M. A novel divalent cation-binding site in the A domain of the beta 2 integrin CR3 (CD11b/CD18) is essential for ligand binding. Cell. 1993;72:857-67
8. Seow K, Xiong J, Arnaout M, Welge J, Rippmann F, Goodman S. Divalent cations and the relationship between αA and βA domains in integrins. Biochem Pharmacol. 2002;64:805-12
9. Lee J, Bankston L, Arnaout M, Liddington R. Two conformations of the integrin A-domain (I-domain): a pathway for activation? Structure. 1995;3:1333-40
10. Xiong J, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott D. et al. Crystal structure of the extracellular segment of integrin αvβ3. Science. 2001;294:339-45
11. Xiong J, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman S. et al. Crystal structure of the extracellular segment of integrin αvβ3 in complex with an Arg-Gly-Asp ligand. Science. 2002;296:151-5
12. Adair B, Yeager M. Three-dimensional model of the human platelet integrin αIIbβ3 based on electron cryomicroscopy and x-ray crystallography. Proc Natl Acad Sci U S A. 2002;99:14059-64
13. Xiong J, Mahalingham B, Alonso J, Borrelli L, Rui X, Anand S. et al. Crystal structure of the complete integrin αvβ3 ectodomain plus an alpha/beta transmembrane fragment. J Cell Biol. 2009;186:589-600
14. Takagi J, Petre B, Walz T, Springer T. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110:599-11
15. Hynes R. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11-25
16. Hawiger J, Timmons S, Kloczewiak M, Strong D, Doolittle R. gamma and alpha chains of human fibrinogen possess sites reactive with human platelet receptors. Proc Natl Acad Sci U S A. 1982;79:2068-71
17. Doolittle R, Watt K, Cottrell B, Strong D, Riley M. The amino acid sequence of the alpha-chain of human fibrinogen. Nature. 1979;280:464-8
18. Phillips D, Charo I, Scarborough R. GPIIb-IIIa: the responsive integrin. Cell. 1991;65:359-62
19. Tam S, Sassoli P, Jordan R, Nakada M. Abciximab (ReoPro, chimeric 7E3 Fab) demonstrates equivalent affinity and functional blockade of glycoprotein IIb/IIIa and αvβ3 integrins. Circulation. 1998;98:1085-91
20. Simon D, Xu H, Ortlepp S, Rogers C, Rao N. 7E3 monoclonal antibody directed against the platelet glycoprotein IIb/IIIa cross-reacts with the leukocyte integrin Mac-1 and blocks adhesion to fibrinogen and ICAM-1. Arterioscler Thromb Vasc Biol. 1997;17:528-35
21. Mager D, Mascelli M, Kleiman N, Fitzgerald D, Abernethy D. Simultaneous modeling of abciximab plasma concentrations and ex vivo pharmacodynamics in patients undergoing coronary angioplasty. J Pharmacol Exp Ther. 2003;307:969-76
22. Mazzaferri EJ, Young J. Abciximab: a review and update for clinicians. Expert Rev Cardiovasc Ther. 2008;6:609-18
23. Dennis M, Henzel W, Pitti R, Lipari M, Napier M, Deisher T. et al. Platelet glycoprotein IIb-IIIa protein antagonists from snake venoms: evidence for a family of platelet-aggregation inhibitors. Proc Natl Acad Sci U S A. 1990;87:2471-5
24. Charo I, Scarborough R, DU MEE C. Therapeutics I: pharmacodynamics of the glycoprotein IIb/ IIIa antagonist Integrilin: Phase I clinical studies in normal healthy volunteers. Circulation. 1992;86:I-260
25. Hartman G, Egbertson M, Halczenko W, Laswell W, Duggan M, Smith R. et al. Non-peptide fibrinogen receptor antagonists. 1. Discovery and design of exosite inhibitors. J Med Chem. 1992;35:4640-2
26. Schrör K, Weber A. Comparative pharmacology of GP IIb/IIIa antagonists. J Thromb Thrombolysis. 2003;15:71-80
