Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Biochemistry of uPAR
Expression of uPAR during normal...
Structure-function relationships...
Development of a potent linear...
Non-invasive radionuclide-based...
Future application
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2013; 3(7):467-476. doi:10.7150/thno.3791 This issue Cite
Review
Structure-Driven Design of Radionuclide Tracers for Non-Invasive Imaging of uPAR and Targeted Radiotherapy. The Tale of a Synthetic Peptide Antagonist
Michael Ploug ![]()
Finsen Laboratory, Rigshospitalet & Biotech Research and Innovation Centre (BRIC), Copenhagen Biocenter, Ole Maaløes Vej 5, DK-2200 Copenhagen N, Denmark.
Published 2013-6-24
Abstract

Research performed during the last two decades has provided a wealth of information to highlight the role of the urokinase-type plasminogen activator receptor (uPAR) in the progression and dissemination of invasive and metastatic cancer. In parallel, our perception of the structure-function relationships in uPAR has been refined to such a level that a rational design of uPAR function as well as compounds specifically targeting defined functions of uPAR are now realistic options. This knowledge opens new avenues for developing therapeutic intervention regimens targeting uPAR as well as for monitoring the effects of such treatments by non-invasive imaging using e.g. positron emission tomography. This mini-review will focus on recent advancements in translational research devoted to non-invasive targeting of uPAR, with a view to molecular imaging of its expression in live individuals as well as specific eradication of these cells by targeted radiotherapy.
Keywords: CD87, urokinase receptor, PLAUR, cancer, plasminogen activation, PET imaging.
Introduction
”I see you” is the greeting used among the Na'vi population, the indigenous species of the moon Pandora, in the science fiction movie “Avatar” by James Cameron. This phrase is being used figuratively to mean "I see into you", or essentially "I understand you". Paraphrasing this statement slightly to “I see uPAR” provides a reasonable projection of the significant advancements in our perception of the structure-function relationships in the urokinase-type plasminogen activator receptor (uPAR) that has emerged during the last decade. This development has primarily been fuelled by the advent of an increasing number of high-resolution crystal structures of uPAR in complex with various bona fide protein ligands [1-4] as well as small, synthetic peptide antagonists [5]. Extending the analogy to the Na'vi greeting even further, research performed in the last couple of years has accordingly revealed that targeted non-invasive molecular imaging using radionuclide-based tracers can be used to visualize uPAR expression in superficial and occult cancer lesions in live animals [6-8]. Instrumental to this advancement of the uPAR-specific imaging by positron emission tomography (PET) is the development of a small, high-affinity peptide antagonist, which enables systemic targeting of uPAR in tissues and organs with high efficacy and specificity, whilst preserving a fast pharmacokinetic clearance profile. The present mini-review is dedicated to tell the tale of this particular peptide antagonist, which among other things proved to be decisive for solving the first crystal structure of human uPAR [5], as well as providing the first non-invasive molecular image of uPAR expression in vivo [6], thereby honouring the Na'vi greeting by transforming it to: “I see uPAR while looking into you”. For a contemporary and comprehensive review on the importance of structure-functional studies on uPAR for translational research aimed at developing intervention regimens specifically targeting uPAR, the reader is referred to consult Kriegbaum et al (2011) [9].
Biochemistry of uPAR
In a historical perspective, the urokinase-type plasminogen activator (uPA) receptor (uPAR) was identified as the single membrane protein responsible for the high-affinity binding of the serine protease uPA to a number of cell lines in culture [10, 11]. The early aspects of the biochemistry of uPAR in promoting activation and focalization of cell surface-associated plasminogen activation have been extensively reviewed [12]. In brief; the high-affinity uPA•uPAR interaction (Kd ~ 0.5 nM) is mediated by the N-terminal growth factor-like domain (GFD) of the modular serine protease ligand uPA [13]. The concomitant binding of the two zymogens, pro-uPA to uPAR and of plasminogen to certain membrane proteins with surface-exposed C-terminal lysines [14, 15], creates a favored microenvironment for focal plasminogen activation. This condition is primarily dependent on two separate conditions. First; the cell surface provides a unique “template effect”, where the proximity of the bound zymogens (pro-uPA and plasminogen) improves the kinetics of the reciprocal zymogen activation cascade. In this context, uPA activates plasminogen and the generated plasmin subsequently feedback activates receptor-bound pro-uPA [16]. Second; as long as the generated plasmin remains bound to the cell surface, it is refractory to inhibition by its cognate inhibitor, α2-antiplasmin. The association of plasmin to cell surfaces is mediated by the lysine binding sites of its kringle domains. As these lysine binding sites also are utilized by α2-antiplasmin, this provides the molecular basis for the lack of inhibition of cell-bound plasmin. As a consequence, the longevity of the catalytic activity of plasmin bound to cell surfaces is significantly prolonged.
Elegant genetic dissections in mice combined with a powerful pro-drug, the cytotoxicity of which is only unleashed when activated by receptor-bound uPA [17], demonstrate that uPAR is indeed the only physiologically relevant cell surface receptor focalizing uPA activity in vivo [18, 19]. The overt phenotypes associated with congenital uPAR deficiency introduced by ablation of the corresponding Plaur gene include chronic hepatic inflammation due to impaired fibrin surveillance. Interestingly, this condition is copied by transgenic mice having a genetic defect that specifically dismantles the uPA•uPAR interaction by introduction of subtle mutations in the receptor-binding module of uPA [3, 19].
The interaction between uPA and uPAR also reversibly modulates integrin-mediated cell adhesion and migration on vitronectin rich matrices in cell culture by stimulation of lamellipodia formation [20-25]. For an exhaustive update on this subject please consult the recent review by Smith and Marshall [26].
From a biochemical perspective, endogenous expression of uPAR thus endows its host cells with the means to control plasminogen activation in the close proximity of their cell surfaces. This may in turn perturb the microenvironment in a number of important aspects. First; the localized plasmin activity will promote a focused degradation of the extracellular matrix (ECM), facilitating the directional migration and invasion of cells into the surrounding tissue. Second; it will cooperate in the maintenance of extravascular fibrin homeostasis by cell-assisted fibrinolysis, as is illustrated by the sinusoidal fibrin accumulation observed in mice with a defective uPA•uPAR interaction [19]. Third; the focal proteolysis and degradation of the ECM components will indirectly modulate the “cytokine microenvironment” sensed by these invading cells by increasing the bioavailability of e.g latent TGF-β, which is sequestered in the ECM as a “passive reservoir” that needs to be mobilized and activated by proteolytic cleavage [27].
