Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2011; 1:290-301. doi:10.7150/thno/v01p0290 This volume Cite
Research Paper
Molecular Imaging and Pharmacokinetic Analysis of Carbon-11 Labeled Antisense Oligonucleotide LY2181308 in Cancer Patients
Azeem Saleem1,2, Julian C. Matthews3, Malcolm Ranson4, Sophie Callies5, Valérie André5, Michael Lahn6, Claire Dickinson1, Christian Prenant3, Gavin Brown3, Adam McMahon3, Denis Talbot7, Terry Jones1, Patricia M. Price1, 4, 8 ![]()
1. Academic Radiation Oncology, The University of Manchester, The Christie Hospital NHS Foundation Trust, Wilmslow Road, Manchester, UK;
2. GlaxoSmithKline Clinical Imaging Centre, Hammersmith Hospital, Du Cane Road, London, UK;
3. Wolfson Molecular Imaging Centre, Manchester Academic Health Sciences Centre (MAHSC), The University of Manchester, Manchester, UK;
4. School of Cancer and Enabling Sciences, The Christie Hospital NHS Foundation Trust, MAHSC; The University of Manchester, Manchester, UK;
5. Eli Lilly and Company Ltd, Earl Wood Research Centre, Windlesham, UK;
6. Eli Lilly and Company, Early Oncology Clinical Investigation, Indianapolis, USA;
7. The University of Oxford Department of Medical Oncology, Oxford Radcliffe Hospitals Trust, UK;
8. Department of Surgery and Cancer, Imperial College Healthcare NHS Trust, Hammersmith Hospital, Du Cane Road, London, UK.
Received 2011-3-8; Accepted 2011-5-27; Published 2011-6-1
Abstract
Antisense oligonucleotides (ASOs) have potential as anti-cancer agents by specifically modulating genes involved in tumorigenesis. However, little is known about ASO biodistribution and tissue pharmacokinetics (PKs) in humans, including whether sufficient delivery to target tumor tissue may be achieved. In this preliminary study in human subjects, we used combined positron emission and computed tomography (PET-CT) imaging and subsequent modeling analysis of acquired dynamic data, to examine the in vivo biodistribution and PK properties of LY2181308 - a second generation ASO which targets the apoptosis inhibitor protein survivin. Following radiolabeling of LY2181308 with methylated carbon-11 ([11C]methylated-LY2181308), micro-doses (<1mg) were administered to three patients with solid tumors enrolled in a phase I trial. Moderate uptake of [11C]methylated-LY2181308 was observed in tumors (mean=32.5ng/mL per mg administered intravenously). Highest uptake was seen in kidney and liver and lowest uptake was seen in lung and muscle. One patient underwent repeat analysis on day 15 of multiple dose therapy, during administration of LY2181308 (750mg), when altered tissue PKs and a favorable change in biodistribution was seen. [11C]methylated-LY2181308 exposure increased in tumor, lung and muscle, whereas renal and hepatic exposure decreased. This suggests that biological barriers to ASO tumor uptake seen at micro-doses were overcome by therapeutic dosing. In addition, 18F-labeled fluorodeoxyglucose (FDG) scans carried out in the same patient before and after treatment showed up to 40% decreased tumor metabolism. For the development of anti-cancer ASOs, the results provide evidence of LY2181308 tumor tissue delivery and add valuable in vivo pharmacological information. For the development of novel therapeutic agents in general, the study exemplifies the merits of applying PET imaging methodology early in clinical investigations.
Keywords: Positron emission tomography (PET) imaging, antisense oligonucleotide (ASO), cancer patients, biodistribution, pharmacokinetic (PK) modeling.
Introduction
Positron Emission Tomography (PET) offers the ability to integrate imaging with the investigation of novel therapeutics in vivo, making it a powerful tool for translational experimental medicine and drug development. Following radiolabeling, a compound of interest may be visualized, measured and tracked to investigate its pharmacological properties in a patients' body. PET in combination with Computed Tomography (PET-CT) allows this to be carried out with high specificity and sensitivity. Subsequent use of modeling techniques to analyze the dynamic PET imaging data enables significant pharmacokinetic (PK) information on the labeled agent to be derived in tissues of interest. This methodology can have considerable advantages compared with conventional blood PK information: compound biodistribution may be investigated in all tissues using only tracer amounts, without the need for invasive biopsies; and meaningful information may be gained without studying large patient groups. Using other radiolabeled tracers, PET studies may also be used to derive valuable pharmacodynamic (PD) tissue functionality information to provide evidence of intended drug activity. For example, 15O-labeled water PET scans or 18F labeled fluorodeoxyglucose (FDG) scans may be used before and after experimental treatment in the investigation of novel anti-cancer agents to assess change in tumor perfusion or metabolism, respectively [1, 2]. Thus, PET analysis early in clinical testing can provide in vivo proof-of-concept evidence and may be particularly beneficial if a compound's pharmacological properties are not well understood, as is the case with antisense oligonucleotides (ASOs) [3].
In the past 20 years, ASO technology has advanced from a laboratory tool to a medicinal chemistry platform [4, 5]. As potential therapeutic agents, the intended activity of ASOs relies on binding specific mRNA to inhibit gene expression associated with pathological disease, such as tumorigenesis. Clinical experiences with first generation anti-tumor ASOs were discouraging [6, 7], which contributed to the development of the more promising second generation ASO molecules. These are characterized by chemical modification of their ribose sugar and phosphodiester backbone. Pre-clinical studies showed reduced rates of nuclease degradation, improved plasma binding protein affinity and rapid tissue biodistribution as a result of the modifications, conferring superior biological potency and stability. Longer half-lives, increased metabolite clearance and improved toxicity profile have also been demonstrated [8-14]. Following study of second generation ASO plasma PKs in other species, clinical phase investigations of second generation ASOs are now well underway in oncology. However, the therapeutic potential of ASOs remains dependent on their successful target cell delivery in vivo without causing dose limiting toxicity due to accumulation in the liver or kidney, yet their tissue distribution and PK/PD properties in humans are not well characterized. In vivo imaging using PET offers the ability to investigate these important aspects of ASOs and accelerate the drug discovery process [15, 16].