27. Roe M, Moliterno D. The EPILOG trial. Abciximab prevents ischemic complications during angioplasty. Evaluation in PTCA to improve long-term outcome with Abciximab GP IIb/IIIa Blockade. Cleve Clin J Med. 1998;65:267-72
28. Sipkins D, Cheresh D, Kazemi M, Nevin L, Bednarski M, Li K. Detection of tumor angiogenesis in vivo by αvβ3-targeted magnetic resonance imaging. Nat Med. 1998;4:623-6
29. Tcheng J, Kereiakes D, Braden G, Jordan R, Mascelli M, Langrall M. et al. Readministration of abciximab: interim report of the ReoPro readministration registry. Am Heart J. 1999;138:S33-8
30. Randomised placebo-controlled trial of abciximab before and during coronary intervention in refractory unstable angina: the CAPTURE Study. Lancet. 1997;349:1429-35
31. Phillips D, Teng W, Arfsten A, Nannizzi-Alaimo L, White M, Longhurst C. et al. Effect of Ca2+ on GP IIb-IIIa interactions with integrilin: enhanced GP IIb-IIIa binding and inhibition of platelet aggregation by reductions in the concentration of ionized calcium in plasma anticoagulated with citrate. Circulation. 1997;96:1488-94
32. Tcheng J, Talley J, O'Shea J, Gilchrist I, Kleiman N, Grines C. et al. Clinical pharmacology of higher dose eptifibatide in percutaneous coronary intervention (the PRIDE study). Am J Cardiol. 2001;88:1097-102
33. Novel dosing regimen of eptifibatide in planned coronary stent implantation (ESPRIT): a randomised, placebo-controlled trial. Lancet. 2000;356:2037-44
34. Gibson C, Dotani M, Murphy S, Marble S, Dauterman K, Michaels A. et al. Correlates of coronary blood flow before and after percutaneous coronary intervention and their relationship to angiographic and clinical outcomes in the RESTORE trial. Randomized Efficacy Study of Tirofiban for Outcomes and REstenosis. Am Heart J. 2002;144:130-5
35. Labinaz M, Ho C, Banerjee S, Martin J, Chen S, Mensinkai S. Meta-analysis of clinical efficacy and bleeding risk with intravenous glycoprotein IIb/IIIa antagonists for percutaneous coronary intervention. Can J Cardiol. 2007;23:963-70
36. Tcheng J. Glycoprotein IIb/IIIa receptor inhibitors: putting the EPIC, IMPACT II, RESTORE, and EPILOG trials into perspective. Am J Cardiol. 1996;78:35-40
37. Renda G, Rocca B, Crocchiolo R, Cristofaro R, Landolfi R. Effect of fibrinogen concentration and platelet count on the inhibitory effect of abciximab and tirofiban. Thromb Haemost. 2003;89:348-54
38. Nurden P, Poujol C, Durrieu-Jais C, Winckler J, Combrié R, Macchi L. et al. Labeling of the internal pool of GP IIb-IIIa in platelets by c7E3 Fab fragments (abciximab): flow and endocytic mechanisms contribute to the transport. Blood. 1999;93:1622-33
39. Use of a monoclonal antibody directed against the platelet glycoprotein IIb/IIIa receptor in high-risk coronary angioplasty. The EPIC Investigation. N Engl J Med. 1994;330:956-61
40. Scarborough R, Kleiman N, Phillips D. Platelet glycoprotein IIb/IIIa antagonists. What are the relevant issues concerning their pharmacology and clinical use? Circulation. 1999;100:437-44
41. Berkowitz S, Harrington R, Rund M, Tcheng J. Acute profound thrombocytopenia after C7E3 Fab (abciximab) therapy. Circulation. 1997;95:809-13
42. Curtis B, Swyers J, Divgi A, McFarland J, Aster R. Thrombocytopenia after second exposure to abciximab is caused by antibodies that recognize abciximab-coated platelets. Blood. 2002;99:2054-9
43. Bassler N, Loeffler C, Mangin P, Yuan Y, Schwarz M, Hagemeyer C. et al. A mechanistic model for paradoxical platelet activation by ligand-mimetic alphaIIb beta3 (GPIIb/IIIa) antagonists. Arterioscler Thromb Vasc Biol. 2007;27:e9-15