Expression of uPAR during normal physiology and in disease
Baseline expression of uPAR is generally low under normal homeostatic conditions. Nevertheless, its expression is often upregulated in tissues undergoing extensive remodeling [28] or in tissues experiencing chronic inflammation, as illustrated by the markedly increased uPAR expression in the synovial tissue isolated from inflamed joints from patients with rheumatoid arthritis [29].
The by far most dominating focus for histological studies on uPAR expression in human pathology is that on invasive and metastatic solid cancers - for a recent review on this issue please consult [30]. These lesions generally present very complex patterns of uPAR expression with varying contributions from neoplastic cancer cells and auxiliary cells of the reactive tumor stroma. Usually, uPAR-positive tumor-associated stromal cells include recruited inflammatory cells (e.g. neutrophils and macrophages), endothelial cells, fibroblasts, and myofibroblasts. Despite this complexity, it is evident that the most prominent uPAR expression observed in solid human tumors generally is confined to those histological fields that contain distinct invasive foci. In the case of colorectal cancer, the reactive tumor-stroma interface is accordingly dominated by infiltrating uPAR-positive macrophages, endothelial cells, and detached, solitary cancer cells (i.e. budding cancer cells). A recent study, which compared resected primary colorectal adenocarcinomas with their corresponding metastatic lesions, showed that uPAR expression correlated to the growth patterns of the liver metastases [31]. Interestingly, the pronounced uPAR expression found in the invasive foci of the primary tumor was only recapitulated by those metastatic lesions that showed extensive desmoplasia, i.e. with significant ECM deposition. Metastases having a pushing growth pattern, where the neoplastic cells are in direct contact with the liver parenchyma, were on the contrary essentially devoid of uPAR expression. This paired correlation between growth patterns of the metastatic hepatic lesions and uPAR expression thus provides a clear example of the importance of contextual signaling for recruitment of non-neoplastic host cells and ECM production in the tumor-stromal microenvironment [32].
In the last two decades, a vast number of reports have demonstrated the prognostic value of measuring uPAR levels in either resected tumor tissues or shed into the circulation as a soluble form, where high uPAR levels generally are correlated to poor patient survival. For a more in-depth discussion on this subject, the reader is referred to the following comprehensive review [30].
Structure-function relationships in the uPA•uPAR interaction
In this section, I will briefly describe selected protein-structural aspects of uPAR function, with special emphasis on subjects pertaining to ligand-binding epitopes and accessible druggable sites in uPAR, which can be targeted by pharmaceuticals or imaging reagents. For a more detailed information on structural aspects of uPAR function, please consult [9].
Human uPAR is encoded on chromosome 19q13 as a 335 residue long polypeptide chain [33], but posttranslational editing excise N- as well as C-terminal signal peptides. The fully processed uPAR thus comprises a 283 residue long single polypeptide chain, which is tethered to the plasma membrane via a C-terminal glycosyl-phosphatidylinositol (GPI) membrane anchor [34]. Utilizing this mode of membrane anchorage renders uPAR expression sensitive to mutations affecting the biosynthesis of GPI-precursors, and uPAR deficiency is consequently observed in leukocytes derived from patients with the hematological bone marrow disorder designated paroxysmal nocturnal hemoglobinuria [35]. Based on homology considerations, it is now clear that the extracellular protein part of uPAR is constructed from three homologous protein modules, which are designated Ly6/uPAR domains (in short LU domains) and are characterized by adopting the archetypical three-finger fold found in snake venom α-neurotoxins [9, 12, 36]. These 3 extracellular LU domains of human uPAR contain no less than 14 intradomain disulfide bonds and 5 potential N-linked glycosylation sites, but only 4 of the latter are generally being utilized [37]. Notwithstanding its relatively small size, uPAR is surprisingly able to choreograph the assembly of a noticeably large and hydrophobic ligand-binding cavity involving all three LU domains. A schematic representation of the multidomain assembly of uPAR attached to the plasma membrane via a GPI-anchor is illustrated in Figure 1A, where the hydrophobic ligand-binding cavity is delineated in Figure 1B on the crystal structure of the ATF•uPAR complex.
Graphical representation of the structure of uPA•uPAR complexes on the cell surface. A composite molecular model representing the structure of human uPAR based on the crystal structure solved for uPAR•ATF complexes is shown to in panel A. Secondary structure elements are depicted as a cartoon representation, where the assembly of the three LU-domains in uPAR is evident from the color coding, yellow (DI), blue (DII), and red (DIII). A hypothetical model for the GPI-anchor, tethering uPAR to the cell surface, is shown in sticks. The molecular shape of uPAR is visualized by a semitransparent surface. In panel B, the corresponding complex with its natural ligand uPA is shown using a solid surface representation for uPAR and a cartoon representation of the receptor-binding fragment of uPA (ATF) used to crystallize the complex. The large hydrophobic ligand-binding cavity of uPAR is highlighted by the grey area delimited by the hatched black line using the following atomic color coding: grey (C), blue (N), red (O), and yellow (S). The inset in the bottom right corner shows a more detailed picture of the tight engagement and burial of the tip of the β-hairpin of GFD in uPA within the deepest region of the central cavity in uPAR.
