Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2012; 2(6):589-596. doi:10.7150/thno.4295 This issue Cite
Research Paper
The Efficient Synthesis and Biological Evaluation of Novel Bi-Functionalized Sarcophagine for 64Cu Radiopharmaceuticals
Shuanglong Liu1, Dan Li1,2, Chiun-Wei Huang1, Li-Peng Yap1, Ryan Park1, Hong Shan2, Zibo Li1 ![]() , Peter S. Conti1
, Peter S. Conti1 ![]()
1. Department of Radiology, Keck School of Medicine, Molecular Imaging Center, University of Southern California, Los Angeles, CA 90033, USA
2. Department of Radiology, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou 510630, China
Received 2012-2-27; Accepted 2012-4-20; Published 2012-6-12
Abstract

Purpose We and others have reported that Sarcophagine-based bifunctional chelators could be effectively used in the syntheses of 64Cu radiopharmaceuticals. The resulted 64Cu-Sarcophagine complexes demonstrated great in vivo stability. The goal of this study was to further derivatize Sarcophagine cage with amino and maleimide functional groups for conjugation with bioligands.
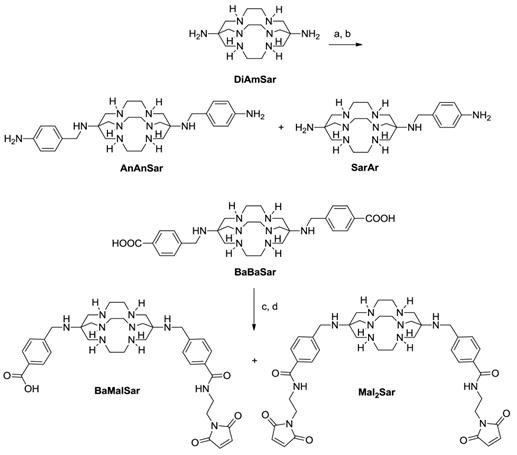
Methods Starting from DiAmSar, three novel chelators (AnAnSar, BaMalSar, and Mal2Sar) with two functional groups have been synthesized. Among those, BaMalSar and Mal2Sar have been conjugated with cyclic peptide c(RGDyC) (denoted as RGD) and the resulted conjugates, BaMalSar-RGD and Mal2Sar-RGD2 have been labeled with 64Cu. The tumor targeting efficacy of 64Cu-labeled RGD peptides were evaluated in a subcutaneous U87MG glioblastoma xenograft model.
Results The conjugates, BaMalSar-RGD and Mal2Sar-RGD2 could be labeled with 64CuCl2 in 10 min with high purity (>98%) and high radiochemical yield (>90%). Both 64Cu-BaMalSar-RGD and 64Cu-Mal2Sar-RGD2 exhibited high tumor uptake and tumor-to-normal tissue ratios.
Conclusion Three novel chelators with two functional groups have been developed based on Sarcophagine cage. The platform developed in this study could have broad applications in the design and synthesis of 64Cu-radiopharmaceuticals.
Keywords: Sarcophagine, 64Cu, microPET, RGD, Integrin αvβ3
Introduction
The physical characteristics of 64Cu (t1/2 = 12.7 h; β+, 0.653MeV [17.8 %]; βˉ, 0.579MeV [38.4 %]) make it a very important isotope in positron emission tomography (PET) imaging, especially for evaluating bioligands with long circulating half lives (> 1 day). In addition, the advanced coordination chemistry of copper has led to a wide variety of efficient chelators that could be linked to antibodies, proteins, peptides, and other biologically active molecules for 64Cu-radiopharmaceutical synthesis. Among all these chelators, 1,4,7,10-tetra-azacyclododecane-N,N',N'',N'''-tetraacetic acid (DOTA) is probably one of the most widely used chelators for 64Cu labeling. However, its moderate in vivo stability could increase the non-targeted organ radiation dosage and lower the tumor-to-nontumor contrast [1-2]. 64Cu-Labeled radiopharmaceuticals with improved stability have been reported including 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA) derivatives [3-4], cross-bridged 1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetraacetic acid (CB-TETA) [1, 5], and 1,4,8,11-tetraazabicyclo[6.6.2]hexadecane (CB-TE2A) [6-8]. For these bifunctional chelators (BFCs), relatively harsh conditions, such as elevated temperature, may be required for 64Cu chelation.
