Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Target proteases
3. Protease-Activatable Prodrugs
4. Perspective on prodrug design
5. Conclusion
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2012; 2(2):156-178. doi:10.7150/thno.4068 This issue Cite
Review
Protease-Activated Drug Development
Ki Young Choi1,3, Magdalena Swierczewska1,2,3, Seulki Lee1 ![]() , Xiaoyuan Chen1
, Xiaoyuan Chen1 ![]()
1. Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH), Bethesda, Maryland, 20892, USA
2. Department of Biomedical Engineering, Stony Brook University, Stony Brook, NY 11794, USA
3. These authors are contributed equally.
Received 2012-1-10; Accepted 2012-1-28; Published 2012-2-8
Abstract

In this extensive review, we elucidate the importance of proteases and their role in drug development in various diseases with an emphasis on cancer. First, key proteases are introduced along with their function in disease progression. Next, we link these proteases as targets for the development of prodrugs and provide clinical examples of protease-activatable prodrugs. Finally, we provide significant design considerations needed for the development of the next generation protease-targeted and protease-activatable prodrugs.
Keywords: Protease, activatable probe, Alzheimer's disease, cancer, caspase, cathepsin, kallikrein, MMP, PSA, serine protease, aspartyl protease
1. Introduction
Proteases play a fundamental and essential role in many biological and pathological processes by the regulatory mechanism, proteolysis. Proteolysis is an irreversible regulatory mechanism and now known to selectively cleave specific substrates. Additionally, multimeric and multicatalytic proteases exist to degrade multiple intracellular proteins, called proteasomes, essential for biological processes [1]. The human degradome, which makes up a complete list of proteases synthesized by human cells, is made up of at least 569 proteases that are distributed into five broad classes (in order from greatest to least number): metalloproteinases, serine, cysteine, threonine, and aspartic proteases [2]. Serine, cysteine and threonine proteases are involved in covalent catalysis. The nucleophile of the catalytic site is part of the specified amino acid. Metalloproteinases and aspartic proteases perform non-covalent catalysis and the nucleophile is an activated water molecule [3].
By their highly controlled actions, proteases play influential roles in DNA replication and transcription, cell proliferation and differentiation, angiogenesis, neurogenesis, ovulation, fertilization, wound repair, stem cell mobilization, hemostasis, blood coagulation, inflammation, immunity, senescence, necrosis and apoptosis [4]. Therefore, deregulated modifications in proteolytic actions underlie many diseases like cancer and neurodegenerative and cardiovascular disorders. Because of proteases' ability to degrade extracellular matrices and proteins, they are strongly associated with cancer progression, specifically invasion and metastasis. Additionally, intracellular proteases, like lysosomal cysteine proteases, are involved in more protective mechanisms, like degrading many endocytosed proteins and foreign bodies.
With strong evidence of protease involvement in diseases, proteases serve an important role in drug development. Some therapies have been formulated to target and inhibit proteases and proteasomes that are dysregulated, especially for tumor suppression. Proteasome inhibitors have shown success in treatment of haematological malignancies and have therefore been tested as therapeutic agents in the clinic for over 10 years [5]. The first inhibitor, bortezomib has been used as a treatment for relapsed multiple myeloma and mantle cell lymphoma. However, the use of such expansive protease inhibitors have shown a lack of success overall. For a comprehensive review of clinical successes and failures of protease inhibitors see reference [3] and of proteasome inhibitors see reference [5]. Therefore, more specific protease-inspired therapies have been attempted. First, the design of recombinant forms of proteases can replace defective protease but are limited by the large doses necessary to achieve this effect. Second, gene-therapy approaches targeting protease genes can intrinsically improve proper protease activity. This approach has been show to work in lentiviral-mediated neprilysin gene transfer to block prostate cancer growth [6]. Third, an indirect approach is to hamper protease inhibitors to reduce protective (anti-tumor) proteases. In this way, proteases are activated that can sensitize cancer cells to drug treatments or induce apoptosis, as seen by Karikari, et al. for the inhibition of caspase inhibitors to induce apoptosis in pancreatic cancer [7]. Fourth, proteases can serve as biomarkers for diagnosis or prognosis of tumors. The presence of protective proteases can predict good clinical prognosis but their absence can indicate the need for different treatments. Dysregulated proteolytic activities can signify the progression of disease. An effective detection technique is the use of protease activatable probes [8-11]. Activatable probes, or molecular beacons, can signal, typically by fluorescence, the detection of proteases after the protease degrades the linkage between the dye and a quencher. In other words, the probe in its natural form gives off no signal; but only once a specific protease is present and degrades its specific substrate, the probe is activated. Using a similar approach, protease-activated prodrugs (PAPs) can be exploited to improve drug delivery to areas where protease expression, like in malignant tissues, is higher than in normal tissues.
Prodrugs are derivatives of drug molecules that can undergo a transformation by an enzyme, chemical or environmental stimuli to release the active parent drug in vivo [12]. Just as activatable probes, prodrugs in their native state are inactive forms of the drug but only after a stimuli release an active drug. In this way, prodrugs are an extremely efficient approach to increase selectivity and efficacy of chemotherapy, reducing the toxic effects on healthy cells. By chemical conjugation, prodrugs improve all pharmaceutical properties of the parent drug, such as its solubility, stability, permeability and distribution [12-13]. Since prodrugs can overcome major hurdles of drug formulations like poor solubility or instability, previously unsuitable drugs for clinical use can now be utilized. It has been estimated that about 5-7% of drugs currently approved worldwide are classified as prodrugs and that an even larger amount of prodrugs are approved every year [14]. Prodrugs are made up of the parent drug conjugated with a promoiety, like a polymer or peptide substrate via a cleavable linkage and/or a targeting moiety for specific delivery like an antibody or aptamer. Common functional groups used to modify prodrugs for superior properties, called promoieties, are listed in ref. [12]. In this review, we will focus on enzyme cleavable prodrugs, specifically protease-cleavable. Using this strategy, the prodrug only achieves its active form when the enzyme of interest, for which the promoiety is its substrate, cleaves it. Therefore, the drug is released at a specific location where the enzyme is overexpressed.
This review will focus on the promising development of PAPs. We will introduce the key target proteases and their involvement in specific diseases and further discuss the development of preclinical and clinical prodrugs utilizing these proteases. Finally, we will give a perspective on design considerations to develop advanced, efficient PAPs. The review will give the reader a background on the important proteases involved in various diseases, the design and outcomes of prodrugs used to target and treat these diseases and an outlook of how this field can advance further.
2. Target proteases
Aberrant protease signaling pathways lead to cancer as well as neurodegenerative, cardiovascular and pulmonary diseases. Specific substrates for upregulated proteases can be used as a promoiety for prodrugs, where the substrate is catalyzed to activate the prodrug into therapeutics. Currently, a large number of proteases have been identified as biomarkers for early diagnostic and prognostic markers, especially for cancer. Here we will discuss which proteases are particularly identified in pathological disorders and their substrates (Table 1 and 2). A protease substrate contains a recognition sequence for the protease to cleave. But in order to be used in prodrug design, the substrate must reach the same location as the protease [3].
Target proteases and diseases associated with overexpressed proteases
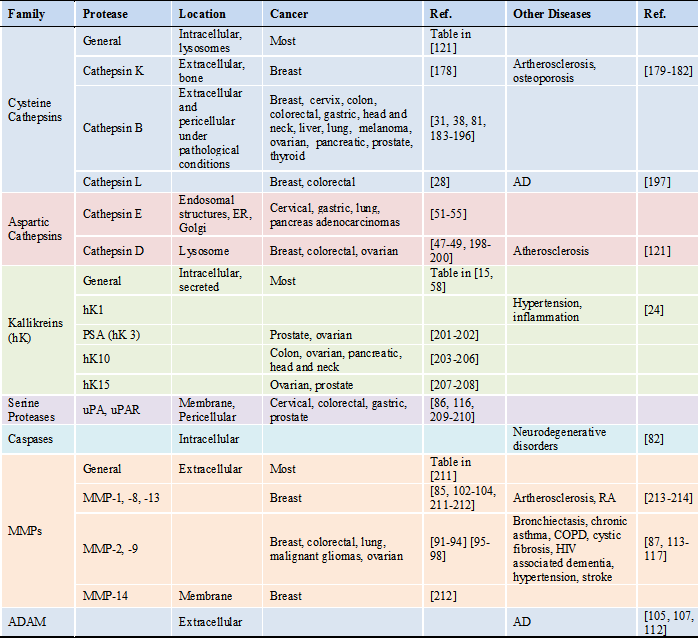
*Abbreviations: AD: Alzheimer's disease; ADAM: a disintegrin and metalloproteinase domain protease; COPD: chronic obstructive pulmonary disease; ER: endoplasmic reticulum; RA: rheumatoid arthritis
Protease-activatable prodrugs
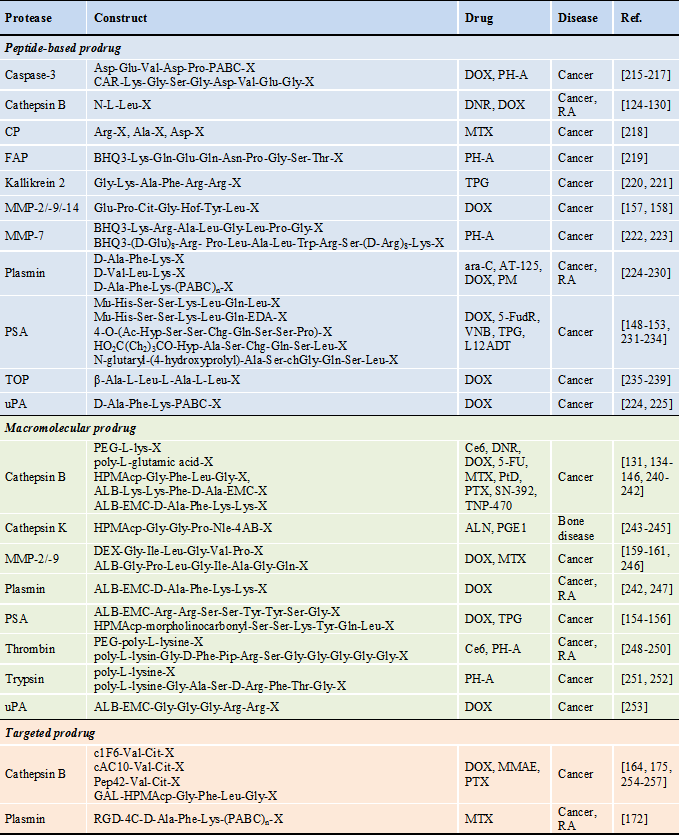
* Abbreviations : 4-AB: 4-aminobenzyl alcohol, ALB: albumin, ALN: alendronate, ara-C: 1-8-D-arabinofuranosylcytosine, AT-125: α-amino-3-chloro-4,5-dihydro-5-isoxazoleacetic acid, BHQ3: black hole quencher 3, cAC10: chimeric anti-CD30 monoclonal antibody, CAR: carotenoid, Ce6: chlorine e6, c1F6: chimeric anti-CD70 monoclonal antibody, chGly: cyclohexaglycyl, Cit: citrulline, CPT: camptothecin, CP: carboxypeptidase, dAc: desacetyl, DEX: dextran, DNR: daunorubicin, DOX: doxorubicin, DTX: docetaxel, EMC: ε-maleimidocaproic acid, FAP: fibroblast activation protein, 5-FU: 5-fluorouracil, 5-FudR: 5-fluorodeoxyuridine, GAL: galactose, Hof: homophenylalanine, HPMAcp: N-(2-hydroxypropyl)methacrylamide copolymer, Hyp: trans-4-hydroxyproline, L12ADT: 8-O-(12[L-leucinoylamino]dodecanoyl)-8-O-debutanoylthapsigargin, MTX: metotrexate, MMAE: monomethyl auristatin E, MMP: matrix metalloproteinase, Mu: morpholinocarbonyl, Nle: norleucyl, PABC: para aminobenzyloxycarboxyl, PEG: polyethylene glycol, Pep42: a cyclic 13-mer oligopeptide, PH-A: pheophorabide a, Pip: piperidine, PM: N,N-bis(2-chloroethyl)-p-phenylenediamine (phenylenediamine mustard), PSA: prostate-specific antigen, PTX: paclitaxel, PtD: platinum-based drug, PGE1: prostaglandin E1, RA: rheumatoid arthritis, RGD-4C: bicyclic Cys-Asp-Cys-Arg-Gly-Asp-Cys-Phe-Cys, SN-392: 10-amino-7-hydroxy camptothecin, TNP-470: O-(chloracetyl-carbamoyl) fumagillol, TOP: thimet oligopeptidase, TPG: thapsigargin, VNB: vinblastine, X: therapeutic agent or its analogue
In terms of cancer, dysregulated intracellular proteases can cause a loss of protective mechanisms and cause overgrowth and overexpression of extracellular proteases, which has been shown to result in tumor metastasis. Currently, it is believed that urokinase plasminogen activator (uPA), cathepsin B, and membrane-type matrix metalloproteinase (MMP) can initiate the activation of pro-MMPs. Then, extracellular matrix (ECM) degrading activities begin by extracellular serine proteases, like uPA, urokinase plasminogen activator receptor (uPAR), plasminogen, and MMPs to initiate cellular motility, invasiveness and a further cascade of tumor growth factors [15-17]. Recently, work has shown that certain extracellular proteases have anti-tumor properties [2]. Yet, overexpressed proteases have been identified in cancerous cells numerous concentration folds higher than in healthy cells [16]. Therefore the development of PAPs allows for specific, active drug delivery to cancer sites. Proteases are produced not only by tumor cells but also by multiple cell types recruited to the tumor site [18]. Therefore, the protease substrate selected in the prodrug can be catalyzed directly in the tumor microenvironment to reduce non-specific toxicity in normal proliferating and healthy cells. Here we introduce key proteases linked in cancer progression - cathepsins, kallikreins, uPA, uPAR, caspase and matrix metalloproteinases. Then we summarize their roles in other diseases.