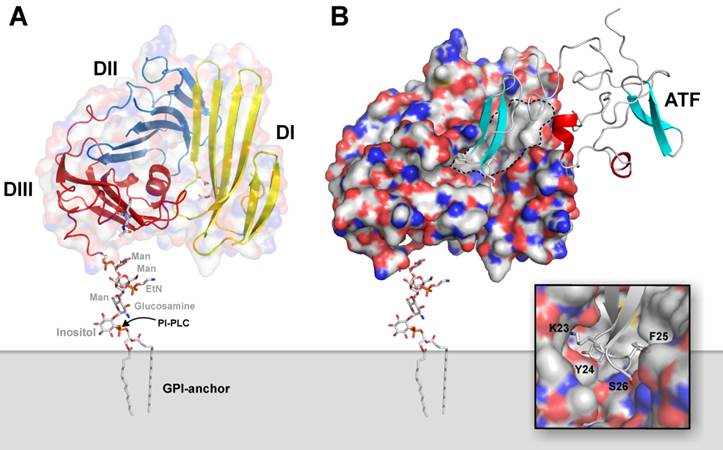
The cognate protease ligand for uPAR is the modular serine proteinase uPA, which, besides the C-terminal serine protease domain, also contains a growth factor-like domain (GFD) followed by a kringle domain in its modular amino-terminal fragment (ATF). Concordant with the observation that the high-affinity interaction between uPA and uPAR (KD ~ 0.5 nM) predominantly is governed by the GFD1-48, a significant portion of this module is indeed buried in the binding interface with uPAR [1-4]. The extensive burial of solvent-accessible surfaces of the GFD module in the uPA•uPAR complex is clearly illustrated in Figure 1B. The close-up of the binding interface, shown in the inset, highlights the complete burial of the tip of the β-hairpin in GFD, where the side chains of Lys23, Tyr24, and Phe25 project deeply into the hydrophobic ligand-binding cavity of uPAR, where they are shielded from the solvent [38]. Site-directed mutagenesis reveals that these buried residues in GFD also contribute significantly to the free energy of binding for uPA•uPAR interaction [3]. This defined architecture of the ligand-binding interface needs self-evidently to be considered, when modifying uPA, ATF or GFD with reporter groups for visualizing the uPA•uPAR interaction in biochemical assays in vitro and for non-invasive imaging in vivo. The importance of such considerations and the consequences of ignoring them are illustrated by the following case-story: In an attempt to develop small molecules as antagonists of the uPA•uPAR interaction, large chemical libraries were originally screened using a peptide surrogate of uPA as a reporter. Unfortunately, this peptide was unconsciously 125I-labeled at a position corresponding to the hotspot residue Tyr24 [39], and the claimed potency of the selected lead compounds could accordingly not be confirmed in subsequent cell-binding assays by an independent group [40]. Along the same lines, some precaution should always be exercised when modifying ATF extensively with reporter groups using traditional N-hydroxysuccinimide-based chemistry, as Lys23 in the β-hairpin of GFD is particularly prone to this modification leading to loss of uPAR binding [41]. Similarly, some concerns should also be exercised, when interpreting imaging data recorded for a particular radiotracer using a peptide surrogate of the β-hairpin of uPA as the uPAR-targeting moiety. In this case, the radionuclide 99mTe is tethered to a large heterocyclic tridentate chelator, which for unknown reasons and ill-advisedly is linked to the ε-amino group corresponding to Lys23 in uPA [42]. Concordant with our structure-function considerations on the uPA•uPAR interaction discussed previously, another group independently states that it is difficult to label this class of compounds for imaging without severely compromising their targeting efficiency [43].
Xenotransplanted cancer models in mice represent a popular first choice for up-front preclinical testing of efficacy and specificity of new lead drug candidates as well as targeted radionuclide-based tracers for non-invasive imaging using e.g. positron emission tomography. One complicating factor in naïvely adopting this workflow is the pronounced species selectivity that is inherent to the uPA•uPAR interaction. The experimental protocols commonly used for such preclinical studies do often not take this selectivity into account, which undoubtedly represents a confounding factor thus limiting the value of such studies. Biochemical studies on purified components reveal the interactions between the mixed orthologs to suffer a 100-300 fold loss in affinity compared to the corresponding unmixed native interactions [3]. Due to this difference, the therapeutic insult exercised on the xenotransplanted tumor and its associated reactive stromal compartment (host-derived) may thus differ immensely for a given uPAR-targeted intervention regimen, and this should be taken into account when evaluating the efficacy of such treatments in these preclinical animal models.
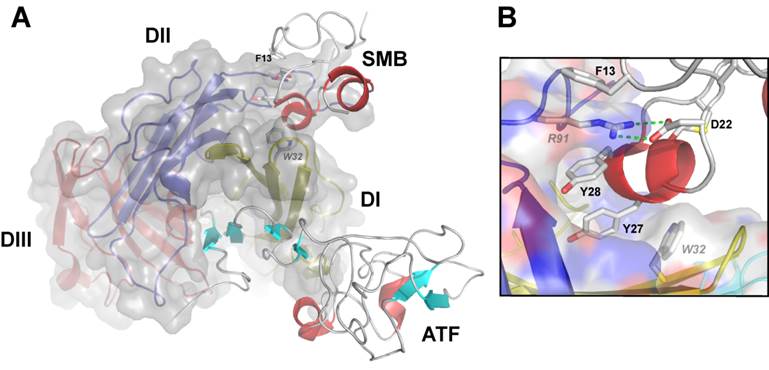
The second established protein ligand for uPAR, which is thoroughly characterized both functionally and structurally, is the matrix protein vitronectin. This interaction is primarily mediated by the small, N-terminal somatomedin B (SMB) domain, which is exposed in matrix-sequestered vitronectin. Compared to uPA binding, the affinity between uPAR and vitronectin is relatively moderate with a Kd of 2-10 µM for the SMB•uPAR interaction in solution [3, 23, 41, 44]. Nonetheless, the multivalent display of both reactants by sequestration in the ECM (vitronectin) or in lipid rafts on the cell surface (uPAR) secures a much higher functional affinity via avidity effects. This interaction between uPAR and matrix-embedded vitronectin may under certain conditions modulate integrin-mediated signaling via e.g. αvβ3 or αvβ5 resulting in a more robust formation of lamellipodia [20-24, 26, 41]. The binding site for SMB on uPAR is located at the domain interface between DI and DII and is topographically distinct from the uPA binding site (Figure 2A). Interestingly, recent data from our laboratory advances the proposition that uPA binding may in fact regulate vitronectin binding by allosteric modulation of the SMB binding site, both biochemically in purified systems as well as on the cell surface [23, 41]. According to this model, unoccupied uPAR exists predominantly in an open conformation with a suboptimal configuration of the SMB binding site in the interface between DI and DII. The mere docking of the β-hairpin of GFD into the hydrophobic binding cavity alters the conformation of uPAR by tightening the assembly of the three LU domains, which is accompanied by the formation of an optimal vitronectin binding site [23]. Corroborating the validity of this model is the finding that uPAR can be genetically engineered into a “constitutively” active receptor variant by covalently tethering DI to DIII via introduction of an artificial disulfide in the corresponding domain interface [41]. This constrained uPAR mutant stimulates lamellipodia formation independently of uPA binding and in this context provides a functional mimicry of uPA ligation [41]. Direct experimental evidence for this inherent conformational flexibility of the unoccupied uPAR is provided by studies using small angle X-ray scattering, hydrogen-deuterium exchange and surface plasmon resonance [45]. An important ramification of this new molecular model with a view to development of small molecule antagonists of uPA binding is the risk that such compounds may actually display undesirable agonist effects on lamellipodia formation by driving uPAR into the closed active conformation [41]. Ultimately, this could in theory stimulate an undesirable increase in cell adhesion and migration on vitronectin rich matrices thus recapitulating the effect observed upon uPA binding [21, 23]. Although the magnitude of this impact remains to be proven in vivo, this precaution should nevertheless be considered when developing and testing new lead drug candidates targeting uPAR for therapeutic intervention.