44. Bougie D, Wilker P, Wuitschick E, Curtis B, Malik M, Levine S. et al. Acute thrombocytopenia after treatment with tirofiban or eptifibatide is associated with antibodies specific for ligand-occupied GPIIb/IIIa. Blood. 2002;100:2071-6
45. Reddy M, Carmody T, Kereiakes D. Severe delayed thrombocytopenia associated with abciximab (ReoPro) therapy. Catheter Cardiovasc Interv. 2001;52:486-8
46. Alexander J, Harrington R. Recent antiplatelet drug trials in the acute coronary syndromes. Clinical interpretation of PRISM, PRISM-PLUS, PARAGON A and PURSUIT. Drugs. 1998;56:965-76
47. Anand S, Yusuf S. Oral anticoagulant therapy in patients with coronary artery disease: a meta-analysis. JAMA. 1999;282:2058-67
48. Chew D, Bhatt D, Topol E. Oral glycoprotein IIb/IIIa inhibitors: why don't they work? Am J Cardiovasc Drugs. 2001;1:421-8
49. Chew D, Bhatt D, Sapp S, Topol E. Increased mortality with oral platelet glycoprotein IIb/IIIa antagonists: a meta-analysis of phase III multicenter randomized trials. Circulation. 2001;103:201-6
50. Stead L, Vaidyanathan L. Evidence-based emergency medicine/systematic review abstract. Role of abciximab in the management of acute ischemic stroke. Ann Emerg Med. 2009;53:392-4
51. Solovey A, Kollander R, Shet A, Milbauer L, Choong S, Panoskaltsis-Mortari A. et al. Endothelial cell expression of tissue factor in sickle mice is augmented by hypoxia/reoxygenation and inhibited by lovastatin. Blood. 2004;104:840-6
52. Hebbel R, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. 2004;11:129-51
53. Platt O. Sickle cell anemia as an inflammatory disease. J Clin Invest. 2000;106:337-8
54. Brittain J, Mlinar K, Anderson C, Orringer E, Parise L. Activation of sickle red blood cell adhesion via integrin-associated protein/CD47-induced signal transduction. J Clin Invest. 2001;107:1555-62
55. Henn V, Slupsky J, Gräfe M, Anagnostopoulos I, Förster R, Müller-Berghaus G. et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391:591-4
56. Dale G, Friese P, Batar P, Hamilton S, Reed G, Jackson K. et al. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature. 2002;415:175-9
57. Frishman W, Grewall P. Serotonin and the heart. Ann Med. 2000;32:195-209
58. Phillips D, Agin P. Platelet membrane defects in Glanzmann's thrombasthenia. Evidence for decreased amounts of two major glycoproteins. J Clin Invest. 1977;60:535-45
59. Lee S, Ataga K, Zayed M, Manganello J, Orringer E, Phillips D. et al. Phase I study of eptifibatide in patients with sickle cell anaemia. Br J Haematol. 2007;139:612-20
60. Bayless K, Meininger G, Scholtz J, Davis G. Osteopontin is a ligand for the alpha4beta1 integrin. J Cell Sci. 1998;111( Pt 9):1165-74
61. Holzmann B, McIntyre B, Weissman I. Identification of a murine Peyer's patch--specific lymphocyte homing receptor as an integrin molecule with an alpha chain homologous to human VLA-4 alpha. Cell. 1989;56:37-46
62. Butcher E, Picker L. Lymphocyte homing and homeostasis. Science. 1996;272:60-6
63. Luster A, Alon R, von Andrian U. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182-90
64. Conlon P, Oksenberg J, Zhang J, Steinman L. The immunobiology of multiple sclerosis: an autoimmune disease of the central nervous system. Neurobiol Dis. 1999;6:149-66
65. Fauci AS. Harrison's Principles of Internal Medicine. 17th ed: McGraw-Hill Medical. 2008
66. Stamper HJ, Woodruff J. Lymphocyte homing into lymph nodes: in vitro demonstration of the selective affinity of recirculating lymphocytes for high-endothelial venules. J Exp Med. 1976;144:828-33
