Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2013; 3(7):507-515. doi:10.7150/thno.5557 This issue Cite
Review
Urokinase Plasminogen Activator Receptor (uPAR) Targeted Nuclear Imaging and Radionuclide Therapy
Dan Li1, 2, Shuanglong Liu1, Hong Shan2, Peter Conti1, Zibo Li1 ![]()
1. Molecular Imaging Center, University of Southern California, Los Angeles, CA 90033, USA.
2. Department of Radiology, the Third Affiliated Hospital of Sun Yat-sen University, Guangzhou 510630, China.
Received 2012-11-16; Accepted 2012-12-17; Published 2013-6-29
Abstract

Urokinase-type plasminogen activator receptor (uPAR) is a glycosylphosphatidylinositol (GPI)-anchored protein. Besides regulating proteolysis, uPAR could also activate many intracellular signaling pathways that promote cell motility, invasion, proliferation, and survival through cooperating with transmembrane receptors. uPAR is overexpressed across a variety of tumors and is associated with cancer invasion and metastasis. In order to meet the demand for a rapid development and potential clinical application of anti-cancer therapy based on uPA/uPAR system, it is desirable to develop non-invasive imaging methods to visualize and quantify uPAR expression in vivo. In this review, we will discuss recent advances in the development of uPAR-targeted nuclear imaging and radionuclide therapy agents. The successful development of molecular imaging probes to visualize uPAR expression in vivo would not only assist preclinical researches on uPAR function, but also eventually impact patient management.
Keywords: uPAR, PET, SPECT, radionuclide therapy.
Introduction
Molecular imaging is a fast growing research area involving the development and evaluation of novel tools, reagents and methods to image specific molecular pathways in vivo; particularly those that are key targets in disease processes. Molecular imaging is rapidly recognized as a tool with the capacity to improve every facet of cancer patient care, such as, assisting early detection and intervention strategies; facilitating the direction of pharmacological, cell-based, and genetic therapeutic regimens; and enhancing the development of emerging theranostics field. In particular, nuclear medicine (including positron emission tomography (PET) and single-photon emission computed tomography (SPECT)) serves as a highly sensitive technology that is ideally suited for pre-clinical and clinical imaging of cancer biological processes non-invasively. The commonly used radionuclides include 99mTc, 123/131I, 111In (γ-emitting isotope for SPECT imaging); 18F, 11C, 64Cu, 68Ga (positron-emitting isotope for PET imaging); and 90Y, 177Lu, 213Bi (for radiation therapy).
This review intends to provide an overview on recent advances in the development of uPAR-targeted nuclear imaging and radionuclide therapy. The successful development of molecular imaging probes to visualize uPAR expression in vivo would not only assist preclinical researches on uPAR function, but also eventually impact patient management.
Biological function of uPAR
Urokinase-type plasminogen activator receptor (uPAR) is a glycosylphosphatidylinositol (GPI)-anchored protein [1]. The receptor binds urokinase-type plasminogen activator (uPA) as well as its proenzyme, pro-uPA, in such a manner that the activation cascade can occur directly on the cell surface. Activated uPA converts inactive plasminogen into active plasmin, which degrades various components of the extracellular matrix. Besides the function of regulating proteolysis, uPAR could also activate many intracellular signaling pathways that promote cell motility, invasion, proliferation and survival through cooperating with transmembrane receptors [2, 3]. uPAR is overexpressed across a variety of tumor cell lines and tissues, including breast, ovary, lung, pancreas, colon, kidney, liver, stomach, endometrium, bone and so on [4-6]. High endogenous level of uPAR was also found to be associated with cancer invasion and metastasis [4, 7]. Therefore, uPAR has become an important target for cancer diagnosis and therapy.
uPAR targeted radiopharmaceuticals
Radiopharmaceuticals are drugs containing radionuclides. A target specific radiopharmaceutical could be constructed by introducing radioactive tag to a targeting ligand. In many cases, radiometal based radiopharmaceuticals rely on the introduction of bifunctional chelators to target binding ligand. In contrast, the non-metallic radionuclides were generally introduced through covalent bond formation. Depending on the ligands to be used, uPAR targeted radiopharmaceuticals could be constructed by introducing corresponding radioactive tag to uPAR binding ligands.
1. Peptide-based ligands
There are two main avenues in the search for peptide-based uPAR ligands. One approach exploits random selection in a phage display library, whereas the other relies on synthesizing peptide derivatives based on uPA, a natural uPAR-binding ligand.
1.1 Ligands discovered by Phage display
A family of 15-mer linear peptide was obtained as antagonists of uPA-uPAR interaction through the selection in a random phage-display library [8]. The selected lead phage peptide was subjected to affinity maturation and stabilization by combinatorial chemistry [9]. The resulting 9-mer core peptide AE105 (D-Cha-F-s-r-Y-L-W-S) [9] demonstrated specific, high-affinity binding to human uPAR (Kd ≈ 0.4 nM). Later on, AE105 and its derivatives, AE120 [(D-Cha-F-s-r-Y-L-W-S)2 -βA-Kc] and AE170 (K-S-D-Cha-F-s-k-CHg-L-W-S-S-K) have been applied in a variety of experimental settings [10]. To date, most uPAR-targeted imaging and therapy studies are based on AE105 and its corresponding derivatives.
