Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Integrin - An ideal therapeutic...
Integrin and RGD-peptide
Integrin targeted delivery of...
Integrin targeted delivery of...
Integrin targeted delivery of...
Conclusion
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2011; 1:189-200. doi:10.7150/thno/v01p0189 This volume Cite
Review
Integrin Targeted Delivery of Chemotherapeutics
Kai Chen1,2 ![]() , Xiaoyuan Chen 1
, Xiaoyuan Chen 1 ![]()
1. National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH), Bethesda, MD;
2. Molecular Imaging Center, Department of Radiology, Keck School of Medicine, University of Southern California, Los Angeles, CA
Published 2011-2-17
Abstract
Targeted delivery of chemotherapeutics is defined in the sense, that is, to maximize the therapeutic index of a chemotherapeutic agent by strictly localizing its pharmacological activity to the site or tissue of action. Integrins are a family of heterodimeric transmembrane glycoproteins involved in a wide range of cell-to-extracellular matrix (ECM) and cell-to-cell interactions. As cell surface receptors, integrins readily interact with extracellular ligands and play a vital role in angiogenesis, leukocytes function and tumor development, which sets up integrins as an excellent target for chemotherapy treatment. The peptide ligands containing the arginine-glycine-aspartic acid (RGD), which displays a strong binding affinity and selectivity to integrins, particularly to integrin αvβ3, have been developed to conjugate with various conventional chemotherapeutic agents, such as small molecules, peptides and proteins, and nanoparticle-carried drugs for integtrin targeted therapeutic studies. This review highlights the recent advances in integrin targeted delivery of chemotherapeutic agents with emphasis on target of integrin αvβ3, and describes the considerations for the design of the diverse RGD peptide-chemotherapeutics conjugates and their major applications.
Keywords: Integrin, Targeted delivery, Chemotherapeutics, RGD, Bioconjugation.
Introduction
Chemotherapy has been the main approach for the systemic treatment of advanced or metastatic diseases for more than half century. During the cells division process, chemotherapeutics is capable of killing proliferating cells, hence affecting aberrant cells. While highly efficient in preventing disease progression, however, chemo-therapeutic agents are often lack of selectivity for aberrant cells. Their toxicity to normal cells and non-diseased tissues, especially to rapidly growing cells such as blood, bone marrow and mucous membrane cells, cause serious side-effects that impose to administrate the chemotherapeutic agents at sub-optimal doses [1]. Because of intrinsic or acquired resistance of aberrant cells, benefits of the chemotherapeutic agents are also often limited [2]. The efficiency of the treatment can be increased by escalating the doses, but this option commonly results in considerable toxicity problem and thus is rarely considered. Selective delivery of chemotherapeutic agents to the disease site therefore represents a major challenge for improving current chemotherapy outcome [3]. Targeted delivery of chemotherapeutics is an emerging therapy strategy endowed with disease-targeting functions and carrying cytotoxic components by enabling the specific delivery of chemotherapeutic agents to aberrant tissues, thereby increasing their local efficacy while limiting their peripheral toxicity. Clearly, the success of this approach is heavily on the rational selection of appropriate biological targets.
Integrin - An ideal therapeutic target
In the past two decades, many efforts have been made to the discovery of cell adhesion molecules (CAMs), and their distinctive functions in cell morphology, locomotion, mitosis, cytokinesis, phagocytosis, and the maintenance of cell polarity [4-6]. It is known that CAMs play important roles in various disease states such as cancer [7-9], thrombosis [10-11], rheumatoid arthritis [12], and diabetes [13]. Numerous studies have been actively performing in investigation of the structure, function, and recycling mechanisms of some CAMs, as well as how to modulate them for controlling disease progression [14]. CAMs are glycoproteins expressed on the cell surface that functionalize as receptors for cell-to-cell and cell-to-extracellular matrix (ECM) adhesion [15-17]. In general, CAMs can be divided into four classes: integrins, selectins, cadherins, and the immunoglobulin superfamily.
Among these CAMs, integrins are a family of heterodimeric transmembrane glycoproteins involving in a wide range of cell-to-ECM and cell-to-cell interactions [18-19]. Each member of this family consists of non-covalently bound transmembrane polypeptide α and β subunits. In mammals, 18 α and 8 β subunits have been identified, which assemble into at least 24 different integrins [20]. The term integrin is derived from the ability of these proteins to link the ECM proteins with the intracellular cytoskeleton [21]. During binding to ECM proteins, integrins form clusters at the cell membrane that are associated with a cytoskeletal complex to promote actin filament assembly [22-23]. In turn, the reorganization of actin filaments into large stress fibers increases integrin clustering followed by enhancement of binding affinity with ECM proteins [24]. During cell locomotion and migration, integrin can undergo endocytosis and exocytosis [25-27]. In the process of cell locomotion, integrins activation can be triggered by vascular endothelial growth factor (VEGF) and controlled by protein kinase C (PKC) to continuously form new integrin-based focal contacts at the front of the cell [28]. While the cell migrates, the integrin interaction is released from extracellular ligands at the rear of the cells after forming new and persistent integrin connections at the front of the cell [29-30]. In migrating neutrophils, the distribution of integrins was observed to be maintained by Ca2+-dependent release of cell adhesion followed by endocytosis of the integrin [31]. Thus, integrin endocytosis has been considered as an attractive mechanism for controlling cell signaling pathways which can be stimulated by ligand binding.
The characteristic that integrins can be internalized by cells on activation with anchoring ligands significantly facilitates delivery of chemotherapeutics into neoplastic cells and leukocytes, while these chemotherapeutics conjugate with integrin-targeted ligands. Additionally, integrins are over-expressed on angiogenetic endothelial cells, whereas they are absent in pre-existing endothelial cells and normal tissues. Moreover, it has been found that certain tumor cells over-express integrins on their cellular membrane surface [32-34], which makes selective delivery of chemotherapeutic agents possible. All of these characteristics set up integrin an ideal target for both cancer and anti-angiogenesis therapy.
Integrin and RGD-peptide
The arginine-glycine-aspartic acid (RGD) cell adhesion sequence was discovered in fibronectin over two decades ago [35-36]. Later on, RGD-recognition sites were found in other ECM proteins [37-40] and the receptors for these ECM proteins were identified in the integrin family. Proteins that contain the RGD attachment site, together with the integrins that serve as receptors for them, constitute a major recognition system for cell adhesion. The RGD sequence is the cell attachment site of a number of adhesive ECM, blood, and cell surface proteins. It has been found that nearly half of the over 20 known integrins, including all five αv integrins, two β1 integrins (α5, α8) and αIIbβ3 integrin, recognize this sequence in their adhesion protein ligands [41].
The integrin αvβ3, consisting a 125-kDa αv subunit and a 105-kDa β3 subunit, binds a wide range of ECM proteins with RGD-containing components of the interstitial matrix such as vitronectin, fibronectin and thrombospondin [42-43]. Crystal structures of integrin αvβ3 complexed with RGD ligands have revealed an atomic basis for this interaction [43]. RGD binds at an interface between the α and β subunits, the R residue fitting into a cleft in a β-propeller module in the subunit, and the D coordinating a cation bound in a von Willebrand factor A - domain in the β subunit. It is well documented that Integrin αvβ3 is expressed on the cell membrane of various tumor cell types such as late stage glioblastoma, melanoma, ovarian, breast, and prostate cancer [32-34]. Integrin αvβ3 also plays critical role in tumor invasion and metastasis arising from its ability to recruit and activate MMP-2 and plasmin, which can degrade components of the basement membrane and interstitial matrix [44]. Among integrins family, integrin αvβ3 is the most intensively studied although many other integrins such as αvβ1, αvβ5, α5β1, and α4β1 also play important roles in regulating angiogenesis [45-49].