67. Steffen B, Butcher E, Engelhardt B. Evidence for involvement of ICAM-1 and VCAM-1 in lymphocyte interaction with endothelium in experimental autoimmune encephalomyelitis in the central nervous system in the SJL/J mouse. Am J Pathol. 1994;145:189-201
68. Hesterberg P, Winsor-Hines D, Briskin M, Soler-Ferran D, Merrill C, Mackay C. et al. Rapid resolution of chronic colitis in the cotton-top tamarin with an antibody to a gut-homing integrin alpha 4 beta 7. Gastroenterology. 1996;111:1373-80
69. Kato S, Hokari R, Matsuzaki K, Iwai A, Kawaguchi A, Nagao S. et al. Amelioration of murine experimental colitis by inhibition of mucosal addressin cell adhesion molecule-1. J Pharmacol Exp Ther. 2000;295:183-9
70. Picarella D, Hurlbut P, Rottman J, Shi X, Butcher E, Ringler D. Monoclonal antibodies specific for β7 integrin and mucosal addressin cell adhesion molecule-1 (MAdCAM-1) reduce inflammation in the colon of scid mice reconstituted with CD45RBhigh CD4+ T cells. J Immunol. 1997;158:2099-106
71. Abraham W, Gill A, Ahmed A, Sielczak M, Lauredo I, Botinnikova Y. et al. A small-molecule, tight-binding inhibitor of the integrin α4β1 blocks antigen-induced airway responses and inflammation in experimental asthma in sheep. Am J Respir Crit Care Med. 2000;162:603-11
72. Sheremata W, Vollmer T, Stone L, Willmer-Hulme A, Koller M. A safety and pharmacokinetic study of intravenous natalizumab in patients with MS. Neurology. 1999;52:1072-4
73. Miller D, Khan O, Sheremata W, Blumhardt L, Rice G, Libonati M. et al. A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2003;348:15-23
74. Stüve O, Marra C, Bar-Or A, Niino M, Cravens P, Cepok S. et al. Altered CD4+/CD8+ T-cell ratios in cerebrospinal fluid of natalizumab-treated patients with multiple sclerosis. Arch Neurol. 2006;63:1383-7
75. Dalton C, Miszkiel K, Barker G, MacManus D, Pepple T, Panzara M. et al. Effect of natalizumab on conversion of gadolinium enhancing lesions to T1 hypointense lesions in relapsing multiple sclerosis. J Neurol. 2004;251:407-13
76. Havrdova E, Galetta S, Hutchinson M, Stefoski D, Bates D, Polman C. et al. Effect of natalizumab on clinical and radiological disease activity in multiple sclerosis: a retrospective analysis of the Natalizumab Safety and Efficacy in Relapsing-Remitting Multiple Sclerosis (AFFIRM) study. Lancet Neurol. 2009;8:254-60
77. Rudick R, Miller D, Hass S, Hutchinson M, Calabresi P, Confavreux C. et al. Health-related quality of life in multiple sclerosis: effects of natalizumab. Ann Neurol. 2007;62:335-46
78. Targan S, Feagan B, Fedorak R, Lashner B, Panaccione R, Present D. et al. Natalizumab for the treatment of active Crohn's disease: results of the ENCORE Trial. Gastroenterology. 2007;132:1672-83
79. Lindå H, von Heijne A, Major E, Ryschkewitsch C, Berg J, Olsson T. et al. Progressive multifocal leukoencephalopathy after natalizumab monotherapy. N Engl J Med. 2009;361:1081-7
80. del Pilar Martin M, Cravens P, Winger R, Frohman E, Racke M, Eagar T. et al. Decrease in the numbers of dendritic cells and CD4+ T cells in cerebral perivascular spaces due to natalizumab. Arch Neurol. 2008;65:1596-603
81. Wenning W, Haghikia A, Laubenberger J, Clifford D, Behrens P, Chan A. et al. Treatment of progressive multifocal leukoencephalopathy associated with natalizumab. N Engl J Med. 2009;361:1075-80
82. Van Assche G, Van Ranst M, Sciot R, Dubois B, Vermeire S, Noman M. et al. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn's disease. N Engl J Med. 2005;353:362-8
83. Kleinschmidt-DeMasters B, Tyler K. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N Engl J Med. 2005;353:369-74
