Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Material and methods
Results
Discussion
CONCLUSIONS
SUPPLEMENTARY MATERIAL
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2011; 1:251-262. doi:10.7150/thno/v01p0251 This volume Cite
Research Paper
PET imaging of CXCR4 using copper-64 labeled peptide antagonist
Orit Jacobson1, Ido D. Weiss2, Lawrence P. Szajek3, Gang Niu1, Ying Ma1, Dale O. Kiesewetter1, Joshua M. Farber2, Xiaoyuan Chen1, ![]()
1. Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH), Bethesda, Maryland
2. Laboratory of Molecular Immunology, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Bethesda, Maryland
3. Positron Emission Tomography Department, Warren Grant Magnuson Clinical Center, National Institutes of Health, Bethesda, Maryland
Received 2011-3-19; Accepted 2011-4-18; Published 2011-4-19
Abstract
Expression of CXCR4 in cancer has been found to correlate with poor prognosis and resistance to chemotherapy. In this study we developed a derivative of the CXCR4 peptide antagonist, T140-2D, that can be labeled easily with the PET isotope copper-64, and thereby enable in vivo visualization of CXCR4 in tumors. T140 was conjugated to 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid mono (N-hydroxysuccinimide ester) (DOTA-NHS) to give T140-2D, which contains a DOTA molecule on each of the two lysine residues. 64Cu-T140-2D was evaluated in vitro by migration and binding experiments, and in vivo by microPET imaging and biodistribution, in mice bearing CXCR4-positive and CXCR4-negative tumor xenografts. T140-2D was labeled with copper-64 to give 64Cu-T140-2D in a high radiochemical yield of 86 ± 3% (not decay-corrected) and a specific activity of 0.28 - 0.30 mCi/µg (10.36 - 11.1 MBq/µg). 64Cu-T140-2D had antagonistic and binding characteristics to CXCR4 that were similar to those of T140. In vivo, 64Cu-T140-2D tended to bind to red blood cells and had to be used in a low specific activity form. In this new form 64Cu-T140-2D enabled specific imaging of CXCR4-positive, but not CXCR4-negative tumors. Undesirably, however, 64Cu-T140-2D also displayed high accumulation in the liver and kidneys. In conclusion, 64Cu-T140-2D was easily labeled and, in its low activity form, enabled imaging of CXCR4 in tumors. It had high uptake, however, in metabolic organs. Further research with imaging tracers targeting CXCR4 is required.
Keywords: T140 peptide, CXCR4 imaging, PET, copper-64
Introduction
CXCR4, a member of the chemokine receptor subfamily of seven-transmembrane-domain G-protein coupled receptors, is a highly conserved chemokine receptor and can be detected on a diverse range of cells, including lymphocytes and monocytes, mast cells, adult CD34+ bone marrow progenitor cells, microglia and neurons [1-6]. CXCR4 and its sole known human natural ligand, SDF-1/CXCL12, were found to be crucial during embryonic growth and development and depletion of either one of them is lethal for the embryo [7].
CXCR4 functions as an HIV co-receptor for entry of T-tropic, but not M-tropic strains of HIV-1 viruses into CD4+ T cells [1, 8]. As such, multiple CXCR4 antagonists have been developed, consisting of small molecules, peptides and antibodies [9]. Further research into the functions of CXCR4 under normal and pathologic conditions revealed that CXCR4 antagonists can be used for stem cell mobilization and anti-tumor therapy [9].
CXCR4 expression was also found in various human cancer types, including breast, prostate, lung, lymphoma, multiple myeloma, melanoma, ovarian, pancreatic, neuroblastoma, esophageal, colorectal, osteosarcoma, and renal cancer [10-18]. Further research on the role of CXCR4 in cancer discovered a correlation between high levels of CXCR4 expression with poor prognosis [19-20], chemotherapy resistance [17-18, 21] and development of metastasis in CXCL12 expressing organs [22-23]. Therefore, CXCR4 became an attractive target for therapy as well as imaging. Development of specific tracers of CXCR4 for positron emission tomography (PET), will allow non-invasive evaluation of the receptor levels in vivo before and after chemotherapies or future anti-CXCR4 therapies.
Several CXCR4 ligands have been radiolabeled for PET and single photon emission computed tomography (SPECT) imaging [24-29], and each displayed different advantages and disadvantages. Nevertheless, an optimal biomarker, in terms of simplicity of synthesis along with selectivity of the tracer, has yet to be found. As an example, we and others reported that [64Cu]AMD3100 showed extremely high specific accumulation in the liver which did not seem to be CXCR4-dependent, suggesting that [64Cu]AMD3100 binds to another unknown target receptor [28-29]. We have also developed a radiosynthesis route to label a peptide-based antagonist 4F-benzoyl-TN14003 (T140, Fig. 1) with the PET isotope fluorine-18, without changing the chemical structure of the peptide [27]. Although 18F-T140 accumulated in CXCR4 positive tumors, it also bound to red blood cells (RBCs), and injection of additional unlabeled mass of the peptide was required to visualize CXCR4 in vivo [27]. Another downside was the long time required for 18F-T140 radiosynthesis and the low yield.
Schematic structure of T140 and T140-2D. For 64Cu-labeling, T140-2D was reacted with [64Cu]Cu(OAc)2 for 20 min at 40 ºC.

In this paper, we have simplified the synthesis of T140-based tracer by conjugation of 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid mono (N-hydroxysuccinimide ester) (DOTA-NHS), and we have labeled it with the longer lived PET isotope copper-64 to give 64Cu-T140-2D derivative. The tendency of 64Cu-T140-2D to bind human CXCR4 receptor and mouse and human RBCs, and its ability to be used as a biomarker for imaging CXCR4 expression in vivo, were assessed.
Material and methods
General
1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetraacetic acid mono (N-hydroxysuccinimide ester) (DOTA-NHS) was purchased from Macrocyclics (Dallas, TX). All other solvents and chemicals were purchased from Sigma-Aldrich (St. Louis, MO). 64Cu was produced at the National Institutes of Health (NIH) by the irradiation of a thin layer of 64Ni (Isoflex, USA) electroplated on a solid gold internal target plate in a CS-30 cyclotron utilizing the nuclear reaction 64Ni(p, n)64Cu, and separated from the target material as [64Cu]CuCl2 by anion chromatography, as previously described [30]. C18 cartridges (Waters Corporation, Milford, MA) were each activated with both 5 mL of EtOH and 10 mL of water. Radio-TLC was performed on an AR-2000 Bioscan scanner (Washington DC), using silica gel plates (LK6DF, 60 Å, 200 mm, Whatman) and 1% ethylenediaminetetraacetic acid (EDTA), 5% NH4OAc in water : methanol (1:1) as a developing solvent. T140 peptide was purchased from C.S. Bio Co. (Menlo Park, CA).