Based on these findings, linear as well as cyclic RGD peptides have been developed as ligands that preferentially bind to integrin αvβ3, related αv integrins, or other types of integrins. Among these RGD peptides, cyclic peptides c(RGDfK) and c(RGDyK), RGD4C, and RGD10 (Figure 1) showed high binding affinity and selectivity for integrin αvβ3 [50]. Thus, these RGD-peptides can be served as vectors for integrin αvβ3 targeted delivery of chemotherapeutics. Cyclic peptides c(RGDfK) and c(RGDyK) are the ones mostly used for the delivery of therapeutic agents. The amino group of the lysine residue on these peptides is an ideal site for further chemical conjugation reactions. Multivalent c(RGDfK) or c(RGDyK) peptide can be used to achieve higher binding affinity. Both of RGD4C and RGD10 peptide were discovered by phage display technology [51-52]. The RGD4C peptide contains two disulfide bonds and displays at least 20-fold more potent than similar peptide with a single disulfide bond. Although the RGD10 peptide contains only one disulfide bond, the residues flanking the CRGDC core display similar physicochemical properties as those in RGD4C. Overall, RGD4C and RGD10 show similar binding affinity to integrin αvβ3.
Chemical Structures of c(RGDyK), c(RGDfK), [c(RGDyK)2], RGD4C, and RGD10. Due to high binding affinity for integrin αvβ3, these RGD-containing peptides are commonly used for integrin αvβ3 targeted delivery of chemotherapeutics.
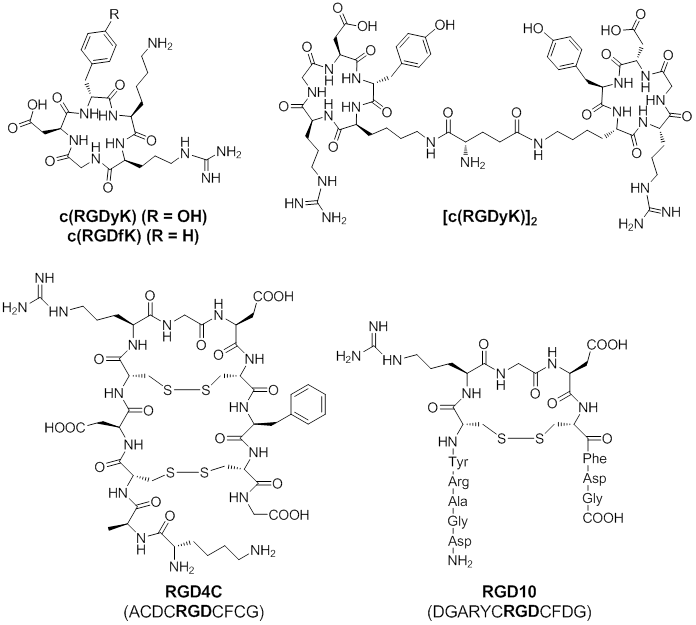
To date, agents that are commonly used to conjugate with RGD-peptides for chemotherapy treatments are: a) therapeutic small molecules, b) therapeutic peptides and proteins, and c) nanoparticle-carried chemotherapeutics.
Integrin targeted delivery of therapeutic small molecules
The first approach in which the RGD-peptide is used for drug targeting purposes focuses on the delivery of cytotoxic drugs for cancer treatments. These treatments are often effective, however, their high toxicity to healthy tissues is difficult to tolerate for patients. Thus, development of alternative vectorization of the cytotoxic drugs without killing healthy cells is critically demanded to circumvent this side effect.
For instance, doxorubicin, commonly used anticancer drug, was conjugated with RGD-peptides for integrin targeted therapy. A doxorubicin-RGD4C conjugate (doxo-RGD4C) is one of good examples. Doxo-RGD4C demonstrated equal efficacy as free doxorubicin in vitro and, more importantly, doxo-RGD4C showed improved inhibition of tumor growth and spreading of pulmonary metastases than free doxorubicin in mouse MDA-MB-435 breast cancer model, in which integrin αvβ3 is expressed by the endothelium in the angiogenic blood vessels and by the tumor cells themselves [53]. In addition, doxo-RGD4C was also found to be less toxic to liver and heart. In another similar study, doxo-RGD4C was tested in mouse αvβ3-negative MH134 hepatoma tumor model. As compared to doxorubicin alone, the doxo-RGD4C conjugate showed less treatment efficacy in vitro; however, the doxo-RGC4C conjugate demonstrated better anti-tumor activity in vivo. Because direct anti-tumor cell effects of the targeted doxorubicin are not expected in this integrin αvβ3-negative tumor model, this study suggests the anti-angiogenetic effect on the endothelial cells induced by doxo-RGD4C conjugate may lead to tumor recession [54].
One of the current strategies for providing substantial increases in the clinical efficacy of chemotherapeutic agents that preferentially kill dividing cells, both normal and tumor, is the use of relatively nontoxic prodrug forms that can be selectively activated in aberrant tissue [55]. Prodrugs are generally defined as agents that are transformed after administration, either by metabolism or spontaneous chemical breakdown, to pharmacologically active species. Several doxo-RGD4C conjugates have been designed as integrin αvβ3 targeted prodrugs that require activation by tumor-secreted enzymes. This strategy would reduce the toxicity of drugs and allow higher doses to be systemically delivered than non-targeted chemotherapeutic agents. A prodrug containing a plasmin-specific cleavage site was developed by de Groot et al. [56]. The release of the parent drug - doxorubicin can be achieved by plasmin cleavage followed by accumulation of the prodrug onto integrin αvβ3. In vitro studies showed the promising activity of prodrug after incubation with plasmin. The RGD-doxo prodrug was further proved to be capable of blocking the adhesion of endothelial cells to vitronectin in submicromolar concentrations, suggesting the prodrug retained antiangiogenic activity. However, poor solubility of doxo-RGD4C prodrug as well as difficulty of synthesizing RGD4C-drug conjugates are problematic for further development of this type of agents [54, 56-57]. In addition, two disulfide bonds in the RGD4C peptide are susceptible to be disrupted under reduced condition in cytoplasm, resulting in inactive form. In a recent study, Burkhart et al. [57] designed two RGD-based (RGD4C and Cilengitide) conjugates with the doxsaliform prodrug, which is spontaneously converted into an active metabolite of doxorubicin, doxorubicinformaldehyde, and leads to more cytotoxicity than doxorubicin [58]. Both conjugates maintained high affinity to integrin αvβ3 with IC50 values of 5-10 nanomolar. The in vitro cytotoxicity studies also revealed good growth inhibition of MDA-MB-435 breast cancer cells, demonstrating their capability of binding to integrin αvβ3. Further experiments showed that the RGD-containing prodrugs cannot significantly penetrate the cell membrane, in contrast to native doxorubicin and the released doxorubicin-formaldehyde. Thus, the proposed mechanism of action of these conjugates is to bind to integrin αvβ3 initially, followed by local release of the more lipophilic doxorubicin species, which then diffuse through cell membranes. Unfortunately, this study only focused on in vitro stage. In vivo anti-tumor efficacy of these two conjugates was not reported.
Besides doxorubicin, several other chemotherapeutic small molecules were also conjugated with RGD-peptides. We recently evaluated the antitumor activity of paclitaxel conjugated with a dimeric RGD peptide E[c(RGDyK)]2 (RGD2) [59]. Paclitaxel (PTX), a prototype of the taxane family of antitumor drugs, is commonly used in the treatment of advanced metastatic breast cancer. The RGD2-PTX conjugate inhibited cell proliferation with activity in comparable with that observed for paclitaxel, both of which are mediated by an arrest of G2/M phase of the cell cycle followed by apoptosis. We then labeled RGD2-PTX with 125I through the tyrosine residue on the RGD peptide. Integrin-specific accumulation of 125I-RGD2-PTX in orthotopic MDA-MB-435 tumor was observed. In a follow-up study, we evaluated the in vivo anti-tumor effect of the RGD2-PTX conjugate [60]. The treatment efficacy of RGD2-PTX was confirmed by size measurement, in vivo PET imaging, and ex vivo histopathology. The tumor growth delay is related to tumor proliferation rather than tumor metabolism, as confirmed by [18F]FDG and [18F]FLT PET imaging.