LY2181308 is a 18-mer 2'-O-methoxyethyl-(MOE) modified second generation ASO which was developed to specifically inhibit survivin. Survivin is a member of the inhibitor of apoptosis protein (IAP) family that is expressed in many types of cancer [17, 18]. High tumor levels are associated with worse prognosis, but there is a relative lack of expression in normal tissues, making it an attractive cancer therapeutic target for molecular inhibition [19]. Encouraging pre-clinical models have led to recent First-in-Human Dose/Phase I trials of LY2181308 [20, 21]. Further, a carbon-11 [11C] positron emitting labeling method which does not require structural modification of the ASO has recently been developed and tested in baboon PET studies [22]. Building upon these studies, we sought to investigate the in vivo biodistribution of labeled LY2181308 in tumor and normal tissues of cancer patients using PET-CT imaging methodologies. Modeling analysis of acquired PET data was subsequently carried out to further measure and understand LY2181308 biodistribution and tissue PKs, including during LY2181308 treatment.
Methods
This study is a companion to the First-in-Human Dose (FHD) study of LY2181308, in which the 750 mg dose was determined to safely reduce survivin in tumor tissue [20]. The primary objectives were to investigate the biodistribution and pharmacokinetic (PK) properties of LY2181308 in normal and tumor tissue of cancer patients.
An external auditing company (Certus, Massachusetts, USA) was employed to assure full regulatory compliance. Prior to administration of radiolabeled LY2181308 to humans, an Administration of Radioactive Substances Advisory Committee (ARSAC) license was obtained, supported by relevant information derived from non-human primate dosimetry studies [22]. Radiolabeled LY2181308 was produced and validated to UK Good Manufacturing Practice (GMP) level and in accordance with Medicines and Health Regulatory Agency (MHRA) regulations. The clinical PET study was carried out with conformance to Good Clinical Practice (GCP) principles and full MHRA and ethical approval (London Multi-Centre Research Ethics Committee 04/MRE02/52). All patients gave written informed consent.
[11C]methylated-LY2181308 was synthesized as previously described by Dence et al [22]. This has the key advantage of not requiring any structural modification of the ASO for the introduction of the positron emitting label. The carbon-11 radioactivity was first produced as [11C]carbon dioxide using a GE PET tracer cyclotron. This was subsequently converted to [11C]methane via a catalytic hydrogenation reaction and then to [11C]methyl iodide by a thermal reaction with iodine in a GE PET Tracer MiI Microlab. The [11C]methyl iodide was then used to randomly [11C]methylate the oligonucleotide substrate under neutral conditions in a GE Tracerlab FX-FE radiochemistry system. Briefly, [11C]methyl iodide (<30nmol) was delivered under a flow of helium and trapped in a reaction vessel containing (in order of addition) 1.4 mg (0.2μmol) LY2181308 (Eli Lilly and Co., Indianapolis, USA), 20 μL of 20 mmol phosphate buffer solution (pH 7.4), 30 μL of ethanol and 250 μL N,N-dimethylformamide. The reaction vessel was sealed and the reaction mixture heated at 80°C for 3 min with stirring then cooled to 30ºC. Heating and cooling was repeated a further 2 times before 1.8 mL phosphate buffered saline (PBS) was added. The ratio of [11C]methyl iodide to LY2181308 set in the labeling reaction, confers the high probability of LY2181308 uni-labeling [22]. The solution resulting from the labeling reaction was purified by size exclusion high-performance liquid chromatography (SE-HPLC) using a Superdex Peptide Gel Filtration Column (HR10/300GL, GE Healthcare, UK) and eluted with PBS at a flow rate of 1 mL/min. The eluent was monitored continuously for both radioactivity and absorbance at 254 nm and the fraction eluting between 11 and 14 min (the retention time of reference LY2181308) was collected in 9mL PBS to recover the final product. This was then purified by passing it through a sterile low protein binding 0.2 μm membrane filter into a sterile vial. The total radiosynthesis time was 40-45 min, giving 0.9-1.5 GBq of formulated product. Quality control was performed by analyzing a 50 uL sample of the formulated product by HPLC. A Polymer Labs PLRPS 100 A, 5 um, 150 x 4.6 mm HPLC column was used with elution at 1.0 mL/min using 200 mM TEAA:acetonitrile (50:50). [11C]methylated-LY2181308 co-eluted with reference LY2181308, with a retention time of 11 min 5s.
All three recruited patients underwent a baseline dynamic [11C]methylated-LY2181308 PET-CT scan for 90 minutes after microdose (<1 mg and <600 MBq) intravenous administration of [11C]methylated-LY2181308 tracer. A Biograph-6 Hi Rez PET-CT scanner (Siemens Medical Solutions Inc. USA) was used for all PET data acquisition and image reconstruction procedures. During the scan, nine venous blood samples were withdrawn at 2, 5, 10, 15, 20, 30, 45, 60 and 90 minutes following [11C]methylated-LY2181308 injection for conventional blood radioactivity assays. Whole blood and plasma samples were immediately analyzed using a calibrated sodium iodide well counter to determine radioactivity concentrations. To calculate partitioning of [11C]methylated LY2181308 into plasma and red blood cells, hematocrit measurements were obtained using a CCX blood gas analyzer (Nova Biomedical Corp. USA). Of the three patients who entered the study, one patient (patient 3) subsequently received multiple doses of 750 mg LY2181308 (Eli Lilly and Co., Indianapolis, USA) as 3-hr infusions on days 1, 2, 3, 8, 15, and 22. A second [11C]methylated-LY2181308 PET-CT scan was carried out on day 15, with the [11C]methylated-LY2181308 injection administered half-way through the 3 hr infusion of 750 mg LY2181308. Patient 3 also underwent [18F]FDG PET-CT scans at baseline and on day 22 following administration of [18F]FDG (≤400MBq) to assess tumor metabolism before and after ASO treatment.
PET image data were reconstructed using an ordered subset expectation maximization (OSEM) algorithm for anatomical information and visualization [23] and discrete inverse Fourier transform (DIFT) reconstruction (similar to filtered back projection) for dynamic quantification of tissue time-concentration curves. Frame durations of 9x10s, 3x30s, 4x60s, 4x120s, 3x300s, and 6x600s were used for the dynamic reconstruction. Regions of interest (ROIs) were delineated on the PET-CT images for the aorta, liver, kidney, muscle, spleen, tumor lesions and vertebral body (as an indicator of marrow uptake). Time-concentration curves for each tissue ROI were generated from the mean concentration within the ROI for each DIFT reconstruction frame. The integral area under the curve during the 90 minutes of data acquisition (AUC0-90: ng*h/mL) was calculated as a comparative measure of tissue ROI exposure to [11C]methylated-LY2181308. Data was corrected for radioactive decay and normalized to an equivalent 1 mg [11C]methylated-LY2181308 dose. Plasma and tissue radioactivity data were interpreted throughout as being from the parent compound only: metabolites were confidently considered negligible in the 90 min post administration time-frame of the study. This is because second generation ASOs have been shown to have long tissue half-lives (>7 days) with no significant build up of metabolites due to their rapid clearance [9, 14, 24].