T140 (C97H144FN33O19S2): observed 2162.48 [M+3H+], calculated: 2159.52.
Radioactive samples were counted on a gamma counter (1480 Wizard 3, Perkin-Elmer, Boston, MA). High performance liquid chromatography (HPLC) was performed on a system with a variable wavelength detector operating at 280 nm and with a radioactivity detector with a NaI crystal. For semi-preparative reversed-phase (RP) HPLC, a Higgins preparative C18 column (5 µm, 20 x 250 mm) was used. The flow was set at 12 mL/min using a gradient system, starting from 95% of solvent A (0.1% TFA in water) and 5% of solvent B (0.1% TFA in acetonitrile (ACN)) and increasing to 35% solvent A and 65% solvent B at 35 min. For analytical RP-HPLC, a Vydac C4 (214TP5415, 5 µm, 4.6 x 150 mm) column was used. The flow was set at 1.5 mL/min with a gradient system starting from 100% of solvent A and 0% of solvent B and increasing to 70% solvent A and 30% solvent B at 30 min. Mass spectrometry analysis employed a Waters LC-MS system (Waters, Milford, MA) that included an Acquity UPLC system coupled to a Waters Q-Tof Premier high resolution mass spectrometer. An Acquity BEH Shield RP18 column (150 x 2.1 mm) was employed for chromatography.
Synthesis of T140-2D
The procedure for conjugation of DOTA-NHS with T140 was conducted by dissolving the peptide (2-3 mg, 0.92-1.38 µmol) in 0.2-0.3 mL dimethylformamide (DMF). Then, 2.5 eq. of DOTA-NHS in 0.1 mL of DMF were added, followed by the addition of 5 eq. of diisopropylethylamine. The reaction was mixed at 4°C overnight. The conjugated peptide was purified by semi-preparative HPLC. The purity of T140-2D was confirmed by analytical HPLC with a retention time of 13.81 min (purity > 99%) and identity confirmed by LC-MS: T140-2D (C129H196FN41O33S2): observed 2934.54 [M+3H+], calculated: 2931.31.
64Cu radiolabeling
[64Cu]CuCl2 was converted to [64Cu]Cu(OAc)2 by adding 0.5 mL of 0.4 M ammonium acetate (NH4OAc) solution (pH = 5.5) to 20 μL [64Cu]CuCl2. [64Cu]Cu(OAc)2 solution (0.2 mL; 10-12 mCi, 370-444 MBq) was added into a solution of conjugated peptide T140-2D (36 µg) in 0.4 M NH4OAc (pH = 5.5). The reaction was stirred for 20 min at 40°C. Complexation of 64Cu and the conjugated peptide was monitored by radio-TLC (Rf [64Cu]T140-2D = 0.06, Rf [64Cu]Cu2+ = 0.9). Radio-TLC showed incorporation of greater than 95%. The reaction vial was then diluted with 10 mL of water and loaded onto an activated C18 Sep-Pak cartridge. The cartridge was washed with water (10 mL), and the desired labeled peptide was eluted with 10 mM HCl in ethanol (1 mL) into a glass test tube. The ethanol was evaporated for 5 min under a stream of argon at 40°C and the labeled peptide was then reformulated with saline. The overall radiochemical yield was 86 ± 3% (not decay-corrected, n = 6), calculated from the start of synthesis to the reformulation of the labeled peptide with saline. [64Cu]T140-2D was achieved with a specific activity of 0.28 - 0.30 mCi/µg (10.36 - 11.1 MBq/µg). Quality control (QC) analysis was performed on an analytical HPLC system. The retention time of [64Cu]T140-2D was 14.02 min. 64Cu-T140-2D was achieved in radiochemical purity greater than 99%.
Cell Culture
Wild-type Chinese hamster ovarian (CHO) cells and CHO cells that were stably transfected with CXCR4 (CHO-CXCR4) were a kind gift from Dr. David McDermott (NIAID, NIH, Bethesda, MD). The CHO cells were grown in F-12K medium (ATCC). Jurkat cells (ATCC) were grown in Roswell Park Memorial Institute (RPMI) medium. All cell culture media were supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 2 mM L-glutamine and non-essential amino acids (GIBCO) at 37°C under an atmosphere containing 5% CO2. CXCR4 level expressed by CHO-CXCR4 cells was evaluated using the FlowCellect kit (Millipore, Bedford, Massachusetts) following the manufacturer's instructions and were found to be 6.8 x 105 receptors per cell [27].
Flow cytometry
Cells were blocked in HBSS buffer (Hanks' balanced salt solution) supplemented with 2% rat serum and 2% mouse serum (Jackson ImmunoResearch labs, West Grove, PA) on ice for 10 min, and thereafter cells were stained with PE-conjugated anti-human CXCR4 (R&D, Minneapolis, MN). Cells from tumors were also stained using FITC-labeled anti-mouse MHC-I H-2Dk (Becton Dickinson, San Jose, CA). Flow data were acquired using an LSR-II cytometer (Becton Dickinson, San Jose, CA) and analyzed using FlowJo (Tree star, Ashland, OR).
Transwell Migration Assay
Six hundred µL of migration medium (RPMI supplemented with 1% fetal bovine serum) containing CXCL12 (PeproTech, Rocky Hill, NJ) at a concentration of 100 ng/mL were placed into the lower chamber of a Costar 24-well Transwell (Corning Inc., Corning, NY). Jurkat cells (105 in 100 µL migration medium) were incubated alone with T140 or with T140-2D at the indicated concentration 15 min before they were placed into the upper chamber (pore size 5 µm). Cells were collected from lower chamber after 3 h of migration at 37 ºC, and counted by flow cytometry using counting beads. Control migrations were performed without the chemokine in the lower chamber.