2.1. Cathepsins
Cysteine cathepsins are primarily intracellular, lysosomal proteases that are responsible for protein turnover but have shown to be upregulated in cancer, often in early lesions [19]. Cathepsin-catalyzed substrate cleavage usually involves nucleophilic cysteine thiol, histidine, and an aspartate in the active site and cleavage is favored by acidic conditions [16]. However, specific substrates of cysteine cathepsins involved in pathological conditions need to be determined [19]. Overexpressed cysteine cathepsins are implicated in glioma [20-21], melanoma [22], cancer of the esophagus[23], stomach [24], colon [25], prostate [26-27], breast [28] and lung [29] and have been used as pathological biomarkers [30]. Active cysteine cathepsins are most commonly found in the acidic conditions of lysosomes with some exceptions. Cathepsin K is normally excreted to the extracellular space between osteoclasts and bone for bone remodeling [31-32]. Although intracellular proteases are less studied in their role in cancer compared to the more common ECM-degrading proteases like MMP and uPA, deregulation in their locations can initiate further extracellular proteolytic cascades [33-34]. For example, secreted cathepsin B and L hydrolyze type IV collagen, fibronectin [35], cell-adhesion proteins [36] and laminin [35] to enable tumor cell proliferation and instigate pro-MMPs. Cysteine cathepsins can also degrade collagen by intracellular proteolysis. Collagen can be taken up by macrophage and tumor cells via uPA and uPAR and then degraded in the lysosomes by cysteine cathepsins [19]. The most researched cathepsins, cathepsins B and L, are shown to be overexpressed via gene amplification, transcript variants expressed by tumor cells, transcription factors and post-transcriptional regulation [37]. Cathepsin B has been identified in membrane caveolae, outside of the lysosome, of human colon cancer cells and hypothesized to mediate further cell-surface and ECM degrading proteolytic events [38]. Recent work has shown that the permeabilization of the lysosomal membrane is implicated in cancer, mostly due to the discharge of many cysteine cathepsins from their innate location [39]. Once the cathepsins exit the cell and into the extracellular environment of tumors they can be activated by the acidic conditions found in the tumor microenvironment [40]. However, different mechanisms of activation are also possible. Cathepsin K is shown to be activated by glycosaminoglycans while cathepsins B, L and S are secreted in active form [41]. Interestingly, cysteine cathepsins, like cathepsins B, H, L, S, and K [42], translocate to the intracellular cytosol without membrane association to initiate apoptosis [43-44]. Some cathepsins have exhibited protective roles [2], like cathepsin L [45]. Therefore, prodrug activation by cathepsins may prove to be a good approach for anticancer drug delivery over therapies inhibiting proteases, which are relevant for normal cell function. Overexpression of the cysteine cathepsin is well-documented in many aggressive forms of cancer [46], thereby giving drug-cathepsin substrate conjugates an opportunity to be activated in cancerous environments and delivering chemotherapeutics directly to the site of need.
Cathepsin E and lysosomal cathepsin D are homologous aspartic proteases that play an equally important role in protein turnover in the cell as cysteine cathepsins. Aspartic proteases are made up of 14 families but all usually exist in highly acidic conditions, like in lysosomes or in the digestive tract. Cathepsin D is found in the lysosome and is implicated in prostate [47], breast [48] and colorectal cancer [49]. Cathepsin D expression and secretion parallel many of the lysosomal cysteine proteases mentioned above. Cathepsin E, on the other hand, is not found in lysosomes but in the intracellular space like endosomal structures, endoplasmic reticulum and Golgi apparatus, as well as in the plasma membrane and predominantly associated with immune cells [50]. Cathepsin E overexpression has been associated in several forms of cancer like human gastric [51], cervical [52], pancreatic [53-54] adenocarcinomas and lung carcinomas [55]. To date, most developed PAPs focus on cathepsin B because of their expression in the intracellular lysosomes.
2.2 Kallikrein and other serine proteases
Kallikreins are serine proteases that are traditionally linked to poor clinical prognosis of human carcinoma. The most popular is prostate specific antigen (PSA), also known as human kallikrein 3 (hK3) that serves as a diagnostic biomarker for prostate cancer. Serine proteases mediate substrate cleavage by histidine, serine and aspartic acid amino acids, which are usually close to one another and hKs additionally contain 10-12 cysteine residues. Tissue kallikrein gene (KLK) expression is highly regulated by sex-steroid hormones [15], like androgen regulation of KLK2 and KLK3 [56], but there are also post-translational regulations to control the irreversible protease action. Proteolytic activity of hKs can be inhibited by interactions with plasma globulin, serpins, tissue inhibitors, zinc ions [57] as well as by its own fragmentation [15]. The pro-enzyme can be activated by intracellular or extracellular hydrolysis or by other hKs [15]. Kallikreins are found in sweat, milk, saliva, seminal plasma and cerebrospinal fluid in humans because they are mainly secreted from epithelial cells in skin, breast, prostate, pancreas and brain. Under diseased conditions like cancer, hKs are dysregulated. In ovarian cancer, for example, twelve KLK genes are upregulated [15]. However, emerging data indicates that hKs can both promote and inhibit tumor progression and is most likely dependent on hormone balances as well as the tissue type. This also makes determining its substrate a challenge as it may be tissue specific [58]. Otin et al. discusses the tumor suppressing roles of hK3, hK8, hK9, hK10, hK13, hK 14 [2]. Yet, it is the overexpressed hKs in different cancers that can serve as viable targets for drug delivery.
A variety of hKs have been implicated in cancer progression like angiogenesis, invasion and metastasis, especially with its interaction with other serine proteases such as uPA and uPAR. But no hK has gotten as much attention as PSA (hK3) and hK2 in their role in prostate cancer. PSA can promote prostate tumor growth by numerous proposed pathways like the initiation of growth factors and proteolytic cascades to degrade the ECM. PSA and hK2 are identified as insulin-like growth factor (IGF) binding protein proteases [59]. When they degrade the binding protein in the tumor microenvironment, they in turn increase the bioavailability of IGF. The growth factor can then easily stimulate the growth of prostate cancer cells. Additionally, hK2 and hK4 can activate the uPA proteolytic cascade by inactivating the plasminogen activator inhibitor 1 [60-62]. Without the inhibitor, uPA can bind to its receptor, uPAR, and convert plasminogen to plasmin, a serine protease. Plasmin then leads to ECM degradation by activation of pro-MMPs and release of growth factors, like endothelial growth factor to promote angiogenesis [63]. Yet, the angiogenic properties of PSA are still debated; works show that PSA can activate tumor growth factor β (TFGβ) to promote angiogenesis [64] but also can block fibroblast growth factor 2 and inhibit angiogenesis [65]. Furthermore, hKs can directly activate ECM degrading MMPs, like type IV collagenases [66]. The uPA-uPAR pathway is a well studied mechanism of cancer progression [67-69], and offers many opportunities to target proteases like PSA, hK2, uPA, plasmin and MMPs, for potential anti-cancer therapy. Elevated levels of uPA and its related serine proteases have been observed in various cancers like colorectal [70], gastric [71], prostate [72], and cervical cancer [73]. Protease-activated receptor (PAR) signaling is another pathway that has been implicated in various cancers. PAR is a G-protein coupled receptor that is expressed in many cancer cells and cells in the microenvironment of a tumor. It can be activated by serine proteases like trypsin [74] and thrombin [75] by cleavage of its extracellular segment and transmit intracellular signals to stimulate cancer cell growth [76-78]. A comprehensive review on hKs and its role in cancer with tabulated clinical implications are found in references [15] and [58], respectively.
2.3 Caspases
Caspases are intracellular cysteine proteases that are fundamental in programmed cell death - apoptosis [79]. They are one of the more specific and efficient proteases [80]. Cancer cells can suppress caspase expression to circumvent apoptosis in order to proliferate. Yet, some aggressive tumors undergo spontaneous apoptosis [81]. Inhibitors of apoptosis proteins are regulators of the caspase cascade, specifically caspase 3 and 7, and have a large role in cancer [82]. Additionally, genetic factors such as deletion of caspase 8 and 10 genes and mutations in caspase 3, 5, 6 and 7 are associated with human tumors [2]. Caspases can serve as interesting proteases to study the mechanisms of cell death and a key hallmark of cancer, but its suppression rather than overexpression is what governs cancer progression.
2.4 Matrix metalloproteinases
The most established extracellular and pericellular proteases identified in cancer metastasis and many other diseases are matrix metalloproteinases (MMPs). MMPs make up a versatile family of proteases that control many physiological functions [83]. But as seen in the previous sections, MMPs are activated by many other proteases and serve as the proteolytic endpoint for tumor progression. As their name suggest, MMPs modulate and regulate the ECM. They are associated with different stages of cancer invasion and metastasis by releasing cancer cells to spread and provide room for cancer cells to invade. Additionally, ECM remodeling is involved in releasing many important proteins such as cytokines, growth factors and chemokines, and in turn MMPs regulate many of these proteins. MMPs can be identified by the presence of Zn2+ ions at the catalytic center that is coordinated by three histidine residues and a serine residue. They are mainly excreted as an inactive proenzyme and activated by a cysteine switch mechanism to expose the catalytic zinc pocket [84]. The main activation system is by the removal of the prodomain of MMPs. As described above, proteases such as plasmin, serine proteases and other MMPs can proteolytically remove the prodomain of MMPs to activate them in the extracellular space [85]. Consisting of at least 23 known human enzymes, the MMP family can be divided into five sub-groups: Collagenases: MMP-1, -8, -13; Gelatinases: MMP-2, -9; Stromelysins: MMP-3, -10, -11; Membrane-type MMPs (MT-MMPs): MMP-15, -16, -17, -24, -25 and the remaining proteases make up a varied group. The family is versatile and some MMPs have shown protective effects like MMP-8, -12,-26 and tumorigenic effects like MMP-9, -11 and more.
Additionally MMPs have been implicated in regulatory functions like survival, angiogenesis, inflammation and signaling as well as other pathologies like multiple sclerosis, stroke, Alzheimer's disease, lung emphysema, arthritis and infections of the central nervous system [86]. The versatility in MMPs comes from their ability to cleave multiple types of elements. For example the gelatinase MMP-2 can cleave gelatin, type I, IV and I collagens, elastin, and vitronectin [87]. MMP-9 can also degrade similar substrates as MMP-2, except collagen I, and release vascular endothelial growth factor to induce neovascularization. Gelatinases, MMP-2 [88] and -9 [89], are implicated in angiogenesis. Many papers have related gelatinases to tumor metastasis and angiogenesis because of their ability to degrade the vascular basal membrane to induce growth of new blood vessels as well as release growth factors responsible for tumor growth [90]. Increased expression of gelatinase has been observed in many various cancers such as breast [91-93], ovarian [94], lung [95] and colorectal [96] as well as malignant gliomas [97-98]. MMP-2 and -9 can be activated by MMP-7, a protease that by itself is associated with cancer cell growth because of its ability to cleave insulin-like growth factor-binding protein. In addition, MMP-7 is a special MMP because it can be produced by cancerous cells [99], which in turn further induces cancer invasiveness by activating other metastasis associated proteases [100]. Although MMP-14 is a membrane-type MMP, it is also associated with similar substrates as gelatinases and release of vascular endothelial growth factor. Furthermore, it aids in decreasing cell adhesion by cleaving membrane proteins like integrins, E-cadherin, and cell surface proteoglycans. MMPs make up a versatile family with broad substrate specificity that are strongly associated with tumor progression. Although many treatments involving MMP inhibitors have historically failed [101], increased MMP expression at tumor sites can play an important role in drug delivery. Excellent reviews on MMPs and their clinical implications are found in references [85, 102-104].
2.5 Proteases in neurodegenerative, pulmonary and cardiovascular diseases
As seen above, irregular protease signaling leads to dysregulation of homeostasis and can lead to cancer. Since proteases are regulatory enzymes and are required for normal cell function, their alterations can also lead to disorders beyond cancer. Proteases have been implicated in diseases such as neurodegenerative, pulmonary and cardiovascular.
Proteases are getting increasing attention in their role in Alzheimer's disease (AD), a neurodegenerative disorder that affects approximately 2% of the population in industrialized countries [105]. The pathological phenotype of AD is characterized by amyloid plague build up and neurofibrillary tangles in various areas of the brain [106]. These plagues are believed to occur due to elevated levels of neurotoxic amyloid beta protein that can damage synapses and neuritis [105]. The proteolytic processing of the amyloid beta protein from amyloid precursor protein (APP) is the predominant abnormality in AD [105]. APP is a large glycoprotein that can be cleaved and activated to amyloid beta protein by two aspartic proteases, β-secretase at the N-terminus and γ-secretase at the C-terminus. Additionally, α-secretase, a disintegrin and metalloproteinase domain protease (ADAM), is known to cleave APP within the important amyloid beta protein sequence [105]. The ADAM family (made up of 21 known human ADAMs [107]) has adhesive and proteolytic properties, which can aid in mediating interaction with other molecules as well as signaling. This family is an emerging field and more research is needed to implicate these MMP-type proteases in various pathologies. An inhibitory and immunization treatment has shown that a removal of amyloid beta protein deposits can recover cognitive defects associated with AD [108-109]. Therefore, proteases that are directly associated with amyloid plaque formation in AD can serve as promising therapeutic targets. Several proteases have shown to be elevated in AD, but their use as diagnostic or prognostic biomarkers are still in preliminary stages [110-111]. Additionally, elevated levels of other MMP types have been seen in AD [112] along with varying neurological conditions like stroke (MMP-9) [113-114] and HIV associated dementia (MMP-2, -7, -9) [115]. Beyond MMPs, caspase overexpression has also been implicated in neurodegenerative disorders [82].
Because of MMPs' role in ECM remodeling and angiogenesis, their dysregulation can lead to pulmonary and cardiovascular disorders. The gelatinase family of MMPs (MMP-2, -9), are implicated in many fibrous pulmonary diseases like chronic asthma [116], cystic fibrosis, bronchiectasis, and chronic obstructive pulmonary disease (COPD) [87]. This may be due to dysregulation of ECM remodeling leading to a thickening of the basement membrane. Protease signaling pathway is involved in the blood coagulation cascade and deregulation of proteases in the pathway can lead to cardiovascular disease. The specific roles that gelatinases play in angiogenesis has lead others to hypothesize their role in vasoconstrictive properties and disorders such as hypertension [117]. Angiotensin-converting enzyme (ACE) is a metalloproteinase that is central to the blood coagulation cascade by converting angiotensin I to angiotensin II, required for angiotensin receptor activation and vessel constriction [118]. The use of ACE inhibitors have been used on the market for over 20 years to treat hypertension, heart failure and heart attack [119]. Outside of the MMP family, hK1 is implicated in kinin production, which can mediate many biological functions like inflammation and hypertension [120]. Additionally, cathepsins have been found to be relevant biomarkers in various diseases beyond cancer, like lung and brain diseases and atherosclerosis [121]. Further reading on the use of proteases as biomarkers and targets in diseases outside of cancer can be found in reference [3] and references within.
