Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
The biology of the uPA system
The uPA system in cancer
Exploiting the biology of the...
Conclusion
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2013; 3(7):496-506. doi:10.7150/thno.4953 This issue Cite
Review
The Many Spaces of uPAR: Delivery of Theranostic Agents and Nanobins to Multiple Tumor Compartments through a Single Target
Thomas V. O'Halloran1,2,3, ![]() , Richard Ahn3, Patrick Hankins3, Elden Swindell4, Andrew P. Mazar1,2,
, Richard Ahn3, Patrick Hankins3, Elden Swindell4, Andrew P. Mazar1,2, ![]()
1. Robert H. Lurie Comprehensive Cancer Center,
2. Department of Molecular Biosciences,
3. Department of Chemistry,
4. Department of Chemical and Biological Engineering, Chemistry of Life Processes Institute, Northwestern University 2145 Sheridan Rd., Evanston, IL 60208, USA.
Received 2012-7-30; Accepted 2013-2-18; Published 2013-6-25
Abstract
The urokinase plasminogen activator (uPA) system is a proteolytic system comprised of uPA, a cell surface receptor for uPA (uPAR), and an inhibitor of uPA (PAI-1) and is implicated in many aspects of tumor growth and metastasis. The uPA system has been identified in nearly all solid tumors examined to date as well as several hematological malignancies. In adults, transient expression of the uPA system is observed during wound healing and inflammatory processes while only limited expression is identified in healthy, quiescent tissue. Members of the uPA system are expressed not only on cancer cells but also on tumor-associated stromal cells. These factors make the uPA system an ideal therapeutic target for cancer therapies. To date most therapeutics targeted at the uPA system have been inhibitors of either the uPA-uPAR interaction or uPA proteolysis but have not shown robust anti-tumor activity. There is now mounting evidence that uPAR participates in a complex signaling network central to its role in cancer progression, which provides a basis for the hypothesis that uPAR may be a marker for cancer stem cells. Several new uPAR-directed therapies have recently been developed based on this new information. A monoclonal antibody has been developed that disrupts the interactions of uPAR with signaling partners and is poised to enter the clinic. In addition, nanoscale drug delivery vehicles targeted to the uPA system using monoclonal antibodies, without disrupting the normal functioning of the system, are also in development. This review will highlight some of these new discoveries and the new uPA system-based therapeutic approaches that have arisen from them.
Keywords: urokinase plasminogen activator, nanobins, theranostics
Introduction
Over the past twenty years, our work and that of others has identified the urokinase plasminogen activator system (uPA, its receptor uPAR, and a specific inhibitor of uPA activity PAI-1) as central mediators of multiple cellular interactions and signaling events that regulate tumor microenvironments [1-3]. The uPA system is involved in multiple processes that contribute to tumor progression (Fig. 1) including tumor cell survival and growth, migration and invasion, angiogenesis, and co-option of inflammatory cells (macrophages, neutrophils) by the tumor [4-6]. The uPA system regulates extracellular proteolysis as well as intracellular signaling mediated by numerous effectors such as integrins and receptor tyrosine kinases [7]. In parallel, the expression of components of the uPA system is regulated at multiple levels. The complex biology and central role of the uPA system in tumor progression have led to the hypothesis that therapeutic targeting of uPA system components would have robust antitumor effects against a broad spectrum of cancer types. However, the development of these agents has been challenging and at times frustrating. The initial strategy of uPA-targeted cancer therapies largely focused on the development of small molecule inhibitors of the enzymatic activity of uPA [8]. A number of inhibitors were identified but most of these did not advance beyond pre-clinical development due to poor pharmacokinetics, bioavailability, and stability. The only small molecule uPA inhibitor (upamostat, Mesupron®, WX-671) advanced to clinical trials was developed by Wilex and several early studies have been completed in patients with advanced cancer. Data from phase II trials in patients with pancreatic cancer and HER-2 negative breast cancer were presented at the 2010 meeting of the American Society of Clinical Oncology, but these results have not yet been published in refereed journals [9, 10]. Wilex recently reported the final data from the HER-2 negative breast cancer study on its web site, and the combination of Mesupron® and capecitabine met its primary endpoint of improved progression free survival [11]. The future of the Mesupron program has yet to be formally determined, and no additional clinical trials appear to be planned. Aside from Mesupron, no other inhibitors of uPA, uPAR or PAI-1 have advanced to the clinic. The lack of clinically successful uPA inhibitory agents suggests that the development of these uPA inhibitors may parallel the difficulties found in the development of matrix-metalloprotease (MMP) inhibitors [12, 13]. The failure of MMP inhibitors in clinic trials and evidence that cell-surface associated MMPs are involved in pro-tumorigenic cell signaling suggest that inhibition of the proteolytic function of these proteins is not sufficient. Thus, the therapeutic potential of targeting the uPA proteolytic system remains an open question.
Targeting uPAR for the treatment of MBC. Proteolysis-targeted approaches, shown in red, have not led to robust antitumor effects in pre-clinical studies. We have focused our therapeutic strategies on targeted delivery of nanoparticles called nanobins using a monoclonal antibody that targets uPA (ATN-291) and induces rapid internalization of the uPA-uPAR complex.
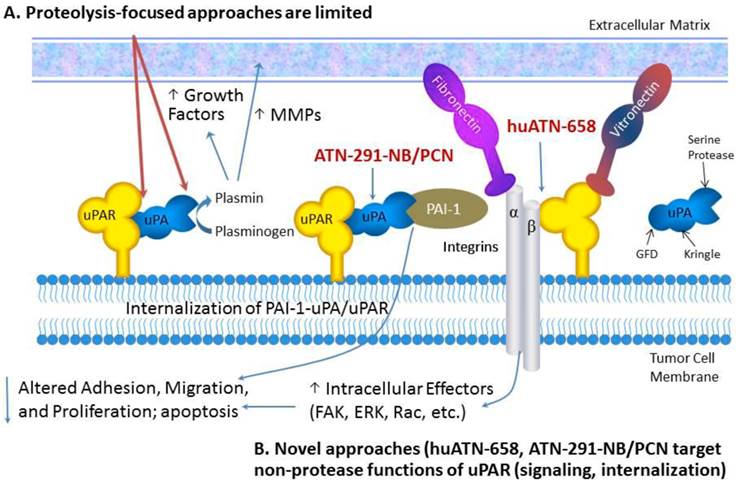
In this review, we will describe new diagnostic and therapeutic approaches targeting the uPA system that take advantage of new insights into the biology and structure-function relationship of uPA and uPAR. These new insights have formed the basis for the development of new uPAR antagonists as well as strategies focused on exploiting the biology of the uPA system for drug delivery. For a comprehensive review of uPA/uPAR directed cancer therapies we refer the reader to the following recently published reviews [1-3]. The selective expression of uPA and uPAR in tumors makes this system one of the most specific targets described, a fact that has been underappreciated by many tumor biologists and oncology drug developers. We will discuss new strategies for targeting the uPA system with a focus on nanotechnology for drug delivery and imaging and uPAR-associated cell signaling. We anticipate several new uPA- and uPAR-targeted agents entering the clinic in the next few years based on these emerging strategies.