The bicyclic chelator, sarcophagine (denoted as “Sar”), is well known for its strong binding of Cu(II) and therefore high stability of its complexes [9-11]. Based on this cage-like hexaazamacrobicyclic sarcophagine, a new class of bifunctional chelators has been synthesized recently. The resulting 64Cu complexes demonstrated great in vivo stability and high radiolabeling efficiency under mild conditions [12-16]. By modifying the inert primary amines of sarcophagine with carboxyl groups, we have successfully developed AmBaSar and BaBaSar chelators in our laboratory [12-14, 17-18]. Biological conjugation is generally achieved through three types of functional groups on biomarkers: amino group, carboxyl group, and sulfhydryl group. Correspondingly, carboxyl group, amino group, or maleimide group are expected to be installed onto the chelators for bioconjugation. To further explore the scope of the applications of Sar chelators, herein we reported the syntheses of three new chelators from the Sar cages: AnAnSar, BaMalSar, and Mal2Sar. AnAnSar, having two reactive amino groups on both sides of the Sar cage could be coupled with the carboxyls of biomarkers by amide bond formation. BaMalSar and Mal2Sar are maleimide-containing Sar chelators, which are designed for biomarkers with free sulfhydryl groups. In order to evaluate the BaMalSar and Mal2Sar in vivo performance, we constructed two integrin αvβ3-specific probes: BaMalSar-RGD and Mal2Sar-RGD2, and evaluated their tumor targeting efficacy in U87MG tumor bearing mice using microPET.
Materials and methods
All chemicals obtained commercially were of analytic grade and used without further purification. The syringe filter and polyethersulfone membranes (pore size, 0.22 μm; diameter, 13 mm) were obtained from Nalge Nunc International (Rochester, NY). The c(RGDyC) peptide were purchased from Peptides International (Louisville, KY). A typical linear gradient HPLC was used for purification and quality control as following. The reversed-phase HPLC using a Vydac protein and peptide column (218TP510; 5µm, 250 × 4.6 mm) was performed on a Dionex 680 chromatography system with a UVD 170U absorbance detector (Sunnyvale, CA) and model 105S single-channel radiation detector (Carroll & Ramsey Associates). At a flow rate of 1 mL/min, the mobile phase was maintained at 95% solvent A [0.1% trifluoroacetic acid (TFA) in water] and 5% B [0.1% TFA in acetonitrile (MeCN)] from 0-2 min and was changed to 35% solvent A and 65% solvent B through 2-32 min. The UV absorbance was monitored at 218 nm and the identification of the peptides was confirmed based on the UV spectrum using a PDA detector.
Preparation of AnAnSar, BaMalSar, and Mal2Sar
DiAmSar was synthesized as reported [12]. tert-Butyl (4-(bromomethyl)phenyl)carbamate (24.0 mg, 84.2 µmol) and sodium carbonate (17.0 mg, 160 µmol) was added to the solution of DiAmSar (20 mg, 63.7 µmol) in N,N-dimethylformamide (DMF). The reaction was incubated at 70 °C for 12 h. After cooling down to room temperature, 1 mL trifluoroacetic acid was added to the crude mixture and the reaction was maintained at room temperature for 30 min. Semipreparative HPLC afforded the AnAnSar as slightly yellow solid (31%, 10.3 mg). The retention time of AnAnSar on analytical HPLC is 6.5 min. Electrospray Ionization Mass Spectrum (ESI-MS): m/z 525.4 for [M+H]+ (Chemical formula: C28H49N10, calculated m/z value: 525.4).
The compound BaBaSar was synthesized using optimized method. In brief, methyl 4-(bromomethyl)benzoate (21.9 mg, 95.6 µmol) and sodium carbonate (17.0 mg, 160 µmol) were added to the solution of DiAmSar (20 mg, 63.7 µmol) in 1:1 tetrahydrofuran and methanol. The reaction was incubated at 70 °C for 8 h. After cooling down to room temperature, 1 mL 1 N sodium hydroxide (NaOH) was added to the mixture and the reaction was maintained at 60 °C for 2 h. Semipreparative HPLC afforded the BaBaSar as slightly yellow solid (44%, 16.3 mg). The retention time of BaBaSar on analytical HPLC is 8.7 min. Electrospray Ionization Mass Spectrum (ESI-MS): m/z 583.4 for [M+H]+ (Chemical formula: C30H47N8O4, calculated m/z value: 583.4). HPLC coinjection with BaBaSar standard further confirmed the identity of the product [18].