Kok's group recently developed a novel linking technique for the conjugation of drug to carriers by applying platinum-coordination chemistry [61]. The so-called universal linker system (ULS) can release the drug via a slow release profile. Thus, a prolonged action of the delivered drug can be achieved. New classes of drug carriers consisted human serum albumin (HSA), cyclic RGD peptides, and polyethylene glycol (PEG). A kinase inhibitor PTK787 binds to the ULS through a coordination linkage at one of the aromatic nitrogen atoms. Drug-targeting conjugates and the respective control conjugates were analyzed for binding affinity to the integrin αvβ3 target receptor on human umbilical vein endothelial cells (HUVEC). All RGD-equipped conjugates displayed reasonable binding affinity to integrin αvβ3, suggesting the conjugated PTK787 did not obstruct binding of the RGD-modified carriers to integrin αvβ3. The highest binding affinity was observed for RGD-PTK787-HSA (IC50 = 4.4 nM) followed by RGD-PTK787-HSA-PEG (IC50 = 65 nM) and RGD-PEG-PTK787-HSA (IC50 = 640 nM). The result implies the additional incorporation of PEG partially obstructed binding in vitro; however, in vivo effect of the PEG moiety for these particular conjugates keeps unknown. Kok's group also developed a series of closely related drug targeting conjugates, consisting of albumins equipped with RGD-peptide, PEG stealth domains, and either the antitubulin agent monomethyl auristatin E (MMAE) or a new F-variant (MMAF) [62-63]. Since MMAF has a C-terminal charge, this compound is potentially less prone to passive redistribution after its release from the carrier. The results demonstrated that RGD-equipped albumin conjugates with MMAF were more potent than MMAE conjugates, in killing both integrin αvβ3-positive tumor cells and proliferating endothelial cells. Efficacy increased more in tumor cells than in endothelial cells, suggesting different drug redistribution behavior for the two cell types. However, in vivo studies need to be performed to further evaluate in vivo efficacy and toxicity of these RGD-drug conjugates.
The various conjugates of RGD-peptides and therapeutic small molecules [53-54,56,57,59,61-66] are summarized in Table 1.
Integrin targeted delivery of therapeutic small molecules
| Therapeutic agent | Targeting motif | Experimental model | Resultsa | Ref. | |
|---|---|---|---|---|---|
| In Vitro | In Vivo | ||||
| Doxorubicin (Dox) | RGD4C | MDA-MB-435 mammary carcinoma | + | + | [53] |
| Doxorubicin | RGD4C | MH134 murine hepatoma | - | + | [54] |
| Plasmin cleavable prodrug | RGD4C | HUVEC | = | ND | [56] |
| Doxsaliform (doxorubicinformaldehyde) | c[RGDf(N-Me)V] | MDA-MB-435 | = | ND | [57] |
| Doxorubicin | RGD-containing tetrameric peptide | H2009 and H1299 | + | ND | [64] |
| Paclitaxel | [c(RGDyK)]2 | MDA-MB-435 | + | ND | [59] |
| Arabinoside C | RGDSK | B16 melanoma | - | = | [66] |
| PTK787 albumin | c(RGDfK)-PEG | HUVEC | + | ND | [61] |
| MMAE (Auristatin E) | c(RGDfK)-PEG | C26 murine colon carcinoma | + | + | [65] |
| MMAF (Auristatin F) | c(RGDfK)-PEG | C26 murine colon carcinoma | + | ND | [62] |
| SB202190 (the p38 MAPkinase Inhibitor) | c(RGDfK)-PEG | HUVEC | + | ND | [63] |
a Efficacy compared to non-targeted chemotherapeutic agent: Improved efficacy (+), comparable efficacy (=), less efficacy (-), and not determined (ND).
Integrin targeted delivery of therapeutic peptide and proteins
The second approach relies on the conjugation of integrin ligands with therapeutic biologically relevant biomolecules, such as peptides and proteins, to target them to the specific site for their action. A good number of therapeutic peptides and proteins are currently becoming available for the treatment of cancer and other diseases. However, poor pharmacokinetics and unacceptable side-effects are often limiting the efficacy of these biomolecules. For instance, cytokines show potent activity in various cancers, but they normally display a range of other actions within the body, which may cause severe adverse events. Cell-specific targeting of a cytokine may significantly improve its selectivity for the target cells within the tumor and, thus, enhance its efficacy. In general, biomolecules, such as therapeutic peptides and proteins, are hydrophilic and they have short plasma half-life. In order to achieve favorable pharmacokinetic properties of therapeutic biomolecules, structural optimizations of RGD-peptides by using PEGylation strategy sometimes are necessary, which could potentially prevent fast renal clearance, enzymatic degradation of the biomolecules, and extend therapeutic windows.
The conjugate of RGD4C peptide with a pro-apoptotic cationic peptide (KLAKLAK)2 has been reported [67]. The in vitro results showed RGD4C-(KLAKLAK)2 induced apoptosis in Kaposi sarcoma tumor cells. In vivo studies, RGD4C-(KLAKLAK)2 reduced tumor weight and size as well as diminish the formation of lung metastases. In addition, intravenous administration of RGD4C-(KLAKLAK)2 to mice subjected to collagen-induced arthritis significantly reduced the arthritic score [68], which is attributed by apoptosis of endothelial cells in the inflamed synovium. This study demonstrated that targeting to integrin αvβ3-positive endothelial cells is not only limited to the treatment of cancer, but also can be a feasible therapeutic approach for chronic inflammatory disease, in which angiogenesis is a hallmark of disease progression as well. In another study [69], RGD4C peptide was conjugated with a naturally occurring antimicrobial cationic peptide, tachyplesin. The in vitro results showed that RGD4C-tachyplesin inhibited the proliferation of both cultured tumor and endothelial cells and reduced the colony formation of TSU prostate cancer cells. The in vivo studies indicated that the RGD4C-tachyplesin could inhibit the growth of tumors on the chorioallantoic membranes of chicken embryos and in syngeneic mice.