CXCR4 Receptor Binding Assay
CHO-CXCR4 cells were trypsinized and resuspended in PBS containing 50 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 1 mM CaCl2, 5 mM MgCl2, 0.5% (w/v) BSA and 0.3 mM NaN3. Incubation was conducted in 200 μL of this solution containing 105 cells, 250 nCi (9.25 KBq) of [64Cu]T140-2D and 0-1000 nM of unlabeled T140-2D for 45 min on a shaker at room temperature. After incubation, cells were spun through 10% sucrose using a desktop Eppendorf centrifuge at 14,000 rpm. Cell bound radioactivity was measured using a gamma counter. Binding results were expressed as percent of total counts, and IC50 values were calculated using Prism software (GraphPad, La Jolla, CA).
Cell Uptake, Internalization and Efflux Studies
For cell uptake studies, CHO-CXCR4 cells were seeded into a 24-well plate at a density of 105 cells per well and incubated with 25 µCi/well (925 kBq/well) of [64Cu]T140-2D at 37°C for 5, 15, 30, 60, 120 and 240 min. Tumor cells were then washed twice with cold PBS and harvested by addition of 250 µL of 0.1 mol/L NaOH. Internalization studies were preformed similarly to the procedure describe above. After 5, 15, 30, 60, 120 and 240 min incubation of CHO-CXCR4 cells with [64Cu]T140-2D at 37°C, the cells were washed twice with cold PBS and then incubated for 1 min with acid-washing-buffer (50 mmol/L glycine, 0.1 mol/L NaCl, pH = 2.8) to remove surface bound radioactive ligand. Thereafter, the cells were washed twice with cold PBS and harvested by addition of 250 µL of 0.1 mol/L NaOH. For efflux studies, 25 µCi/well of [64Cu]T140-2D (925 kBq/well) were added to CHO-CXCR4 cells in a 24-well plate and incubated for 2 h at 37°C. Then cells were washed twice with cold PBS, and incubated with F-12K medium for 5, 15, 30, 60, 120 and 240 min. After washing twice with PBS, cells were harvested by addition of 250 µL of 0.1 mol/L NaOH. The cell suspensions were collected and measured in a gamma counter. Each data point is an average of triplicate wells.
Binding Assay of Human and Mouse Red Blood Cells (RBCs)
Mice were euthanized and blood was collected using heparin (Abbott laboratories, Chicago, IL). Mouse RBCs (mRBCs) were separated by a Histopaque (Sigma-Aldrich) gradient, collected from the bottom and washed three times with cold saline (Quality Biological, Gaithersburg, MD). Human RBCs (hRBCs) were acquired from the NIH blood bank (NIH, Bethesda, MD).
mRBCs and hRBCs were incubated in 200 µL of saline containing 2 x 105 cells, 250 nCi (9.25 KBq) of [64Cu]T140-2D and 0-1000 nM of unlabeled T140-2D for 45 min on a shaker at room temperature. After incubation, cells were washed three times with cold saline. Thereafter, the plates were heated to 40°C and dried. The dried filter membranes were punched off from the wells and each collected in a polystyrene culture test tube (12 x 75 mm). Cell bound radioactivity was measured using a gamma counter. The IC50 values were calculated by nonlinear regression analysis using the GraphPad Prism computer-fitting program. Each data point is a result of the average of triplicate wells.
Animals
C57BL/6 and athymic nude mice at age of 4-6 weeks were purchased from Taconic (Germantown, NY) and housed under specific-pathogen free conditions. All animal studies were conducted in accordance with the principles and procedures outlined in the National Institutes of Health Guide for the Care and Use of Animals using protocols approved by the NIH Institutional Animal Care and Use Committee. Duffy antigen/receptor for chemokines (DARC) knockout (KO) mice were kindly provided by Drs. Philip Murphy and Eric Schneider (NIAID, NIH, Bethesda, MD).
Athymic nude mice were injected subcutaneously (s.c.) at two sites on the shoulder with 107 CXCR4-positive or CXCR4-negative CHO tumor cells per site. The tumors were allowed to develop for 2 weeks prior to imaging or biodistribution studies. For metastatic tumor model, mice were injected intravenously via the tail vein with 106 CHO-CXCR4 cells and underwent imaging studies 2 weeks later.
Biodistribution
Fifty µCi (1.85 MBq) of [64Cu]T140-2D in a volume of 100 µL saline were injected through the tail veins of tumor-bearing mice. For a blocking experiment, 2, 5, 10 or 400 µg of unlabeled T140-2D were co-injected with the labeled peptide. At 4 h postinjection, blood was drawn from the heart under anesthesia and the mice were then sacrificed. Spleen, liver, muscle, kidneys, intestine, bone marrow, and tumors were removed. Bone marrow was flushed from within the bones, and the remaining organs were weighed. All organs were assayed for radioactivity using a gamma counter. The results were calculated as percent injected dose per gram tissue (%ID/g). Each group contained 4-5 mice.
PET Studies
Tumor-bearing mice were anesthetized using isoflurane/O2 (1.5-2% v/v) and injected with 100 µCi (3.7 MBq) of [64Cu]T140-2D in a volume of 100 µL saline. PET scans were performed using an Inveon scanner (Siemens Medical Solutions) at 1, 2, 4 and 24 h post-injection. For blocking experiments relating to red blood cells (RBC), 100 µCi (3.7 MBq) of [64Cu]T140-2D were co-injected with 5 µg of unlabeled T140-2D.
The images were reconstructed by a three-dimensional ordered subsets expectation maximization (3D-OSEM) algorithm, and no correction was applied for attenuation or scatter. Image analysis was done using ASI Pro VMTM software. The %ID/g for the various tissues was determined by drawing regions of interest (ROIs) surrounding an entire organ on the coronal images. The radioactivity contained in the ROI divided by the dose administered to the animal gave the %ID and the volume of the ROI was converted to mass, assuming a density of 1 for all tissues. Each group contained 4-5 mice.
Statistical Analysis
Results were expressed as mean and SD. Two-tailed paired and unpaired Student's t tests were used to determine differences within groups and between groups, respectively. P values <0.05 were considered statistically significant.
Results
Chemistry and Radiochemistry
Conjugation of an excess of DOTA with T140 peptide yielded two molecules of DOTA per T140 peptide, one on each free lysine residue of the peptide (Fig. 1). The conjugated peptide was achieved in high chemical yield of 80-86% after HPLC purification, and was analyzed by both analytical HPLC and mass spectroscopy to confirm the identity of the product.