Proteases are important regulatory molecules in our body. Their proper function is needed for appropriate protein turnover and signaling. However, when these proteases are unbalanced their irreversible actions can greatly affect normal cellular function and lead to pathologies like cancer and neurodegenerative, pulmonary and cardiovascular diseases. Many studies have suggested the use of different proteases as biomarkers and prognostic markers for such diseases. Furthermore, protease inhibitors have been developed to stabilize protease function. Here we briefly introduced key human proteases that have been found to be overexpressed in various pathologies, specifically cancer. Furthermore, we describe how they are deregulated and list the proteases commonly implicated in various forms of disorders (Table 1). We hope that this protease introduction can serve to educate readers on potential drug targets and how their overexpression can be utilized for specific drug delivery by PAPs.
3. Protease-Activatable Prodrugs
Prodrugs are chemically-caged derivatives of therapeutic agents that can be transformed by an enzyme, chemical or physiological stimuli to release the active parent drug in vivo [12]. From the time the term prodrug was first coined by Adrien Albert in 1958 [122], a wide variety of prodrugs has been extensively investigated. The prodrug approach generally aims to i) improve the physicochemical properties of parent drug molecules, such as low chemical stability or poor water-solubility; ii) improve their pharmacokinetic/ pharmacologic properties, like insufficient oral absorption, unwanted rapid metabolism or low selectivity; and also iii) reduce undesirable irritation, pain or non-specific toxicity to normal tissues [12-13].
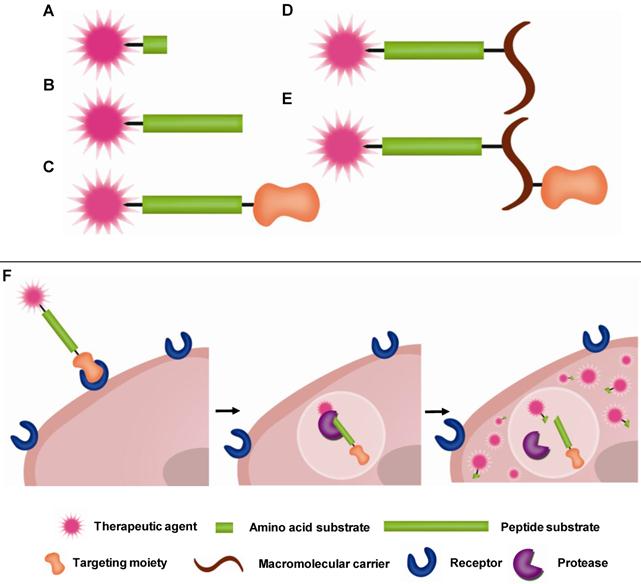
Prodrugs have minimal pharmacological effects in its native state. However, pharmacological activity is recovered when they are transformed into the active parent drug or its derivative by disease-specific or environmental stimuli. Among various targets of prodrugs, proteases are considered an important target because proteases are highly involved in diseases as described in Section 2 above. For PAPs to be effective, the linkage between the drug and promoiety should remain stable in the bloodstream but degrade once it reaches its target protease. Peptide sequences, which are stable in the blood but highly specific towards target proteases, have been developed. To facilitate delivery of therapeutic agents to the target site, targeting moieties (e.g. antibody) or macromolecular carriers (e.g. albumin or polymers) are coupled to the peptide-based prodrugs. Therefore, in most cases, prodrugs are composed of two major components: i) an active therapeutic agent and ii) peptide substrates and/or macromolecular carriers, i.e. promoiety. To endow prodrugs with target-specificity, they can also be modified with additional iii) targeting ligands [12, 123]. In other words, the parent therapeutic agent is caged by the promoiety and/or modified with the targeting ligand via target-specific, cleavable linkages (Fig. 1). Prodrugs activated by proteolysis have attracted much attention in the field of drug discovery and development. In this section, representative PAP approaches including peptide-based prodrugs and macromolecular prodrugs will be briefly exemplified.
Schematic diagram of (A-E) prodrug constructs and (F) hypothetical pathway of prodrug activation
3.1. Cathepsin-Activatable Prodrugs
Proteases are known to selectively cleave specific substrates, e.g. an amino acid or peptide sequences. The fact that proteases can recognize and degrade specific substrates sheds light on the development of prodrugs that can be activated at target disease sites. Among proteases, cathepsins are well-known proteases that are upregulated in several tumor tissues (Table 1). Overexpressed cathepsins are considered important biomarkers for cancer and can serve as key targets for prodrugs to induce anticancer drug release to tumor tissues or inside tumor cells.
Initial works on protease prodrugs focused on conjugating single amino acids or dipeptides onto common cancer therapeutic drugs like daunorubicin (DNR) or doxorubicin (DOX) to study their increased therapeutic effects.
For example, amino acid- and dipeptide-daunorubicin conjugates (L-Leu-DNR, Val-DNR, Ile-DNR, Ala-Leu-DNR and Leu-Leu-DNR) were prepared to investigate the effect of modification of DNR on its toxicity and antitumor efficacy toward murine L1210 leukemia [124]. When intravenously administrated into L1210 leukemia xenografts, Leu-DNR, Ala-Leu-DNR and Leu-Leu-DNR exhibited superior results on the suppression of tumor growth to DNR and on the survival rate. Leu-DNR accumulated in the heart muscle much less than DNR at equitoxic doses in the rabbit, and therefore exhibited reduced cardiotoxicity, a major side effect of anthracycline derivatives. Similarly, Leu-DOX prodrugs were also developed to lower the cardiotoxicity and improve the therapeutic index of DOX [125-130]. Compared to DOX alone, Leu-DOX had higher antitumor efficacy together with lower toxicity in various tumor-bearing mice including human ovarian, breast and lung carcinoma models. Notably, it was suggested that superior antitumor efficacy of Leu-DOX to DOX is due to their enhanced hydrophobicity and also to their proteolysis in tumor tissue by proteases such as cathepsins, which are known to be highly expressed in several tumor tissues (Table 1). However, the studies did not show any direct evidence of proteolysis of the leucyl group by cathepsins.
Follow up studies investigated the peptide substrates, proteolysis mechanisms and prodrug delivery agents like albumin and macromolecules on cancer therapeutic effects. DNR or DNR derivatives (DNR, Leu-DNR, Ala-Leu-DNR, Leu-Ala-Leu-DNR or Ala-Leu-Ala-Leu-DNR) were further conjugated onto albumin (ALB) [131]. Remarkably, when the DNR prodrugs were incubated with lysosomal enzymes, active DNR molecules were released from ALB-Leu-Ala-Leu-DNR and ALB-Ala-Leu-Ala-Leu-DNR but not from ALB-DNR, ALB-Leu-DNR or ALB-Ala-Leu-DNR. However, in the presence of 95% calf serum, ALB-Leu-Ala-Leu-DNR and ALB-Ala-Leu-Ala-Leu-DNR were over 97% stable after 24 h incubation. Likewise, in the intraperitoneal L1210 leukemia model, the ALB-tri-/tetra-peptide-DNR prodrugs were proven to be superior to mono-peptide linkers, with improved anticancer activity and prolonged survivals. From the results, it was proposed that the ALB-tri-/tetra-peptide-DNR prodrugs internalize into leukemia cells and release parent DNR molecules inside the cells because of proteolysis of the peptide substrates by lysosomal proteases, like cathepsin B.
Besides albumin, macromolecules have been vigorously investigated as potent drug-delivery carriers. Owing to the abnormal characteristics of tumors like leaky blood vessels and lack of lymphatic drainage, macromolecules are known to preferentially accumulate in tumor tissue and remain for a prolonged period of time, the so called enhanced permeable and retention (EPR) effect [132-133]. In an effort to improve blood stability and tumor targetability of prodrugs, macromolecule-based prodrugs have been developed. As an example, N-(2-hydroxypropyl) methacrylamide copolymer (HPMAcp) was exploited as a macromolecular carrier to deliver therapeutic agents or their derivatives. Onto the HPMAcp backbone, DOX was conjugated via a cathepsin B-specific tetrapeptide (Gly-Phe-Leu-Gly) linker [134-140]. After intravenous administration, the macromolecular prodrug circulated approximately 15-times longer than free DOX. Furthermore, the prodrug exhibited 100-times less heart uptake, which resulted in decreased toxicity and improved therapeutic efficacy compared with free DOX. Interestingly, the pharmacokinetics and amount of released DOX was found to be correlated with both lysosomal activity of cathepsin B and vascular properties (Fig. 2) [135].
(A) Chemical structure of PK1 (HPMAcp-Gly-Phe-Leu-Gly-DOX). DOX concentrations in MAC15A (◯) and MAC26 (□) plasma (dotted line) and tumor (solid line) after administration of 10 mg/kg DOX i.v (B) or 490 mg/kg PK1, i.v. (40 mg/kg DOX equivalent) (C). Antitumor effects of DOX or PK1 against MAC26 tumors (D), MAC15A (E and F). ◯, PK1 (40 mg/kg); □, PK1 (20 mg/kg); △, PK1 (10 mg/kg); ▽, dox (10 Mg/kg); ◇, controls. Adapted with permission from Loadman et al [135]. Copyright 1999, American Association for Cancer Research
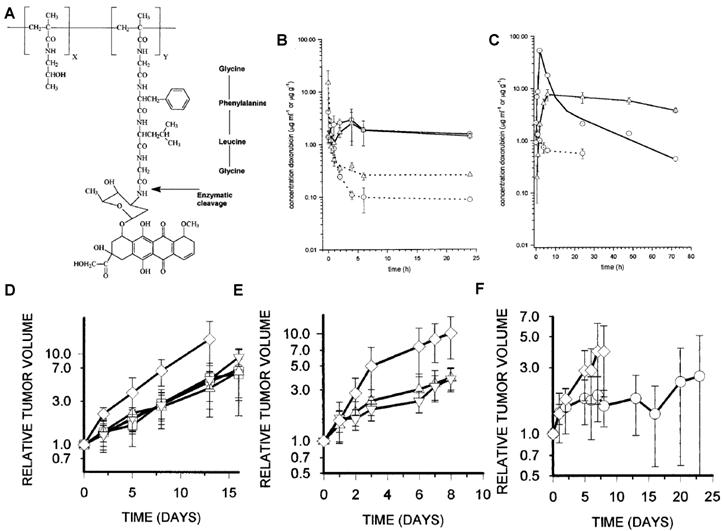
This study implies that highly-activated cathepsin B can induce vascularization around tumors and accelerate DOX release. Most importantly, the study exemplifies that a protease can enhance the therapeutic efficacy of a macromolecular prodrug. However, the study could not elucidate whether the excellent therapeutic efficacy was attributed to DOX release activated by cathepsin B or a higher accumulation of the prodrug through highly vascularized tumor vessels. With the apparent enhanced drug efficacy, HPMAcp was also conjugated to DNR [141], 5-fluorouracil (5-FU)[142], platinum (II) [143] and O-(chloracetyl-carbamoyl) fumagillol (TNP-470) [144] via the same peptide linker (Gly-Phe-Leu-Gly). Just as the DOX conjugate, these formulations also showed significant therapeutic activity to free drugs.
A significant example of cathepsin B-activatable prodrugs utilized a polymerized substrate to reverse cancer progression, emphasizing the use of macromolecular protease substrates for efficient drug delivery and effective therapy. The cathepsin B-activatable prodrug, poly(L-glutamic acid) conjugated with paclitaxel (PTX), exerted impressive antitumor activity in OCA-1 ovarian carcinoma models [145]. Notably, a single intravenous injection of the macromolecular prodrug resulted in the disappearance of 13762F adenocarcinoma implanted in rats and OCA-1 ovarian carcinoma inoculated in rats. The excellent antitumor activity possibly resulted from its prolonged half-life in plasma and preferential accumulation of released PTX in tumor tissue. In a follow-up study, the metabolic pathway of the macromolecular prodrug, poly(L-glutamic acid)-PTX was investigated [146]. The study demonstrated that it can be metabolized into mono- or di-glutamyl-PTX form after it internalizes into cancer cells. The release rate of PTX is correlated with the proteolytic activity of cathepsin B. The metabolism of poly(L-glutamic acid)-PTX was inhibited in cathepsin B deficient mice or by administration of cathepsin B inhibitor in mice.
Although cathepsin B is a common protease target for prodrugs (Table 2), its proteolytic activity needs to be examined more closely. For example, previous works have shown that cathepsin B can be released to the extracellular space in active form under certain pathologies (Section 3.1). This may cause premature activity of the prodrug and reduce its efficacy inside tumor cells. However, cathepsin activatable prodrugs are exemplary in their roles for therapeutic delivery because these PAPs are shown to be much more efficient than equitoxic doses of free drugs. It is important to consider additional molecules for proper drug delivery, especially when the protease of interest in found intracellularly. In the above examples, the use of macromolecular substrates or the addition of albumin greatly increased the delivery of the prodrug to its target site.