Tumor necrosis factor α (TNF-α) is a multi-functional cytokine playing a key role in apoptosis and cell survival, as well as in inflammation and immunity. Although TNF-α treatment on tumor progression shows beneficial effects in animal studies, clinical use of TNF-α in treatment of cancer is limited due to systemic toxicity. It has been found that the full potential of TNF-α could be exploited while systemic distribution of TNF-α was prevented by local treatment protocols such as isolated limb perfusion [70-71]. Thus, specific targeting of TNF-α to a selective cell population may improve its cytostatic potential. One example is the conjugate of RGD peptide with the TNF mutant V29, where Arginine 29 was replaced by Valine. TNF-V29 selectively binds to TNF-R1 rather than TNF-R2 [72], and TNF-V29 showed potent antitumor activity in mice, combined with a reduced systemic toxicity [73]. In addition, it has been reported that RGD-motif in wild-type TNF reduced its gastrointestinal toxicity [74] and improved its anti-metastatic activity [75]. Thus, the RGD-TNF-V29 mutant F4614 was expected to achieve better overall therapeutic efficacy as compared to the RGD and wild-type TNF conjugate. Indeed, RGD-TNF-V29 displayed considerably lower cytotoxicity in normal endothelial cells, while culturing in tumor-conditioned medium restored the TNF-responsiveness of endothelial cells to the level of wild type TNF-α [76]. RGD-TNF-V29 also showed an increased tumor selectivity of up to 460-fold, whereas wild-type TNF displayed only a 4-fold difference in tumor-endothelial selectivity. In in vivo studies, RGD-TNF-V29 demonstrated a 2-fold improved therapeutic index (LD50/MED = 4.8), which may be contributed by the improved accumulation in the tumor tissue [73]. In another example [77], RGD4C peptide was conjugated with a TNF-α fusion protein, mouse-TNF-α (mTNF-α). Because mTNF is a compact homotrimeric protein, RGD4C-TNF actually contains three binding domains per protein. Due to this multivalent effect, RGD4C-TNF showed a 10-fold higher affinity than free RGD4C peptide for integrin αvβ3 in the endothelial cell line EA.hy926. It was also found that subnanogram doses of RGD4C-TNF are sufficient to induce antitumor effects in tumor-bearing mice as combined with melphalan, a chemotherapeutic drug. Cell adhesion assays and competitive binding experiments with anti-integrin antibodies further demonstrated that showed that the RGD moiety interacts with integrin αvβ3. In addition, RGD4C-TNF conjugate induced cytotoxic effects in standard cytolytic assays, implying that RGD4C-TNF conjugate can also bind TNF receptors and trigger death signals. These results suggested that coupling TNF with RGD4C improves its anti-neoplastic activity. We recently developed an RGD4C-TNF fusion protein for tumor-specific delivery of TNF [78-79]. RGD4C-TNF was significantly more potent than TNF in inhibiting orthotopic MDA-MB-435 tumor growth. Intramuscular administration of plasmid DNA encoding RGD4C-TNF also was found to inhibit the growth of melanomas and lymphomas implanted at sites distant from the plasmid injection site [80]. Tumor necrosis factor (TNF)-related apoptosis inducing ligand (TRAIL) is an apoptosis-inducing member of the TNF gene family, which acts as a homotrimer interacting with five cognate receptors: TRAIL-R1, TRAIL-R2, TRAIL-R3, TRAIL-R4, and osteoprotegerin (OPG). Unlike the other TNF family members TNF-α and CD95L, TRAIL has the unique ability to elicit apoptotic cell death in a variety of cancer cells, with minimal cytoxicity for normal cells and tissues. The conjugate of the fusion of TRAIL with the RGD-based peptide has been reported [81]. The results showed that RGD4C led to enhanced apoptosis inducing activity (caspase-3 and caspase-8 activation) in integrin αvβ3 and αvβ5-positive cancer cells.
Tissue Factor (TF) is a cell membrane receptor protein that is the initiator of the extrinsic pathway of the blood coagulation cascade and normally released from damaged tissues [82]. It is expected that this potent thrombogenic protein in its truncated form (tTF) can be targeted to the tumor, occlude the tumor's blood supply and, thus cause rapid tumor destruction. To test this hypothesis, three fusion proteins, chTNT-3/tTF, chTV-1/tTF, and RGD/tTF, which target DNA exposed in degenerative areas of tumors, fibronectin on the tumor vascular basement membrane, and integrin αvβ3 on the luminal side of tumor vessels, respectively, were developed and tested for their antitumor effects [82]. In vitro, all fusion proteins retained similar thrombotic activity. In MAD109 mouse lung tumor model, RGD-tTF was found to be localized mainly in capillaries and small vessels of the tumor. In vivo, daily injections of RGD-tTF resulted in thrombosis of about 40% of the tumor blood vessels, but no significant inhibition of tumor growth was observed. In contrast, the other two fusion proteins showed thrombosis in up to 80% of the scored blood vessels, leading to massive tumor necrosis and more than 50% reduction in tumor volume versus the RGD-tTF group. Similar results were obtained in the C26 colon carcinoma model. The data implied that these thrombogenic agents had to occlude medium and large vessels within the tumor in order to attain a significant antitumor effect. Interestingly, most impressive tumor suppression was observed for the combination therapy of all three-fusion proteins, suggesting the delivery of tTF to all available targets produced an additive thrombotic effect.
Interleukin 12 (IL-12) plays an important role in the activities of natural killer (NK) cells and T lymphocytes. It mediates enhancement of the cytotoxic activity of NK cells and CD8+ cytotoxic T lymphocytes. However, systemic administration of IL-12 was associated with dose-limiting toxicity, thus preventing IL-12 from attaining its full therapeutic potential [83]. A fusion protein was synthesized by conjugating mouse IL-12 with RGD4C [84]. The results showed that RGD4C-IL-12 retained the immunostimulatory activity of IL-12. In corneal angiogenesis assay, RGD4C-IL-12 demonstrated excellent inhibition of bFGF-induced vessel growth, whereas native mIL-12 only partially inhibited neovascularization. In addition, in a neuroblastoma model (NXS2 model), RGD4C-IL-12 showed an improved antitumor effect, whereas native IL-12 was not effective. Moreover, RGD4C-mediated targeting prevented IL-12 induced hepatic necrosis, which was observed after continuous subcutaneous infusion for two weeks via surgically implanted osmotic pumps. While RGD-IL-12 was tested in knockout mice lacking the IL-12 receptor, RGD4C-IL-12 showed the neovascularization inhibition for up to 25%, whereas mIL-12 was completely ineffective. The enhanced antiangiogenic effect of mrIL-12vp may involve several mechanisms, including increased IL-12 concentrations delivered directly to angiogenic endothelial cells, activation of immune cells within the angiogenic site, and contribution of RGD4C in suppressing endothelial cell survival pathways.
The various conjugates of RGD-peptides and therapeutic peptides and proteins [67,69,73,77-78,80-82,84-88] are summarized in Table 2.
Integrin targeted delivery of therapeutic peptides and proteins
| Therapeutic agent | Targeting motif | Experimental model | Resultsa | Ref. | |
|---|---|---|---|---|---|
| In Vitro | In Vivo | ||||
| (KLAKLAK)2 | RGD4C | MDA-MB-435 mammary carcinoma | + | + | [67] |
| Tachyplesin | CRGDC | TSU prostate cancer cell lines and B16 melanoma | + | + | [69] |
| Truncated tissue factor (tTF) | RGD4C | MAD109 and COLON 26 | + | + | [82] |
| mIL12 | RGD4C | NXS2 neuroblastoma | - | + | [84] |
| Endostatin | RGDS | LS174T colon carcinoma | + | + | [85] |
| Fc fragment of IgG | RGD4C | DU145 prostate carcinoma | ND | + | [86] |
| Hu-TNFα | RGD4C | B16F1 melanoma | ND | + | [80] |
| Hu-TNFα mutant selective for TNFR1 (V29) | RGDSD | Meth A fibrosarcoma and Mqnu-1 lung cancer | - | + | [73] |
| mTNFα | RGD4C | RMA | = | + | [77] |
| TNF | RGD4C | MDA-MB-435 | = | + | [78] |
| Anti-CD3-mAB | c(RGDfK) | HUVEC | + | ND | [88] |
| TRAIL | c(RGDfK)-PEG | Jurkat T cells and HUVEC | = | ND | [87] |
| TRAIL | RGD | COLO-205 and HT-29 human colon carcinoma | + | + | [81] |
a Efficacy compared to non-targeted chemotherapeutic agent: Improved efficacy (+), comparable efficacy (=), less efficacy (-), and not determined (ND).