64Cu complexation into the desired T140-2D peptide was very efficient. Following incubation at 40°C for 20 min, almost no free 64Cu was detected by radio-TLC. The overall radiochemical yield was 86 ± 3% (not decay-corrected, n = 6), calculated from the start of synthesis to the reformulation of the labeled peptide with saline. [64Cu]T140-2D was achieved with a total radiosynthesis time of 50 min, a specific activity of 0.28 - 0.30 mCi/µg (10.36 - 11.1 MBq/µg) and a radiochemical purity greater than 99%.
Migration Properties of T140-2D
In order to verify that the CXCR4 inhibitory characteristics of T140 were not affected by the addition of two DOTA molecules, we evaluated the ability of T140-2D to inhibit migration of Jurkat cells towards CXCL12. Jurkat T-cells are known to express high levels of CXCR4 [28]. The migration experiment was performed using 5 µm pore membrane in a standard assay format. We used CXCL12 in the lower wells at 100 ng/mL, the lowest concentration that induces maximal migration [28], and tested various concentrations of T140-2D and T140 in the upper wells. Both T140-2D and T140 were found to inhibit Jurkat migration to CXCL12 in a similar manner. In concentrations of 10 and 100 nM, there was almost no migration of cells with either peptide (Fig. 2A), which indicated that T140-2D retained the ability to inhibit CXCL12-induced chemotaxis through CXCR4.
(A) Inhibition of SDF-1 induced migration of Jurkat cells by T140 and T140-2D. (B) Homologous displacement binding assay of T140-2D with [64Cu]T140-2D using CHO-CXCR4 cells. (C) Cell uptake and internalization assays of [64Cu]T140-2D in CHO-CXCR4 cells. (D) Cell efflux assay of [64Cu]T140-2D in CHO-CXCR4 cells. (E) Homologous displacement binding assay of T140-2D with [64Cu]T140-2D using mouse and human red blood cells (RBCs).
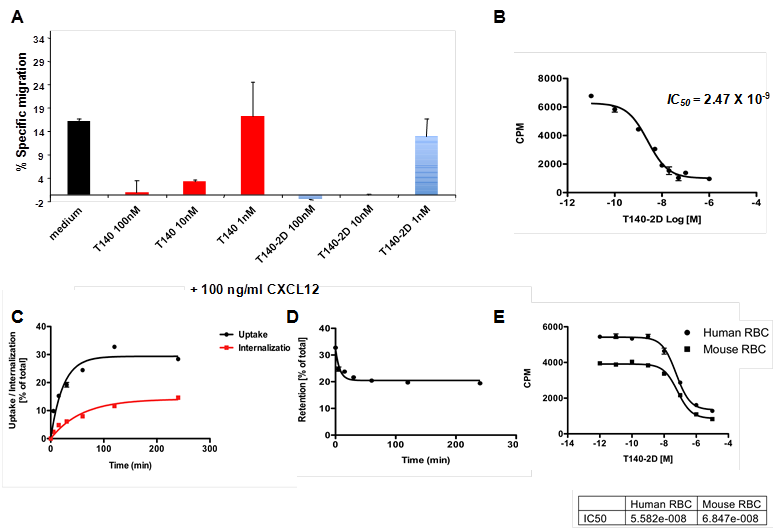
Homologous Displacement Binding Assay
The affinity of T140-2D to CHO cells transfected with human CXCR4 was evaluated in a homologous displacement competitive binding assay using [64Cu]T140-2D. The IC50 of T140-2D binding to CHO-CXCR4 cells was found to be 2.47 ± 0.08 nM (Fig. 2B), which was similar to the reported IC50 of T140 (2.5 nM) that was determined in a competitive binding assay with 125I-CXCL12 [27]. This result implied that introduction of two DOTA molecules into the peptide did not change its binding affinity to CXCR4.
Uptake, Internalization and Efflux Studies
The cell uptake, internalization and efflux of [64Cu]T140-2D were evaluated in CHO-CXCR4 cells. [64Cu]T140-2D uptake increased between 5 min to 2 h (Fig. 2C), reached its peak at 2 h (average of 32.76 ± 0.57 % of total) and then slightly decreased at 4 h (average of 28.38 ± 0.63 % of total, Fig. 2C). In order to evaluate the internalization of the tracer, cells were treated with acid buffer (pH 2.8) before the final wash. Approximately 1/3-1/2 of the observed uptake at all time points was due to the internalization of [64Cu]T140-2D into the cells. The internalization did not reach a full plateau. At 2 h, an average of 11.65 ± 0.99 % of the total peptide was internalized, which was a third of the uptake that was observed (Fig. 2C). At 4 h, there was a slight increase in the internalized peptide (average of 14.61 ± 0.54 % of total), which was about half of the uptake that was observed at this time point (Fig. 2C). Retention studies showed slow release of [64Cu]T140-2D by CHO-CXCR4, which reached a plateau after 1 h (Fig. 2D), suggesting that the radioactivity was retained within the cells.
Binding Assays to Mouse RBCs (mRBCs) and Human RBCs (hRBCs)
We have previously reported that 18F-T140 binds in vivo to red blood cells (RBCs) [27]. To test whether the addition of two DOTA molecules had any effect on this unwanted binding to RBCs, we evaluated the binding of [64Cu]T140-2D to mRBCs. Since the [64Cu]T140-2D tracer was being developed for human imaging, binding of [64Cu]T140-2D was also evaluated using hRBCs. Both types of RBCs bound [64Cu]T140-2D with similar IC50 values of 68.4 and 55.8 nM for mRBCs and hRBCs, respectively (Fig. 2E), which was more than 20-fold higher than the IC50 value for binding to human CXCR4 (Fig. 2B).
Biodistribution
CHO cells and CHO-CXCR4 cells were evaluated for CXCR4 expression by flow cytometry before injection. In vivo, >98% of the CHO-CXCR4 tumor cells retained CXCR4 expression levels similar to the levels before injection (data not shown). Biodistribution of [64Cu]T140-2D was analyzed by organ dissection, followed by gamma counting, in female nude mice that had been inoculated subcutaneously with CHO-CXCR4 and CHO tumors. Data were obtained at 4 h postinjection. To determine the minimal amount of unlabeled peptide needed to block binding to RBCs and to achieve the highest tumor-to-blood and tumor-to-muscle ratios, we preformed the biodistribution experiment with different amounts of unlabeled T140-2D (0, 2, 5, 10 and 400 µg, Fig. 3).