Kinetic modeling analysis was carried out using the dynamic blood and tissue ROI radioactivity data recorded, with the blood input function derived from the aortic PET-CT ROI imaging data. Both the high spatial resolution of the PET scanner, and the advantage of not requiring correction for delay and dispersion (as with withdrawn arterial blood data), are expected to benefit their accuracy. Indeed, comparison of image derived aortic concentrations and independently derived venous concentrations showed excellent agreement for patients 1 and 3 (Table 2), indicating adequate signal-to-noise ratio and negligible degrading effects such as partial volume. For patient 2, reliable aortic blood vessel regions could not be adequately delineated on the pelvic PET-CT scan and so modeling analysis was not carried out on data from this patient. Two models were subsequently fitted to the PET derived data. The first was an irreversible two-tissue compartmental model with a blood volume term [25]. This model is relatively rigid and dependent on a number of assumptions regarding radiotracer partitioning. The transfer of the radiotracer between compartments is described by rate constants, K1 - the rate of uptake of radiotracer from blood into tissue, k2 - the rate of efflux of radiotracer from tissue into blood and k3 - the rate of irreversible binding within tissues. K1/k2 is the distribution volume, and represents the ratio of free and reversibly bound radiotracer in tissue to blood at equilibrium. The second model used was a more general spectral analysis model with a term for blood volume [26]. This is more flexible as it is not restricted to predefined 'compartments' and requires few a priori assumptions about the distribution of the radiotracer. It is based on identifying a spectrum of kinetic components within a given data set which are involved in the regional uptake and portioning of radiotracer from blood to tissue, allowing for the distinction between transient components associated with radiotracer in the tissue vasculature, and reversible and irreversible components comprising the tissue response to the administered radiotracer. The parameter Ki was estimated from this model using the value of the impulse response function at 90 minutes, which represents the rate of irreversible uptake of radiotracer from blood. All models were implemented using Matlab® r2008a (The MathworksTM Inc., Massachusetts, USA).
Results
Two female (71 and 66 years) and one male (78 years) patient, who had exhausted all standard anti-cancer therapies, were enrolled between June and November 2008. All patients had solid tumors: patient 1 had pancreatic carcinoma, patient 2 had colon cancer, and patient 3 had mesothelioma. All PET-CT scanning and treatment procedures were well tolerated by patients.
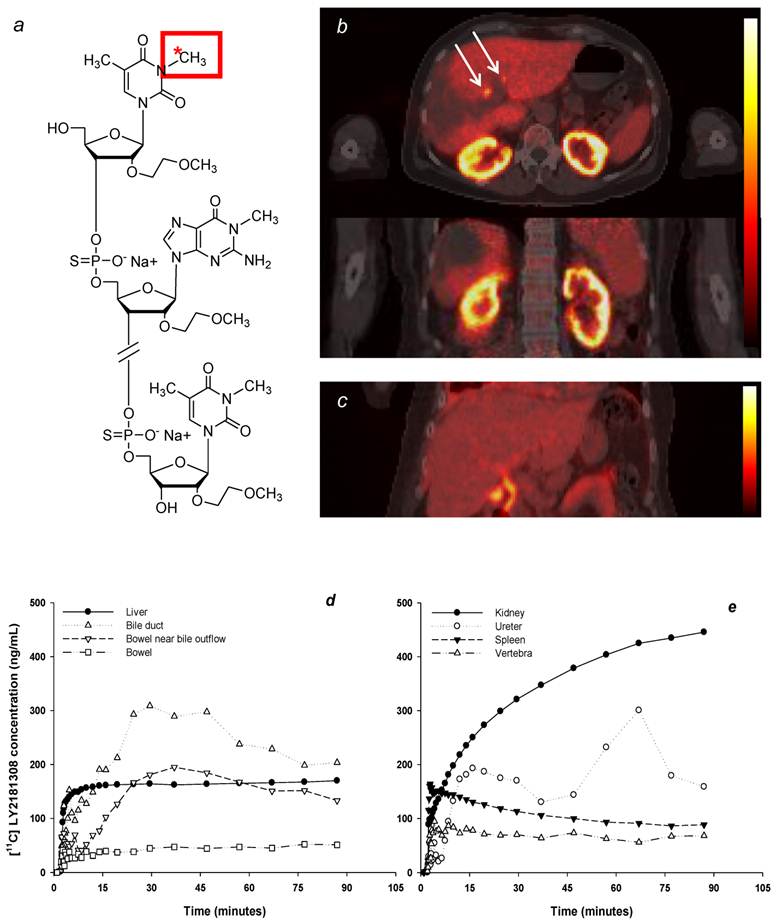
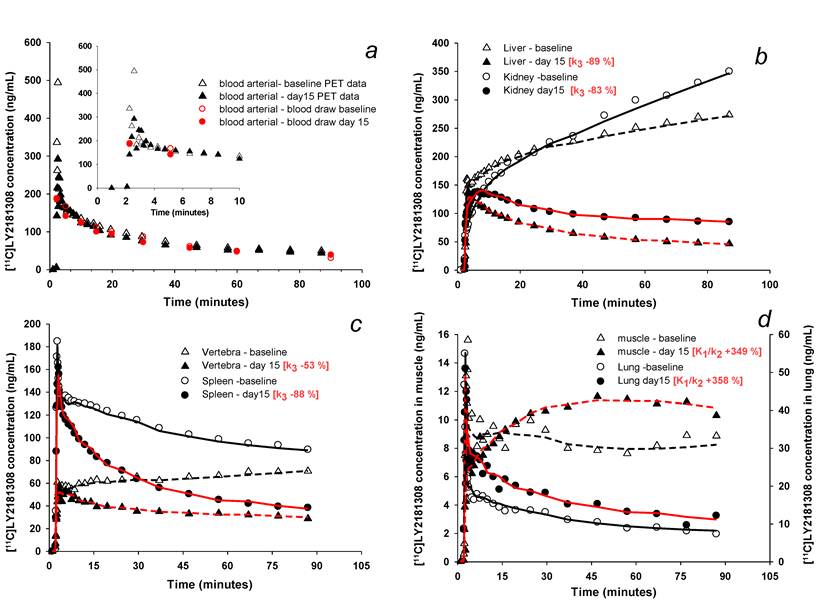
PET imaging data showed highest levels of [11C]methylated-LY2181308 in the renal cortex, liver, and spleen: lowest levels were seen in lung and muscle (Table 1 and Fig. 1b-e). Moderate levels of [11C]methylated-LY2181308 were observed in tumor tissue. At baseline, tumor [11C]methylated-LY2181308 exposure (AUC0-90), normalized to 1mg intravenous administration, ranged between 20.8 to 79.2 ng*h/mL (Table 1). Mean tumor exposure was 32.5 ng*h/mL. Biliary elimination of [11C]methylated-LY2181308 was observed in PET-CT scans, with relatively high radioactivity signals in the gall bladder and bile duct (Fig 1c-d), which was more clearly seen when adjacent liver uptake was lower in patient 3 on day 15 (Fig 1d). High exposure of gall bladder and bile duct was measured from tissue-concentration curves. Bowel adjacent to the opening of the biliary duct on the right side showed high exposure values, relative to that measured in the bowel on the left side of the abdomen (Table 1). Renal elimination was also visible, with lower concentrations of [11C]methylated-LY2181308 detected in the renal pelvis, ureter and bladder, compared with the renal cortex (Table 1 and Fig 1e). A repeat PET scan on day 15 during LY2181308 therapy in patient 3 showed a marked decrease of [11C]methylated-LY2181308 signal in high baseline-uptake organs (kidney, liver and spleen: Figs. 2b-c and Table 1). Conversely, tissue exposure increased in tumor tissue and in low baseline-uptake organs (Fig 2d and Table 1). No concurrent changes in blood time-concentration curves were detected (Fig. 2a).
