Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
INTRODUCTION
MATERIALS AND METHODS
RESULTS
DISCUSSION
CONCLUSION
SUPPLEMENTARY MATERIAL
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2011; 1:341-353. doi:10.7150/thno/v01p0341 This volume Cite
Research Paper
New Methods for Labeling RGD Peptides with Bromine-76
Lixin Lang1, Weihua Li1, 2, Hong-Mei Jia3, De-Cai Fang3, Shushu Zhang1, Xilin Sun2, Lei Zhu1, Ying Ma1, Baozhong Shen2, Dale O. Kiesewetter1, Gang Niu1, 4, Xiaoyuan Chen1 ![]()
1. Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH), Bethesda, Maryland, 20892, USA
2. Department of Medical Imaging and Nuclear Medicine, the Fourth Affiliated Hospital, Harbin Medical University, Harbin 150001, China
3. Key Laboratory of Radiopharmaceuticals, College of Chemistry, Beijing Normal University, Beijing 100875, China
4. Imaging Sciences Training Program, Radiology and Imaging Sciences, Clinical Center and National Institute Biomedical Imaging and Bioengineering, NIH, Bethesda, Maryland, 20892, USA
Received 2011-7-23; Accepted 2011-8-15; Published 2011-8-22
Abstract
Direct bromination of the tyrosine residues of peptides and antibodies with bromine-76, to create probes for PET imaging, has been reported. For peptides that do not contain tyrosine residues, however, a prosthetic group is required to achieve labeling via conjugation to other functional groups such as terminal α-amines or lysine ε-amines. The goal of this study was to develop new strategies for labeling small peptides with Br-76 using either a direct labeling method or a prosthetic group, depending on the available functional group on the peptides. A new labeling agent, N-succinimidyl-3-[76Br]bromo-2,6-dimethoxybenzoate ([76Br]SBDMB) was prepared for cyclic RGD peptide labeling. N-succinimidyl-2, 6-dimethoxybenzoate was also used to pre-attach a 2, 6-dimethoxybenzoyl (DMB) moiety to the peptide, which could then be labeled with Br-76. A competitive cell binding assay was performed to determine the binding affinity of the brominated peptides. PET imaging of U87MG human glioblastoma xenografted mice was performed using [76Br]-BrE[c(RGDyK)]2 and [76Br]-BrDMB-E[c(RGDyK)]2. An ex vivo biodistribution assay was performed to confirm PET quantification. The mechanisms of bromination reaction between DMB-c(RGDyK) and the brominating agent CH3COOBr were investigated with the SCRF-B3LYP/6-31G* method with the Gaussian 09 program package. The yield for direct labeling of c(RGDyK) and E[c(RGDyK)]2 using chloramine-T and peracetic acid at ambient temperature was greater than 50%. The yield for [76Br]SBDMB was over 60% using peracetic acid. The conjugation yields for labeling c(RGDfK) and c(RGDyK) were over 70% using the prosthetic group at room temperature. Labeling yield for pre-conjugated peptides was over 60%. SDMB conjugation and bromination did not affect the binding affinity of the peptides with integrin receptors. Both [76Br]Br-E[c(RGDyK)]2 and [76Br]BrDMB-E[c(RGDyK)]2 showed high tumor uptake in U87MG tumor bearing mice. The specificity of the imaging tracers was confirmed by decreased tumor uptake after co-administration of unlabeled dimeric RGD peptides. The energy barrier of the transition state of bromination for the dimethoxybenzoyl group was about 9 kcal/mol lower than that for the tyrosine residue. In conclusion, the newly developed N-succinimidyl-2, 6-dimethoxybenzoate molecule can be used either for one step labeling through pre-conjugation or as the precursor for a Br-76 labeled prosthetic group for indirect labeling. Radiobromination on a dimethoxybenzoyl group has selectivity over radiobromination on tyrosine. The energy barrier difference of the transition states of bromination between the dimethoxybenzoyl group and the tyrosine residue may account for the reaction selectivity when both groups are present in the same molecule.
Keywords: Bromine-76, Arg-Gly-Asp (RGD) peptides, positron emission tomography (PET), N-succinimidyl-2, 6-dimethoxybenzoate
INTRODUCTION
Bromine-76, a positron emitter with 57% positron emission and 43% electron capture, has been used to label antibodies and small molecules for positron emission tomography (PET) imaging.[1] The relatively long half-life (16.2 h) and high positron energy (3.4 MeV maximum) also make it possible to use 76Br for internal radiotherapy. The therapeutic effect can also be attributed to the electron capture component of the Br-76 radionuclidic decay.[2] High specific activity 76Br can be produced with a high energy cyclotron (>50 MeV) using the 75As(3He, 2n)76Br nuclear reaction[3] or with a low energy cyclotron (~16 MeV) using the 76Se(p, n)76Br nuclear reaction.[4]
The radiochemistry of bromine, analogous to that of iodine, relies primarily on oxidative electrophilic substitution to provide a relatively stable radiolabel. The radiobromination of small molecules typically involves the oxidative substitution of a trialkyltin leaving group using various oxidizing agents. Large molecular weight proteins, such as antibodies, can be labeled directly on their tyrosine residues or through a prosthetic group that reacts with the amines of lysine residues. The direct labeling method involves either enzymatic oxidation[5] or chemical oxidizing agents such as chloramine-T.[6] Prosthetic methods using N-succinimidyl 3-(4-hydroxyphenyl) propionate,[7] N-succinimidyl p-(4-tri-n-butylstannyl)benzoate[8] or N-succinimidyl p-(4-trimethystannyl)benzoate[9] have also been reported.
The cyclic Arg-Gly-Asp (RGD) peptides have been labeled with various radioisotopes, including 18F, 64Cu, 89Zr, 99mTc and 111In,[10-12] for imaging αvβ3 integrin expression in tumors. 125I has also been used to directly label RGD peptides using iodogen[13] or chloramine-T.[14] For labeling peptides that contain no tyrosine residue, such as c(RGDfK), labeled N-succinimidyl 4-[76Br]-bromobenzoate (SBB) has been used with satisfactory results. However, the drawback of this indirect method is that it usually requires two HPLC purifications, one for purifying SBB and the other for purifying the peptide conjugate. Alternatively, direct labeling may be achieved by first attaching a prosthetic group to the peptides.
We have previously labeled small molecules with 76Br and observed that an electron rich 2, 6-dimethoxybenzene moiety can easily attach a bromine to its 3 position ortho to the methoxy group without the need of a trialkyltin leaving group.[15] Based on this dimethoxybenzene structure, we developed a new labeling agent, N-succinimidyl-2,6-dimethoxybenzoate that can be used either for one step labeling through pre-conjugation or as the precursor for a 76Br-labeled prosthetic group for indirect labeling. Moreover, we investigated the mechanism of bromination reaction with the SCRF-B3LYP/6-31G* method.
