Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Characteristics and limitations...
Development of a peptide-based...
Radiolabeling of peptides
Peptide radiopharmaceuticals for...
Conclusions
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2012; 2(5):481-501. doi:10.7150/thno.4024 This issue Cite
Review
Radiolabeled Peptides: Valuable Tools for the Detection and Treatment of Cancer
M. Fani1, H. R. Maecke1, S. M. Okarvi2 ![]()
1. Department of Nuclear Medicine, University Hospital Freiburg, Freiburg, Germany
2. Cyclotron and Radiopharmaceuticals Department, King Faisal Specialist Hospital and Research Centre, Riyadh 11211, Saudi Arabia
Received 2011-12-29; Accepted 2012-3-31; Published 2012-5-16
Abstract
Human cancer cells overexpress many peptide receptors as molecular targets. Radiolabeled peptides that bind with high affinity and specificity to the receptors on tumor cells hold great potential for both diagnostic imaging and targeted radionuclide therapy. The advantage of solid-phase peptide synthesis, the availability of different chelating agents and prosthetic groups and bioconjugation techniques permit the facile preparation of a wide variety of peptide-based targeting molecules with diverse biological and tumor targeting properties. Some of these peptides, including somatostatin, bombesin, vasoactive intestinal peptide, gastrin, neurotensin, exendin and RGD are currently under investigation. It is anticipated that in the near future many of these peptides may find applications in nuclear oncology. This article presents recent developments in the field of small peptides, and their applications in the diagnosis and treatment of cancer.
Keywords: Radiolabeled peptides, tumor imaging, radionuclide therapy, radionuclides
Introduction
Molecular imaging techniques are increasingly being used in the localization of disease, the staging of disease and for therapy control. The most sensitive imaging methods are those using nuclear probes for single photon emission computed tomography (SPECT) and positron emission tomography (PET). A variety of imaging probes have been developed for different molecular targets. Radiolabeled peptides are valuable biological tools for tumor receptor imaging and targeted radionuclide therapy. This attributed to the favorable pharmacokinetics and specific tumor targeting characteristics, together with the overexpression of their receptors on the tumor cells, making these peptides attractive agents for imaging and therapy [1-4]. Peptides primarily synthesized in the brain, especially in neurons, are called “neuropeptides” [2, 3]. However, since most of these peptides are also found in the gut, lymphatic tissue, endocrine system, etc., the terminology “regulatory peptide” is often used [3]. Some of the most important regulatory peptides and their receptors overexpressed on tumors are listed in Table 1. The action of neuropeptides is mediated by their binding to specific, membrane-associated receptors. The majority of these receptors belong to the family of G-protein-coupled receptors [2]. These receptors are consisting of a single polypeptide chain, with seven transmembrane domains, an extracellular domain with the ligand binding site, and an intracellular domain linked to G-proteins and arrestin for the activation of second messengers and internalization [5]. These receptors represent useful molecular targets for the detection and treatment of cancer because they are located on the plasma membrane and upon binding of the radioligand, the receptor-ligand complex is internalized, allowing long retention of radioactivity in tumor cells [6].
Peptide receptor expression patterns. The main receptor types overexpressed in human tumor cells are in bold.
| Peptide | Receptor types/subtypes | Tumor expression |
|---|---|---|
| Somatostatin | sst1, sst2, sst3, sst4, sst5 | Neuroendocrine tumors (gastroenteropancreatic tumors), lymphoma, paraganglioma, carcinoids, breast, brain, renal, small cell lung cancer, medullary thyroid cancer |
| Bombesin/GRP | BB1 (NMB-R), BB2 (GRP-R), BB3, BB4 | Prostate, breast, pancreas, gastric, colorectal, small cell lung cancer |
| VIP | VPAC1, VPAC2 | Adenocarcinomas of breast, prostate, stomach and liver; neuroendocrine tumors |
| α-M2 | α-M2-R | Breast cancer |
| α-MSH | MC1-5R | Melanomas |
| CCK/gastrin | CCK1, CCK2 | Medullary thyroid cancer, small cell lung cancer, gastrointestinal stromal tumor, stromal ovarian cancer, astrocytomas |
| Neurotensin | NTR1, NTR2, NTR3 | Small cell lung cancer, colon, exocrine ductal pancreatic cancer, Ewing sarcoma, meningioma, astrocytoma, breast, prostate cancer |
| LHRH | LHRH-R | Prostate, breast cancer |
| Substance P | NK1, NK2, NK3 | Glial tumors (glioblastoma, medullary thyroid cancer), pancreas, breast, small cell lung cancer |
| Exendin | GLP-1 | Insulinomas, gastrinomas, pheochromocytomas, paragangliomas and medullary thyroid carcinomas |
| RGD | αvβ3-integrin | Glioma, breast, prostate cancer |
GRP, gastrin-releasing peptide; VIP, vasoactive intestinal peptide; α-MSH, α-melanocyte-stimulating hormone; CCK, cholecystokinin; LHRH, luteinizing hormone-releasing hormone; GLP, glucagon-like peptide; RGD, Arg-Gly-Asp.
Peptide-based radiopharmaceuticals were introduced into the clinic more than two decades ago [7, 8]. The first and most successful registered imaging agent to date is the somatostatin (SST) analog, 111In-DTPA0-octreotide (111In-OctreoScan, 111In-pentetreotide) (Figure 1) [9, 10]. The Food and Drug Administration (FDA) approved 111In-DTPA0-octreotide is proven to be effective for imaging SST receptor-positive lesions, such as neuroendocrine tumors (NETs), mammary cancer and small cell lung cancer [9-11]. Another important application of radiolabeled octreotide and other SST peptide analogs is peptide receptor-mediated radionuclide therapy (PRRT) [11-14]. The thriving advent of 111In-DTPA0-octreotide raised interest in the development of radiolabeled peptides to target other tumor-related peptide receptor systems [1, 4]. This interest resulted in the discovery of bombesin peptide analogs to target bombesin/gastrin-releasing peptide receptors, which are overexpressed in many common human cancers [2, 15-18]. Other radiolabeled peptides, such as the analogs of vasoactive intestinal peptide [19-21], neurotensin [22-25], cholecystokinin/gastrin [26-29], exendin [30-33] and RGD (Arg-Gly-Asp) [34-37], have been developed and are currently under preclinical or clinical evaluation to establish their applicability for the diagnosis or treatment of cancers [2, 4].
Structures of somatostatin-14 and -28 and [DTPA0-D-Phe1]octreotide (OctreoScan).
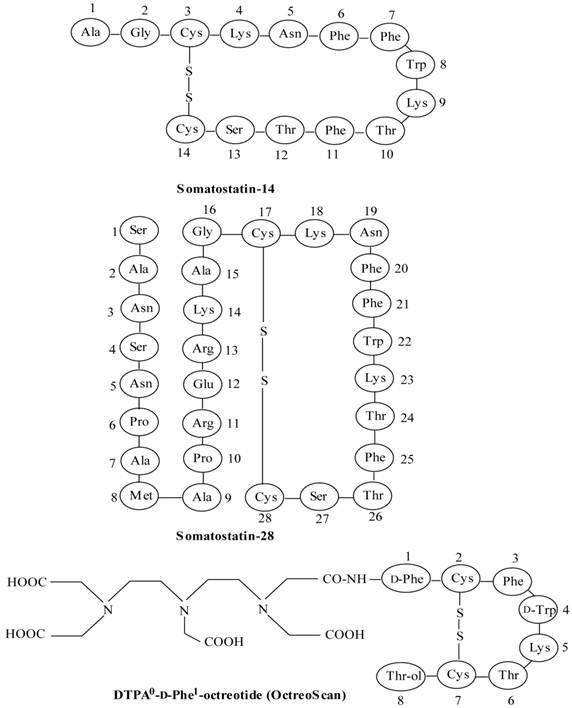
Because of rapid progress in the field of radiolabeled peptides it is difficult to cover all aspects pertaining to the subject. The focus of this article remains the chief applications of radiolabeled peptides in cancer research and development, with major emphasis on recent development of small peptides as targeting agents in nuclear oncology.
Characteristics and limitations of small radiopeptides
The distinctive advantages of small radiopeptides over other biologically active molecules, such as proteins and antibodies, are summarized in Table 2. Receptors for peptides are often found in higher density on tumor cells than in normal tissues; hence specifically designed receptor-binding radiolabeled peptides could enable efficient visualization of tumors. Because of their small size, peptides usually exhibit rapid pharmacokinetics, and good tumor targeting characteristics, with the ability to penetrate into tumors efficiently [1-3]. Peptides can easily be synthesized using conventional peptide synthesizers and the desired pharmacokinetic characteristics can be molecularly engineered (by making appropriate changes in the peptide sequence) during synthesis and/or by adding a biomodifying molecule [38, 39]. Automated peptide synthesizers are available for parallel synthesis procedures allowing the synthesis of peptide libraries in short time.
Requirements of small peptides.
| - Small in size |
| - Easy to synthesize |
| - Easy to radiolabel |
| - feasibility of kit formulation - Amenable to chemical/molecular modifications |
| - Ability to attach a chelating agent at the C- or N-terminus of the peptide |
| - High receptor binding affinity |
| - High tumor penetration |
| - Favorable pharmacokinetics |
| - Attain a high concentration in the target tissues |
| - Rapid clearance from the blood and non-target tissues |
| - Rate and route of excretion can be modified |
| - Few side effects |
| - Not immunogenic |
| - Many biologically important targets |
Tumor receptor imaging creates distinct challenges for the design of peptide-based radiopharmaceuticals and imaging approaches [40]. Most receptors have high affinities for their ligands and are active at nanomolar concentrations of the ligands. Therefore, radiopharmaceutical preparations with high specific radioactivity are crucial. Even small molar quantities of imaging agents may saturate a receptor, limiting the ability to visualize receptor expression and increasing the background of nonspecific binding. For this reason, molecular imaging of tumor receptors has been mainly confined to radionuclide imaging (SPECT and PET), with which it is possible to generate medical images with micromolar to picomolar concentrations of imaging probes [40]. It is important to use a minimum possible amount of a peptide in humans to reduce any adverse pharmacologic effects. For instance, it has been shown that VIP is pharmacologically very potent peptide molecule and doses even in the submicrogram range will produce toxic effects, including hypertension, bronchospasm and diarrhea [4], requiring efficient purification step before administration in order to reduce the administered dose to subpharmacologic levels.
For smaller biomolecules, such as peptides the radioisotope label may significantly affect binding to the receptor and in vivo metabolism. In this situation, the choices of radionuclide, labeling position or location of the radiolabel can be critical [40]. Radiolabeling at a specific-site (chelation-site) remote from the binding region is important to prevent the loss of binding affinity and biological activity of the radiolabeled peptides [1, 3].
The biologic role of most receptor systems important in cancer is derived from their role in the tissue of cancer origin. In general, tumor receptors are expressed in the parent cell lineage and have an established physiologic function. For example, estrogen receptor expression is vital to the function of normal mammary gland epithelial cells. Many tumor receptors also play an important role in promoting carcinogenesis or tumor growth, as is the case for steroid receptors in breast and prostate cancer. The dependence on the receptor pathway for tumor growth makes the receptor a suitable target for therapy, because interruption of the receptor-initiated signal will result in a cessation of tumor growth and often tumor cell death. Thus, knowledge of the levels of receptor expression, which may vary significantly in different types of tumors and even in different sites in the same tumor, is required to infer the possibility that receptor-directed targeting will be effective [40].
One major problem associated with unmodified linear peptides is their often short biological half-life due to rapid proteolysis in plasma. A short half-life in blood is a major obstacle for the successful in vivo application as radiopharmaceuticals since they may be degraded before reaching the intended target. Therefore, most peptides have to be modified synthetically to minimize rapid enzymatic degradation [1, 41]. Great efforts have been focused on developing metabolically-stable peptides suitable for clinical use by carrying appropriate molecular modifications, such as the use of more stable D-amino acids for L-amino acids, the use of pseudo-peptide bonds, the inclusion of amino alcohols and the insertion of unnatural amino acids or amino acid residues with modified side-chains without compromising the receptor binding affinity and biological activity of the peptide [1]. It is the specific amino acid sequence of a peptide and usually the nature and type of particular amino acid side-chains that determine resistance to enzymatic degradation. For example, native SST, has a plasma half-life of approximately 2-3 min, but its molecularly modified synthetic peptide derivative, octreotide has a half-life of 1.5-2 h, making it suitable for clinical application [40].
Another issue related to radiolabeled peptides is often their high uptake and retention by the kidneys, which is of a concern, particularly for radionuclide therapy because of the potential nephrotoxicity [14, 42-44]. Though procedures have been developed and applied successfully in the clinics to manage nephrotoxicity that includes the infusion of a mixture of amino acids, such as lysine and arginine [45]; there is a need to reduce kidney uptake and/or enhanced renal clearance of the radiopeptide. Recently, it has been shown that the use of the cytoprotective drug amifostine [46], and low doses of the plasma expander succinylated gelatin and gelatin-based gelofusine can inhibit the renal uptake of radiolabeled octreotide analogs [47, 48].