Integrin targeted delivery of nanoparticle-carried chemotherapeutic agents
The third approach is to employ a nanoparticle-carried delivery system. Integrin targeted ligands, such as RGD peptides, can be coated on the surface of nanoparticle carrier system to selectively deliver chemotherapeutic agents to cancer cells or diseased tissues. There are several advantages for the nanoparticle carrier system over conventional drug delivery system. First, multivalent ligands can be conjugated on the surface of the nanoparticle. Multivalency not only significantly improves the binding affinity to the integrin, but also facilitates internalization. Carrier systems, such as liposomes and nanoparticles, after bearing multiple RGD-peptides, can be more readily internalized via receptor-mediated endocytosis. As a result, larger amount of chemotherapeutic agents can be delivered through internalizing receptor-targeted ligand. In addition, high molecular weight of nanoparticle carriers may lead to passive retention in a tumor. Leaky blood vessels and poor lymphatic drainage are commonly presented in tumor. Nanoparticles can penetrate and accumulate in the tumor via those leaky vessels by the enhanced penetration and retention (EPR) effect [89]. In order to protect nanoparticles from protein absorption and prolong the circulation time after administration, polyethylene glycol (PEG) is normally incorporated into the delivery system. The most commonly used nanoparticulate delivery systems include liposomes, polymeric nanosphere, micelles, and polymersomes [90].
The RGD-containing peptide-decorated nanoparticulate delivery system has been extensively investigated. For example, RGD-peptides were coupled to the distal end of poly(ethylene glycol)-coated liposomes (LCL) to obtain a stable long-circulating drug delivery system functioning as a platform for multivalent interaction with integrin αvβ3 [91]. The results showed that RGD-peptide-modified LCL exhibited increased binding to endothelial cells in vitro. Moreover, intravital microscopy demonstrated a specific interaction of these liposomes with tumor vasculature, a characteristic not observed for LCL. In in vivo study, RGD-LCL encapsulating doxorubicin inhibited tumor growth in a doxorubicin-insensitive murine C26 colon carcinoma model, whereas doxorubicin in LCL failed to decelerate tumor growth. Overall, RGD-LCL containing doxorubicin showed superior efficacy over non-targeted LCL in inhibiting C26 doxorubicin-insensitive tumor outgrowth. Likely, these RGD-LCL-doxorubicin antitumor effects are brought about through direct effects on tumor endothelial cells [91]. Recently, cRGDfK peptide was coupled to PEGylated liposome encapsulating anticancer drug (doxorubicin) to target integrin αvβ3-expressing tumor vasculature [92]. The results showed that delivery of targeted nanoparticles inhibited the growth of metastases while eliminating the toxicity and weight loss associated with systemic administration of doxorubicin. The delivery resulted in a 15-fold improvement in tumor and anti-metastatic activity when compared with administration of the free drug. The preferential activity of these nanoparticles on metastases implies that growing metastatic tumors may have a greater dependence on angiogenic vessels and thus could be more susceptible to integrin αvβ3-targeted therapy [93]. In another example, Xiong et al. achieved high tumor accumulation and intercellular delivery of doxorubicin after conjugating synthetic RGD mimic compound with the drug-loading liposome in syngeneic B16 melanoma mouse model. Administration of RGD mimic-modified nanoparticle resulted in retarded tumor growth and prolonged lifespan compared with the non-modified one [94-95]. Similar RGD liposome modification strategies have also been used to deliver other anticancer drugs, such as 5-fluorouracil (5-FU) and paclitaxel, to malignant tumor-bearing animals. The significant anti-primary tumor and antimetastatic activities can also be achieved [96, 52, 97]. The liposomal delivery of a new snake venom disintegrin, contortrostatin (CN) has been reported in an orthotopic human breast tumor xenograft model [98]. This disintegrin modulates its interaction with integrins on tumor cells and angiogenic vascular endothelial cells.
RGD-modified liposomes have also been applied for other disease treatments in addition to cancer therapy. The effect of RGD-modified liposomes on platelet activation and aggregation was investigated in vitro, with and without platelet agonists [99]. RGD-liposomes were found to bind activated platelets at levels significantly greater than the control RGE-liposomes. The RGD-liposomes did not exhibit any statistically significant effect on platelet activation or aggregation. The results demonstrate the ability of the RGD-modified liposomes to target and bind activated platelets without causing significant platelet aggregation and suggests a feasible way for the development of a platelet-targeted anti-thrombogenic drug delivery system. In another study, RGD-coated liposomes were developed for selective cell targeting in cardiovascular drug delivery [100].
Except for using liposome as a carried system, biodegradable polymeric micelles, a type of self-assembled nanoparticle from amphiphilic copolymers, were utilized as a carrier system for integrin targeted doxorubicin delivery [101]. The results showed that attachment of the cyclic RGD ligand greatly enhanced internalization of the micelles in tumor endothelial cells that overexpress integrin αvβ3, apparently through receptor-mediated endocytosis. Biodegradable nanosphere is another attractive carrier system for drug delivery. A RGD-PEG-PLGA nanosphere delivery system was developed for intracellular delivery of doxorubicin to different malignant cancer cells [102]. The results showed this new nanosphere can alleviate the burst drug release effect commonly associated with PLGA nanosphere systems. Recently, self-assembled glycol chitosan nanoparticles were developed to explore whether this construct might function as a prolonged and sustained drug delivery system for RGD peptide [103]. In vitro work demonstrated that RGD-HGC can prolong and sustain release of RGD, lasting for 1 week. RGD-HGC also inhibited HUVEC adhesion to a beta ig-h3 protein-coated surface, indicating an antiangiogenic effect of the RGD peptide in the HGC nanoparticles. In an in vivo study, the antiangiogenic peptide drug formulation of RGD-HGC markedly inhibited bFGF-induced angiogenesis and decreased hemoglobin content in Matrigel plugs. Intratumoral administration of RGD-HGC significantly decreased tumor growth and microvessel density compared to native RGD peptide.
The various conjugates of RGD-peptides and nanoparticle-carried chemotherapeutic agents [52,91-92,94-96,104-105] are summarized in Table 3.
Integrin targeted delivery of nanoparticle-carried chemotherapeutic agents
| Nanocarriers | Therapeutic agent | Targeting motif | Experimental model | Resultsa | Ref. | |
|---|---|---|---|---|---|---|
| In Vitro | In Vivo | |||||
| Liposome | Doxorubicin | c(RGDfK)-PEG | C26 murine colon carcinoma | ND | + | [91] |
| Liposome | Doxorubicin | RGD10 | C26 murine colon carcinoma | ND | + | [52] |
| Liposome | Doxorubicin | RGD-PEG | B16 murine melanoma | - | + | [94,95] |
| Nanoparticle | Doxorubicin | c(RGDfC)-PEG | Cl-66 mammary carcinoma | ND | ND | [104] |
| Liposome | Doxorubicin | c(RGDfK)-PEG | Orthotopic mouse R40P pancreatic carcinoma, human M21L-GFP melanoma, and human SN12C-RFP renal carcinoma | + | + | [92] |
| Liposome | 5-Fluorouracil | c(RGDfK)-PEG | B16F10 murine melanoma | + | + | [96] |
| Nanoparticle | Fumagillin | Intergrin αvβ3 peptidomimetic antagonist | Vx-2 rabbit adenocarcinoma | ND | + | [105] |
a Efficacy compared to non-targeted chemotherapeutic agent: Improved efficacy (+), comparable efficacy (=), less efficacy (-), and not determined (ND).
Conclusion
Development of molecular devices endowed with disease-targeting functions and carrying cytotoxic components enables the specific delivery of chemotherapeutic agents to aberrant tissues, thus increasing their local efficacy while limiting their peripheral toxicity. A large number of studies demonstrated that integrin receptor, in particular integrin αvβ3, is an ideal target of specific delivery of chemotherapeutic agents, because not only integrin αvβ3 can be internalized by cells on activation with anchoring ligands, but also integrin αvβ3 is overexpressed on angiogenetic endothelial cells and absent in pre-existing endothelial cells and normal tissues. Therefore, integrin targeted delivery of chemotherapeutic agents represents a valuable approach and the numerous results obtained to date using different systems open up interesting prospect to future chemotherapy in treatment of various diseases.