3.2. PSA-activatable prodrugs
The peptide sequence (Ser-Ser-Lys-Leu-Gln) is highly specific for PSA [147]. The peptide substrate has been coupled with DOX to develop a PSA-activatable prodrug [148-149]. The resulting prodrug (morpholinocarbonyl(Mu)-Ser-Ser-Lys-Leu-Gln-DOX) exerted severe cytotoxicity not only to PC-82 human prostate cancer cells, that secrete high levels of PSA, but also to LNCaP human prostate cancer cells, that secrete 30 fold less active PSA. Yet following 72-h incubation with LNCaP cells, over 90% of the prodrug was activated to release Leu-DOX and killed 50% of cells at a low concentration (IC50 = 70 nM). Importantly, the drug did not show detectable toxicity to PSA-nonproducing cells, TSU human prostate cancer cells, which indicates great sensitivity of the PSA-activatable prodrug. When the prodrug was given by continuous infusion into PC-82 human prostate cancer xenografts at a high dose (four times higher than the 100% lethal dose of free DOX), no noticeable toxicity was observed. Moreover, an intraperitoneal injection of the prodrug suppressed tumor growth up to 57% in tumor weight compared with the non-treated control group. The continuous infusion with the prodrug also led to similar suppression activity on tumor growth (47% lower than control group in tumor weight) at 60% lower dose than the dose administrated intraperitoneally. In the same way, thapsigargin (TPG) [150-151] or 5-fluorodeoxyuridine (5-FudR) [152] were coupled with the same PSA-specific peptide substrate. The PSA-activatable prodrugs also exhibited remarkable inhibitory effects on tumor growth without discernible toxicity after intravenous administration.
Another peptide substrate (N-glutaryl-(4-hydroxyprolyl)-Ala-Ser-chGly-Gln-Ser-Leu) specific for PSA was also discovered and exploited for the development of PSA-activatable prodrugs (Fig. 3) [153]. In this research, DOX was covalently incorporated with PSA-specific peptide sequence. The resulting conjugate was hydrolyzed specifically by PSA and transformed into cytotoxic form of DOX, leucine-DOX and DOX. Like other prodrugs, it was much less toxic than free DOX to PSA-negative cells; however, it led to remarkable reduction in tumor burden in human prostate cancer xenografts inoculated with either LNCaP or CWR22.
(A) Chemical structure of L-377,202. Changes in concentrations of leucine-DOX and DOX in tumor tissue (B) and heart tissue (C). ◆, DOX from L-377,202; ●, leucine-DOX from L-377,202; ◇, DOX from administration of conventional DOX. (D) Reduction in LNCaP tumor weights in nude mice treated with DOX or L-377,202. Adapted with permission from DeFeo-Jones et al [153]. Copyright 2000, Nature Publishing Group
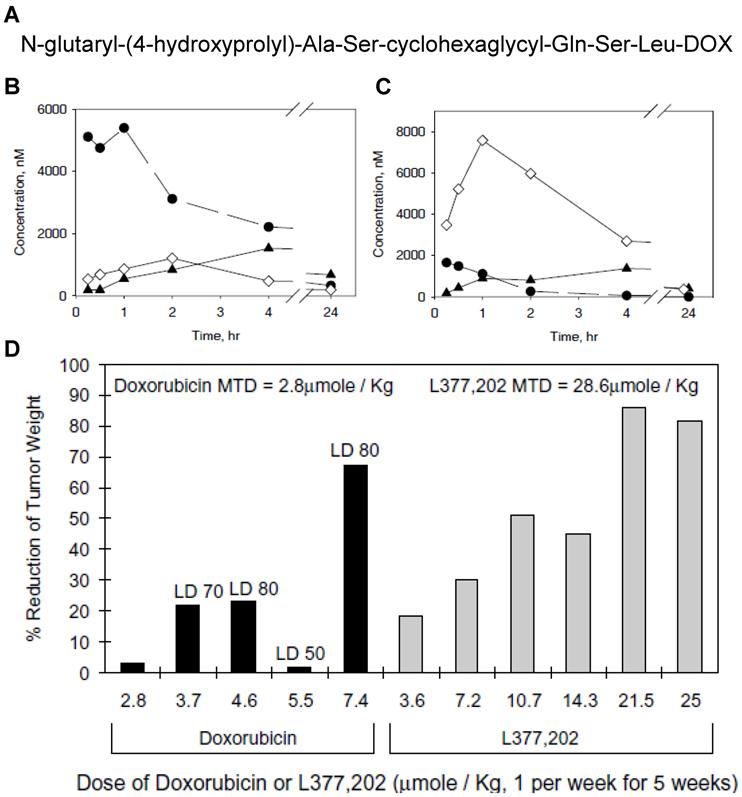
Macromolecules, such as ALB and HPMAcp, were also combined with therapeutic agents like TPG or DOX via PSA-specific substrates. In a macromolecular prodrug system, the same peptide sequence previously described (Mu-Ser-Ser-Lys-Leu-Gln), was used as a linker to conjugate TPG derivative named L12ADT onto the HPMAcp backbone [154]. In another system, DOX was coupled with ALB via a similar peptide substrate (ε-maleimidocaproic acid (EMC)-Arg-Arg-Ser-Ser-Tyr-Tyr-Ser-Gly or -Arg) [155-156]. PSA selectively cleaves the substrate, converting inactive prodrugs into active forms of parent drugs or their derivatives. As an example, ALB-EMC-Arg-Arg-Ser-Ser-Tyr-Tyr-Ser-Arg-DOX exerted enhanced suppression of tumor growth by 62% in tumor size over free DOX. Additionally, the prodrug showed no sign of detectable toxicity after administration of equimolar DOX into a prostate cancer model orthotopically implanted with LNCaP LN prostate cancer cells. More interestingly, administration of the prodrug reduced the metastatic burden in the lungs by 50%.
3.3. MMP-activatable prodrugs
MMPs are representative target protease for prodrugs that may arguably be the most studied proteases. Because they are found in the extracellular and pericellular areas of the cell, MMPs can serve as excellent protease targets for prodrug delivery to the tumor microenvironment. Although a versatile family, specific substrates have been discovered that can be cleaved by a limited amount of MMPs. In particular, the progression of various malignancies can be characterized by the overexpression of MMP-2 [2].
Like the other prodrugs described above, MMP-activatable prodrugs have been developed by chemical conjugation of MMP-cleavable peptide substrate (Glu-Pro-Cit-Gly-Hof-Tyr-Leu) to DOX [157-158]. The peptide substrate was specifically cleaved by MMP-2, -9, -14. Interestingly, it was found that the peptide substrate was initially metabolized by neprilysin, a membrane-bound metalloproteinase, when they were incubated with an MMP-expressing fibrosarcoma cell line, HT1080. In HT1080 xenografts, the substrate was preferentially metabolized in the tumor tissue, resulting in a 10-fold higher accumulation of released DOX at the tumor than that in the heart. When this MMP-substrate prodrug was administered to mice, 80% of mice were cured and no significant toxicity was detected, such as weight loss or marrow toxicity.
Since MMP-2, -9 specifically cleave the peptide sequence Gly-Pro-Leu-Gly-Ile-Ala-Gly-Gln, the peptide substrate was also used to design a DOX prodrug that can be specifically activated by MMP-2 [159]. ALB was incorporated with the peptide-DOX conjugate as a carrier. The intact albumin-peptide-DOX conjugate was not significantly toxic. However, following addition of MMP-2, the albumin-peptide-DOX conjugate released tetrapeptide-DOX and was further transformed into the parent anticancer drug, DOX. The prodrug was 4-fold less toxic than free DOX in vivo, whereas it showed superior anticancer effect to DOX at an equitoxic dose.
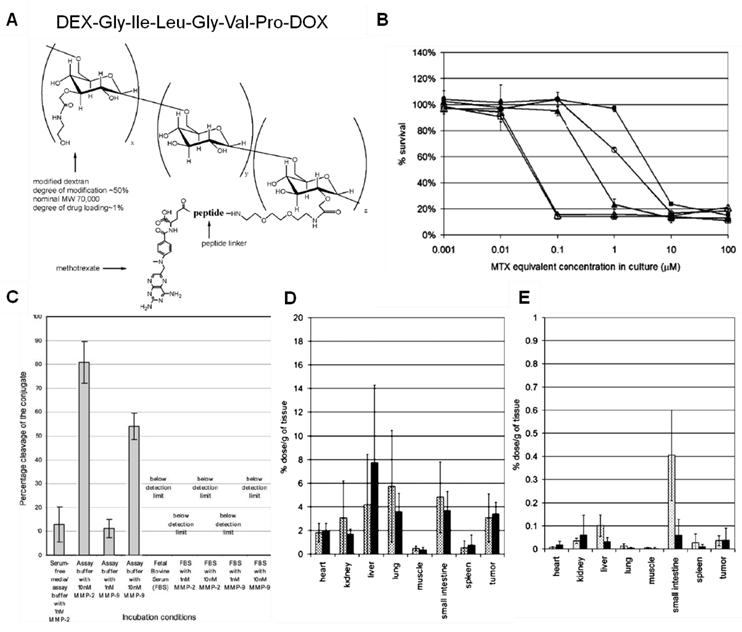
Just as tumor-specific chemotherapeutics, MMP-activatable prodrugs can be improved by utilizing a macromolecular carrier. For example, metotraxate (MTX) was conjugated onto a macromolecular carrier, dextran (DEX) via an MMP-specific peptide linker (Pro-Val-Gly-Leu-Ile-Gly) (Fig. 4) [160-161]. This macromolecular prodrug shows specificity to MMP-2 and -9 and releases peptidyl-MTX when incubated with targeted proteases; however, it remained stable in serum. This study illustrated that macromolecular prodrugs preferentially accumulate in tumor tissues by the EPR effect more than free MTX and then release MTX derivatives close to or within tumor tissues. Importantly, the amount of tumor accumulation in vivo was not significantly correlated with the specificity of peptide substrates, even though the peptide sequence was proven to be highly specific in vitro. The unexpected results suggest that since there is a great number of endo- and exopeptidases in the human body, the in vivo properties, such as stability in the bloodstream and the specificity to the target protease, should be carefully considered.
(A) Chemical structure of the MTX-PVGLIG-DEX. (B) Cytotoxicity of MTX and different types of MTX-peptide analogs: MTX (△), MTX-G (□), MTX-GI (◯), MTX-GIV (▲), and MTXPVG (■). (C) Stability of the conjugate (MTX-PVGLIG-DEX) in various media conditions. Tissue distribution of MTX equivalent in mice receiving MTX-PVGLIG-DEX (D) and free MTX (E), at 5 h (▩) and 24 h (■) post injection. Adapted with permission from Chau et al [160-161]. Copyright 2004, American Chemical Society and Copyright 2006, John Wiley & Sons, Inc
MMP overexpression is linked to numerous cancers (Table 1). Their role in ECM remodeling is required for cancer cells to metastasize and invade different locations. Therefore MMP-specific prodrugs can especially target metastasized forms of cancer. A drawback to MMP targeting is their cleavage of various substrates by multiple types of MMPs. The exemplified prodrugs in this section utilized unique peptide sequences but still targeted more than one MMP. Although this may be a limitation for molecular biology in order to identify a specific form of MMP, multiple-MMP targeted prodrugs can be used to deliver anticancer drugs to especially harmful forms of cancer.
3.4. Targeted protease-activatable prodrugs
As we reviewed in the previous sections, PAPs are generally inactive but become pharmacologically active when exposed to target proteases. Owing to its improved stability and target-specific drug release, prodrugs have a wide therapeutic window when compared to free chemotherapeutic agents. However, in terms of the targeting mechanism, most conventional prodrugs are still limited because they mainly rely on passive accumulation pathways, e.g. EPR effect. In an effort to bestow disease-specific targetability upon prodrugs, various targeting moieties, e.g. antibody, peptide or oligonucleic acid molecules, have been employed.
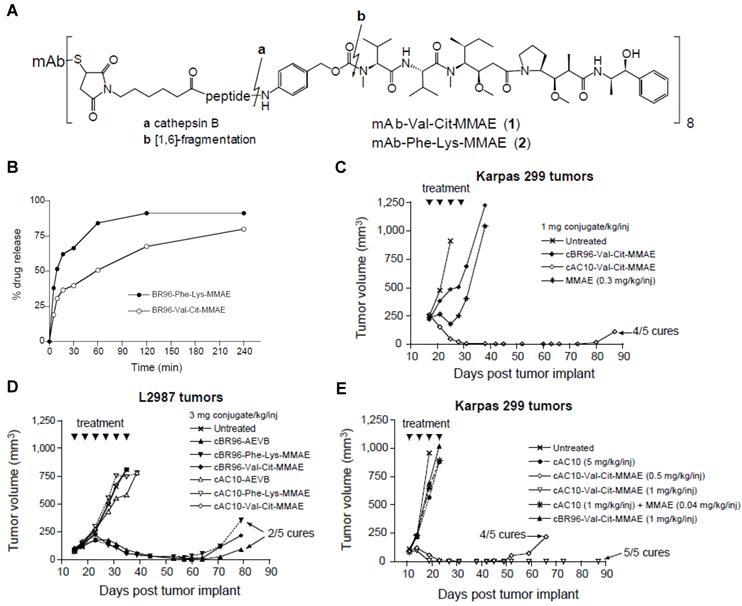
For instance, therapeutic agents have been covalently conjugated with disease-specific antibodies via protease-cleavable linkages to incorporate both a tumor-specific recognition site and a tumor selective enzymatic activation sequence in a single prodrug platform. The antibody-drug conjugates (ADCs) have come a long way and shown significant therapeutic efficacy against various diseases [162-163]. An interesting study demonstrates the development and in vivo applications of a cathepsin activated ADC. A synthetic dolastatin 10 analog, monomethyl auristatin E (MMAE), was incorporated onto an antibody cAC10 with specificity for CD30 on hematological malignancies via a dipeptide substrate (Val-Cit) as a cathepsin B-specific linker (Fig. 5) [164]. The mAb-Val-Cit-MMAE conjugate is highly stable in plasma but released active MMAE in lysosomes of CD30-positive tumor cells. The ADC showed higher specificity in vitro, lower toxicity in vivo and improved antitumor effect than control ADCs containing hydrazone linker. Notably, with the use of this ADC, the therapeutic index significantly increased up to 60-fold leading to its extensive investigation in the clinic. Recently, the conjugate, named Adcetris (brentuximab vedotin) has been approved by The U.S. Food and Drug Administration (FDA) for the treatment of Hodgkin lymphoma (HL) and a rare lymphoma known as systemic anaplastic large cell lymphoma (ALCL). [165]
(A) Chemical structure of mAb-drug conjugates. (B) Hydrolysis of cBR96-Phe-Lys-MMAE and cBR96-Val-Cit-MMAE (eight drug-mAb combinations) with human cathepsin B. In vivo therapeutic efficacy of the conjugates in immunocompromised mice with subcutaneous human tumor xenografts. (C) Athymic mice with subcutaneous L2987 human lung adenocarcinoma tumors (cBR96 Ag+, cAC10 Ag-) were treated with the conjugates. (D) SCID mice with subcutaneous Karpas 299 human ALCL tumors (cAC10 Ag+, cBR96 Ag-) were treated with MMAE or with the mAb-Val-Cit-MMAE. (E) SCID mice with Karpas 299 tumors were treated with cAC10, cAC10 + unconjugated MMAE, cAC10-Val-Cit-MMAE or cBR96-Val-Cit-MMAE. Adapted with permission from Doronina et al [164]. Copyright 2003, Nature Publishing Group
Another example of targeted PAPs is macromolecular prodrugs, PK1 and PK2. As we described, HPMAcp-Gly-Phe-Leu-Gly-DOX, called PK1, is a macromolecular prodrug targeting cathepsin B [134-135, 137-140]. When compared with free DOX, PK1 showed prolonged circulation, less cardiotoxicity and improved therapeutic activity. In addition, it was proposed that the excellent therapeutic efficacy of PK1 is due to the passive accumulation into the tumor tissue by the EPR effect and the target-specific drug release by lysosomal degradation of the peptide linkage. PK2 was further modified with galactose (GAL) to facilitate liver targeting, GAL-HPMAcp-Gly-Phe-Leu-Gly-DOX. After intravenous injection of PK2, acute toxicity decreased three times over free DOX without noticeable signs of cardiotoxicity. With these promising results, PK1 and PK2 are now under clinical trials [166-171].