Development of a peptide-based radiopharmaceutical
The main steps involved in developing a radiolabeled peptide for clinical application are as follows: (i) identification of the molecular target (receptor) with relevance to human disease and search for a lead peptide, which may be a natural or synthetic peptide, (ii) solid-phase peptide synthesis of a peptide or its analogs. In general, the design of a peptide is based on the structural composition of the endogenous ligand (natural peptide ligand), which exhibits high affinity for the corresponding receptor system. The natural peptide molecule is often structurally modified to produce a metabolically stabilized analog, which preserves most of the biological activity and receptor affinity of the original peptide molecule, (iii) covalent attachment of a chelating agent or a prosthetic group to the peptide either directly or through a linker/spacer group, (iv) radiolabeling that allow high labeling efficiency and high specific activity radiolabeled peptide preparation, (v) in vitro characterization, such as the binding of a radiopeptide with tumor cells, determination of receptor binding affinity, internalization into the tumor cells and dissociation from the tumor cells, (vi) in vivo evaluation to assess the biological behavior, biokinetics and tumor targeting capacity of the radiolabeled peptide in animal models. Many aspects should be considered for further development, such as the accumulation of radiolabeled peptide in target and non-target tissues, the rate and extent of the clearance of radioactivity from the body, the excretory pathway and in vivo stability of the radiolabeled peptide. (vii) The radiolabeled peptides, which successfully passed all the preclinical tests, after toxicological studies and established radiopharmaceutical preparation, may enter clinical studies in humans [10].
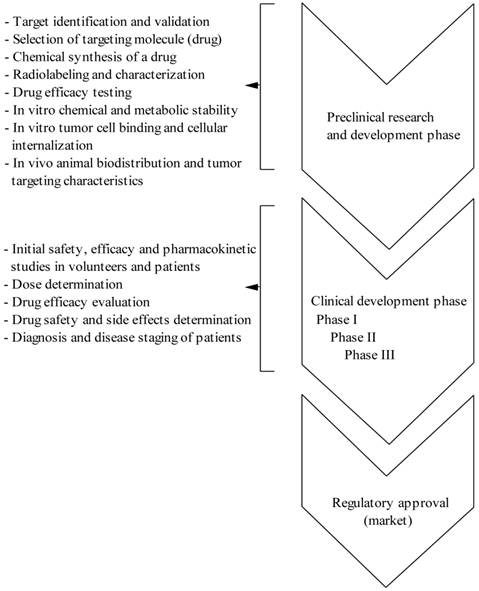
From the design of a new peptide molecule until the use in the clinical settings is a long way and from a huge number of developed radiolabeled peptides only few meet the criteria of a radiopharmaceutical for clinical application. Thus, in the drug development process, considerable preclinical work and the need for validation at each step of development is require in order to transfer a drug from the laboratory bench to the clinical bedside and ultimately obtaining the regulatory approval (see Figure S1/Scheme 1). As mentioned above, several characteristics are desirable when developing a molecularly engineered peptide to be used as a tumor imaging agent: it must bind with high affinity to the target receptor that is found predominantly on cancer cells and not on normal tissues, be specific for its target and not bind to the nontarget tissues, be sufficiently stable to reach the tumor lesions in an intact state, and be cleared rapidly from the blood and nontarget tissues in order to minimize the background radioactivity.
(Scheme 1) Different phases of drug (peptide radiopharmaceutical) development process.
Radiolabeling of peptides
Peptide-based targeted agents either design for diagnostic imaging or radionuclide therapy involve the use of a radiometal. This requires that the radiometal be stably attached to the peptide using a bifunctional chelating agent (BFCA). An important property of BFCA is that it chelates radiometals with high in vivo stability, resulting in minimal deposition of free radiometal in normal tissues. A wide variety of BFCAs and prosthetic groups have been developed in recent years, allowing rapid and convenient radiolabeling of peptides with different radionuclides. Several clinically relevant radionuclides have been used for labeling bioactive peptides either for diagnostic imaging (99mTc, 111In, 68/66Ga, 18F, 123I, 64Cu), or for therapy (111In, 64/67Cu, 90Y, 177Lu, 213Bi) are listed in Table 3.
Methods for labeling peptides with different diagnostic and therapeutic radionuclides.
| Radionuclide | Half-life | BFCA/prosthetic group | Application |
|---|---|---|---|
| Technetium-99m (99mTc) | 6.02 h | MAG3, DADT, HYNIC | diagnosis |
| Fluorine-18 (18F) | 1.83 h | SFB | diagnosis |
| Iodine-123 (123I) | 13.2 h | SIB, SIPC | diagnosis |
| Gallium-68 (68Ga) | 1.13 h | NOTA, DOTA | diagnosis |
| Copper-64 (64Cu) | 12.7 h | TETA, DOTA, NOTA | diagnosis/therapy |
| Indium-111 (111In) | 67.2 h | DTPA, DOTA | diagnosis/therapy |
| Lutetium-177 (177Lu) | 160.8 h | DOTA | therapy |
| Yttrium-90 (90Y) | 64.1 h | DOTA | therapy |
| Bismuth-213 (213Bi) | 45.6 min | DOTA | therapy |
BFCA, bifunctional chelating agent; MAG3, mercaptoacetyltriglycine; DADT, diaminedithiol; HYNIC, 2-hydrazinonicotinic acid; SFB, N-succinimidyl-4-[18F]fluorobenzoate; TETA, 1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetraacetic acid; SIB, N-succinimidyl-3-iodobenzoate; SIPC, N-succinimidyl-5-iodo-3-pyridinecarboxylate; DTPA, diethylenetriaminepentaacetic acid; DOTA, 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid; NOTA, 1,4,7-triazacyclononane-1,4,7-triacetic acid.
Peptide radiopharmaceuticals for tumor imaging
Tumor receptor imaging poses unique challenges for the design and development of a peptide-based agent. The biological actions of the peptides are mediated upon binding with high affinity receptors. The high overexpression of these receptors on various tumor cells as compared to their low density in normal tissues has provided the molecular basis for the clinical use of radiolabeled peptides as tumor receptor imaging and therapeutic agents [40]. After intravenous injection, the radiolabeled peptide will extravascate and bind to sites with high receptor density, e.g. tumor. Imaging and/or therapy follows depending on the radionuclide used. In recent years, many radiolabeled peptide analogs, such as somatostatin, bombesin, vasoactive intestinal peptide, cholecystokinin/gastrin, neurotensin, exendin and RGD derivatives, have been developed for scintigraphic detection of different tumor types [1-4]. These are described below.
Somatostatin peptide analogs
Somatostatin (SST) is a naturally occurring cyclic disulphide-containing peptide with either 14 or 28 amino acids (Figure 1), which binds to SST receptors. Both natural SST-14 and SST-28 bind with high affinity to five different receptor subtypes (sst1-5), but have a short plasma half-life (~3 min), owing to rapid enzymatic degradation by endogenous peptidases, preventing their in vivo use [9, 49-51]. Several synthetic SST peptide analogs that are more resistant to enzymatic degradation have been prepared by molecular modifications preserving most of the biological activity of the original SST peptide [9, 42, 49, 50]. Introduction of the D-amino acids, and decreasing the ring size to the bioactive core sequence, resulted in an 8-amino acid-containing SST analog, “octreotide” (Figures 1 and 2), that preserves the 4-amino acid motif (Phe-D-Trp-Lys-Thr) of native SST-14 involved in receptor binding and has a significantly longer plasma half-life as compared to endogenous SST. Other SST receptor-targeting analogs developed include lanreotide and vapreotide (RC-160) (Figure 2), all with enhanced metabolic stability [9, 50]. These analogs preserved the cyclic form via a disulfide bond. These stabilized SST peptide analogs have different binding profiles for SST receptor subtypes, but all show high affinity for sst2, which is the most prominent SST receptor on human tumors [2, 52, 53].
Structures of DOTA-coupled somatostatin analogs.
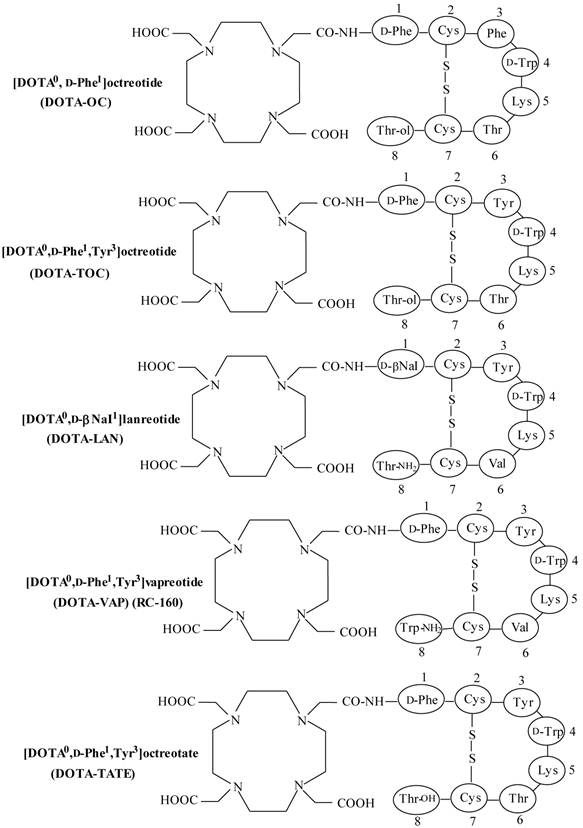
Somatostatin receptors have been identified on the surface of many NETs and represent a valid target for in vivo tumor imaging [2, 55]. SPECT and PET-based agents have been applied successfully to image and quantify the uptake of receptor-specific radiopeptides in SST receptor-positive tumors for cancer staging, treatment planning as well as follow-up response to therapy. Also, SST receptor binding peptides allows the detection of small metastatic sites, which underline the importance and strength of molecular imaging in nuclear oncology [55]. In addition to its usefulness as a diagnostic imaging agent, non-radioactive SST peptide analogs have been used for symptomatic treatment of hormone-secreting NETs [2, 11, 42]. It has been shown that SST peptide analogs reduce the symptoms associated with excess hormone secretion and may also have direct antitumor effects [11, 40]. Several SST-derived peptides labeled with radiohalogens, such as 18F via prosthetic groups, or linked to the chelating agents to facilitate labeling with radiometals, such as 99mTc, 111In, 64Cu and 67/68Ga have been used for in vivo imaging of SST receptor-expressing tumors. When labeled with β-emitters (90Y or 177Lu), these SST peptide analogs can be utilized for receptor-mediated radionuclide therapy [1, 11-14]. A few of SST peptide analogs are summarized in Table 4.
Radiolabeled somatostatin analogs under preclinical/clinical evaluation.
| Peptide | Major application | Reference |
|---|---|---|
| Gluc/Cel-S- Dpr([18F]FBOA)TOCA | - Targeting of sstr-positive AR42J tumor in mice | [114] |
| 64Cu-CB-TE2A-Y3-TATE | - microPET imaging of sstr-positive AR42J tumor in rats | [115] |
| (DOTA0),1-Nal3,Thr8]-octreotide (111In-DOTA-NOC-ATE)/ [DOTA0,BzThi3,Thr8]-octreotide (111In-DOTA-BOC-ATE) | - Targeting of sstr-expressing tumors in AR4-2J tumor- bearing rats | [116] |
| 99mTc-EDDA/HYNIC-TOC (99mTc-TOC) | - Diagnostic imaging of sstr-positive tumors in patients | [117] |
| 99mTc-EDDA/HYNIC-octreotate | - Imaging of sstr-expressing carcinoid tumors in patients | [118] |
| Nα-(1-deoxy-D-fructosyl)-Nε-(2-[18F]fluoropropionyl)-Lys0-Tyr3-octreotate (Glu-Lys([18F] FP)-TOCA) | - PET imaging of sstr-expressing tumors in patients | [119] |
| [18F]FP-Glu-TOCA | - PET imaging of sstr-positive tumors in patient with metastatic carcinoid in the liver | [120] |
| 68Ga-DOTA-D-Phe1-Tyr3-octreotide (68Ga-DOTA-TOC) | - PET imaging of sstr-positive meningioma tumors in Patients | [121] |
| [DOTA]-1-Nal3-octreotide (68Ga-DOTA-NOC) | - PET imaging of sstr (subtypes 2 and 5) expressing tumors in patients | [122] |
| 111In-DOTA-TATE | - Imaging of sstr-positive tumors in patients | [123] |
| [111In-DOTA]-lanreotide (111In-DOTA-LAN) | - Detection of sstr-expressing tumors in patients | [124] |
| Maltotriose-[123I] Tyr3-octreotate ([123I]Mtr-TOCA) | - Scintigraphic detection of sstr-positive tumors in patients | [125] |
sstr, somatostatin receptor; Glu, glucose; Cel, cellobiose; CB-TE2A, cross-bridged-tetraazamacrocycle 4,11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo [6.6.2]hexadecane; BzThi, benzothienyl-Ala. 99mTc-TOC, 99mTc-hydrazinonicotinyl-Tyr3-octreotide; EDDA, ethylenediamine-N,N'-diacetic acid; [18F]FP-Gluc-TOCA, Nα-(1-deoxy-D-fructosyl)-Nε-(2-[18F]fluoropropionyl)-Lys0-Tyr3-octreotate.
There are a number of factors that usually determine the tumor uptake capacity of radiolabeled SST peptide analogs. These include: (i) high specific radioactivity preparation of the radioligand, (ii) the in vivo stability of the radioligand, (iii) the density of SST receptor-expression in the tumor, (iv) the type/subtype of SST receptors expression by the tumor, (v) the receptor binding affinity of the radiolabeled SST analog for the particular SST receptor type, (vi) the efficiency of SST receptor-mediated internalization and recycling of radiopeptide, (vii) the final trapping of the radiolabeled SST peptide analog within tumor cells, and (viii) the amount of the peptide administered [5, 11, 54].