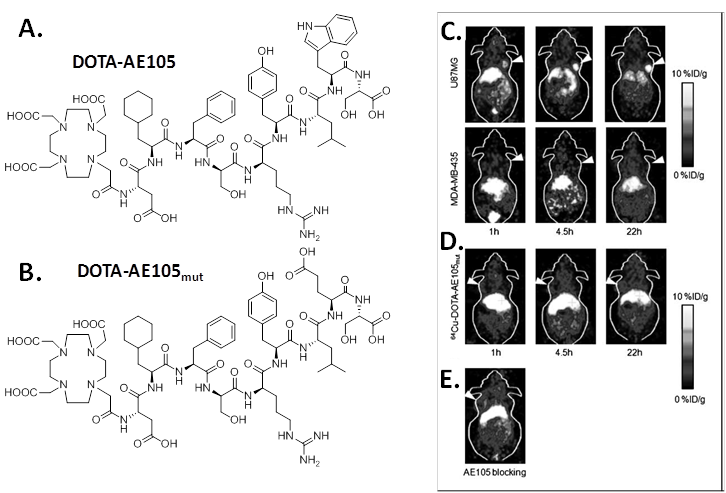
Targeted copper-64 labeled molecules have shown promising results for diagnostic PET imaging and radionuclide therapy, due to the favorable nuclear characteristics of this isotope (t1/2 = 12.7 h, β+ 17.4%, Emax = 0.656 MeV, β- 39%, Emax = 0.573 MeV) and its availability with high specific activity. In 2007, Li et al first developed 64Cu labeled AE105 for noninvasive microPET imaging of uPAR expression [11]. In that study, uPAR-binding peptide AE105 was conjugated with 1,4,7,10-tetraazadodecane-N,N',N'',N'''-tetraacetic acid (DOTA) (DOTA-AE105, Figure 1) and labeled with 64Cu. MicroPET imaging revealed that 64Cu-DOTA-AE105 accumulated rapidly in uPAR-positive U87MG human glioblastoma tumors (10.8 ± 1.5 percentage of injected dose per gram of tissue (%ID/g) at 4.5 h) but not in uPAR-negative MDA-MB-435 human breast tumors (1.2 ± 0.6 %ID/g at 4.5 h ) (Figure 1). In the presence of excess amount of unlabeled peptide, the U87MG tumor uptake was successfully reduced to 3.7 ± 1.3 %ID/g at 4.5 hours post injection (p.i.), which further demonstrated the receptor specificity of this tracer. A nonbinding variant of AE105 was also tested, the uptake in U87MG tumors was 1.7 ± 0.4, 2.2 ± 0.5, and 3.1 ± 0.6 %ID/g at 1, 4.5, and 22 hours p.i., which was significantly lower than that of the original 64Cu-DOTA-AE105 (P < 0.005).
A, chemical structure of DOTA-conjugated AE105 peptide (DOTA-D-Cha-F-s-r-Y-L-W-S). B, chemical structure of DOTA-conjugated AE105-mutant peptide (DOTA-D-Cha-F-s-r-Y-L-E-S, where capitals denote the single letter code for amino acids in the L-configuration, whereas lowercase denote D-configurations. Cha is L-cyclohexylalanine). C, decay-corrected whole-body coronal microPET images of athymic female nude mice bearing U87MG tumors or MDA-MB-435 tumors at 1, 4.5, and 22 h p.i. of ~9 MBq 64Cu-DOTA-AE105. D, decay-corrected whole-body coronal microPET images of athymic female nude mice bearing U87MG tumors at 1, 4.5, and 22 h p.i. of ~9 MBq 64Cu-DOTA-AE105mut. E, decay-corrected whole-body coronal microPET images of a U87MG tumor-bearing mouse at 4.5 hours after injection of 64Cu-DOTA-AE105 and a blocking dose of AE105 (15 mg/kg). Images shown are of 5- or 10-min static scans of a single mouse but are representative for the three mice tested in each group. Tumors are indicated by arrows. Reproduced with permission from ref. [11].
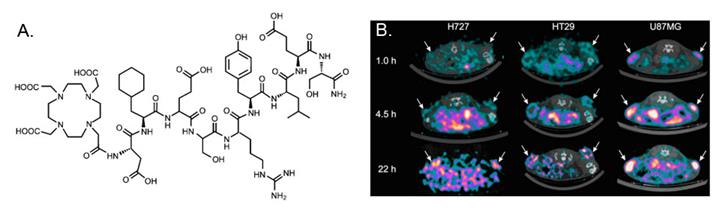
In a recent study, Persson et al [12] evaluated 64Cu-DOTA-AE105-NH2 in a quantitative PET study. The major difference between 64Cu-DOTA-AE105-NH2 and 64Cu-DOTA-AE105 is the C-terminal amidation. Although it is not validated in the manuscript, C-terminal amidation would generally make peptide ends uncharged (compared to standard synthetic peptides). In vivo stability of the probe could be improved in some extend by increasing the stability toward digestions by aminopeptidases and blocking activities towards synthetase. In this research, a significant correlation between tumor uptake of 64Cu-DOTA-AE105-NH2 and uPAR expression was found (R2 = 0.73; P < 0.0001) across 3 cancer xenografts (H727, HT-29, and U87MG) (Figure 2). For uPAR positive U87MG tumor, tumor uptake was 5.9 ± 0.7%ID/g at 4.5 h p.i., which was lower than the number reported in the first study [11] (10.8 ± 1.5 %ID/g at 4.5 h), although the same cell line was used. The discrepancy may be related to the basic properties of the chelated radiopharmaceuticals as they are two different compounds. 18F-FDG (2-deoxy-2-18F-fluoro-D-glucose) PET was also performed on U87MG and H727 tumors. As expected, no difference in tumor uptake was observed in 18F-FDG PET, which clearly demonstrated that additional information can be obtained on tumor biology using 64Cu-DOTA-AE105-NH2 PET. Furthermore, primary tumor uptake of 64Cu-DOTA-AE105-NH2 apparently correlates to the efficacy of 5-fluorouracil-based chemotherapies, illustrating the potentials of using uPAR PET in a clinical setting to monitor treatment response. Despite the initial success of these two examples, both probes demonstrated prominent liver uptake that could not be blocked by cold AE105 ligand. Previously, it has been demonstrated that the 64Cu2+ complexes with DOTA, TETA (1,4,8,11-tetraazacyclotetradecane-N,N',N”,N'”-tetraacetic acid), and their derivatives have limited stability in vivo due to the dissociation of 64Cu2+ from these bifunctional chelators (BFCs), leading to high retention in the liver. It would be interesting to conjugate other stable chelators, such as NOTA (1,4,7-triazacyclononane-1,4,7-triacetic acid), cross-bridged TETA, and sarcophagine agents, to AE105 and compare the liver uptake among these PET imaging agents. The pharmacokinetic of uPAR PET may also be improved by using these newly developed chelators.
A, chemical structures of control peptide inactive DOTA-AE105-NH2 (DOTA-D-Cha-E-s-r-Y-L-E-S) in which two essential amino acids for uPAR binding (Phe → Glu and Trp → Glu) are substituted as compared to DOTA-AE105-NH2. B, Decay-corrected transverse images of subcutaneously xenotransplanted U87MG, HT-29, and H727 mice at 1, 4.5, and 22 h after injection of 64Cu-DOTA-AE105-NH2. Images shown are static scans of single mouse, which is representative of 4 mice tested in each group. Arrows indicate tumors. Reproduced with permission from ref. [12].