In addition to mAb or GAL, a bicyclic peptide sequence, RGD-4C (Cys-Asp-Cys-Arg-Gly-Asp-Cys-Phe-Cys) was exploited as a targeting moiety [172]. RGD-4C selectively binds αvβ3 and αvβ5 integrins, which are known to highly overexpress on invading tumor endothelial cells. The targeting peptide was coupled with DOX via a plasmin-specific peptide substrate (D-Ala-Phe-Lys). As we mentioned, plasmin is highly associated with tumor progression (Section 2.2). The resulting conjugate exerted cytotoxicity against plasmin-positive HT1080 fibrosarcoma cells but not to endothelial cells, showing significant specificity to the target protease plasmin. Likewise, a cyclic 13-mer oligopeptide named pep 42 was exploited as a targeting moiety to develop tumor targeted PAPs [173]. Pep 42 can bind to glucose-regulated protein GRP78 and facilitate cancer cell-specific uptake. Anticancer drug, PTX or DOX was conjugated with pep 42 via a cathepsin B-specific cleavable linker (Val-Cit). The conjugates were designed to localize at lysosomes and to release therapeutic agents when taken up by the cancer cells. The targeted PAP system exhibited improved anticancer activity against SJSA-1 osteosarcoma cells known to express GRP78.
Overall, PAPs have shown superior pharmacokinetic properties to free drug molecules because chemical modification can improve their chemical stability, hydrophilicity and circulation time in the bloodstream. More importantly, the PAP approach improves pharmacological properties owing to their preferential accumulation and release of active forms of therapeutic agents in target tissues or cells by proteolysis. Additional modification of PAP with targeting molecules facilitates the accumulation and release of active therapeutic agents at the target site. Over the past half-century, a great number of PAPs have been invented and used in clinical settings. Based on the successful examples of the PAP approach, the following section will elucidate imperative design factors in the development of PAP.
4. Perspective on prodrug design
Although a few PAPs have entered clinical development already (Table 3), we further identify key design elements that are needed to develop highly efficient therapeutic agents that are released in protease overexpressed environments. We acknowledge many proteases require further research in their physiological roles, but the overwhelming research showing their overexpression in pathologies cannot be ignored (Table 1). PAPs do not hamper or inhibit normal protease activity directly, but instead use the protease deregulation to identify an area that requires a specific form of drug. In this way, unnecessary toxicity to healthy cells is eliminated. The three design elements to consider when developing PAPs are: 1) stability and specificity of peptide substrate, 2) location of drug activation, 3) addition of a delivery system.
Protease-activatable prodrugs in the clinic
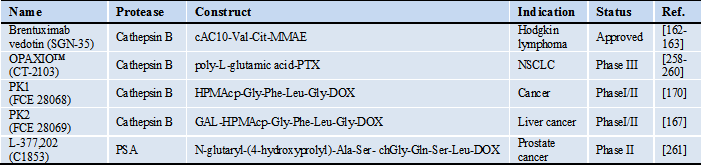
*Abbreviations: cAC10: chimeric anti-CD30 monoclonal antibody, chGly: cyclohexaglycyl, Cit: citrulline, GAL: galactose, DOX: doxorubicin, HPMAcp: N-(2-hydroxypropyl)methacrylamide copolymer, NSCLC: non-small-cell lung cancer, MMAE: monomethyl auristatin E, PTX: paclitaxel
First, the stability and specificity of a peptide substrate is by far the most important element to efficiently target proteases. The peptide substrate must remain stable in blood and plasma until it reaches its target protease upon which, it is cleaved to release the parent drug. If the peptide is not stable in these conditions, the prodrug cannot be intravenously administered and does not provide any benefit over administration of the parent drug directly. The stability of peptide substrates can be enhanced by the use of macromolecular structures, as seen in the example above of the polymerized peptide substrate for cathepsin B. Furthermore, the prodrug should target a specific protease that is associated with the disease. This is an area that requires further research, as many specific substrates for proteases linked to cancer have not been identified. For example, MMP substrates target many different MMPs and therefore have low specificity for one member of the MMP family. Although this is not necessarily a disadvantage, it may still reduce the efficacy of the drug and increase toxicity to areas that do not require the therapy. Caspases, on the other hand, have more specific substrates but they are generally not seen as good targets for cancer because they are key signal molecules for apoptosis, a process normally found in healthy cells but circumvented in cancer cells.
Second, location of target protease expression is important to recognize in order to design drug conjugates that can reach the specific target. In most cases, drugs exert their therapeutic effect when delivered specifically to the intracellular space of the unhealthy cell. But certain important target proteases are expressed extracellularly, especially under pathological conditions, and the PAP can be activated prematurely outside of the cell. In this view, prodrug constructs including peptide substrates, therapeutic agent and targeting moiety should be designed together with the target protease and its active site in mind. For example, PEG improves chemical stability and biocompatibility of drug molecules and also prolongs circulation time when coupled with hydrophobic drug molecules, but also reduces cellular uptake of drug molecules. In that case, the linkage between drug molecules and PEG should be cleaved by a specific protease that is active in extracellular conditions prior to cellular uptake as shown in ref [174]. On the other hand, if a therapeutic agent works well inside target cells but is not stable outside the cells, the drug should be caged by a specific promoiety that can protect it until it is degraded at target compartments of the cells [164, 175-176]. In such cases, various targeting moieties, like aptamers or antibodies and additional promoieties may be helpful to target the prodrug into the intracellular space. Furthermore, function of the protease in the type and stage of tumors is significant when designing PAPs. Most enzymes are temporal, for examples caspase 2 and 3 are markers of apoptosis but caspase 3 is only present at late stages. This may explain why many caspase-based probes have mainly served as sensors for apoptosis over drug delivery agents. Emerging data, such as for hKs, indicate that their function is dependent on hormone balances as well as tissue type. Therefore, it is beneficial to target enzymes that are present throughout the entire disease progression and to understand the protease role in the specific tissue targeted. A solution is to use combinatorial delivery approaches as described in the final design consideration.
Thirdly, a delivery carrier system can be utilized to target the prodrug more efficiently. As seen in the current prodrug examples, albumin served as a stabilizing agent for the therapeutic and also aided in prodrug accumulation by the EPR effect. Nanoparticles, as seen in many drug delivery applications, can accumulate in the tumor by EPR efficiently [177]. More importantly, nanoparticles can serve as platforms for numerous moieties that can aid in PAP design like protease substrates, targeting molecules, stabilizing agents, and imaging agents. In this way, a nanoparticle based approach for protease targeted therapeutic delivery can act as multimodal imaging and drug delivery agent - a true theranostics agent.
5. Conclusion
We have introduced key proteases that are involved in various pathologies, especially cancer, and their potential as targets for prodrugs. With a brief introduction on the roles of cathepsins, kallikreins, serine proteases, caspases and MMPs, we show that their dysregulation can be implicated in many forms of cancer, as well as in neurodegenerative, pulmonary and cardiovascular diseases. Furthermore, we provide few examples of PAPs that have utilized specific proteases to target intracellular cathepsins, PSA (a type of human kallikrein), and MMPs. In this way, we hope to illuminate the important roles that proteases can play in targeted therapies and go beyond their functional role in molecular imaging. With our experience in engineering protease activatable probes, we have identified important design considerations required for efficient development of PAPs and further imply that such conjugates can play an important role in theranostics.
Acknowledgements
This work was supported by the Intramural Research Program (IRP) of the National Institutes of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH). S.L. is partially supported by the NIH Pathway to Independence (K99/R00) Award.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Lopez-Otin C, Hunter T. The regulatory crosstalk between kinases and proteases in cancer. Nat Rev Cancer. 2010;10:278-92
2. Lopez-Otin C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer. 2007;7:800-8
3. Turk B. Targeting proteases: Successes, failures and future prospects. Nat Rev Drug Discov. 2006;5:785-99
4. Lopez-Otin C, Bond JS. Proteases: Multifunctional enzymes in life and disease. J Biol Chem. 2008;283:30433-7
5. Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: Lessons from the first decade. Clin Cancer Res. 2008;14:1649-57
6. Horiguchi A, Zheng R, Goodman OB Jr. et al. Lentiviral vector neutral endopeptidase gene transfer suppresses prostate cancer tumor growth. Cancer Gene Ther. 2007;14:583-9
7. Karikari CA, Roy I, Tryggestad E. et al. Targeting the apoptotic machinery in pancreatic cancers using small-molecule antagonists of the x-linked inhibitor of apoptosis protein. Mol Cancer Ther. 2007;6:957-66
8. Jang B, Choi Y. Photosensitizer-conjugated gold nanorods for enzyme-activatable fluorescence imaging and photodynamic therapy. Theranostics. 2012;2:190-7
9. Kim GB, Kim Y-P. Analysis of protease activity using quantum dots. Theranostics. 2012;2:127-38
10. Yhee JY, Kinm SA, Koo HB. et al. Optical imaging of cancer-related proteases using near-infrared fluorescence matrix metalloproteinase-sensitive and cathepsin b-sensitive probes. Theranostics. 2012;2:179-89
11. Zhu L, Xie J, Swierczewska M. et al. Real-time video imaging of protease expression in vivo. Theranostics. 2011;1:18-27
12. Rautio J, Kumpulainen H, Heimbach T. et al. Prodrugs: Design and clinical applications. Nat Rev Drug Discov. 2008;7:255-70
13. Mahato R, Tai W, Cheng K. Prodrugs for improving tumor targetability and efficiency. Adv Drug Deliv Rev. 2011;63:659-70
14. Stella VJ. Prodrugs as therapeutics. Expert Opin Ther Pat. 2004;14:277-80
15. Borgono CA, Diamandis EP. The emerging roles of human tissue kallikreins in cancer. Nat Rev Cancer. 2004;4:876-90
16. Gabriel D, Zuluaga MF, Van Den Bergh H. et al. It is all about proteases: From drug delivery to in vivo imaging and photomedicine. Curr Med Chem. 2011;18:1785-805
17. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860-7
18. Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71-8
19. Mohamed MM, Sloane BF. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat Rev Cancer. 2006;6:764-75
20. Flannery T, Gibson D, Mirakhur M. et al. The clinical significance of cathepsin s expression in human astrocytomas. Am J Pathol. 2003;163:175-82
21. Mikkelsen T, Yan PS, Ho KL. et al. Immunolocalization of cathepsin b in human glioma: Implications for tumor invasion and angiogenesis. J Neurosurg. 1995;83:285-90
22. Sloane BF, Honn KV, Sadler JG. et al. Cathepsin-b activity in b-16 melanoma-cells - a possible marker for metastatic potential. Cancer Res. 1982;42:980-6
23. Hughes SJ, Glover TW, Zhu X-X. et al. A novel amplicon at 8p22-23 results in overexpression of cathepsin b in esophageal adenocarcinoma. Proc Natl Acad Sci U S A. 1998;95:12410-5
24. Krueger S, Kalinski T, Hundertmark T. et al. Up-regulation of cathepsin x in helicobacter pylori gastritis and gastric cancer. J Pathol. 2005;207:32-42
25. Campo E, Munoz J, Miquel R. et al. Cathepsin b expression in colorectal carcinomas correlates with tumor progression and shortened patient survival. Am J Pathol. 1994;145:301-9
26. Fernández PL, Farré X, Nadal A. et al. Expression of cathepsins b and s in the progression of prostate carcinoma. Int J Cancer. 2001;95:51-5
27. Nägler DK, Krüger S, Kellner A. et al. Up-regulation of cathepsin x in prostate cancer and prostatic intraepithelial neoplasia. Prostate. 2004;60:109-19
28. Santamaría I, Velasco G, Cazorla M. et al. Cathepsin l2, a novel human cysteine proteinase produced by breast and colorectal carcinomas. Cancer Res. 1998;58:1624-30
29. Linnerth NM, Sirbovan K, Moorehead RA. Use of a transgenic mouse model to identify markers of human lung tumors. Int J Cancer. 2005;114:977-82
30. Paik S, Shak S, Tang G. et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817-26