It is worth mentioning here that common to all receptors is the interaction of a ligand and the receptor, in which specific binding of the ligand to the receptor results in downstream biochemical or physiologic changes. Ligands that cause physiological changes with receptor binding, typically the naturally occurring ligands, are called “agonists”. Ligands that bind to the receptor and block the binding of agonists but that do not activate changes are known as “antagonists” [40]. It is generally believed that receptor agonist radioligands are more suited for tumor targeting as they exhibit good receptor-mediated internalization into the tumor cell upon binding to the respective cell-surface receptors, thus promoting active accumulation in the target results in optimal visualization [3]. Interestingly, however, a recent study performed with two potent SST receptor-selective antagonists demonstrated that the high-affinity SST receptor antagonists that poorly internalized into tumor cells, can also be effective in terms of in vivo tumor uptake characteristics in animal models as compared to the corresponding agonists, which highly internalized into tumor cells. This observation which was made both for sst2 and sst3-selective SST peptide analogs, demonstrates that the SST receptor antagonists are preferable to SST receptor agonists for in vivo tumor targeting [56]. In another recent study, 64Cu-CB-TE2A-sst2-ANT, a SST antagonist was evaluated for in vivo PET imaging of sst2-positive tumors and compared to 64Cu-CB-TE2A-TATE [57]. The pharmacokinetic characteristics indicated the slight superiority of the radioantagonists over receptor agonists.
The most recent study on the newly developed sst2-antagonist, namely LM3, demonstrated high and persistent tumor uptake of radiolabeled SST antagonists. Also, profound influence of the chelator and the radiometal was observed on the receptor binding affinity of the radiolabeled conjugates [58]. The first clinical study of SST antagonists confirmed the preclinical data as it showed higher tumor uptake of the antagonist 111In-DOTA-sst2-ANT compared to the agonist 111In-DTPA0-octreotide and improved tumor-to-background contrast, in particular tumor-to-kidney [59].
Bombesin peptide analogs
Bombesin (BN), an amphibian 14-amino acid peptide (Figure 3), is a homolog of the 27-amino acid mammalian gastrin-releasing peptide (GRP) (Figure 3). Bombesin and GRP share a highly conserved 7-amino acid C-terminal sequence (Trp-Ala-Val-Gly-His-Leu-Met-NH2), which is required for immunogenicity and for high-affinity binding to the BN/GRP-preferring receptor [1, 2, 60]. Both BN and GRP show high affinity binding to the human GRP receptor, which is overexpressed by a variety of cancers, including prostate, breast, gastrointestinal and small cell lung cancer [2, 15, 16]. The BN receptor family comprises four subtypes of G-protein-coupled receptors, including the neuromedin B (NMB) receptor (BB1), the GRP receptor (BB2), the orphan receptor (BB3), and the amphibian receptor (BB4) [2, 15, 16]. Following activation of these receptors, BN and GRP possess a variety of physiological and pharmacological functions and also play an important role in stimulating the growth of different types of cancers [2]. Of the four receptors, three receptor subtypes (BB1, BB2, and BB3) have shown to be expressed in variable degrees on various cancers. Of particular interest is the GRP receptor (BB2) that has been found to be overexpressed in a variety of tumors, including lung, breast, GIST, brain and prostate [2, 15, 16], and hence is an attractive target for the detection and treatment of these cancers. Radiolabeled BN-like peptides, which bind to BN/GRP receptors with high affinity and specificity, have potential to be used for site-directed diagnostic and/or therapeutic purposes [1, 3, 4]. A recent study with 68Ga-labeled Pan-bombesin analog, DOTA-PEG2-[D-Tyr6, β-Ala11, Thi13, Nle14] BN(6-14)amide (68Ga-BZH3), was performed on patients with gastrointestinal stromal tumors in order to determine the impact of peptide receptor scintigraphy on the diagnosis and the potential therapy [17]. Also in vivo kinetics of the BN peptide was compared with 18F-fluorodeoxyglucose (18F-FDG) on the same patients. The study demonstrates that 68Ga-BZH3 showed poor tumor targeting potential as compared to 18F-FDG. Further studies on larger number of patients are required to determine the real potential of 68Ga-BZH3 for targeting BN receptor-expressing tumors.
Amino acid sequences of gastrin-releasing peptide, bombesin-14, vasoactive intestinal peptide, cholecystokinin and neurotensin.

One major problem that often associated with BN-like peptides is their tendency to accumulate in the liver and intestines due to high hepatobiliary clearance despite their hydrophilic nature, making scintigraphic exploration of the abdominal area problematic [6]. The presence of GRP receptor in gastrointestinal tissues might be responsible for high hepatobiliary clearance of BN-like peptides [60]. Several new BN peptide derivatives with the low liver and intestinal uptake have been designed and developed [1, 10, 61]. A number of BN peptide analogs radiolabeled with 99mTc, 68Ga, 111In, 64Cu, 18F and 177Lu have been prepared and investigated for targeting of BN receptor-expressing tumors including prostate and breast cancer and are summarized in Tables 5 and 6. Among these analogs, BN peptide antagonists have also been developed in order to further validate the “shift” from agonists to antagonists [62-64]. The data suggest that the GRP antagonists may be superior targeting agents to GRP receptor agonists, which indicate a possible change of paradigm in the field of BN-like peptides. In addition, the antagonists have clearly less side-effects compared to the agonists. The encouraging results obtained with these radiolabeled BN analogs indicate that some of them may find applications in clinical oncology in the near future.
Radiolabeled bombesin peptide analogs.
| Radiopeptide | Major application | Reference |
|---|---|---|
| 111In-DTPA-GABA-[D-Tyr6-Gln7-Trp8-Ala9-Val10-β-Ala11-His12-Thi13-Nle14-NH2 BN(6-14) (BZH1) | - Targeting of BN/GRP receptor subtypes expressing tumors in rats | [126] |
| [111In/177Lu]-DOTA-GABA-[D-Tyr6-Gln7-Trp8- Ala9-Val10-β-Ala11-His12-Thi13-Nle14-NH2 BN(6-14) (BZH2) | ||
| [67Ga/177Lu]-DOTA-PEG4-BN(7-14) (DOTA-PESIN) | - Imaging and therapy of BN/GRP receptor-positive PC-3 tumors in mice | [127] |
| 177Lu-DO3A-CH2CO-G-4-aminobenzoyl- Q-W-A-V-G-H-L-M-NH2 (177Lu-AMBA) | - Imaging and therapy of GRP receptor-positive tumors in mouse models | [128] |
| 177Lu-DOTA-8-Aoc-BBN(7-14)NH2 / 64Cu- NOTA-8-Aoc-BBN(7-14)NH2 | - Targeting of BN/GRP receptor-positive PC-3 prostate cancer cells in mice | [129, 130] |
| 111In-[DTPA-Pro1,Tyr4]BN | - Targeting of androgen-dependent BN/GRP receptor in human prostate tumor xenografts | [131] |
| [111In-DTPA-ACMpip5,Tha6,βAla11,Tha13,Nle14]BN(5-14) | - Imaging of BN/GRP receptor-expressing prostate cancer in PC-3 tumor-bearing mice | [132] |
| [64Cu-NO2A-8-Aoc-BBN(7-14)NH2]/ [64Cu-NO2A-(X)-BBN(7-14)NH2] | - microPET imaging of GRP receptor-positive T47-D breast and PC-3 prostate cancer xenografts in mice | [133] |
| 64Cu-CB-TE2A-8-AOC-BBN(7-14)NH2 64Cu-DOTA-8-AOC-BBN(7-14)NH2 | - microPET/CT imaging of BB2 receptor-positive prostate tumor in nude mice | [134] |
| 64Cu-DOTA-linker-BN(7-14) | - microPET imaging of BN/GRP receptor-positive T47-D breast cancer in mice. | [135] |
| 64Cu-SarAr-SA-AoC-GSG-BN(7-14)/ 64Cu-SarAr-SA-AoC-BN(7-14) | - Targeting of GRP receptor-expressing prostate cancer in mice bearing PC-3 xenografts using PET/CT. | [136] |
| 111In/68Ga-RM2 | - SPECT/PET-based imaging of GRP receptor-positive PC-3 and LNCaP tumor-bearing nude mice. | [137] |
| 68Ga-AMBA | - GRP receptor-based imaging of prostate cancer in mice bearing human prostate VCaP xenografts. | [138] |
GABA, γ-aminobutyric acid; PEG4, 15-amino-4,7,10,13-tetraoxapentadecanoic acid; X, 0, β-Ala, 5-Ava, 8-Aoc, or 11-Aun; CB-TE2A, 1,4,8,11-tetraazabicyclo[6.6.2]hexadecane-4,11-diacetic acid. linker, Aba, Ava, Ahx, Aoc, or Ado.; 8-Aoc, 8-aminooctanoic acid; GRP, gastrin-releasing peptide; Aca, aminocaproic acid; 8-Aoc, 8-aminooctanoic acid.
Bombesin peptide analogs radiolabeled with F-18 and Tc-99m radionuclides.
| Radiopeptide | Major application | Reference |
|---|---|---|
| 18F-FB-[Lys3]BBN/18F-FB-Aca-BBN(7-14) | - microPET imaging of BN/GRP receptor-positive prostate cancer in nude mice | [139] |
| 18F-BAY 86-4367 | - Detection of BN/GRP receptor-positive PC-3 xenografts using PET/CT. | [140] |
| 99mTc(CO)3-BBS(7-14) | - Targeting of BN/GRP receptor-positive PC-3 prostate cancer in nude mice | [141] |
| 99mTc(CO)3-DTMA-(X)-BBN(7-14)NH2 | - Targeting of GRP receptor-positive prostate cancer in mice bearing PC-3 xenografs | [142] |
| 99mTc-(CO)3-Lys-PEG-BN | - Small-animal SPECT imaging of BN2/GRP receptor-positive prostate cancers in nude mice with PC-3 tumor xenografts. | [143] |
| 99mTc-(CO)3-X-Y-BN(7-14)NH2 | - Targeting of GRP receptor-positive T47-D breast cancer in xenografted SCID mice. | [144] |
FB, 4-Fluorobenzoyl; BBN, bombesin; GRP, gastrin-releasing peptide; Aca, aminocaproic acid; DTMA, 2-(N,N'-Bis(tert-butoxycarbonyl)diethylenetriamine) acetic acid.
Vasoactive intestinal peptide analogs
Vasoactive intestinal peptide (VIP) is a 28-amino acid neuropeptide (Figure 3) with a broad range of biologic activities. In addition to its vasodilatory properties, it promotes growth and proliferation of normal and cancer cells mediated by cell-surface receptors [1, 2, 20]. The actions of VIP are mediated by two receptor subtypes (VPAC1 and VPAC2). Each of these receptor subtypes have different pharmacology and distributions, but VIP has high-affinity for both VIP receptor subtypes [65]. VIP is pharmacologically very potent molecule and doses in the submicrogram range will produce toxic effects thus requiring an efficient purification step of the radiolabeled peptide before administration in order to reduce the administration dose to subpharmacologic levels [6]. As with other natural peptides, VIP also undergoes rapid degradation in vivo with a half-life less than 1 min [66]. Thus, the development of synthetic VIP analogs that are metabolically stable and retain high affinity and selectivity for VIP receptors or a VIP receptor subtype can be useful for targeting VIP receptor-expressing tumors. A high number of VIP receptors are expressed on various tumors, including neuroendocrine tumors, brain tumors, adenocarcinomas of the pancreas, prostate, breast, colon, stomach and liver [2, 67]. Clinical studies of VIP analogs radiolabeled with 123I and 99mTc for scintigraphic imaging of VIP receptor-positive tumors (pancreatic adenocarcinomas, endocrine tumors, colorectal cancer, etc.) demonstrate the usefulness of these radiolabeled VIP analogs [19-21]. Unfortunately, the overexpression of VIP receptors on normal tissues, particularly in the lungs, central nervous system, liver, intestine etc., combined with the complex radiolabeling protocol limit the widespread clinical application of radiolabeled VIP analogs.
Cholecystokinin/gastrin peptide analogs
The peptides gastrin and cholecystokinin (CCK), which are highly expressed in the intestines and also in brain, exert their functions through interaction with two receptors, CCK2/gastrin and CCK1 receptors [2, 26]. High expressions of CCK2/gastrin receptors are found in human medullary thyroid cancer (MTC), small cell lung cancer (SCLC), astrocytomas, stromal ovarian cancer, gastroenteropancreatic NETs and gastrointestinal stromal tumors [2, 67]. The peptides of the gastrin and CCK family are characterized by the C-terminal amidated receptor binding sequence Trp-Met-Asp-Phe-NH2. It has been shown that the C-terminal octapeptide portion of CCK (Figure 3) has the same efficacy and potency as the full length 33-amino acid residue peptide [2, 28]. The development of radiolabeled CCK/gastrin peptide analogs is beneficial for targeting the CCK/gastrin expressing tumors in vivo. In a recent study [27], the diagnostic and therapeutic value of three promising CCK-2 receptor-binding peptides, 99mTc-N4-Gly-D-Glu-(Glu)5-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2 (99mTc-demogastrin 2), 111In-DOTA-D-Asp-Tyr-Nle-Gly-Trp-Nle-Asp-Phe-NH2 (111In-DOTA-CCK), and 111In-DOTA-D-Glu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2 (111In-DOTA-MG11) were evaluated in patients with MTCs. The results of this study suggest that 99mTc-demogastrin 2 showed better detectability and visualized more tumor lesions in patients than with 111In-DOTA-CCK and 111In-DOTA-MG11. Also, the uptake of the 111In-labeled peptides in patients with MTCs and metastases was found to be lower than that of 99mTc-demogastrin 2. All three radiolabeled peptides showed comparable renal uptake profiles. These findings indicate the potential 99mTc-demogastrin 2 for targeting of the CCK/gastrin expressing tumors in patients and deserve further evaluation.