Approaches based on multivalent presentation of the RGD ligand will likely continue to receive attention in the future since they provide therapeutic agents with higher binding affinity, prolonged half-life, and passive retention in diseased tissues and are compulsory for cellular uptake. In this regard, the employment of nanoparticle will certainly represents a promising area of continuing investigation. However, the toxicity of novel nanoparticle carriers will have to be clearly addressed in future clinical application. In addition, since integrin αvβ3 expression is not homogeneous all over diseased tissues, strategies that combine multiple biomarkers, such as vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), epidermal growth factor (EGF), and other integrin receptors are expected to generate significantly greater benefits for optimal treatment. Moreover, in order to maximize the therapeutic index, therapeutic multi-modalities, the combination of chemotherapy, radiotherapy, and gene therapy, will be likely more effective than single modality alone. However, the specific synergistic interactions among different modalities have to be investigated and clearly understood. Finally, non-invasive imaging technologies provide precise ways to visualize biological processes of disease at the cellular and molecular level in real time. Thus, imaging-guided therapy should be routinely applied in clinical settings to improve the overall patient outcome and help further steps toward personalized medicine.
Acknowledgements
The authors would like to acknowledge the Intramural Research Program at the National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH) for financial support.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Garanger E, Boturyn D, Dumy P. Tumor targeting with RGD peptide ligands-design of new molecular conjugates for imaging and therapy of cancers. Anticancer Agents Med Chem. 2007;7(5):552-8
2. Broxterman HJ, Lankelma J, Hoekman K. Resistance to cytotoxic and anti-angiogenic anticancer agents: similarities and differences. Drug Resist Updat. 2003;6(3):111-27
3. Magrath IT. Targeted approaches to cancer therapy. Int J Cancer. 1994;56(2):163-6
4. Turowski P, Adamson P, Greenwood J. Pharmacological targeting of ICAM-1 signaling in brain endothelial cells: potential for treating neuroinflammation. Cell Mol Neurobiol. 2005;25(1):153-70
5. Pavalko FM, Otey CA. Role of adhesion molecule cytoplasmic domains in mediating interactions with the cytoskeleton. Proc Soc Exp Biol Med. 1994;205(4):282-93
6. Haass NK, Smalley KS, Li L, Herlyn M. Adhesion, migration and communication in melanocytes and melanoma. Pigment Cell Res. 2005;18(3):150-9
7. Christofori G. Changing neighbours, changing behaviour: cell adhesion molecule-mediated signalling during tumour progression. EMBO J. 2003;22(10):2318-23
8. Maaser K, Wolf K, Klein CE, Niggemann B, Zanker KS, Brocker EB. et al. Functional hierarchy of simultaneously expressed adhesion receptors: integrin α2β1 but not CD44 mediates MV3 melanoma cell migration and matrix reorganization within three-dimensional hyaluronan-containing collagen matrices. Mol Biol Cell. 1999;10(10):3067-79
9. Ruoslahti E. Specialization of tumour vasculature. Nat Rev Cancer. 2002;2(2):83-90
10. Andrews RK, Berndt MC. Platelet physiology and thrombosis. Thromb Res. 2004;114(5-6):447-53
11. Gibbins JM. Platelet adhesion signalling and the regulation of thrombus formation. J Cell Sci. 2004;117(Pt 16):3415-25
12. Banquy X, Leclair G, Rabanel JM, Argaw A, Bouchard JF, Hildgen P. et al. Selectins ligand decorated drug carriers for activated endothelial cell targeting. Bioconjug Chem. 2008;19(10):2030-9
13. Ichinose K, Kawasaki E, Eguchi K. Recent advancement of understanding pathogenesis of type 1 diabetes and potential relevance to diabetic nephropathy. Am J Nephrol. 2007;27(6):554-64
14. Shimaoka M, Springer TA. Therapeutic antagonists and the conformational regulation of the β2 integrins. Curr Top Med Chem. 2004;4(14):1485-95
15. Hynes RO. A reevaluation of integrins as regulators of angiogenesis. Nat Med. 2002;8(9):918-21
16. Hynes RO, Zhao Q. The evolution of cell adhesion. J Cell Biol. 2000;150(2):F89-96
17. Ruoslahti E. Fibronectin and its integrin receptors in cancer. Adv Cancer Res. 1999;76:1-20
18. Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin αvβ3 for angiogenesis. Science. 1994;264(5158):569-71
19. Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2(2):91-100
20. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673-87
21. Tamkun JW, DeSimone DW, Fonda D, Patel RS, Buck C, Horwitz AF. et al. Structure of integrin, a glycoprotein involved in the transmembrane linkage between fibronectin and actin. Cell. 1986;46(2):271-82
22. Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol. 1996;12:463-518
23. Takagi J, Erickson HP, Springer TA. C-terminal opening mimics 'inside-out' activation of integrin alpha5beta1. Nat Struct Biol. 2001;8(5):412-6
24. Dunehoo AL, Anderson M, Majumdar S, Kobayashi N, Berkland C, Siahaan TJ. Cell adhesion molecules for targeted drug delivery. J Pharm Sci. 2006;95(9):1856-72
25. Bretscher MS. Endocytosis and recycling of the fibronectin receptor in CHO cells. EMBO J. 1989;8(5):1341-8
26. Bretscher MS. Circulating integrins: α5β1, α6β4 and Mac-1, but not α3β1, α4β1 or LFA-1. EMBO J. 1992;11(2):405-10
27. Vicente-Manzanares M, Sancho D, Yanez-Mo M, Sanchez-Madrid F. The leukocyte cytoskeleton in cell migration and immune interactions. Int Rev Cytol. 2002;216:233-89
28. Podar K, Tai YT, Lin BK, Narsimhan RP, Sattler M, Kijima T. et al. Vascular endothelial growth factor-induced migration of multiple myeloma cells is associated with beta 1 integrin- and phosphatidylinositol 3-kinase-dependent PKC alpha activation. J Biol Chem. 2002;277(10):7875-81
29. Felsenfeld DP, Choquet D, Sheetz MP. Ligand binding regulates the directed movement of beta1 integrins on fibroblasts. Nature. 1996;383(6599):438-40
30. Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84(3):359-69
31. Lawson MA, Maxfield FR. Ca2+- and calcineurin-dependent recycling of an integrin to the front of migrating neutrophils. Nature. 1995;377(6544):75-9
32. Cai W, Chen X. Anti-angiogenic cancer therapy based on integrin αvβ3 antagonism. Anticancer Agents Med Chem. 2006;6(5):407-28
33. Mizejewski GJ. Role of integrins in cancer: survey of expression patterns. Proc Soc Exp Biol Med. 1999;222(2):124-38
34. Jin H, Varner J. Integrins: roles in cancer development and as treatment targets. Br J Cancer. 2004;90(3):561-5
35. Pierschbacher MD, Ruoslahti E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature. 1984;309(5963):30-3
36. Ruoslahti E. The RGD story: a personal account. Matrix Biol. 2003;22(6):459-65
37. Grant DS, Tashiro K, Segui-Real B, Yamada Y, Martin GR, Kleinman HK. Two different laminin domains mediate the differentiation of human endothelial cells into capillary-like structures in vitro. Cell. 1989;58(5):933-43