Biodistribution of [64Cu]T140-2D in female athymic nude mice bearing CHO and CHO-CXCR4 tumors at 4 h post-injection of the labeled peptide in the presence of different amounts of unlabeled peptide (0, 2, 5, 10 and 400 µg). Results shown are averages of 4-5 mice ± SD. *P < 0.01 vs. [64Cu]T140-2D alone.
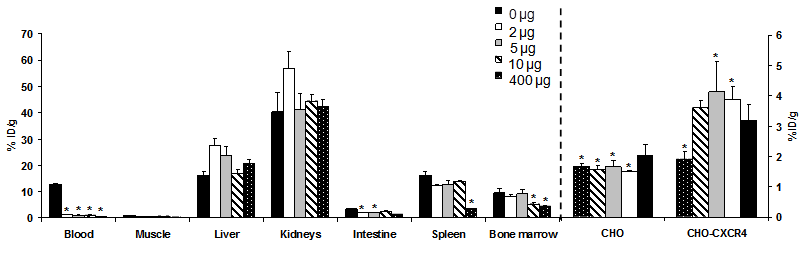
As expected, when no unlabeled mass was added, [64Cu]T140-2D bound to RBCs (12.73 ± 0.23 %ID/g). [64Cu]T140-2D had relatively high uptake in CXCR4-expressing organs, such as spleen (16.15 ± 1.69 %ID/g) and bone marrow (9.45 ± 1.53 %ID/g, Fig. 3) [31-32] and high accumulation in metabolic organs, such as liver (16.34 ± 1.40 %ID/g), intestine (3.36 ± 0.31 %ID/g), and kidneys (40.40 ± 7.25 %ID/g). [64Cu]T140-2D showed slightly higher uptake, but was not statistically significant, in CHO-CXCR4 tumors (3.18 ± 0.51 %ID/g) than in CXCR4 negative CHO tumors (2.01 ± 0.37 %ID/g, Fig. 3).
When only 2 µg of unlabeled T140-2D were added, the binding of [64Cu]T140-2D to RBCs was significantly decreased, with a more than 90% reduction of radioactivity in the blood, to a value of 1.11 ± 0.01 %ID/g. Due to the blocking of [64Cu]T140-2D binding to RBCs, the uptake of [64Cu]T140-2D was also significantly decreased in the CHO negative tumor (1.51 ± 0.03 %ID/g, p < 0.001) and increased in the CHO-CXCR4 positive tumor (3.87 ± 0.42 %ID/g, p < 0.01, Fig. 3). [64Cu]T140-2D uptake was also slightly decreased in the spleen and bone marrow to values of 12.29 ± 0.24 %ID/g and 8.16 ± 0.76 %ID/g, respectively. The uptake in the intestine was also significantly decreased (1.86 ± 0.17 %ID/g, p < 0.01), probably due to the blocking of uptake in the blood. The uptake of [64Cu]T140-2D in liver and kidneys was increased to 27.42 ± 2.68 %ID/g and 57.03 ± 6.42 %ID/g, respectively.
Co-injection of [64Cu]T140-2D with 5 or 10 µg of unlabeled T140-2D gave patterns to the addition of 2 µg (Fig. 3). The uptake in the blood was decreased to approximately 0.85 %ID/g. The uptake in the liver and kidneys was also decreased. Addition of 10 µg blocked some of the uptake in the bone marrow as well (Fig. 3). The highest uptake in the CHO-CXCR4 tumor (4.12 ± 1.02 %ID/g) was achieved with co-injection of [64Cu]T140-2D and 5 µg of unlabeled T140-2D (Fig. 3). Co-injection of [64Cu]T140-2D and 400 µg of unlabeled T140-2D further decreased the uptake in the blood to 0.50 ± 0.10 %ID/g. In addition, administration of 400 µg of unlabeled T140-2D blocked the accumulation in CXCR4-expressing organs, such as spleen (decreased to 3.36 ± 0.25 %ID/g), bone marrow (decreased to 4.31 ± 0.28 %ID/g) and CHO-CXCR4 positive tumor (decreased to 1.91 ± 0.25 %ID/g). These results suggested a specific binding of this peptide to CXCR4 (Fig. 3).
Blocking the uptake in the RBCs by adding different amounts of unlabeled T140-2D (2, 5, 10 and 400 µg) significantly increased the tumor-to-blood ratios in comparison to the result achieved without cold mass (Table 1). Co-injection of 5 µg T140-2D gave the highest tumor-to-blood and tumor-to-muscle ratios (4.86 ± 0.89 and 12.56 ± 3.66, respectively).
CHO-CXCR4 tumor-to-muscle and tumor-to-blood ratios at 4 h postinjection of 64Cu-T140-2D in the presence of different amounts of T140-2D (n = 4 or 5 mice per group).
| Tumor-to-blood | Tumor-to-muscle | |
|---|---|---|
| 64Cu-T140-2D | 0.25 ± 0.01 | 3.75 ± 0.50 |
| 64Cu-T140-2D + 2 µg T140-2D | 3.46 ± 0.32 | 10.41 ± 1.15 |
| 64Cu-T140-2D + 5 µg T140-2D | 4.86 ± 0.89 | 12.56 ± 3.66 |
| 64Cu-T140-2D + 10 µg T140-2D | 4.19 ± 0.11 | 5.37 ± 0.51 |
| 64Cu-T140-2D + 400 µg T140-2D | 3.77 ± 0.46 | 7.60 ± 0.62 |
To test whether the usage of different metals, such as Ga-68, for complexation with the DOTA would provide better in vivo kinetics, these experiments were repeated using [68Ga]T140-2D. However, the results obtained were similar to those of [64Cu]T140-2D with the liver and kidneys displaying high uptake (Supplemental Fig. 1).