The biology of the uPA system
uPA is a three domain serine protease comprised of a growth factor-like domain (GFD), a kringle domain, and a serine protease domain (Fig. 1) [2]. The proteolytic activity of uPA is highly specific for its preferred substrate, plasminogen. uPA activation of plasminogen to plasmin unleashes a promiscuous cascade of proteolysis wherein plasmin cleaves and activates a number of substrates including matrix metalloprotease pro-enzymes (e.g. proMMP-2 and -9) [14]. These proteases can then degrade components of the extracellular matrix (ECM) (e.g. fibronectin, vitronectin) and can release latent growth factors that are bound to glycosaminoglycans in the ECM [15]. This proteolysis leads to ECM remodeling and triggers cell signaling events that result in increased cell motility, invasion and survival [16]. The efficient activation of plasminogen to plasmin requires the localization of uPA to the cell surface via its specific receptor, uPAR, which enhances the catalytic efficiency of this activation by almost two orders of magnitude [17]. For many years, therapeutic development targeting the uPA system was focused on either inhibiting the catalytic activity of uPA directly or blocking the binding of uPA to uPAR, thereby significantly attenuating the ability of the system to activate plasminogen to plasmin [8]. However, in general, these approaches failed to exhibit significant antitumor effects in pre-clinical studies and were not advanced to clinical trials.
The last decade has brought about a paradigm shift in how the uPA system is viewed largely based on a number of structure-function studies which demonstrated that uPAR interacts with a number of cell-surface molecules and thereby alters cell adhesion and signaling. These interactions are the basis for many of the new approaches for uPAR-targeted therapies [8]. The recognition of uPAR as a signaling receptor was not intuitively obvious because uPAR has no transmembrane signaling domain. uPAR is a three-domain protein (D1, D2, D3) that is covalently attached to the outer layer of the cytoplasmic membrane by a glycosylphosphatidylinositol (GPI) anchor. Although all three uPAR domains contribute to uPAR binding to uPA, the D2D3 domains are thought to primarily control uPAR interactions with other cell surface proteins. Reports of uPAR interactions with numerous cell-surface molecules including a variety of integrins (including α5β1, α3β1, αvβ3 and αvβ5) [18, 19], receptor tyrosine kinases (RTK) such as the epithelial growth factor receptor (EGFR) and platelet-derived growth factor receptor (PDGFR) [20], G-protein coupled receptors (GPCR) [21], very low density lipoprotein receptor (VLDLR) family members [22] have been reported though there are likely more proteins interacting with uPAR that have yet to be identified. The demonstration of these interactions has often been inferred or indirectly demonstrated by immunoprecipitation. Direct binding of uPAR to EGFR, PDGFR, GPCRs, or VLDLR using purified proteins or cross-linking has not been achieved, unlike the more well-understood interaction of uPAR with uPA [23]. This suggests that the interactions of uPAR are copious and complex and several groups have hypothesized based on the cumulative uPAR literature that uPAR actually exists as part of a large, multi-protein complex called a signalosome that contains integrins, RTKs, caveolin, VLDLR members, and signaling effectors such as focal adhesion kinase (FAK), Src, and Akt [24, 25]. The fact that many of the same proteins that bind to uPAR have also been implicated in tumor progression provides an opportunity to explore therapeutically targeting these interactions and may be more important than the uPA-uPAR interaction for the inhibition of tumor growth and progression [8, 26, 27]. Recent advances in that regard will be discussed later in this review.
Part of the regulation of uPAR function involves the internalization of the uPA-PAI-1-uPAR complex, degradation of uPA-PAI-1, and recycling of unoccupied uPAR to the cell surface. When uPA bound to uPAR is inactivated by PAI-1, internalization via the low-density lipoprotein receptor related protein (LRP), a VLDLR family member, is initiated leading to endocytosis of the tripartite uPAR-uPA-PAI-1 complex via a clathrin-mediated mechanism. Once internalized, the uPA-PAI-1 complex dissociates from uPAR. The uPA-PAI-1 complex is trafficked to the lysosome for degradation and the unoccupied uPAR is recycled to the cell surface [8, 28]. This recycling likely regulates uPAR signaling as well as cell motility. The exact order of events and their organization at the molecular level, including the mechanism by which internalization of uPAR-uPA-PAI-1 is initiated, as well how the dissociation of the uPA-PAI-1 complex from uPAR after internalization results in a difference in subcellular trafficking, remain to be elucidated. We view internalization as an opportunity to develop new uPA- and uPAR-targeted agents that selectively deliver therapeutic agents to the inside of cells within a tumor.
The uPA system in cancer
Expression of uPA and uPAR has been demonstrated in almost every solid tumor type evaluated to date, as well as in certain hematologic malignancies (e.g. myeloma, AML) [3, 29, 30]. These include breast, prostate, ovarian, renal cell, lung, colon, endometrial, liver, thyroid, pancreatic and gastric cancers as well as in gliomas [3, 8, 31-37]. uPA and uPAR expression often correlates with poor prognosis and is associated with high-grade and progressive disease including metastasis. The expression of uPAR falls into two general categories: tumors where both tumor cells and tumor-associated (stromal) cells (e.g. angiogenic endothelial cells, fibroblasts, and macrophages) express uPA and uPAR [31, 32] and tumors in which only the tumor-associated cells express uPAR [37]. The expression of uPA and uPAR has prognostic value even if it is only expressed in the stromal cells, emphasizing the importance of the tumor stromal cell compartment to tumor progression [33]. We recently evaluated uPAR expression in a panel of normal tissues obtained from human adult cadaver donors and observed that uPAR is rarely expressed in normal quiescent tissue. In fact, uPAR expression was only reproducibly observed in histiocytes/monocytes in that study [8]. Although selective expression of uPA and uPAR has been alluded to in many previous studies, this was the first comprehensive analysis of adult human tissue and was in contrast to previous studies in mouse, where uPAR expression as observed in several tissues albeit in tissues undergoing remodeling.