DOTA-conjugates of MG11 have been developed by introducing hydrophilic but uncharged spacers of different lengths, such as PEG or D-amino acids (D-Ser or D-Gln) [29], where it was shown that the number of D-Gln residues played an important role and the length of the spacer determined the metabolic stability as this was significantly improving from (D-Gln)2 to (D-Gln)6. Recently, several radiolabeled CCK/gastrin peptide analogs have been prepared and evaluated for diagnostic imaging or radionuclide therapy of CCK/gastrin receptor-expressing tumors. Based on side-by-side comparative studies among twelve DOTA-conjugated analogs, developed by different research groups, optimal peptide analogs were found to be the linear minigastrin analog, with six D-Glu residues (PP-F11), a divalent analog (MGD5) and a cyclic peptide cyclo-MG1, combining the high tumor uptake with low kidney retention [68].
Neurotensin peptide analogs
Neurotensin (NT) is a tridecapeptide (Figure 3) found in the brain and gut and has many growth regulatory functions. The biological effects of NT are mediated through the action of three different NT receptors: NTR1, NTR2 and NTR3 [2, 69]. The pharmacological effects of NT initiated from the specific interaction of the peptide with cell-surface receptors. But the pharmacology and mode of action of NT receptors are not well understood [2]. NTR1 and NTR2 belong to the family of G-protein-coupled receptors, whereas the NTR3 is a single transmembrane domain receptor and is similar to sortilin, a protein involved in receptor sorting [70]. Overexpression of NTR1 has been found in several human cancers including Ewing sarcomas, meningiomas, astrocytomas, medulloblastomas and pancreatic carcinomas [2, 67]. Neurotensin is rapidly degraded in blood by endogenous peptidases. Recently, different cleavage-sites in the natural sequence have been identified which provided the future direction for the development of metabolically-stable NT analogs [23]. Based on the favorable preclinical animal studies, one of the 99mTc-labeled NT analogs, [99mTc(CO)3](NαHis)Ac-Lys-(ψCH2-NH)-Arg-Pro-Tyr-Tle-Leu (99mTc-NT-XI), was evaluated for its tumor targeting characteristics in ductal pancreatic adenocarcinoma patients [22]. The initial clinical findings are not very favorable due to the high nonspecific uptake of radioactivity in the intestinal region and in the kidneys. More clinical data is required to determine the real potential of this radiolabeled peptide as a tumor imaging agent. Some of the NT analogs developed for targeting NT receptor-expressing tumors are summarized in Table 7.
Tumor imaging peptide radiopharmaceuticals based on neurotensin or α-MSH.
| Radiopeptide | Major application | Reference |
|---|---|---|
| 99mTc/188Re-NT-XIX | - Imaging and therapy of NTR1-positive tumors in nude mice with HT-29 xenografts | [23] |
| [99mTc-N40,(β)Ala7,Dab9,XAA12] NT(7-13) | - Imaging of NTR1-positive tumors in WiDr tumor-bearing mice | [25] |
| [Cys3,4,10,D-Phe7,Arg11]α-MSH3-13 (99mTc-(Arg11)CCMSH)/ [68Ga/111In]-DOTA-Re(Arg11)CCMSH | - Targeting of primary and metastatic melanoma in B16/F1 melanoma-bearing mice | [71, 78] |
| 64Cu-CBTE2A-ReCCMSH(Arg11) | - microPET imaging of malignant melanoma in mice bearing B16/F1 melanoma tumor | [72] |
| [Ac-Nle4,Asp5,D-Phe5,Lys11(pz-99mTc(CO)3]-α-MSH4-11 | - Melanoma tumor targeting in melanoma-bearing C57BL6 mice | [145] |
| [111In/90Y/68Ga]-CHX-A″-Re(Arg11)CCSMSH | - SPECT/PET imaging of melanoma tumors in B16/F1 melanoma-bearing mice | [73] |
| Ac-Nle-Asp-His-D-Phe-Arg-Trp-Gly-Lys-NH2 (18F-FB-NAPamide) | - PET imaging of MC1R expression in melanoma xenografted mouse models | [74] |
NT, neurotensin; NTR1, neurotensin receptor 1; XAA, Ile or Tle.
Alpha-melanocyte stimulating hormone peptide analogs
In recent years, alpha-melanocyte-stimulating hormone (α-MSH) peptide analogs were prepared and evaluated for both melanoma imaging and radiotherapy [71-75]. Alpha-MSH is a 13-amino acid peptide (Ac-Ser1-Tyr2-Ser3-Met4-Glu5-His6-Phe7-Arg8-Trp9-Gly10-Lys11-Pro12-Val13-NH2) and is mainly responsible for the regulation of skin pigmentation [76]. Alpha-MSH peptide targets the α-MSH receptor (melanocortin type 1 receptor (MC1R)). The MC1R is overexpressed in most murine and human melanoma metastases [2, 77], thus making it an attractive molecular target for imaging and radiotherapy of melanomas. Recently, several α-MSH peptide analogs radiolabeled with 99mTc [71], 64Cu [72], 68Ga [73], 18F [74], and 111In [71, 75] have been prepared and evaluated for melanoma targeting (see Table 7). In addition, an α-MSH analog, ReCCMSH(Arg11), radiolabeled with 188Re has shown good therapeutic potential in animals [78]. The data demonstrates that radiolabeled α-MSH analogs are potential candidates for diagnostic imaging or radiotherapy of melanoma tumors.
RGD peptides for targeting integrin αvβ3 expression
In addition to target G-protein-coupled receptors on tumor cell membranes, there has been a growing interest in targeting integrin receptors for in vivo tumor imaging [6, 79, 80]. The vitronectin receptor, αvβ3 integrin receptor is known to play an important role in tumor-induced angiogenesis and tumor metastasis. This receptor is expressed on various malignant human tumors (ovarian, breast, neuroblastoma, melanoma, etc.) and is highly expressed on activated endothelial cells during angiogenesis [34, 81]. In contrast, expression of αvβ3 is weak in resting endothelial cells and most normal tissues. The restricted expression of integrin αvβ3 during tumor growth, invasion and metastasis present an attractive molecular target for diagnosis and therapy of the rapidly growing and metastatic tumors [79]. The inhibition of αvβ3 is currently being evaluated as a new strategy for tumor-specific anticancer therapy [34]. RGD family of peptides containing the RGD sequences preferentially bind to the αvβ3 integrin receptor have been used for noninvasive imaging of the integrin αvβ3-positive tumors by SPECT and PET. A number of synthetic peptides containing the RGD sequences have been radiolabeled with 99mTc, 111In, 123I, 68Ga, 64Cu, 18F and 90Y and evaluated for their ability to target the αvβ3 receptor (see Table 8). Most of them showed in vitro affinity for αvβ3 and allowed targeting of receptor-positive tumors in vivo. However, major differences were found in the pharmacokinetic profiles of different radiolabeled RGD peptide analogs [34-37, 79, 82-92]. One of the peptide tested clinically is [18F]Galacto-RGD, a glycosylated cyclic pentapeptide, which demonstrated noninvasive quantitative assessment of the αvβ3 expression pattern in patients with malignant tumors [34, 80]. However, the facile preparation of labeled RGD with radiometals, such as 68Ga-NODAGA-c(RGDfK) makes it an attractive alternative to 18F-labelled RGD peptides. Moreover, NODAGA has as well been used for radiolabeling of RGD with 64Cu showing improved characteristics compared to the corresponding 64Cu-DOTA-c(RGDfK) [36].
RGD peptide-based radiopharmaceuticals.
| Radiopeptide | Major application | Reference |
|---|---|---|
| Cyclo(RGDfK(DOTA)) [68Ga/111In-DOTA-RGD] | - Targeting of αvβ3-positive human melanoma M21 tumor cells in nude mice | [35] |
| c(RGDyK), (68Ga-NOTA-RGD1) E[c(RGDyK)]2, (68Ga-NOTA-RGD2) E{E[c(RGDyK]2}2 (68Ga-NOTA-RGD4) | - microPET imaging of integrin αvβ3 expression in nude mice bearing U87MG human glioblastoma xenografts | [85] |
| PEG4-E[PEG4-c(RGDfK)]2, P-PRGD2) (18F-FP-P-PRGD2) | - Imaging of tumor angiogenesis in nude mice bearing αvβ3-positive tumors | [86] |
| 64Cu-DOTA-E{E[c(RGDfK)]2}2 | - microPET imaging of glioma integrin αvβ3 expression in mice with glioma xenografts | [87] |
| 64Cu-CB-TE2A-c(RGDyK) 64Cu-diamsar-c(RGDfD) | - Targeting of tumor angiogenesis in nude mice with αvβ3-positive M21melanoma | [146] |
| [111In]-DOTA-E-[c(RGDfk)]2 [111In]-DOTA-PEG4-E-[c(RGDfk)]2 [111In]-DOTA-E-E-[c(RGDfK)]2 [111In]-DOTA-K-E-[c(RGDfK)]2 | - Targeting of αvβ3-expressing tumors in mice carrying SK-RC-52 cells | [88] |
| 111In-DOTA-E-c(RGDfK) | - αvβ3 integrin-targeting in mice with OVCAR-3 human ovarian carcinoma cells | [89] |
| [111In/90Y]-DOTA-E-[c(RGDfK)]2 | - Diagnosis/therapy of αvβ3-expressing tumors in mice bearing αvβ3-positive OVCAR-3 cells | [90] |
| 111In-DOTA-E-c(RGDfK) 111In-DOTA-E-[c(RGDfK)]2 111In-DOTA-E-{E[c(RGDfK)]2}2 | - Targeting of αvβ3 inegrin expression in mice with SK-RC-52 xenografts | [37] |
| 68Ga-NOTA-E[PEG4-c(RGDfK)]2 68Ga-NOTA-E[Gly3-c(RGDfK)]2 | - microPET imaging of integrin αvβ3 expression in U87MG tumor xenograft models | [91] |
| 68Ga/64Cu-NODAGA-c(RGDfK) | - microPET imaging of integrin αvβ3 expression in U87MG tumor xenograft models | [36] |
| [99mTc]HYNIC-RGD | - Imaging integrin αvβ3 expression in nude mice bearing M21, M21L melanoma and A549 small cell lung tumor cells | [92] |
| 99mTc-HPMA-RGD4C-DPK | - Targeting tumor angiogenesis in mice bearing DU145 or PC-3 prostate tumor xenografts | [147] |
| 99mTcO(MAG2-3G3-dimer) | - Imaging of tumor integrin αvβ3 expression in nude mice bearing U87MG xenografts | [148] |
RGD, Arg-Gly-Asp; DOTA, 1,4,7,10-tetraazadodecane-N,N',N'', N'''-tetraacetic acid; PEG4, 15-amino-4,7,10,13-tetraoxapentadecanoic acid; NOTA, 1,4,7-triazacyclononane-1,4,7-triacetic acid; 18F-FP, 4-nitrophenyl 2-18F-fluoropropionate; HPMA, N-(2-hydroxypropyl)methacrylamide).
In a recent study, a RGD-SST hybrid peptide, RGD-DTPA-octreotate [c(Arg-Gly-Asp-D-Tyr-Asp)-Lys(DTPA)-D-Phe-c(Cys-Tyr-D-Trp-Lys-Thr-Cys)-Thr], composed of the SST receptor-targeting peptide, Tyr3-octreotate peptide [D-Phe-c(Cys-Tyr-D-Trp-Lys-Thr-Cys)-Thr], the DTPA chelator and the apoptosis-inducing RGD peptide sequence was evaluated for targeting SST and αvβ3-receptor-positive tumors. In rats bearing CA20948 rat pancreatic tumors, RGD-111In-DTPA-octreotate showed a high tumor uptake and retention. A significantly high renal uptake was observed in rats making this peptide unsuitable for radiotherapy. The results highlight the benefit of using RGD-SST hybrid peptide for targeting of tumors [79, 82]. It was also found that the 111In-labeled hybrid peptide, RGD-DTPA-octreotate, is more powerful in promoting apoptosis than the two monopeptides (the RGD and octreotate), thus the hybrid peptide can greatly improve the therapeutic efficacy of SST peptide analogs [82, 83].