MicroPET Imaging Studies
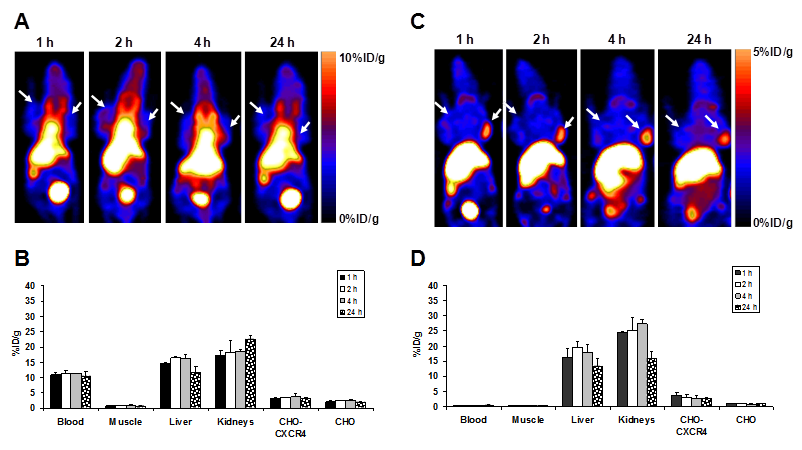
The tumor-targeting efficacy of [64Cu]T140-2D was evaluated by static microPET scans using mice bearing subcutaneous CHO-CXCR4 and CHO tumors (Fig. 4) and mice injected intravenously with CHO-CXCR4 cells (Fig. 5). The %ID/g was calculated from PET images for the blood, muscle, liver, kidneys, CHO-CXCR4 tumor and CHO negative tumor at different time points (Fig. 4).
Representative coronal PET images of an athymic nude mouse bearing CHO-CXCR4 (right shoulder) and CHO (left shoulder) after injection with 100 µCi of [64Cu]T140-2D without (A) and with (C) 5 μg of unlabeled peptide (low specific activity tracer). Arrows indicate tumors. Uptake of [64Cu]T140-2D without (B) and with (D) 5 μg of unlabeled peptide in tumor-bearing mice over time. Results were calculated from PET scans and are shown as averages of 4-5 mice ± SD.
Representative PET images of mouse bearing internal tumors at 2 and 24 h post-injection of [64Cu]T140-2D with 5 μg of T140-2D. Left - coronal ventral slices, Middle - coronal dorsal slices, Right - sagittal view. Arrows indicate CHO-CXCR4 tumors.
Mice that were injected with [64Cu]T140-2D had high uptake in the blood and the abdominal region (Fig. 4A&B). This high uptake in the blood remained stable over time, to give high background throughout the image and specifically in the heart, liver and kidneys, masking some of the uptake in the tumors.
Since the biodistribution results demonstrated that the highest tumor-to-background ratio and the highest tumor uptake were achieved by the addition of 5 µg of unlabeled T140-2D, the mice used for micro-PET studies were co-injected with [64Cu]T140-2D and 5 µg of unlabeled T140-2D. At all time points, CXCR4-positive, but not CXCR4-negative tumors, were clearly visualized (Fig. 4C). The uptake in CHO-CXCR4 tumor was steady over time (Figs. 4C&D). High uptake was also seen in the liver and kidneys, but decreased at 24 h post-injection (Figs. 4C&D).
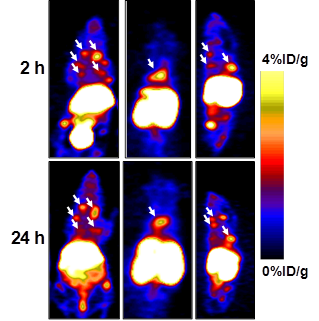
In order to evaluate whether [64Cu]T140-2D is suitable for imaging tumors at internal sites, mice were injected intravenously with CHO-CXCR4 cells. PET scanning detected CHO-CXCR4 tumors in the chest and spine (Fig. 5) at 2 h and 24 h postinjection of the tracer. Accumulation of [64Cu]T140-2D was approximately 2.5 %ID/g in the small tumor lesions in the chest and 4 %ID/g in the lesion on the spine (Fig. 5) and was stable for 24 h.
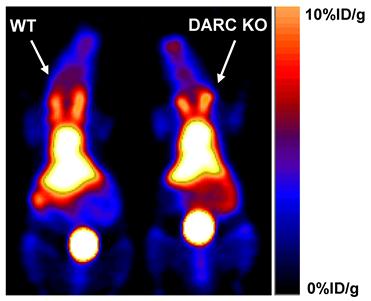
RBCs were previously reported to express several scavenger receptors, including one chemokine scavenger receptor--the Duffy antigen receptor of chemokine (DARC) [33]. The fact that T140 binds to CXCR4 highlighted DARC as a possible receptor that binds T140. To test whether T140 peptide binds to DARC, DARC knockout mice were injected with [64Cu]T140-2D and scanned by microPET side by side with the wild-type mice. However, similar uptake of [64Cu]T140-2D was observed in the blood of both mice strains (Fig. 6), eliminating the possibility of DARC as a candidate for [64Cu]T140-2D binding on RBCs. The high uptake in the blood of DARC knockout mice was confirmed by biodistribution (data not shown).
Representative coronal PET images of wild-type (WT) C57BL/6 mice (Left) and Duffy antigen/receptor for chemokines (DARC) knockout (KO) mice (Right) at 2 h after injection with 100 µCi of [64Cu]T140-2D.
Discussion
T140 peptide binds CXCR4 with high affinity and high inhibition potency. It was shown to increase in vivo mobilization of stem cells from the bone marrow more than the approved drug Mozobil (Plerixafor/AMD3100) [34]. Moreover, it was found to reduce the growth of human acute myeloid leukemia and multiple myeloma xenografts in mice, and to directly induce apoptosis of malignant cells [35].
We have recently reported on the labeling of T140 with fluorine-18 and its evaluation in-vivo as an imaging agent [27]. We have shown that when injected in high specific activity (with no addition of unlabeled mass of peptide), [18F]T140 bound to mouse RBCs and gave high background [27]. We were able to block the binding to RBCs by co-injection of the tracer with unlabeled mass of T140 peptide (low specific activity tracer), resulting in elevated accumulation in CXCR4 positive tumor, but not CXCR4 negative tumor, and high tumor-to-background ratios [27]. However, radiosynthesis of 18F-T140 requires several steps, a long reaction time, and results in low yield, limiting its potential for clinical use.