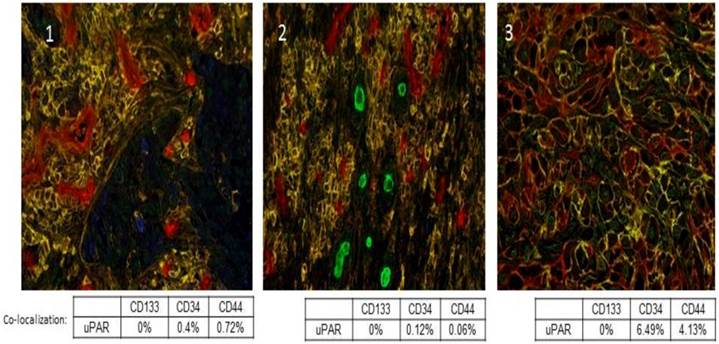
uPA and uPAR expression is heterogeneous within tumors, and they are not expressed by all tumor cells. Further, uPA and uPAR expression tends to be associated with the invasive edge of a tumor, usually at the tumor-vasculature or tumor-benign tissue interface [38]. In addition, uPAR expression has been described in cancer stem cells and this has now been shown in pancreatic, colon, small cell lung cancer, and breast cancer [39-42]. We recently evaluated a panel of tumor sections from patients with Stage IV non-small cell lung cancer (NSCLC) and observed co-expression of uPAR with CD44 in a small sub-population of cells, which we hypothesize may represent the cancer stem cell population in those tumors (Fig. 2). Functionally, uPAR positive tumor cells embody many of the characteristics of a cancer stem cell including resistance to chemotherapy [43], high tumor initiating potential, self-renewal, and plasticity [44]. Further, many tumor cell lines that do not express uPAR or express only low levels of uPAR (e.g. MCF-7, LNCaP) are poorly tumorigenic in mice. uPA and uPAR expression regulate and are regulated by the Wnt and Notch signaling pathways, which are also implicated in cancer stem cell proliferation [45, 46].
Co-localization of uPAR with previously described cancer stem cell markers CD133, CD34 and CD44. A multiplex IHC assay was developed for uPAR, CD133, CD34, and CD44. uPAR immunostaining of Stage IV NSCLC sections was performed using the ATN-658 antibody from Attenuon using DAB as a chromogen, which produces brown staining. CD133 staining was first optimized using two antibodies. The rabbit polyclonal antibody from AbCam demonstrated staining in formalin-fixed, paraffin-embedded tissue. Vulcan Red was used as a chromogen, and produced red staining in antigenic cells. Staining appeared nuclear and cytoplasmic, unlike the expected membrane localization. In order to produce specific staining of endothelia and rare cells, the antibody had to be used a titer that was high enough to produce week to moderate non-specific nuclear staining, especially around the edge of tissue. CD34 staining was performed using antibody clone QBEnd/10, and was localized using BCIP/NBT, a purple chromogen. Staining was membranous in nature and was localized to endothelia and, in a few specimens, cancer cells. CD44 staining was performed using antibody clone G44-26, and was localized using Vector SG, a gray chromogen. Staining was membranous and was localized to inflammatory cells and cancer cells. Red-Green-Blue color images were collected and spectrally unmixed to generate colored images for easier quantitation as described [74]. In the spectrally unmixed images, uPAR appears blue; CD133: green; CD34: red and CD44: yellow. Panels 1, 2, and 3 are three separate Stage IV NSCLC cases that demonstrate the co-localization of uPAR with a small percentage of CD34+ and CD44+ tumor cells.
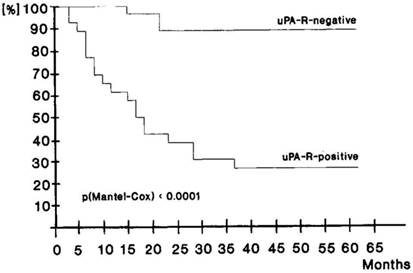
There is clinical data that supports uPAR as a mediator of cancer stem cell function. Only a few CK18+ uPAR+ tumor cells that had disseminated to bone in gastric cancer patients undergoing curative resection were sufficient to lead to tumor recurrence and a poor clinical outcome [47]. Over 90% of patients that had disseminated CK18+ uPAR- tumor cells in their bone marrow survived for 5 years whereas the 5-year survival of patients with disseminated CK18+ uPAR+ tumor cells was <30% (Fig. 3).
Disease free survival of 44 curatively resected patients according to uPAR detection on disseminated tumor cells in bone marrow taken intraopertaively. Upper curve, patients without uPAR evidence on tumor cells. MRT 60.88 ± 0.96 months, for 24 cases and 2 events. Lower curve, patients with uPAR expression on tumor cells. MRT 28.06 ± 2.44 months, for 20 cases and 14 events. Used by permission Nature Publishing Group, license number 2940350215579.
Autocrine and paracrine regulation of uPA and uPAR expression is also frequently observed in tumor and tumor stromal cells by mediators such as EGFR, IGF-1R, integrins, Akt, ERK, Src and c-Met/HGF. uPA and uPAR in turn also regulate the expression and functions of these same pathways. For example, there is now substantial evidence demonstrating cross-talk between uPAR and ErbB signaling [48]. How uPA and uPAR can regulate the function and expression of such a diverse set of mediators remains an open question. However, the signalosome hypothesis described above would account for these functions and would have significant implications for therapeutic development. For example, the interaction and cross-talk of uPAR with a number of molecules involved in breast cancer progression and metastasis including EGFR1, HER2, and HER3 has been described and uPAR has been demonstrated to mediate signaling though various signaling effectors implicated in breast cancer progression including Ras/ERK, Src, integrins/FAK, and Akt [16]. Tumor co-amplification of the genes for uPAR and HER2 has also been observed in patients with metastatic breast cancer (MBC) and predicts poor outcome [49]. In addition, the expression of uPAR in breast cancer cells primes these cells to, and may compensate for, the inhibition of ErbB signaling [50]. Thus, the inhibition of uPAR signaling may potentiate the effects of ErbB signaling inhibitors such as erlotonib, lapatanib and trastuzumab and may overcome resistance to these agents, which often occurs through feedback regulation of a downstream signaling mediator in the same signaling pathway or up-regulation of other ErbB family members. Several recent publications of clinical data support this notion in cancer patients where inhibition of EGFR signaling in metastatic colon cancer at the level of ligand binding and RTK signaling using cetuximab and erlotinib led to significant clinical responses in patients that had failed chemotherapy [51]. In the adjuvant setting in breast cancer, clinical benefit was also observed when trastuzumab was combined with erlotinib [52]. Given the cross-talk between uPAR signaling and other signaling pathways, understanding the way these pathways interact will be critical to designing rational drug combinations that can be used with uPAR-targeted therapy. However, the selective expression of uPAR in tumor tissue suggests that uPAR-targeted therapy may be well-tolerated as a component of many different types of drug combinations.
Exploiting the biology of the uPA system for targeting
Given the selective expression of uPA and uPAR by tumor tissue compared to normal tissue and the broad expression of uPA and uPAR by a variety of different cancer types, the uPA system is an ideal target to utilize for drug delivery. Several groups including ours have generated various constructs that target the uPA system for the delivery of anticancer therapeutics or imaging agents [8, 42, 53]. The internalization of uPA and uPAR is especially attractive from a targeting perspective and provides a mechanism to deliver large boluses of cytotoxic drugs or imaging agents that can selectively accumulate within a tumor. The expression of uPA and uPAR by tumor stromal cells will also allow the therapeutic targeting of this tumor compartment, which we hypothesize will lead to improved therapeutic effects compared to targeting the tumor cells alone.