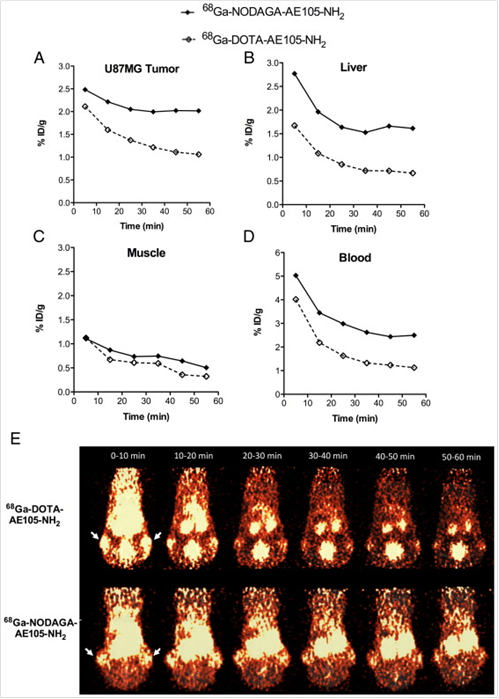
In addition to 64Cu, AE105 has been labeled with 68Ga for noninvasive PET imaging of uPAR expression. 68Ga decays by 89% through positron emission of 1.92 MeV (maximum energy) and is available from an in-house 68Ge/68Ga generator (68Ge, t 1/2=270.8 day), which renders it independent of an onsite cyclotron. With a half-life of 68 min, it is suitable for studying the pharmacokinetics of many peptides. Gallium-68 labeled uPAR radiotracers was first reported as 68Ga-DOTA-AE105-NH2 and 68Ga-NODAGA (N, N-di(2-hydroxybenzyl) ethylene-diamine-N,N-diacetic acid)-AE105-NH2 [13]. The tumor uptake in U87MG glioblastoma xenograft model was 2.1 % ID/g and 1.3 % ID/g (30 min p.i.) and 2.0 % ID/g and 1.1 % ID/g (60 min p.i.) for 68Ga-NODAGA-AE105-NH2 and 68Ga-DOTA-AE105-NH2, respectively (Figure 3). Although both 68Ga-based tracers could be used for tumor imaging, 68Ga-DOTA-AE105-NH2 demonstrated significantly lower Tumor/Muscle ratio (7.4) than that of 64Cu-DOTA-AE105-NH2 (15.9) at 60 min p.i. (P < 0 .05). The same pattern was found when comparing tumor to blood, liver or kidney ratios. Although these 68Ga-tracers were developed as close analogs of 64Cu-DOTA-AE105, their absolute tumor uptake was significantly reduced. Apparently, both tumor uptake and tumor-to-background ratios need to be improved when developing 68Ga-based tracers for uPAR targeted imaging in the future.
Quantitative results based on manually drawn ROI analysis for U87MG tumor (A), liver (B), muscle (C) and blood (D) during the 1 h dynamic PET scan. E, Representative 10 min PET images during 1 h dynamic PET recording. Tumor could clearly be visualized for both labeled peptides (white arrows indicate tumors), with a clear washout of the tracer over time. Each mouse received 5-7 MBq tracer (approx. 0.5 nmol peptide). Reproduced with permission from ref. [13].
SPECT agents were also developed for uPAR imaging in vivo. In 2009, a dimeric peptide [(NAc-dD-CHA-F-dS-dR-Y-L-W-S-βAla)2-K-K] (most closely related to AE120, named as AE120 analogue) was conjugated with DOTA chelator and labeled with 111In [14]. The IC50-value of (NAc-dD-CHA-F-dS-dR-Y-L-W-S-βAla)2-K-K(DOTA)-NH2 (AE120 analogue-DOTA) was 240 nM, which was higher than that of DOTA-AE105 (IC50 = 130 nM) [11]. AE120 analogue-DOTA is structurally different from DOTA-AE105 and related peptide [9] in being N-terminally acetylated and C-terminally amidated; the appended DOTA moiety at C-terminal in the branching lysine residue; and the chirality of the N-terminal aspartate residue [14]. These structural differences could lead to the limited free conformational space in the region of the corresponding uPAR complex. The IC50-value of AE120 analogue was not get at the same time in this study. Therefore, it is not clear whether DOTA conjugation would affect this peptide's binding affinity.
Biodistribution data was collected at 1, 4 and 24 h p.i. of 111In-DOTA-AE120 analogue on MDA-MB-231 human breast tumor-bearing mice. It was shown that the elimination of the 111In-labeled AE120 analogue from the blood pool was rapid, with 0.12 ± 0.06 % ID/g remaining in blood at 4 h p.i. Tumor uptake at this time point was 0.53 ± 0.11 %ID/g; and tumor-to-blood and tumor-to-muscle ratios were 4.2 and 9.4, respectively. 111In-DOTA- AE120 analogue cleared through both liver and kidney. Liver and kidney retention was higher than 125I-labeled ATF (Amino-terminal fragment of uPA). In vivo biodistribution data of 111In-DOTA-AE120 analogue and 125I-ATF was shown in Table 1 and Table 2. The comparison of these two tracers is discussed in the following Section 1.2.
In vivo biodistribution of (NAc-dD-CHA-F-dS-dR-Y-LW-S-Ala)2-K-K(111In-DOTA)-NH2 (Test) at 1, 4, and 24 h p.i. and scrambled negative control peptide (NAc-dD-W-dS-L-Y-dR-F-S-CHA-Ala)2-K-K(111In-DOTA)-NH2 (control) at 4 h p.i. in SCID mice bearing MDA-MB-231 human breast cancer tumor xenografts (n = 3, mean % ID/g ± SD). Reproduced with permission from ref. [14].
