Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Aberrant receptor activity...
Somatostatin receptors mediate...
The PI3K-pathway is frequently...
The MAPK pathways promotes...
PI3K and MAPK signaling are...
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2014; 4(4):336-365. doi:10.7150/thno.7851 This issue Cite
Review
PI3K-AKT-mTOR-Signaling and beyond: the Complex Network in Gastroenteropancreatic Neuroendocrine Neoplasms
Franziska Briest1,2 ![]() , Patricia Grabowski1,3
, Patricia Grabowski1,3
1. Medizinische Klinik 1 CBF, Dept. of Gastroenterology, Infectious Diseases, Rheumatology CC13, Charité Berlin, Germany;
2. Freie Universität Berlin, Institute for Chemistry and Biochemistry, Berlin, Germany;
3. Department of Hematology and Internal Oncology and Center for Neuroendocrine Tumours Bad Berka - ENETS Center of Excellence, Zentralklinik Bad Berka, Germany.
Received 2013-10-9; Accepted 2013-12-16; Published 2014-1-29
Abstract

Gastroenteropancreatic neuroendocrine neoplasms are heterogeneous in their clinical behavior and require therapies specially tailored according to staging, grading, origin and expression of peptide receptors. Despite extensive scientific efforts, the therapy options are still not satisfactory. The main reasons are due to the lack of a broad mechanistic knowledge, an insufficient classification of specific diagnostic sub-groups, and predictive markers. GEP-NEN tumors evade early diagnosis because of slow asymptomatic growth behavior and are frequently not detected until metastasized. How signaling networks contribute to tumor progression and how these networks interact remains unclear in large parts. In this review we summarize the knowledge on the growth factor responsive non-angiogenetic pathways in sporadic GEP-NENs, highlight promising mechanistic research approaches, and describe important therapy targets.
Keywords: Gastroenteropancreatic neuroendocrine neoplasms, signal transduction, growth factors, kinases, biotherapy, molecular biology, inhibitor.
Introduction
GEP-NENs (Gastroenteropancreatic neuroendocrine neoplasms) emerge from various neuroendocrine cells of the gastroenteropancreatic system and represent the largest subgroup of neuroendocrine neoplasms. This heterogenic entity of solid tumors, formerly termed GEP-NETs or GEP-NECs (gastroenteropancreatic neuroendocrine tumors and carcinomas) or “carcinoids”, displays a broad spectrum of characteristics concerning behavior during growth and differentiation, functional aspects, localization and prognosis.
Although they are ranked among rare neoplastic diseases in general, their incidence has increased exponentially throughout the last decade. Currently, GEP-NENs state the second most common gastrointestinal malignancy after colorectal cancer [1].
The majority of GEP-NENs is characterized by slow proliferating, well differentiated G1 phenotypes (WHO/ENETS classification 2010, refer to table 1), which are often diagnosed late in the developmental course by the occurrence of metastases (NEN G1, previously termed WDNET: well differentiated neuroendocrine tumor). In contrast, a small G3 subgroup of rapidly growing and poorly differentiated GEP-NENs display a behavior that is comparable to those of prevalent solid carcinoma entities (NEN G3 or NEC, previously called PDNEC: poorly differentiated neuroendocrine carcinoma). The third group, characterized by an intermediate malignancy or unclear behavior, NEN G2, approximates the former description of a well differentiated neuroendocrine carcinoma. The tumor grade is dependent on the proliferative behavior marked by mitoses and by the Ki‑67 antigen, which is a proliferation-related antigen and is immunohistochemically analyzed during diagnosis by default (refer to table 1). In contrast to previous classifications or the TNM staging, the differentiation of the tumor cells is not involved in grading decisions, which still hampers the evaluation of slowly growing poorly differentiated or highly proliferating well differentiated tumors and does not provide insights regarding invasive behavior. However, the main advantage of the current grading is the possibility to predict proliferation behavior and thus facilitate decisions regarding surgical resection, chemo-, radio- and biotherapy or monitoring.
WHO 2010 grading for (neuro-)endocrine tumors.
| Grade | Mitotic count (10 HPF) | Ki-67 index (%) |
|---|---|---|
| G1 | <2 | ≤2 |
| G2 | 2-20 | 3-20 |
| G3 | >20 | >20 |
Several signaling cascades influence malignant transformation, progression and metastasis in neuroendocrine cancers including RTKs (receptor tyrosine kinases) and GPCRs (G-protein coupled receptors) downstream signaling, which regulate Ras/Raf, MAPK, PI3K-Akt-mTOR and JNK and lead to DNA synthesis and cell proliferation. Pancreatic NENs are highly vascularized and nourished. Accordingly, they exhibit a vast expression of growth factors such as VEGF (vascular endothelial growth factor), PDGF (platelet-derived growth factor), IGF-1 (insulin-like growth factor 1), bFGF (basic fibroblast growth factor), TGF-α and -β (transforming growth factor) and PIGF (placental growth factor). Not surprisingly, aberrant receptor activity, including those of the IGF-1R (IGF-1 receptor) and FGFR3 (FGF receptor 3), and highly activated downstream signaling is frequent in pNENs [2-5].
Analogous to the pancreatic subgroup, gastrointestinal NENs overexpress VEGF, bFGF, TGF α and -β, PDGF, IGF-1 and its corresponding receptors PDGFR (PDGF receptor), IGF-IR, EGFR (epidermal growth factor receptor), VEGFR (VEGF receptor), and c-kit (stem cell factor receptor). They thus exhibit increased growth factor, pro-angiogenic and typically pro-secretory signaling [6-16]. This work will focus on the major growth-factor related signaling networks, namely the PI3 kinase and the MAP kinase cascades and their importance for GEP-NEN therapy and diagnosis today and for future therapy approaches.
Aberrant receptor activity induces sustained growth factor signaling in GEP-NENs and activates the PI3K and MAPK signaling network
IGF receptors
The IGF-1R is one of the crucial RTKs in gastroenteropancreatic neuroendocrine tumor growth factor biology. The intrinsic RTK activity of IGF-1R is activated upon binding of its respective ligand and leading to auto-phosphorylation of intracellular tyrosine residues in the juxtamembrane and C-terminal domains. Those phosphorylated tyrosines serve as docking stations for insulin receptor substrates, such as IRS-1 and Src resulting in PI3K signaling via Grb2/SOS and Ras and MAPK pathways [17].
NEN cells have been shown to secrete high amounts of IGF‑1. In gastrinomas, increased levels of both IGF-1 and the corresponding IGF-1R were associated with tumor growth, aggressiveness, and progression [18]. Furthermore, other studies have displayed that the expression of IGF-1R is decreased in functionally inactive neuroendocrine tumors of different offspring in relation to their functional analogues. These findings and further in vitro experiments suggest that IGF-1 is not only a major autocrine regulator of neuroendocrine tumor growth but also of neuroendocrine secretion itself. Inhibition of IGF-1R activity, e.g. by direct inhibition or by blocking its regulators, such as HSP90 (heat shock protein 90), resulted in decreased PI3K and ERK1/2 (extracellular signal-regulated kinase) signaling and induction of cell cycle arrest and apoptosis [14, 18-26]. Additionally, an alternatively spliced IGF-1R mRNA transcript could be detected with a higher abundance in neuroendocrine tumors of different offspring, suggesting that post-transcriptional mechanisms may cause regulatory aberrations [19].
In addition to aberrant receptor and ligand abundance, an important regulator of IGF signaling was found to be significantly up-regulated in metastatic NENs in two gene expression studies: IGFBP3 (IGF binding protein 3), which is considered to maintain the serum level of IGF-1 in a tissue specific pro- or antiproliferative manner. IGFBP3 was overexpressed in >80% of lymph node or distant metastases versus <60% in primary pNEN lesions [27-29]. Those data might indicate a stoma or tumor cell-controlled regulation of a distinct IGF-1 homeostasis and allocation even in target tissues with a completely different composition. Adaptive and cooperative behavior of metastasizing NEN cells in the context of circulation and homing should be further explored in the future.
Therefore, IGF-1 and its receptor IGF-R1 are highly expressed in GEP-NENs with an altered abundance which depends on IGF binding factors and the relative ratio of specific receptor isoforms. IGF-1 has been shown to be a major autocrine regulator of neuroendocrine tumor growth and of neuroendocrine secretion.
EGF receptors and FGF
The EGFR belongs to the HER receptor family that consists of EGFR (HER1 or erbB1), erbB2 (HER2), erbB3 (HER3) and erb4 (HER4). Gastrointestinal and pancreatic NENs express and activate EGFRs. In immunohistochemical analyses of NENs located in different primary locations, 96% of the specimens were positive for EGFR expression and 63% were positive for phosphorylated EGFR [6]. Another study demonstrated a significantly higher expression (> 91%) in metastatic and non-metastatic gastrointestinal NENs in contrast to <25% in primary and metastatic pNEN [30]. A third study retrospectively evaluated the expression of EGFR and one of its ligands, TGF-α (transforming growth factor alpha), in pNENs, demonstrating that 63% of the tumors were positive for TGF-alpha and 65% were positive for the intracellular and/or extracellular domain of EGFR, but failed to prove a correlation with size, functional status, secretory profile, or biologic behavior [31]. These data were confirmed by Nilsson and colleagues, who showed that several human neuroendocrine tumors express both TGF-alpha and EGF receptors in vivo and in vitro, suggesting an autocrine mechanism [9].
Di Florio and colleagues recently demonstrated that gastrointestinal hormones and neurotransmitters stimulate the growth of the human BON, QGP-1 and the rat Rin-14B-cell lines in an EGFR dependent manner through activation of PKC and Src kinases, matrix metalloproteinase activation and the generation of reactive oxygen species [32].
Although a recent analysis of pancreatic neuroendocrine tumors uncovered frequent single nucleotide polymorphisms (SNPs) in PDGFRA and EGFR, no druggable EGFR, KIT or PDGFRA mutation could be found in these receptors to date. Nevertheless, elevated copy number of the EGFR and HER-2/neu loci could be detected in 38% and 33% of the cases and high expression of PDGFRA in 65%, respectively [21].