MATERIALS AND METHODS
General materials
Cyclic RGD peptides were purchased from Peptides International (Louisville, KY), and all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). The chemical structures of cyclic RGD peptides c(RGDfK), c(RGDyK) and their glutamic acid linked dimeric forms E[c(RGDfK)]2 and E[c(RGDyK)]2 are shown in Figure 1A. 1H and 13C NMR spectra were recorded at 300 MHz on a Bruker 300 Ultra-Shield spectrometer in CDCl3 with tetramethylsilane (TMS) as the internal standard. Mass spectra (MS) were obtained using a Thermo-Finnigan Trace DSQ GC-MS or Waters Acquity UPLC system coupled with a Waters Qtof Premier MS (LC-MS). Preparative reversed-phase HPLC was performed on a Waters 600 gradient system with a Waters 996 PDA detector using a Higgins PROTO 300 C18 column (5 µm, 250 x 20 mm). Analytical reversed-phase HPLC was performed on a separate Waters 600 gradient system also with a 996 PDA detector, plus a Beckman 170 radioisotope detector using a Vydac protein and peptide column (5 µm, 250 x 4.6 mm). The flow rate was 10 mL/min for the preparative column and 1 mL/min for the analytical column running the same linear gradient starting from 5% A (0.1% TFA in acetonitrile) and 95% B (0.1% in water) for 5 min and increasing A to 65% at 35 min. A Varian BOND ELUT C18 column (50 mg) was used for solid phase extraction of labeled peptides.
(A) Chemical structures of RGD peptides. (B) Synthesis of brominated RGD peptide standards and pre-conjugated RGD peptides.
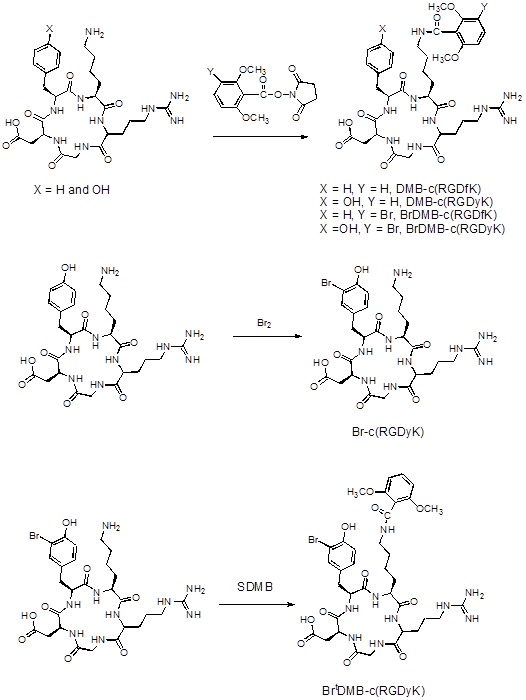
Preparation of N-succinimidyl 2, 6-dimethoxybenzoate (SDMB)
1.0 g of 1, 3-dicyclohexylcarbodiimide (DCC) was added to a mixture of 0.9 g of 2, 6-dimethoxybenzoic acid and 0.58 g of N-hydroxysuccinimide in 20 mL of methylene chloride (CH2Cl2), and the reaction mixture was stirred at room temperature overnight. The precipitate was filtered and the product was purified with silica gel column flash chromatography with 30% ethyl acetate. The fractions containing the desired product (based on TLC and GC-MS analysis) were combined, and the solvent was evaporated with reduced pressure to give 0.7 g of product (51% yield) with a 99% purity based on GC-MS analysis (Rt = 7.7 min). 1H NMR (CDCl3) δ 2.88 (s, 4H), 3.89 (s, 6H), 6.69 (d, 2H), 7.39 (t, 1H). 13C NMR (CDCl3) δ 25.7, 56.4, 104.0, 106.9, 133,4, 159.0, 161.5, 169.0. GC-MS (EI) 280 (M+), 193, 165 (calc: 279.1).
Preparation of N-succinimidyl 3-bromo-2, 6-dimethoxybenzoate (SBDMB)
The 3-bromo-2, 6-dimethoxybenzoic acid was prepared in a 100 mL round bottom flask containing 2.0 g of 2, 6-dimethoxybenzoic acid in 20 mL acetonitrile by adding 0.9 g of bromine. The reaction was monitored with GC-MS. After 30 min, all the starting material was converted to the desired product. The solvent was removed with a rotary evaporation apparatus to give 2.7 g of product with a 95% yield and 96% purity based on GC-MS analysis (Rt = 6.2 min), and the solid product was used without further purification. 1H NMR (CDCl3) δ 3.88 (s, 3H), 3.96 (s, 3H), 6.66 (d, 1H), 7.56 (d, 1H). GC-MS (EI) 260, 262 (M+), 214, 185, 157 (calc: 260.0).
The following were added to a 50 mL flask: 0.68 g of 3-bromo-2,6-dimethoxybenzoic acid, 0.33 g of N-hydroxysuccimide, 0.59 g of DCC and 20 mL of CH2Cl2. The mixture was stirred at room temperature overnight. At the end of the reaction, the solid was removed by filtration and the filtrate was passed through a small silica gel column. Upon standing at room temperature overnight, the solvent was evaporated to give 0.5 g of pale yellow solid (54% yield), and the product was checked with GC-MS and used without further purification. The GC-MS analysis showed that the product had over 95% purity with a 8.3 min retention time. 1H NMR (CDCl3) δ 2.90 (s, 4H), 3.89 (s, 3H), 3.99 (s, 3H), 6.67 (d, 1H), 7.63 (d, 1H). 13C NMR (CDCl3) δ 25.7, 56.7, 62.7, 107.9, 108.5, 114.0, 136,7, 156.2, 158.0, 160.5, 168.8. GC-MS (EI) 357, 359 (M+), 227, 184, 156 (calc: 356.9).
Preparation of Bromo(Tyr)-c(RGDyK)
2 µL of acetic acid and 1 µL of bromine were added to a 4 mL glass vial containing 10.4 mg of c(RGDyK) in 0.3 mL of water.. After 30 min, the reaction was quenched with 10 mg of sodium metabisulfite in 100 µL of water and injected onto a preparative RP HPLC. The product was collected (Rt = 17.1 min) and lyophilized to give 4.5 mg of product (yield 35%). Bromo(Tyr)-c(RGDyK) was denoted as Brt-c(RGDyK). LC-MS: [MH]+ = 698.2899 (m/z), calc: 698.2261.