For example, disulfide cyclized peptide derived from the growth factor domain (GFD) of uPA (amino acids 19-31)-DOTA conjugates bound to 213-Bi (an α emitter) were cytotoxic to OV-MZ-6 uPAR expressing ovarian cancer cells in vitro and localized to OV-MZ-6 tumors in xenograft models [53]. A recent study used a uPA GFD mimetic peptide that binds to human uPAR with high affinity conjugated to DOTA loaded with 64Cu to image experimental colon cancer tumors in mice [54]. In addition to the detection of these experimental tumors, demonstrating the proof of concept for this approach, this imaging technique was able to correlate uPAR expression levels with response to 5-FU and showed that higher uPAR expression rendered the tumors less sensitive to 5-FU. This is the first study that suggests that there may be a threshold effect for uPAR expression in tumor growth and progression and that the level of uPAR expression may mediate drug effect. This will be important to explore further with uPAR targeted therapy in order to understand whether a similar threshold will be required for response to uPAR targeted therapy similar to what has been observed with other cell-surface tumor targets such as c-MET and HER2 [55]. Several groups have also focused on using the amino terminal fragment of uPA (ATF, which contains the GFD) to deliver novel therapeutic payloads. The ATF binds to uPAR with an affinity that is similar to full size uPA [56] and provides a scaffold for the conjugation of payloads. Several ATF-toxin fusions have been reported. For example, a fusion protein (ATF-PE) comprised of the ATF and the Pseudomonas exotoxin (PE) retained the binding affinity of wild-type ATF and was cytotoxic to a number of cell lines in vitro with IC50 values as low as 0.3 pM [57]. ATF-PE required internalization for its cytotoxic activity but this internalization was not mediated by uPAR alone. Experiments using radiolabeled ATF and ATF-PE demonstrated a ~2 fold greater internalization of ATF-PE, compared to ATF alone. In addition, adding unlabeled ATF as a competitor to the radiolabeled ATF-PE blocked internalization of ATF-PE, which demonstrates that ATF played an important role in the toxicity of ATF-PE. It is likely that the PE moiety itself was responsible for the enhanced internalization of ATF-PE, possibly through interactions with other lipoprotein receptors (e.g. the α2-macroglobulin receptor) [58]. In our hands, free ATF is generally not endocytosed via uPAR and trafficked to the lysosome although other mechanisms of internalization, as described above, may be possible. An ATF-diphteria toxin (DTAT) fusion protein has also been described. Similar to ATF-PE, DTAT retained the binding activity of wild type ATF and was cytotoxic to U87 glioma cells in vitro with an IC50 similar to the Kd for binding [59]. DTAT was also evaluated in a sub-cutaneous U87 model in vivo where treatment with DTAT significantly delayed tumor growth, more than doubling the time it took for tumors to achieve 2000 mm3 [60]. DTAT also demonstrated activity in a model of metastatic NSCLC to the brain [61]. Intracerebral infusion using convection-enhanced delivery in mice with established brain metastases significantly prolonged survival in treated vs control mice (*87 vs 63 days, p=0.006). In these xenograft studies it should be again noted that the targeting effects were solely based on targeting human uPAR on the tumor cells, due the lack of cross-reactivity of human uPA and mouse uPAR. Therefore, DTAT would not be expected to target the tumor stroma in these studies.
More recently, several groups have exploited ATF-mediated delivery to target various nanoparticles to uPAR and describe ATF-mediated targeting to tumor cells. Yang et al. describe the conjugation of iron oxide (IO) nanoparticles to ATF for delivery of ATF-IO to uPAR expressing breast cancer cells [42]. ATF-IO binds to tumor cells in vitro and localizes to tumor in vivo. Abdalla et al. have also described theranostic ATF conjugates containing IO and noscapine [61]. Cytotoxicity of these conjugates against prostate cancer cells was observed in the μM range. Huang et al. have used the ATF to target gold nanorods in xenograft models though the total tumor accumulation of these rods was not greatly enhanced compared to non-targeted nanorods [62]. A notable result is that more of the targeted nanorods were internalized by tumor cells compared to tumor macrophages, which is consistent with other targeted nanoparticle delivery vehicles [63]. In a report from Wang and coworkers, a targeting ligand for stealth liposomes was developed from a linear peptide sequence in the GFD; in this system, modest selectivity was presented [64]. One limitation to all xenograft and biodistribution studies with uPA and uPA-binding ligands is the species specificity of the uPA-uPAR interaction; human uPA has a 100-1000-fold lower binding affinity for mouse uPAR than human [65]. This has several implications: 1) treatment response may be underestimated in xenograft models, since mouse stromal uPAR is not affected; 2) the toxicity profile of uPA directed therapies may not be apparent until tested in humans; and 3) murine biodistribution studies may not be representative of the biodistribution in humans. However, as described earlier, toxicity concerns are mitigated since a thorough analysis of cadaveric human tissue showed a narrow tissue distribution of uPAR [8]. While these reports show the potential of uPA/uPAR targeting for cancer therapy, to our knowledge no uPA-targeted nanoparticles have demonstrated therapeutic efficacy in vivo.
Various studies have shown that nanoscale drug delivery vehicles can increase the therapeutic index of cytotoxic drugs by altering their pharmacokinetics and biodistribution [66, 67]. Enhanced tumor accumulation via extravasation of nanoliposomes through fenestrated tumor vasculature may also lead to preferential accumulation in tumor through an “enhanced permeability and retention effect” (EPR effect) [68]. We have developed a novel liposomal nanoparticle drug encapsulation and formulation platform, that we named the nanobin. The nanobin is based on nanoliposomes, which have been formulated with cytotoxic payloads, including arsenic trioxide, cisplatin, and doxorubicin [69-71]. We have used two strategies to modify the surface characteristics and drug release properties of these particles: PEGylation to reduce opsonization and a pH-responsive cross-linked polymer shell. The PEGylated nanobins retain the reduced opsonization of other PEGylated nanoliposome formulations, and are fully able to take advantage of the EPR effect in highly vascularized tumors. In the second approach, the addition of a pH-responsive cross-linked polymer cage to the surface of the nanobins helps to improve the stability of the nanobins, even after freeze-drying. The polymer cage also increases the drug release rate at low pH, which could potentially improve the therapeutic efficacy of these nanoparticles in the low-pH of the tumor microenvironment.