To the solution of BaBaSar (2.9 mg, 5µmol) was added 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide, hydrochloric acid (EDC) (1.5 mg, 8 µmol) and N-hydroxysulfosuccinimide (SNHS) (1.5 mg, 7 µmol). After the mixture was adjusted to pH 5.5-6.0 using 0.1 N NaOH, the reaction was maintained at room temperature for 1 h. Then, N-(2-aminoethyl)maleimide trifluoroacetate salt (1.7 mg, 6.5 µmol) in 500 µL borate buffer (pH 8.5) was added to the BaBaSar solution. The reaction stayed at 4 °C overnight. HPLC purification afforded the product BaMalSar (1.3 mg, 37%) and Mal2Sar (1.0 mg, 25%) as white powder. The retention time of BaMalSar on analytical HPLC is 14.1 min. Electrospray Ionization Mass Spectrum (ESI-MS): m/z 705.4 for [M+H]+ (Chemical formula: C36H53N10O5, calculated m/z value: 705.4). The retention time of Mal2Sar on analytical HPLC is 14.9 min. Electrospray Ionization Mass Spectrum (ESI-MS): m/z 827.5 for [M+H]+ (Chemical formula: C42H59N12O6, calculated m/z value: 827.5).
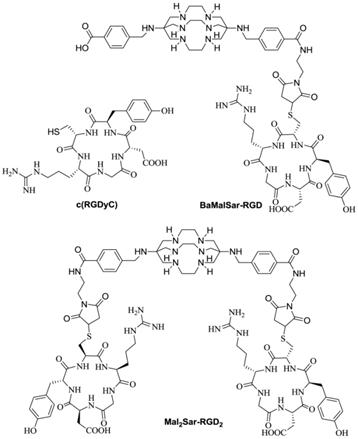
Preparation of BaMalSar-RGD and Mal2Sar-RGD2
To BaMalSar (2.4 mg, 3.4 µmol) in phosphate buffer (pH 6.5-7.0) was added c(RGDyC) (1.8 mg, 3.0 µmol). The reaction stayed at room temperature for 10 min and HPLC afforded BaMalSar-RGD as white powder (yield 92%, 3.6 mg). The retention time of BaMalSar-RGD on analytical HPLC is 15.5 min. ESI-MS: m/z 1299.7 for [M+H]+ (Chemical formula: C60H87N18O13S, calculated m/z value: 1299.6). To Mal2Sar (2.8 mg, 3.4 µmol) in phosphate buffer (pH 6.5-7.0) was added c(RGDyC) (5.2 mg, 8.8 µmol). The reaction stayed at room temperature for 2 h and HPLC afforded Mal2Sar-RGD2 as white powder (yield 85%, 5.8 mg). The retention time of Mal2Sar-RGD2 on analytical HPLC is 14.6 min. ESI-MS: m/z 1008.5 for [M+2H]2+ (Chemical formula: C90H128N28O22S2, calculated m/z2+ value: 1008.5).
Radiochemistry
The labeling was performed similar to the reported procedure [18]. In brief, 20 µL 64CuCl2 (74 MBq, 2.0 mCi in 0.1 N HCl) was diluted in 200 µL of 0.1 N ammonium acetate (NH4OAc, pH 5.5) and added to BaMalSar-RGD or Mal2Sar-RGD2 (5-10 µg per mCi 64Cu). The reaction mixture was kept at 37 °C for 10 min. 64Cu-labeled peptide was subsequently purified by analytical HPLC and the radioactive peak containing the desired product was collected. After removal of the solvent by rotary evaporation, the conjugated peptide tracer was reconstituted in 1 mL phosphate buffer saline (PBS) and passed through a 0.22 µm syringe filter for in vivo animal experiments. The decay-corrected radiochemical yield (RCY) for 64Cu-BaMalSar-RGD and 64Cu-Mal2Sar-RGD2 was 95% and 92% respectively.
Cell Culture
Human glioblastoma cell line U87MG was obtained from the American Type Culture Collection (Manassas, VA) and were cultured in DMEM containing high glucose (GIBCO, Carlsbad, CA), which was supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. The cells were expanded in tissue culture dishes and kept in a humidified atmosphere of 5% CO2 at 37 °C. The medium was changed every other day. A confluent monolayer was detached with 0.05% Trypsin-EDTA, 0.01M PBS (pH 7.4) and dissociated into a single-cell suspension for further cell culture.