| Organ | Test 1 h | Test 4 h | Control 4 h | Test 24 h |
|---|---|---|---|---|
| blood | 2.85 ± 0.57 | 0.12 ± 0.06 | 0.15 ± 0.04 | 0.07 ± 0.02 |
| heart | 0.93 ± 0.22 | 0.16 ± 0.02 | 0.55 ± 0.08 | 0.10 ± 0.03 |
| lung | 2.58 ± 0.51 | 0.42 ± 0.18 | 0.32 ± 0.06 | 0.32 ± 0.05 |
| liver | 7.97 ± 0.70 | 5.52 ± 0.95 | 13.44 ± 1.57 | 4.06 ± 0.34 |
| spleen | 0.72 ± 0.18 | 0.39 ± 0.34 | 0.37 ± 0.09 | 0.41 ± 0.04 |
| intestines | 1.07 ± 0.06 | 1.74 ± 1.09 | 1.71 ± 0.38 | 0.23 ± 0.07 |
| kidney | 19.09 ± 0.97 | 13.27 ± 2.61 | 13.19 ± 3.18 | 10.23 ± 2.29 |
| muscle | 0.30 ± 0.08 | 0.06 ± 0.02 | 0.12 ± 0.02 | 0.03 ± 0.01 |
| bone | 0.49 ± 0.22 | 0.06 ± 0.04 | 0.09 ± 0.02 | 0.06 ± 0.05 |
| pancreas | 0.83 ± 0.26 | 0.15 ± 0.07 | 0.25 ± 0.03 | 0.07 ± 0.06 |
| tumor | 1.48 ± 0.28 | 0.53 ± 0.11 | 0.36 ± 0.05 | 0.23 ± 0.07 |
In vivo biodistribution (n = 3, mean %ID/g ± SD) of 125I-ATF in SCID mice bearing MDA-MB-231 human breast cancer tumor xenografts at 1, 4, and 24 h p.i.. Reproduced with permission from ref. [14].
| Organ | 1 h | 4 h | 24 h | 24 h (blocked) |
|---|---|---|---|---|
| blood | 7.75 ±1.00 | 3.41 ± 0.86 | 0.26 ± 0.06 | 0.60 ± 0.69 |
| heart | 2.56 ± 0.12 | 1.19 ± 0.16 | 0.08 ± 0.02 | 0.06 ± 0.01 |
| lung | 5.39 ± 0.35 | 2.67 ± 0.51 | 0.16 ± 0.05 | 0.17 ± 0.02 |
| liver | 3.01 ± 0.45 | 1.81 ± 0.42 | 0.25 ± 0.06 | 0.23 ± 0.02 |
| spleen | 2.92 ± 0.39 | 1.72 ± 0.35 | 0.09 ± 0.02 | 0.10 ± 0.03 |
| intestines | 3.27 ± 0.40 | 2.53 ± 0.28 | 0.16 ± 0.03 | 0.13 ± 0.02 |
| kidney | 16.72 ± 1.59 | 4.65 ± 0.84 | 0.88 ± 0.21 | 0.81 ± 0.09 |
| muscle | 1.21 ± 0.08 | 0.61 ± 0.10 | 0.03 ± 0.01 | 0.02 ± 0.01 |
| bone | 1.55 ± 0.13 | 0.87 ± 0.11 | 0.04 ± 0.01 | 0.03 ± 0.001 |
| pancreas | 3.98 ± 0.77 | 2.18 ± 0.65 | 0.06 ± 0.03 | 0.05 ± 0.01 |
| tumor | 4.60 ± 0.63 | 3.09 ± 0.29 | 0.29 ± 0.06 | 0.19 ± 0.05 |
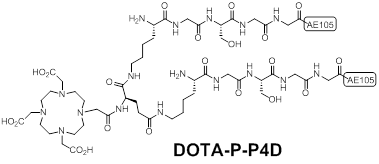
Sebastian et al synthesized a pseudo-symmetrical covalent dimer of AE105 (P-P4D) with high affinity to uPAR [15]. The spacer Kb-G-S-G-G was used to link two AE105 motifs. P-P4D was conjugated with DOTA and labeled with 213Bi with the aim to perform α-emitter therapy for advanced ovarian cancer. Chemical structure of DOTA-P-P4D was shown in Figure 4. Organ distribution studies of 213Bi-P-P4D showed that tumor uptake were 9 ± 0.9%, 4.8 ± 1.5% and 2.2 ± 0.4% ID/g at 20, 45 and 90 min respectively after intraperitoneal (i.p.) administration of the tracer in peritoneal disseminated OV-MZ-6 human ovarian tumor model. Although 213Bi is α -emitter for therapy, no therapeutic efficacy studies were carried out in this report. Moreover, the pharmacokinetic changes caused by i.p. injection were unknown as above imaging probes were evaluated after intravenous (i.v.) injection instead of i.p.
Chemical structures of a pseudo-symmetrical covalent dimer of AE105 conjugated with DOTA (DOTA-P-P4D).
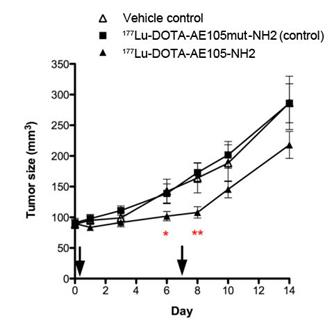
uPAR-targeted radionuclide therapeutic intervention was evaluated by Persson et al [16]. In their report, uPAR-positive HT-29 xenografts were treated twice with 177Lu-DOTA-AE105-NH2 through tail vein injection. 177Lu-DOTA-AE105mut-NH2 (non-binding analog) and vehicle were used as controls. First treatment was administrated on day 1 with a dose of (18.4 ± 3.1 MBq) and (19.9 ± 4.3 MBq) for 177Lu-DOTA-AE105-NH2 and 177Lu-DOTA-AE105mut-NH2, respectively. Second treatment was administrated on day 7 with a dose of (28.3 ± 2.2 MBq) and (26.7 ± 5.8 MBq) for 177Lu-DOTA-AE105-NH2 and 177Lu-DOTA-AE105mut-NH2, respectively. A significant difference in tumor size was observed (n = 12) at day 6 (P = 0.04) and day 8 (P = 0.002) after receiving 177Lu-DOTA-AE105-NH2, compared with the control groups. However, at day 14, the average tumor sizes were 287 ± 44 mm3, 286 ± 32 mm3 and 218±22 mm3 for vehicle controls, 177Lu-DOTA-AE105mut-NH2 and 177Lu-DOTA-AE105-NH2, respectively (Figure 5). The tumor size different is not significant (P = 0.09) (Figure 5). Apparently, the treatment conditions need to be further optimized in order to demonstrate that uPAR could be a valid target for radionuclide therapy.
Mean tumor volume in animals after 2 doses of 177Lu-DOTA-AE105-NH2, control peptide 177Lu-DOTA-AE105mut-NH2 or vehicle on day 0 and day 7. A significant effect of 177Lu-DOTA-AE105 on tumor volume was found on day 6 (P < 0.05) and day 8 (P < 0.01) compared with control groups. No significant differences in tumor volumes were found at the end of study day 14. Mean tumor volume was calculated by manual drawing of tumor lesions on CT images. Results presented as mean±SEM, n = 12 tumor/group. Reproduced with permission from ref. [16].