The non-secretory protein FGF13 has recently been described as new progression marker in pNENs. Although FGF13 is insufficient to stimulate FGF receptors, it was demonstrated to be an independent predictor of a shorter progression-free survival associated with positive Ki-67 staining. Furthermore FGF overexpression correlates with the occurrence of liver metastases and shortened disease-free survival in patients that underwent complete tumor resection [33]. FGF13 has recently been identified as microtubule-stabilizing protein in neuronal cells that promotes neuronal migration in the cerebral cortex. These processes might also facilitate dissemination of neuroendocrine tumor cells that share at least some characteristics with neuronal cells, but the underlying mechanisms remain unknown [34].
Taken together, these studies have accounted for high growth factor abundance in GEP-NENs. Although SNPs in several growth factor receptors have been demonstrated, no druggable mutation could be found to date. Therefore growth factor receptors might serve as targets for anti-growth receptor therapy in patients with GEP‑NENs, as several IGF-1R and EGFR inhibitors are currently under clinical assessment (refer to Supplementary Material: suppl. 1). Nevertheless further subgroup-specific genetic and post-transcriptional analyses are necessary to clarify the role of the distinct receptor aberrations and improve the therapeutic potential of growth factor receptors as therapeutic targets in GEP-NENs.
Somatostatin receptors mediate anti-proliferative signals, inhibit PI3K and MAPK signaling and are important therapeutic targets in GEP-NENs
Somatostatin signaling
The cyclopeptide family of SSTs (somatostatins), which function as somatotropin-release inhibiting factors, is distributed throughout the central nervous system and peripheral organs and can be found in endocrine, immune and neuronal cells, as well as in certain tumors. Its preserved peptide structure indicates a fundamental regulatory function in vertebrate hormone homeostasis.
The human genome includes five non-allelic genes that encode for five or six distinct transmembrane domain G-protein-coupled SSTRs (somatostatin receptors). The gene encoding SSTR2 produces two splice variants (SSTR2A and SSTR2B) in mouse and presumably in humans as well [35-38], whereas the other genes are intronless and generate one receptor in each case [39-42]. The natural ligands of SSTR1-5 (SST-14, SST-28 and cortistatin) are bound with a high affinity. Nevertheless the majority of (longer acting) synthetic peptide analogues, namely MS201-995 (octreotide), RC-160 (vapreotide), BIM 23014 (lanreotide) and MK 678 (Seglitide), only interact with the subtypes SSTR 2, 3 and 5 to a satisfactory extent. Moreover, Pasireotide (SOM 230) shows higher binding capacity towards SSTR1 [43, 44].
Somatostatin receptor signaling is complex. Its activation mediates cell cycle arrest, apoptosis and is involved in the regulation of hormone secretion in endocrine target cells. Appropriately, binding of somatostatin and its analogues to SSTRs triggers a number of intracellular signaling events, initiated by specific G-Protein activation. These G-proteins function as transducers and in turn modulate the activity of several key enzymes including adenylyl cyclase, phospho-tyrosine phosphatases (PTPs) and MAPKs (mitogen activated kinases) [45-47]. Inhibitory effects of SST on adenylyl cyclase, cAMP production and several distinct ion channels regulate both endocrine and exocrine secretion [39, 48, 49]. Intracellular PTPs exert anti-proliferative activity by deactivation of intracellular PI3K and MAPK signaling. Phosphatases, such as SHP-1 and SHP-2, mediate SST-induced cell cycle arrest in many cell lines in vitro. SHP-2 is also considered to inactivate insulin and epidermal growth factor binding RTKs and to repress C-Raf and MAPK signaling, whereas SHP-1 is involved in dephosphorylation of the p85 PI3K regulatory subunit and IRS-1 and thus, decreased PDK-1 (phosphoinositide-dependent kinase 1) and Akt activity. It furthermore inhibits S phase entry through the overexpression of p27kip1 and increase of hypo-phosphorylated retinoblastoma gene product (Rb) [47, 50-61]. A third PTP is involved in the SST-induced proliferation arrest: DEP-1 (density enhanced phosphatase-1, also termed PTPη in rats) is a negative regulator of Src-mediated growth factor receptor activity and participates in cellular differentiation [62-66].
The mechanism of SST signaling has been confirmed in different cellular models indicating that this modular multi-effector pathway, which is induced by several SSTRs, transmitted by similar kinases and PTPs and resulting in the activation of final effector PTPs, is a common mechanism in various cells [67, 68]. Furthermore the downstream signaling of the distinct SSTRs differs in certain subtype specific functions: e.g. whereas both SSTR2 and 5 regulate GH secretion, insulin secretion is predominantly controlled by SSTR5. Glucagon secretion and immune response are SSTR2 dependent (reviewed in [39]).
Somatostatin receptors in GEP-NEN therapy
Gastroenteropancreatic neuroendocrine neoplasms express all five subtypes of SSTRs, although at a variable extent and in various combinations. The most prevalent receptor is SSTR2A, which has been immunohistochemically detected in 84% of the GEP-NEN specimens in a recent study. SSTR3, 4, 5 and 1 have been found expressed in 84, 44, 32 and 32%, respectively [69]. This data agree with previous studies in insulinomas where the SSTR2, 1 and 3 have been found to be predominantly expressed [70]. In midgut NENs SSTR2 is the most prominent as well, but followed by SSTR1 and SSTR5, and, with a less frequency, by SSTR3 and SSTR4. [70-73]. Comparable findings have been recently published in a cohort of 67 NENs, with an expression of SSTR1, 2a, 3, 4 and 5 in 42, 63, 6, 32 and 65% of the cases, respectively. Interestingly, the SSTR5 immunoreactivity was correlated with the presence of metastases and angioinvasion [74]. Other studies have previously demonstrated a positive correlation of SSTR2 and SSTR5 expression with a better prognosis and higher differentiation [75-80].
Although corresponding data is rare, some studies have demonstrated a frequent co-expression of SSTRs and the D2R (dopamine D2 receptor) in GEP-NENs [38, 77, 81]. Co-expression of SSTR5 and D2R is assumed to contribute to a hetero-oligomerization and creation of a novel receptor with enhanced functional activity [82]. A Co-expression of SSTR2 and SSTR5 with D2R and its correlation with low tumor grade has been described in mixed NENs [77]. The high frequency of SSTR1, SSTR2, and SSTR5 expression and inverse correlation with COX2, a cytochrome C oxidase and component of the respiratory chain, was recently demonstrated. The study also accounted for a better prognosis concomitant with a generally high SSTR expression [76].
Additionally, one group was also able to demonstrate a high expression of the human SSTR2B which raises further questions, whether this splice variant exists in humans [38].
Data concerning the SSTR receptor distribution pattern in different subgroups was published for SSTR2A, which was present in a high frequency in carcinoids and gastrinomas, but detectable in only half of the analyzed insulinomas, suggesting one reason for the high number of octreotide insensitive insulinomas [83, 84]. Another study demonstrated a higher SSTR expression in non-pancreatic NENs versus pancreatic NENs for all receptor isoforms [78].
This high heterogeneity of data, beyond the fact that SSTR2 exerts to be the most prevalent of the generally highly expressed SSTRs in GEP-NENs, is summarized in table 2 which highlights the need for better defined criteria. For example, a discussion about scoring systems as criteria in the use of mono- versus polyclonal antibodies is still ongoing [69, 85-88].
SSTR expression in GEP-NENs. Several studies have been conducted to date, but with very heterogeneous results. SSTR2 is presumed to be the most prevalently expressed isoform.
| Reference | Tissue | Method | SSTR1 | SSTR2 | SSTR3 | SSTR4 | SSTR5 |
|---|---|---|---|---|---|---|---|
| Papotti 2002 [73] | GEP-NENs | RT-PCR | 90,1% | 84,8% | 78,8% | 24,2% | 42,4% |
| IHC pAb | 68,2% | 36,4% | 63,6% | ||||
| Reubi 2003 [70] | Midgut NENs | receptor auto-radiography | 50-60% | 90-100% | 10-20% | < 10% | ~ 50% |
| Srirajaskanthan 2009 [77] | Low grade | IHC pAb | 100% | 100% | |||
| Interm. gr. | 94,4% | 94,4% | |||||
| High grade | 66,7% | 66,7% | |||||
| Corleto 2009 [75] | functioning NEN | RT-PCR | 73% | 100% | 64% | 9% | 64% |
| Zamora 2010 [78] | GEP-NENs | IHC | 46% | 86% | 26% | 24% | 62% |
| Kim 2011 [76] | GEP-NENs | IHC m/pAb | 84% | 72% | 55% | ||
| Diakatou 2011 [38] | GEP-NENS | IHC pAb | 39% | 62% (2A) 49% (2B) | 38% | 15% | 38% |
| Kaemmerer 2012 [69] | GEP-NENs | IHC mAb | 32% | 84% | 84% | 44% | 32% |
| Schmid 2012 [74] | GEP-NENs | IHC mAb | 42% | 63% | 6% | 32% | 65% |
Furthermore the trafficking of SSTRs has become more and more interesting in somatostatin receptor-positive cancer entities and GEP-NENs. Concerning the internalization of SSTRs and its regulation in NENs, mainly controversially discussed immunohistochemical data is available. The involved agonists, phosphorylation sites and phosphatases in NENs are almost unknown, but basic neuroscience research has increasingly focused on this fundamental aspect of SSTR regulation. [89-95].
Throughout two decades, efforts have already been made to translate the knowledge of neuroendocrine SSTR expression into effective GEP-NEN therapies. During this process a multitude of somatostatin peptide agonists and antagonists and non-peptide agonists and antagonists have been developed, of which the agonists have found their way into clinical applications (refer to Supplementary Material: suppl. 2).