Normal tissue and tumor uptake of [11C]methylatedLY2181308, normalized to 1 mg bolus administration (AUC0-90: ng*h/mL)
| Tissue | Baseline | Day 15 | ||
|---|---|---|---|---|
| Patient 1 | Patient 2 | Patient 3 | Patient 3 | |
| Kidney (left / right) | 525.8 / 516.7 | NA / 566.1 | 376.7 / NA | 149.4 / NA |
| Liver | 242.7 | 340 | 98.9 | |
| Gall bladder | 187.2 | |||
| Bile duct | 343.7 | 195.5 | ||
| Bowel (near liver or bile outflow) | 218.9 | 151.0 | ||
| Spleen | 158.9 | 156.1 | 83.3 | |
| Bladder | 199.4 | |||
| Ureter (left / right) | NA / 262.8 | 116.9 / 135.8 | ||
| Vertebra | 101.4 | 50.8 | 95.0 | 52.8 |
| Tumor (see table footnotes) | 79.2* 73.6† 36.6‡ | 20.8§ | 57.5†† 49.7** | 71.4†† 84.4** |
| Bowel (left side) | 65.8 | 89.6 | ||
| Ribs | 42.5 | |||
| Ilium (bone) | 35.2 | |||
| Spinal cord | 25.3 | 18.9 | 35 | 24.7 |
| Sternum | 20.3 | |||
| Lung (left / right) | 18.1 / 16.9 | NAa /23.9 | ||
| Muscle | 10.3 | 14.4 | 12.5 | 15.6 |
*Pancreas. †Metastatic lymph node. ‡Necrotic tumor core. §Colon. ††Mesothothelioma adjacent to liver. **Mesothelioma adjacent to lung. NA - data not available. aNormal left lung data was not available for patient 3 on day 15 as there was insufficient tissue for reliable ROI data in the field of view of the post-treatment scan.
The blood time-concentration curves derived from the PET-CT images (aortic ROI data) were typical of rapid distribution into tissue (Fig. 2a). Blood concentrations (AUC0-90 values) of [11C]methylated-LY2181308 derived from the PET-CT aortic ROI are compared with those obtained from discrete venous blood samples in Table 2. The values available for patients 1 and 3 agree well, verifying the accuracy of the PET-CT image derived aortic ROI data used as input function for subsequent kinetic modeling. Additional analysis of discrete blood samples withdrawn during scanning showed plasma radioactivity concentrations were 1.5 times higher than whole blood, with corresponding hematocrit values of 35% (patient 2) and 32% (patient 3), demonstrating [11C]methylated-LY2181308 was almost entirely partitioned into plasma rather than other blood components.
Comparison of blood [11C]methylated-LY2181308 AUC0-90 values, determined from PET-CT images and venous samples withdrawn during scanning.
| Patients | Aortic PET ROI | Blood Samples | |
|---|---|---|---|
| Blood ng*h/mL | Blood ng*h/mL | Plasma ng*h/mL | |
| 1a | 95 | NA | NA |
| 2b | NA | 94.4 | 141.4 |
| 3 (Baseline) 3 (Day15 ) | 118.8 108.2 | 111.7 98.5 | 163.4 145.0 |
NA - data not available. aNo discrete venous samples were taken for patient 1. bReliable arterial blood vessel regions could not be adequately delineated on pelvic scan obtained for patient 2.
Kinetic models used to interpret the dynamic [11C]methylated-LY2181308 PET-CT image data accurately fitted the time-concentration data for most tissues (Figs. 2b-d), with the only exceptions regions of biliary or renal elimination. Both models fitted well and showed similar patterns of derived parameters for the tissues analysed (Table 3). Modeling showed highest baseline irreversible uptake rates (Ki) in the kidney and liver, with moderate Ki values determined for spleen and bone marrow of vertebrae (Table 3). During LY2181308 therapy on day 15 (patient 3), a marked decrease in the rate of irreversible uptake (Ki) and rate of irreversible tissue binding (k3) was observed in the liver, spleen and kidney. No corresponding large changes in K1/k2 were observed (Table 3). In muscle and lung, modeling confirmed low baseline tissue exposure in these tissues (Fig 2d), with very low distribution volume (K1/k2) values (Table 3). In these organs, most of the radioactivity concentration measured in the ROI may be attributed to [11C]methylated-LY2181308 within the blood, as quantified by the blood volume terms of the kinetic models. This reflects very low tissue extraction during transit of [11C]methylated-LY2181308 through the capillary bed. During LY2181308 therapy on day 15 (patient 3), the distribution volumes (K1/k2) in muscle and lung substantially increased to measurable values. A decrease in irreversible binding (k3) was also observed (Table 3). These changes most likely reflect a combination of increased extraction but also decreased binding.