Untargeted nanobins (NB) containing arsenic trioxide packaged with nickel [NB(Ni,As)] tested in the MDA-MB-231 orthotopic breast cancer model exhibited improved pharmacokinetics, increased tolerability, and greater antitumor effects in vivo compared to free drug [69]. Nanobins encapsulating arsenic trioxide packaged with cisplatin [NB(Pt,As)] have also been developed and also show activity in the MDA-MB-231 model. In addition, doxorubicin (DXR) encapsulated in the polymer-coated nanobin [PCN(DXR)] demonstrated increased antitumor activity in this model [72]. Further, cisplatin-conjugated PCN(DXR) [Pt-PCN(DXR)], which can be modified to deliver different cisplatin:DXR ratios, is highly synergistic compared to the combination of free cisplatin and free DXR when tested against multiple tumor cell lines in vitro including MDA-MB-231 [73]. The observed synergistic activity of DXR and cisplatin against the cisplatin-resistant breast cancer cell line MDA-MB-231 emphasizes another potential advantage of nanoparticle drug delivery, namely the ability to circumvent certain drug resistance mechanisms such as MDR1-dependent drug efflux, which may not recognize encapsulated drugs. These properties have led us to hypothesize that uPA- or uPAR-targeted nanobins that can be internalized into tumor and tumor-associated cells could have even greater therapeutic efficacy. We have developed a panel of uPA-targeted monoclonals as well as a panel of uPAR targeted monoclonals (Table 1), several of which are internalized. The majority of our current work has focused on the uPA-targeted monoclonal antibody, ATN-291. ATN-291 binds to the kringle domain of uPA and can bind to uPA even if it is already bound to uPAR. Although the internalization mediated by uPAR depends on the presence of both uPA and its inhibitor, PAI-1, in most cell lines evaluated [28], ATN-291 appears to trigger internalization when it binds to uPA irrespective of whether PAI-1 is present.
Description and characterization of a novel panel of uPA and uPAR targeted monoclonal antibodies with therapeutic potential. A panel of uPA and uPAR targeted antibodies with different epitope specificities has been developed. The biological and targeting activity of this panel is presently being evaluated in our laboratory. We hypothesize that the different epitope specificities and targeting either uPA or uPAR will allow us to dissect some of the basic biology of this system as well as identify antibodies that may not affect the biology of uPA/uPAR but may have utility as targeting agents. For example, ATN-292 inhibits the binding of uPA to uPAR by binding to the GFD of uPA but is not internalized. ATN-291 binds to the kringle domain of uPA, can bind to uPA when it is bound to uPAR and is internalized. Various targeting studies using ATN-291 are ongoing. ATN-616 and ATN-617 block the binding of uPA to uPAR but have little antitumor activity as monotherapies. ATN-615 and ATN-658 do not block the binding of uPA to uPAR and can bind to uPAR even when uPA is bound. Both of these antibodies are also internalized. The epitope for ATN-615 has been described [23] and the antitumor activity of ATN-658 in multiple tumor models has also been published [34, 75-77]. ATN-615 and ATN-658 are also being evaluated as targeting agents in our laboratory.
| Clone # | Antibody# | Specificity | Isotype | Kd, nM (ELISA) | Kd, nM (HeLa) |
|---|---|---|---|---|---|
| ATF-392 | ATN-291 | uPA (Kringle) | IgG1 κ | 0.3 | N.A. |
| ATF-1091 | ATN-292 | uPA (GFD) | IgG1 κ | 0.5 | N.A. |
| 234E-33 | ATN-615 | D2D3 | IgG1 κ | 2 | 1.3 |
| 234E-151 | ATN-658 | D2D3 | IgG1 κ | 1 | 5.4 |
| 234E-174 | ATN-616 | D2D3 | IgG1 κ | 5 | 0.6 |
| 234E-180 | ATN-617 | D2D3 | IgG1 κ | 29 | 1.3 |
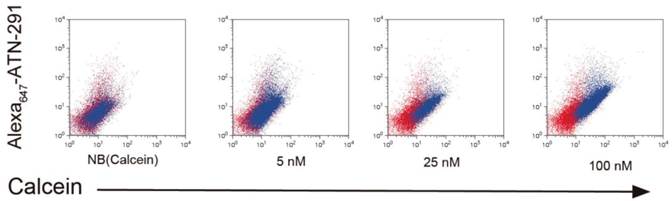
NB and PCN have now been conjugated to ATN-291 to generate uPA-targeted NB and PCN conjugates. We hypothesize that directed uptake of ATN-291-NB/PCN by uPA/uPAR-expressing tumors will allow targeting of metastatic lesions and further improve the antitumor efficacy and tolerability of the NB- and PCN-encapsulated chemotherapeutic agents. We predict that ATN-291-dependent endocytosis-mediated drug delivery will circumvent the MDR1 drug resistance efflux pump, allowing the delivery of chemotherapy to tumors that exhibit MDR-dependent drug resistance. Currently, we are evaluating ATN-291-NB and ATN-291-PCN containing various therapeutic payloads in several xenograft models and plan to identify at least one clinical candidate to move forward into pre-clinical development in the near future. We have observed the binding and internalization of ATN-291-NB(calcein) conjugates in a variety of tumor cell lines that express uPA in vitro (Fig. 4), whereas no binding or internalization has been observed in cell lines that are either uPA or uPAR null.
Internalization of ATN-291-NB(Calcein) by cells. Uptake experiments of ATN-291-NB(Calcein) in uPA positive PC3 prostate cancer cells and as determined by flow cytometry. Red dots represent untreated cells, while blue dots represent NB(Calcein), or ATN-291-NB(Calcein). The fluorescence intensity of the calcein fluorescence is plotted on the x-axis and the fluorescence intensity of the Alexa647 is plotted on the y-axis. With increasing uptake the distribution of the blue dots will shift right on the x-axis and up the y-axis. MDA-MB-435 uPA-negative breast cancer cells did not show uptake (data not shown).
Moving forward, several questions remain to be answered. First, it is not clear whether interrupting uPAR signaling will contribute positively to the delivery of anti-tumor drugs to cancer cells. Second, it must be shown whether interrupting uPAR signaling will improve patient outcomes any more than directly inhibiting uPA-uPAR binding. Finally, there is no clear indication if targeting nanoparticles to the uPA-uPAR system will perturb this signaling. These questions must be answered conclusively to fully understand the utility of uPA-uPAR for nanoparticle targeting.
Conclusion
The uPA system remains an attractive target for the treatment of cancer that has not yet been extensively explored in the clinic. Recent advances in the understanding of uPA and uPAR structure-function and biology have now provided a basis for the development of new therapeutics targeting this system. Several new molecules that target uPAR directly or utilize the uPA system to deliver therapeutic payloads have been described and are advancing toward the clinic. The next few years should see several new uPA and uPAR targeted agents enter clinical trials where the importance and potential of the uPA system in tumor progression will finally be explored.