31. Bossard MJ, Tomaszek TA, Thompson SK. et al. Proteolytic activity of human osteoclast cathepsin k. J Biol Chem. 1996;271:12517-24
32. Xia L, Kilb J, Wex H. et al. Localization of rat cathepsin k in osteoclasts and resorption pits: Inhibition of bone resorption and cathepsin k-activity by peptidyl vinyl sulfones. Biol Chem. 1999;380:679-87
33. Gocheva V, Joyce JA. Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle. 2007;6:60-4
34. Palermo C, Joyce JA. Cysteine cathepsin proteases as pharmacological targets in cancer. Trends Pharmacol Sci. 2008;29:22-8
35. Buck MR, Karustis DG, Day NA. et al. Degradation of extracellular-matrix proteins by human cathepsin b from normal and tumour tissues. Biochem J. 1992;282( Pt 1):273-8
36. Gocheva V, Zeng W, Ke D. et al. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006;20:543-56
37. Yan S, Sloane BF. Molecular regulation of human cathepsin b: Implication in pathologies. Biol Chem. 2003;384:845-54
38. Cavallo-Medved D, Mai J, Dosescu J. et al. Caveolin-1 mediates the expression and localization of cathepsin b, pro-urokinase plasminogen activator and their cell-surface receptors in human colorectal carcinoma cells. J Cell Sci. 2005;118:1493-503
39. Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27:6434-51
40. Cardone RA, Casavola V, Reshkin SJ. The role of disturbed ph dynamics and the na+/h+ exchanger in metastasis. Nat Rev Cancer. 2005;5:786-95
41. Reddy VY, Zhang QY, Weiss SJ. Pericellular mobilization of the tissue-destructive cysteine proteinases, cathepsins b, l, and s, by human monocyte-derived macrophages. Proc Natl Acad Sci U S A. 1995;92:3849-53
42. Cirman T, Orešić K, Mazovec GD. et al. Selective disruption of lysosomes in hela cells triggers apoptosis mediated by cleavage of bid by multiple papain-like lysosomal cathepsins. J Biol Chem. 2004;279:3578-87
43. Stoka V, Turk B, Schendel SL. et al. Lysosomal protease pathways to apoptosis - cleavage of bid, not pro-caspases, is the most likely route. J Biol Chem. 2001;276:3149-57
44. Vasiljeva O, Turk B. Dual contrasting roles of cysteine cathepsins in cancer progression: Apoptosis versus tumour invasion. Biochimie. 2008;90:380-6
45. Reinheckel T, Hagemann S, Dollwet-Mack S. et al. The lysosomal cysteine protease cathepsin l regulates keratinocyte proliferation by control of growth factor recycling. J Cell Sci. 2005;118:3387-95
46. Joyce JA, Baruch A, Chehade K. et al. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell. 2004;5:443-53
47. Conover CA, Perry JE, Tindall DJ. Endogenous cathepsin d-mediated hydrolysis of insulin-like growth factor-binding proteins in cultured human prostatic-carcinoma cells. J Clin Endocrinol Metab. 1995;80:987-93
48. Tandon AK, Clark GM, Chamness GC. et al. Cathepsin-d and prognosis in breast-cancer. N Engl J Med. 1990;322:297-302
49. Ma YM, Zhao M, Zhong JL. et al. Proteomic profiling of proteins associated with lymph node metastasis in colorectal cancer. J Cell Biochem. 2010;110:1512-9
50. Zaidi N, Hermann C, Herrmann T. et al. Emerging functional roles of cathepsin e. Biochem Biophys Res Commun. 2008;377:327-30
51. Matsuo K, Kobayashi I, Tsukuba T. et al. Immunohistochemical localization of cathepsins d and e in human gastric cancer: A possible correlation with local invasive and metastatic activities of carcinoma cells. Hum Pathol. 1996;27:184-90
52. Tenti P, Romagnoli S, Silini E. et al. Cervical adenocarcinomas express markers common to gastric, intestinal, and pancreatobiliary epithelial cells. Pathol Res Pract. 1994;190:342-9
53. Uno K, Azuma T, Nakajima M. et al. Clinical significance of cathepsin e in pancreatic juice in the diagnosis of pancreatic ductal adenocarcinoma. J Gastroenterol Hepatol. 2000;15:1333-8
54. Terris B, Blaveri E, Crnogorac-Jurcevic T. et al. Characterization of gene expression profiles in intraductal papillary-mucinous tumors of the pancreas. Am J Pathol. 2002;160:1745-54
55. Ullmann R, Morbini P, Halbwedl I. et al. Protein expression profiles in adenocarcinomas and squamous cell carcinomas of the lung generated using tissue microarrays. J Pathol. 2004;203:798-807
56. Cleutjens KBJM, Van Eekelen CCEM, Van Der Korput HaGM. et al. Two androgen response regions cooperate in steroid hormone regulated activity of the prostate-specific antigen promoter. J Biol Chem. 1996;271:6379-88
57. Malm J, Hellman J, Hogg P. et al. Enzymatic action of prostate-specific antigen (psa or hk3): Substrate specificity and regulation by Zn2+, a tight-binding inhibitor. Prostate. 2000;45:132-9
58. Yousef GM, Diamandis EP. The new human tissue kallikrein gene family: Structure, function, and association to disease. Endocr Rev. 2001;22:184-204
59. Réhault S, Monget P, Mazerbourg S. et al. Insulin-like growth factor binding proteins (igfbps) as potential physiological substrates for human kallikreins hk2 and hk3. Eur J Biochem. 2001;268:2960-8
60. Mikolajczyk SD, Millar LS, Kumar A. et al. Prostatic human kallikrein 2 inactivates and complexes with plasminogen activator inhibitor-1. Int J Cancer. 1999;81:438-42
61. Takayama TK, Mcmullen BA, Nelson PS. et al. Characterization of hk4 (prostase), a prostate-specific serine protease: Activation of the precursor of prostate specific antigen (pro-psa) and single-chain urokinase-type plasminogen activator and degradation of prostatic acid phosphatase. Biochemistry. 2001;40:15341-8
62. Yoshida E, Ohmura S, Sugiki M. et al. Prostate-specific antigen activates single-chain urokinase-type plasminogen activator. Int J Cancer. 1995;63:863-5
63. Pepper MS. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler Thromb Vasc Biol. 2001;21:1104-17
64. Killian CS, Corral DA, Kawinski E. et al. Mitogenic response of osteoblast cells to prostate-specific antigen suggests an activation of latent tgf-β and a proteolytic modulation of cell adhesion receptors. Biochem Biophys Res Commun. 1993;192:940-7
65. Fortier AH, Nelson BJ, Grella DK. et al. Antiangiogenic activity of prostate-specific antigen. J Natl Cancer Inst. 1999;91:1635-40
66. Desriviéres S, Lu H, Peyri N. et al. Activation of the 92 kda type iv collagenase by tissue kallikrein. J Cell Physiol. 1993;157:587-93
67. Andreasen PA, Egelund R, Petersen HH. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol Life Sci. 2000;57:25-40
68. Foekens JA, Peters HA, Look MP. et al. The urokinase system of plasminogen activation and prognosis in 2780 breast cancer patients. Cancer Res. 2000;60:636-43
69. Pyke C, Kristensen P, Ralfkiaer E. et al. Urokinase-type plasminogen-activator is expressed in stromal cells and its receptor in cancer-cells at invasive foci in human colon adenocarcinomas. Am J Pathol. 1991;138:1059-67
70. Stephens RW, Nielsen HJ, Christensen IJ. et al. Plasma urokinase receptor levels in patients with colorectal cancer: Relationship to prognosis. J Natl Cancer Inst. 1999;91:869-74
71. Duffy MJ, Maguire TM, Mcdermott EW. et al. Urokinase plasminogen activator: A prognostic marker in multiple types of cancer. J Surg Oncol. 1999;71:130-5
72. Miyake H, Hara I, Yamanaka K. et al. Elevation of serum levels of urokinase-type plasminogen activator and its receptor is associated with disease progression and prognosis in patients with prostate cancer. Prostate. 1999;39:123-9
73. Dass K, Ahmad A, Azmi AS. et al. Evolving role of upa/upar system in human cancers. Cancer Treat Rev. 2008;34:122-36
74. Jin E, Fujiwara M, Pan X. et al. Protease-activated receptor (par)-1 and par-2 participate in the cell growth of alveolar capillary endothelium in primary lung adenocarcinomas. Cancer. 2003;97:703-13
75. Takeuchi T, Harris JL, Huang W. et al. Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J Biol Chem. 2000;275:26333-42
76. Darmoul D, Gratio V, Devaud H. et al. Protease-activated receptor 2 in colon cancer. J Biol Chem. 2004;279:20927-34
77. Morris DR, Ding Y, Ricks TK. et al. Protease-activated receptor-2 is essential for factor viia and xa-induced signaling, migration, and invasion of breast cancer cells. Cancer Res. 2006;66:307-14
78. Shi X, Gangadharan B, Brass LF. et al. Protease-activated receptors (par1 and par2) contribute to tumor cell motility and metastasis. Mol Cancer Res. 2004;2:395-402
79. Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770-6
80. Thornberry NA, Lazebnik Y. Caspases: Enemies within. Science. 1998;281:1312-6
81. Leist M, Jaattela M. Four deaths and a funeral: From caspases to alternative mechanisms. Nat Rev Mol Cell Biol. 2001;2:589-98
82. Hunter AM, Lacasse EC, Korneluk RG. The inhibitors of apoptosis (iaps) as cancer targets. Apoptosis. 2007;12:1543-68
83. Klein T, Bischoff R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids. 2011;41:271-90
84. Ra H-J, Parks WC. Control of matrix metalloproteinase catalytic activity. Matrix Biol. 2007;26:587-96
85. Nagase H. Activation mechanisms of matrix metalloproteinases. Biol Chem. 1997;378:151-60
86. Page-Mccaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8:221-33
87. Chakrabarti S, Patel KD. Matrix metalloproteinase-2 (mmp-2) and mmp-9 in pulmonary pathology. Exp Lung Res. 2005;31:599-621
88. Nguyen M, Arkell J, Jackson CJ. Human endothelial gelatinases and angiogenesis. Int J Biochem Cell Biol. 2001;33:960-70
89. Bergers G, Brekken R, Mcmahon G. et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737-44
90. Van Den Steen PE, Dubois B, Nelissen I. et al. Biochemistry and molecular biology of gelatinase b or matrix metalloproteinase-9 (mmp-9). Crit Rev Biochem Mol Biol. 2002;37:375-536
91. Somiari SB, Somiari RI, Heckman CM. et al. Circulating mmp2 and mmp9 in breast cancer - potential role in classification of patients into low risk, high risk, benign disease and breast cancer categories. Int J Cancer. 2006;119:1403-11
92. Turpeenniemi-Hujanen T. Gelatinases (mmp-2 and-9) and their natural inhibitors as prognostic indicators in solid cancers. Biochimie. 2005;87:287-97
93. Ranuncolo SM, Armanasco E, Cresta C. et al. Plasma mmp-9 (92 kda-mmp) activity is useful in the follow-up and in the assessment of prognosis in breast cancer patients. Int J Cancer. 2003;106:745-51
94. Schmalfeldt B, Prechtel D, Harting K. et al. Increased expression of matrix metalloproteinases (mmp)-2, mmp-9, and the urokinase-type plasminogen activator is associated with progression from benign to advanced ovarian cancer. Clin Cancer Res. 2001;7:2396-404
95. Nawrocki B, Polette M, Marchand V. et al. Expression of matrix metalloproteinases and their inhibitors in human bronchopulmonary carcinomas: Quantificative and morphological analyses. Int J Cancer. 1997;72:556-64
96. Zucker S, Vacirca J. Role of matrix metalloproteinases (mmps) in colorectal cancer. Cancer Metastasis Rev. 2004;23:101-17
97. Forsyth PA, Wong H, Laing TD. et al. Gelatinase-a (mmp-2), gelatinase-b (mmp-9) and membrane type matrix metalloproteinase-1 (mt1-mmp) are involved in different aspects of the pathophysiology of malignant gliomas. Br J Cancer. 1999;79:1828-35
98. Yong VW, Power C, Forsyth P. et al. Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci. 2001;2:502-11
99. Ii M, Yamamoto H, Adachi Y. et al. Role of matrix metalloproteinase-7 (matrilysin) in human cancer invasion, apoptosis, growth, and angiogenesis. Exp Biol Med. 2006;231:20-7
100. Wang F, Reierstad S, Fishman DA. Matrilysin over-expression in mcf-7 cells enhances cellular invasiveness and pro-gelatinase activation. Cancer Lett. 2006;236:292-301
101. Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science. 2002;295:2387-92
102. Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and timps. Cardiovasc Res. 2006;69:562-73
103. Nagase H, Woessner JF. Matrix metalloproteinases. J Biol Chem. 1999;274:21491-4
104. Nelson AR, Fingleton B, Rothenberg ML. et al. Matrix metalloproteinases: Biologic activity and clinical implications. J Clin Oncol. 2000;18:1135
105. Mattson MP. Pathways towards and away from alzheimer's disease. Nature. 2004;430:631-9
106. Selkoe DJ, Schenk D. Alzheimer's disease: Molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol and Toxicol. 2003;43:545-84
107. Lichtenthaler SF, Haass C, Steiner H. Regulated intramembrane proteolysis - lessons from amyloid precursor protein processing. J Neurochem. 2011;117:779-96
108. Janus C, Pearson J, Mclaurin J. et al. A[beta] peptide immunization reduces behavioural impairment and plaques in a model of alzheimer's disease. Nature. 2000;408:979-82
109. Nicoll JaR, Wilkinson D, Holmes C. et al. Neuropathology of human alzheimer disease after immunization with amyloid-[beta] peptide: A case report. Nat Med. 2003;9:448-52
110. Diamandis EP, Yousef GM, Petraki C. et al. Human kallikrein 6 as a biomarker of alzheimer's disease. Clin Biochem. 2000;33:663-7
111. Blennow K, Hampel H, Weiner M. et al. Cerebrospinal fluid and plasma biomarkers in alzheimer disease. Nat Rev Neurol. 2010;6:131-44
112. Edwards DR, Handsley MM, Pennington CJ. The adam metalloproteinases. Mol Asp Med. 2008;29:258-89