MicroPET Imaging
Animal procedures were performed according to a protocol approved by the University of Southern California Institutional Animal Care and Use Committee. MicroPET scans were performed on a microPET R4 rodent model scanner (Siemens Medical Solutions USA, Inc., Knoxville, TN). The scanner has a computer-controlled bed and 10.8-cm transaxial and 8-cm axial fields of view (FOVs). It has no septa and operates exclusively in the 3-dimensional (3D) list mode. Animals were placed near the center of the FOV of the scanner. For static microPET scans, the mice bearing U87MG xenografts were injected with about 3.7 MBq (100 μCi) of 64Cu-BaMalSar-RGD or 64Cu-Mal2Sar-RGD2 via the tail vein (n = 3 for each group). Similarly, the blocking study was performed by injecting the probe with c(RGDyC) (10 mg/kg body weight) through the tail vein (n = 3). At 1 h, 4 h, and 20 h post injection (p.i.), the mice were anesthetized with isoflurane (5% for induction and 2% for maintenance in 100% O2) using a knock-down box. With the help of a laser beam attached to the scanner, the mice were placed in the prone position and near the center of the field of view of the scanner. The 3-min static scans were then obtained. Images were reconstructed by use of a 2-dimensional ordered-subsets expectation maximization (OSEM) algorithm. No background correction was performed. Regions of interest (ROIs; 5 pixels for coronal and transaxial slices) were drawn over the tumor on decay-corrected whole-body coronal images. The maximum counts per pixel per minute were obtained from the ROI and converted to counts per milliliter per minute by using a calibration constant. With the assumption of a tissue density of 1 g/ml, the ROIs were converted to counts per gram per min. Image ROI-derived %ID/g values were determined by dividing counts per gram per minute by injected dose. No attenuation correction was performed.
Statistical Analysis
Quantitative data are expressed as mean ± SD. Means were compared using 1-way ANOVA and the Student t test. P values of < 0.05 were considered statistically significant.
Results
Chemistry and Radiochemistry
The chelator An(Boc)An(Boc)Sar was prepared by direct alkylation of DiAmSar using tert-butyl (4-(bromomethyl)phenyl)carbamate (Fig. 1). Deprotection with TFA afforded AnAnSar in 31% yield. BaBaSar was activated with EDC/SNHS, followed by the conjugation with N-(2-aminoethyl)maleimide to afford BaMalSar and Mal2Sar (Fig. 1). The yields of BaMalSar and Mal2Sar were 37% and 25%, respectively. BaMalSar and Mal2Sar were conjugated with c(RGDyC) to give BaMalSar-RGD and Mal2Sar-RGD2 in 92% and 85% yields, respectively (Fig. 2). The products were purified by HPLC and characterized by ESI-MS. The purity of each RGD conjugate was determined to be > 95% by HPLC. The 64Cu labeling procedure was done within 1 h (including the radioisotope incorporation, HPLC purification, rotary evaporation, and formulation in PBS) with a decay-corrected yield higher than 90% and more than 98% radiochemical purity. The specific activity of purified 64Cu-BaMalSar-RGD and 64Cu-Mal2Sar-RGD2 were about 200-300 mCi/µmol.
BFC syntheses. (a). tert-butyl (4-(bromomethyl)phenyl)carbamate, DMF, Na2CO3, 12 h. (b). TFA, 30 min. (c). EDC, SNHS, pH 5.5, 1h. (d). 2-aminoethylmaleimide, borate buffer, pH 8.5.
Structures of c(RGDyC), BaMalSar-RGD, and Mal2Sar-RGD2.
MicroPET imaging study
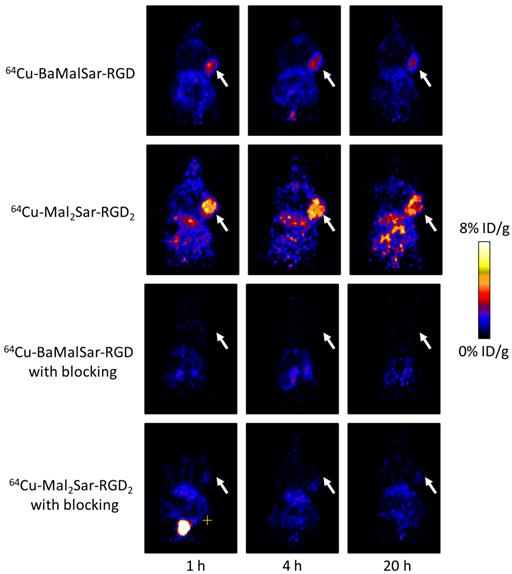
Static microPET scans were performed on a U87MG tumor model and representative decay-corrected coronal images at 1, 4, and 20 h after tail vein injection of 64Cu-BaMalSar-RGD and 64Cu-Mal2Sar-RGD2 are shown in Fig. 3 (n = 3 per group). The U87MG tumors were clearly visualized with good tumor-to-background contrast for both tracers. The uptake in the tumor or other organs was measured from the ROI analysis and shown in Fig. 3. For 64Cu-BaMalSar-RGD, the tumor uptake was 3.02 ± 0.20, 2.55 ± 0.43, and 2.05 ± 0.18 %ID/g, at 1, 4, and 20 h p.i., respectively. For 64Cu-Mal2Sar-RGD2, the tumor uptake was 5.56 ± 0.38, 5.01 ± 0.19, and 3.81 ± 0.14 %ID/g, at 1, 4, and 20 h p.i., respectively. The tumor uptake of 64Cu-Mal2Sar-RGD2 was significantly higher than that of 64Cu-BaMalSar-RGD (P < 0.01, Fig. 4) at all three time points examined. Both of the tracers cleared rapidly from the blood, and were excreted mainly through the kidneys as evidenced by the high kidney uptake. The liver uptakes of 64Cu-BaMalSar-RGD and 64Cu-Mal2Sar-RGD2 were significantly lower than tumor and kidneys at all time-points indicating the good in vivo stability of the 64Cu chelation indirectly. The nonspecific uptake in the muscle and lung was very low for both tracers.