In this paper we tried to address this problem and simplify the radiosynthesis by introduction of two DOTA molecules, one on each of the free lysine residues of T140, for labeling with PET metal isotopes, such as copper-64, to give a new T140 derivative, [64Cu]T140-2D (Fig. 1). Complexation of copper-64 into the peptide was very efficient and straightforward, resulting in an overall short reaction time, high yield and no need of semi-preparative HPLC purification. Labeling with copper-64 also allowed scanning the mice for longer time periods. PET scans with low specific activity tracer gave a clear visualization of CXCR4-positive tumor, but not CXCR4 negative tumor (Fig. 4C) and enabled imaging of small internal lesions of CXCR4-expressing tumors in the chest and spine (Fig. 5).
Introduction of two DOTA into T140 did not change its high binding affinity to CXCR4 or its high inhibition potency (Fig. 2A&B), in accordance with the results reported by Hanaoka and Tamamura et al. that Arg2, NaI3, Tyr5 and Arg14 are the amino acid residues indispensable for the antagonistic activity [24, 36]. We also found that approximately half of the bound tracer was internalized into the cells and retained within the cells over time (Fig. 2C&D).
[64Cu]T140-2D showed similar behavior as [18F]T140 in terms of binding to mouse RBCs and human RBCs (Fig. 2E). This can be problematic for clinical applications, in terms of quantification of the receptor levels in vivo. Similar to the circumstance for [18F]T140, binding of [64Cu]T140-2D to RBCs can be blocked by addition of unlabeled peptide. We hypothesize that injection of the peptide i.v. primarily interact with RBCs because this is the first binding site the peptide encounters in the circulation. Once the binding sites on RBCs are blocked, the remaining peptide can bind to CXCR4.
Co-injection of [64Cu]T140-2D with 5 µg of T140-2D gave the highest tumor-to-blood and tumor-to muscle ratios (4.86 ± 0.89 and 12.56 ± 3.66 respectively, Table 1). When different masses of T140-2D were added, the uptake of [64Cu]T140-2D in the blood was significantly decreased, however it was much higher than that achieved with [18F]T140 and 10 µg of T140 [27]. For example, in a co-injection of [64Cu]T140-2D and 10 µg of T140-2D, the uptake in the blood was 0.86 ± 0.09 %ID/g, while co-injection of [18F]T140 and 10 µg of T140 gave a value of 0.12 ± 0.04 %, which was 7 times lower than the [64Cu]T140-2D blood value. Blocking experiments by co-injection of [64Cu]T140-2D and 400 µg of T140-2D still gave a higher value (0.50 ± 0.1%ID/g). Overall, the accumulation of low specific activity [64Cu]T140-2D in the blood resulted in tumor-to-blood ratios that were always lower than the ratios achieved with the fluorine-18 derivative.
RBCs do not express CXCR4, however they express scavenger receptors such as Duffy antigen receptor of chemokine (DARC), which is able to bind to various chemokines [33] and seems to be a reasonable candidate to bind T140. However, no difference of [64Cu]T140-2D in blood uptake was found between the DARC knockout mice and wild-type mice (Fig. 6), eliminating DARC as a candidate for binding 64Cu-T140-2D. Hence, the binding site for T140 on RBC is yet to be found.
One downside of [64Cu]T140-2D imaging, when compared to [18F]T140, is the significantly higher uptake in the liver and kidneys that remained high over time (Fig. 4). Labeling T140-2D with another PET metal isotope, 68Ga, gave similar results with persistent activity accumulation in the liver and kidneys (Supplemental Fig. 1). The uptake in the metabolic organs was likely a result of transchelation of the metal ion from the DOTA chelator [37].
CONCLUSIONS
Tumor CXCR4 expression can be imaged using [64Cu]T140-2D but with high uptakes in the blood and metabolic organs. The addition of unlabeled peptide, viz, low specific activity tracer, significantly reduced the red blood cell binding without compromising tumor uptake leading to clear tumor contrast. The high liver and kidney uptake of [64Cu]T140-2D may be reduced by using chelators that form complexes with 64Cu that are theramodynamically and kinetically more inert complexes with 64Cu.
SUPPLEMENTARY MATERIAL
Supplementary Figure 1Acknowledgements
We thank Dr. Henry S. Eden for proof-reading the manuscript. This research was supported in part by the Intramural Research Program (IRP) of the National Institute of Biomedical Imaging and Bioengineering (NIBIB), and the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), and the International Cooperative Program of the National Science Foundation of China (NSFC) (81028009).
Conflict of Interest
The authors declare that they have no conflicts of interest.