The clinical use of SST analogues is limited in clinical application due to a poor oral bioavailability, short half-life and immunogenicity. Non-peptide analogues are considered to be more advantageous as synthesis can be directed upon a higher specificity, bioavailability and less immunogenicity. Furthermore, somatostatin analogue-conjugated radioligands are applied in PRRT (peptide receptor radionuclide therapy) and scintigraphy-based diagnosis (Reviewed in [96-106]).
The two octapeptide analogues octreotide and lanreotide, which are also available as long-acting repeatable (LAR) depot formulation, bind to SSTR2 with a high and to SSTR5 with a moderate affinity. They inhibit the release of neuroendocrine hormones and have been shown to prolong disease stability in >50% of patients with progressive GEP-NEN disease. Although no objective tumor responses were observed in several studies, including the randomized, prospective Phase III PROMID trial in patients with metastatic neuroendocrine midgut tumors, patients profit from prolonged time to progression [107-117]. The results of the Phase III CLARINET-study using lanreotide in progressive neuroendocrine tumors of various sites are awaited later on this year. Further SST analogues, binding to the other SSTRs, such as Pasireotide, which binds to SSTR1, 2, 3 and 5, have been clinically assessed so far and chimeric molecules directed towards SSTRs and DRs (dopastatins) are under development and preclinical assessment in GEP‑NENs [45, 118-123].
In summary, the SSTRs are highly expressed in GEP-NENs. They have evolved into establishing therapy targets, although to date the individual expression patterns of the patients have not been related to therapeutic outcome.
The PI3K-pathway is frequently deregulated in many human cancers entities including neuroendocrine neoplasms of the gastroenteropancreatic system
PI3 Kinases
The lipid kinase family of PI3Ks (Phosphatidylinositol 3-kinases) consists of three classes (I-III) that promote the phosphorylation of 3-hydroxyl-phosphoinositides. Whereas heterodimeric class I PI3Ks influence cellular proliferation, insulin signaling and inflammation, the monomeric class II PI3Ks determine the regulation of membrane trafficking. The sole class III member is involved in autophagy. The most important subclass in human cancers is formed by class IA PI3Ks [124, 125].
Extracellular growth factor signaling such as by VEGF, PDGF, IGF-1, FGF and TGF- α and -β is transmitted by RTKs to PI3Ks and results in their activation [2, 3]. Activated class I PI3Ks convert their substrate PI(4,5)P2 (phosphatidylinositol 4,5-biphosphate) into its triple-phosphorylated form PI(3,4,5)P3 (phosphatidylinositol (3,4,5)-triphosphate). Subsequently, PI(3,4,5)P3 recruits proteins that contain a PH-domain (Pleckstrin homology domain) into proximity and thus functions as a docking site for Akt and PDK-1, allowing the latter to phosphorylate Akt at T308 (Threonin-308) [125, 126]. Phosphorylation on both, T308 by PDK-1 and on S473 (Serin-473) by mTORC2 is required for the full activation of Akt kinase activity [127].
The PIK3CA gene, which encodes for p110α (the catalytic subunit of class I PI3K) and is considered as the only relevant catalytic subunit in the context of cancer associated mutations, was found mutated in only 1.4% and 8% of pNENs, respectively [128, 129]. Data about PI3K-p85α subunit mutation nor PI3K amplification in NENs have not been published to date.
The regulatory impact of PI3K could be validated by preclinical studies with PI3K inhibitors. LY294002, a quercetin analogue and PI3K inhibitor, decreased cell proliferation in non-gastrointestinal neuroendocrine cell lines when applied as single agent or combined with rapamycin [130, 131]. Studies with LY294002 treatment of rat-derived GEP-NEN cell lines propose an inhibitory effect of LY294002 on the VEGF secretion by neoplastic endocrine cells [132]. The mTORC2-PI3K-mediated activation of the ERK cascade during mTOR inhibition of NENs was demonstrated through stimulation of human neuroendocrine BON (pNEN), GOT-1 (ileal NEN), KRJ-I (ileal NEN), H-STS (hepatic metastasis of ileal NEN) and NCI-H727 (bronchial carcinoid) cell lines with single and dual inhibitors [133-135]. Previous studies on BON cells have demonstrated that LY294002 blocks the constitutive activation of PI3K and ERKs, respectively. PI3K, but not the ERK cascade, regulates expression of cyclin D1 and p27kip1, induced by an autocrine IGF-I loop, in BON cells [136]. Not least, PI3K signaling is negatively involved in NE secretion, as demonstrated by PI3K subunit p110α-inhibition in vitro. Li et al. demonstrated that inhibition of p110α increases neurotensin granule trafficking by up-regulating α-tubulin acetylation and regulating Ras-related protein Rab27A in BON and QGP-1 cells [137].
A vast number of agents and inhibitors interfering with PI3 kinases and upstream receptors have been developed so far and are currently in different stages of clinical testing (refer to Supplementary Material: suppl. 3). The majority have not been assessed in GEP-NENs thus far.
The physiological inhibition and termination of PI3K signaling by degradation of PI(3,4,5)P3 is mediated by two major types of phosphatases. The SH2 domain-containing inositol phosphatases SHIP1 and SHIP2 dephosphorylate position 5 of the inositol ring and produce PI (3,4)P2. The loss of SHIP2 results in a significant increase of insulin sensitivity, indicating that this phosphatase is a crucial regulator of PI3K signaling downstream of insulin [138].
In summary, PI3 Kinases show up to be rarely mutated in GEP-NENs but contribute to several feedback loops as signal transduction mediators. They are therefore prominent targets for dual and combined targeting therapy approaches but their importance for GEP-NEN disease remains to be further explored in clinical and in vivo contexts.
The tumor suppressor PTEN
Another crucial phosphatase and tumor suppressor protein involved in growth factor signaling is PTEN (phosphatase and tensin homologue). After recruitment from cytosol to plasma membrane, PTEN dephosphorylates position 3 of PI(3,4,5)P3 and produces PI(4,5)P2. Loss of PTEN is frequently observed in a wide variety of human cancers, with the highest incidence found in endometrium, central nervous system, skin, and prostate cancers [139, 140]. Beyond its function as upstream regulator of PI3K signaling, loss of PTEN was linked to occurrence of metastases and is regarded as critical marker for therapy resistance and sensitivity towards mTOR inhibition [139, 141-144].
The activity of PTEN is lost through diverse mechanisms in many different entities of cancer, but the majority of PTEN mutations induce truncations of the protein. PTEN is often mutated in tumor-prone germ line diseases and in cancer-associated somatic mutations [145-147].
The impact of PTEN towards cellular integrity is not limited to its cytoplasm-located lipid phosphatase activity. PTEN is localized in the nucleus under various conditions, such as cell differentiation and cell cycle arrest under stress and apoptotic stimuli, e.g. by regulating the APC/C (anaphase-promoting complex/cyclosome) [148-152].
Nuclear localization has been linked to maintenance of chromosome stability. The absence of nuclear PTEN is thus linked to tumor aggression, which suggests that the nuclear localization of PTEN is tumor-suppressive [148, 153-155]. PTEN also influences cytoskeletal remodeling processes and thereby controls cell size, cell invasion and migration [156, 157].
Interesting work with regard to the sequestration of PTEN was published by Putz and colleagues in 2012. They demonstrated that mono-ubiquitinated PTEN can be exosomally trafficked between cells. In target cells, internalized PTEN had functional activity, which led to a reduction in the abundance of pAkt and a decrease in the extent of target cell proliferation [158]. These findings disclose new insight into tumor-stroma interactions and highlight the unlimited influence of tumor-associated cells on tumor cell signaling.
In analogy to many other cancers, PTEN is frequently affected in pancreatic neuroendocrine neoplasms (pNENs). PTEN gene mutations are rare events in 7 and 9% of the cases respectively, but reduced PTEN expression is a frequently observed phenomenon in sporadic pNENs. Allelic loss of heterozygosity of PTEN, which is located at 10q23.3, was identified in one-third of sporadic pNENs. In 25% of pNENs a somatic deletion of 10q occurs [33, 128, 154, 159-162].
In nonfunctioning endocrine tumors of the pancreas alterations in microRNA expression have been related to endocrine and acinar neoplastic transformation and progression of malignancy. Assuming that MicroRNA-21 regulates the expression of PTEN in hepatocellular cancer, this scenario is as well discussed as a potential inhibitory mechanism of PTEN down-regulation in pNENs [163-165].
In primary pNENs, low expression of PTEN correlates with advanced WHO phenotype and higher proliferation index. Moreover, hyper-phosphorylation of mTOR is associated with significantly lower 5-year overall survival [166].
Several studies have demonstrated that the subcellular localization of PTEN is altered in both sporadic and VHL-associated pNENs, although there are contradictory conclusions whether cytoplasmic or nuclear localization is sufficient to predict therapy outcome or whether an increased or decreased nuclear to cytoplasmatic ratio is associated with worse prognosis [33, 154, 167, 168].
In vitro data concerning the response of neuroendocrine pancreatic (BON) and insulinoma (CM) cell lines towards triciribine inhibition suggest that PTEN might also influence the sensitivity of pNENs cells to Akt inhibition [169-171].
In general, in GEP-NENs the expression of PTEN was found to correlate with response to streptozotocin-based cytostatic therapy and to systemic therapy with streptozotocin and doxorubicin [168]. As observed in the pNENs subclass study, PTEN expression in overall GEP-NENs was prevalently effected in tumors with lower differentiation in contrast to those with well differentiated phenotype [172].
PTEN might therefore serve as therapy response and malignancy marker, especially in combination with TSC2 expression analyses (see below). The mechanisms that lead to the down regulation of PTEN remain almost unexplored.
The role of Akt
The Akt family of serine/threonine kinases (Akt1/2/3, also known as protein kinase B, PKBα/β/γ) is the key mediator of PI3K signaling and connector to several interrelated pathways. It is involved in the majority of cellular processes, such as protein synthesis and cell growth, survival, proliferation and metabolism. Presuming that these processes constitute the backbone of cancer development and progression [173], Akt isoforms represent a highly prominent target for GEP-NET therapy research and for drug development in general.