To find out whether dual receptor-targeting is advantageous over a single receptor-binding probe, recently BBN-RGD hybrid peptide was evaluated for its ability to target an androgen-independent prostate cancer cell line (PC-3), which expresses receptors for both GRP and integrin αvβ3 [84]. A heterodimer BBN-RGD conjugate, that binds to both GRP and integrin αvβ3 receptor, was synthesized by coupling the cyclic RGD peptide c(RGDyK) with BBN(7-14) through a glutamate linker and then labeled with 18F. The dual receptor-targeting (GRP and integrin αvβ3) study showed the significantly higher tumor targeting and better pharmacokinetic characteristics of 18F-BBN-RGD as compared to the corresponding monomeric 18F-BBN and 18F-RGD analogs. Thus, there is a need to develop heterodimeric ligands for efficient targeting of multiple-receptor systems and this interesting approach should also be tested on other peptide receptor systems. Based on the RGD peptide sequence, a number of small cyclic peptides that possess high and specific affinity for the αvβ3 integrin have been synthesized and radiolabeled with SPECT/PET-based radionuclides (see Table 8). The abilities of these peptides to target αvβ3-expressing tumors have made radiolabeled cyclic RGD peptides powerful tools for diagnostic imaging and targeted therapy of αvβ3-expressing tumors.
Exendin peptide analogs
The Glucagone-like peptide receptor (GLP-1R) is a member of the G-protein-coupled receptor family and is mainly expressed in the pancreatic islet cells, the intestine, lung, kidney, breast and the brainstem. GLP-1R were found to be overexpressed in insulinomas, an usually benign insulin secreting neuroendocrine tumor of the pancreas [53]. The natural ligand GLP-1 consists of 30-amino acids and has a half-life of <2 min. Exendin-4 and Exendin-3 are two 39-amino acid natural peptides which are metabolically resistant and share nearly 50% homology with the human GLP-1. Exendin-4 and Exendin-3 differ by only two amino acid residues near the amino terminus.
Exendin-4 has been radiolabeled with 111In initially using DTPA coupled to Lys40 side-chain with aminohexanoic acid (Ahx) as a spacer ([Lys40(Ahx-DTPA)NH2]-exendin-4) [30, 31]. 111In-DTPA-exendin-4 was evaluated for its therapeutic efficiency, based on the the Auger electrons of 111In in tumor bearing transgenic Rip1Tag2 mice [93]. Clinically, 111In-DTPA-exendin-4 scans were able to distinguish between benign and malignant insulinomas [94]. Recently, 111In-DTPA-exendin-4 was used to follow β-cell viability of an islet transplantation into the forearm of a human patient, where one year after the transplantation, focal accumulation of the tracer was observed in planar images [95]. The corresponding 111In-DOTA-exendin-4 conjugate has also been used in patients with very promising results in the localization of insulinomas and detection of pancreatic and ectopic insulinomas [96, 97]. Exendin-4 and exendin-3 have been radiolabeled with 99mTc and 68Ga [32, 33]. Among these tracers, 99mTc-labeled exendin-4 had the lowest estimated effective dose and it is currently under clinical evaluation [98, 99].
Receptor-mediated radionuclide therapy with peptide radiopharmaceuticals
Receptor-binding peptides radiolabeled with diagnostic radionuclides can be used for identification of receptor-positive tumor lesions, treatment planning and dosimetry. When labeled with a therapeutic radionuclide, the same peptide can be utilized for targeted radionuclide therapy [9-14]. Typically, peptide receptor radionuclide therapy carried out with a peptide labeled with a therapeutic radionuclide, able to bind specifically to the target receptor on tumor cells and be able to deliver an effective radiation dose to the tumor cells with minimal damage to normal tissues. The success of this approach depends upon the amount of radioligand accumulated within tumor cells and the rates of internalization, degradation and recycling of both radioligand and receptor. The location and the size of tumor/tumor type are the important factors to be considered for effective radiotherapy. The in vivo stability and construction of a peptide radiopharmaceutical is crucial as it may influence tumor uptake, retention, and clearance via the renal pathway [100]. The radionuclide's physical properties, such as the type of radiation emitted, the emission energies, the distance over which the energy is deposited, and the physical half-life of the radionuclide are important. Some of the therapeutic radionuclides used for labeling peptides include yttrium-90 (90Y) and lutetium-177 (177Lu). Both are β-particle emitters; 90Y has a maximum energy of 2.3 MeV and a maximum range of 12 mm in tissue and 177Lu has a maximum energy of 0.5 MeV and a maximum tissue range of approximately 2 mm [13, 14, 101]. Lu-177 is also a γ-emitter, which enables γ-scintigraphy and thus facilitates dosimetry and disease staging. A low-energy β-emitter, such as 177Lu, with a shorter range is more appropriate for smaller tumors or metastases while the high-energy β-emitter such as 90Y is suited for larger tumors, with a heterogenous receptor distribution [11, 13, 51, 101, 102]. The selection of a radionuclide for therapy is thus dependent on the potential clinical applications and on the chemistry and physical properties of the therapeutic radionuclide [103]. Indeed, it has been shown that the combination of radionuclides with different β-energies and particle ranges can achieve higher cure rates in tumors of different sizes. Clinical trials are underway to support the experimental findings.
A number of radiolabeled SST peptide analogs are currently in use to treat patients with SST receptor-expressing tumors. These conjugates consist of a SST peptide analog (i.e. octreotide or octreotate), a chelating moiety (i.e. DOTA) and, a therapeutic radionuclide (i.e. 90Y or 177Lu). The chelator, which is linked to the SST peptide analog, forms a stable complex with the radionuclide. The basic principle of tumor-targeting after systemic administration of the peptide involves binding to the SST receptors, which are overexpressed on the cell surface of the tumor cells, followed by internalization of the radionuclide-peptide complex into the tumor cells. The emitted radiation can damage the DNA, which may cause the cell death. In clinical practice, different combinations of radionuclides and SST peptide analogs are used in order to achieve optimal targeting of SST receptor-positive tumors. These analogs differ from each other in their affinity profiles for the various SST receptor types. This variable receptor affinity is important because it can greatly affect the clinical usefulness of radiolabeled SST peptide analogs. The development of a “universal SST ligand” capable of targeting multireceptor system will be advantageous for the efficient targeting of SST receptor-expressing tumors having different SST receptor types. The commonly available radionuclides and SST peptide analogs are described briefly in the following sections.
Somatostatin receptor radiotherapy with 111In-DTPA0-octreotide (111In-OctreoScan)
After the successful use of radiolabeled SST analogs (e.g., 111In-OctreoScan) for the diagnosis of SST receptor-expressing tumors in clinical practice, the next rational step was to label these peptides with therapeutic radionuclides (emitting α- or β-particles or Auger or conversion electrons) [104] and to carry out radionuclide therapy with these radiopeptides. Initial therapeutic studies were performed with high radioactivity doses of 111In-DTPA0-octreotide (which emits Auger and internal conversion electrons that exert an anti-proliferative effect in malignant tumors) in patients with metastatic NETs, have been associated with significant symptomatic improvement but relatively few and short-lived objective tumor responses [11]. Despite encouraging results with substantial symptomatic improvement in the patients, 111In-coupled peptides are not ideal for radiotherapy because of their small particle range, short tissue penetration (~10 μm) and radiotoxicity [11, 105].
To develop a more effective radiotherapeutic peptide, DTPA0-D-Phe1-octreotide (OctreoScan) (Figure 1) was modified by replacing Phe3 with Tyr3 in the octapeptide sequence to increase its hydrophilicity and to improve its affinity for sst2 as compared to octreotide and the DTPA chelator was replaced with DOTA (Figure 2) [1, 2, 53], in order to improve radionuclide-chelator complex stability for peptides radiolabeled with more effectual therapeutic radionuclides, such as 90Y and 177Lu. The macrocyclic chelator DOTA-linked SST peptide analogs can also be radiolabeled stably with 111In, making them suitable for both diagnosis and radiotherapy [9, 10]. Peptides radiolabeled with β-emitting radionuclides (90Y or 177Lu) are preferred for radionuclide therapy since the particles they emit have sufficient energy to cause cell damage without penetrating very far into surrounding tissue [11]. This underlines the importance of the radiometal used for radiolabeling of peptides (see the following sections), and its influence on the pharmacokinetic and tumor targeting characteristics [103].
Somatostatin receptor radiotherapy with [90Y-DOTA0,Tyr3]octreotide (90Y-DOTA-TOC)
Somatostatin receptor binding peptides radiolabeled with β-emitters have superior therapeutic potential since a high-energy β-emitter (e.g. 90Y) attached to a SST peptide analog can potentially deliver a high radiation dose specifically to SST receptor-expressing tumor cells. The same SST analog (D-Phe1, Tyr3-octreotide), was coupled with DOTA (instead of DTPA) (Figure 2), for complexing with 90Y for radiotherapy application. Targeted radionuclide therapy studies conducted in different institutions with this SST peptide analog (90Y-DOTA-TOC or OctreoTher) [8, 44, 106-108] showed complete or partial remissions (10-34%) in patients with NETs. These ranges were higher than those obtained with 111In-DTPA0-octreotide. In the most recent clinical study [44], the outcome of 90Y-DOTA-TOC therapy (i.e. response, survival, and safety profile) was investigated in a large cohort of patients with a wide spectrum of NETs. This study documents the long-term outcome of 90Y-DOTA-TOC treatment in patients; response to 90Y-DOTA-TOC was found to be associated with longer survival. SST receptor imaging is found to be predictive for both survivals after 90Y-DOTA-TOC treatment and occurrence of renal toxicity. The high tumor uptake in the pretherapeutic diagnostic scan with [111In-DTPA]octreotide was significantly associated and predictive of longer survival whereas high kidney uptake in the diagnostic scan predicted the risk of severe kidney toxicity.
Somatostatin receptor radiotherapy with [177Lu-DOTA0,Tyr3]octreotate (DOTA-TATE)
Another SST peptide analog, Tyr3-octreotate, in which the C-terminal Thr(ol) (threoninol), as used in octreotide, is replaced with the natural amino acid Thr (threonine) (Figure 2), is shown to have significantly higher affinity for the sst2 compared with [DOTA0,Tyr3]octreotide in in vitro investigation [52, 109]. Targeted radionuclide therapy with 177Lu-DOTA-TATE [102, 105, 110, 111] showed an overall response of 30-38% and a significantly high median overall survival of 48 months has been reported. It was observed that the quality of life improved significantly after treatment with 177Lu-DOTA-TATE [111].
The kidney is the dose-limiting organ in these peptide receptor radionuclide studies, as the small peptides are generally reabsorbed and retained in the negatively charged proximal tubule cells after glomerular filtration. In order to reduce the uptake of a radiopeptide in the kidney, infusions containing positively charged lysine and arginine are used during therapy [44, 45, 102]. A recent study regarding long term renal toxicity after different doses of 177Lu-DOTA-TATE together with the effects of dose fractionation and lysine co-injection in two tumor-bearing rat models [112] revealed that the administration of high doses of 177Lu-DOTA-TATE resulted in severe renal damage in rats. The data confirms that the fractionation of doses together with co-injection of lysine has beneficial effects in preventing the kidney damage.
The overall results obtained with 90Y-DOTA-TOC and 177Lu-DOTA-TATE is impressive in terms tumor regression. It has been shown that by using the proper kidney protection procedures during radionuclide therapy, renal toxicity can be minimized.
Combination radionuclide therapy with 90Y-DOTA-TOC and 177Lu-DOTA-TATE
In order to treat patients with tumors of different sizes and of non-homogenous receptor distribution, the “combination radiotherapy” using a high-energy β-emitter 90Y for larger tumor lesions and the low-energy β-emitter 177Lu for smaller lesions and metastases seems to be an attractive approach [11, 101]. Animal studies have shown this combination of different radionuclides (90Y and 177Lu) provides different β-energies and particle ranges and achieves higher cure rates in tumors of various sizes than either radionuclide alone [11]. It has been shown that tumor-bearing rats treated with a combination of 90Y-DOTA-TOC and 177Lu-DOTA-TATE 1:1 have longer survival than those treated with either 100% 90Y-DOTA-TOC or 177Lu-DOTA-TATE conjugate alone [14, 101]. These results highlight the benefit of using combination of radionuclides with different energies and particle ranges to achieve maximum therapeutic effects.
Recently, a combined protocol was applied by Kunikowska et al. [113] in 50 patients with disseminated NETs in a prospective study, where one group (A) was treated with 90Y-DOTA-TATE, the second group (B) received 1:1 90Y/177Lu-DOTA-TATE. The median survival time in group A was 26.2 months while in group B it was not reached during the observation period (54 months). Median event-free survival was also superior in group B (29.4 months) vs. group A (21.4 months). The safety of both protocols was comparable and the side effects were mild and rare.
Conclusions
Peptides are important biological tools in the fight against cancer. The importance of peptides is due to the overexpression of their receptors in many human cancers that can be effectively targeted using these biological targeting vectors. In addition to their diagnostic value, radiolabeled peptides have shown enormous strength in targeted radionuclide therapy. The peptides are used as transport vehicles to guide the radionuclides to the tissues expressing a particular receptor. The effectiveness of radionuclide therapy can be greatly enhanced when performed in combinations of different peptides (peptides cocktail) or radionuclides, possibly because of more homogenous receptor targeting.
The presence of multiple peptide receptors in selected cancers can be the basis for multireceptor targeting using two or more radiopeptides in parallel. The co-expression of multiple receptors in human tumors appears to be a distinct feature of peptide receptors. Many of the peptide receptors exist as multiple subtypes. A major challenge is to take full advantage of the co-expression of peptide receptors for multireceptor tumor targeting. With rapid advancement in technology, it is possible to identify the presence of new peptide receptor types/subtypes for efficient tumor targeting in order to obtain a more efficient and powerful means of diagnosis and therapy.