Preparation of Bromo(Tyr)-2, 6-dimethoxybenzoyl-c(RGDyK)
0.5 mg of SDMB in 0.1 mL of acetonitrile was added to a 4 mL glass vial containing 1.5 mg of Br-c(RGDyK) in 0.1 mL of 0.1 M borate buffer (pH 8.4), and the mixture was reacted at 4 ºC overnight. The reaction was quenched with 10 µL of acetic acid and purified with preparative HPLC to give 0.9 mg of product (50% yield, Rt = 22.5 min). Bromo(Tyr)-2, 6-dimethoxybenzoyl-c(RGDyK) was denoted as BrtDMB-c(RGDyK). LC-MS: [MH]+= 862.2984 (m/z), calc: 862.2735.
Synthesis of 2, 6-dimethoxybenzoyl-c(RGDfK), 2, 6-dimethoxybenzoyl-c(RGDyK), 2, 6-dimethoxybenzoyl-E[c(RGDfK)]2 and 2, 6-dimethoxybenzoyl-E[c(RGDyK)]2
20 µL of diisopropylethylamine (DIPEA) and 5 mg of N-succinimidyl 2, 6-dimethoxybenzoate in 100 µL of DMF were added to a 4 mL glass vial containing 14.0 mg of c(RGDfK) in 100 µL of DMF, and the mixture was stirred at room temperature for 1 hr. The reaction was quenched with 25 µL of acetic acid and the reaction mixture was diluted with 0.5 mL of water and injected onto a preparative RP HPLC for purification. The peak containing desired product was collected (Rt = 22.8 min) and the solution was frozen over dry-ice and lyophilized overnight to give 11.8 mg of product denoted as DMB-c(RGDfK), (66% yield). LC-MS: [MH]+ = 768.4506 (m/z), calc: 768.3681.
2, 6-Dimethoxybenzoyl-c(RGDyK), 2,6-dimethoxybenzoyl-E[c(RGDfK)]2 and 2,6-dimethoxybenzoyl-E[c(RGDyK)]2 were prepared in a similar manner and were denoted as DMB-c(RGDyK), DMB-E[c(RGDfK)]2, and DMB-E[c(RGDyK)]2, respectively. DMB-c(RGDyK) (yield 63%, Rt = 20.2 min), LC-MS: [MH]+ = 784.2714 (m/z), calc: 784.3630; DMB-E[c(RGDfK)]2 (yield 39%, Rt = 23.2 min), LC-MS: [MH]+ = 1482.7344 (m/z), calc: 1482.7130; DMB-E[c(RGDyK)]2 (yield 49%, Rt = 19.9 min), LC-MS: [MH]+=1514.5557 (m/z), calc: 1514.7028.
Synthesis of 3-bromo-2, 6-dimethoxybenzoyl-c(RGDfK), 3-bromo-2, 6-dimethoxybenzoyl-c(RGDyK), 3-bromo-2, 6-dimethoxybenzoyl-E[c(RGDfK)]2 and 3-bromo-2, 6-dimethoxybenzoyl-E[c(RGDyK)]2
N-succinimidyl 3-bromo-2, 6-dimethoxybenzoate (41 mg) was dissolved in 200 µL of acetonitrile and 50 µL of this solution was added to each of 4 separate vials containing variously 11.0 mg of c(RGDfK), 12.5 mg of c(RGDyK), 24.0 mg of E[c(RGDfK)]2 or 20.0 mg of E[c(RGDyK)]2. Each vial also contained 200 µL of DMF and 20 µL of DIPEA. After two hours, 25 µL of acetic acid and 0.5 mL of water were added to each vial. Each sample was purified with preparative RP HPLC and lyophilized to give the desired product. 3-Bromo-2, 6-dimethoxybenzoyl-c(RGDfK), 3-bromo-2, 6-dimethoxybenzoyl-c(RGDyK), 3-bromo-2, 6-dimethoxybenzoyl-E[c(RGDfK)]2 and 3-bromo-2, 6-dimethoxybenzoyl-E[c(RGDyK)]2 were denoted as BrDMB-c(RGDfK), BrDMB-c(RGDyK), BrDMB-E[c(RGDfK)]2, and BrDMB-E[c(RGDyK)]2, respectively. BrDMB-c(RGDfK), (yield 33%, Rt = 25.0 min), LC-MS: [MH]+ = 846.3982 (m/z), calc: 846.2786; BrDMB-c(RGDyK), (yield 22%, 22.8 min), LC-MS: [MH]+ = 862.4221 (m/z), calc: 862.2735; BrDMB-E[c(RGDfK)]2 (yield 21%, Rt = 25.4 min), LC-MS: [MH]+ = 1560.8088 (m/z), calc:1560.6235; BrDMB-E[c(RGDyK)]2 (yield 14%, Rt = 22.1 min), LC-MS: [MH]+=1592.8000 (m/z), calc: 1592.6133.
Radiochemistry
Bromine-76 was produced by cyclotron irradiation of high-purity arsenic targets through a 75As(3He, 2n)76Br nuclear reaction and isolated by chromic acid dissolution and oxidation followed by simple distillation of [76Br]HBr. The radiobromine was trapped in an aqueous solution of NH4OH. The solution was evaporated to produce dry [76Br]NH4Br.
Direct labeling of peptides with chloramine-T
5-10 µL of [76Br]NH4Br in water was added to about 100 µg of either c(RGDyK) or E[c(RGDyK)]2 in 0.1 mL of pH 6.8 acetate buffer in a 1 mL polypropylene tube, and followed by adding 100 µg of chloramine-T in 10 µL of water. After 10 min, the reaction mixture was quenched by adding 200 µg of sodium metabisulfite in 10 µL of water and then injected onto an analytical RP HPLC column. Radioactivity peaks were collected and measured using a dose calibrator. Most reactions were performed at room temperature and a few selected reactions were performed at 80 °C to test the temperature effect.
Direct labeling of peptides with peracetic acid
5-10 µL of [76Br]NH4Br (~0.5-1.0 mCi) in water was added to about 100 µg of c(RGDyK) or E[c(RGDyK)]2 in 0.1 mL of water in a 1 mL polypropylene tube, followed by adding either 2 µL or 0.2 µL of anhydrous peracetic acid as the oxidizing agent. After 10 min, the reaction mixture was injected onto an analytical RP HPLC column. Radioactivity peaks were collected and measured using a dose calibrator. The reactions were performed at room temperature when 2 µL of peracetic acid was used, and reactions were performed at room temperature and 80 °C when 0.2 µL of peracetic acid was used.