Abbreviations
uPA: urokinase plasminogen activator; uPAR: urokinase plasminogen activator receptor; PAI-1: plasminogen activator inhibitor -1; GFD: growth factor domain; ECM: extracellular matrix; GAG: glycosaminoglycans; RTK: receptor tyrosine kinase; EGFR: epidermal growth factor receptor; PDGFR: platelet derived growth factor receptor; GPCR: G-protein coupled receptor; VLDL: very low density lipoprotein; VLDR: very low density lipoprotein receptor; FAK: focal adhesion kinase; AML: acute myeloid leukemia; IGF-1R: insulin like growth factor receptor 1; MBC: metastatic breast cancer; DOTA: 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid; 5-FU: fluorouracil; ATF: amino terminal fragment; PE: Pseudomonas exotoxin; DTAT: a fusion protein of the amino-terminal fragment of uPA and diphtheria toxin; NSCLC: non-small cell lung cancer; IO: Iron oxide; NB(Ni,As): nanobins encapsulating arsenic trioxide stabilized by nickel ions; NB(Pt,As): nanobins co-encapsulating cisplatin and arsenic trioxide; PCN: polymer caged nanobins; DXR: doxorubicin; MDR1: multidrug resistance protein 1.
Acknowledgements
The authors thank Mosaic Laboratories (Lake Forest, CA) for IHC and spectral analysis. Studies described herein were partially supported by funding from Lynn Sage Breast Cancer Foundation and the H Foundation (APM) as well as NIH U01CA151461 (TVO, APM), the NU MSTP (RWA) and the Biotechnology Cluster program at Northwestern (EPS, PLH).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol. 2002;3:932-43
2. Dass K, Ahmad A, Azmi AS, Sarkar SH, Sarkar FH. Evolving role of uPA/uPAR system in human cancers. Cancer Treat Rev. 2008;34:122-36 doi:S0305-7372(07)00181-8 [pii]10.1016/j.ctrv.2007.10.005
3. Mazar AP. Urokinase plasminogen activator receptor choreographs multiple ligand interactions: implications for tumor progression and therapy. Clin Cancer Res. 2008;14:5649-55
4. Binder BR, Mihaly J, Prager GW. uPAR-uPA-PAI-1 interactions and signaling: a vascular biologist's view. Thromb Haemost. 2007;97:336-42
5. Hildenbrand R, Gandhari M, Stroebel P, Marx A, Allgayer H, Arens N. The urokinase-system--role of cell proliferation and apoptosis. Histol Histopathol. 2008;23:227-36
6. Mekkawy AH, Morris DL, Pourgholami MH. Urokinase plasminogen activator system as a potential target for cancer therapy. Future Oncol. 2009;5:1487-99
7. Eden G, Archinti M, Furlan F, Murphy R, Degryse B. The urokinase receptor interactome. Curr Pharm Des. 2011;17:1874-89
8. Mazar AP, Ahn RW, O'Halloran TV. Development of novel therapeutics targeting the urokinase plasminogen activator receptor (uPAR) and their translation toward the clinic. Curr Pharm Des. 2011;17:1970-8
9. Goldstein LJ, Stemmer SM, Schmalfeldt B, Gottschalk N, Cardoso F, Dushkin H. et al. Phase II, two-arm, double-blind, multicenter, randomized study of the combination of oral WX-671 plus capecitabine versus capecitabine in first-line HER2-negative metastatic breast cancer (MBC). J Clin Oncol. 2010;28(Suppl 15s):abstrTPS131
10. Heinemann V, Ebert MP, Pinter T, Bevan P, Neville NG, Mala C. Randomized phase II trial with an uPA inhibitor (WX-671) in patients with locally advanced nonmetastatic pancreatic cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010:28
11. Wilex. MESUPRON® met its primary objective of demonstrating efficacy in the proof of concept phase II breast cancer trial. 2012.
12. Zucker S, Cao J, Chen WT. Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene. 2000;19:6642-50 doi:10.1038/sj.onc.1204097
13. Noel A, Gutierrez-Fernandez A, Sounni NE, Behrendt N, Maquoi E, Lund IK. et al. New and paradoxical roles of matrix metalloproteinases in the tumor microenvironment. Frontiers in pharmacology. 2012;3:140. doi:10.3389/fphar.2012.00140
14. Baramova EN, Bajou K, Remacle A, L'Hoir C, Krell HW, Weidle UH. et al. Involvement of PA/plasmin system in the processing of pro-MMP-9 and in the second step of pro-MMP-2 activation. FEBS Letters. 1997;405:157-62 doi:10.1016/s0014-5793(97)00175-0
15. Werb Z. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 1997;91:439-42
16. Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362-74 doi:10.1038/nrc1075
17. Ellis V, Behrendt N, Dano K. Plasminogen activation by receptor-bound urokinase. A kinetic study with both cell-associated and isolated receptor. J Biol Chem. 1991;266:12752-8
18. Kugler MC, Wei Y, Chapman HA. Urokinase receptor and integrin interactions. Curr Pharm Des. 2003;9:1565-74
19. Tarui T, Mazar AP, Cines DB, Takada Y. Urokinase-type plasminogen activator receptor (CD87) is a ligand for integrins and mediates cell-cell interaction. J Biol Chem. 2001;276:3983-90 doi:10.1074/jbc.M008220200
20. Hu J, Jo M, Cavenee WK, Furnari F, VandenBerg SR, Gonias SL. Crosstalk between the urokinase-type plasminogen activator receptor and EGF receptor variant III supports survival and growth of glioblastoma cells. Proc Natl Acad Sci U S A. 2011;108:15984-9 doi:10.1073/pnas.1113416108
21. Furlan F, Orlando S, Laudanna C, Resnati M, Basso V, Blasi F. et al. The soluble D2D3(88-274) fragment of the urokinase receptor inhibits monocyte chemotaxis and integrin-dependent cell adhesion. J Cell Sci. 2004;117:2909-16 doi:10.1242/jcs.01149
22. Webb DJ, Nguyen DH, Gonias SL. Extracellular signal-regulated kinase functions in the urokinase receptor-dependent pathway by which neutralization of low density lipoprotein receptor-related protein promotes fibrosarcoma cell migration and matrigel invasion. J Cell Sci. 2000;113( Pt 1):123-34
23. Huai Q, Mazar AP, Kuo A, Parry GC, Shaw DE, Callahan J, Li Y, Yuan C, Bian C, Chen L, Furie B, Furie BC, Cines DB, Huang M. Structure of human urokinase plasminogen activator in complex with its receptor. Science. 2006;311:656-9
24. Degryse B. The Urokinase Receptor and Integrins Constitute a Cell Migration Signalosome The Cancer Degradome. In: Edwards D, Høyer-Hansen G, Blasi F, Sloane BF, editors.: Springer New York. 2008:451-74