Despite the availability of an FDA-approved SST peptide analog, 111In-DTPA0-octreotide, potential improvements in the field of SST receptor targeting are still necessary. One important step would be the development of SST analogs having broader affinity profiles for SST receptor types, which may enhance the tumor uptake characteristic due to the presence of several receptor subtypes on the same tumor cell but also on the development of antagonistic peptides, based on the most recent findings.
In spite of the remarkable advancements in the field of small peptides as targeting agents, there is a need to find new peptide receptors overexpressed (i.e., suitable peptide targets) preferentially on common human cancers (breast and prostate, etc.), and to develop appropriate radiolabeled peptides for respective targets. More important, is the development of peptide-based therapeutic compounds capable of obtaining regulatory approval.
An upcoming challenge is the transfer from preclinical animal trials to clinical human trials. With the commitments of different disciplines and support from the industries, it is possible to translate preclinical research into clinical settings. The application of small peptides will continue to expand as new discoveries are made.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Okarvi SM. Peptide-based radiopharmaceuticals: future tools for diagnostic imaging of cancers and other diseases. Med Res Rev. 2004;24:357-97
2. Reubi JC. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr Rev. 2003;24:389-427
3. Langer M, Beck-Sickinger AG. Peptides as carrier for tumor diagnosis and treatment. Curr Med Chem Anticancer Agents. 2001;1:71-93
4. Van Den Bossche B, Van de Wiele C. Receptor imaging in oncology by means of nuclear medicine: current status. J Clin Oncol. 2004;22:3593-607
5. Reubi JC, Macke HR, Krenning EP. Candidates for peptide receptor radiotherapy today and in the future. J Nucl Med. 2005;46(Suppl 1):67S-75S
6. Mariani G, Erba PA, Signore A. Receptor-mediated tumor targeting with radiolabeled peptides: there is more to it than somatostatin analogs. J Nucl Med. 2006;47:1904-7
7. Krenning EP, Bakker WH, Breeman WA, Koper JW, Kooij PP, Ausema L. et al. Localisation of endocrine-related tumours with radioiodinated analogue of somatostatin. Lancet. 1989;1:242-4
8. Otte A, Mueller-Brand J, Dellas S, Nitzsche EU, Herrmann R, Maecke HR. Yttrium-90-labelled somatostatin-analogue for cancer treatment. Lancet. 1998;351:417-8
9. Rufini V, Calcagni ML, Baum RP. Imaging of neuroendocrine tumors. Semin Nucl Med. 2006;36:228-47
10. Ambrosini V, Fani M, Fanti S, Forrer F, Maecke HR. Radiopeptide Imaging and Therapy in Europe. J Nucl Med. 2011;52:42S-55S
11. Kaltsas GA, Papadogias D, Makras P, Grossman AB. Treatment of advanced neuroendocrine tumours with radiolabelled somatostatin analogues. Endocr Relat Cancer. 2005;12:683-99
12. Bodei L, Paganelli G, Mariani G. Receptor radionuclide therapy of tumors: a road from basic research to clinical applications. J Nucl Med. 2006;47:375-7
13. Nicolas G, Giovacchini G, Muller-Brand J, Forrer F. Targeted radiotherapy with radiolabeled somatostatin analogs. Endocrinol Metab Clin North Am. 2011;40:187-204
14. Teunissen JJ, Kwekkeboom DJ, de Jong M, Esser JP, Valkema R, Krenning EP. Endocrine tumours of the gastrointestinal tract. Peptide receptor radionuclide therapy. Best Pract Res Clin Gastroenterol. 2005;19:595-616
15. Gonzalez N, Moody TW, Igarashi H, Ito T, Jensen RT. Bombesin-related peptides and their receptors: recent advances in their role in physiology and disease states. Curr Opin Endocrinol Diabetes Obes. 2008;15:58-64
16. Ohki-Hamazaki H, Iwabuchi M, Maekawa F. Development and function of bombesin-like peptides and their receptors. Int J Dev Biol. 2005;49:293-300
17. Dimitrakopoulou-Strauss A, Hohenberger P, Haberkorn U, Macke HR, Eisenhut M, Strauss LG. 68Ga-labeled bombesin studies in patients with gastrointestinal stromal tumors: comparison with 18F-FDG. J Nucl Med. 2007;48:1245-50
18. Scopinaro F, De Vincentis G, Varvarigou AD, Laurenti C, Iori F, Remediani S. et al. 99mTc-bombesin detects prostate cancer and invasion of pelvic lymph nodes. Eur J Nucl Med Mol Imaging. 2003;30:1378-82
19. Hessenius C, Bader M, Meinhold H, Bohmig M, Faiss S, Reubi JC. et al. Vasoactive intestinal peptide receptor scintigraphy in patients with pancreatic adenocarcinomas or neuroendocrine tumours. Eur J Nucl Med. 2000;27:1684-93
20. Raderer M, Kurtaran A, Leimer M, Angelberger P, Niederle B, Vierhapper H. et al. Value of peptide receptor scintigraphy using 123I-vasoactive intestinal peptide and 111In-DTPA-D-Phe1-octreotide in 194 carcinoid patients: Vienna University Experience, 1993 to 1998. J Clin Oncol. 2000;18:1331-6
21. Rao PS, Thakur ML, Pallela V, Patti R, Reddy K, Li H. et al. 99mTc labeled VIP analog: evaluation for imaging colorectal cancer. Nucl Med Biol. 2001;28:445-50
22. Buchegger F, Bonvin F, Kosinski M, Schaffland AO, Prior J, Reubi JC. et al. Radiolabeled neurotensin analog, 99mTc-NT-XI, evaluated in ductal pancreatic adenocarcinoma patients. J Nucl Med. 2003;44:1649-54
23. Garcia-Garayoa E, Blauenstein P, Blanc A, Maes V, Tourwe D, Schubiger PA. A stable neurotensin-based radiopharmaceutical for targeted imaging and therapy of neurotensin receptor-positive tumours. Eur J Nucl Med Mol Imaging. 2009;36:37-47
24. de Visser M, Janssen PJ, Srinivasan A, Reubi JC, Waser B, Erion JL. et al. Stabilised 111In-labelled DTPA- and DOTA-conjugated neurotensin analogues for imaging and therapy of exocrine pancreatic cancer. Eur J Nucl Med Mol Imaging. 2003;30:1134-9
25. Maina T, Nikolopoulou A, Stathopoulou E, Galanis AS, Cordopatis P, Nock BA. [99mTc]Demotensin 5 and 6 in the NTS1-R-targeted imaging of tumours: synthesis and preclinical results. Eur J Nucl Med Mol Imaging. 2007;34:1804-14
26. Nock BA, Maina T, Behe M, Nikolopoulou A, Gotthardt M, Schmitt JS. et al. CCK-2/gastrin receptor-targeted tumor imaging with 99mTc-labeled minigastrin analogs. J Nucl Med. 2005;46:1727-36
27. Froberg AC, de Jong M, Nock BA, Breeman WA, Erion JL, Maina T. et al. Comparison of three radiolabelled peptide analogues for CCK-2 receptor scintigraphy in medullary thyroid carcinoma. Eur J Nucl Med Mol Imaging. 2009;36:1265-72
28. Laverman P, Behe M, Oyen WJ, Willems PH, Corstens FH, Behr TM. et al. Two technetium-99m-labeled cholecystokinin-8 (CCK8) peptides for scintigraphic imaging of CCK receptors. Bioconjug Chem. 2004;15:561-8
29. Kolenc-Peitl P, Mansi R, Tamma M, Gmeiner-Stopar T, Sollner-Dolenc M, Waser B. et al. Highly improved metabolic stability and pharmacokinetics of indium-111-DOTA-gastrin conjugates for targeting of the gastrin receptor. J Med Chem. 2011;54:2602-9
30. Wild D, Behe M, Wicki A, Storch D, Waser B, Gotthardt M. et al. [Lys40(Ahx-DTPA-111In)NH2]exendin-4, a very promising ligand for glucagon-like peptide-1 (GLP-1) receptor targeting. J Nucl Med. 2006;47:2025-33
31. Gotthardt M, Lalyko G, van Eerd-Vismale J, Keil B, Schurrat T, Hower M. et al. A new technique for in vivo imaging of specific GLP-1 binding sites: first results in small rodents. Regul Pept. 2006;137:162-7
32. Wild D, Wicki A, Mansi R, Behe M, Keil B, Bernhardt P. et al. Exendin-4-based radiopharmaceuticals for glucagon like peptide-1 receptor PET/CT and SPECT/CT. J Nucl Med. 2010;51:1059-67
33. Brom M, Oyen WJ, Joosten L, Gotthardt M, Boerman OC. 68Ga-labelled exendin-3, a new agent for the detection of insulinomas with PET. Eur J Nucl Med Mol Imaging. 2010;37:1345-55
34. Haubner R, Weber WA, Beer AJ, Vabuliene E, Reim D, Sarbia M. et al. Noninvasive visualization of the activated αvβ3 integrin in cancer patients by positron emission tomography and [18F]Galacto-RGD. PLoS Med. 2005;2:e70
35. Decristoforo C, Hernandez Gonzalez I, Carlsen J, Rupprich M, Huisman M, Virgolini I. et al. 68Ga- and 111In-labelled DOTA-RGD peptides for imaging of αvβ3 integrin expression. Eur J Nucl Med Mol Imaging. 2008;35:1507-15
36. Dumont RA, Deininger F, Haubner R, Maecke HR, Weber WA, Fani M. Novel 64Cu- and 68Ga-labeled RGD conjugates show improved PET imaging of αvβ3 integrin expression and facile radiosynthesis. J Nucl Med. 2011;52:1276-84
37. Dijkgraaf I, Kruijtzer JA, Liu S, Soede AC, Oyen WJ, Corstens FH. et al. Improved targeting of the αvβ3 integrin by multimerisation of RGD peptides. Eur J Nucl Med Mol Imaging. 2007;34:267-73
38. Antunes P, Ginj M, Walter MA, Chen J, Reubi JC, Maecke HR. Influence of different spacers on the biological profile of a DOTA-somatostatin analogue. Bioconjug Chem. 2007;18:84-92
39. Garcia Garayoa E, Schweinsberg C, Maes V, Brans L, Blauenstein P, Tourwe DA. et al. Influence of the molecular charge on the biodistribution of bombesin analogues labeled with the [99mTc(CO)3]-core. Bioconjug Chem. 2008;19:2409-16
40. Mankoff DA, Link JM, Linden HM, Sundararajan L, Krohn KA. Tumor receptor imaging. J Nucl Med. 2008;49:149S-163S
41. Behr TM, Gotthardt M, Barth A, Behe M. Imaging tumors with peptide-based radioligands. Q J Nucl Med. 2001;45:189-200
42. Virgolini I, Traub T, Novotny C, Leimer M, Fuger B, Li SR. et al. New trends in peptide receptor radioligands. Q J Nucl Med. 2001;45:153-9
43. Valkema R, Pauwels SA, Kvols LK, Kwekkeboom DJ, Jamar F, de Jong M. et al. Long-term follow-up of renal function after peptide receptor radiation therapy with 90Y-DOTA0,Tyr3-octreotide and 177Lu-DOTA0, Tyr3-octreotate. J Nucl Med. 2005;46(Suppl 1):83S-91S
44. Imhof A, Brunner P, Marincek N, Briel M, Schindler C, Rasch H. et al. Response, survival, and long-term toxicity after therapy with the radiolabeled somatostatin analogue [90Y-DOTA]-TOC in metastasized neuroendocrine cancers. J Clin Oncol. 2011;29:2416-23
45. Rolleman EJ, Valkema R, de Jong M, Kooij PP, Krenning EP. Safe and effective inhibition of renal uptake of radiolabelled octreotide by a combination of lysine and arginine. Eur J Nucl Med Mol Imaging. 2003;30:9-15
46. Rolleman EJ, Forrer F, Bernard B, Bijster M, Vermeij M, Valkema R. et al. Amifostine protects rat kidneys during peptide receptor radionuclide therapy with [177Lu-DOTA0,Tyr3]octreotate. Eur J Nucl Med Mol Imaging. 2007;34:763-71
47. Vegt E, Wetzels JF, Russel FG, Masereeuw R, Boerman OC, van Eerd JE. et al. Renal uptake of radiolabeled octreotide in human subjects is efficiently inhibited by succinylated gelatin. J Nucl Med. 2006;47:432-6
48. van Eerd JE, Vegt E, Wetzels JF, Russel FG, Masereeuw R, Corstens FH. et al. Gelatin-based plasma expander effectively reduces renal uptake of 111In-octreotide in mice and rats. J Nucl Med. 2006;47:528-33
49. Susini C, Buscail L. Rationale for the use of somatostatin analogs as antitumor agents. Ann Oncol. 2006;17:1733-42
50. Breeman WA, de Jong M, Kwekkeboom DJ, Valkema R, Bakker WH, Kooij PP. et al. Somatostatin receptor-mediated imaging and therapy: basic science, current knowledge, limitations and future perspectives. Eur J Nucl Med. 2001;28:1421-9