38. Pytela R. Amino acid sequence of the murine Mac-1 alpha chain reveals homology with the integrin family and an additional domain related to von Willebrand factor. EMBO J. 1988;7(5):1371-8
39. Miyauchi A, Alvarez J, Greenfield EM, Teti A, Grano M, Colucci S. et al. Recognition of osteopontin and related peptides by an αvβ3 integrin stimulates immediate cell signals in osteoclasts. J Biol Chem. 1991;266(30):20369-74
40. Lawler J, Weinstein R, Hynes RO. Cell attachment to thrombospondin: the role of ARG-GLY-ASP, calcium, and integrin receptors. J Cell Biol. 1988;107(6 Pt 1):2351-61
41. Ruoslahti E. RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol. 1996;12:697-715
42. Ruoslahti E, Pierschbacher MD. New perspectives in cell adhesion: RGD and integrins. Science. 1987;238(4826):491-7
43. Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL. et al. Crystal structure of the extracellular segment of integrin αvβ3 in complex with an Arg-Gly-Asp ligand. Science. 2002;296(5565):151-5
44. Brooks PC, Stromblad S, Sanders LC, von Schalscha TL, Aimes RT, Stetler-Stevenson WG. et al. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin αvβ3. Cell. 1996;85(5):683-93
45. Friedlander M, Brooks PC, Shaffer RW, Kincaid CM, Varner JA, Cheresh DA. Definition of two angiogenic pathways by distinct αv integrins. Science. 1995;270(5241):1500-2
46. Goh KL, Yang JT, Hynes RO. Mesodermal defects and cranial neural crest apoptosis in α5 integrin-null embryos. Development. 1997;124(21):4309-19
47. Taverna D, Hynes RO. Reduced blood vessel formation and tumor growth in alpha5-integrin-negative teratocarcinomas and embryoid bodies. Cancer Res. 2001;61(13):5255-61
48. Yang JT, Rayburn H, Hynes RO. Cell adhesion events mediated by α4 integrins are essential in placental and cardiac development. Development. 1995;121(2):549-60
49. Yang JT, Rayburn H, Hynes RO. Embryonic mesodermal defects in α5 integrin-deficient mice. Development. 1993;119(4):1093-105
50. Temming K, Schiffelers RM, Molema G, Kok RJ. RGD-based strategies for selective delivery of therapeutics and imaging agents to the tumour vasculature. Drug Resist Updat. 2005;8(6):381-402
51. Koivunen E, Wang B, Ruoslahti E. Phage libraries displaying cyclic peptides with different ring sizes: ligand specificities of the RGD-directed integrins. Biotechnology (N Y). 1995;13(3):265-70
52. Holig P, Bach M, Volkel T, Nahde T, Hoffmann S, Muller R. et al. Novel RGD lipopeptides for the targeting of liposomes to integrin-expressing endothelial and melanoma cells. Protein Eng Des Sel. 2004;17(5):433-41
53. Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279(5349):377-80
54. Kim JW, Lee HS. Tumor targeting by doxorubicin-RGD-4C peptide conjugate in an orthotopic mouse hepatoma model. Int J Mol Med. 2004;14(4):529-35
55. Denny WA. Tumor-activated prodrugs--a new approach to cancer therapy. Cancer Invest. 2004;22(4):604-19
56. de Groot FM, Broxterman HJ, Adams HP, van Vliet A, Tesser GI, Elderkamp YW. et al. Design, synthesis, and biological evaluation of a dual tumor-specific motive containing integrin-targeted plasmin-cleavable doxorubicin prodrug. Mol Cancer Ther. 2002;1(11):901-11
57. Burkhart DJ, Kalet BT, Coleman MP, Post GC, Koch TH. Doxorubicin-formaldehyde conjugates targeting αvβ3 integrin. Mol Cancer Ther. 2004;3(12):1593-604
58. Fenick DJ, Taatjes DJ, Koch TH. Doxoform and Daunoform: anthracycline-formaldehyde conjugates toxic to resistant tumor cells. J Med Chem. 1997;40(16):2452-61
59. Chen X, Plasencia C, Hou Y, Neamati N. Synthesis and biological evaluation of dimeric RGD peptide-paclitaxel conjugate as a model for integrin-targeted drug delivery. J Med Chem. 2005;48(4):1098-106
60. Cao Q, Li ZB, Chen K, Wu Z, He L, Neamati N. et al. Evaluation of biodistribution and anti-tumor effect of a dimeric RGD peptide-paclitaxel conjugate in mice with breast cancer. Eur J Nucl Med Mol Imaging. 2008;35(8):1489-98
61. Temming K, Lacombe M, Schaapveld RQ, Orfi L, Keri G, Poelstra K. et al. Rational design of RGD-albumin conjugates for targeted delivery of the VEGF-R kinase inhibitor PTK787 to angiogenic endothelium. ChemMedChem. 2006;1(11):1200-3
62. Temming K, Meyer DL, Zabinski R, Senter PD, Poelstra K, Molema G. et al. Improved efficacy of αvβ3-targeted albumin conjugates by conjugation of a novel auristatin derivative. Mol Pharm. 2007;4(5):686-94
63. Temming K, Lacombe M, van der Hoeven P, Prakash J, Gonzalo T, Dijkers EC. et al. Delivery of the p38 MAPkinase inhibitor SB202190 to angiogenic endothelial cells: development of novel RGD-equipped and PEGylated drug-albumin conjugates using platinum(II)-based drug linker technology. Bioconjug Chem. 2006;17(5):1246-55
64. Guan H, McGuire MJ, Li S, Brown KC. Peptide-targeted polyglutamic acid doxorubicin conjugates for the treatment of αvβ6-positive cancers. Bioconjug Chem. 2008;19(9):1813-21
65. Temming K, Meyer DL, Zabinski R, Dijkers EC, Poelstra K, Molema G. et al. Evaluation of RGD-targeted albumin carriers for specific delivery of auristatin E to tumor blood vessels. Bioconjug Chem. 2006;17(6):1385-94
66. Jasseron S, Contino-Pepin C, Maurizis JC, Rapp M, Pucci B. In vitro and in vivo evaluations of THAM derived telomers bearing RGD and Ara-C for tumour neovasculature targeting. Eur J Med Chem. 2003;38(9):825-36
67. Ellerby HM, Arap W, Ellerby LM, Kain R, Andrusiak R, Rio GD. et al. Anti-cancer activity of targeted pro-apoptotic peptides. Nat Med. 1999;5(9):1032-8
68. Gerlag DM, Borges E, Tak PP, Ellerby HM, Bredesen DE, Pasqualini R. et al. Suppression of murine collagen-induced arthritis by targeted apoptosis of synovial neovasculature. Arthritis Res. 2001;3(6):357-61
69. Chen Y, Xu X, Hong S, Chen J, Liu N, Underhill CB. et al. RGD-Tachyplesin inhibits tumor growth. Cancer Res. 2001;61(6):2434-8
70. Eggermont AM, Schraffordt Koops H, Lienard D, Kroon BB, van Geel AN, Hoekstra HJ. et al. Isolated limb perfusion with high-dose tumor necrosis factor-alpha in combination with interferon-gamma and melphalan for nonresectable extremity soft tissue sarcomas: a multicenter trial. J Clin Oncol. 1996;14(10):2653-65
71. Grunhagen DJ, Brunstein F, ten Hagen TL, van Geel AN, de Wilt JH, Eggermont AM. TNF-based isolated limb perfusion: a decade of experience with antivascular therapy in the management of locally advanced extremity soft tissue sarcomas. Cancer Treat Res. 2004;120:65-79
72. Kuroda K, Miyata K, Shikama H, Kawagoe T, Nishimura K, Takeda K. et al. Novel muteins of human tumor necrosis factor with potent antitumor activity and less lethal toxicity in mice. Int J Cancer. 1995;63(1):152-7
73. Kuroda K, Miyata K, Fujita F, Koike M, Fujita M, Nomura M. et al. Human tumor necrosis factor-alpha mutant RGD-V29 (F4614) shows potent antitumor activity and reduced toxicity against human tumor xenografted nude mice. Cancer Lett. 2000;159(1):33-41