25. Monaghan-Benson E, McKeown-Longo PJ. Urokinase-type plasminogen activator receptor regulates a novel pathway of fibronectin matrix assembly requiring Src-dependent transactivation of epidermal growth factor receptor. J Biol Chem. 2006;281:9450-9 doi:10.1074/jbc.M501901200
26. Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516-23
27. Hutchinson J, Jin J, Cardiff RD, Woodgett JR, Muller WJ. Activation of Akt (protein kinase B) in mammary epithelium provides a critical cell survival signal required for tumor progression. Mol Cell Biol. 2001;21:2203-12
28. Nykjaer A, Conese M, Christensen EI, Olson D, Cremona O, Gliemann J. et al. Recycling of the urokinase receptor upon internalization of the uPA:serpin complexes. Embo J. 1997;16:2610-20 doi:10.1093/emboj/16.10.2610
29. Bene MC, Castoldi G, Knapp W, Rigolin GM, Escribano L, Lemez P. et al. CD87 (urokinase-type plasminogen activator receptor), function and pathology in hematological disorders: a review. Leukemia. 2004;18:394-400 doi:10.1038/sj.leu.2403250
30. Hjertner O, Qvigstad G, Hjorth-Hansen H, Seidel C, Woodliff J, Epstein J. et al. Expression of urokinase plasminogen activator and the urokinase plasminogen activator receptor in myeloma cells. Br J Haematol. 2000;109:815-22
31. Bhuvarahamurthy V, Schroeder J, Denkert C, Kristiansen G, Schnorr D, Loening SA. et al. In situ gene expression of urokinase-type plasminogen activator and its receptor in transitional cell carcinoma of the human bladder. Oncol Rep. 2004;12:909-13
32. Cantero D, Friess H, Deflorin J, Zimmermann A, Brundler MA, Riesle E. et al. Enhanced expression of urokinase plasminogen activator and its receptor in pancreatic carcinoma. Br J Cancer. 1997;75:388-95
33. Giannopoulou I, Mylona E, Kapranou A, Mavrommatis J, Markaki S, Zoumbouli C. et al. The prognostic value of the topographic distribution of uPAR expression in invasive breast carcinomas. Cancer Lett. 2007;246:262-7
34. Kenny HA, Leonhardt P, Ladanyi A, Yamada SD, Montag A, Im HK. et al. Targeting the urokinase plasminogen activator receptor inhibits ovarian cancer metastasis. Clin Cancer Res. 2011;17:459-71 doi:1078-0432.CCR-10-2258 [pii]10.1158/1078-0432.CCR-10-2258
35. Li Y, Cozzi PJ. Targeting uPA/uPAR in prostate cancer. Cancer Treat Rev. 2007;33:521-7 doi:10.1016/j.ctrv.2007.06.003
36. Li Z-B, Niu G, Wang H, He L, Yang L, Ploug M. et al. Imaging of Urokinase-Type Plasminogen Activator Receptor Expression Using a 64Cu-Labeled Linear Peptide Antagonist by microPET. Clin Cancer Res. 2008;14:4758-66
37. Pyke C, Ralfkiaer E, Ronne E, Hoyer-Hansen G, Kirkeby L, Dano K. Immunohistochemical detection of the receptor for urokinase plasminogen activator in human colon cancer. Histopathology. 1994;24:131-8
38. Sullivan R, Graham CH. Hypoxia-driven selection of the metastatic phenotype. Cancer Metastasis Rev. 2007;26:319-31 doi:10.1007/s10555-007-9062-2
39. Gutova M, Najbauer J, Gevorgyan A, Metz MZ, Weng Y, Shih CC. et al. Identification of uPAR-positive chemoresistant cells in small cell lung cancer. PLoS One. 2007;2:e243. doi:10.1371/journal.pone.0000243
40. Jo M, Eastman BM, Webb DL, Stoletov K, Klemke R, Gonias SL. Cell signaling by urokinase-type plasminogen activator receptor induces stem cell-like properties in breast cancer cells. Cancer Res. 2010;70:8948-58 doi:10.1158/0008-5472.CAN-10-1936
41. Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS. et al. Acquisition of Epithelial-Mesenchymal Transition Phenotype of Gemcitabine-Resistant Pancreatic Cancer Cells Is Linked with Activation of the Notch Signaling Pathway. Cancer Res. 2009;69:2400-7 doi:10.1158/0008-5472.can-08-4312
42. Yang L, Peng XH, Wang YA, Wang X, Cao Z, Ni C. et al. Receptor-targeted nanoparticles for in vivo imaging of breast cancer. Clin Cancer Res. 2009;15:4722-32 doi:10.1158/1078-0432.CCR-08-3289
43. Cortes-Dericks L, Carboni GL, Schmid RA, Karoubi G. Putative cancer stem cells in malignant pleural mesothelioma show resistance to cisplatin and pemetrexed. Int J Oncol. 2010;37:437-44
44. Yang L, Avila H, Wang H, Trevino J, Gallick GE, Kitadai Y. et al. Plasticity in Urokinase-Type Plasminogen Activator Receptor (uPAR) Display in Colon Cancer Yields Metastable Subpopulations Oscillating in Cell Surface uPAR Density—Implications in Tumor Progression. Cancer Res. 2006;66:7957-67 doi:10.1158/0008-5472.can-05-3208
45. Bin Hafeez B, Adhami VM, Asim M, Siddiqui IA, Bhat KM, Zhong W. et al. Targeted knockdown of Notch1 inhibits invasion of human prostate cancer cells concomitant with inhibition of matrix metalloproteinase-9 and urokinase plasminogen activator. Clin Cancer Res. 2009;15:452-9 doi:10.1158/1078-0432.CCR-08-1631
46. Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11:23-36
47. Heiss MM, Allgayer H, Gruetzner KU, Funke I, Babic R, Jauch KW. et al. Individual development and uPA-receptor expression of disseminated tumour cells in bone marrow: a reference to early systemic disease in solid cancer. Nat Med. 1995;1:1035-9
48. Meng S, Tripathy D, Shete S, Ashfaq R, Saboorian H, Haley B. et al. uPAR and HER-2 gene status in individual breast cancer cells from blood and tissues. Proc Natl Acad Sci U S A. 2006;103:17361-5 doi:10.1073/pnas.0608113103
49. Braun S, Schlimok G, Heumos I, Schaller G, Riethdorf L, Riethmuller G. et al. ErbB2 overexpression on occult metastatic cells in bone marrow predicts poor clinical outcome of stage I-III breast cancer patients. Cancer Res. 2001;61:1890-5
50. Li C, Cao S, Liu Z, Ye X, Chen L, Meng S. RNAi-mediated downregulation of uPAR synergizes with targeting of HER2 through the ERK pathway in breast cancer cells. International journal of cancer Journal international du cancer. 2010;127:1507-16 doi:10.1002/ijc.25159
51. Weickhardt AJ, Price TJ, Chong G, Gebski V, Pavlakis N, Johns TG. et al. Dual Targeting of the Epidermal Growth Factor Receptor Using the Combination of Cetuximab and Erlotinib: Preclinical Evaluation and Results of the Phase II DUX Study in Chemotherapy-Refractory, Advanced Colorectal Cancer. Journal of Clinical Oncology. 2012;30:1505-12 doi:10.1200/jco.2011.38.6599