The most prevalent member of the Akt family of protein kinases is Akt1, which is implicated in cell growth and survival. Akt2 is predominantly expressed in muscle and adipocytes and is in involved in the insulin-mediated regulation of glucose homeostasis. The distribution of Akt3 is almost limited to testes and brain [174-178].
Full Akt activation depends on the concomitant phosphorylation of two distinct sites that can be activated independently: the PDK-1-catalyzed T308 phosphorylation inside of the activation loop serves as readout of PI3K activation. In contrast, the phosphorylation of S473 in the hydrophobic motif of the C-terminal tail indicates a mTORC2 to Akt feedback signaling activity or is induced by PIKK (PI3 kinase-related kinase) superfamily or DNA-PK. Activity of Akt is detected by a fivefold increase upon S473 phosphorylation [179-184]. Furthermore the role of the Akt phosphorylation site is not limited to activation enhancement, but also required for target specification. One of the most prevalent downstream targets of Akt T308 is TSC2 (tuberous sclerosis complex 2). Akt mediated phosphorylation of the TSC2 subunit hinders TSC1/2 complex formation and activates the GAP function of TSC2 toward the small GTPase Rheb. Hereupon Rheb activates mTORC1 at S2448, leading to the phosphorylation of the downstream effectors 4EBP1 (Eukaryotic translation initiation factor 4E-binding protein 1) and p70S6K (70 kDa ribosomal protein S6 kinase 1, also termed S6K) [185]. Phospho-(T308)-Akt mediated PI3K signaling also determines cellular metabolism, cell cycle progression and insulin signaling by affecting the subcellular localization of GSK3 (glycogen synthase kinase 3) [125, 186-189]. Interestingly, in vitro and in vivo studies on deletion of Rictor, mSIN1 (stress-activated map kinase interacting protein 1) or mLST8 (mammalian lethal with SEC13 protein 8) suggest, that the inhibition of mTORC2 mediated S473 phosphorylation selectively affects Akt substrates FOXO1 and FOXO3a (O-Family Forkheadbox proteins), with little effect on Akt substrates GSK3 and TSC2 [183, 190]. The discrepancy between S473 phosphorylation and resulting Akt activity indicates that S473 phosphorylation might not be the major regulator of Akt activity [191].
Nevertheless, activated Akt promotes survival signaling by inhibition of pro-apoptotic proteins, such as BIM (Bcl-2 interacting mediator of cell death) and BAD (BCL2-associated agonist of cell death) by phosphorylation, leading to their sequestration and degradation. Akt also inhibits the expression of pro-apoptotic genes and cell cycle inhibitors such as p21cip1 and p27kip1 by targeting their transcription factors and stimulates cyclin D1- and c-Myc-controlled cell cycle progression by inhibiting their negative regulators, e.g. FOXOs and GSK3.
Relatively few studies are available on Akt phosphorylation and expression in GEP-NENs. Weak expression of Akt or presence of the PI3-signaling antagonist PTEN was associated with response to systemic chemotherapy and Akt overexpression has been correlated to shorter median survival rates for patients with well-differentiated and poorly differentiated tumors [168].
Shah and colleagues demonstrated a phospho-S473 activation of Akt in 76% of 98 differently graded NEN-tissue samples, indicating that activation of Akt may contribute to GEP-NEN tumorigenesis. Unfortunately the data failed to correlate Akt S473 phosphorylation to tumor grade by multi-variant statistical analysis [6]. These findings were supported by further immunohistochemical analysis of enteropancreatic NENs respecting Akt S473 phosphorylation. In a subsequent study S473-activated Akt was found in 61 % of the tissues, but also failed to correlate with tumor grade, tumor size or the presence of metastases [192].
Nevertheless, NEN patients treated with everolimus and octreotide in an open-label phase II trial (NCT00113360) showed a significant longer PFS in the case of T308-phosphorylated Akt in pretreatment and on-treatment tumor biopsies. Even partial responders were more likely to have an increased Akt T308 phosphorylation in comparison to non-responders. Therefore, although baseline Akt activation is associated with a more aggressive clinical course, increase of Akt activation has been considered a predictor of rapamycin response. These data have been supported by several preclinical studies on rapamycin treated cell lines of various offspring in vitro and xenografted [193-195]. In vitro experiments on neuroendocrine cell lines have furthermore evaluated an isoform specific impact of Akt on NEN cancerogenesis that might be important for therapeutic approaches. Only the directed down-regulation of Akt1 and Akt3 decreased the phosphorylation of classical Akt downstream targets such as GSK3α/β, MDM2 and p70S6K and suppressed cell viability. Inhibition and knockdown of Akt1 and Akt3 resulted in decreased ERK1/2 phosphorylation and Akt3 ablation induced apoptosis in BON cells, indicating that this isoform is particularly relevant to neuroendocrine cell survival. Akt1 and Akt3 seem to be important for NEN cell viability while Akt2 may have antitumor activity as demonstrated by pan and isoform-selective knockdown of Akt. The data suggest a particular role for these Akt isoforms in NEN activity [196].
Several specific inhibitors of pan-Akt, Akt isoforms and PDK-1, an Akt regulator, are commercially available. Only a few of them are already under clinical and preclinical investigation in GEP-NENs (refer to Supplementary Material: suppl. 4).
In conclusion, the Akt proteins serve as crucial feedback mediators within the growth factor response network. Nevertheless, the sole focus on pan-Akt S473 phosphorylation might not suffice to understand the complex interplay between various isoforms, phosphorylation sites and scaffold regulators. Akt activation and subtype distribution has a high potential to predict therapy response, but the importance of the specific patterns in vitro and in clinical settings remains to be elucidated.
mTOR complexes and feedback activation
The most important downstream mediator of PI3K-Akt signaling is the serine/threonine kinase mTOR mammalian target of rapamycin. It is encoded by the FRAP 1 gene and contributes to a multitude of cancer associated cellular processes. It forms two distinct protein complexes: mTORC1 and mTORC2. Whereas mTORC1 is sensitive to rapalogues such as rapamycin, everolimus and temsirolimus, mTORC2 is considered resistant to rapamycin and insensitive to nutrient signals [197].
The mTOR complex 1 is formed by the mTOR protein itself, associated with RAPTOR (Regulatory associated protein of mTOR), and two negative regulators, PRAS40 (proline rich Akt substrate 40 kDa) and DEPTOR (DEP domain containing mTOR interacting protein) [198-201]. RAPTOR functions as a scaffold and positively modulates the mTOR kinase reaction towards the mTORC1 key targets 4EBP1 and p70S6K in vivo [198]. The second subunit of the mTORC1 complex, mLST8, is considered to bind to the kinase domain of mTOR and to regulate its kinase activity positively. It is also considered to maintain the interaction between mTOR and either RAPTOR or RICTOR (Rapamycin-insensitive companion of mTOR), which is part of the mTORC2 complex, and thus, to be important for shuttling mTOR between the two complexes and for sustaining the intracellular equilibrium of mTORC1 and mTORC2 [183, 202, 203]. Whereas PRAS40 inhibits the mTORC1 activity via raptor, DEPTOR was identified to interact directly with mTOR in both mTORC1 and mTORC2 complexes. However, DEPTOR overexpression leads to decreased p70S6K phosphorylation and to mTORC2-mediated signaling back to Akt, monitored by S473 phosphorylation [201, 204].
PI3K-mTORC1 signaling controls the transcription of many genes, some of which are involved in metabolic pathways and regulate nutrient-responsive transcription programs [205-208]. Its major downstream effectors, including p70S6K and 4EBP1 are regulated by phosphorylation [209]. Subsequently, activated p70S6K phosphorylates S6 (40S ribosomal protein S6) and other ribosomal proteins and elongation factors and therefore enhances the translation of mRNAs and promotes proteins synthesis [210]. Although p70S6K can also be activated by mTOR independent pathways such as PDK-1, MAPK and SAPK (stress-activated protein kinase), the mTORC1-mediated phosphorylation at T389 is required for its complete activation [211].
Hypo-phosphorylated 4EBP1 inhibits the initiation of protein translation by binding and inactivating eIF4E (eukaryotic translation initiation factor 4E). Phosphorylation of 4EBP1 by mTORC1 promotes dissociation from eIF4E and thereby facilitates eIF4E-dependent translation initiation [212].
Furthermore, mTORC1 controls the activity of many other proteins, such as ODC (ornithine decarboxylase), glycogen synthase, HIF-1α (hypoxia-inducible factor 1α), eEF2 kinase (eukaryotic elongation factor 2 kinase), PKCδ and PKCɛ (protein kinases C delta and epsilon), PP2A (protein phosphatase 2A), p21cip1 and p27Kip1 cyclin-dependent kinase inhibitors and STAT3 (signal transducer and activator of transcription 3) [213-223]. Accordingly, the impact of the mTORC1 complex spans a multitude of cellular processes that modulate cellular behavior in response to local circumstances and links availability of growth factors, nutrients and energy to cell growth, survival, proliferation, angiogenesis and motility.
The rapamycin-insensitive mTOR Complex 2 consists of mTOR mLST8, mSin1, PROTOR1/PRR5 and 5 PROTOR2/PRR5L (proline-rich protein), HSP70 (heat shock 70 kDa protein) and DEPTOR.
The mSin1 protein contains a Ras binding domain and a PH-domain, which allows localizing the protein near the plasma membrane. It is therefore considered to be important for the assembly and dynamic localization of mTORC2 and to the phosphorylation of Akt at S473. HSP70 assures proper assembly of the protein complex under physiological conditions and following heat shock [190, 224-227]. HSP70, which has various cellular functions beyond maintaining proper protein structures, has been found to exist in a neuroendocrine tumor specific truncated isoform in NENs. The authors conclude, that the altered HSP70 isoform equilibrium might contribute to apoptosis inhibition or might be based on similarities with neuronal cells, as protein folding and protection against aggregate formation is of particular importance in the nervous system [228].