84. Khatri B, Man S, Giovannoni G, Koo A, Lee J, Tucky B. et al. Effect of plasma exchange in accelerating natalizumab clearance and restoring leukocyte function. Neurology. 2009;72:402-9
85. Stüve O, Cravens P, Frohman E, Phillips J, Remington G, von Geldern G. et al. Immunologic, clinical, and radiologic status 14 months after cessation of natalizumab therapy. Neurology. 2009;72:396-401
86. Wehner N, Shopp G, Oneda S, Clarke J. Embryo/fetal development in cynomolgus monkeys exposed to natalizumab, an alpha4 integrin inhibitor. Birth Defects Res B Dev Reprod Toxicol. 2009;86:117-30
87. Wehner N, Shopp G, Osterburg I, Fuchs A, Buse E, Clarke J. Postnatal development in cynomolgus monkeys following prenatal exposure to natalizumab, an alpha4 integrin inhibitor. Birth Defects Res B Dev Reprod Toxicol. 2009;86:144-56
88. Spence S, Vetter C, Hagmann W, Van Riper G, Williams H, Mumford R. et al. Effects of VLA-4 antagonists in rat whole embryo culture. Teratology. 2002;65:26-37
89. Feagan B, Greenberg G, Wild G, Fedorak R, Paré P, McDonald J. et al. Treatment of active Crohn's disease with MLN0002, a humanized antibody to the alpha4beta7 integrin. Clin Gastroenterol Hepatol. 2008;6:1370-7
90. Crofts F, Pino M, DeLise B, Guittin P, Barbellion S, Brunel P. et al. Different embryo-fetal toxicity effects for three VLA-4 antagonists. Birth Defects Res B Dev Reprod Toxicol. 2004;71:55-68
91. Rohatagi S, Shah B, Zhang J, Zeng Z, Kirkesseli S, Jensen B. Estimating mass balance for inhaled drugs in humans: an example with a VLA-4 antagonist, IVL745. J Clin Pharmacol. 2004;44:348-58
92. Norris V, Choong L, Tran D, Corden Z, Boyce M, Arshad H. et al. Effect of IVL745, a VLA-4 antagonist, on allergen-induced bronchoconstriction in patients with asthma. J Allergy Clin Immunol. 2005;116:761-7
93. Diamant Z, Kuperus J, Baan R, Nietzmann K, Millet S, Mendes P. et al. Effect of a very late antigen-4 receptor antagonist on allergen-induced airway responses and inflammation in asthma. Clin Exp Allergy. 2005;35:1080-7
94. Ravensberg A, Luijk B, Westers P, Hiemstra P, Sterk P, Lammers J. et al. The effect of a single inhaled dose of a VLA-4 antagonist on allergen-induced airway responses and airway inflammation in patients with asthma. Allergy. 2006;61:1097-103
95. Avraamides C, Garmy-Susini B, Varner J. Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008;8:604-17
96. Saegusa J, Yamaji S, Ieguchi K, Wu C, Lam K, Liu F. et al. The direct binding of insulin-like growth factor-1 (IGF-1) to integrin alphavbeta3 is involved in IGF-1 signaling. J Biol Chem. 2009;284:24106-14
97. Varner J, Nakada M, Jordan R, Coller B. Inhibition of angiogenesis and tumor growth by murine 7E3, the parent antibody of c7E3 Fab (abciximab; ReoPro). Angiogenesis. 1999;3:53-60
98. Cheresh D, Spiro R. Biosynthetic and functional properties of an Arg-Gly-Asp-directed receptor involved in human melanoma cell attachment to vitronectin, fibrinogen, and von Willebrand factor. J Biol Chem. 1987;262:17703-11
99. Jain R. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58-62
100. Mikkelsen T, Brodie C, Finniss S, Berens M, Rennert J, Nelson K. et al. Radiation sensitization of glioblastoma by cilengitide has unanticipated schedule-dependency. Int J Cancer. 2009;124:2719-27
101. Masarachia P, Yamamoto M, Leu C, Rodan G, Duong L. Histomorphometric evidence for echistatin inhibition of bone resorption in mice with secondary hyperparathyroidism. Endocrinology. 1998;139:1401-10