Graphical representation of the crystal structure of the ternary uPA•uPAR•vitronectin complex. A composite molecular model of the ATF•uPAR•SMB complex solved by X-ray crystallography is shown in panel A. The structure is rotated 90° compared to Figure 1, providing a “top view” of uPAR and moving the cell surface to the back of the picture. As in Figure 1, uPAR is shown in a composite semitransparent surface and cartoon representation. The bound ligands ATF (representing uPA) and SMB (representing vitronectin) are depicted in cartoon representations. The picture to the right shows the detailed binding interface between uPAR and SMB highlighting the key residues for this interaction in uPAR (R91 and W32) and SMB (F13, D22, Y27 and Y28).
Choosing the large hydrophobic binding cavity in uPAR as target site for the development of small molecules may at first sight seem obvious, but it does nevertheless confer some inherent limitations on targeting selectivity. The endogenous level of receptor occupancy with uPA in vivo may thus represent an additional confounding factor, particularly for antagonists targeting this site, as they selectively detect unoccupied uPAR. The severity of this limitation is at present unknown, but under normal homeostatic conditions the blood level of uPA is only 20 pmol [46], which will leave the majority of uPAR unoccupied (KD ~ 0.5 nM). In pathophysiology, this scenario is, however, likely to change as uPA levels in many cancer lesions increase by local synthesis or release from activated neutrophils [47].
Development of a potent linear peptide antagonist
In the last decade, academia and industry have sought to develop new and specific pharmaceuticals targeting uPAR, which could ideally enter a translational pipeline, entailing their implementation as new treatment modalities in cancer patient management. These endeavors include the assessment of inhibitory recombinant proteins, monoclonal antibodies, protease-activated pro-drugs, synthetic antagonist peptides, low molecular weight compounds, and various means of gene silencing [9, 48, 49]. Disappointingly, none of these approaches has, however, so far reached clinical testing. Despite the accumulation of a vast literature documenting this research effort, the present short mini-review will, nevertheless, only focus on the development of a single class of promising linear peptide antagonists of the uPA•uPAR interaction. The founding member of this class of compounds is a 9-mer core peptide, which has proven highly versatile and is currently being used in a number of different settings as discussed below.
Originally, a number of 15-mer precursor peptides of this class of uPAR antagonists were identified by an unbiased selection in a naïve M13 phage display library, using transfected cell lines expressing high levels of uPAR as panning “reagents” [50]. The corresponding synthetic peptides inhibit the uPA•uPAR interaction with IC50-values in the range of 0.010-10 µM. The corresponding lead peptide (clone 20) was subsequently synthesized and optimized by consecutive truncations, alanine replacement synthesis, photoaffinity labeling, and affinity maturation using combinatorial chemistry [51-53]. The resultant 9-mer core peptide is denoted AE105 and has the following sequence: Asp1-Cha2-Phe3-ser4-arg5-Tyr6-Leu7-Trp8-Ser9, where Cha is cyclohexyl-(L)-alanine, ser and arg are both present in the D-configuration, and the underlined residues are hot spots for the interaction with uPAR - as illustrated in Figure 3 [52]. Due to the presence of unnatural amino acids, this peptide is remarkably stable in serum, particularly after blocking its C-terminus e.g. by synthesis of a pseudosymmetrical dimer on a lysine scaffold - designated AE120 [52]. This peptide binds uPAR with high affinity (Kd < 1 nM), competes uPA binding to breast cancer cell lines with an IC50-value of 2 nM, and inhibits intravasation of HEp3 cells into the chick chorioallantois membrane [52]. It is, however, strictly species selective as it does not inhibit the mouse uPA•uPAR interaction, which in some settings represents a complicating factor for its preclinical testing in mouse model systems using transplanted human xenografts.
Graphical representation of uPAR in complex with the core peptide providing the targeting specificity of the PET imaging probes in vivo. The first X-ray crystal structure solved for human uPAR is shown in panel A using a similar graphical setup as in the previous figures. In this case, uPAR is co-crystallized with a small peptide antagonist derived from AE105 (panel B), which is used as the uPAR-targeting moiety in the PET tracers discussed in this review. A more detailed representation of the peptide-uPAR binding interface is shown in panel C, where the helical nature of the bound peptide is evident (hydrogen bonds in yellow). The hotspot residues Cha2, F3, and W8 are shown, whereas the important L7 is partly hidden behind the α-helix of the bound peptide. Y6 represents the non-essential position to which the covalently linked mercury was attached for structure determination [5]. The positions in uPAR that are covalently modified when W8 or F3 in the peptide antagonist is replaced by different photoaffinity labeling probes [51] are also depicted, illustrating the proximity of W8 to H251 (cyan) and of F3 to R53 and L66 (green).