74. Shikama H, Miyata K, Sakae N, Kuroda K, Nishimura K, Yotsuya S. et al. A novel Mutein of TNFalpha Containing the Arg-Gly-Asp Sequence Shows Reduced Toxicity in Intestine. Mediators Inflamm. 1994;3(2):111-6
75. Miyata K, Mitsuishi Y, Shikama H, Kuroda K, Nishimura K, Sakae N. et al. Overcoming the metastasis-enhancing potential of human tumor necrosis factor alpha by introducing the cell-adhesive Arg-Gly-Asp sequence. J Interferon Cytokine Res. 1995;15(2):161-9
76. Shikama H, Miyata K, Sakae N, Mitsuishi Y, Nishimura K, Kuroda K. et al. Novel mutein of tumor necrosis factor alpha (F4614) with reduced hypotensive effect. J Interferon Cytokine Res. 1995;15(8):677-84
77. Curnis F, Gasparri A, Sacchi A, Longhi R, Corti A. Coupling tumor necrosis factor-alpha with alphaV integrin ligands improves its antineoplastic activity. Cancer Res. 2004;64(2):565-71
78. Wang H, Chen K, Cai W, Li Z, He L, Kashefi A. et al. Integrin-targeted imaging and therapy with RGD4C-TNF fusion protein. Mol Cancer Ther. 2008;7(5):1044-53
79. Liu Z WF, Chen X. Integrin avb3-targeted cancer therapy. Drug Development Research. 2008;69:329-39
80. Zarovni N, Monaco L, Corti A. Inhibition of tumor growth by intramuscular injection of cDNA encoding tumor necrosis factor alpha coupled to NGR and RGD tumor-homing peptides. Hum Gene Ther. 2004;15(4):373-82
81. Cao L, Du P, Jiang SH, Jin GH, Huang QL, Hua ZC. Enhancement of antitumor properties of TRAIL by targeted delivery to the tumor neovasculature. Mol Cancer Ther. 2008;7(4):851-61
82. Hu P, Yan J, Sharifi J, Bai T, Khawli LA, Epstein AL. Comparison of three different targeted tissue factor fusion proteins for inducing tumor vessel thrombosis. Cancer Res. 2003;63(16):5046-53
83. Car BD, Eng VM, Lipman JM, Anderson TD. The toxicology of interleukin-12: a review. Toxicol Pathol. 1999;27(1):58-63
84. Dickerson EB, Akhtar N, Steinberg H, Wang ZY, Lindstrom MJ, Padilla ML. et al. Enhancement of the antiangiogenic activity of interleukin-12 by peptide targeted delivery of the cytokine to αvβ3 integrin. Mol Cancer Res. 2004;2(12):663-73
85. Yokoyama Y, Ramakrishnan S. Addition of integrin binding sequence to a mutant human endostatin improves inhibition of tumor growth. Int J Cancer. 2004;111(6):839-48
86. Li J, Ji J, Holmes LM, Burgin KE, Barton LB, Yu X. et al. Fusion protein from RGD peptide and Fc fragment of mouse immunoglobulin G inhibits angiogenesis in tumor. Cancer Gene Ther. 2004;11(5):363-70
87. Tarrus M, van der Sloot AM, Temming K, Lacombe M, Opdam F, Quax WJ. et al. RGD-avidin-biotin pretargeting to αvβ3 integrin enhances the proapoptotic activity of TNF alpha related apoptosis inducing ligand (TRAIL). Apoptosis. 2008;13(2):225-35
88. Schraa AJ, Kok RJ, Botter SM, Withoff S, Meijer DK, de Leij LF. et al. RGD-modified anti-CD3 antibodies redirect cytolytic capacity of cytotoxic T lymphocytes toward alphavbeta3-expressing endothelial cells. Int J Cancer. 2004;112(2):279-85
89. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387-92
90. Wang Z, Chui WK, Ho PC. Integrin targeted drug and gene delivery. Expert Opin Drug Deliv. 2010;7(2):159-71
91. Schiffelers RM, Koning GA, ten Hagen TL, Fens MH, Schraa AJ, Janssen AP. et al. Anti-tumor efficacy of tumor vasculature-targeted liposomal doxorubicin. J Control Release. 2003;91(1-2):115-22
92. Murphy EA, Majeti BK, Barnes LA, Makale M, Weis SM, Lutu-Fuga K. et al. Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proc Natl Acad Sci U S A. 2008;105(27):9343-8
93. Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10(1):9-22
94. Xiong XB, Huang Y, Lu WL, Zhang X, Zhang H, Nagai T. et al. Intracellular delivery of doxorubicin with RGD-modified sterically stabilized liposomes for an improved antitumor efficacy: in vitro and in vivo. J Pharm Sci. 2005;94(8):1782-93
95. Xiong XB, Huang Y, Lu WL, Zhang X, Zhang H, Nagai T. et al. Enhanced intracellular delivery and improved antitumor efficacy of doxorubicin by sterically stabilized liposomes modified with a synthetic RGD mimetic. J Control Release. 2005;107(2):262-75
96. Dubey PK, Mishra V, Jain S, Mahor S, Vyas SP. Liposomes modified with cyclic RGD peptide for tumor targeting. J Drug Target. 2004;12(5):257-64
97. Zhao H, Wang JC, Sun QS, Luo CL, Zhang Q. RGD-based strategies for improving antitumor activity of paclitaxel-loaded liposomes in nude mice xenografted with human ovarian cancer. J Drug Target. 2009;17(1):10-8
98. Swenson S, Costa F, Minea R, Sherwin RP, Ernst W, Fujii G. et al. Intravenous liposomal delivery of the snake venom disintegrin contortrostatin limits breast cancer progression. Mol Cancer Ther. 2004;3(4):499-511
99. Gupta AS, Huang G, Lestini BJ, Sagnella S, Kottke-Marchant K, Marchant RE. RGD-modified liposomes targeted to activated platelets as a potential vascular drug delivery system. Thromb Haemost. 2005;93(1):106-14
100. Lestini BJ, Sagnella SM, Xu Z, Shive MS, Richter NJ, Jayaseharan J. et al. Surface modification of liposomes for selective cell targeting in cardiovascular drug delivery. J Control Release. 2002;78(1-3):235-47
101. Nasongkla N, Shuai X, Ai H, Weinberg BD, Pink J, Boothman DA. et al. cRGD-functionalized polymer micelles for targeted doxorubicin delivery. Angew Chem Int Ed Engl. 2004;43(46):6323-7
102. Wang Z, Chui WK, Ho PC. Design of a multifunctional PLGA nanoparticulate drug delivery system: evaluation of its physicochemical properties and anticancer activity to malignant cancer cells. Pharm Res. 2009;26(5):1162-71
103. Kim JH, Kim YS, Park K, Kang E, Lee S, Nam HY. et al. Self-assembled glycol chitosan nanoparticles for the sustained and prolonged delivery of antiangiogenic small peptide drugs in cancer therapy. Biomaterials. 2008;29(12):1920-30
104. Bibby DC, Talmadge JE, Dalal MK, Kurz SG, Chytil KM, Barry SE. et al. Pharmacokinetics and biodistribution of RGD-targeted doxorubicin-loaded nanoparticles in tumor-bearing mice. Int J Pharm. 2005;293(1-2):281-90
105. Winter PM, Schmieder AH, Caruthers SD, Keene JL, Zhang H, Wickline SA. et al. Minute dosages of αvβ3-targeted fumagillin nanoparticles impair Vx-2 tumor angiogenesis and development in rabbits. FASEB J. 2008;22(8):2758-67
Author contact
![]() Corresponding author: Dr. Xiaoyuan Chen, Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH), 31 Center Dr, 31/1C22, Bethesda, MD 20892, USA; Tel: 301-451-4246; Email: shawn.chengov; Dr. Kai Chen, Molecular Imaging Center, Department of Radiology, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA; Tel: 323-442-3582; Email: chenkaiedu
Corresponding author: Dr. Xiaoyuan Chen, Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH), 31 Center Dr, 31/1C22, Bethesda, MD 20892, USA; Tel: 301-451-4246; Email: shawn.chengov; Dr. Kai Chen, Molecular Imaging Center, Department of Radiology, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA; Tel: 323-442-3582; Email: chenkaiedu