Decay-corrected whole-body coronal microPET images of athymic female nude mice bearing U87MG tumor from a static scan at 1 h, 4 h, and 20 h after the injection of 64Cu-BaMalSar-RGD, and 64Cu-Mal2Sar-RGD2, with or without c(RGDyC) as the blocking agent (10 mg/kg body weight). Tumors are indicated by arrows.
MicroPET quantification of tumors and major organs at 1 h, 4 h, and 20 h after the injection of 64Cu-BaMalSar-RGD, and 64Cu-Mal2Sar-RGD2, with or without c(RGDyC) as the blocking agent (10 mg/kg body weight).
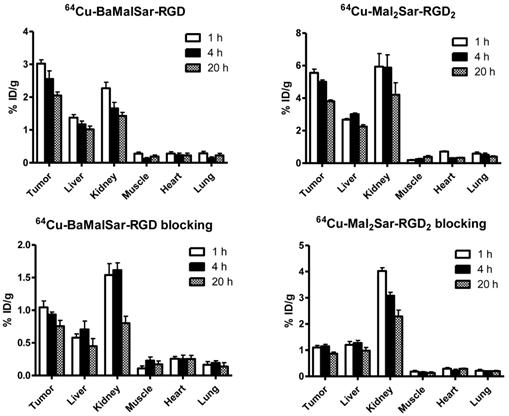
The integrin αvβ3 targeting specificities of 64Cu-BaMalSar-RGD and 64Cu-Mal2Sar-RGD2 were demonstrated by co-injection of excess RGD as the blocking agent (Fig. 4). Co-injection of excess dose of RGD significantly lower the tumor uptake for 64Cu-BaMalSar-RGD and 64Cu-Mal2Sar-RGD2. For example, at 1 h p.i., the tumor uptake of 64Cu-BaMalSar-RGD with blocking RGD agent is 1.04 ± 0.17 %ID/g (vs. 3.02 ± 0.20 %ID/g without RGD) and the tumor uptake of 64Cu-Mal2Sar-RGD2 is 1.10 ± 0.13 %ID/g with the blocking RGD agent (vs. 5.56 ± 0.38 %ID/g without RGD) (Fig. 4).
Discussion
Although sarcophagine 64Cu2+ complexes have been demonstrated to be superior to other chelators such as DOTA with respect to in vivo stability, attempts to attach the DiAmSar directly to protein using EDC activation has been unsuccessful due to its relatively inert primary amine [15]. In order to overcome this limitation, it is necessary to further derivatize these hexaazamacrobicyclic caged-like BFCs, which would allow the conjugation of sarcophagine with bioligands using conventional synthetic strategies such as amide bond formation. For example, monofunctionalized AmBaSar and SarAr have been developed for 64Cu radiopharmaceuticals [14, 16, 19]. As sarcophagine has two relatively inert primary amines on either end of its cage, we have focused on developing novel Sar cage derivatives with multifunctional groups introduced to both ends. Recently, we have successfully improved the functionalization approach of the Sar cage through a direct alkylation (SN2) reaction [18]. The bi-functionalized BaBaSar chelator demonstrated the superior properties compared with the mono-functionalized AmBaSar for constructing 64Cu radiopharmaceuticals. Here, we further extended our effort and developed three new BFCs with two functional groups for the 64Cu radiopharmaceutical synthesis.
In our first design, we proposed to introduce two aromatic amines to the DiAmSar backbone through the direct alkylation (SN2) reaction [18]. As expected, An(Boc)An(Boc)Sar was successfully synthesized through the alkylation between tert-butyl (4-(bromomethyl)phenyl)carbamate and DiAmSar. After deprotection, AnAnSar was obtained in 31% isolation yield. At the same time, the monoalkylated product, SarAr, was afforded as a side product, which could also be useful for bioligands conjugation. Similar to the bifunctional BaBaSar, the free amino groups from AnAnSar should be able to react with the activated carboxylate groups to construct multivalent or multimodality imaging agent. Moreover, the aromatic amine could be selectively reacted without interference from a crosslink reaction between the activated carboxylate group and lysine amines [20].