113. Montaner J, Molina CA, Monasterio J. et al. Matrix metalloproteinase-9 pretreatment level predicts intracranial hemorrhagic complications after thrombolysis in human stroke. Circulation. 2003;107:598-603
114. Romanic AM, White RF, Arleth AJ. et al. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats - inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998;29:1020-30
115. Zhang KY, Mcquibban GA, Silva C. et al. Hiv-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat Neurosci. 2003;6:1064-71
116. Lee CG, Homer R, Zhou Z. et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med. 2001;194:809-21
117. Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: Influence on cardiac form and function. Physiol Rev. 2007;87:1285-342
118. Zaman MA, Oparil S, Calhoun DA. Drugs targeting the renin-angiotensin-aldosterone system. Nat Rev Drug Discov. 2002;1:621-36
119. Jessup M, Brozena S. Heart failure. N Engl J Med. 2003;348:2007-18
120. Holland OB, Chud JM, Braunstein H. Urinary kallikrein excretion in essential and mineralocorticoid hypertension. J Clin Invest. 1980;65:347-56
121. Berdowska I. Cysteine proteases as disease markers. Clin Chim Acta. 2004;342:41-69
122. Albert A. Chemical aspects of selective toxicity. Nature. 1958;182:421-2
123. Law B, Tung CH. Proteolysis: A biological process adapted in drug delivery, therapy, and imaging. Bioconjug Chem. 2009;20:1683-95
124. Baurain R, Masquelier M, Deprez-De Campeneere D. et al. Amino acid and dipeptide derivatives of daunorubicin. 2. Cellular pharmacology and antitumor activity on l1210 leukemic cells in vitro and in vivo. J Med Chem. 1980;23:1171-4
125. Boven E, Hendriks HR, Erkelens CA. et al. The anti-tumour effects of the prodrugs n-l-leucyl-doxorubicin and vinblastine-isoleucinate in human ovarian cancer xenografts. Br J Cancer. 1992;66:1044-7
126. Breistol K, Hendriks HR, Berger DP. et al. The antitumour activity of the prodrug n-l-leucyl-doxorubicin and its parent compound doxorubicin in human tumour xenografts. Eur J Cancer. 1998;34:1602-6
127. Breistol K, Hendriks HR, Fodstad O. Superior therapeutic efficacy of n-l-leucyl-doxorubicin versus doxorubicin in human melanoma xenografts correlates with higher tumour concentrations of free drug. Eur J Cancer. 1999;35:1143-9
128. De Jong J, Klein I, Bast A. et al. Analysis and pharmacokinetics of n-l-leucyldoxorubucin and metabolites in tissues of tumor-bearing balb/c mice. Cancer Chemother Pharmacol. 1992;31:156-60
129. Deprez-De Campeneere D, Baurain R, Trouet A. Accumulation and metabolism of new anthracycline derivatives in the heart after iv injection into mice. Cancer Chemother Pharmacol. 1982;8:193-7
130. Zbinden G, Decampeenere D, Baurain R. Preclinical assessment of the cardiotoxic potential of anthracycline antibiotics: N-l-leucyl-doxorubicin. Arch Toxicol Suppl. 1991;14:107-17
131. Trouet A, Masquelier M, Baurain R. et al. A covalent linkage between daunorubicin and proteins that is stable in serum and reversible by lysosomal hydrolases, as required for a lysosomotropic drug-carrier conjugate: In vitro and in vivo studies. Proc Natl Acad Sci U S A. 1982;79:626-9
132. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387-92
133. Maeda H, Matsumura Y. Epr effect based drug design and clinical outlook for enhanced cancer chemotherapy. Adv Drug Deliv Rev. 2011;63:129-30
134. Duncan R, Coatsworth JK, Burtles S. Preclinical toxicology of a novel polymeric antitumour agent: Hpma copolymer-doxorubicin (pk1). Hum Exp Toxicol. 1998;17:93-104
135. Loadman PM, Bibby MC, Double JA. et al. Pharmacokinetics of pk1 and doxorubicin in experimental colon tumor models with differing responses to pk1. Clin Cancer Res. 1999;5:3682-8
136. Malugin A, Kopeckova P, Kopecek J. Liberation of doxorubicin from hpma copolymer conjugate is essential for the induction of cell cycle arrest and nuclear fragmentation in ovarian carcinoma cells. J Control Release. 2007;124:6-10
137. Minko T, Kopeckova P, Kopecek J. Comparison of the anticancer effect of free and hpma copolymer-bound adriamycin in human ovarian carcinoma cells. Pharm Res. 1999;16:986-96
138. Minko T, Kopeckova P, Pozharov V. et al. Hpma copolymer bound adriamycin overcomes mdr1 gene encoded resistance in a human ovarian carcinoma cell line. J Control Release. 1998;54:223-33
139. Seymour LW, Ulbrich K, Strohalm J. et al. The pharmacokinetics of polymer-bound adriamycin. Biochem Pharmacol. 1990;39:1125-31
140. Yeung TK, Hopewell JW, Simmonds RH. et al. Reduced cardiotoxicity of doxorubicin given in the form of n-(2-hydroxypropyl)methacrylamide conjugates: And experimental study in the rat. Cancer Chemother Pharmacol. 1991;29:105-11
141. Duncan R, Kopeckova P, Strohalm J. et al. Anticancer agents coupled to n-(2-hydroxypropyl)methacrylamide copolymers. Ii. Evaluation of daunomycin conjugates in vivo against l1210 leukaemia. Br J Cancer. 1988;57:147-56
142. Putnam D, Kopecek J. Enantioselective release of 5-fluorouracil from n-(2-hydroxypropyl)methacrylamide-based copolymers via lysosomal enzymes. Bioconjug Chem. 1995;6:483-92
143. Gianasi E, Buckley RG, Latigo J. et al. Hpma copolymers platinates containing dicarboxylato ligands. Preparation, characterisation and in vitro and in vivo evaluation. J Drug Target. 2002;10:549-56
144. Satchi-Fainaro R, Puder M, Davies JW. et al. Targeting angiogenesis with a conjugate of hpma copolymer and tnp-470. Nat Med. 2004;10:255-61
145. Li C, Yu DF, Newman RA. et al. Complete regression of well-established tumors using a novel water-soluble poly(l-glutamic acid)-paclitaxel conjugate. Cancer Res. 1998;58:2404-9
146. Shaffer SA, Baker-Lee C, Kennedy J. et al. In vitro and in vivo metabolism of paclitaxel poliglumex: Identification of metabolites and active proteases. Cancer Chemother Pharmacol. 2007;59:537-48
147. Denmeade SR, Lou W, Lovgren J. et al. Specific and efficient peptide substrates for assaying the proteolytic activity of prostate-specific antigen. Cancer Res. 1997;57:4924-30
148. Denmeade SR, Nagy A, Gao J. et al. Enzymatic activation of a doxorubicin-peptide prodrug by prostate-specific antigen. Cancer Res. 1998;58:2537-40
149. Khan SR, Denmeade SR. In vivo activity of a psa-activated doxorubicin prodrug against psa-producing human prostate cancer xenografts. Prostate. 2000;45:80-3
150. Denmeade SR, Jakobsen CM, Janssen S. et al. Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. J Natl Cancer Inst. 2003;95:990-1000
151. Jakobsen CM, Denmeade SR, Isaacs JT. et al. Design, synthesis, and pharmacological evaluation of thapsigargin analogues for targeting apoptosis to prostatic cancer cells. J Med Chem. 2001;44:4696-703
152. Mhaka A, Denmeade SR, Yao W. et al. A 5-fluorodeoxyuridine prodrug as targeted therapy for prostate cancer. Bioorg Med Chem Lett. 2002;12:2459-61
153. Defeo-Jones D, Garsky VM, Wong BK. et al. A peptide-doxorubicin 'prodrug' activated by prostate-specific antigen selectively kills prostate tumor cells positive for prostate-specific antigen in vivo. Nat Med. 2000;6:1248-52
154. Chandran SS, Nan A, Rosen DM. et al. A prostate-specific antigen activated n-(2-hydroxypropyl) methacrylamide copolymer prodrug as dual-targeted therapy for prostate cancer. Mol Cancer Ther. 2007;6:2928-37
155. Kratz F, Mansour A, Soltau J. et al. Development of albumin-binding doxorubicin prodrugs that are cleaved by prostate-specific antigen. Arch Pharm (Weinheim). 2005;338:462-72
156. Graeser R, Chung DE, Esser N. et al. Synthesis and biological evaluation of an albumin-binding prodrug of doxorubicin that is cleaved by prostate-specific antigen (psa) in a psa-positive orthotopic prostate carcinoma model (lncap). Int J Cancer. 2008;122:1145-54
157. Albright CF, Graciani N, Han W. et al. Matrix metalloproteinase-activated doxorubicin prodrugs inhibit ht1080 xenograft growth better than doxorubicin with less toxicity. Mol Cancer Ther. 2005;4:751-60
158. Kline T, Torgov MY, Mendelsohn BA. et al. Novel antitumor prodrugs designed for activation by matrix metalloproteinases-2 and -9. Mol Pharm. 2004;1:9-22
159. Mansour AM, Drevs J, Esser N. et al. A new approach for the treatment of malignant melanoma: Enhanced antitumor efficacy of an albumin-binding doxorubicin prodrug that is cleaved by matrix metalloproteinase 2. Cancer Res. 2003;63:4062-6
160. Chau Y, Tan FE, Langer R. Synthesis and characterization of dextran-peptide-methotrexate conjugates for tumor targeting via mediation by matrix metalloproteinase ii and matrix metalloproteinase ix. Bioconjug Chem. 2004;15:931-41
161. Chau Y, Dang NM, Tan FE. et al. Investigation of targeting mechanism of new dextran-peptide-methotrexate conjugates using biodistribution study in matrix-metalloproteinase-overexpressing tumor xenograft model. J Pharm Sci. 2006;95:542-51
162. Alley SC, Okeley NM, Senter PD. Antibody-drug conjugates: Targeted drug delivery for cancer. Curr Opin Chem Biol. 2010;14:529-37
163. Senter PD. Potent antibody drug conjugates for cancer therapy. Curr Opin Chem Biol. 2009;13:235-44
164. Doronina SO, Toki BE, Torgov MY. et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol. 2003;21:778-84
165. Fda approves adcetris to treat two types of lympoma. FDA. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm268781.htm
166. Vasey PA, Kaye SB, Morrison R. et al. Phase I clinical and pharmacokinetic study of pk1 [n-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: First member of a new class of chemotherapeutic agents-drug-polymer conjugates. Cancer research campaign phase I/II committee. Clin Cancer Res. 1999;5:83-94
167. Seymour LW, Ferry DR, Anderson D. et al. Hepatic drug targeting: Phase I evaluation of polymer-bound doxorubicin. J Clin Oncol. 2002;20:1668-76
168. Duncan R. Development of hpma copolymer-anticancer conjugates: Clinical experience and lessons learnt. Adv Drug Deliv Rev. 2009;61:1131-48
169. Greco F, Vicent MJ. Combination therapy: Opportunities and challenges for polymer-drug conjugates as anticancer nanomedicines. Adv Drug Deliv Rev. 2009;61:1203-13
170. Seymour LW, Ferry DR, Kerr DJ. et al. Phase II studies of polymer-doxorubicin (pk1, fce28068) in the treatment of breast, lung and colorectal cancer. Int J Oncol. 2009;34:1629-36
171. Duncan R, Vicent MJ. Do hpma copolymer conjugates have a future as clinically useful nanomedicines? A critical overview of current status and future opportunities. Adv Drug Deliv Rev. 2010;62:272-82
172. De Groot FM, Broxterman HJ, Adams HP. et al. Design, synthesis, and biological evaluation of a dual tumor-specific motive containing integrin-targeted plasmin-cleavable doxorubicin prodrug. Mol Cancer Ther. 2002;1:901-11
173. Yoneda Y, Steiniger SC, Capkova K. et al. A cell-penetrating peptidic grp78 ligand for tumor cell-specific prodrug therapy. Bioorg Med Chem Lett. 2008;18:1632-6
174. Kawakami S, Munakata C, Fumoto S. et al. Novel galactosylated liposomes for hepatocyte-selective targeting of lipophilic drugs. J Pharm Sci. 2001;90:105-13
175. Sutherland MS, Sanderson RJ, Gordon KA. et al. Lysosomal trafficking and cysteine protease metabolism confer target-specific cytotoxicity by peptide-linked anti-cd30-auristatin conjugates. J Biol Chem. 2006;281:10540-7
176. Erickson HK, Park PU, Widdison WC. et al. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006;66:4426-33
177. Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy and diagnosis. Adv Drug Deliv Rev. 2002;54:631-51
178. Littlewood-Evans AJ, Bilbe G, Bowler WB. et al. The osteoclast-associated protease cathepsin k is expressed in human breast carcinoma. Cancer Res. 1997;57:5386-90
179. Liu JA, Sukhova GK, Sun JS. et al. Lysosomal cysteine proteases in atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:1359-66
180. Sukhova GK, Shi GP, Simon DI. et al. Expression of the elastolytic cathepsins s and k in human atheroma and regulation of their production in smooth muscle cells. J Clin Invest. 1998;102:576-83
181. Bromme D, Lecaille F. Cathepsin k inhibitors for osteoporosis and potential off-target effects. Expert Opin Invest Drugs. 2009;18:585-600
182. Costa AG, Cusano NE, Silva BC. et al. Cathepsin k: Its skeletal actions and role as a therapeutic target in osteoporosis. Nat Rev Rheumatol. 2011;7:447-56
183. Sinha AA, Wilson MJ, Gleason DF. et al. Immunohistochemical localization of cathepsin-b in neoplastic human prostate. Prostate. 1995;26:171-8
184. Bongers V, Konings CH, Grijpma AM. et al. Serum proteinase activities in head and neck squamous cell carcinoma patients. Anticancer Res. 1995;15:2763-6
185. Lopez Couto E, Tersariol ILS, Pinhal MaS. et al. Cathepsin b and heparanase activity and expression in head and neck squamous cell carcinoma. Oral Oncol. 2007:96
186. Roshy S, Sloane BF, Moin K. Pericellular cathepsin b and malignant progression. Cancer Metastasis Rev. 2003;22:271-86
187. Zhang H, Fu T, Mcgettigan S. et al. Il8 and cathepsin b as melanoma serum biomarkers. Int J Mol Sci. 2011;12:1505-18