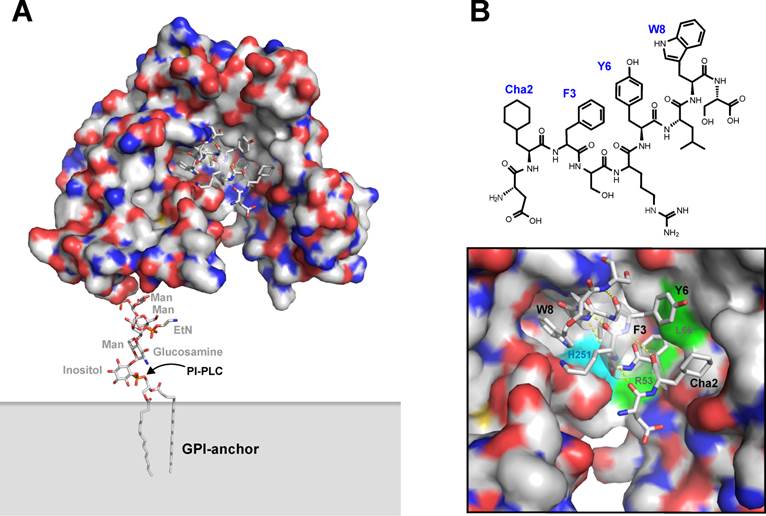
From a mechanistic point of view, this small linear peptide played a major role in delineating the molecular basis underlying the high affinity of the uPA•uPAR interaction. First and foremost, it played an instrumental part in solving the first three-dimensional structure of human uPAR by X-ray crystallography, where it undertook a dual role in both facilitating crystallization of uPAR and in solving the phases of the recorded diffraction patterns after modifying Tyr6 with covalently bound mercury [5]. The structure solved for the uPAR•peptide antagonist complex is shown in Figure 3, which bears a close resemblance to the structure subsequently solved for the corresponding uPAR•ATF complex (Figure 1). Nonetheless, a closer examination reveals that significant differences exist between these structures, notably in the assembly of the DI-DIII domain interface. However, additional biochemical studies performed on this interaction in solution further validated this molecular model of human uPAR derived from the X-ray data, including e.g. hydrogen-deuterium exchange [54] and photoaffinity labeling [51, 53]. These studies were in fact conducted before the crystal structure of uPAR was solved, and they reassuringly demonstrate the pairwise proximity of Trp8 and Phe3 in the bound peptide to His251 and Arg53/Leu66 in uPAR. This relationship is clearly highlighted in Figure 3B. The noticeable differences in the uPAR structures presented in Figures 1A and 3A are therefore real, and they were recently reconciled in a molecular model for uPAR, where the significant conformational flexibility of unoccupied uPAR plays a functional role [45]. According to this model, uPA binding to the central cavity drives the receptor into a closed conformation with an accompanying increase in its vitronectin-dependent signaling [23, 41].
The versatility and specificity of this peptide antagonist is further illustrated by the very different applications it supports. In our laboratory, we are e.g using it extensively for affinity purification of high quality preparations of recombinant human uPAR [55]. Other groups have successfully used it for various analytical assays. These include using AE120 as an additive in a time-resolved immunofluorescence assay to enable the specific measurement of shed uPAR domain I in blood samples from cancer patients [56]. In yet another setting, the N-terminus of this peptide is conjugated to fluorescein, and the labeled peptide is used as a surrogate reporter of the uPA•uPAR interaction in a fluorescence polarization assay for high-throughput screening of low molecular weight antagonists [57]. Despite these virtues, it is, nevertheless, within non-invasive imaging by PET scanning and in theranostics, I expect we in the future will experience the full impact of the potential of this particular uPAR targeting peptide.
Non-invasive radionuclide-based imaging
Examination of the crystal structure of human uPAR in complex with this 9-mer core peptide antagonist (Figure 3) clearly discloses the ample free conformational space present in the vicinity of the N-terminus of the bound peptide, whereas the C-terminal is more tightly constrained by the receptor surface. Conjugating AE105 or N-terminal extended versions thereof to tetraazamacrocyclic chelators with pendant carboxylate arms covalently via their α-amino group is accordingly feasible without paying an unacceptable penalty on affinity and specificity [6,7]. This strategy has so far been used successfully for conjugating DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) in complex with the radionuclides 64Cu [6, 7], 111In [58], 177Lu [59], and 213Bi [43], and NODAGA (1-(1-carboxy-3-carboxy-propyl)-4,7-(carboxy-methyl)-1,4,7-triazacyclononane) in complex with 68Ga [8]. Importantly, our extensive knowledge on the biochemistry, dynamics, and molecular basis for the uPA•uPAR and peptide•uPAR interactions enables us now to implement rigorous control experiments, which are required to reveal any unspecific targeting properties of the tracer systems employed.
One of the persisting and annoying confounding factors in targeted non-invasive imaging of tumour lesions in vivo is the substantial passive tumour accumulation of the tracer that is often encountered due to the enhanced permeability and retention effect (EPR). Another complication factor for targeted imaging is the well-established in vivo instability of particularly 64Cu-DOTA-based tracers [60]. Transchelation of 64Cu from these chelators to abundant bona fide copper-containing proteins, such as superoxide dismutase, may therefore cause an undesirable and unspecific baseline tracer accumulation in the liver and tumour tissues [61]. With the advent of structure-based design, we now have the option of constructing a radionuclide-labelled non-functional targeting analogue, which should ideally be included as a reliable sensor of unacceptable high levels of non-target-mediated tracer accumulation [7]. In the case of uPAR imaging with 64Cu-DOTA-AE105, the levels of unspecific tracer accumulation in the tumour lesions are, reassuringly, comparable whether the mice receive the labelled dysfunctional targeting construct, or they receive a bolus of “cold” competitor peptide just prior to infusion of the labelled functional targeting construct [7]. Independent evidence further substantiating the specificity of the present uPAR imaging in vivo is provided by two additional observations. First, quantitative PET-based evaluation of the tracer accumulation in xenotransplants in vivo correlates to the actual levels of uPAR measured by ELISA in lysates of the corresponding resected tumours [7]. Second, low-medium resolution reconstructed computer images of the accumulation of the uPAR-specific PET tracer in vivo by and large recapitulate the actual uPAR protein expression as defined by high-resolution immunohistochemistry on the corresponding resected tumour tissue [7].
Future application
Although the present uPAR-targeting PET probe already has proved its diligence as imaging agent in preclinical mouse models, there is nevertheless still room for improvements before attempting the first studies in man. At present we are exploring various new and optimized macrobicyclic chelators [60] to reduce the levels of non-specific baseline tracer accumulation of 64Cu in liver and tumor. Bearing in mind that the present PET tracer only targets human uPAR, leaving murine uPAR undetected [52], these preclinical mouse models are therefore by far perfect surrogates for the partitioning of this PET tracer in patients.