Preparing Br-76 labeled prosthetic group [76Br]SBDMB
About 100 µg of SDMB in 50 µL of acetonitroile was added to a 1 mL polypropylene tube, followed by adding 5-10 µL of [76Br]NH4Br (~0.5-1.0 mCi) and 100 µg of chloramine-T in 10 µL of water or 2 µL anhydrous peracetic acid. The mixture was heated at 80 °C for 10 min and injected onto a Waters Symmetry C18 column (5 µm, 3.9 x 150 mm) using 40% acetonitrile/water as the mobile phase containing 0.1% of acetic acid with a 1 mL/min flow rate. The radioactive product was collected and trapped on a Waters C18 Sep-Pak column. The radioactivity on the C18 column was eluted with 1 mL of CH2Cl2 to a 1 mL polypropylene tube, the small amount of water on top of the organic phase was removed by a needle syringe, and CH2Cl2 was removed by argon flow. The radioactivity was redissolved in 5 µL of acetonitrile for further use.
Labeling RGD peptides with Br-76 labeled prosthetic group
About 300 to 500 µg of RGD peptide in 100 µL of 1 M borate buffer (pH 8.4) was added to the 1 mL polypropylene tube containing Br-76 radioactivity as described above. The mixture was vortexed and allowed to react at room temperature for 10 min and then was loaded onto the RP HPLC for purification. The radioactive peak was collected, diluted with 10 mL water and then trapped on a Varian Bond-Elute C18 column. The column was washed with 5 mL of water and the radioactivity eluted with 200 µL of 10 mM HCl ethanol solution. The ethanol was removed using argon flow and radioactivity re-dissolved in saline for further use.
Br-76 labeling of pre-conjugated RGD peptides
5-10 µL of [76Br]NH4Br in water was added to about 100 µg of pre-conjugated RGD peptide in 100 µL of water in a 1 mL polypropylene tube, followed by adding 0.2 µL of 32% peracetic acid in 10 µL of acetonitrile. The mixture was vortexed and heated at 80 °C for 10 min. The reactions were performed at room temperature when 2 µL of peracetic acid was used (see section on preparing Br-76 labeled prosthetic group [76Br]SBDMB, above). The product was purified with a procedure similar to that described above for the indirect labeling procedure.
Cell binding assay
In vitro integrin binding affinity and specificity of RGD peptide derivatives were assessed via a competitive cell-binding assay using 125I-echistatin (Perkin-Elmer, Waltham, MA) as the integrin αvβ3 specific radioligand. Experiments were performed on human glioblastoma U87MG cells with high integrin αvβ3 expression.[16] In brief, U87MG cells were trypsinized and resuspended in binding buffer containing 20 mM Tris (pH 7.4), 150 mM NaCl, 2 mM CaCl2, 1 mM MgCl2, 1 mM MnCl2 and 0.1% BSA. Incubation was conducted with 96 well MultiScreen filter plates (Millipore, MA). For each well, the reaction volume was 200 µL containing 105 cells, 20 nCi (0.78 KBq) of 125I-echistatin, and 0-20,000 nM of RGD compounds. The reaction was incubated for 60 min on a shaker at room temperature. After incubation, the filters were washed three times with the binding buffer. Filter bound radioactivity was measured using a gamma counter (1480 Wizard 3, Perkin-Elmer). Binding results were expressed as percent of total counts, and IC50 values were calculated using Prism software (GraphPad, La Jolla, CA).
Animal model and PET imaging
All animal studies were conducted in accordance with the principles and procedures outlined in the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the Clinical Center, NIH. The human glioblastoma cell line U87MG was grown in DMEM medium supplemented with 10% fetal bovine serum (FBS), 100 IU/mL penicillin, and 100 μg/mL streptomycin (Invitrogen), and in a humidified atmosphere containing 5% CO2 at 37°C. The cells were harvested by trypsinization with trypsin/EDTA. The U87MG tumor model was developed in 5 to 6‑week old female athymic nude mice (Harlan Laboratories) by injection of 5×106 cells into the right shoulders. Tumor growth was monitored using caliper measurements of perpendicular axes of the tumor. The mice underwent small-animal PET studies when the tumor volume reached 100-300 mm3 (3 - 4 weeks after inoculation).
PET scans and image analysis were performed using an Inveon microPET scanner (Siemens Medical Solutions). About 3.7 MBq (100 μCi) of 76Br-labeled RGD peptide was administered via tail vein injection under isoflurane anesthesia. For a blocking study, about 300 µg of unlabeled dimeric RGD peptide E[c(RGDyK)]2 was injected 30 min before the 76Br-labeled tracer. Five-minute static PET images were acquired at 30 min, 1 h and 2 h postinjection (n = 3 per group). The images were reconstructed using a two-dimensional ordered-subset expectation maximum (2D OSEM) algorithm, and no correction was applied for attenuation or scatter. For each scan, regions of interest (ROIs) were drawn using vendor software (ASI Pro 5.2.4.0) on decay-corrected whole-body coronal images. The radioactivity concentrations (accumulation) within the tumor, muscle, liver, and kidneys were obtained from mean pixel values within the multiple ROI volume and then converted to MBq per milliliter. These values were then divided by the administered activity to obtain (assuming a tissue density of 1 g/mL) an image-ROI-derived percent injected dose per gram (%ID/g).
Biodistribution assay
Immediately after PET imaging, the tumor-bearing mice were sacrificed and dissected. Blood, tumor, major organs, and tissues were collected and wet-weighed. The radioactivity in the wet whole tissue was measured with a γ-counter (Packard). The results were expressed as percentage of injected dose per gram of tissue (%ID/g). Values were expressed as mean ± SD (n = 3 per group).
Theoretical Calculation
All calculations included in this work were performed with the Gaussian 09 program package.[17] CH3COOBr molecule was used as the brominating agent for the no-carrier-added electrophilic radiobromination reaction. SCRF-B3LYP/6-31G* calculations were carried out for bromination reactions between DMB-c(RGDyK) peptide and CH3COOBr, where SCRF calculations were performed with the default PCM model within G09 program[18] with water (dielectric constant ε = 78.39) as solvent at a temperature of 298 K. The geometric parameters of the reactant, transition states and products have been optimized and characterized with SCRF-B3LYP/6-31G* method. The relative energies of all stationary points were corrected with zero-point vibrational energies to account for the thermal effect.
Statistical Analysis
Quantitative data were expressed as mean ± SD. Means were compared using one-way analysis of variance and Student's t test. P values of < 0.05 were considered statistically significant.
RESULTS
Pre-conjugation and bromination
The syntheses of brominated RGD peptide standards and pre-conjugated precursors are shown in Figure 1B using c(RGDyK) as an example. For dimeric peptides, the coupling was on the α-amine of the glutamic acid used for linking the two monomers. A similar procedure was followed for all coupling reactions and the yields were generally higher for the monomeric RGD peptides than the dimeric counterparts, which can be explained by that fact that ε-amine on lysine is more reactive than the α-amine on glutamic acid.