51. Reubi JC, Schar JC, Waser B, Wenger S, Heppeler A, Schmitt JS. et al. Affinity profiles for human somatostatin receptor subtypes SST1-SST5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur J Nucl Med. 2000;27:273-82
52. Reubi JC, Waser B. Concomitant expression of several peptide receptors in neuroendocrine tumours: molecular basis for in vivo multireceptor tumour targeting. Eur J Nucl Med Mol Imaging. 2003;30:781-93
53. de Jong M, Breeman WA, Bernard BF, van Gameren A, de Bruin E, Bakker WH. et al. Tumour uptake of the radiolabelled somatostatin analogue [DOTA0, TYR3]octreotide is dependent on the peptide amount. Eur J Nucl Med. 1999;26:693-8
54. Burdette JE. In vivo imaging of molecular targets and their function in endocrinology. J Mol Endocrinol. 2008;40:253-61
55. Ginj M, Zhang H, Waser B, Cescato R, Wild D, Wang X. et al. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc Natl Acad Sci U S A. 2006;103:16436-41
56. Wadas TJ, Eiblmaier M, Zheleznyak A, Sherman CD, Ferdani R, Liang K. et al. Preparation and biological evaluation of 64Cu-CB-TE2A-sst2-ANT, a somatostatin antagonist for PET imaging of somatostatin receptor-positive tumors. J Nucl Med. 2008;49:1819-27
57. Fani M, Del Pozzo L, Abiraj K, Mansi R, Tamma ML, Cescato R. et al. PET of somatostatin receptor-positive tumors using 64Cu- and 68Ga-somatostatin antagonists: the chelate makes the difference. J Nucl Med. 2011;52:1110-8
58. Wild D, Fani M, Behe M, Brink I, Rivier JE, Reubi JC. et al. First clinical evidence that imaging with somatostatin receptor antagonists is feasible. J Nucl Med. 2011;52:1412-7
59. Shipp MA, Tarr GE, Chen CY, Switzer SN, Hersh LB, Stein H. et al. CD10/neutral endopeptidase 24.11 hydrolyzes bombesin-like peptides and regulates the growth of small cell carcinomas of the lung. Proc Natl Acad Sci U S A. 1991;88:10662-6
60. Pansky P, De Weerth A, Fasler-Kan E, Boulay J-L, Schulz M. et al. Gastrin releasing peptide-preferring bombesin receptors mediate growth of human renal cell carcinoma. J Am Soc Nephrol. 2000;11:1409-1418
61. Lin KS, Luu A, Baidoo KE, Hashemzadeh-Gargari H, Chen MK, Brenneman K. et al. A new high affinity technetium-99m-bombesin analogue with low abdominal accumulation. Bioconjug Chem. 2005;16:43-50
62. Cescato R, Maina T, Nock B, Nikolopoulou A, Charalambidis D, Piccand V. et al. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J Nucl Med. 2008;49:318-26
63. Mansi R, Wang X, Forrer F, Kneifel S, Tamma ML, Waser B. et al. Evaluation of a 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid-conjugated bombesin-based radioantagonist for the labeling with single-photon emission computed tomography, positron emission tomography, and therapeutic radionuclides. Clin Cancer Res. 2009;15:5240-9
64. Abiraj K, Mansi R, Tamma ML, Fani M, Forrer F, Nicolas G. et al. Bombesin antagonist-based radioligands for translational nuclear imaging of gastrin-releasing Peptide receptor-positive tumors. J Nucl Med. 2011;52:1970-8
65. Igarashi H, Ito T, Mantey SA, Pradhan TK, Hou W, Coy DH. et al. Development of simplified vasoactive intestinal peptide analogs with receptor selectivity and stability for human vasoactive intestinal peptide/pituitary adenylate cyclase-activating polypeptide receptors. J Pharmacol Exp Ther. 2005;315:370-81
66. Domschke S, Domschke W, Bloom SR, Mitznegg P, Mitchell SJ, Lux G. et al. Vasoactive intestinal peptide in man: pharmacokinetics, metabolic and circulatory effects. Gut. 1978;19:1049-53
67. Brans B, Linden O, Giammarile F, Tennvall J, Punt C. Clinical applications of newer radionuclide therapies. Eur J Cancer. 2006;42:994-1003
68. Laverman P, Joosten L, Eek A, Roosenburg S, Peitl PK, Maina T. et al. Comparative biodistribution of 12 111In-labelled gastrin/CCK2 receptor-targeting peptides. Eur J Nucl Med Mol Imaging. 2011;38:1410-6
69. Tanaka K, Masu M, Nakanishi S. Structure and functional expression of the cloned rat neurotensin receptor. Neuron. 1990;4:847-54
70. Vincent JP, Mazella J, Kitabgi P. Neurotensin and neurotensin receptors. Trends Pharmacol Sci. 1999;20:302-9
71. Miao Y, Benwell K, Quinn TP. 99mTc- and 111In-labeled α-melanocyte-stimulating hormone peptides as imaging probes for primary and pulmonary metastatic melanoma detection. J Nucl Med. 2007;48:73-80
72. Wei L, Butcher C, Miao Y, Gallazzi F, Quinn TP, Welch MJ. et al. Synthesis and biologic evaluation of 64Cu-labeled rhenium-cyclized α-MSH peptide analog using a cross-bridged cyclam chelator. J Nucl Med. 2007;48:64-72
73. Wei L, Miao Y, Gallazzi F, Quinn TP, Welch MJ, Vavere AL. et al. Gallium-68-labeled DOTA-rhenium-cyclized α-melanocyte-stimulating hormone analog for imaging of malignant melanoma. Nucl Med Biol. 2007;34:945-53
74. Cheng Z, Zhang L, Graves E, Xiong Z, Dandekar M, Chen X. et al. Small-animal PET of melanocortin 1 receptor expression using a 18F-labeled α-melanocyte-stimulating hormone analog. J Nucl Med. 2007;48:987-94
75. Froidevaux S, Calame-Christe M, Tanner H, Eberle AN. Melanoma targeting with DOTA-α-melanocyte-stimulating hormone analogs: structural parameters affecting tumor uptake and kidney uptake. J Nucl Med. 2005;46:887-95
76. Cone RD, Mountjoy KG, Robbins LS, Nadeau JH, Johnson KR, Roselli-Rehfuss L. et al. Cloning and functional characterization of a family of receptors for the melanotropic peptides. Ann N Y Acad Sci. 1993;680:342-63
77. Siegrist W, Solca F, Stutz S, Giuffre L, Carrel S, Girard J. et al. Characterization of receptors for alpha-melanocyte-stimulating hormone on human melanoma cells. Cancer Res. 1989;49:6352-8
78. Miao Y, Owen NK, Fisher DR, Hoffman TJ, Quinn TP. Therapeutic efficacy of a 188Re-labeled α-melanocyte-stimulating hormone peptide analog in murine and human melanoma-bearing mouse models. J Nucl Med. 2005;46:121-9
79. Capello A, Krenning EP, Bernard BF, Breeman WA, Erion JL, de Jong M. Anticancer activity of targeted proapoptotic peptides. J Nucl Med. 2006;47:122-9
80. DeNardo SJ. Combined molecular targeting for cancer therapy: a new paradigm in need of molecular imaging. J Nucl Med. 2006;47:4-5
81. Brooks PC. Role of integrins in angiogenesis. Eur J Cancer. 1996;32A:2423-9
82. Hofland LJ, Capello A, Krenning EP, de Jong M, van Hagen MP. Induction of apoptosis with hybrids of Arg-Gly-Asp molecules and peptides and antimitotic effects of hybrids of cytostatic drugs and peptides. J Nucl Med. 2005;46(Suppl 1):191S-8S
83. Capello A, Krenning EP, Bernard BF, Breeman WA, van Hagen MP, de Jong M. Increased cell death after therapy with an Arg-Gly-Asp-linked somatostatin analog. J Nucl Med. 2004;45:1716-20
84. Li ZB, Wu Z, Chen K, Ryu EK, Chen X. 18F-labeled BBN-RGD heterodimer for prostate cancer imaging. J Nucl Med. 2008;49:453-61
85. Li ZB, Chen K, Chen X. 68Ga-labeled multimeric RGD peptides for microPET imaging of integrin αvβ3 expression. Eur J Nucl Med Mol Imaging. 2008;35:1100-8
86. Liu Z, Liu S, Wang F, Chen X. Noninvasive imaging of tumor integrin expression using 18F-labeled RGD dimer peptide with PEG4 linkers. Eur J Nucl Med Mol Imaging. 2009;36:1296-307
87. Wu Y, Zhang X, Xiong Z, Cheng Z, Fisher DR, Liu S. et al. microPET imaging of glioma integrin αvβ3 expression using 64Cu-labeled tetrameric RGD peptide. J Nucl Med. 2005;46:1707-18
88. Dijkgraaf I, Liu S, Kruijtzer JA, Soede AC, Oyen WJ, Liskamp RM. et al. Effects of linker variation on the in vitro and in vivo characteristics of an 111In-labeled RGD peptide. Nucl Med Biol. 2007;34:29-35
89. Dijkgraaf I, Kruijtzer JA, Frielink C, Corstens FH, Oyen WJ, Liskamp RM. et al. αvβ3 integrin-targeting of intraperitoneally growing tumors with a radiolabeled RGD peptide. Int J Cancer. 2007;120:605-10
90. Janssen M, Frielink C, Dijkgraaf I, Oyen W, Edwards DS, Liu S. et al. Improved tumor targeting of radiolabeled RGD peptides using rapid dose fractionation. Cancer Biother Radiopharm. 2004;19:399-404
91. Liu Z, Niu G, Shi J, Liu S, Wang F, Chen X. 68Ga-labeled cyclic RGD dimers with Gly3 and PEG4 linkers: promising agents for tumor integrin αvβ3 PET imaging. Eur J Nucl Med Mol Imaging. 2009;36:947-57
92. Decristoforo C, Faintuch-Linkowski B, Rey A, von Guggenberg E, Rupprich M, Hernandez-Gonzales I. et al. [99mTc]HYNIC-RGD for imaging integrin αvβ3 expression. Nucl Med Biol. 2006;33:945-52
93. Wicki A, Wild D, Storch D, Seemayer C, Gotthardt M, Behe M. et al. [Lys40(Ahx-DTPA-111In)NH2]-Exendin-4 is a highly efficient radiotherapeutic for glucagon-like peptide-1 receptor-targeted therapy for insulinoma. Clin Cancer Res. 2007;13:3696-705
94. Wild D, Christ E, Caplin ME, Kurzawinski TR, Forrer F, Brandle M. et al. Glucagon-like peptide-1 versus somatostatin receptor targeting reveals 2 distinct forms of malignant insulinomas. J Nucl Med. 2011;52:1073-8
95. Pattou F, Kerr-Conte J, Wild D. GLP-1-receptor scanning for imaging of human beta cells transplanted in muscle. N Engl J Med. 2010;363:1289-90
96. Wild D, Macke H, Christ E, Gloor B, Reubi JC. Glucagon-like peptide 1-receptor scans to localize occult insulinomas. N Engl J Med. 2008;359:766-8
97. Christ E, Wild D, Forrer F, Brandle M, Sahli R, Clerici T. et al. Glucagon-like peptide-1 receptor imaging for localization of insulinomas. J Clin Endocrinol Metab. 2009;94:4398-405
98. Hubalewska-Dydejczyk A, Sowa-Staszczak A, Mikolajczak R, Pach D, Janota B, Tomaszuk M. et al. 99mTc labeled GLP-1 scintigraphy with the use of [Lys40-(Ahx-HYNIC/EDDA)NH2]-Exendin-4 in the insulinoma localization. J Nucl Med. 2011;52(Suppl 1):561
99. Sowa-Staszczak A, Stefanska A, Pach D, Tomaszuk M, Jabrocka-Hybel A, Glowa B. et al. First clinical application of 99mTc labelled long-acting agonist of GLP-1 (Exendin-4) in endocrine diagnosis. Eur J Nucl Med Mol Imaging. 2011;38(Suppl 2):S206
100. Liu S, Edwards DS. Bifunctional chelators for therapeutic lanthanide radiopharmaceuticals. Bioconjug Chem. 2001;12:7-34
101. de Jong M, Breeman WA, Valkema R, Bernard BF, Krenning EP. Combination radionuclide therapy using 177Lu- and 90Y-labeled somatostatin analogs. J Nucl Med. 2005;46(Suppl 1):13S-7S
102. Kwekkeboom DJ, de Herder WW, Kam BL, van Eijck CH, van Essen M, Kooij PP. et al. Treatment with the radiolabeled somatostatin analog [177Lu-DOTA0,Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol. 2008;26:2124-30
103. Sosabowsky J, Melendez-Alafort L, Mather S. Radiolabelling of peptides for diagnosis and therapy of non-oncological diseases. Q J Nucl Med. 2003;47:223-37
104. Norenberg JP, Krenning BJ, Konings IR, Kusewitt DF, Nayak TK, Anderson TL. et al. 213Bi-[DOTA0, Tyr3]octreotide peptide receptor radionuclide therapy of pancreatic tumors in a preclinical animal model. Clin Cancer Res. 2006;12:897-903
105. Kwekkeboom DJ, Teunissen JJ, Bakker WH, Kooij PP, de Herder WW, Feelders RA. et al. Radiolabeled somatostatin analog [177Lu-DOTA0,Tyr3]octreotate in patients with endocrine gastroenteropancreatic tumors. J Clin Oncol. 2005;23:2754-62