1.2 Ligands based on uPA fragment
uPAR targeted peptide ligands were also constructed on the basis of uPA, the natural ligand of uPAR. The high affinity of uPA for uPAR is primarily governed by β-hairpin in NH2-terminal growth factor-like domain (GFD, residues 1-44) [17, 18]. Amino-terminal fragment (ATF) of uPA, containing the first 135 amino acids including GFD, was developed in 1987 as the first uPAR targeting peptide [19]. ATF has high affinity for uPAR and competes with uPA, reducing the endogenous enzymatic activity [18, 20, 21]. ATF has been applied to uPAR-targeted magnetic resonance imaging (MRI), near-infrared (NIR) imaging and drug delivery [22-24]. The biodistribution of ATF was studied on SCID mice bearing MDA-MB-231 human breast cancer tumor xenografts using 125I labeled ATF [14]. This study also compared the biodistribution of 125I-ATF with that of 111In-DOTA-AE120 analogue (see Table 1 and Table 2). Overall, 125I-ATF showed higher tumor uptake (3.09 ± 0.29 %ID/g at 4 h p.i.) and slower elimination from the blood pool. Tumor-to-blood and tumor-to-muscle ratios at 4 h p.i. were 0.9 and 5 for 125I-ATF, respectively, which were lower than that of 111In-DOTA-AE120 analogue. Binding affinity of 125I-ATF to MDA-MB-231 cells is increased when they have either had endogenous uPA removed by acid stripping, or have had endogenous expression of uPA reduced by the application of specific siRNA [14]. This result suggested that autocrine saturation could be responsible for reduced tumor uptake in vivo. Unfortunately, the influence of uPAR occupancy by endogenous uPA on the tumor uptake of the AE120 analogue, which is an antagonist of the uPA-uPAR interaction, was not carried out.
Based on GFD of uPA, a number of related peptide ligands for uPAR were further designed, such as uPA19-31 [25], cyclo19,31 uPA 19-31 [25], cyclo19,31[D-Cys(19)]uPA19-31 [26], cyclo21,29[Cys(21,29)]uPA21-30 [27], cyclo21,29[D-Cys(21)Cys(29)]uPA21-30 [27], cyclo21,29[D-Cys(21)Nle(23)Cys(29)]uPA21-30 [27]. Among these ligands, cyclo21,29[D-Cys(21)Cys(29)] uPA21-30 [27] is the lead compound that displays an IC50 of 40 nM for the uPA-uPAR interaction. In the meanwhile, cyclo21,29[D-Cys(21)Nle(23)Cys(29)]uPA21-30 synthesized from systematic point mutations, can still binds to uPAR but is resistant to proteolytic cleavage[27].
Andrea et al [28] evaluated a series of single amino acid chelate (SAAC) for their ability to incorporate 99mTc into peptides. They applied the lead SAAC to synthesize 99mTc-labeled cyclo19,31[D-Cys(19)]uPA19-31. Unfortunately, the ligand's binding affinity towards uPAR decreased 30-fold after conjugation with the SAAC. SPECT scanning studies were not carried out to validate its applicability as a radiotracer for uPAR imaging.
In addition, it was reported that cyclo19,31 uPA 19-31 elicited mitogenic effect upon binding to the ovarian cancer cells OV-MG-6 [29]. The biological effects of this kind of cyclic peptide mimetics are expected to be thoroughly studied before being used for uPAR-targeted nuclear imaging and therapy. In addition to uPA, uPAR was found to bind the somatomedin B (SMB) domain of vitronectin [30, 31]. Rationally, peptides mimic the somatomedin B (SMB) domain may be potential ligands for uPAR as well.
2. Protein and Antibody-based ligands
The half-life of peptide ligands is generally short due to its small molecular weight and stability. To improve the pharmaceutical property, ATF of uPA has been fused of human serum albumin (ATF-HSA) [32]. Similarly, engineered antibodies targeting human uPAR were developed through fusing ATF or GFD of uPA to Fc portion of human IgG [33]. These hybrid molecules could dramatically blocks tumor growth by disrupting uPA/uPAR interaction. To date, no imaging study based on these molecules has been reported.
A Large number of anti-uPAR antibodies against various epitopes have been developed [34-36]. Rabbani et al [37] developed a polyclonal antibody against rat uPAR (ruPAR). To study the bio-distribution of this anti-ruPAR Ab, this antibody was labeled with 125I and injected into the Mat B-III-uPAR rat breast tumor metastases model via tail vein. Significantly higher levels of anti-ruPAR Ab were observed in primary tumors and in tissues commonly affected by tumor metastases (liver, spleen, lungs, and lymph nodes) as compared with normal IgG at 12 h p.i.. The 125I-Anti-ruPAR Ab uptake in primary tumor and blood were about 0.75 and 0.9 %ID/g respectively. Compared to peptide-based probes, this antibody-based probe showed longer circulation. As the antibody based probe is closely related to the potential immuno- or radioimmunotherapies, an antibody based imaging probe would be ideal for patient stratification and screening.
Epitope-specific monoclonal antibodies against human uPAR would be superior for pre-clinical study. Another antibody, ATN-658 [35], is in line with this criteria. ATN-658 could bind to domains 3 (D3) of uPAR with high affinity (Kd ≈ 1 nM) [35]. It is specific for human uPAR and does not cross-react with mouse uPAR [35]. In human cancer xenograft models, ATN-658 could inhibit uPAR-positive cancer invasion, migration, growth and metastasis [35, 38, 39]. ATN-658 was also humanized (huATN-658) and is expected to enter a phase I clinical cancer study [39]. ATN-658 did not interfere with uPA/uPAR binding, and could bind to uPAR even when uPA was also bound to the receptor, which would make it suitable for monitoring therapies focused on uPAR suppression [35]. Recently, 12 unique human Fabs that bind human uPAR were identified through a highly diverse and naive human fragment of the antigen binding (Fab) phage display library [36]. Among them, two Fabs (2E9 and 2G10) could compete with uPA for uPAR binding and one Fab (3C6) competes α5β1 integrin for uPAR binding [36]. Both ATN-658 and these Fabs are suitable for uPAR-targeted imaging and therapy. Dullin et al [40] used Cy5.5-labeled uPAR-specific monoclonal antibody to visualize mammary carcinomas in an orthotopic mouse model in vivo, showing tumor specificity compared to the control antibody. The major disadvantage of optical imaging is the limited depth penetration and intense light scattering, which allows only for demarcation of superficial tumors and tissues accessible by endoscopy, as well as intraoperative imaging [41]. To obtain quantitative information about tumor targeting and distribution patterns of this imaging agent, PET or SPECT will be required for further studies.