Another avenue the research on this uPAR antagonist peptide may take in the coming years is to explore its applicability in targeted radiotherapy. In fact, two studies have already briefly touched upon this issue by labeling a DOTA-AE105 derivative with the α-emitter 213Bi [43] or DOTA-AE105 with the β-emitter 177Lu [59]. The former tracer binds specifically to uPAR-expressing cell lines in culture and has biodistribution profiles in mice bearing intraperitoneal xenografts of human OV-MZ-6 ovarian cancer cell-lines, which are comparable to those we observe with the uPAR specific PET probe 64Cu-DOTA-AE105. A dose-dependent cytotoxicity was furthermore observed in vitro in cell culture [43]. Unfortunately, the proper controls using a dysfunctional targeting peptide or competition by excess “cold” peptide as mentioned previously are lacking. As a consequence, it cannot be firmly excluded that the observed radionuclide-dependent insult is caused by the general radiation from unbound compound in the medium rather than a targeted effect per se. In the latter study, 177Lu-DOTA-AE105 [59] was also found to exhibit comparable biodistribution profiles in mice bearing subcutaneous xenografts of human HT-29 colon cancer cells. Importantly, uPAR expressing cancer cells were completely eradicated from the xenografts in those mice receiving the uPAR targeting radionuclide, whereas the frequency of uPAR expressing cancer cells remain unaltered in the xenografts of mice receiving a similar dose of a non-targeting control peptide [59]. Despite these encouraging findings, the full theranostic potential of this targeting system still needs further validation, since the species-selectivity inherent to this targeting peptide [52] precludes bona fide toxicity assessment as it leaves the host stromal compartment essentially untouched in these xenograft mouse models thereby attenuating the general toxicity of this targeted radiotherapy. A similar precaution should, nonetheless, also have been applied to a recent study, where the efficacy and translational potential of a 177Lu-conjugated recombinant antibody, which binds human uPAR with moderate affinity (KD ~ 10-40 nM) but not mouse uPAR, was evaluated as a uPAR-targeting radiotherapeutic in an orthotopic mammary carcinoma xenograft model in nude mice [62, 63]. Unfortunately, this study also lacks the imperative control using an irrelevant 177Lu-labelled mAb to demonstrate the specificity of the targeted ionization-induced insult in vivo.
Acknowledgements
The photographic artwork was generated with the excellent assistance of John Post (Finsen Laboratory). All molecular models were visualized using PyMol (Schrödinger, Portland, OR). Benedikte Jacobsen (Finsen Laboratory) is thanked for constructive comments on the manuscript.
Conflict of interest
The author declares that no conflict of interest exists.
References
1. Huai Q, Mazar AP, Kuo A; Parry GC, Shaw DE, Callahan J, Li Y, Yuan C, Bian C. et al. Structure of human urokinase plasminogen activator in complex with its receptor. Science. 2006;311:656-9
2. Huai Q, Zhou A, Lin L, Mazar AP, Parry GC, Callahan J, Shaw DE, Furie B, Furie BC. et al. Crystal structures of two human vitronectin, urokinase and urokinase receptor complexes. Nat. Struct. Mol. Biol. 2008;15:422-3
3. Lin L, Gårdsvoll H, Huai Q, Huang M, Ploug M. Structure-based engineering of species selectivity in the interaction between urokinase and its receptor: implication for preclinical cancer therapy. J. Biol. Chem. 2010;285:10982-92
4. Barinka C, Parry G, Callahan J, Shaw DE, Kuo A, Bdeir K, Cines DB, Mazar A, Lubkowski J. Structural basis of interaction between urokinase-type plasminogen activator and its receptor. J. Mol. Biol. 2006;363:482-5
5. Llinas P, Le Du MH, Gårdsvoll H, Danø K, Ploug M, Gilquin B, Stura EA, Ménez A. Crystal structure of the human urokinase plasminogen activator receptor bound to an antagonist peptide. EMBO J. 2005;24:1655-63
6. Li ZB, Niu G, Wang H, He L, Yang L, Ploug M, Chen X. Imaging of urokinase-type plasminogen activator receptor expression using a 64Cu-labeled linear peptide antagonist by microPET. Clin. Cancer Res. 2008;14:4758-66
7. Persson M, Madsen J, Østergård S, Jensen MM, Jørgensen JT, Juhl K, Lehman C, Ploug M, Kjaer A. Quantitative PET Imaging of human uPAR with 64Cu-DOTA-AE105: Implications for Localizing Cancer Invasion. J. Nuclear Med. 2012;53:138-45
8. Persson M, Madsen J, Østergård S, Ploug M, Kjaer A. 68Ga-labeling and in vivo evaluation of a uPAR binding DOTA- and NODAGA-conjugated peptide for PET imaging of invasive cancers. Nucl. Med. Biol. 2012;39:560-69
9. Kriegbaum MC, Persson M, Haldager L, Alpizár-Alpizár W, Jacobsen B, Gårdsvoll H, Kjær A, Ploug M. Rational targeting of the urokinase receptor (uPAR): Development of antagonists and non-invasive imaging probes. Current Drug Targets. 2011;12:1711-28
10. Stoppelli MP, Corti A, Soffientini A, Cassani G, Blasi F, Assoian RK. Differentiation-enhanced binding of the amino-terminal fragment of human urokinase plasminogen activator to a specific receptor on U937 monocytes. Proc. Natl. Acad. Sci. USA. 1985;82:4939-43
11. Vassalli JD, Baccino D, Belin D. A cellular binding site for the Mr 55,000 form of the human plasminogen activator, urokinase. J. Cell Biol. 1985;100:86-92
12. Ploug M. Structure-function relationships in the interaction between the urokinase-type plasminogen activator and its receptor. Curr. Pharm. Des. 2003;9:1499-528