The mTOR complex 2 is activated by growth factors, G protein-coupled receptor ligands and cytokines. It phosphorylates PKC-α and paxillin (a focal adhesion-associated adaptor protein) and regulates the activity of the small GTPases Rac and Rho which control motility, invasion and cytoskeletal assembly. Beside motility aspects that play distinct roles in metastasis, the Akt S473 feedback activation, that is mediated by mTORC2 following mTORC1 inhibition, is one of the crucial mechanisms for PI3K cancer signaling [181, 229-231].
In summary, two major feedback loops control PI3K-signaling, which could potentially impact therapy approaches, especially with regard to monotherapy (refer to figure 1): Activation of p70S6K by mTORC1 causes feedback inhibition of IGF-1/insulin signaling by phosphorylating IRS-1 (insulin receptor substrate 1), causing IRS-1 degradation, and leads to decreased PI3K signaling and reduced Akt T308 phosphorylation. Reciprocally, rapalogue-induced inhibition of mTORC1 consequently inhibits p70S6K phosphorylation, but relieves this feedback and induces Akt T308 re-phosphorylation and thus increased mTORC2 activation. Subsequent Akt S473 phosphorylation follows in an mTORC2-dependent manner, which attenuates the therapeutic effects of rapalogues in tumors, in model systems and in patients as well [204, 232, 233]. This feedback activation could be obviated in serum free in vitro conditions, due to the known requirement for growth factors for mTORC1 to PI3K feedback loops [204]. For some cancer entities it is evident that even inhibition of both, mTORC1 and mTORC2, e.g. by ATP-competitive mTOR kinase inhibitor AZD8055, did not result in a persistent inhibition of PI3K signaling: although sustained inhibition of mTORC2 activity and AKT S473 phosphorylation was detectable, a distinct activation of RTK signaling occurred, which induced PI3K signaling and reinduction of T308 phosphorylation [234].
PI3K signaling feedbacks: PI3K signaling is highly activated and de-regulated in GEP‑NENs. Several feedback loops contribute to this effect [330, 331]. RTK activation by ligand binding triggers a phosphorylation cascade onto IRS-1 and PI3K. The latter phosphorylates PIP2 and thereby activates PDK-1 and Akt at T308. Akt in turn inhibits TSC2 by phosphorylation, which leads to mTOR activation. The mTOR complex 1 activates translation via eIF4A and p70S6K and cell cycle progression, whereas mTORC2 relieves a feedback loop by fully-activating Akt at S273. S6K promotes a negative feedback onto IRS‑1 and thereby counteracts mTORC1 inhibition. The two major negative regulators, PTEN and TSC2 are frequently down regulated in GEP-NENs which lead to a highly deregulated Akt activation [33, 128].
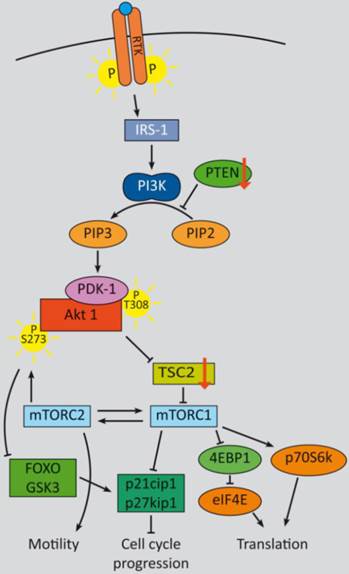
Expression and activation level of mTOR in GEP-NENs has been analyzed in a vast number of studies and experiments but mainly failed to correlate to clinical outcome in a clear statistically significant matter. Nevertheless, a distinct tendency of high malignant NEN to show highly activated mTOR expression was obvious. Catena and colleagues demonstrated that mTOR was expressed in the majority (80%) of poorly differentiated neuroendocrine carcinoma patients (WHO 2000 classification) however with no relationship to tumor origin or proliferation rate determined by MIB-1 (Ki-67 antibody for paraffin-embedded tissue specimens) [235]. Another immunhistochemical analysis detected high levels of S2448 phosphorylated mTOR in 67% of poorly differentiated neuroendocrine carcinomas (including positive staining of all large cell neuroendocrine carcinomas) in contrast to 27% mTOR activation in well-differentiated tumors and carcinomas (WHO 2000 classification). Further statistical validation failed due to the low number of analyzed patients [236].
Significant correlation of mTOR expression with primary tumor location and metastatic status in GEP-NENs could be demonstrated by Kasajima and colleagues. Expression of mTOR, 4EBP4 and phosphorylated p70S6K was found to be higher in foregut than in midgut tumors. Furthermore, higher proliferation indices (indicated by Ki-67) were associated with significantly higher mTOR and 4EBP1 expression, as well as with higher activation of the mTOR targets 4EBP1, p70S6K and eIF4E (indicated by phosphorylation) [237].
One of the major regulatory proteins of mTOR activation, TSC2, is frequently down-regulated in pNENs and correlates with worse prognosis concerning overall survival, time to progression and disease-free survival. Low levels of TSC2 significantly correlate with functional tumor status and aggressiveness [33]. Although neither TSC2 nor PTEN were significant independent prognostic indicators regarding occurrence of metastases in a multivariate analysis by Missiaglia and colleagues, conjoint low levels of PTEN and TSC2 are assumed to be related to liver metastasis and might predict response to PI3K-pathway inhibitors. While somatic mutation of TSC2 and PTEN was found in only less than 10% of pNENs, 85% of primary pNENs showed altered protein levels of TSC2, PTEN or both. [33, 128].
Germline mutations in TSC1 and TSC2 tumor suppressor genes, resulting in the activation of the mTORC1 pathway, have been described in patients with tuberous sclerosis complex. TSC is an autosomal dominant genetic disorder and most of the occurring neoplasms are benign with an early onset [238]. A study on 219 TSC patients revealed that the incidence of pNENs was 1.8% as compared to the rate of 0,002% in the general population. Therefore it seems likely that pNENs are a dominant pancreatic pathology in the setting of TSC and aberrant TSC1 and TSC2 proteins may play a crucial role in the development of pancreatic neuroendocrine lesions [238, 239, 114, 240-242].
In summary, several studies have highlighted the importance of high mTOR expression and activation, activation of its targets as well as the functional impact of the low expression of its direct negative regulator, TSC2. Relevant somatic mutations that influence the expression of either TSC2 or mTOR have not been detected thus far. Additional regulatory mechanisms such as transcriptional or post-transcriptional gene silencing might be involved in their regulation and require further investigation.
Inhibition of mTOR as therapeutic strategy
Analyses of mTOR aberrations have furnished the rationale for use of inhibitors of mTOR and its downstream targets (refer to Supplementary Material: suppl. 5) [33, 165]: The mTORC1 inhibitor and rapalogue everolimus (RAD001, Afinitor®) is the most clinically advanced and the furthest developed target directed therapy in GEP-NENs. It was approved by the FDA for advanced well-differentiated pNENs in 2011.
Studies on genetic determinants of rapalogue response have demonstrated that drug-resistant cells (such as HT-29, HCT116, and DLD-1) carried mutations in both PIK3CA and KRAS/BRAF. Everolimus-sensitive cells displayed PI3K pathway alterations but no mutations in the KRAS/BRAF genes, rendering neuroendocrine tumors a favorable target for everolimus treatment [239]. This effect is assumed to be dependent on mTOR-Ras/Raf crosstalk and its outcome depends on Raf mutational status (paradoxical activation, see below).
Nevertheless in preclinical studies of everolimus, inhibition of mTORC1 in neuroendocrine cell lines was demonstrated to induce growth inhibition and induction of apoptosis [240]. However it also led to a global upregulation of upstream PI3K signaling and to cross-activation of Ras/Raf/Erk signaling via mTORC1-p70S6K-IRS-1 mediated negative feedback loops. This cross-activation resulted in upregulation of VEGF secretion, through a raise of NF-κB (nuclear factor-κB)-mediated VEGF expression and HIF-1α induction [134, 135, 195]. In vivo, Rapamycin monotherapy was notably efficacious in pNEN bearing transgenic mice and prolonged survival concomitant with stable disease. Nevertheless, the tumors developed resistance [241]. Preclinical xenograft studies on mouse cells that mimic PDNEC revealed a significant deduction of tumor mass and Ki-67 in response to everolimus treatment, suggesting further exploration in poorly differentiated neuroendocrine cancers, that particularly lack therapy options to date [242].
Three human phase II or III studies have been conducted, including 600 patients with advanced pNENs [114, 243, 244]. The response rate as assessed by conventional Response Evaluation Criteria in Solid Tumors (RECIST) criteria has been shown to be very low; however, everolimus significantly affected progression-free survival. The RADIANT-3 clinical study, which led to FDA approval, resulted in a stable disease of 73% and 51% in the everolimus and placebo arms respectively, rather than a partial response with 5% and 2%.
Concluding, everolimus delayed tumor progression without changing the pattern of progression among patients with advanced pNENs [244, 245]. Recent therapeutic approaches have therefore focused on inhibitors of alternative components of the PI3K pathway, dual target inhibitors and effective combinatory bio/chemotherapies.
Outlook: PI3 signaling in diagnosis and therapy
The multiple results of PI3K signaling analyzes in GEP-NENs are summarized in table 3. Noticeably, with few exceptions, no study detected serious activating mutations of PI3K-pathway mediators. In contrast to other cancer entities, where activating mutations in receptors or the PI3 kinase itself are frequent, GEP-NENs thus lack appropriate druggable targets. Nevertheless, a high level of deregulation (as indicated by extensive activation of kinases) triggers proliferation, neoangiogenesis and a secretory phenotype. The complex interactions within the autoregulatory network bypass the current therapeutic approaches. It is therefore all the more important to identify bottle neck factors that might not have been recognized as key players to date. Evaluating the role of regulator non-coding RNAs might also answer some of the questions, why the neuroendocrine growth factors pathways are deregulated to that high extent although almost no appreciable mutations could be detected responsible to date. Furthermore, the tumor stroma and tumor-associated cells also contribute to a growth factor-saturated microenvironment in general. Studying their importance for GEP-NEN cancerogenesis and metastasis might also reveal a high potential of prognostic factors and therapeutic interventions.