3. Small molecule ligands
Small molecule ligands binding to uPAR are rather limited. N-substituted glycine-based peptidomimetics [42], O-substituted hydroxycumaranones [43], oligothiophenes [44], porphyrin derivatives [45], suramin [46], and amino-biphenyl-based peptidomimetics [47] have been reported to inhibit uPA-uPAR interaction (Figure 6). As these small molecules would be very sensitive to modifications, 18F and 11C labeling might be preferred because the structure of imaging probes may vary slightly compared with the small molecule ligands or inhibitors. Nonetheless, in vivo nuclear imaging studies based on these small molecules are relatively challenging and less studied.
Chemical structures of small molecule ligands to uPAR. A, N-substituted glycine-based peptidomimetics [42]; B, O-substituted hydroxycumaranones [43]; C, aminobiphenyl-based peptidomimetics [47]. D, E, Chemical structures of non-protein inhibitors of the uPA•uPAR interaction.
Summary
uPAR is overexpressed across a variety of tumors and is associated with cancer invasion and metastasis. In order to meet the demand for a rapid development and potential clinical application of anti-cancer therapy based on uPA/uPAR system, various molecular imaging probes have been successfully developed to visualize uPAR expression in vivo. Despite the recent progress on uPAR targeted imaging, this field is less studied compared with other well established systems, such as integrin αvβ3 receptor and the corresponding RGD ligands. In fact, several aspects need to be considered in order to develop a valid imaging probe for uPAR.
First, in many cancers, uPAR up-regulation coincides with a rise in uPA [4]. Therefore, it is highly possible that majority of uPAR in tumors may be occupied with uPA, which would make it rather challenging if the imaging probe binds to the same site of uPAR as uPA. Unfortunately, detailed information about uPAR occupancy in tumors is not available. Additional information on this aspect will provide important guidance on the design of uPAR targeted imaging probes. Nonetheless, ligands, such as ATN-658 [35], that do not interfere with uPA-uPAR binding, may be advantageous for uPAR-targeted imaging in this aspect. Second, the binding affinity of imaging probes should be carefully studied after modifying the high binding affinity ligands. In fact, several studies have indicated that binding affinity towards uPAR could be reduced dramatically after conjugation with various chelates [14, 28]. Therefore, detailed knowledge on the molecule structure of uPAR and its ligands will provide key information on developing uPAR targeted ligands for both imaging and therapy purpose. Third, for uPAR targeted imaging, proper ligands should be chosen according to disease types of interest. Generally, the use of small hydrophilic peptides would be a specific problem for kidney tumors in term of background. In fact, kidney also displays a high baseline expression of uPAR [48, 49]. Kidney uptake of different uPAR-targeting radiopharmaceuticals varied dramatically depending on their chemical characteristics. For example, kidney uptake was 4.9 %ID/g and 19.09 %ID/g for 64Cu-DOTA-AE105 [11] and 64Cu-DOTA-AE120 analogue [14] respectively at 1 h p.i., compared with 31.9 %ID/g for 213Bi-P-P4D [15] at 90 min p.i.. Therefore, nephrotoxicity should be considered during the application of radio-labeled ligands that are mainly metabolized through kidney. To reduce the kidney uptake of radiopeptides, various compounds are currently being explored including spironolactone [50], probenecid [51] and the plasma expander gelofusine [52]. A study on uPAR-targeted α-emitter therapy has demonstrated that kidney uptake of 213Bi-P-P4D was reduced from 31.9 %ID/g to 18.28 %ID/g at 90 min p.i. through the administration of gelofusine [15]. At last, uPAR binding ligands have species selectivity. For example, AE105 has extreme selectivity toward human uPAR compared to mouse, and hamster uPAR [9]. Even within primates, several species are actually insensitive to AE105 [53]. uPAR expression is not confined to the tumor cells. uPAR expression is also found on several tumor-associated cell types, including macrophages, mast cells, endothelial cells, NK cells and fibroblasts [54, 55]. This may cause problems when using nude mouse bearing human tumor xenograft for research. Accurate evaluation of uPAR expression profiles and therapeutic efficiency can't be accomplished when the ligands only bind to human uPAR. Ligands that can bind to both human and mouse uPAR are better choice for these kinds of research.
Acknowledgements
This publication was funded by American Cancer Society (MSRG-12-034-01-CCE).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ploug M, Ronne E, Behrendt N, Jensen AL, Blasi F, Dano K. Cellular receptor for urokinase plasminogen activator. Carboxyl-terminal processing and membrane anchoring by glycosyl-phosphatidylinositol. J Biol Chem. 1991;266:1926-1933
2. Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11:23-36
3. D'Alessio S, Blasi F. The urokinase receptor as an entertainer of signal transduction. Front Biosci. 2009;14:4575-4587
4. Dass K, Ahmad A, Azmi AS, Sarkar SH, Sarkar FH. Evolving role of uPA/uPAR system in human cancers. Cancer Treat Rev. 2008;34:122-136
5. Ge Y, Elghetany MT. Urokinase plasminogen activator receptor (CD87): something old, something new. Lab Hematol. 2003;9:67-71
6. de Bock CE, Wang Y. Clinical significance of urokinase-type plasminogen activator receptor (uPAR) expression in cancer. Med Res Rev. 2004;24:13-39
7. Wang Y. The role and regulation of urokinase-type plasminogen activator receptor gene expression in cancer invasion and metastasis. Med Res Rev. 2001;21:146-170