102. Fisher J, Caulfield M, Sato M, Quartuccio H, Gould R, Garsky V. et al. Inhibition of osteoclastic bone resorption in vivo by echistatin, an "arginyl-glycyl-aspartyl" (RGD)-containing protein. Endocrinology. 1993;132:1411-3
103. Yamamoto M, Fisher J, Gentile M, Seedor J, Leu C, Rodan S. et al. The integrin ligand echistatin prevents bone loss in ovariectomized mice and rats. Endocrinology. 1998;139:1411-9
104. Crippes B, Engleman V, Settle S, Delarco J, Ornberg R, Helfrich M. et al. Antibody to beta3 integrin inhibits osteoclast-mediated bone resorption in the thyroparathyroidectomized rat. Endocrinology. 1996;137:918-24
105. McHugh K, Hodivala-Dilke K, Zheng M, Namba N, Lam J, Novack D. et al. Mice lacking β3 integrins are osteosclerotic because of dysfunctional osteoclasts. J Clin Invest. 2000;105:433-40
106. Holt I, Marshall M. Integrin subunit β3 plays a crucial role in the movement of osteoclasts from the periosteum to the bone surface. J Cell Physiol. 1998;175:1-9
107. Nakamura I, Tanaka H, Rodan G, Duong L. Echistatin inhibits the migration of murine prefusion osteoclasts and the formation of multinucleated osteoclast-like cells. Endocrinology. 1998;139:5182-93
108. Cheresh D. Human endothelial cells synthesize and express an Arg-Gly-Asp-directed adhesion receptor involved in attachment to fibrinogen and von Willebrand factor. Proc Natl Acad Sci U S A. 1987;84:6471-5
109. Miyauchi A, Alvarez J, Greenfield E, Teti A, Grano M, Colucci S. et al. Binding of osteopontin to the osteoclast integrin αvβ3. Osteoporos Int. 1993;3(Suppl 1):132-5
110. Drake C, Cheresh D, Little C. An antagonist of integrin alpha v beta 3 prevents maturation of blood vessels during embryonic neovascularization. J Cell Sci. 1995;108(Pt 7):2655-61
111. Brooks P, Strömblad S, Klemke R, Visscher D, Sarkar F, Cheresh D. Antiintegrin alpha v beta 3 blocks human breast cancer growth and angiogenesis in human skin. J Clin Invest. 1995;96:1815-22
112. Rader C, Cheresh D, Barbas Cr. A phage display approach for rapid antibody humanization: designed combinatorial V gene libraries. Proc Natl Acad Sci U S A. 1998;95:8910-5
113. Patel S, Jenkins J, Papadopolous N, Burgess M, Plager C, Gutterman J. et al. Pilot study of vitaxin--an angiogenesis inhibitor-in patients with advanced leiomyosarcomas. Cancer. 2001;92:1347-8
114. Posey J, Khazaeli M, DelGrosso A, Saleh M, Lin C, Huse W. et al. A pilot trial of Vitaxin, a humanized anti-vitronectin receptor (anti-αvβ3) antibody in patients with metastatic cancer. Cancer Biother Radiopharm. 2001;16:125-32
115. Cai W, Wu Y, Chen K, Cao Q, Tice D, Chen X. In vitro and in vivo characterization of 64Cu-labeled Abegrin, a humanized monoclonal antibody against integrin αvβ3. Cancer Res. 2006;66:9673-81
116. Delbaldo C, Raymond E, Vera K, Hammershaimb L, Kaucic K, Lozahic S. et al. Phase I and pharmacokinetic study of etaracizumab (Abegrin), a humanized monoclonal antibody against αvβ3 integrin receptor, in patients with advanced solid tumors. Invest New Drugs. 2008;26:35-43
117. Hersey P, Sosman J, O'Day S, Richards J, Bedikian A, Gonzalez R. et al. A randomized phase 2 study of etaracizumab, a monoclonal antibody against integrin αvβ3, +/- dacarbazine in patients with stage IV metastatic melanoma. Cancer. 2010;116(6):1526-34
118. Trikha M, Zhou Z, Nemeth J, Chen Q, Sharp C, Emmell E. et al. CNTO 95, a fully human monoclonal antibody that inhibits alphav integrins, has antitumor and antiangiogenic activity in vivo. Int J Cancer. 2004;110:326-35