Summary of studies that analyzed the role of PI3K signaling in GEP-NENs.
| Study results | Reference |
|---|---|
| Expression of multiple growth factors is very high in GEP-NENs | [2-16]. |
| Expression of IGF-1 and IGF-1R is elevated in GEP-NENs | [14, 18-26] |
| Elevated copy number of the EGFR and HER-2/neu loci might contribute to high receptor expression | [21, 246] |
| EGFR expression is high in GEP-NENs and correlates with metastasis in pNENs | [6, 9, 30, 31] |
| PIK3CA mutations are rare events | [128, 129]. |
| PI3K signaling stimulates the ERK pathway of NE cells in vitro | [133-135] |
| PI3K signaling triggers secretion in vitro | [132, 136, 137] |
| PTEN mutations are rare events, but LOH may contribute to malignant transformation in (pancreatic) NENs | [33, 128, 154, 161, 162] |
| Low PTEN expression correlates with prognosis and response to therapy | [166, 168, 169, 172] |
| Subcellular localization of PTEN might serve as prognosis marker | [33, 154, 167, 168] |
| Akt overexpression associated with shorter median survival in with well- and poorly differentiated tumors | [168] |
| Phospho-T308 Akt might serve as marker of rapamycin response | [193-195] |
| Phospho-S473 Akt might not serve as a valuable marker | [6, 192] |
| Subtype specific activity: Akt1 and Akt3 pro-survival; Akt2 pro-apoptotic | [196] |
| Expression and activation of mTOR, 4EBP4 and p70S6K is associated with higher proliferation index and might correlate with low differentiation | [235-237] |
| Low level of TSC2 is correlated with tumor status and aggressiveness | [33, 128] |
The MAPK pathways promotes growth and cancer progression as well as feedback bypasses to PI3K signaling
The MAPK cascades
Alteration of the Ras-MAPK cascades has frequently been described in human cancer. These pathways comprise several kinases that transmit extracellular signals from growth factors, chemokines and ECM signals and regulate cell growth, differentiation, proliferation, apoptosis and migration. They are classified in several interconnected branches constructed of functional analogues of MAPKs (mitogen-activated kinases), their MAPK kinases and the latter's MAPKK kinases. The four most important branches are (1) the ERK/MAPK, (2) the JNK (c-Jun amino-terminal kinase)/SAPK pathway, (3) the p38 pathway and (4) the BMK (big mitogen-activated protein kinase)/ERK 5 pathway [247]. We will therefore focus on the classical ERK/MAPK cascade and glance to other important members.
The classical cascade is induced by ligand binding to RTKs, such as VEGFR or EGFR, leading to its dimerization and auto-phosphorylation of the intracellular c-terminal region. Thereby binding sites for adaptor proteins are generated that in turn recruit GEFs (guanine nucleotide exchange factors) to the plasma membrane. GEFs facilitate the binding of GTP to Ras proteins. GTP-bound Ras recruits Raf kinases and activates the latter's serine/threonine kinase function to phosphorylate MEK and induce the phosphorylation of ERK effector kinases [248].
Abnormal activation of receptor tyrosine kinases or gain-of-function mutations in the RAS and BRAF genes have been frequently identified in a vast number of cancers. Multiple cross links and feedback loops to other mitogen pathways induce therapy resistance and bypass inhibitory approaches. The MAPK pathway contributes to neuroendocrine cancerogenesis, although many aspects in GEP-NENs remain to be explored.
The role of Ras
The Ras superfamily is divided into five main families which in total comprise more than 150 members in humans: Ras, Rho, Rab, Arf, and Ran. The Ras family members are the most intensively studied. Three human RAS genes encode four Ras protein isoforms, designated as H-Ras, N‑Ras, K-Ras4A and K-Ras4B. Ras proteins exhibit GTPase function and a structure that is related to the Gα subunit of heterotrimeric G proteins. G proteins are molecular switches that oscillate from inactive GDP-bound to active GTP-bound states [249]. The cyclic process of GDP/GTP is facilitated by GEF and GAP (GTPase activating proteins) classes of regulatory proteins that build a three-protein-complex with Ras.
The Ras proteins are anchored in close proximity to adaptor proteins such as Grb2 (growth factor receptor bound protein 2) and GEFs in the plasma membrane by farnesylation. The SOS family (son of sevenless) of RasGEFs, facilitates the exchange of Ras bound GDP with GTP and thereby activates Ras by conformational change [250].
Activating point mutations of the three Ras family genes are common in human cancers (30%) with a very high incidence in pancreatic adenocarcinoma, colorectal and lung cancers. Mutations in other Ras superfamily GTPases are rare and thus, their hyper-activation requires alternative induction mechanisms [251-255]. Deregulation of their regulators is a common event.
Aberrant signaling from growth factor receptors, in particular, RTKs and GPCRs (G protein-coupled receptors), or up-regulated gene expression can lead to aberrant GEF regulation and thus to enhanced activity of small GTPase proteins of the Ras superfamily.
GAPs, which return the GTPase to its GDP-bound inactive state, have been shown to exert crucial roles in curtailing GTPase activity in cancer. Since activation of Ras superfamily GEFs has been frequently described in human cancers, loss of GAP activity permits uncontrolled GTPase activity and can promote cancer [256-262].
In contrast to other entities such as pulmonary neuroendocrine carcinomas, mutations in the Ras genes are uncommon and rarely documented in GEP-NENs. Early studies could demonstrate that Ras mutations are virtually absent in gastroenteropancreatic neuroendocrine neoplasms [21, 263-265]. H‑Ras and K‑Ras expression could be detected in 65% and 10%, respectively. However, further information regarding its activity has not been generated to date [266], but much more comprehensive data is available for the Ras downstream targets, especially for B-Raf.
Wild type Raf and the importance of paradoxical activation
Activation of Raf family members (A-Raf, B-Raf and C-Raf or Raf-1) is initiated by binding of their Ras binding domain to Ras‑GTP and release from a 14-3-3 dimer bound to the N-terminal phosphorylation site. Concomitant conformational changes stimulate their serine/threonine kinase activity, dimerization and trigger sequential phosphorylation and activation of their targets MEK and ERK [267, 268].
B-Raf is the family member most easily activated by Ras, since both A-Raf and C-Raf need additional steps, such as phosphorylation of activating residues and dephosphorylation of negative regulatory residues, to reach maximal activation [269, 270]. Furthermore the kinase activity of B-Raf is higher than those of the other family members [271].
Recent studies discovered that besides its involvement in oncogenic Ras signaling, BRAF itself is also mutated at a high frequency in human cancers, especially in melanoma (30-60%), thyroid cancer (30-50%) and ovarian cancer (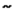 30%) [272]. A very common BRAF mutation, B-RAFV600E, is involved in the expression of hypoxia-inducible factor-1α and VEGF and thus contributes to neoangiogenesis in those entities [273-275].
30%) [272]. A very common BRAF mutation, B-RAFV600E, is involved in the expression of hypoxia-inducible factor-1α and VEGF and thus contributes to neoangiogenesis in those entities [273-275].
Overexpression or gain of function mutation of full-length Raf (or the truncated catalytic domain) leads to the activation of the ERK pathway and increases proliferation and tumor growth. Although there is no evidence that Raf activation participates in senescence evasion to date, it has been demonstrated to retrain apoptosis by regulating the expression and/or the activity of Bcl-2 family members [276].
Activated Raf is also involved in EMT (epithelial to mesenchymal transition), invasion and metastasis by promoting the production of TGFβ. Additionally, both B-Raf and C-Raf antagonistically control cell contractility and migration: B-Raf increases Rho-dependent contractility and opposes migration in an ERK-dependent manner, whereas C-Raf reduces contractility and increases migration by interfering with the activity of the cytoskeleton-based Rho effector ROCK2 (Rok-α) [277-284].
The C-Raf protein appears as part of a multiprotein complex composed of HSP90, p50, and several scaffold proteins, such as 14-3-3. This complex is required for controlling its stability and activation status as well as its activity [285-287]. The phosphorylation state of C-Raf is influenced by multiple further protein kinases, including Src, PKC (protein kinase C) family members, the p21cip1-activated protein kinase PAK, and Akt [288-291].
In the case of the ubiquitous C-Raf, other targets potentially contributing to tumor progression have been identified such as the NF-κB, Rb and BAD [292-294]. C-Raf also contributes to genomic instability. Loss of RKIP (Raf kinase inhibitor protein) or C-Raf overexpression lowers the activity of the Aurora-B kinase, allowing cells to bypass the spindle assembly checkpoint [295].
The serine/threonine phosphorylation of MEK proteins as an intermediate step of the MAPK cascade features two special objectives: to enhance the cooperativity of activation of the MAPK and to allow modulation by other signaling events. In the case of the ERK1/2 pathway, amplification occurs at the Raf-MEK step, because MEK1 is much more abundant than Raf. Another controlling feature of this step in the MAPK cascade depends on the dual phosphorylation of the MAPK by MEK. The tyrosine residues of ERK1/2 are phosphorylated with a higher affinity than threonine, leading to a nonprocessive phosphorylation and to the establishment of a threshold. The tyrosine phosphorylated proteins remain in an inactive state and accumulate until the threshold is reached. Subsequently the kinases are rapidly converted to the active state by threonine phosphorylation [296-303].
MEK mutations are rare events in human cancers with an incidence of 3% in melanomas and 2% in colon carcinomas [304].
The data concerning Raf and MEK downstream signaling and the discussion whether raf inhibition is reasonable or not is very contradictory and is still in the focus of preclinical investigation. Several further inhibitors of Raf and MEK are under preclinical assessment in GEP-NENs however with not very promising results to date (refer to Supplementary Material: suppl. 6).