The major concern for protein labeling through amide bond formation is the possible interference with biological activity; modification of one or more lysines or carboxylic acids located at or near the active site could reduce the binding affinity. Thiol-reactive agents have also been used to modify peptides and proteins at specific sites, providing high chemoselectivity as compared with amine or carboxylate-reactive reagents [21-22]. Starting from BaBaSar, the thiol reactive chelators BaMalSar and Mal2Sar were synthesized through a two-step reaction: (1) activation of the benzoic acid moieties in BaBaSar with EDC/SNHS; (2) installment of maleimide functional group via an amide bond formation. During the preparation of starting material BaBaSar, it was found that the reaction yield could be significantly improved if methyl-4-bromomethylbenzoate was used as the alkylation agent for DiAmSar (instead of using 4-bromomethylbenzoic acid). Although this modification did require one extra deprotection step, the reaction yield could be as high as 91% based on both AmBaSar and BaBaSar. This improved method was used for AmBaSar and BaBaSar synthesis thereafter. At this stage, we have developed a library of multifunctional Sar chelators that could be used for amino, carboxyl, and sulfhydryl conjugation with bioligands.
To demonstrate the application of our Sar chelators in 64Cu radiopharmaceuticals, both BaMalSar and Mal2Sar were selected and conjugated with c(RGDyC) [23-25]. The free sulfhydryl group on c(RGDyC) reacted with both chelators in high yield under mild condition (neutral pH and room temperature) to afford BaMalSar-RGD and Mal2Sar-RGD2. This conjugation could be ideal when the bioligands are susceptible to acid, base, or high reaction temperature. It also provides an excellent alternative to the widely used DOTA-NHS. Furthermore, this sulfhydryl-specific reaction could avoid reducing the target binding affinity if the lysine or terminal amino groups are at or near the active site of a peptide or protein.
Both of the BaMalSar-RGD and Mal2Sar-RGD2 were labeled with 64Cu very efficiently in 0.1 M NH4OAc buffer within 10 min in high yield. Although the specific activity of 64Cu-BaMalSar-RGD and 64Cu-Mal2SarSar-RGD2 was about 200-300 mCi/µmol, decreasing the loading of sarcophagine RGD and using more 64Cu for the labeling will increase the specific activity some extent. These probes demonstrated the high tumor to background ratios. For example, the tumor to muscle ratio reached 20.46 ± 4.47 at 4 h post injection of 64Cu-Mal2Sar-RGD2, which is consistent with high contrast shown in Fig. 3. 64Cu-Mal2Sar-RGD2 also had significantly increased tumor uptake compared with 64Cu-BaMalSar-RGD, which could be caused by a multivalency effect as has been observed before [8, 26-28]. Similar to previous reports, increased tumor uptake was accompanied with an elevated kidney uptake [27-28]. In the blocking study, the tumor uptakes of 64Cu-BaMalSar-RGD and 64Cu-Mal2Sar-RGD2 were reduced to the same level (no significant difference, P > 0.05) after saturating the integrin receptors by large excess of unlabeled RGD peptide. The residual background uptake may be related to passive targeting of the probes. Nonetheless, the efficient blocking results clearly demonstrated the receptor specificity of our probes.
Previously, 64Cu labeled DOTA-c(RGDyK) had been tested in U87MG human glioma tumors [29]. The observed tumor to muscle ratios were 7.10 ± 1.30, and 6.30 ± 1.28 at 1, and 4 h post injection. In this study, the tumor to muscle ratios reached 11.71 ± 3.58, and 26.78 ± 15.18 for 64Cu-BaMalSar-RGD at 1, and 4 h post injection. The improved in vivo kinetics are extremely important for 64Cu based PET imaging and therapy. It has been pointed out that the transchelation of 64Cu from DOTA to superoxide dismutase in the liver and the persistent localization of the final radiometal metabolite 64Cu-DOTA lysine within the tissue are disadvantageous for 64Cu-DOTA therapy application [30]. The demonstrated high in vivo stability of 64Cu-sarcophagine [16, 18], plus the ease of conjugation reported herein (such as maleimido-chelators with free sulfhydryl containing biomarkers), will expedite the 64/67Cu application in both imaging and therapy fields. We also need to point out that BaMalSar is a heterofunctional chelator. In this proof of principle study, we only modified one side with c(RGDyC) using the site-specific sulfhydryl-maleimide Michael addition. Conjugation of a second biomarker on the benzoic acid moiety of BaMalSar, will allow us to readily contruct novel multinodality or heterofunctional probes.