References
1. Horuk R. Chemokine receptors. Cytokine Growth Factor Rev. 2001;12:313-35
2. Bleul CC, Wu L, Hoxie JA, Springer TA, Mackay CR. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc Natl Acad Sci U S A. 1997;94:1925-30
3. Gupta SK, Lysko PG, Pillarisetti K, Ohlstein E, Stadel JM. Chemokine receptors in human endothelial cells. Functional expression of CXCR4 and its transcriptional regulation by inflammatory cytokines. J Biol Chem. 1998;273:4282-7
4. Hesselgesser J, Halks-Miller M, DelVecchio V, Peiper SC, Hoxie J, Kolson DL. et al. CD4-independent association between HIV-1 gp120 and CXCR4: functional chemokine receptors are expressed in human neurons. Curr Biol. 1997;7:112-21
5. Patrussi L, Baldari CT. Intracellular mediators of CXCR4-dependent signaling in T cells. Immunol Lett. 2008;115:75-82
6. Chen WJ, Jayawickreme C, Watson C, Wolfe L, Holmes W, Ferris R. et al. Recombinant human CXC-chemokine receptor-4 in melanophores are linked to Gi protein: seven transmembrane coreceptors for human immunodeficiency virus entry into cells. Mol Pharmacol. 1998;53:177-81
7. Klein RS, Rubin JB. Immune and nervous system CXCL12 and CXCR4: parallel roles in patterning and plasticity. Trends Immunol. 2004;25:306-14
8. Phillips RJ, Burdick MD, Lutz M, Belperio JA, Keane MP, Strieter RM. The stromal derived factor-1/CXCL12-CXC chemokine receptor 4 biological axis in non-small cell lung cancer metastases. Am J Respir Crit Care Med. 2003;167:1676-86
9. Burger JA, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2009;23:43-52
10. Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME. et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50-6
11. Redjal N, Chan JA, Segal RA, Kung AL. CXCR4 inhibition synergizes with cytotoxic chemotherapy in gliomas. Clin Cancer Res. 2006;12:6765-71
12. Balkwill F. The significance of cancer cell expression of the chemokine receptor CXCR4. Semin Cancer Biol. 2004;14:171-9
13. Tanaka T, Bai Z, Srinoulprasert Y, Yang BG, Hayasaka H, Miyasaka M. Chemokines in tumor progression and metastasis. Cancer Sci. 2005;96:317-22
14. Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62:1832-7
15. Vicari AP, Caux C. Chemokines in cancer. Cytokine Growth Factor Rev. 2002;13:143-54
16. Rubin JB, Kung AL, Klein RS, Chan JA, Sun Y, Schmidt K. et al. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc Natl Acad Sci U S A. 2003;100:13513-8
17. Azab AK, Runnels JM, Pitsillides C, Moreau AS, Azab F, Leleu X. et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009;113:4341-51
18. Kurtova AV, Tamayo AT, Ford RJ, Burger JA. Mantle cell lymphoma cells express high levels of CXCR4, CXCR5, and VLA-4 (CD49d): importance for interactions with the stromal microenvironment and specific targeting. Blood. 2009;113:4604-13
19. Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature. 2003;425:307-11
20. Helbig G, Christopherson KW2nd, Bhat-Nakshatri P, Kumar S, Kishimoto H, Miller KD. et al. NF-kappaB promotes breast cancer cell migration and metastasis by inducing the expression of the chemokine receptor CXCR4. J Biol Chem. 2003;278:21631-8
21. Tomescu O, Xia SJ, Strezlecki D, Bennicelli JL, Ginsberg J, Pawel B. et al. Inducible short-term and stable long-term cell culture systems reveal that the PAX3-FKHR fusion oncoprotein regulates CXCR4, PAX3, and PAX7 expression. Lab Invest. 2004;84:1060-70
22. Li JK, Yu L, Shen Y, Zhou LS, Wang YC, Zhang JH. Inhibition of CXCR4 activity with AMD3100 decreases invasion of human colorectal cancer cells in vitro. World J Gastroenterol. 2008;14:2308-13
23. Yoon Y, Liang Z, Zhang X, Choe M, Zhu A, Cho HT. et al. CXC chemokine receptor-4 antagonist blocks both growth of primary tumor and metastasis of head and neck cancer in xenograft mouse models. Cancer Res. 2007;67:7518-24
24. Hanaoka H, Mukai T, Tamamura H, Mori T, Ishino S, Ogawa K. et al. Development of a 111In-labeled peptide derivative targeting a chemokine receptor, CXCR4, for imaging tumors. Nucl Med Biol. 2006;33:489-94
25. Nimmagadda S, Pullambhatla M, Pomper MG. Immunoimaging of CXCR4 expression in brain tumor xenografts using SPECT/CT. J Nucl Med. 2009;50:1124-30
26. Misra P, Lebeche D, Ly H, Schwarzkopf M, Diaz G, Hajjar RJ. et al. Quantitation of CXCR4 expression in myocardial infarction using 99mTc-labeled SDF-1alpha. J Nucl Med. 2008;49:963-9
27. Jacobson O, Weiss ID, Kiesewetter DO, Farber JM, Chen X. PET of tumor CXCR4 expression with 4-18F-T140. J Nucl Med. 2010;51:1796-804
28. Jacobson O, Weiss ID, Szajek L, Farber JM, Kiesewetter DO. 64Cu-AMD3100--a novel imaging agent for targeting chemokine receptor CXCR4. Bioorg Med Chem. 2009;17:1486-93
29. Nimmagadda S, Pullambhatla M, Stone K, Green G, Bhujwalla ZM, Pomper MG. Molecular imaging of CXCR4 receptor expression in human cancer xenografts with [64Cu]AMD3100 positron emission tomography. Cancer Res. 2010;70:3935-44
30. Szajek LP MW, Plascjak P, Eckelman WC. Semi-remote production of [64Cu]CuCl2 and preparation of high specific activity [64Cu]Cu-ATSM for PET studies. Radiochim Acta. 2005;93:239
31. Tashiro K, Tada H, Heilker R, Shirozu M, Nakano T, Honjo T. Signal sequence trap: a cloning strategy for secreted proteins and type I membrane proteins. Science. 1993;261:600-3
32. Federsppiel B, Melhado IG, Duncan AM, Delaney A, Schappert K, Clark-Lewis I. et al. Molecular cloning of the cDNA and chromosomal localization of the gene for a putative seven-transmembrane segment (7-TMS) receptor isolated from human spleen. Genomics. 1993;16:707-12
33. Rot A. Contribution of Duffy antigen to chemokine function. Cytokine Growth Factor Rev. 2005;16:687-94
34. Abraham M, Beider K, Wald H, Weiss ID, Zipori D, Galun E. et al. The CXCR4 antagonist 4F-benzoyl-TN14003 stimulates the recovery of the bone marrow after transplantation. Leukemia. 2009;23:1378-88
35. Beider K, Begin M, Abraham M, Wald H, Weiss ID, Wald O. et al. CXCR4 antagonist 4F-benzoyl-TN14003 inhibits leukemia and multiple myeloma tumor growth. Exp Hematol. 2011;39:282-92
36. Tamamura H, Omagari A, Oishi S, Kanamoto T, Yamamoto N, Peiper SC. et al. Pharmacophore identification of a specific CXCR4 inhibitor, T140, leads to development of effective anti-HIV agents with very high selectivity indexes. Bioorg Med Chem Lett. 2000;10:2633-7
37. Boswell CA, Sun X, Niu W, Weisman GR, Wong EH, Rheingold AL. et al. Comparative in vivo stability of copper-64-labeled cross-bridged and conventional tetraazamacrocyclic complexes. J Med Chem. 2004;47:1465-74
Author contact
![]() Corresponding author: Dr. Xiaoyuan Chen, Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institute of Health (NIH), 31 Center Dr, 31/1C22, Bethesda, MD 20892, USA; Tel: 301-451-4246; Email: shawn.chengov
Corresponding author: Dr. Xiaoyuan Chen, Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institute of Health (NIH), 31 Center Dr, 31/1C22, Bethesda, MD 20892, USA; Tel: 301-451-4246; Email: shawn.chengov