Kinetic model parameters
| Organs | Patient | Compartmental analysis | Spectral analysis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K1/k2a (mL blood / mL tissue) | k3 b (rate/min ) | Ki c (mL blood /min / mL tissue) | ||||||||||
| Baseline | Day 15 | % change | Baseline | Day15 | % change f | Baseline | Day 15 | % change | ||||
| Kidney | Patient 3 Patient 1 | 0.98 4.20 | 0.79 | -19.1 | 0.0591 0.0275 | 0.0100 | -83.0 | 0.0435 0.0563 | 0.0043 | -90.2 | ||
| Liver | Patient 3 Patient 1 | 1.05 1.24 | 0.74 | -30.1 | 0.0358 0.0220 | 0.0040 | -88.8 | 0.0258 0.0158 | 0.0008 | -97.1 | ||
| Spleen | Patient 3 Patient 1 | 0.73 1.12 | 0.71 | -2.43 | 0.0126 0.009 | 0.0015 | -87.9 | 0.0031 0.0035 | 0.000028 | -99.1 | ||
| Vertebra (bone marrow) | Patient 3 Patient 1 | 0.32 0.59 | 0.24 | -24.7 | 0.0291 0.0148 | 0.0136 | -53.3 | 0.0062 0.0052 | 0.0013 | -79.4 | ||
| Tumor (in liver*) | Patient 3 Patient 1 | 0.40 0.20 | 0.68 | +71.1 | 0.0180 0.0609 | 0.0070 | -61.1 | 0.0037 0.0030 | 0.0034 | -6.91 | ||
| Tumor (in lung*) | Patient 3 Patient 1 | 0.12 0.29 | 0.76 | +531 | 0.0401 0.0338 | 0.0024 | -94.0 | 0.0023 0.004 | 0.0019 | -17.9 | ||
| Lung | Patient 3 Patient 1 | 0.03 NA | 0.13 | +358 | 0.0603 NA | 0.0058 | -90.4 | 0.00006 NA | 0.0001 | +89.0 | ||
| Muscle | Patient 3 Patient 1 | 0.01 0.01 | 0.05 | +349 | 0.0605 0.0605 | 0.0361 | -40.3 | 0.0007 0.0005 | 0.0006 | -3.64 | ||
| % change across all organs/tissues for patient 3 | ||||||||||||
| K1/k2a | k3 b | Ki c | ||||||||||
| % | % | % | ||||||||||
| median | 34.4 | -85.5 | -48.6 | |||||||||
| range | -30.0 to +531 | -40.3 to -94.0 | -99.0 to +89.0 | |||||||||
aK1/k2 is the distribution volume (the ratio of free and reversibly bound [11C]methylated LY2181308 in tissue to blood at equilibrium); bk3 is the rate of irreversible binding of [11C]methylated LY2181308 within tissues and cKi is the rate of irreversible uptake of [11C]methylated LY2181308 from blood. *Tumors located in liver and lung refers to metastases from primary tumors. NA - data not available.
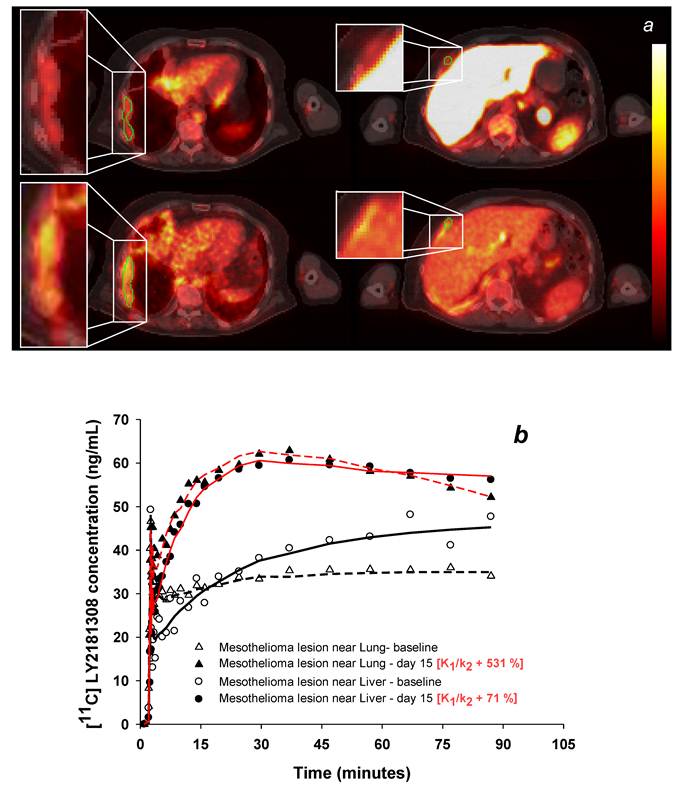
In tumor tissue, kinetic modeling confirmed moderate exposure at baseline (Fig 3b) and showed moderately low distribution volume (K1/k2) values (Table 3). The latter may be partially attributed to the presence of poorly perfused/necrotic areas within the sampled tumor ROI. During LY2181308 therapy on day 15 (patient 3), modeling confirmed increased tumor uptake of [11C]methylated-LY2181308 (Figs 3a and b). Correspondingly, the distribution volume (K1/k2) increased, similar to low baseline-uptake tissues, whereas the rate of irreversible binding (k3) decreased, as observed for all other tissues (Table 3). Concomitantly, [18F]FDG-PET carried out in patient 3 following LY2181308 therapy showed a reduction of up to 40% in tumor metabolism, indicating proof-of-concept of LY2181308 as an anti-tumor agent [20].
Biodistribution of radiolabeled [11C]methylated-LY2181308. The molecular structure depicts LY2181308 labeled at the N3 position of the thymine residue (panel a); note that [11C]methylation at the N1 position of the guanine residue may also be possible. Representative transverse and coronal AUC30-90 images of [11C]methylated-LY2181308 uptake (scaled between 0 and 555 ng*h/mL) in patient 1 prior to LY2181308 therapy (panel b). Coronal AUC30-90 images of [11C]methylated-LY2181308 uptake (scaled between 0 and 278 ng*h/mL) in patient 3 during day 15 LY2181308 infusion (panel c). Both images show relatively high signal in the biliary tree (white arrows), but this is more clearly seen in the image from patient 3 following the reduced uptake in the adjacent liver on day 15 (panel c). Time-concentration curves for ROIs in patient 1 (panel d-e).