8. Goodson RJ, Doyle MV, Kaufman SE, Rosenberg S. High-affinity urokinase receptor antagonists identified with bacteriophage peptide display. Proc Natl Acad Sci U S A. 1994;91:7129-7133
9. Ploug M, Ostergaard S, Gardsvoll H, Kovalski K, Holst-Hansen C, Holm A, Ossowski L, Dano K. Peptide-derived antagonists of the urokinase receptor. affinity maturation by combinatorial chemistry, identification of functional epitopes, and inhibitory effect on cancer cell intravasation. Biochemistry. 2001;40:12157-12168
10. Kriegbaum MC, Persson M, Haldager L, Alpizar-Alpizar W, Jacobsen B, Gardsvoll H, Kjaer A, Ploug M. Rational Targeting of the Urokinase Receptor (uPAR): Development of Antagonists and Non-Invasive Imaging Probes. Curr Drug Targets. 2011Nov;12(12):1711-28
11. Li ZB, Niu G, Wang H, He L, Yang L, Ploug M, Chen X. Imaging of urokinase-type plasminogen activator receptor expression using a 64Cu-labeled linear peptide antagonist by microPET. Clin Cancer Res. 2008;14:4758-4766
12. Persson M, Madsen J, Ostergaard S, Jensen MM, Jorgensen JT, Juhl K, Lehmann C, Ploug M, Kjaer A. Quantitative PET of human urokinase-type plasminogen activator receptor with 64Cu-DOTA-AE105: implications for visualizing cancer invasion. J Nucl Med. 2012;53:138-145
13. Persson M, Madsen J, Ostergaard S, Ploug M, Kjaer A. 68Ga-labeling and in vivo evaluation of a uPAR binding DOTA- and NODAGA-conjugated peptide for PET imaging of invasive cancers. Nucl Med Biol. 2012;39:560-569
14. Liu D, Overbey D, Watkinson L, Giblin MF. Synthesis and characterization of an (111)In-labeled peptide for the in vivo localization of human cancers expressing the urokinase-type plasminogen activator receptor (uPAR). Bioconjug Chem. 2009;20:888-894
15. Knor S, Sato S, Huber T, Morgenstern A, Bruchertseifer F, Schmitt M, Kessler H, Senekowitsch-Schmidtke R, Magdolen V, Seidl C. Development and evaluation of peptidic ligands targeting tumour-associated urokinase plasminogen activator receptor (uPAR) for use in alpha-emitter therapy for disseminated ovarian cancer. Eur J Nucl Med Mol Imaging. 2008;35:53-64
16. Persson M, Rasmussen P, Madsen J, Ploug M, Kjaer A. New peptide receptor radionuclide therapy of invasive cancer cells: in vivo studies using (177)Lu-DOTA-AE105 targeting uPAR in human colorectal cancer xenografts. Nucl Med Biol. 2012;39:962-969
17. Lin L, Gardsvoll H, Huai Q, Huang M, Ploug M. Structure-based engineering of species selectivity in the interaction between urokinase and its receptor: implication for preclinical cancer therapy. J Biol Chem. 2010;285:10982-10992
18. Appella E, Robinson EA, Ullrich SJ, Stoppelli MP, Corti A, Cassani G, Blasi F. The receptor-binding sequence of urokinase. A biological function for the growth-factor module of proteases. J Biol Chem. 1987;262:4437-4440
19. Appella E, Blasi F. The growth factor module of urokinase is the binding sequence for its receptor. Ann N Y Acad Sci. 1987;511:192-195
20. Quax PH, Pedersen N, Masucci MT, Weening-Verhoeff EJ, Dano K, Verheijen JH, Blasi F. Complementation between urokinase-producing and receptor-producing cells in extracellular matrix degradation. Cell Regul. 1991;2:793-803
21. Li H, Soria C, Griscelli F, Opolon P, Soria J, Yeh P, Legrand C, Vannier JP, Belin D, Perricaudet M, Lu H. Amino-terminal fragment of urokinase inhibits tumor cell invasion in vitro and in vivo: respective contribution of the urokinase plasminogen activator receptor-dependent or -independent pathway. Hum Gene Ther. 2005;16:1157-1167
22. Yang L, Peng XH, Wang YA, Wang X, Cao Z, Ni C, Karna P, Zhang X, Wood WC, Gao X, Nie S, Mao H. Receptor-targeted nanoparticles for in vivo imaging of breast cancer. Clin Cancer Res. 2009;15:4722-4732
23. Yang L, Mao H, Cao Z, Wang YA, Peng X, Wang X, Sajja HK, Wang L, Duan H, Ni C, Staley CA, Wood WC, Gao X, Nie S. Molecular imaging of pancreatic cancer in an animal model using targeted multifunctional nanoparticles. Gastroenterology. 2009;136:1514-1525
24. Abdalla MO, Karna P, Sajja HK, Mao H, Yates C, Turner T, Aneja R. Enhanced noscapine delivery using uPAR-targeted optical-MR imaging trackable nanoparticles for prostate cancer therapy. J Control Release. 2011;149:314-322
25. Burgle M, Koppitz M, Riemer C, Kessler H, Konig B, Weidle UH, Kellermann J, Lottspeich F, Graeff H, Schmitt M, Goretzki L, Reuning U, Wilhelm O, Magdolen V. Inhibition of the interaction of urokinase-type plasminogen activator (uPA) with its receptor (uPAR) by synthetic peptides. Biol Chem. 1997;378:231-237
26. Magdolen V, Burgle M, de Prada NA, Schmiedeberg N, Riemer C, Schroeck F, Kellermann J, Degitz K, Wilhelm OG, Schmitt M, Kessler H. Cyclo19,31[D-Cys19]-uPA19-31 is a potent competitive antagonist of the interaction of urokinase-type plasminogen activator with its receptor (CD87). Biol Chem. 2001;382:1197-1205
27. Schmiedeberg N, Schmitt M, Rolz C, Truffault V, Sukopp M, Burgle M, Wilhelm OG, Schmalix W, Magdolen V, Kessler H. Synthesis, solution structure, and biological evaluation of urokinase type plasminogen activator (uPA)-derived receptor binding domain mimetics. J Med Chem. 2002;45:4984-4994
28. Armstrong AF, Lemon JA, Czorny SK, Singh G, Valliant JF. Evaluation of single amino acid chelate derivatives and regioselective radiolabelling of a cyclic peptide for the urokinase plasminogen activator receptor. Nucl Med Biol. 2009;36:907-917
29. Fischer K, Lutz V, Wilhelm O, Schmitt M, Graeff H, Heiss P, Nishiguchi T, Harbeck N, Kessler H, Luther T, Magdolen V, Reuning U. Urokinase induces proliferation of human ovarian cancer cells: characterization of structural elements required for growth factor function. FEBS Lett. 1998;438:101-105