13. Appella E, Robinson EA, Ullrich SJ, Stoppelli MP, Corti A, Cassani G, Blasi F. The receptor-binding sequence of urokinase. A biological function for the growth-factor module of proteases. J. Biol. Chem. 1987;262:4437-40
14. Andronicos NM, Chen EI, Baik N, Bai H, Parmer CM, Kiosses WB, Kamps MP, Yates JR, Parmer RJ. et al. Proteomics-based discovery of a novel, structurally unique, and developmentally regulated plasminogen receptor, Plg-RKT, a major regulator of cell surface plasminogen activation. Blood. 2010;115:1319-30
15. Madureira PA, Surette AP, Phipps KD, Taboski MA, Miller VA, Waisman DM. The role of the annexin A2 heterotetramer (AIIt) in vascular fibrinolysis. Blood. 2011;8:8
16. Ellis V, Behrendt N, Danø K. Plasminogen activation by receptor-bound urokinase. A kinetic study with both cell-associated and isolated receptor. J Biol Chem. 1991;266:12752-58
17. Liu S, Bugge TH, Leppla SH. Targeting of tumor cells by cell surface urokinase plasminogen activator-dependent anthrax toxin. J. Biol. Chem. 2001;276:17976-84
18. Liu S, Aaronson H, Mitola DJ, Leppla SH, Bugge TH. Potent antitumor activity of a urokinase-activated engineered anthrax toxin. Proc. Natl. Acad. Sci. USA. 2003;100:657-62
19. Connolly BM, Choi EY, Gårdsvoll H, Bey AL, Currie BM, Chavakis T, Liu S, Molinolo A, Ploug M. et al. Selective abrogation of the uPA-uPAR interaction in vivo reveals a novel role in suppression of fibrin-associated inflammation. Blood. 2010;116:1593-603
20. Kjøller L, Hall A. Rac mediates cytoskeletal rearrangements and increased cell motility induced by urokinase-type plasminogen activator receptor binding to vitronectin. J. Cell Biol. 2001;152:1145-57
21. Madsen CD, Ferraris GM, Andolfo A, Cunningham O, Sidenius N. uPAR-induced cell adhesion and migration: vitronectin provides the key. J. Cell Biol. 2007;177:927-39
22. Hillig T, Engelholm LH, Ingvarsen S, Madsen DH, Gårdsvoll H, Larsen JK, Ploug M, Danø K, Kjøller L. et al. A composite role of vitronectin and urokinase in the modulation of cell morphology upon expression of the urokinase receptor. J Biol Chem. 2008;283:15217-23
23. Gårdsvoll H, Jacobsen B, Kriegbaum MC, Behrendt N, Engelholm LH, Østergaard S, Ploug M. Conformational regulation of urokinase receptor (uPAR) function. Impact of receptor occupancy and epitope-mapped monoclonal antibodies on lamellipodia induction. J Biol Chem. 2011;286:33544-56
24. Salasznyk RM, Zappala M, Zheng M, Yu L, Wilkins-Port C, McKeown-Longo PJ. The uPA receptor and the somatomedin B region of vitronectin direct the localization of uPA to focal adhesions in microvessel endothelial cells. Matrix Biol. 2007;26:359-70
25. Waltz DA, Chapman HA. Reversible cellular adhesion to vitronectin linked to urokinase receptor occupancy. J. Biol. Chem. 1994;269:14746-50
26. Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell. Biol. 2010;11:23-36
27. Jenkins G. The role of proteases in transforming growth factor-beta activation. Int. J. Biochem. Cell. Biol. 2008;40:1068-78
28. Solberg H, Ploug M, Høyer-Hansen G, Nielsen BS, Lund LR. The murine receptor for urokinase-type plasminogen activator is primarily expressed in tissues actively undergoing remodeling. J. Histochem. Cytochem. 2001;49:237-46
29. Busso N, Peclat V, So A, Sappino AP. Plasminogen activation in synovial tissues: differences between normal, osteoarthritis, and rheumatoid arthritis joints. Ann. Rheum. Dis. 1997;56:550-57
30. Lund IK, Illemann M, Thurison T, Christensen IJ, Høyer-Hansen G. uPAR as Anti-Cancer Target: Evaluation of Biomarker Potential, Histological Localization, and Antibody-Based Therapy. Current Drug Targets. 2011;12:1744-60
31. Illemann M, Bird N, Majeed A, Laerum OD, Lund LR, Danø K, Nielsen BS. Two distinct expression patterns of urokinase, urokinase receptor and plasminogen activator inhibitor-1 in colon cancer liver metastases. Int. J. Cancer. 2009;124:1860-70
32. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
33. Børglum AD, Byskov A, Ragno P, Roldan AL, Tripputi P, Cassani G, Danø K, Blasi F, Bolund L. et al. Assignment of the urokinase-type plasminogen activator receptor gene (PLAUR) to chromosome 19q13.1-q13.2. Am. J. Hum. Genet. 1992;50:492-97
34. Ploug M, Rønne E, Behrendt N, Jensen AL, Blasi F, Danø K. Cellular receptor for urokinase plasminogen activator. Carboxyl-terminal processing and membrane anchoring by glycosyl-phosphatidylinositol. J Biol Chem. 1991;266:1926-33
35. Ploug M, Plesner T, Rønne E, Ellis V, Høyer-Hansen G, Hansen NE, Danø K. The receptor for urokinase-type plasminogen activator is deficient on peripheral blood leukocytes in patients with paroxysmal nocturnal hemoglobinuria. Blood. 1992;79:1447-55
36. Ploug M, Ellis V. Structure-function relationships in the receptor for urokinase-type plasminogen activator. Comparison to other members of the Ly-6 family and snake venom alpha-neurotoxins. FEBS Lett. 1994;349:163-68
37. Ploug M, Rahbek-Nielsen H, Nielsen PF, Roepstorff P, Danø K. Glycosylation profile of a recombinant urokinase-type plasminogen activator receptor expressed in Chinese hamster ovary cells. J. Biol. Chem. 1998;273:13933-43
38. Ploug M, Rahbek-Nielsen H, Ellis V, Roepstorff P, Danø K. Chemical modification of the urokinase-type plasminogen activator and its receptor using tetranitromethane. Evidence for the involvement of specific tyrosine residues in both molecules during receptor-ligand interaction. Biochemistry. 1995;34:12524-34