Changes in [11C]methylated-LY2181308 time-concentration curves (ng/mL) with therapeutic LY2181308 dosing (750mg) in patient 3. Symbols show observed data. Lines show model fits using the spectral analysis model. Open symbols and black lines show baseline microdose [11C]methylated-LY2181308 data. Solid symbols and red lines show [11C]methylated-LY2181308 data during LY2181308 therapy on day 15. There is no change in concentration in blood (panel a). High and medium baseline uptake tissues (liver, kidney, vertebra and spleen) show similar early concentrations, but marked decreased concentrations by 90 minutes (panel b and c). Low-baseline uptake tissues of muscle and lung show increased [11C]methylated-LY2181308 concentrations (panel d).
Changes in tumor [11C]methylated-LY2181308 uptake with LY2181308 therapy in patient 3 (scaled between 0 and 110 ng*h/mL). Panel a: Images of AUC30- 90 of [11C]methylated-LY2181308 prior to therapy (top row) and with day 15 infusion (bottom row), showing increased concentrations within mesothelial tumor adjacent to the lung (left column) and liver (right column). Panel b: Time-concentration curves from ROIs delineated on the mesothelial tumor adjacent to lung and adjacent to the liver (shown in green on panel a). Symbols show observed data. Lines show model fits using the spectral analysis model. Open symbols and black lines show baseline microdose [11C]methylated-LY2181308 data. Solid symbols and red lines show [11C]methylated-LY2181308 data with 750 mg infusion of LY2181308 on day 15.
Discussion
This study contains novel imaging data which describes for the first time biodistribution of a second generation MOE modified ASO LY2181308 in cancer patients. This was made possible by radiolabeling the ASO molecule with carbon-11 [22], allowing PET studies in human subjects during a Phase I trial. The adherence to quality standards throughout allowed consistent and accurate measurement of the distribution of [11C]methylated-LY2181308. This was observed to distribute rapidly into tissue, despite its size of 6778 Da [27], which enabled precise evaluation of tissue kinetics within the short 20-min half-life of carbon-11. The baseline PET data was collected following microdose infusion of [11C]methylated-LY2181308 in three patients. This experimental approach is on a similar scale to pre-clinical in-vivo animal biodistribution assessments [22] and has the advantage of only requiring tracer amounts of compound. The results yielded unique and valuable human biodistribution and tissue PK information of labeled LY2181308 early in its clinical development. In addition, analysis during multiple, pharmacologically relevant doses of LY2181308 in one patient showed substantial and favorable changes in the biodistribution of [11C]methylated LY2181308. This suggests that biological barriers to tumor ASO uptake may be overcome, and normal tissue toxicity minimised, by ASO dosing levels/schedules.
Overall, the biodistribution information and PK data obtained is generally consistent with pre-clinical assessments of second generation ASOs, which have shown remarkably similar properties across different species [9, 14]. Importantly, moderate yet comparable uptake of [11C]methylated-LY2181308 was seen in tumor tissue in all three patients showing successful ASO tumor penetration. This is consistent with the detection of unlabeled LY2181308 in tumor tissue by immunohistochemistry [28] and predictive models [29]. As expected from other preclinical studies of unlabeled second generation ASOs with phosphodiester backbone and MOE modification, [11C]methylated-LY2181308 exposure was highest in the kidney followed by liver, spleen, and bone marrow of vertebrae [30]. The results also agree with prior biodistribution and tumor uptake studies of [11C]methylated LY2181308 in rodents [22].
Image derived arterial blood concentrations of [11C]methylated LY2181308 were comparable with blood concentrations conventionally determined from discrete venous blood samples (Table 2). This indicates robust image arterial blood concentration data, which was subsequently used as the blood input function in PET kinetic modeling analysis. For most organs, both the analytical models used here described the PET data well, with slightly better fits with the spectral analysis model. Neither models fitted well in regions of biliary or renal elimination. This is because both models are based on radioactivity being delivered to tissues via the arterial blood, and do not model activity being transferred within bile and urine.
A key limitation of the study is that the biodistribution and PK properties of [11C]methylated-LY2181308 could not be directly compared to unlabeled LY2181308. It is possible that labeling the molecule could affect its stability, solubility and 'cellular penetration' properties. However, one of the key advantages of the 11C labeling method is that no structural modification of the ASO is required to introduce the positron emitting precursor - the sugar and phosphodiester backbone already offers a handle for the introduction of the carbon-11 label. The labeled and unlabeled molecules also elute together when purified with high performance gel filtration, demonstrating close molecular size and similar solubility. In general, MOE modified ASOs also show very similar biodistribution and PK properties across various animal species, despite molecular differences [9, 14, 30]. Our PET-CT biodistribution studies of labeled LY2181308 in humans, and those carried out using PET in baboons [22], are also very similar. In addition, both image derived blood concentrations and venous blood derived blood concentrations (Table 2) agreed with the predicted plasma concentration for the 750mg dose of unlabeled LY2181308 from pre-clinical modeling [24] and conventional plasma PK analysis of unlabeled LY2181308 from phase 1 studies [20, 21]. Further, second generation ASO molecules are known to be very stable in vivo, with half-lives longer than 7 days (1-4 weeks depending on chemistry and sequence). Very little breakdown product is detected in tissue compared with parent compound, and even using sensitive techniques, metabolites of MOE modified ASOs are not detected in plasma or tissue within several hours post-dose administration [9, 14]. Thus, it appears that there are no major size stability or functional differences in the labeled ASO molecule when compared with prior studies of unlabeled LY2181308 and other similar unlabeled ASO molecules. However, a change in 'cellular penetration properties' due to the introduction of the 11C label cannot be ruled out. It would therefore be beneficial to investigate this in future studies, and the potential differences between unlabeled and labeled molecules should be borne in mind in the interpretation of results.
Several factors may affect tissue uptake of ASOs from blood plasma, including blood flow/perfusion, plasma concentration, and binding affinity gradient between plasma and tissue. Second generation phosphorothioate backbone ASOs are known to bind to plasma proteins, largely as a function of non-specific low-affinity binding to hydrophilic sites. This prevents rapid loss of ASOs through urinary excretion and promotes systemic uptake, but many biological barriers may subsequently impede penetration of ASOs from plasma proteins into target cells [4, 9, 14, 31]. While the PET data from the ROI analyzed does not provide information on what determines LY2181308 tissue uptake, retention or subcellular distribution, our results support prior evidence that indicates ASO tissue biodistribution is primarily dependent on the differential expression, availability and avidity of cell surface binding sites that promote ASO uptake [4, 9]. For example, receptors present on phagocytically active cells of the reticuloendothelial system, such as active histiocytes, and von Kuppfer macrophages, are known to predispose liver and spleen to high ASO uptake [4, 9, 32-34].