Conclusion
Three novel bifunctional chelators for the prepareation of 64Cu radiopharmaceuticals have been successfully synthesized. BaMalSar and Mal2Sar were conjugated with cyclic c(RGDyC) through free sulfhydryl and maleimide reactions. The conjugates, BaMalSar-RGD and Mal2Sar-RGD2 could be labeled with 64CuCl2, and exhibited high tumor uptake and tumor-to-normal tissue ratios. In the future, two different bioligands will be installed into the two pedant arms of AnAnSar, MalBaSar, or Mal2Sar for constructing dual targeting probes. Furthermore, the two reactive sites of AnAnSar, BaMalSar and Mal2Sar could be used to attach a targeting moiety on one side and an additional label (for secondary imaging modality) on the other. We anticipate that this newly developed method will offer a novel way to construct multimodality imaging probes as well.
Acknowledgements
This work was supported by the USC Department of Radiology, the Department of Energy (DE-SC0002353), the National Cancer Institute (P30CA014089), and the USC Provost's Biomedical Imaging Science Initiative.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Boswell CA, Sun X, Niu W, Weisman GR, Wong EH, Rheingold AL. et al. Comparative in vivo stability of copper-64-labeled cross-bridged and conventional tetraazamacrocyclic complexes. J Med Chem. 2004;47:1465-74
2. Niu G, Li Z, Cao Q, Chen X. Monitoring therapeutic response of human ovarian cancer to 17-DMAG by noninvasive PET imaging with 64Cu-DOTA-trastuzumab. Eur J Nucl Med Mol Imaging. 2009;36:1510-9
3. Chong HS, Mhaske S, Lin M, Bhuniya S, Song HA, Brechbiel MW. et al. Novel synthetic ligands for targeted PET imaging and radiotherapy of copper. Bioorg Med Chem Lett. 2007;17:6107-10
4. Prasanphanich AF, Nanda PK, Rold TL, Ma L, Lewis MR, Garrison JC. et al. [64Cu-NOTA-8-Aoc-BBN(7-14)NH2] targeting vector for positron-emission tomography imaging of gastrin-releasing peptide receptor-expressing tissues. Proc Natl Acad Sci U S A. 2007;104:12462-7
5. Sprague JE, Peng Y, Fiamengo AL, Woodin KS, Southwick EA, Weisman GR. et al. Synthesis, characterization and in vivo studies of Cu(II)-64-labeled cross-bridged tetraazamacrocycle-amide complexes as models of peptide conjugate imaging agents. J Med Chem. 2007;50:2527-35
6. Sun X, Wuest M, Weisman GR, Wong EH, Reed DP, Boswell CA. et al. Radiolabeling and in vivo behavior of copper-64-labeled cross-bridged cyclam ligands. J Med Chem. 2002;45:469-77
7. Woodin KS, Heroux KJ, Boswell CA, Wong EH, Weisman GR, Niu WJ. et al. Kinetic inertness and electrochemical behavior of copper(II) tetraazamacrocyclic complexes: Possible implications for in vivo stability. Eur J Inorg Chem. 2005:4829-33
8. Liu W, Hao G, Long MA, Anthony T, Hsieh JT, Sun X. Imparting multivalency to a bifunctional chelator: a scaffold design for targeted PET imaging probes. Angew Chem Int Ed Engl. 2009;48:7346-9
9. Bernhardt PV, Dyahningtyas TE, Harrowfield JM, Kim JY, Kim Y, Rukmini E. Chiral resolution of hexaamine cobalt(III) cages: Substituent effects on chiral discrimination. Aust J Chem. 2003;56:1187-91
10. Bottomley GA, Clark IJ, Creaser II, Engelhardt LM, Geue RJ, Hagen KS. et al. The Synthesis and Structure of Encapsulating Ligands - Properties of Bicyclic Hexamines. Aust J Chem. 1994;47:143-79
11. Sargeson A. Encapsulated metal ions. Pure Appl Chem. 1984;56:1603-19
12. Cai H, Fissekis J, Conti PS. Synthesis of a novel bifunctional chelator AmBaSar based on sarcophagine for peptide conjugation and 64Cu radiolabelling. Dalton Trans. 2009:5395-400