188. Hara H, Friedlander RM, Gagliardini V. et al. Inhibition of interleukin 1 beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci U S A. 1997;94:2007-12
189. Schellmann N, Deckert PM, Bachran D. et al. Targeted enzyme prodrug therapies. Mini Rev Med Chem. 2010;10:887-904
190. Martinon F, Tschopp J. Inflammatory caspases: Linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561-74
191. Huttunen KM, Rautio J. Prodrugs - an efficient way to breach delivery and targeting barriers. Curr Top Med Chem. 2011;11:2265-87
192. Hartmann A, Hunot S, Michel PP. et al. Caspase-3: A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in parkinson's disease. Proc Natl Acad Sci U S A. 2000;97:2875-80
193. Li MW, Ona VO, Guegan C. et al. Functional role of caspase-1 and caspase-3 in an als transgenic mouse model. Science. 2000;288:335-9
194. Huttunen KM, Raunio H, Rautio J. Prodrugs--from serendipity to rational design. Pharmacol Rev. 2011;63:750-71
195. Ona VO, Li MW, Vonsattel JPG. et al. Inhibition of caspase-1 slows disease progression in a mouse model of huntington's disease. Nature. 1999;399:263-7
196. Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431-568
197. Yoshiyama Y, Arai K, Oki T. et al. Expression of invariant chain and pro-cathepsin l in alzheimer's brain. Neurosci Lett. 2000;290:125-8
198. Johnson MD, Torri JA, Lippman ME. et al. The role of cathepsin-d in the invasiveness of human breast-cancer cells. Cancer Res. 1993;53:873-7
199. Losch A, Schindl M, Kohlberger P. et al. Cathepsin d in ovarian cancer: Prognostic value and correlation with p53 expression and microvessel density. Gynecol Oncol. 2004;92:545-52
200. Tedone T, Correale M, Barbarossa G. et al. Release of the aspartyl protease cathepsin d is associated with and facilitates human breast cancer cell invasion. FASEB J. 1997;11:785-92
201. Reynolds MA, Kastury K, Groskopf J. et al. Molecular markers for prostate cancer. Cancer Lett. 2007;249:5-13
202. Adib TR, Henderson S, Perrett C. et al. Predicting biomarkers for ovarian cancer using gene-expression microarrays. Br J Cancer. 2004;90:686-92
203. Yousef GM, Borgono CA, Popalis C. et al. In-silico analysis of kallikrein gene expression in pancreatic and colon cancers. Anticancer Res. 2004;24:43-52
204. Lu KH, Patterson AP, Wang L. et al. Selection of potential markers for epithelial ovarian cancer with gene expression arrays and recursive descent partition analysis. Clin Cancer Res. 2004;10:3291-300
205. Iacobuzio-Donahue CA, Ashfaq R, Maitra A. et al. Highly expressed genes in pancreatic ductal adenocarcinomas. Cancer Res. 2003;63:8614-22
206. Chung CH, Parker JS, Karaca G. et al. Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell. 2004;5:489-500
207. Yousef GM, Scorilas A, Katsaros D. et al. Prognostic value of the human kallikrein gene 15 expression in ovarian cancer. J Clin Oncol. 2003;21:3119-26
208. Yousef GM, Scorilas A, Jung K. et al. Molecular cloning of the human kallikrein 15 gene (klk15). J Biol Chem. 2001;276:53-61
209. Trengove NJ, Stacey MC, Macauley S. et al. Analysis of the acute and chronic wound environments: The role of proteases and their inhibitors. Wound Repair Regen. 1999;7:442-52
210. Rosenberg GA. Matrix metalloproteinases in neuroinflammation. Glia. 2002;39:279-91
211. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161-74
212. Heppner KJ, Matrisian LM, Jensen RA. et al. Expression of most matrix metalloproteinase family members in breast cancer represents a tumor-induced host response. Am J Pathol. 1996;149:273-82
213. Peake NJ, Khawaja K, Myers A. et al. Levels of matrix metalloproteinase (mmp)-1 in paired sera and synovial fluids of juvenile idiopathic arthritis patients: Relationship to inflammatory activity, mmp-3 and tissue inhibitor of metalloproteinases-1 in a longitudinal study. Rheumatology. 2005;44:1383-9
214. Molloy KJ, Thompson MM, Jones JL. et al. Unstable carotid plaques exhibit raised matrix metalloproteinase-8 activity. Circulation. 2004;110:337-43
215. Chen J, Stefflova K, Niedre MJ. et al. Protease-triggered photosensitizing beacon based on singlet oxygen quenching and activation. J Am Chem Soc. 2004;126:11450-1
216. Stefflova K, Chen J, Marotta D. et al. Photodynamic therapy agent with a built-in apoptosis sensor for evaluating its own therapeutic outcome in situ. J Med Chem. 2006;49:3850-6
217. Carter PJ, Gazzard L. Caspase activated prodrugs therapy. Wip organization. 2001
218. Kuefner U, Lohrmann U, Montejano YD. et al. Carboxypeptidase-mediated release of methotrexate from methotrexate alpha-peptides. Biochemistry. 1989;28:2288-97
219. Lo PC, Chen J, Stefflova K. et al. Photodynamic molecular beacon triggered by fibroblast activation protein on cancer-associated fibroblasts for diagnosis and treatment of epithelial cancers. J Med Chem. 2009;52:358-68
220. Janssen S, Jakobsen CM, Rosen DM. et al. Screening a combinatorial peptide library to develop a human glandular kallikrein 2-activated prodrug as targeted therapy for prostate cancer. Mol Cancer Ther. 2004;3:1439-50
221. Janssen S, Rosen DM, Ricklis RM. et al. Pharmacokinetics, biodistribution, and antitumor efficacy of a human glandular kallikrein 2 (hk2)-activated thapsigargin prodrug. Prostate. 2006;66:358-68
222. Chen J, Liu TW, Lo PC. et al. "Zipper" molecular beacons: A generalized strategy to optimize the performance of activatable protease probes. Bioconjug Chem. 2009;20:1836-42
223. Zheng G, Chen J, Stefflova K. et al. Photodynamic molecular beacon as an activatable photosensitizer based on protease-controlled singlet oxygen quenching and activation. Proc Natl Acad Sci U S A. 2007;104:8989-94
224. De Groot FM, De Bart AC, Verheijen JH. et al. Synthesis and biological evaluation of novel prodrugs of anthracyclines for selective activation by the tumor-associated protease plasmin. J Med Chem. 1999;42:5277-83
225. Carl PL, Chakravarty PK, Katzenellenbogen JA. A novel connector linkage applicable in prodrug design. J Med Chem. 1981;24:479-80
226. Chakravarty PK, Carl PL, Weber MJ. et al. Plasmin-activated prodrugs for cancer chemotherapy. 1. Synthesis and biological activity of peptidylacivicin and peptidylphenylenediamine mustard. J Med Chem. 1983;26:633-8
227. Carl PL, Chakravarty PK, Katzenellenbogen JA. et al. Protease-activated "prodrugs" for cancer chemotherapy. Proc Natl Acad Sci U S A. 1980;77:2224-8
228. Balajthy Z, Aradi J, Kiss IT. et al. Synthesis and functional evaluation of a peptide derivative of 1-beta-d-arabinofuranosylcytosine. J Med Chem. 1992;35:3344-9
229. Devy L, De Groot FM, Blacher S. et al. Plasmin-activated doxorubicin prodrugs containing a spacer reduce tumor growth and angiogenesis without systemic toxicity. FASEB J. 2004;18:565-7
230. De Groot FM, Loos WJ, Koekkoek R. et al. Elongated multiple electronic cascade and cyclization spacer systems in activatible anticancer prodrugs for enhanced drug release. J Org Chem. 2001;66:8815-30
231. Wong BK, Defeo-Jones D, Jones RE. et al. Psa-specific and non-psa-specific conversion of a psa-targeted peptide conjugate of doxorubicin to its active metabolites. Drug Metab Dispos. 2001;29:313-8
232. Kumar SK, Williams SA, Isaacs JT. et al. Modulating paclitaxel bioavailability for targeting prostate cancer. Bioorg Med Chem. 2007;15:4973-84
233. Defeo-Jones D, Brady SF, Feng DM. et al. A prostate-specific antigen (psa)-activated vinblastine prodrug selectively kills psa-secreting cells in vivo. Mol Cancer Ther. 2002;1:451-9
234. Brady SF, Pawluczyk JM, Lumma PK. et al. Design and synthesis of a pro-drug of vinblastine targeted at treatment of prostate cancer with enhanced efficacy and reduced systemic toxicity. J Med Chem. 2002;45:4706-15
235. Dubois V, Dasnois L, Lebtahi K. et al. Cpi-0004na, a new extracellularly tumor-activated prodrug of doxorubicin: In vivo toxicity, activity, and tissue distribution confirm tumor cell selectivity. Cancer Res. 2002;62:2327-31
236. Fernandez AM, Van Derpoorten K, Dasnois L. et al. N-succinyl-(beta-alanyl-l-leucyl-l-alanyl-l-leucyl)doxorubicin: An extracellularly tumor-activated prodrug devoid of intravenous acute toxicity. J Med Chem. 2001;44:3750-3
237. Pan C, Cardarelli PM, Nieder MH. et al. Cd10 is a key enzyme involved in the activation of tumor-activated peptide prodrug cpi-0004na and novel analogues: Implications for the design of novel peptide prodrugs for the therapy of cd10+ tumors. Cancer Res. 2003;63:5526-31
238. Trouet A, Passioukov A, Van Derpoorten K. et al. Extracellularly tumor-activated prodrugs for the selective chemotherapy of cancer: Application to doxorubicin and preliminary in vitro and in vivo studies. Cancer Res. 2001;61:2843-6
239. Dubois V, Nieder M, Collot F. et al. Thimet oligopeptidase (ec 3.4.24.15) activates cpi-0004na, an extracellularly tumour-activated prodrug of doxorubicin. Eur J Cancer. 2006;42:3049-56
240. Choi Y, Weissleder R, Tung CH. Selective antitumor effect of novel protease-mediated photodynamic agent. Cancer Res. 2006;66:7225-9
241. Choi Y, Weissleder R, Tung CH. Protease-mediated phototoxicity of a polylysine-chlorin(e6) conjugate. Chem Med Chem. 2006;1:698-701
242. Fiehn C, Kratz F, Sass G. et al. Targeted drug delivery by in vivo coupling to endogenous albumin: An albumin-binding prodrug of methotrexate (mtx) is better than mtx in the treatment of murine collagen-induced arthritis. Ann Rheum Dis. 2008;67:1188-91
243. Pan H, Liu J, Dong Y. et al. Release of prostaglandin e(1) from n-(2-hydroxypropyl)methacrylamide copolymer conjugates by bone cells. Macromol Biosci. 2008;8:599-605
244. Pan H, Kopeckova P, Wang D. et al. Water-soluble hpma copolymer--prostaglandin e1 conjugates containing a cathepsin k sensitive spacer. J Drug Target. 2006;14:425-35
245. Pan H, Sima M, Kopeckova P. et al. Biodistribution and pharmacokinetic studies of bone-targeting n-(2-hydroxypropyl)methacrylamide copolymer-alendronate conjugates. Mol Pharm. 2008;5:548-58
246. Kratz F, Drevs J, Bing G. et al. Development and in vitro efficacy of novel mmp2 and mmp9 specific doxorubicin albumin conjugates. Bioorg Med Chem Lett. 2001;11:2001-6
247. Warnecke A, Fichtner I, Sass G. et al. Synthesis, cleavage profile, and antitumor efficacy of an albumin-binding prodrug of methotrexate that is cleaved by plasmin and cathepsin b. Arch Pharm (Weinheim). 2007;340:389-95
248. Gabriel D, Busso N, So A. et al. Thrombin-sensitive photodynamic agents: A novel strategy for selective synovectomy in rheumatoid arthritis. J Control Release. 2009;138:225-34
249. Hamblin MR, Miller JL, Rizvi I. et al. Pegylation of a chlorin(e6) polymer conjugate increases tumor targeting of photosensitizer. Cancer Res. 2001;61:7155-62
250. Hamblin MR, Miller JL, Rizvi I. et al. Pegylation of charged polymer-photosensitiser conjugates: Effects on photodynamic efficacy. Br J Cancer. 2003;89:937-43
251. Gabriel D, Campo MA, Gurny R. et al. Tailoring protease-sensitive photodynamic agents to specific disease-associated enzymes. Bioconjug Chem. 2007;18:1070-7
252. Campo MA, Gabriel D, Kucera P. et al. Polymeric photosensitizer prodrugs for photodynamic therapy. Photochem Photobiol. 2007;83:958-65
253. Chung DE, Kratz F. Development of a novel albumin-binding prodrug that is cleaved by urokinase-type-plasminogen activator (upa). Bioorg Med Chem Lett. 2006;16:5157-63
254. Jeffrey SC, Nguyen MT, Andreyka JB. et al. Dipeptide-based highly potent doxorubicin antibody conjugates. Bioorg Med Chem Lett. 2006;16:358-62
255. Francisco JA, Cerveny CG, Meyer DL. et al. Cac10-vcmmae, an anti-cd30-monomethyl auristatin e conjugate with potent and selective antitumor activity. Blood. 2003;102:1458-65
256. Hopewel JW, Duncan R, Wilding D. et al. Preclinical evaluation of the cardiotoxicity of pk2: A novel hpma copolymer-doxorubicin-galactosamine conjugate antitumour agent. Hum Exp Toxicol. 2001;20:461-70
257. Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis - the good, the bad, and the ugly. Circ Res. 2002;90:251-62
258. Chipman SD, Oldham FB, Pezzoni G. et al. Biological and clinical characterization of paclitaxel poliglumex (ppx, ct-2103), a macromolecular polymer-drug conjugate. Int J Nanomedicine. 2006;1:375-83
259. O'brien ME, Socinski MA, Popovich AY. et al. Randomized phase III trial comparing single-agent paclitaxel poliglumex (ct-2103, ppx) with single-agent gemcitabine or vinorelbine for the treatment of ps 2 patients with chemotherapy-naive advanced non-small cell lung cancer. J Thorac Oncol. 2008;3:728-34
260. Paz-Ares L, Ross H, O'brien M. et al. Phase III trial comparing paclitaxel poliglumex vs docetaxel in the second-line treatment of non-small-cell lung cancer. Br J Cancer. 2008;98:1608-13
261. Dipaola RS, Rinehart J, Nemunaitis J. et al. Characterization of a novel prostate-specific antigen-activated peptide-doxorubicin conjugate in patients with prostate cancer. J Clin Oncol. 2002;20:1874-9
Author contact
![]() Corresponding author: E-mail: Shawn.Chengov and Seulki.Leegov
Corresponding author: E-mail: Shawn.Chengov and Seulki.Leegov