Although only performed in one patient, clear changes in tissue distribution and kinetics were observed during therapeutic dosing of 750mg LY2181308, despite negligible changes in [11C]methylated-LY2181308 blood concentration. The changes were therapeutically advantageous, as tumor uptake increased whereas kidney, liver and spleen uptake reduced. This provides additional data describing non-linear ASO tissue distribution kinetics [9]. It also indicates that dosing levels/schedules may overcome biological barriers to ASO target tissue delivery, and enable minimization of toxicity in normal organs, thereby supporting the concept of repeated infusions of 750 mg LY2181308. Previous studies have shown that redistribution of ASO at the cellular and subcellular level may lead to changes in the uptake pattern over time [35, 36]. However, our modeling analysis results suggest saturation or down-regulation of high-affinity tissue/plasma binding sites are most likely to account for the observed changes in biodistribution following higher drug concentrations/multiple administrations. For example, saturation or down-regulation of high-affinity plasma binding sites could result in an increase of either unbound ASO, or ASO which is bound to lower affinity plasma proteins such as albumin [37]. In turn, this could change the affinity gradient between plasma and tissue, thereby promoting ASO delivery to tissues via lower affinity receptor-mediated endocytosis, or enhanced pinocytosis or endothelial permeation. At the same time, saturation or down-regulation of high-affinity tissue binding sites would result in decreased high-affinity ASO-protein binding transit. Both these proposed mechanisms are consistent with the observed reduced retention (k3) of all tissues during LY2181308 therapy. The former plasma saturation mechanism appeared to have the greater effect upon tissues with negligible baseline extraction, such as muscle and lung, whereas the latter tissue saturation mechanism appeared to have the greater effect upon tissues with high baseline uptake, such as the kidney, liver and spleen. Notably, similar changes to high baseline uptake organs have been observed in rodents, where uptake of [11C]methylated-LY2181308 was partially blocked in the presence of higher amounts of unlabeled ASO [22]. For tumor tissue, the plasma binding protein saturation mechanism is proposed as the major influence to account for increased uptake of [11C]methylated-LY2181308 during therapy. But the increases in extraction are potentially more complex in tumor. This is due to the physiological changes that may occur in tumor following tumor cell arrest, apoptosis or necrosis as a result of LY2181308 treatment, as supported by the partial metabolic responses measured with FDG-PET. For example, reduction in tumor pressure or an increase in perfusion volume could independently increase distribution volume (K1/k2) with unpredictable changes in the rate of tumor uptake (K1).
In summary, the data obtained here adds to the knowledge of ASO biodistribution and tissue PKs. It shows successful ASO tumor penetration, but indicates that target tissue uptake may be confined by the affinity gradient between plasma and tumor tissue and/or the processes of endothelial permeation or endocytosis. Although not definitive as the data is limited, the observation that ASO biodistribution favorably changed during therapy suggests that ASO dosing levels/schedules may influence the biological factors controlling tissue uptake. The results also exemplify how PET-CT imaging and modeling methodology may be applied to enhance the clinical development of novel therapeutic compounds.
Acknowledgements
This work was supported by Cancer Research UK and the UK Department of Health Experimental Cancer Medicine Centre. Additional sponsorship was provided by Eli Lilly and Co. We would like to thank Michael Burgess, Joseph Birkett, Helen Desmier, Maria Forbes, Stacey Maxwell, Judith Grimes from Lilly Oncology, and Angela Ball, Paula Goodege, Helen Ferns from the Christie Hospital. From Academic Radiation Oncology, we acknowledge general project management aid from Jo Cresswell and manuscript preparation and revision by Melanie Green. We also appreciate the help and expertise of Peter Julyan, Jose Anton, Campbell McCullough and other supporting staff from the Wolfson Molecular Imaging Centre.
Conflict of Interest
All academic authors have no conflict of interest to declare in relation to this work. Authors employed by Lilly and Co. hold shares in the company.
References
1. Anderson H, Price P. Clinical measurement of blood flow in tumours using positron emission tomography: a review. Nucl Med Commun. 2002;23:131-8
2. Young H, Baum R, Cremerius U, Herholz K, Hoekstra O, Lammertsma AA. et al. Measurement of clinical and subclinical tumour response using [18F]-fluorodeoxyglucose and positron emission tomography: review and 1999 EORTC recommendations. European Organization for Research and Treatment of Cancer (EORTC) PET Study Group. Eur J Cancer. 1999;35:1773-82
3. Lendvai G, Estrada S, Bergstrom M. Radiolabelled oligonucleotides for imaging of gene expression with PET. Curr Med Chem. 2009;16:4445-61
4. Crooke ST. Antisense Drug Technology: principles, strategies, and application. 2nd ed. Bocan Raton, FL: CRC Press (Taylor&Francis Group). 2008
5. Aboul-Fadl T. Antisense oligonucleotides: the state of the art. Curr Med Chem. 2005;12:2193-214
6. Paz-Ares L, Douillard J-Y, Koralewski P, Manegold C, Smit EF, Reyes JM. et al. Phase III Study of Gemcitabine and Cisplatin With or Without Aprinocarsen, a Protein Kinase C-Alpha Antisense Oligonucleotide, in Patients With Advanced-Stage Non-Small-Cell Lung Cancer. J Clin Oncol. 2006;24:1428-34
7. Genta. Oblimersen: Augmerosen, BCL-2 antisense oligonucleotide - Genta, G 3139, GC 3139, oblimersen sodium. Drugs R D. 2007;8:321-34