13. Cai H, Li Z, Huang CW, Park R, Shahinian AH, Conti PS. An improved synthesis and biological evaluation of a new cage-like bifunctional chelator, 4-((8-amino-3,6,10,13,16,19-hexaazabicyclo[6.6.6]icosane-1-ylamino)methyl) benzoic acid, for 64Cu radiopharmaceuticals. Nucl Med Biol. 2010;37:57-65
14. Cai H, Li Z, Huang CW, Shahinian AH, Wang H, Park R. et al. Evaluation of copper-64 labeled AmBaSar conjugated cyclic RGD peptide for improved microPET imaging of integrin αvβ3 expression. Bioconjug Chem. 2010;21:1417-24
15. Di Bartolo NM, Sargeson AM, Donlevy TM, Smith SV. Synthesis of a new cage ligand, SarAr, and its complexation with selected transition metal ions for potential use in radioimaging. J Chem Soc Dalton. 2001:2303-9
16. Voss SD, Smith SV, DiBartolo N, McIntosh LJ, Cyr EM, Bonab AA. et al. Positron emission tomography (PET) imaging of neuroblastoma and melanoma with 64Cu-SarAr immunoconjugates. Proc Natl Acad Sci U S A. 2007;104:17489-93
17. Li Z, Jin Q, Huang CW, Dasa S, Chen L, Yap LP. et al. Trackable and Targeted Phage as Positron Emission Tomography (PET) Agent for Cancer Imaging. Theranostics. 2011;1:371-80
18. Liu S, Li Z, Yap LP, Huang CW, Park R, Conti PS. Efficient preparation and biological evaluation of a novel multivalency bifunctional chelator for 64Cu radiopharmaceuticals. Chemistry. 2011;17:10222-5
19. Cai H, Li Z, Huang CW, Park R, Shahinian AH, Conti PS. An improved synthesis and biological evaluation of a new cage-like bifunctional chelator, 4-((8-amino-3,6,10,13,16,19-hexaazabicyclo[6.6.6]icosane-1-ylamino)methyl)benzoic acid, for 64Cu radiopharmaceuticals. Nucl Med Biol. 2010;37:57-65
20. Cline GW, Hanna SB. The Aminolysis of N-Hydroxysuccinimide Esters. A Structure-Reactivity Study. J Am Chem Soc. 1987;109:3087-91
21. Brinkley M. A brief survey of methods for preparing protein conjugates with dyes, haptens, and cross-linking reagents. Bioconjug Chem. 1992;3:2-13
22. Wilbur DS. Radiohalogenation of proteins: an overview of radionuclides, labeling methods, and reagents for conjugate labeling. Bioconjug Chem. 1992;3:433-70
23. Beer AJ, Kessler H, Wester HJ, Schwaiger M. PET Imaging of Integrin αvβ3 Expression. Theranostics. 2011;1:48-57
24. Ye Y, Chen X. Integrin targeting for tumor optical imaging. Theranostics. 2011;1:102-26
25. Zhang Y, Yang Y, Cai W. Multimodality Imaging of Integrin αvβ3 Expression. Theranostics. 2011;1:135-48
26. Zhang X, Liu H, Miao Z, Kimura R, Fan F, Cheng Z. Macrocyclic chelator assembled RGD multimers for tumor targeting. Bioorg Med Chem Lett. 2011;21:3423-6
27. Li ZB, Chen K, Chen X. 68Ga-labeled multimeric RGD peptides for microPET imaging of integrin αvβ3 expression. Eur J Nucl Med Mol Imaging. 2008;35:1100-8
28. Wu Y, Zhang X, Xiong Z, Cheng Z, Fisher DR, Liu S. et al. microPET imaging of glioma integrin αvβ3 expression using 64Cu-labeled tetrameric RGD peptide. J Nucl Med. 2005;46:1707-18
29. Chen X, Park R, Tohme M, Shahinian AH, Bading JR, Conti PS. MicroPET and autoradiographic imaging of breast cancer αv-integrin expression using 18F- and 64Cu-labeled RGD peptide. Bioconjug Chem. 2004;15:41-9
30. Bass LA, Wang M, Welch MJ, Anderson CJ. In vivo transchelation of copper-64 from TETA-octreotide to superoxide dismutase in rat liver. Bioconjug Chem. 2000;11:527-32
Author contact
![]() Corresponding author: Tel.: +1 323 442 3858; fax: +1 323 442 3253. Email: pcontiedu (P.S. Conti). Tel.: +1 323 442 3252; fax: +1 323 442 3253. Email: ziboliedu (Z. Li)
Corresponding author: Tel.: +1 323 442 3858; fax: +1 323 442 3253. Email: pcontiedu (P.S. Conti). Tel.: +1 323 442 3252; fax: +1 323 442 3253. Email: ziboliedu (Z. Li)