30. Deng G, Royle G, Wang S, Crain K, Loskutoff DJ. Structural and functional analysis of the plasminogen activator inhibitor-1 binding motif in the somatomedin B domain of vitronectin. J Biol Chem. 1996;271:12716-12723
31. Wei Y, Waltz DA, Rao N, Drummond RJ, Rosenberg S, Chapman HA. Identification of the urokinase receptor as an adhesion receptor for vitronectin. J Biol Chem. 1994;269:32380-32388
32. Lu H, Yeh P, Guitton JD, Mabilat C, Desanlis F, Maury I, Legrand Y, Soria J, Soria C. Blockage of the urokinase receptor on the cell surface: construction and characterization of a hybrid protein consisting of the N-terminal fragment of human urokinase and human albumin. FEBS Lett. 1994;356:56-59
33. Hu XW, Duan HF, Gao LH, Pan SY, Li YM, Xi Y, Zhao SR, Yin L, Li JF, Chen HP, Wu CT. Inhibition of tumor growth and metastasis by ATF-Fc, an engineered antibody targeting urokinase receptor. Cancer Biol Ther. 2008;7:651-659
34. Luther T, Magdolen V, Albrecht S, Kasper M, Riemer C, Kessler H, Graeff H, Muller M, Schmitt M. Epitope-mapped monoclonal antibodies as tools for functional and morphological analyses of the human urokinase receptor in tumor tissue. Am J Pathol. 1997;150:1231-1244
35. Bauer TW, Liu W, Fan F, Camp ER, Yang A, Somcio RJ, Bucana CD, Callahan J, Parry GC, Evans DB, Boyd DD, Mazar AP, Ellis LM. Targeting of urokinase plasminogen activator receptor in human pancreatic carcinoma cells inhibits c-Met- and insulin-like growth factor-I receptor-mediated migration and invasion and orthotopic tumor growth in mice. Cancer Res. 2005;65:7775-7781
36. Duriseti S, Goetz DH, Hostetter DR, LeBeau AM, Wei Y, Craik CS. Antagonistic anti-urokinase plasminogen activator receptor (uPAR) antibodies significantly inhibit uPAR-mediated cellular signaling and migration. J Biol Chem. 2010;285:26878-26888
37. Rabbani SA, Gladu J. Urokinase receptor antibody can reduce tumor volume and detect the presence of occult tumor metastases in vivo. Cancer Res. 2002;62:2390-2397
38. Kenny HA, Leonhardt P, Ladanyi A, Yamada SD, Montag A, Im HK, Jagadeeswaran S, Shaw DE, Mazar AP, Lengyel E. Targeting the urokinase plasminogen activator receptor inhibits ovarian cancer metastasis. Clin Cancer Res. 2011;17:459-471
39. Rabbani SA, Ateeq B, Arakelian A, Valentino ML, Shaw DE, Dauffenbach LM, Kerfoot CA, Mazar AP. An anti-urokinase plasminogen activator receptor antibody (ATN-658) blocks prostate cancer invasion, migration, growth, and experimental skeletal metastasis in vitro and in vivo. Neoplasia. 2010;12:778-788
40. Dullin C, Zientkowska M, Napp J, Missbach-Guentner J, Krell HW, Muller F, Grabbe E, Tietze LF, Alves F. Semiautomatic landmark-based two-dimensional-three-dimensional image fusion in living mice: correlation of near-infrared fluorescence imaging of Cy5.5-labeled antibodies with flat-panel volume computed tomography. Mol Imaging. 2009;8:2-14
41. van Dam GM, Themelis G, Crane LM, Harlaar NJ, Pleijhuis RG, Kelder W, Sarantopoulos A, de Jong JS, Arts HJ, van der Zee AG, Bart J, Low PS, Ntziachristos V. Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-alpha targeting: first in-human results. Nat Med. 2011;17:1315-1319
42. Rosenberg J SK, Martin EJ. US19985747458. 1998.
43. Bauer S ER, Fertig G, Friebe W, Koerner M, Krell H. WO 04076444. 2004.
44. di Domenico R DCG, König B, Zimmermann G. WO 999906393. 1999.
45. König B WU, Krell H, Kilpert C, Knipp B. WO0979731645. 1997.
46. Behrendt N, Ronne E, Dano K. Binding of the urokinase-type plasminogen activator to its cell surface receptor is inhibited by low doses of suramin. J Biol Chem. 1993;268:5985-5989
47. Blood CH NB, Smith EM. US20016228985. 2001.
48. Wei C, Moller CC, Altintas MM, Li J, Schwarz K, Zacchigna S, Xie L, Henger A, Schmid H, Rastaldi MP, Cowan P, Kretzler M, Parrilla R, Bendayan M, Gupta V, Nikolic B, Kalluri R, Carmeliet P, Mundel P, Reiser J. Modification of kidney barrier function by the urokinase receptor. Nat Med. 2008;14:55-63
49. Solberg H, Ploug M, Hoyer-Hansen G, Nielsen BS, Lund LR. The murine receptor for urokinase-type plasminogen activator is primarily expressed in tissues actively undergoing remodeling. J Histochem Cytochem. 2001;49:237-246
50. Jaggi JS, Seshan SV, McDevitt MR, Sgouros G, Hyjek E, Scheinberg DA. Mitigation of radiation nephropathy after internal alpha-particle irradiation of kidneys. Int J Radiat Oncol Biol Phys. 2006;64:1503-1512
51. Kakizaki T, Yokoyama Y, Natsuhori M, Yamada N, Hashimoto M, Sato K, Ito N, Daniel GB. Quantitative analysis of the effect of probenecid on pharmacokinetics of 99mTc-mercaptoacetyltriglycine in dogs. J Vet Pharmacol Ther. 2005;28:559-564
52. Vegt E, Wetzels JF, Russel FG, Masereeuw R, Boerman OC, van Eerd JE, Corstens FH, Oyen WJ. Renal uptake of radiolabeled octreotide in human subjects is efficiently inhibited by succinylated gelatin. J Nucl Med. 2006;47:432-436
53. Engelholm LH, Behrendt N. Differential binding of urokinase and peptide antagonists to the urokinase receptor: evidence from characterization of the receptor in four primate species. Biol Chem. 2001;382:435-442
54. Mazar AP. The urokinase plasminogen activator receptor (uPAR) as a target for the diagnosis and therapy of cancer. Anticancer Drugs. 2001;12:387-400
55. Mazar AP, Henkin J, Goldfarb RH. The urokinase plasminogen activator system in cancer: implications for tumor angiogenesis and metastasis. Angiogenesis. 1999;3:15-32
Author contact
![]() Corresponding author: Dr. Zibo Li, Molecular Imaging Center, University of Southern California, Los Angeles, CA 90033, USA; Email: ziboliedu; Phone: +1 323 4423252; Fax: +1 323 4423253.
Corresponding author: Dr. Zibo Li, Molecular Imaging Center, University of Southern California, Los Angeles, CA 90033, USA; Email: ziboliedu; Phone: +1 323 4423252; Fax: +1 323 4423253.