Genetic analyses demonstrated that B-Raf mutations are rare in GEP-NENs [263, 305-307], with the exception of colorectal NENs, which have been demonstrated to harbor ~ 60% KRAS and BRAF mutations, but presumably due to its high content of adenoma and/or an adenocarcinoma cells [308].
Nevertheless the small GTPase Rap1 and B-Raf are highly expressed in GEP-NEN specimens, and both contribute to ERK1/2 and E26-like kinase (Elk-1) activation in NE cell lines [309]. Disrupting Raf-MEK-Erk signaling by the B-Raf inhibitor Raf265 significantly decreased Bcl-2 level and sensitized to TRAIL signaling in neuroendocrine cell lines. Raf265 inhibited Erk1/2 phosphorylation but in turn induced Akt phosphorylation and VEGF secretion, suggesting the existence of a compensatory feedback loop onto PI3K-Akt signaling [135, 310, 311].
In vitro experiments demonstrated the crucial functional role of another Raf isoform: C-Raf activating retroviral transduction of BON cells resulted in high levels of phosphorylated ERK1/2 as well and caused remarkable morphologic and functional changes, such as reductions in 5-HT (5-hydroxytryptamine, the neurotransmitter serotonin), chromogranin A (marker of large dense core vesicles), and synaptophysin (marker of small synaptic vesicles) levels, which are strong neuroendocrine-related secretion markers. Accordingly, treatment of C-Raf-transducted BON cells with MEK inhibitors blocked morphological changes and hormone suppression but not ERK1/2 phosphorylation, indicating a dependency of NE dedifferentiation on MEK-mediated Raf to ERK signaling. Furthermore, activation of the C-Raf signaling cascade in BON cells resulted in significant decrease in cellular adhesion and migration in a FAK (focal adhesion kinase) dependent manner [312-314]. Although a reduction of NE marker production, especially those of chromogranin A and synaptophysin, might be generally referred to worse differentiation in GEP-NENs in vitro and in vivo [315-317], cellular adhesion and migration are a prerequisite for invasion and metastatic dissemination and thus, markers of malignant phenotypes. Paradoxically, studies with several ATP-competitive C‑Raf activators could demonstrate, that wild type Raf activation can not only suppress the expression of chromogranin A, but also the proliferation via p21cip1 upregulation in p21 wild type NEN cells [318-321]. These data conform with several publications that have proved an insensitivity of wild type B‑Raf cell lines from several cancer entities to ATP-competitive Raf inhibitors. On the other hand, the exposure to Raf inhibitors resulted in a dose-dependent and sustained paradox activation of mitogen-activated protein kinase signaling in cells and tumors with wild type B-Raf. These paradoxical effects of Raf inhibition were seen in various malignant and normal cells in vitro, xenografted and in vivo, leading to entry into the cell cycle, enhanced proliferation, and significantly stimulated tumor growth in vivo (refer to figure 2). Moreover, even in the clinical setting this paradoxical ERK-activation by the B-raf-Inhibitor Vemurafenib could be observed and led to a restriction on use of the drug [322]. The mechanism for this paradox activation is assumed to depend on an inhibitor-induced C-Raf/C-Raf or B-Raf/C-Raf homo- and heterodimerization, respectively, following sustained upstream signaling via Ras or enhanced RTK activation such as via IGF-1R or HER2 [20, 323-329].
Paradoxical activation of Raf downstream signaling following treatment with ATP-competitive Raf inhibitors lead to an enhanced proliferation of B-Raf wild type cells in vitro and in vivo (adapted from Cichowski et al. [327]): a) mutant B-Raf constitutively activates MAPK signaling in cancer cells; b) ATP-competitive inhibition of mutant B-Raf counteracts ERK activation and leads to tumor growth reduction; c) ATP-competitive wild type Raf inhibition in normal cells results in increased ERK activation and proliferation in vitro, and in hyperplasia in normal tissues of mice in vivo [323]; d) and e) sustained RTK or Ras dependent upstream signaling activates MAPK signaling under ATP-competitive wild type Raf inhibition [323, 324, 330].
Assuming that ATP-competitive wild type Raf inhibition indeed enhances paradoxical mitogen ERK downstream signaling and C-Raf activation in GEP-NENs (under the prerequisite of wild type Ras) and thus inhibits the neuroendocrine phenotype [318], the p70S6K/PI3K/RAS crosstalk under rapamycin treatment [330, 331] might emerge as an desirable side effect. This might e.g. comprise the reduced secretion of the bioactive hormones and thus alleviate the symptoms of functional active neuroendocrine tumors, but better understanding requires further mechanistic exploration.
Cross-regulation of Raf by inhibition of the PI3K-Akt axis might lead to unexpected and (under certain circumstances) harmful therapeutic outcome. Although Raf mutations are rare events in GEP-NENs tumors, subgroup-specific assessment of Raf and Akt inhibitors might improve therapeutic precision.
The MAP Kinases ERK1/2 and their downstream effectors
ERK1/2 and other MAP kinases target a vast number of effector proteins, including membrane proteins, such as phospholipase A2, cytoplasmic proteins, such as downstream kinases and cytoskeletal proteins, and nuclear proteins, such as transcription factors. In response to cytokines, stress and chemotactic factors, the MAPK p38 and ERK2 regulate transcription by phosphorylation of MAPKAP kinase-2, which induces phosphorylation of the transcriptions factors CREB (c-AMP response element binding Protein) and ATF-1 (activating transcription factor) in cells and thus regulate the transcription of a large variety of genes.
Furthermore a recent publication demonstrated that p38-ATF/CREB signal transduction pathway can coordinately induce (promote transcription and RNA stability) and repress (promote RNA decay) transcript levels for distinct sets of genes, without shutting off transcription itself. This Stress-Activated RNA decay provides a mechanism to reduce the expression of target genes as it is required for cellular decisions in response to stress and other stimuli [332].
Ras responsive genes can be transcriptionally activated by ETS/AP-1 transcription factors. Hereby AP-1 comprises c-Jun, c-Fos and ATF-2 and controls the early transcriptional response to extracellular signals [333-340]. The TCFs (ternary complex factors) are substrates of the MAPKs ERK1/2, JNK/SAPK and p38. These targets, such as Elk-1, mediate transcription of genes containing SREs (serum response elements) in their promoters [341-343].
ERK2 has been shown to phosphorylate SRC-1 (steroid receptor coactivator-1), which shows histone acetyltransferase activity and is a coactivator of steroid nuclear receptors. SRC-1 also interacts with CREB to enhance estrogen and progesterone receptor-mediated gene activation [344-346].
Not least, MAP kinase pathways are considered to phosphorylate STAT3 (signal transducers and activators of transcription) and thus stimulate cytokine production, induction of pro-angiogenetic factors and invasion in tumor cells [347-353].
MAPKs also contribute to chromatin remodeling, for instance by phosphorylating Rsk2 (ribosomal protein S6 kinase), which can subsequently phosphorylate histone H3, or by interacting with topoisomerase II. Further targets, Msk1 and 2 (mitogen and stress-activated protein kinase), are able to phosphorylate CREB and its co-factors, but also are very potent histone H3 and HMG-14 kinases [334, 354-360].
These mitogen effector functions of MAPK has put forward the development of several MAPK inhibitors in recent years (refer to Supplementary Material: suppl. 7). Few of those are under preclinical assessment for GEP-NEN therapy.
The activation of MAP Kinases in GEP-NENs is complex and due to multifunctional effects (such as differentiation and proliferation) not yet fully understood. Phosphorylated and thus activated ERK could be detected very frequently in an early immunohistochemically analysis of the MAPK pathway in GEP-NENs [263]. These data were supported by a second study where 96% of the analyzed specimens were positive for phospho-ERK1/2 and ERK activation could be related to EGFR and Akt phosphorylation, suggesting a simultaneous induction of Akt and ERK mediated pathways in NENs under EGFR kinase activity [6]. Consistently, in vitro treatment of neuroendocrine cell lines with EGFR or Raf inhibitors resulted in a time and dose-dependent dephosphorylation of ERK1/2 [309, 361].
In summary, analogous to PI3K signaling, the MAPK pathway is highly activated but the triggering mechanisms remain unclear. It is highly involved in the generation of the NE phenotype as inhibition of MAPK signaling results in impaired secretion and migration (summarized in table 4).
Summary of study results concerning MAPK signaling in GEP-NENs.
| Study results | Reference |
|---|---|
| Ras is expressed in GEP-NENs but mutations are rare | [21, 263-266] |
| Raf mutations are rare events | [263, 305-307] |
| Rap1 and B-Raf are highly expressed in GEP-NEN | [309] |
| Raf inhibition triggers feedback activation of Akt | [135, 310, 311] |
| MAPK signaling triggers NE secretion and migration in vitro | [312-314] |
| Raf activation induces a paradox inhibition of proliferation and NE secretion | [318-321] |
| ERK activation is frequent in GEP-NENs | [6, 263, 309, 361] |
PI3K and MAPK signaling are highly interwoven pathways and cooperate in therapy resistance in GEP-NENs
Although both, the PI3K and the MAPK pathway can be activated by the same RTKs, the agonists only partially overlap, since e.g. insulin, and IGF-1 are weak Ras-ERK activators, but strong PI3K-mTORC1 activators. Nevertheless the response of a certain pathway to specific growth factors depends on the factor's abundance, the receptor status in relation to expression and localization and the expression level of pathway mediators as well as of required docking and scaffold proteins [362-364].