8. Fattal E, Bochot A. State of the art and perspectives for the delivery of antisense oligonucleotides and siRNA by polymeric nanocarriers. Int J Pharm. 2008;364:237-48
9. Geary RS. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. 2009;5:381-91
10. Yu RZ, Geary RS, Monteith DK, Matson J, Truong L, Fitchett J. et al. Tissue disposition of 2'-O-(2-methoxy) ethyl modified antisense oligonucleotides in monkeys. J Pharm Sci. 2004;93:48-59
11. Zhao X, Pan F, Holt CM, Lewis AL, Lu JR. Controlled delivery of antisense oligonucleotides: a brief review of current strategies. Expert Opin Drug Deliv. 2009;6:673-86
12. Henry SP, Geary RS, Yu R, Levin AA. Drug properties of second-generation antisense oligonucleotides: how do they measure up to their predecessors? Curr Opin Investig Drugs. 2001;2:1444-9
13. Jason TL, Koropatnick J, Berg RW. Toxicology of antisense therapeutics. Toxicol Appl Pharmacol. 2004;201:66-83
14. Levin AA, Yu RZ, Geary RS. Basic Principles of the Pharmacokinetics of Antisense Oligonucleotide Drugs. In: (ed.) Crooke ST. Antisense Drug Technology Principles, Strategies, and Applications; Second ed. US: CRC Press. 2007:183-216
15. Saleem A, Ranson M, Callies S, Lahn M, Prenant C, Brown G. et al. Microdosing imaging pharmacokinetic (PK) study of the anti-sense oligonucleotide (ASO) to Survivin (LY2181308) using positron emission tomography (PET): a novel paradigm in clinical drug development. J Clin Oncol. 2009;27:3578
16. Tavitian B, Terrazzino S, Kuhnast B, Marzabal S, Stettler O, Dolle F. et al. In vivo imaging of oligonucleotides with positron emission tomography. Nat Med. 1998;4:467-71
17. Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917-21
18. Li F, Ackermann EJ, Bennett CF, Rothermel AL, Plescia J, Tognin S. et al. Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat Cell Biol. 1999;1:461-6
19. Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer. 2003;3:46-54
20. Talbot DC, Ranson M, Davies J, Lahn M, Callies S, Andre V. et al. Tumor survivin is downregulated by the antisense oligonucleotide LY2181308: a proof-of-concept, first-in-human dose study. Clin Cancer Res. 2010;16:6150-8
21. Tanioka M, Nokihara H, Yamamoto N, Yamada Y, Yamada K, Goto Y. et al. Phase I study of LY2181308, an antisense oligonucleotide against survivin, in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2010 epub
22. Dence CS, Laforest R, Sun X, Sharp TL, Welch MJ, Mach RH. Radiochemical Synthesis, Rodent Biodistribution and Tumor Uptake, and Dosimetry Calculations of [11C] Methylated LY2181308. Mol Imaging Biol. 2010;12:608-15
23. Hudson HM, Larkin RS. Accelerated image reconstruction using ordered subsets of projection data. IEEE Trans Med Imaging. 1994;13:601-9
24. Callies S, Andre V, Patel B, Waters D, Francis P, Burgess M. et al. Integrated analysis of preclinical data to support the design of the first in man study of LY2181308, a second generation antisense oligonucleotide. Br J Clin Pharmacol. 2011;71:416-28
25. Gunn RN, Gunn SR, Turkheimer FE, Aston JA, Cunningham VJ. Positron emission tomography compartmental models: a basis pursuit strategy for kinetic modeling. J Cereb Blood Flow Metab. 2002;22:1425-39
26. Cunningham VJ, Jones T. Spectral analysis of dynamic PET studies. J Cereb Blood Flow Metab. 1993;13:15-23
27. Bennett FC, Ackermann EJ, Cowsert LM. Antisense Modulation of Survivin Expression. United States Patent Office. 2007 10/983392
28. Talbot DC, Davies J, Olsen A, Andre V, Lahn M, Powell E. et al. Pharmacodynamic (PD) evaluation of LY2181308 in patients with metastatic malignancies. J Clin Oncol. 2009;27:3507
29. Callies S, Andre V, Vick A-M, Graff J, Patel B, Brail L. et al. Modelling pharmacokinetic and pharmacodynamic properties of second generation antisense-oligonucleotides (ASOs). Annual Meeting of the Population Approach Group in Europe. 2007:1132
30. Geary RS, Watanabe TA, Truong L, Freier S, Lesnik EA, Sioufi NB. et al. Pharmacokinetic properties of 2'-O-(2-methoxyethyl)-modified oligonucleotide analogs in rats. J Pharmacol Exp Ther. 2001;296:890-7
31. Juliano R, Bauman J, Kang H, Ming X. Biological barriers to therapy with antisense and siRNA oligonucleotides. Mol Pharm. 2009;6:686-95
32. Butler M, Crooke RM, Graham MJ, Lemonidis KM, Lougheed M, Murray SF. et al. Phosphorothioate oligodeoxynucleotides distribute similarly in class A scavenger receptor knockout and wild-type mice. J Pharmacol Exp Ther. 2000;292:489-96
33. Benimetskaya L, Tonkinson JL, Koziolkiewicz M, Karwowski B, Guga P, Zeltser R. et al. Binding of phosphorothioate oligodeoxynucleotides to basic fibroblast growth factor, recombinant soluble CD4, laminin and fibronectin is P-chirality independent. Nucleic Acids Res. 1995;23:4239-45
34. Butler M, Stecker K, Bennett CF. Cellular distribution of phosphorothioate oligodeoxynucleotides in normal rodent tissues. Lab Invest. 1997;77:379-88
35. Peng B, Andrews J, Nestorov I, Brennan B, Nicklin P, Rowland M. Tissue distribution and physiologically based pharmacokinetics of antisense phosphorothioate oligonucleotide ISIS 1082 in rat. Antisense Nucleic Acid Drug Dev. 2001;11:15-27
36. Lorenz P, Misteli T, Baker BF, Bennett CF, Spector DL. Nucleocytoplasmic shuttling: a novel in vivo property of antisense phosphorothioate oligodeoxynucleotides. Nucleic Acids Res. 2000;28:582-92
37. Watanabe TA, Geary RS, Levin AA. Plasma protein binding of an antisense oligonucleotide targeting human ICAM-1 (ISIS 2302). Oligonucleotides. 2006;16:169-80
Author contact
![]() Corresponding author: Professor Pat Price, Department of Surgery and Cancer, Imperial College Healthcare NHS Trust, Hammersmith Hospital, Du Cane Road, London, W12 ONN. E-mail:ppriceac.uk. Tel: +44 (0)203 313 2738.
Corresponding author: Professor Pat Price, Department of Surgery and Cancer, Imperial College Healthcare NHS Trust, Hammersmith Hospital, Du Cane Road, London, W12 ONN. E-mail:ppriceac.uk. Tel: +44 (0)203 313 2738.