52. Blackwell KL, Burstein HJ, Storniolo AM, Rugo H, Sledge G, Koehler M. et al. Randomized Study of Lapatinib Alone or in Combination With Trastuzumab in Women With ErbB2-Positive, Trastuzumab-Refractory Metastatic Breast Cancer. Journal of Clinical Oncology. 2010;28:1124-30 doi:10.1200/jco.2008.21.4437
53. Knor S, Sato S, Huber T, Morgenstern A, Bruchertseifer F, Schmitt M. et al. Development and evaluation of peptidic ligands targeting tumour-associated urokinase plasminogen activator receptor (uPAR) for use in alpha-emitter therapy for disseminated ovarian cancer. Eur J Nucl Med Mol Imaging. 2008;35:53-64 doi:10.1007/s00259-007-0582-3
54. Persson M, Madsen J, Ostergaard S, Jensen MM, Jorgensen JT, Juhl K. et al. Quantitative PET of human urokinase-type plasminogen activator receptor with 64Cu-DOTA-AE105: implications for visualizing cancer invasion. J Nucl Med. 2012;53:138-45 doi:10.2967/jnumed.110.083386
55. Duffy MJ, O'Donovan N, Crown J. Use of molecular markers for predicting therapy response in cancer patients. Cancer Treat Rev. 2011;37:151-9 doi:10.1016/j.ctrv.2010.07.004
56. Mazar AP, Buko A, Petros AM, Barnathan ES, Henkin J. Domain analysis of urokinase plasminogen activator (u-PA): Preparation and characterization of intact A-chain molecules. Fibrinolysis. 1992;1:49-55 doi:10.1016/0268-9499(92)90094-x
57. Rajagopal V, Kreitman RJ. Recombinant toxins that bind to the urokinase receptor are cytotoxic without requiring binding to the alpha(2)-macroglobulin receptor. J Biol Chem. 2000;275:7566-73
58. Kounnas MZ, Morris RE, Thompson MR, FitzGerald DJ, Strickland DK, Saelinger CB. The alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein binds and internalizes Pseudomonas exotoxin A. J Biol Chem. 1992;267:12420-3
59. Hall WA, Vallera DA. Efficacy of antiangiogenic targeted toxins against glioblastoma multiforme. Neurosurg Focus. 2006;20:E23. doi:10.3171/foc.2006.20.4.15
60. Huang J, Li YM, Massague J, Sicheneder A, Vallera DA, Hall WA. Intracerebral infusion of the bispecific targeted toxin DTATEGF in a mouse xenograft model of a human metastatic non-small cell lung cancer. J Neurooncol. 2012 doi:10.1007/s11060-012-0904-6
61. Abdalla MO, Karna P, Sajja HK, Mao H, Yates C, Turner T. et al. Enhanced noscapine delivery using uPAR-targeted optical-MR imaging trackable nanoparticles for prostate cancer therapy. J Control Release. 2011;149:314-22 doi:10.1016/j.jconrel.2010.10.030
62. Huang X, Peng X, Wang Y, Wang Y, Shin DM, El-Sayed MA. et al. A Reexamination of Active and Passive Tumor Targeting by Using Rod-Shaped Gold Nanocrystals and Covalently Conjugated Peptide Ligands. ACS Nano. 2010;4:5887-96 doi:10.1021/nn102055s
63. Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB. et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66:6732-40 doi:10.1158/0008-5472.CAN-05-4199
64. Wang M, LoÃàwik DWPM, Miller AD, Thanou M. Targeting the Urokinase Plasminogen Activator Receptor with Synthetic Self-Assembly Nanoparticles. Bioconjugate Chemistry. 2008;20:32-40 doi:10.1021/bc8001908
65. Lin L, Gardsvoll H, Huai Q, Huang M, Ploug M. Structure-based engineering of species selectivity in the interaction between urokinase and its receptor: implication for preclinical cancer therapy. J Biol Chem. 2010;285:10982-92 doi:10.1074/jbc.M109.093492
66. Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science (New York, NY. 2004;303:1818-22
67. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nano. 2007;2:751-60
68. Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271-84 doi:10.1016/s0168-3659(99)00248-5
69. Ahn RW, Chen F, Chen H, Stern ST, Clogston JD, Patri AK. et al. A Novel Nanoparticulate Formulation of Arsenic Trioxide with Enhanced Therapeutic Efficacy in a Murine Model of Breast Cancer. Clin Cancer Res. 2010;16:3607-17 doi:10.1158/1078-0432.ccr-10-0068
70. Chen H, Pazicni S, Krett NL, Ahn RW, Penner-Hahn JE, Rosen ST. et al. Coencapsulation of Arsenic- and Platinum-based Drugs for Targeted Cancer Treatment. Angew Chem Int Ed. 2009;121:9459-63
71. Lee S-M, Chen H, Dettmer CM, O'Halloran TV, Nguyen ST. Polymer-caged liposomes: a pH-responsive delivery system with high stability. J Am Chem Soc. 2007;129:15096-7
72. Lee S-M, Ahn RW, Chen F, Fought AJ, O'Halloran TV, Cryns VL. et al. Biological evaluation of pH-responsive polymer-caged nanobins for breast cancer therapy. ACS Nano. 2010;4:4971-8 doi:10.1021/nn100560p
73. Lee S-M, O'Halloran TV, Nguyen ST. Polymer-Caged Nanobins for Synergistic Cisplatin-Doxorubicin Combination Chemotherapy. J Am Chem Soc. 2010 doi:10.1021/ja107333g
74. Levenson RM. Spectral imaging perspective on cytomics. Cytometry A. 2006;69:592-600 doi:10.1002/cyto.a.20292
75. Bauer TW, Liu W, Fan F, Camp ER, Yang A, Somcio RJ, Bucana CD, Callahan J, Parry GC, Evans DB, Boyd DD, Mazar AP, Ellis LM. Targeting of urokinase plasminogen activator receptor in human pancreatic carcinoma cells inhibits c-Met- and insulin-like growth factor-I receptor-mediated migration and invasion and orthotopic tumor growth in mice. Cancer Res. 2005;65:7775-81
76. Van Buren G, Gray MJ, Dallas NA, Xia L, Lim SJ, Fan F, Mazar AP, Ellis LM. Targeting the urokinase plasminogen activator receptor with a monoclonal antibody impairs the growth of human colorectal cancer in the liver. Cancer. 2009;115:3360-8 doi: 10.1002/cncr.24371
77. Rabbani SA, Ateeq B, Arakelian A, Valentino ML, Shaw DE, Dauffenbach LM, Kerfoot CA, Mazar AP. An anti-urokinase plasminogen activator receptor antibody (ATN-658) blocks prostate cancer invasion, migration, growth, and experimental skeletal metastasis in vitro and in vivo. Neoplasia. 2010;12:778-88
Author contact
![]() Corresponding author: Andrew P. Mazar, Email: a-mazaredu, or Thomas V. O'Halloran, Email: t-ohalloranedu.
Corresponding author: Andrew P. Mazar, Email: a-mazaredu, or Thomas V. O'Halloran, Email: t-ohalloranedu.