In order to confirm that pre-conjugation and bromination do not affect the binding affinity of the peptides to integrin receptors, we performed competitive cell binding assays with 125I-echistatin as the radioligand in integrin αvβ3 positive U87MG cells. Results of the cell-binding assays were plotted and fitted to sigmoid curves for the displacement of 125I-echistatin from U87MG cells (Figure 2A). The IC50 values are listed in Figure 2B. Consistent with the data reported previously,[19] the dimers showed similar binding affinity with FPPRGD2 and higher binding affinity than the monomers. The results indicated that the preconjugation and bromination had little effect on the binding affinity of the peptides.
(A) Cell binding assay of RGD peptides in U87MG cells; (B) IC50 of each RGD compound (n = 3).
Br-76 labeling of RGD peptides
The radiolabeling schemes are shown in Figure 3. The yields for direct radiobromination of c(RGDyK) and E[c(RDGyK)]2 using chloramine-T were over 50% at room temperature with minor radioactive impurities. At 80 °C, the desired products were diminished and undesired multiple radioactive peaks were formed. When peracetic acid was used as the oxidizing agent, the labeling yields were also over 50% at room temperature with less radioactive impurity. When diluted peracetic acid (10% of normal concentration) was used, the radiochemical yield was less than 5% even at 80 °C. However, at 80 °C the yield for Br-76 SBDMB and pre-conjugated RGD peptides was over 60% using the diluted peracetic acid. High labeling yields were also achieved using more concentrated peracetic acid at room temperature for these reactions. The coupling yields of [76Br]SBDMB with c(RGDfK) and c(RGDyK) were over 70% and coupling yield with their dimeric forms were less than 10% under similar conditions. The specific activity at the end of synthesis (EOS) for [76Br]BrDMB-E[c(RGDyK)]2 used for imaging studies was 262 mCi/µmol. We assumed the specific activity for [76Br]Br-E[c(RDGyK)]2 was in the same range from the same batch of Br-76.
(A) Radiolabeling with Br-76 prosthetic group; (B) Direct labeling vs. labeling on pre-conjugated prosthetic group.

MicroPET imaging
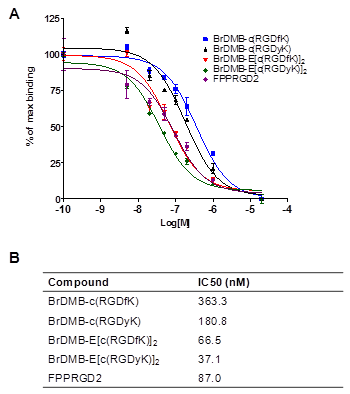
After pre-conjugation and bromination, we conducted in vivo imaging of [76Br]Br-E[c(RDGyK)]2 and [76Br]BrDMB-E[c(RGDyK)]2 in a U87MG xenograft mouse model. Representative coronal microPET images of U87MG tumor bearing mice at different times after intravenous injection of 3.7 MBq (100 μCi) of the imaging tracers are shown in Figure 4A-C. The U87MG tumors are clearly visible with high contrast in relation to the contralateral background. The tumor uptake of [76Br]Br-E[c(RDGyK)]2 was 2.94 ± 0.55, 2.86 ± 0.52 and 2.23 ± 0.19 %ID/g at 30, 60 and 120 min p.i., respectively (Fig. 4D). The tumor uptake of [76Br]BrDMB-E[c(RGDyK)]2 was 3.07 ± 0.73, 2.50 ± 0.62 and 2.05 ± 0.41 %ID/g at 30, 60 and 120 min p.i., respectively (Fig. 4E). The receptor specificity of the tracer accumulation was confirmed by a blocking assay, in which about 300 µg of unlabeled dimeric RGD peptide was injected 30 min before the tracer injection. As shown in Figure 4C&F, the tumor uptake of [76Br]BrDMB-E[c(RGDyK)]2 was significantly lower (p < 0.01) than that of unblocked tumors (1.04 ± 0.22 vs. 2.94 ± 0.55 %ID/g at 30 min, 0.62 ± 0.25 vs. 2.86 ± 0.52 %ID/g at 60 min, and 0.38 ± 0.30 vs. 2.23 ± 0.19 %ID/g at 120 min). Prominent uptake of [76Br]BrDMB-E[c(RGDyK)]2 was also observed in the kidneys, suggesting that this tracer is mainly excreted through the renal-urinary routes as is common with this class of integrin binding peptides.
In vivo PET imaging of U87MG xenograft mice by 76Br-labeled RGD peptides. Decay-corrected whole-body coronal microPET images of U87MG tumor-bearing mice at 30, 60, and 120 min after injection of 3.7 MBq (100 μCi) of [76Br]Br-E[c(RGDyK)]2 (A) and [76Br]BrDMB-E[c(RGDyK)]2 (B). (C) Decay-corrected whole-body coronal microPET images of U87MG tumor-bearing mice at 30, 60 and 120 min after injection of 3.7 MBq (100 μCi) of [76Br] BrDMB-E[c(RGDyK)]2 with 300 µg of unlabled RGD peptide as blocking agents. (D, E & F) Quantification of [76Br] Br-E[c(RGDyK)]2 (D), [76Br]BrDMB-E[c(RGDyK)]2 (E) and [76Br]BrDMB-E[c(RGDyK)]2 (Blocking, F) in U87MG tumor, liver, kidneys and muscle. ROIs are shown as mean %ID/g ± SD.
Biodistribution Studies
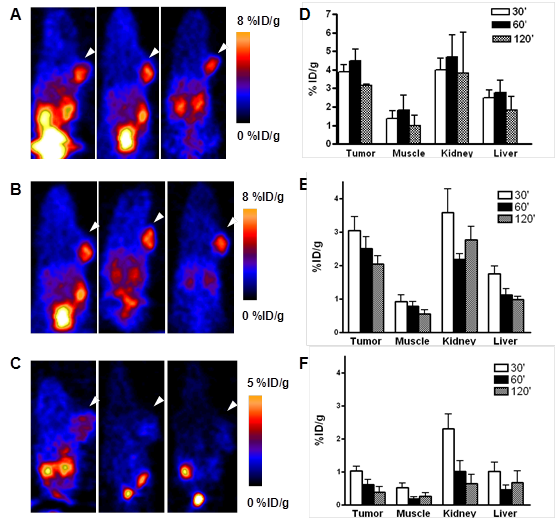
In order to further confirm the PET imaging quantification, the biodistribution of [76Br]BrDMB-E[c(RGDyK)]2 was evaluated in U87MG tumor-bearing athymic nude mice immediately after PET imaging (Fig. 5). Consistent with PET imaging data, the tumor uptake measured by direct tissue sampling and gamma-counting was 1.76 ± 0.61 %ID/g at the 2 h time point. The tracer accumulations in the kidneys and liver were 2.69 ± 0.17 and 0.64 ± 0.13 %ID/g, respectively, at 2 h p.i. In the blocking experiment, U87MG tumor showed significantly decreased accumulation of [76Br]BrDMB-E[c(RGDyK)]2 with an uptake value of 0.53 ± 0.15 %ID/g (p < 0.001).