39. Blood CH, Neustadt BR, Smith EM. US20016228985, 2001.
40. De Souza M, Matthews H, Lee JA, Ranson M, Kelso MJ. Small molecule antagonists of the urokinase (uPA): urokinase receptor (uPAR) interaction with high reported potencies show only weak effects in cell-based competition assays employing the native uPAR ligand. Bioorg. Med. Chem. 2011;19:2549-56
41. Gårdsvoll H, Kjaergaard M, Jacobsen B, Kriegbaum MC, Huang M, Ploug M. Mimicry of the regulatory role of urokinase in lamellipodia formation by introduction of a non-native interdomain disulfide bond in its receptor. J. Biol. Chem. 2011;286:43515-26
42. Armstrong AF, Lemon JA, Czorny SK, Singh G, Valliant JF. Evaluation of single amino acid chelate derivatives and regioselective radiolabelling of a cyclic peptide for the urokinase plasminogen activator receptor. Nucl. Med. Biol. 2009;36:907-17
43. Knör S, Sato S, Huber T, Morgenstern A, Bruchertseifer F, Schmitt M, Kessler H, Senekowitsch-Schmidtke R, Magdolen V. et al. Development and evaluation of peptidic ligands targeting tumour-associated urokinase plasminogen activator receptor (uPAR) for use in alpha-emitter therapy for disseminated ovarian cancer. Eur. J. Nucl. Med. Mol. Imaging. 2008;35:53-64
44. Gårdsvoll H, Ploug M. Mapping of the vitronectin-binding site on the urokinase receptor: involvement of a coherent receptor interface consisting of residues from both domain I and the flanking interdomain linker region. J. Biol. Chem. 2007;282:13561-72
45. Mertens HD, Kjaergaard M, Mysling S, Gårdsvoll H, Jørgensen TJD, Svergun DI, Ploug M. A flexible multidomain structure drives the function of the urokinase-type plasminogen activator receptor (uPAR). J. Biol. Chem. 2012;287:34304-15
46. Grøndahl-Hansen J, Agerlin N, Munkholm-Larsen P, Bach F, Nielsen LS, Dombernowsky P, Danø K. Sensitive and specific enzyme-linked immunosorbent assay for urokinase-type plasminogen activator and its application to plasma from patients with breast cancer. J. Lab. Clin. Med. 1988;111:42-51
47. Schmitt M, Mengele K, Napieralski R, Magdolen V, Reuning U, Gkazepis A, Sweep FC, Brunner N, Foekens J. et al. Clinical utility of level-of-evidence-1 disease forecast cancer biomarkers uPA and its inhibitor PAI-1. Expert Rev. Mol. Diagn. 2010;10:1051-67
48. Pillay V, Dass CR. et al. The urokinase plasminogen activator receptor as a gene therapy target for cancer. Trends Biotechnol. 2007;25:33-39
49. Mazar AP, Ahn RW, O'Halloran TV. Development of novel therapeutics targeting the urokinase plasminogen activator receptor (uPAR) and their translation toward the clinic. Curr. Pharm. Des. 2011;17:1970-78
50. Goodson RJ, Doyle MV, Kaufman SE, Rosenberg S. High-affinity urokinase receptor antagonists identified with bacteriophage peptide display. Proc. Natl. Acad. Sci. USA. 1994;91:7129-33
51. Ploug M. Identification of specific sites involved in ligand binding by photoaffinity labeling of the receptor for the urokinase-type plasminogen activator. Residues located at equivalent positions in uPAR domains I and III participate in the assembly of a composite ligand-binding site. Biochemistry. 1998;37:16494-505
52. Ploug M, Østergaard S, Gårdsvoll H, Kovalski K, Holst-Hansen C, Holm A, Ossowski L, Danø K. Peptide-derived antagonists of the urokinase receptor. affinity maturation by combinatorial chemistry, identification of functional epitopes, and inhibitory effect on cancer cell intravasation. Biochemistry. 2001;40:12157-168
53. Ploug M, Østergaard S, Hansen LB, Holm A, Danø K. Photoaffinity labeling of the human receptor for urokinase-type plasminogen activator using a decapeptide antagonist. Evidence for a composite ligand-binding site and a short interdomain separation. Biochemistry. 1998;37:3612-22
54. Jørgensen TJ, Gårdsvoll H, Danø K, Roepstorff P, Ploug M. Dynamics of urokinase receptor interaction with Peptide antagonists studied by amide hydrogen exchange and mass spectrometry. Biochemistry. 2004;43:15044-57
55. Jacobsen B, Gårdsvoll H, Juhl Funch G, Østergaard S, Barkholt V, Ploug M. One-step affinity purification of recombinant urokinase-type plasminogen activator receptor using a synthetic peptide developed by combinatorial chemistry. Protein Expr. Purif. 2007;52:286-96
56. Piironen T, Laursen B, Pass J, List K, Gårdsvoll H, Ploug M, Danø K, Høyer-Hansen G. Specific immunoassays for detection of intact and cleaved forms of the urokinase receptor. Clin. Chem. 2004;50:2059-68
57. Wang F, Li J, Sinn AL, Knabe WE, Khanna M, Jo I, Silver JM, Oh K, Li L. et al. Virtual Screening Targeting the Urokinase Receptor, Biochemical and Cell-Based Studies, Synthesis, Pharmacokinetic Characterization, and Effect on Breast Tumor Metastasis. J. Med. Chem. 2011;54:7193-05
58. Liu D, Overbey D, Watkinson L, Giblin MF. Synthesis and characterization of an 111In-labeled peptide for the in vivo localization of human cancers expressing the urokinase-type plasminogen activator receptor (uPAR). Bioconjug. Chem. 2009;20:888-94
59. Persson M, Rasmussen P, Madsen J, Ploug M, Kjaer A. New peptide receptor radionuclide therapy of invasive cancer cells: in vivo studies using 177Lu-DOTA-AE105 targeting uPAR in human colorectal cancer xenografts. Nucl. Med. Biol. 2012;25:962-69
60. Wadas TJ, Wong EH, Weisman GR, Anderson CJ. Coordinating radiometals of copper, gallium, indium, yttrium, and zirconium for PET and SPECT imaging of disease. Chem. Rev. 2010;110:2858-902
61. Boswell C, Sun X, Niu W, Weisman G, Wong E, Rheingold A, CJ Andersson. Comparative in vivo stability of copper-64-labeled cross-bridged and conventional tetraazamacrocyclic complexes. J. Med. Chem. 2004;47:1465-74
62. Duriseti S, Goetz DH, Hostetter DR, LeBeau AM, Wei Y, Craik CS. Antagonistic anti-urokinase plasminogen activator receptor (uPAR) antibodies significantly inhibit uPAR-mediated cellular signalling and migration. J. Biol. Chem. 2010;285:26878-888
63. LeBeau AM, Duriseti S, Murphy ST, Pepin F, Hann B, Gray JW, van Brocklin HF, Craik CS. Targeting uPAR with antagonistic recombinant human antibodies in aggressive breast cancer. Cancer Res. 2013 DOI:10.1158/0008-5472
Author contact
![]() Corresponding author: Michael Ploug, Finsen Laboratory, Rigshospitalet & BRIC, Copenhagen Biocenter room 3.3.31, Ole Maaløes Vej 5, DK-2200 Copenhagen N, Denmark, FAX: +45-35453797, E-mail: m-plougdk.
Corresponding author: Michael Ploug, Finsen Laboratory, Rigshospitalet & BRIC, Copenhagen Biocenter room 3.3.31, Ole Maaløes Vej 5, DK-2200 Copenhagen N, Denmark, FAX: +45-35453797, E-mail: m-plougdk.