119. Blue R, Murcia M, Karan C, Jirousková M, Coller B. Application of high-throughput screening to identify a novel alphaIIb-specific small- molecule inhibitor of αIIbβ3-mediated platelet interaction with fibrinogen. Blood. 2008;111:1248-56
120. Martin P, Jiao Q, Cornacoff J, Hall W, Saville B, Nemeth J. et al. Absence of adverse effects in cynomolgus macaques treated with CNTO 95, a fully human anti-αv integrin monoclonal antibody, despite widespread tissue binding. Clin Cancer Res. 2005;11:6959-65
121. Mullamitha S, Ton N, Parker G, Jackson A, Julyan P, Roberts C. et al. Phase I evaluation of a fully human anti-alphav integrin monoclonal antibody (CNTO 95) in patients with advanced solid tumors. Clin Cancer Res. 2007;13:2128-35
122. Heckmann D, Kessler H. Design and chemical synthesis of integrin ligands. Methods Enzymol. 2007;426:463-503
123. Nisato R, Tille J, Jonczyk A, Goodman S, Pepper M. αvβ3 and αvβ5 integrin antagonists inhibit angiogenesis in vitro. Angiogenesis. 2003;6:105-19
124. Taga T, Suzuki A, Gonzalez-Gomez I, Gilles F, Stins M, Shimada H. et al. αv-integrin antagonist EMD 121974 induces apoptosis in brain tumor cells growing on vitronectin and tenascin. Int J Cancer. 2002;98:690-7
125. Burke P, DeNardo S, Miers L, Lamborn K, Matzku S, DeNardo G. Cilengitide targeting of αvβ3 integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts. Cancer Res. 2002;62:4263-72
126. Tentori L, Dorio A, Muzi A, Lacal P, Ruffini F, Navarra P. et al. The integrin antagonist cilengitide increases the antitumor activity of temozolomide against malignant melanoma. Oncol Rep. 2008;19:1039-43
127. Albert J, Cao C, Geng L, Leavitt L, Hallahan D, Lu B. Integrin αvβ3 antagonist Cilengitide enhances efficacy of radiotherapy in endothelial cell and non-small-cell lung cancer models. Int J Radiat Oncol Biol Phys. 2006;65:1536-43
128. Yamada S, Bu X, Khankaldyyan V, Gonzales-Gomez I, McComb J, Laug W. Effect of the angiogenesis inhibitor Cilengitide (EMD 121974) on glioblastoma growth in nude mice. Neurosurgery. 2006;59:1304-12
129. Oliveira-Ferrer L, Hauschild J, Fiedler W, Bokemeyer C, Nippgen J, Celik I. et al. Cilengitide induces cellular detachment and apoptosis in endothelial and glioma cells mediated by inhibition of FAK/src/AKT pathway. J Exp Clin Cancer Res. 2008;27:86-98
130. MacDonald T, Taga T, Shimada H, Tabrizi P, Zlokovic B, Cheresh D. et al. Preferential susceptibility of brain tumors to the antiangiogenic effects of an αv integrin antagonist. Neurosurgery. 2001;48:151-7
131. Hariharan S, Gustafson D, Holden S, McConkey D, Davis D, Morrow M. et al. Assessment of the biological and pharmacological effects of the αvβ3 and αvβ5 integrin receptor antagonist, cilengitide (EMD 121974), in patients with advanced solid tumors. Ann Oncol. 2007;18:1400-7
132. Eskens F, Dumez H, Hoekstra R, Perschl A, Brindley C, Böttcher S. et al. Phase I and pharmacokinetic study of continuous twice weekly intravenous administration of Cilengitide (EMD 121974), a novel inhibitor of the integrins αvβ3 and αvβ5 in patients with advanced solid tumours. Eur J Cancer. 2003;39:917-26
133. Nabors L, Mikkelsen T, Rosenfeld S, Hochberg F, Akella N, Fisher J. et al. Phase I and correlative biology study of cilengitide in patients with recurrent malignant glioma. J Clin Oncol. 2007;25:1651-7
134. MacDonald T, Stewart C, Kocak M, Goldman S, Ellenbogen R, Phillips P. et al. Phase I clinical trial of cilengitide in children with refractory brain tumors: Pediatric Brain Tumor Consortium Study PBTC-012. J Clin Oncol. 2008;26:919-24