Ex vivo biodistribution of [76Br]BrDMB-E[c(RGDyK)]2 (3.7 MBq per mouse) in U87MG tumor bearing nude mice at 2 h time points after microPET scans with or without unlabeled RGD peptide as blocking agent. Columns, mean %ID/g (n = 3 per group); bars, SD.
Reaction Mechanism Study
Grovenstein et al.[20] proposed an iodination mechanism of phenol with acetyl hypoiodite as an iodinating agent at very low iodine concentration. Acetyl hypoiodite could in principle both donate I+ and remove H+ in a single transition state. Based on the Br-76 radiolabeling condition, we used CH3COOBr as the effective brominating agent. The four carbon atoms of DMB-c(RGDyK) peptide labeled by * could be attacked by Br of CH3COOBr, respectively. The proposed brominating process included the attack of Br on C1* and the leaving of H2 from the aromatic ring of DMB-c(RGDyK) peptide. When the brominating reaction took place on the dimethoxybenzoyl group, the transition state was defined as TS1a and TS1a'. On the other hand, when the brominating reaction took place on the tyrosine residue, the transition state was defined as TS1b and TS1b'. The main atomic numbering systems and geometric parameters of the possible stationary points are shown in Figure 6, and the full geometric parameters and the frequencies of stationary points optimized are listed in Supplementary Material: Table S1-S4 in supporting information. The normal mode of vibration of the imaginary frequency of TS1a is given in Figure 7, from which one can see that the imaginary vibration mode in transition state is connected to the reactants and the products. Moreover, the sum of Mulliken atomic charges of CH3COO group is -0.825, while the other part of the TS1a is +0.825, indicating that the transition state looks like δ+…δ- form, a σ-type complex.
The possible brominating reaction channels for DMB-c(RGDyK) peptide, along with the main geometric parameters for transition states optimized with SCRF-B3LYP/6-31G*.

The normal mode (in arrows) of imaginary vibration and group charge (e) in TS1a.
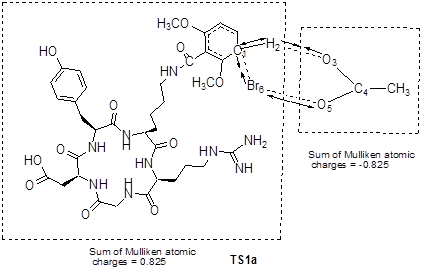
As to the potential energy surface for the brominating process, the energy of TS1a, TS1a', TS1b and TS1b' are 12.7, 13.3, 21.8 and 22.1 kcal/mol (with ZPE correction), respectively, above the sum of the energies for the reactants. The significant difference in the energy barriers of TS1a and TS1b may account for the difference in the bromination reactivity of the dimethoxybenzoyl group and the tyrosine residue of DMB-c(RGDyK) peptide.
DISCUSSION
Integrins are a family of heterodimeric glycoproteins responsible for the regulation of cellular activation, migration, proliferation, survival, and differentiation.[21-22] One of the most important members of this receptor class is integrin αvβ3 which is preferentially expressed on several types of cancer cells, including melanoma, glioma, and ovarian and breast cancers.[23] Integrin αvβ3 is also expressed on proliferating endothelial cells associated with neovascularization in both malignant tumors and normal tissue, but not in quiescent blood vessels.[24-25] Because arginine-glycine-aspartic acid (RGD) containing peptides can bind strongly to integrin αvβ3, many RGD peptide probes have been developed for multimodal imaging of integrin expression.[13, 26-32] In this study, we chose RGD peptides as model compounds to test the novel bromine labeling strategy, which potentially can be applied to bromination of other proteins and peptides. Our preliminary results of using chloramine-T as the oxidizing agent indicated that direct labeling of RGD peptides with 76Br is possible, but the applications are limited to stable peptides containing tyrosine residues such as c(RGDyK) and E[c(RGDyK)]2. Peptides such as c(RGDfK) and E[c(RGDfK)]2 contain phenylalanine residues instead of tyrosine residues, thus makes direct labeling impossible. In order to solve this problem, we introduced N-succinimidyl 2, 6-dimethoxybenzoate as either a prosthetic group for indirect labeling or a preconjugated moiety on peptides for direct labeling.
Based on the iodination mechanism proposed by Grovenstein et al.,[20] we conducted theoretical calculations of the transition state energies for the four reactive sites on DMB-c(RGDyK) peptide using acetyl hypobromite as the brominating agent. The energy of the transition state for bromination on the dimethoxybenzoyl group (TS1a) and that on tyrosine (TS1b) are about 13 kcal/mol and 22 kcal/mol, respectively, above the sum of the energies for the reactants. The energy of TS1a is about 9 kcal/mol lower than that of TS1b, from which one can conclude that acetyl hypobromite could selectively brominate the dimethoxybenzoyl in the presence of a tyrosine residue. The above calculation results are in good agreement with the experimental selectivity of second reaction in Figure 3B. The reaction rate and selectivity seem to be determined by the obtained transition state (TS1a and TS1b). If the concentration of peracetic acid is relatively high, the formation of acetyl hypobromite will become more rapid. That is why the reaction can occur at room temperature in the presence of 2 μL of peracetic acid, whereas heating to 80 ºC is required when only 0.2 μL of peracetic acid was used.
Both chloramine-T and peracetic acid have been used as the oxidizing agents for halogenation reaction.[33] We found out that peracetic acid was a better oxidizing agent than chloramine-T for bromination of RGD peptides. For direct labeling of c(RGDyK), both chloramine-T and peracetic acid gave satisfactory results. However, fewer undesired radioactive peaks were formed using peracetic acid. When peracetic acid was used, very few side products were formed even at elevated temperature, suggesting that it is a milder oxidizing agent than chloramine-T. The radioactive brominating species was [76Br]bromine chloride when chloramine-T was used. On the other hand, [76Br]acetyl hypobromite was the brominating species when peracetic acid was used. When choramine-T was used, a large excess of N-chloro species was also present and would compete with [76Br] bromide chlorine to consume the substrate. This could explain the existence of multiple radioactive peaks due to the chlorination of the substrate on other positions of the substrate. This was also evident when chloramine-T was used to prepare 3-bromo-2, 6-dimethoxybenzoic acid. When equal molar amount of ammonium bromide was used, little desired product was detected on GC-MS, and the main product was 3-chloro-2, 6 dimethoxybenzoic acid. We also detected the formation of chlorinated c(RGDyK) by LC-MS when chloramine-T was used for direct labeling.