106. Waldherr C, Pless M, Maecke HR, Schumacher T, Crazzolara A, Nitzsche EU. et al. Tumor response and clinical benefit in neuroendocrine tumors after 7.4 GBq 90Y-DOTATOC. J Nucl Med. 2002;43:610-6
107. Bodei L, Cremonesi M, Grana C, Rocca P, Bartolomei M, Chinol M. et al. Receptor radionuclide therapy with 90Y-[DOTA]0-Tyr3-octreotide (90Y-DOTATOC) in neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2004;31:1038-46
108. Valkema R, Pauwels S, Kvols LK, Barone R, Jamar F, Bakker WH. et al. Survival and response after peptide receptor radionuclide therapy with [90Y-DOTA0,Tyr3]octreotide in patients with advanced gastroenteropancreatic neuroendocrine tumors. Semin Nucl Med. 2006;36:147-56
109. Capello A, Krenning E, Bernard B, Reubi JC, Breeman W, de Jong M. 111In-labelled somatostatin analogues in a rat tumour model: somatostatin receptor status and effects of peptide receptor radionuclide therapy. Eur J Nucl Med Mol Imaging. 2005;32:1288-95
110. van Essen M, Krenning EP, Bakker WH, de Herder WW, van Aken MO, Kwekkeboom DJ. Peptide receptor radionuclide therapy with 177Lu-octreotate in patients with foregut carcinoid tumours of bronchial, gastric and thymic origin. Eur J Nucl Med Mol Imaging. 2007;34:1219-27
111. Van Essen M, Krenning EP, De Jong M, Valkema R, Kwekkeboom DJ. Peptide receptor radionuclide therapy with radiolabelled somatostatin analogues in patients with somatostatin receptor positive tumours. Acta Oncol. 2007;46:723-34
112. Rolleman EJ, Krenning EP, Bernard BF, de Visser M, Bijster M, Visser TJ. et al. Long-term toxicity of [177Lu-DOTA0,Tyr3]octreotate in rats. Eur J Nucl Med Mol Imaging. 2007;34:219-27
113. Kunikowska J, Krolicki L, Hubalewska-Dydejczyk A, Mikolajczak R, Sowa-Staszczak A, Pawlak D. Clinical results of radionuclide therapy of neuroendocrine tumours with 90Y-DOTATATE and tandem 90Y/177Lu-DOTATATE: which is a better therapy option? Eur J Nucl Med Mol Imaging. 2011;38:1788-97
114. Schottelius M, Poethko T, Herz M, Reubi JC, Kessler H, Schwaiger M. et al. First 18F-labeled tracer suitable for routine clinical imaging of sst receptor-expressing tumors using positron emission tomography. Clin Cancer Res. 2004;10:3593-606
115. Sprague JE, Peng Y, Sun X, Weisman GR, Wong EH, Achilefu S. et al. Preparation and biological evaluation of copper-64-labeled Tyr3-octreotate using a cross-bridged macrocyclic chelator. Clin Cancer Res. 2004;10:8674-82
116. Ginj M, Chen J, Walter MA, Eltschinger V, Reubi JC, Maecke HR. Preclinical evaluation of new and highly potent analogues of octreotide for predictive imaging and targeted radiotherapy. Clin Cancer Res. 2005;11:1136-45
117. Gabriel M, Decristoforo C, Donnemiller E, Ulmer H, Watfah Rychlinski C, Mather SJ. et al. An intrapatient comparison of 99mTc-EDDA/HYNIC-TOC with 111In-DTPA-octreotide for diagnosis of somatostatin receptor-expressing tumors. J Nucl Med. 2003;44:708-16
118. Hubalewska-Dydejczyk A, Fross-Baron K, Mikolajczak R, Maecke HR, Huszno B, Pach D. et al. 99mTc-EDDA/HYNIC-octreotate scintigraphy, an efficient method for the detection and staging of carcinoid tumours: results of 3 years' experience. Eur J Nucl Med Mol Imaging. 2006;33:1123-33
119. Meisetschlager G, Poethko T, Stahl A, Wolf I, Scheidhauer K, Schottelius M. et al. Gluc-Lys([18F]FP)-TOCA PET in patients with SSTR-positive tumors: biodistribution and diagnostic evaluation compared with [111In]DTPA-octreotide. J Nucl Med. 2006;47:566-73
120. Wester HJ, Schottelius M, Scheidhauer K, Meisetschlager G, Herz M, Rau FC. et al. PET imaging of somatostatin receptors: design, synthesis and preclinical evaluation of a novel 18F-labelled, carbohydrated analogue of octreotide. Eur J Nucl Med Mol Imaging. 2003;30:117-22
121. Koukouraki S, Strauss LG, Georgoulias V, Schuhmacher J, Haberkorn U, Karkavitsas N. et al. Evaluation of the pharmacokinetics of 68Ga-DOTATOC in patients with metastatic neuroendocrine tumours scheduled for 90Y-DOTATOC therapy. Eur J Nucl Med Mol Imaging. 2006;33:460-6
122. Wild D, Macke HR, Waser B, Reubi JC, Ginj M, Rasch H. et al. 68Ga-DOTANOC: a first compound for PET imaging with high affinity for somatostatin receptor subtypes 2 and 5. Eur J Nucl Med Mol Imaging. 2005;32:724
123. Forrer F, Uusijarvi H, Waldherr C, Cremonesi M, Bernhardt P, Mueller-Brand J. et al. A comparison of 111In-DOTATOC and 111In-DOTATATE: biodistribution and dosimetry in the same patients with metastatic neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2004;31:1257-62
124. Rodrigues M, Traub-Weidinger T, Li S, Ibi B, Virgolini I. Comparison of 111In-DOTA-DPhe1-Tyr3-octreotide and 111In-DOTA-lanreotide scintigraphy and dosimetry in patients with neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2006;33:532-40
125. Stahl A, Meisetschlager G, Schottelius M, Bruus-Jensen K, Wolf I, Scheidhauer K. et al. [123I]Mtr-TOCA, a radioiodinated and carbohydrated analogue of octreotide: scintigraphic comparison with [111In]octreotide. Eur J Nucl Med Mol Imaging. 2006;33:45-52
126. Zhang H, Chen J, Waldherr C, Hinni K, Waser B, Reubi JC. et al. Synthesis and evaluation of bombesin derivatives on the basis of pan-bombesin peptides labeled with indium-111, lutetium-177, and yttrium-90 for targeting bombesin receptor-expressing tumors. Cancer Res. 2004;64:6707-15
127. Zhang H, Schuhmacher J, Waser B, Wild D, Eisenhut M, Reubi JC. et al. DOTA-PESIN, a DOTA-conjugated bombesin derivative designed for the imaging and targeted radionuclide treatment of bombesin receptor-positive tumours. Eur J Nucl Med Mol Imaging. 2007;34:1198-208
128. Maddalena ME, Fox J, Chen J, Feng W, Cagnolini A, Linder KE. et al. 177Lu-AMBA biodistribution, radiotherapeutic efficacy, imaging, and autoradiography in prostate cancer models with low GRP-R expression. J Nucl Med. 2009;50:2017-24
129. Smith CJ, Gali H, Sieckman GL, Hayes DL, Owen NK, Mazuru DG. et al. Radiochemical investigations of 177Lu-DOTA-8-Aoc-BBN[7-14]NH2: an in vitro/in vivo assessment of the targeting ability of this new radiopharmaceutical for PC-3 human prostate cancer cells. Nucl Med Biol. 2003;30:101-9
130. Prasanphanich AF, Nanda PK, Rold TL, Ma L, Lewis MR, Garrison JC. et al. [64Cu-NOTA-8-Aoc-BBN(7-14)NH2] targeting vector for positron-emission tomography imaging of gastrin-releasing peptide receptor-expressing tissues. Proc Natl Acad Sci U S A. 2007;104:12462-7
131. de Visser M, van Weerden WM, de Ridder CM, Reneman S, Melis M, Krenning EP. et al. Androgen-dependent expression of the gastrin-releasing peptide receptor in human prostate tumor xenografts. J Nucl Med. 2007;48:88-93
132. de Visser M, Bernard HF, Erion JL, Schmidt MA, Srinivasan A, Waser B. et al. Novel 111In-labelled bombesin analogues for molecular imaging of prostate tumours. Eur J Nucl Med Mol Imaging. 2007;34:1228-38
133. Lane SR, Nanda P, Rold TL, Sieckman GL, Figueroa SD, Hoffman TJ. et al. Optimization, biological evaluation and microPET imaging of copper-64-labeled bombesin agonists, [64Cu-NO2A-(X)-BBN(7-14)NH2], in a prostate tumor xenografted mouse model. Nucl Med Biol. 2010;37:751-61
134. Garrison JC, Rold TL, Sieckman GL, Figueroa SD, Volkert WA, Jurisson SS. et al. In vivo evaluation and small-animal PET/CT of a prostate cancer mouse model using 64Cu bombesin analogs: side-by-side comparison of the CB-TE2A and DOTA chelation systems. J Nucl Med. 2007;48:1327-37
135. Parry JJ, Andrews R, Rogers BE. MicroPET imaging of breast cancer using radiolabeled bombesin analogs targeting the gastrin-releasing peptide receptor. Breast Cancer Res Treat. 2007;101:175-83
136. Lears KA, Ferdani R, Liang K, Zheleznyak A, Andrews R, Sherman CD. et al. In vitro and in vivo evaluation of 64Cu-labeled SarAr-bombesin analogs in gastrin-releasing peptide receptor-expressing prostate cancer. J Nucl Med. 2011;52:470-7
137. Mansi R, Wang X, Forrer F, Waser B, Cescato R, Graham K. et al. Development of a potent DOTA-conjugated bombesin antagonist for targeting GRPr-positive tumours. Eur J Nucl Med Mol Imaging. 2011;38:97-107
138. Schroeder RP, van Weerden WM, Krenning EP, Bangma CH, Berndsen S, Grievink-de Ligt CH. et al. Gastrin-releasing peptide receptor-based targeting using bombesin analogues is superior to metabolism-based targeting using choline for in vivo imaging of human prostate cancer xenografts. Eur J Nucl Med Mol Imaging. 2011;38:1257-66
139. Zhang X, Cai W, Cao F, Schreibmann E, Wu Y, Wu JC. et al. 18F-labeled bombesin analogs for targeting GRP receptor-expressing prostate cancer. J Nucl Med. 2006;47:492-501
140. Honer M, Mu L, Stellfeld T, Graham K, Martic M, Fischer CR. et al. 18F-labeled bombesin analog for specific and effective targeting of prostate tumors expressing gastrin-releasing peptide receptors. J Nucl Med. 2011;52:270-8
141. Garcia Garayoa E, Schweinsberg C, Maes V, Ruegg D, Blanc A, Blauenstein P. et al. New [99mTc]bombesin analogues with improved biodistribution for targeting gastrin releasing-peptide receptor-positive tumors. Q J Nucl Med Mol Imaging. 2007;51:42-50
142. Lane SR, Veerendra B, Rold TL, Sieckman GL, Hoffman TJ, Jurisson SS. et al. 99mTc(CO)3-DTMA bombesin conjugates having high affinity for the GRP receptor. Nucl Med Biol. 2008;35:263-72
143. Dapp S, Garcia Garayoa E, Maes V, Brans L, Tourwe DA, Muller C. et al. PEGylation of 99mTc-labeled bombesin analogues improves their pharmacokinetic properties. Nucl Med Biol. 2011;38:997-1009
144. Retzloff LB, Heinzke L, Figureoa SD, Sublett SV, Ma L, Sieckman GL. et al. Evaluation of [99mTc-(CO)3-X-Y-bombesin(7-14)NH2] conjugates for targeting gastrin-releasing peptide receptors overexpressed on breast carcinoma. Anticancer Res. 2010;30:19-30
145. Raposinho PD, Correia JD, Alves S, Botelho MF, Santos AC, Santos I. A 99mTc(CO)3-labeled pyrazolyl-α-melanocyte-stimulating hormone analog conjugate for melanoma targeting. Nucl Med Biol. 2008;35:91-9
146. Wei L, Ye Y, Wadas TJ, Lewis JS, Welch MJ, Achilefu S. et al. 64Cu-labeled CB-TE2A and diamsar-conjugated RGD peptide analogs for targeting angiogenesis: comparison of their biological activity. Nucl Med Biol. 2009;36:277-85
147. Line BR, Mitra A, Nan A, Ghandehari H. Targeting tumor angiogenesis: comparison of peptide and polymer-peptide conjugates. J Nucl Med. 2005;46:1552-60
148. Shi J, Wang L, Kim YS, Zhai S, Jia B, Wang F. et al. 99mTcO(MAG2-3G3-dimer): a new integrin αvβ3-targeted SPECT radiotracer with high tumor uptake and favorable pharmacokinetics. Eur J Nucl Med Mol Imaging. 2009;36:1874-84
Author contact
![]() Corresponding author: S. M. Okarvi, Cyclotron and Radiopharmaceuticals Department. King Faisal Specialist Hospital and Research Centre. MBC-03, PO Box 3354, Riyadh 11211, Saudi Arabia. Tel: +9661-442-4812, Fax: +9661-442-4743. E-mail: sokarviedu.sa
Corresponding author: S. M. Okarvi, Cyclotron and Radiopharmaceuticals Department. King Faisal Specialist Hospital and Research Centre. MBC-03, PO Box 3354, Riyadh 11211, Saudi Arabia. Tel: +9661-442-4812, Fax: +9661-442-4743. E-mail: sokarviedu.sa