135. Reardon D, Fink K, Mikkelsen T, Cloughesy T, O'Neill A, Plotkin S. et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J Clin Oncol. 2008;26:5610-7
136. Bradley DA, Dunn R, Ryan C. et al. EMD121974 (NSC 707544, cilengitide) in asymptomatic metastatic androgen independent prostate cancer (AIPCa) patients (pts): A randomized trial by the Prostate Cancer Clinical Trials Consortium (NCI 6372). 2007 ASCO Annual Meeting Proceedings Part I. J Clin Oncol. 2007:5137
137. Friess H, Langrehr J, Oettle H, Raedle J, Niedergethmann M, Dittrich C. et al. A randomized multi-center phase II trial of the angiogenesis inhibitor Cilengitide (EMD 121974) and gemcitabine compared with gemcitabine alone in advanced unresectable pancreatic cancer. BMC Cancer. 2006;6:285
138. Raguse J, Gath H, Bier J, Riess H, Oettle H. Cilengitide (EMD 121974) arrests the growth of a heavily pretreated highly vascularised head and neck tumour. Oral Oncol. 2004;40:228-30
139. Stupp R, Hegi M, Gilbert M, Chakravarti A. Chemoradiotherapy in malignant glioma: standard of care and future directions. J Clin Oncol. 2007;25:4127-36
140. Gilbert M, Lamborn K, Lassman A. Tumor tissue delivery of cilengitide after intravenous administration to patients with recurrent glioblastoma (GBM): preliminary data from NABTC protocol 03-02. Dallas, Texas: 12th Annual Meeting for the Society of Neuro-Oncology. 2007:525
141. Hutchinson J, Halczenko W, Brashear K, Breslin M, Coleman P, Duong lT. et al. Nonpeptide αvβ3 antagonists. 8. In vitro and in vivo evaluation of a potent alphavbeta3 antagonist for the prevention and treatment of osteoporosis. J Med Chem. 2003;46:4790-8
142. Murphy M, Cerchio K, Stoch S, Gottesdiener K, Wu M, Recker R. Effect of L-000845704, an αvβ3 integrin antagonist, on markers of bone turnover and bone mineral density in postmenopausal osteoporotic women. J Clin Endocrinol Metab. 2005;90:2022-8
143. Lark M, Stroup G, Dodds R, Kapadia R, Hoffman S, Hwang S. et al. Antagonism of the osteoclast vitronectin receptor with an orally active nonpeptide inhibitor prevents cancellous bone loss in the ovariectomized rat. J Bone Miner Res. 2001;16:319-27
144. Ramakrishnan V, Bhaskar V, Law D, Wong M, DuBridge R, Breinberg D. et al. Preclinical evaluation of an anti-α5β1 integrin antibody as a novel anti-angiogenic agent. J Exp Ther Oncol. 2006;5:273-86
145. Bhaskar V, Fox M, Breinberg D, Wong M, Wales P, Rhodes S. et al. Volociximab, a chimeric integrin α5β1 antibody, inhibits the growth of VX2 tumors in rabbits. Invest New Drugs. 2008;26:7-12
146. Ricart A, Tolcher A, Liu G, Holen K, Schwartz G, Albertini M. et al. Volociximab, a chimeric monoclonal antibody that specifically binds α5β1 integrin: a phase I, pharmacokinetic, and biological correlative study. Clin Cancer Res. 2008;14:7924-9
147. Kuwada S. Drug evaluation: Volociximab, an angiogenesis-inhibiting chimeric monoclonal antibody. Curr Opin Mol Ther. 2007;9:92-8
148. Umeda N, Kachi S, Akiyama H, Zahn G, Vossmeyer D, Stragies R. et al. Suppression and regression of choroidal neovascularization by systemic administration of an alpha5beta1 integrin antagonist. Mol Pharmacol. 2006;69:1820-8
Author contact
![]() Corresponding author: Dr. Nouri Neamati, Department of Pharmacology and Pharmaceutical Sciences, University of Southern California, School of Pharmacy, PSC304 1985 Zonal Avenue, Los Angeles, CA 90089; E-mail: neamatiedu
Corresponding author: Dr. Nouri Neamati, Department of Pharmacology and Pharmaceutical Sciences, University of Southern California, School of Pharmacy, PSC304 1985 Zonal Avenue, Los Angeles, CA 90089; E-mail: neamatiedu