[76Br]Acetyl hypobromite appears to be less reactive towards the tyrosine residue than the dimethoxybenzoyl group, as confirmed by both the experimental data and theoretical calculations. While being less reactive towards hydroxyphenyl and almost not reactive towards the 4-methoxybenzoyl group, [76Br]acetyl hypobromite could easily brominate the dimethoxybenzoyl moiety. For example, when labeling pre-conjugated DMB-c(RGDyK), two radioactive peaks were formed with HPLC retention times of 18.2 min and 18.6 min respectively. The 18.2 min peak corresponded to BrtDMB-c(RGDyK) with the Br-76 on tyrosine, whereas the 18.6 min peak corresponded to BrDMB-c(RDGyK) with the Br-76 on dimethoxybenzoyl moiety with the peak ratio of 1 to 9.
The peak assignments were based on the HPLC retention time of cold standards. When labeling pre-conjugated dimeric DMB-E[c(RGDyK)]2, two radioactive peaks were also formed with the peak ratio of 1 to 3, since there were two tyrosine residues to compete with one dimethoxybenzoyl group. Alternatively the same [76Br]BrDMB-E[c(RGDyK)]2 can be prepared by coupling E[c(RGDyK)]2 with [76Br]SBDMB to give a single radioactive product. [76Br]SBDMB can be easily prepared from SDMB using peracetic acid without a trialkyltin leaving group. The drawback of this procedure is that it requires two HPLC purifications. When labeling pre-conjugated DMB-c(RDGfK) and DMB-E[c(RDGfK)]2, only a single radioactive product was observed since there were no other competing groups present.
The cell binding results with compound containing non-radioactive bromine demonstrated that the preconjugation had little effect on RGD peptide binding affinity with αvβ3 integrin receptor. In the cell binding assay, we also included FPPRGD2, which is a fluorine-labeled dimeric RGD peptide recently developed in our lab that has higher receptor binding affinity and enhanced tumor uptake than monomeric RGD peptide counterparts.[34-36] Brominated RGD dimers showed cell binding affinity comparable to FPPRGD2.
In order to further evaluate the pharmacodynamics and pharmacokinetics of 76Br labeled RGD compounds, we performed in vivo PET imaging and biodistribution studies with two dimeric RGD peptides, since the dimers have been shown to have higher binding affinities for αvβ3 integrin receptor than the monomeric analogs. For direct comparison, E[c(RGDyK)]2 and pre-conjugated E[c(RGDyK)]2 were chosen. Both [76Br]Br-E[c(RGDyK)]2 and [76Br]BrDMB-E[c(RGDyK)]2 were prepared using the direct labeling method. 76Br was attached on the tyrosine residue in [76Br]Br-E[c(RGDyK)]2. In [76Br]BrDMB-E[c(RGDyK)]2, 76Br was labeled on the dimethoxybenzoyl group. Both tracers showed rapid and high tumor accumulation in U87MG xenografted mice. There was no significant difference between [76Br]Br-E[c(RGDyK)]2 and [76Br]BrDMB-E[c(RGDyK)]2 as to tumor uptake (p > 0.1) at all time points examined. In addition, both tracers showed similar excretion patterns through renal-urinary routes. The integrin αvβ3 binding specificity of [76Br]BrDMB-E[c(RGDyK)]2 was also confirmed by effective blocking of tumor accumulation with excess amounts of unlabeled dimeric RGD peptide.
Bromination using N-succinimidyl-2, 6-dimethoxybenzoate as a preconjugated group has several advantages over direct labeling on tyrosine residues. First, this method can be used to label most proteins or peptides that have available ε- or α- amine groups. Second, the labeling reaction occurs under mild conditions to help preserve the biological activity of protein molecules. Finally, bromine on the dimethoxybenzoyl group may be more resistant to potential enzymatic dehalogenation [37] after administration. Such dehalogenation could lead to loss of some of the radioactivity from a protein or peptide before it reaches its target region and, concurrently, contribute to background radioactivity.
CONCLUSION
The newly developed N-succinimidyl-2, 6-dimethoxybenzoate can be used either for one-step labeling through pre-conjugation or as the precursor for Br-76 labeled prosthetic group for indirect labeling. High labeling yields were achieved under mild reaction conditions without the need of a trialkyltin leaving-group. Suitably brominated RGD peptides showed integrin-specific tumor uptake. The methods presented here for RGD peptide labeling can also be applied to radiobromination of other bioactive proteins and peptides.
SUPPLEMENTARY MATERIAL
Table S1. The optimized Cartesian coordinates of stationary points for the reaction 1 + CH3COOBr with the SCRF-B3LYP/6-31G* method in water with the Gaussian 09 program package. Table S2. Frequencies of stationary points for the reactions of 1 + CH3COOBr with the SCRF-B3LYP/6-31G* method in water with the Gaussian 09 program package.Table S3. The total energies of reactants, products, and transition states of the reaction 1 + CH3COOBr with the SCRF-B3LYP/6-31G* method in water with the Gaussian 09 program package. Table S4. The relative energies of reactants, products, and transition states of the reaction 1 + CH3COOBr with the SCRF-B3LYP/6-31G* method in water with the Gaussian 09 program package.
Acknowledgements
This work was supported by the Intramural Research Program (IRP) of the National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH), and the International Cooperative Program of the National Science Foundation of China (NSFC) (81028009). G. Niu holds an Imaging Sciences Training Fellowship, which is jointly supported by the Radiology and Imaging Sciences Department, NIH Clinical Center and the Intramural Research Program, NIBIB, NIH. We acknowledge the NIH/CC cyclotron facility for isotope production and Dr. Henry S. Eden for proof-reading the manuscript.
CONFLICT OF INTEREST
The authors have declared that no conflict of interest exists.
References
1. Ribeiro MJ, Almeida P, Strul D, Ferreira N, Loc'h C, Brulon V. et al. Comparison of fluorine-18 and bromine-76 imaging in positron emission tomography. Eur J Nucl Med. 1999;26:758-66
2. Zhou D, Sharp TL, Fettig NM, Lee H, Lewis JS, Katzenellenbogen JA. et al. Evaluation of a bromine-76-labeled progestin 16α,17α-dioxolane for breast tumor imaging and radiotherapy: in vivo biodistribution and metabolic stability studies. Nucl Med Biol. 2008;35:655-63
3. Loc'h C, Mardon K, Valette H, Brutesco C, Merlet P, Syrota A. et al. Preparation and pharmacological characterization of [76Br]-meta-bromobenzylguanidine ([76Br]MBBG). Nucl Med Biol. 1994;21:49-55
4. Tolmachev V, Lovqvist A, Einarsson L, Schultz J, Lundqvist H. Production of Br-76 by a low-energy cyclotron. Appl Radiat Isot. 1998;49:1537-40
5. Mcelvany KD, Welch MJ. Characterization of Bromine-77-Labeled Proteins Prepared Using Bromoperoxidase. J Nucl Med. 1980;21:953-60
6. Sundin J, Tolmachev V, Koziorowski J, Carlsson J, Lundqvist H, Welt S. et al. High yield direct Br-76-bromination of monoclonal antibodies using chloramine-T. Nucl Med Biol. 1999;26:923-9
7. Knight LC, Harwig SSL, Welch MJ. Invitro stability and in vivo clearance of fibrinogen or serum-albumin labeled with Br-77, I-131, or I-125 by direct or indirect synthetic methods. J Nucl Med. 1977;18:282-8
8. Wilbur DS, Hylarides MD. Radiolabeling of a Monoclonal-Antibody with N-Succinimidyl Para-[Br-77]Bromobenzoate. Nucl Med Biol. 1991;18:363-5
9. Hoglund J, Tolmachev V, Orlova A, Lundqvist H, Sundin A. Optimized indirect Br-76-bromination of antibodies using N-succinimidyl para-[Br-76]bromobenzoate for radioimmuno PET. Nucl Med Biol. 2000;27:837-43
10. Beer AJ, Kessler H, Wester HJ, Schwaiger M. PET imaging of integrin αvβ3 expression. Theranostics. 2011;1:48-57
11. Zhou Y, Chakraborty S, Liu S. Radiolabeled cyclic RGD peptides as radiotracers for imaging tumors and thrombosis by SPECT. Theranostics. 2011;1:58-82
12. Chen X. Integrin targeted imaging and therapy. Theranostics. 2011;1:28-9
13. Haubner R, Wester HJ, Reuning U, Senekowitsch-Schmidtke R, Diefenbach B, Kessler H. et al. Radiolabeled αvβ3 integrin antagonists: a new class of tracers for tumor targeting. J Nucl Med. 1999;40:1061-71
14. Chen X, Park R, Shahinian AH, Bading JR, Conti PS. Pharmacokinetics and tumor retention of 125I-labeled RGD peptide are improved by PEGylation. Nucl Med Biol. 2004;31:11-9
15. Lang L, Ma Y, Kim BM, Jagoda EM, Rice KC, Szajek LP. et al. [Br-76]BMK-I-152, a non-peptide analogue for PET imaging of corticotropin-releasing hormone type 1 receptor (CRHR1). J Labelled Compd Radiopharm. 2009;52:394-400
16. Chen X, Park R, Shahinian AH, Tohme M, Khankaldyyan V, Bozorgzadeh MH. et al. 18F-labeled RGD peptide: initial evaluation for imaging brain tumor angiogenesis. Nucl Med Biol. 2004;31:179-89
17. Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR. et al. Gaussian 09; A.02 ed. Wallingford, CT: Gaussian Inc. 2009
18. Scalmani G, Frisch MJ. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J Chem Phys. 2010;132:114110
19. Li ZB, Chen K, Chen X. 68Ga-labeled multimeric RGD peptides for microPET imaging of integrin αvβ3 expression. Eur J Nucl Med Mol Imaging. 2008;35:1100-8
20. Grovenstein E, Aprahamian NS, Bryan CJ, Gnanapragasam NS, Kilby DC, McKelvey JM. et al. Aromatic halogenation. IV. Kinetics and mechanism of iodination of phenol and 2'6-dibromophenol. J Am Chem Soc. 1973;95:4261-70
21. Ruegg C, Alghisi GC. Vascular integrins: therapeutic and imaging targets of tumor angiogenesis. Recent Results Cancer Res. 2010;180:83-101
22. Niu G, Chen X. Why integrin as a primary target for imaging and therapy. Theranostics. 2011;1:30-47
23. Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9-22
24. Kassmeyer S, Plendl J, Custodis P, Bahramsoltani M. New insights in vascular development: vasculogenesis and endothelial progenitor cells. Anat Histol Embryol. 2009;38:1-11
25. Weis SM. Evaluating integrin function in models of angiogenesis and vascular permeability. Methods Enzymol. 2007;426:505-28
26. Liu Z, Yan Y, Liu S, Wang F, Chen X. 18F, 64Cu, and 68Ga labeled RGD-bombesin heterodimeric peptides for PET imaging of breast cancer. Bioconjug Chem. 2009;20:1016-25
27. Chen X. Multimodality imaging of tumor integrin αvβ3 expression. Mini Rev Med Chem. 2006;6:227-34
28. Chen X, Hou Y, Tohme M, Park R, Khankaldyyan V, Gonzales-Gomez I. et al. Pegylated Arg-Gly-Asp peptide: 64Cu labeling and PET imaging of brain tumor αvβ3-integrin expression. J Nucl Med. 2004;45:1776-83
29. Tan M, Lu Z-R. Integrin targeted MR imaging. Theranostics. 2011;1:83-101
30. Ye Y, Chen X. Integrin targeting for tumor optical imaging. Theranostics. 2011;1:102-26
31. Kiessling F, Gaetjens J, Palmowski M. Application of molecular ultrasound for imaging integrin expression. Theranostics. 2011;1:127-34
32. Zhang Y, Yang Y, Cai W. Multimodality imaging of integrin αvβ3 expression. Theranostics. 2011;1:135-48
33. Van Dort ME, Jung YW, Gildersleeve DL, Hagen CA, Kuhl DE, Wieland DM. Synthesis of the 123I- and 125I-labeled cholinergic nerve marker (-)-5-iodobenzovesamicol. Nucl Med Biol. 1993;20:929-37
34. Liu S, Liu Z, Chen K, Yan Y, Watzlowik P, Wester HJ. et al. 18F-labeled galacto and PEGylated RGD dimers for PET imaging of αvβ3 integrin expression. Mol Imaging Biol. 2010;12:530-8
35. Mittra ES, Goris ML, Iagaru AH, Kardan A, Burton L, Berganos R. et al. Pilot pharmacokinetic and dosimetric studies of F-FPPRGD2: A PET radiopharmaceutical agent for imaging αvβ3 integrin levels. Radiology. 2011;260:182-91
36. Chin FT, Shen B, Liu S, Berganos RA, Chang E, Mittra E. et al. First experience with clinical-grade [18F]FPP(RGD)2: An automated multi-step radiosynthesis for clinical PET studies. Mol Imaging Biol. 2011 [Epub ahead of print]
37. Niu G, Cai W, Chen X. Molecular imaging of human epidermal growth factor receptor 2 (HER-2) expression. Front Biosci. 2008;13:790-805
Author contact
![]() Corresponding author: Xiaoyuan Chen, PhD, 31 Center Dr, 1C22, Bethesda, MD 20892-2281. Email: shawn.chengov
Corresponding author: Xiaoyuan Chen, PhD, 31 Center Dr, 1C22, Bethesda, MD 20892-2281. Email: shawn.chengov