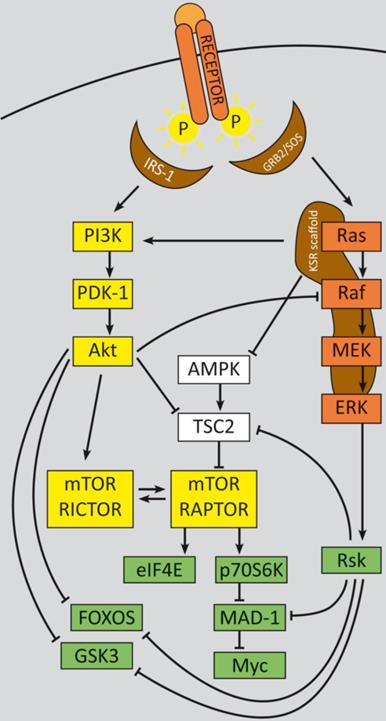
The PI3K pathway can be cross-activated upstream by direct Ras-GTP interaction with PI3 Kinases or downstream by ERK and Rsk mediated phosphorylation of TSC2 and RAPTOR. In the latter case, phosphorylation inhibits the GAP function of TSC and promotes mTORC1 activity [365-369].
Furthermore, studies have demonstrated that also the KSR (kinase suppressor of Ras) scaffold, which maintains the co-localization of RAF, MEK, and ERK during ERK activation, interacts with mTOR, RAPTOR, RICTOR, and the TSC2-activating kinases AMPK and GSK3 [366, 370-372]. Consequently both, the PI3K and the MAPK pathways frequently converge on the same downstream targets and promote cell survival, proliferation, metabolism, and motility (refer to figure 3). Prominent proteins that are regulated by both pathways are the FOXOs and the c-Myc early transcription factors, as well as BAD and GSK3. FOXOs suppress cell survival and proliferation and once phosphorylated they undergo nuclear export and ubiquitin-proteasome-mediated degradation, whereas c-Myc induces pro-survival genes [373-378].
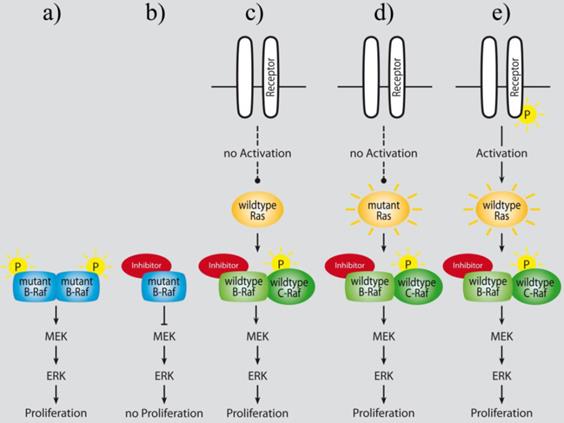
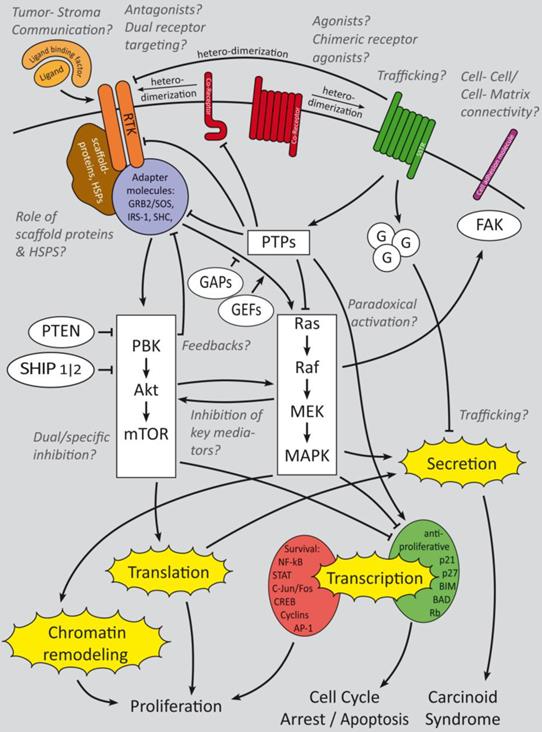
Summary of growth factor-induced cellular mechanisms and inhibitory effects of somatostatin signaling in GEP-NENs. The major receptors of interest are RTKs, SSTRs and their Co-receptors. Cell adhesion molecules are of minor research interest but might have an interesting potential. Whereas RTK activate downstream PI3K and mitogen activated signaling that triggers gene regulation and secretion, SSTRs activate G-protein coupled signaling which has a predominant regulatory effect onto growth factor signaling cascades in GEP-NENs. Beside the main questions, which crosstalks are relevant for GEP-NEN cancerogenesis and how to inhibit their key mediators, the effect of paradoxical activation has not been clinically evaluated. Trafficking, intercellular and cell-matrix interactions have been analyzed only in a small number of publications [90, 92, 94] but might have a high potential for therapeutic and prognostic issues. Ligand binding factors and receptor agonists and antagonists are of high research interest and the focus is on dual receptor targeting to develop a more subgroup specific therapy approach [27-29]. Italic: major fields of interest in current research and remaining open questions.
Furthermore, Rsk, Akt and p70S6K share the ability to induce protein translation by regulating initiation and elongation factors, such as eIF4B (eukaryotic initiation factor 4B), eEF2K (eukaryotic elongation factor 2 kinase), RPS6 (ribosomal protein S6) and eEF2 [365, 379-381].
A very important cross-inhibitory mechanism in the context of therapy resistance is the crosstalk of C‑Raf and Akt. Highly activated Akt suppresses Raf kinase activity by phosphorylation of S259. Phospho-S259 results in binding of the 14-3-3 protein and in Raf inactivation [289, 331, 382-386]. Consequently, inhibition of PI3K signaling can activate Raf as demonstrated for everolimus in GEP-NETs [204]. This mechanism is discussed to be one reason why mTOR inhibition has limited impact in GEP-NET therapy. Nevertheless the effect of paradoxical MAPK pathway activation has not been integrated in this debate to date and requires further investigation.
In summary, the PI3K and the MAPK pathways share overlapping activation patterns and converge on a number of common targets (refer to figure 4), although with varying affinities. The role of distinct posttranscriptional modification sites and multi-protein-complex assembly is analyzed in a large number of more detailed publications. Inhibiting one of those non-specific pathway mediators thus harbors the danger of activating other cascades and states the rationale for many dual inhibition approaches, unless with little success in GEP-NENs so far.
Major crosstalks between the PI3K and the MAPK cascades. The PI3K and the MAPK pathway are highly interconnected and can be activated by the same RTKs, dependent on the triggering ligand. Direct Ras-GTP interaction with PI3 Kinases can trigger Ras-activated PI3K signaling [365-369]. The KSR scaffold is also a potent regulator of key PI3K signaling mediators. Several cross-activations and -inhibitions have been assessed to date. For instance, Akt can inhibit Ras function [289, 331, 382-386] and Rsk can interfere with TSC2 [365-369]. Furthermore, both pathways converge on the same downstream targets, such as the FOXO proteins, GSK3, Myc or p70S6K [373-378], and thus promote cancer associated-phenotypes.
Conclusion
In sporadic GEP-NENs, growth factor receptor downstream signaling is ensured by large networks of triggering kinases, stabilizing scaffold proteins and regulating phosphatases, as well as by various transcription factors and co-factors. This network is highly upregulated and triggers cell growth, proliferation and secretion. Although basic cell-biological research has elucidated a vast number of mechanisms and interrelations that might explain observations that have been made in GEP-NENs, the majority of molecular information still remains to be elucidated. In contrast to cancer entities, where the oncogenic input can be “translated” in a manageable number of mutated proteins which facilitates the development of “target-directed” therapies, main sources of growth factor deregulation in sporadic GEP-NENs have not been identified to date. Genomic studies have revealed a number of chromosomal alterations but their relation to specific genes remains unclear. Subgroup specific data is rare due to the already limited number of cases, but might be important to understand differences between distinct groups of patients. Moreover, the “crosstalk” of different signaling pathways in a NEN cell may be much more complex and interactive as it is already known and the inhibition of one pathway can easily activate others by feedback loops, as described for mTOR inhibition.
In this review we have compiled several studies on the molecular basis for growth factor induced mitogen downstream-signaling in GEP-NENs. The two most promising and approved therapies in GEP-NENs are target-directed approaches, namely targeting the mTOR protein and somatostatin receptors. Recommendations for the diagnostics and therapy of gastroenteropancreatic neuroendocrine neoplasms are given by experts of the European Neuroendocrine Tumor Society in consensus conferences and are updated regularly [387]. Certainly, somatostatin analogues have been introduced as anti-proliferative agents after the positive results of the PROMID study [116] and are used in midgut tumors, whereas Everolimus and Sunitinib have their indication in metastatic pNENs [244, 388]. Everolimus might possibly also evolve as therapeutic option in midgut NENs [389] as results of RADIANT-4 study are awaited.
Nevertheless the therapy outcome depends on the abundance and activation pattern of these targets. Subgrouping data by therapy predictive biological markers, such as the phosphorylation status of Akt or the SSTR expression pattern, might improve the therapeutic precision for choosing the right therapy. Here we provide some evidence that even the most advanced therapeutic agent, everolimus, which is widely used in GEP-NENs therapy and was approved by the FDA two years ago, may still hold some drawbacks which may diminish its success.
In preclinical models Everolimus leads to a global upregulation of upstream PI3K signaling and cross-activation of Ras/Raf/Erk signaling via negative feedback loops. Therefore, recent therapeutic approaches focus on inhibitors of alternative components of the PI3K pathway, dual target inhibitors and effective combinatory bio/chemotherapies. Another example is the crosstalk between B-Raf/C-Raf and Ras/MEK/ERK-pathway. In the clinical setting, the use of a B-Raf-inhibitor in a melanoma patient led to the (paradoxical) activation and progression of a second malignancy (n-Ras mutated AML) in the same patient [322]. This effect was reversed by withdrawing the inhibitor - but it impressively demonstrated the need for further investigations on the crosstalks and the complexity of different pathways.
There is also a need for entity-specific research data concerning processes that trigger GEP-NEN-specific cellular behavior, such as intra- and intercellular trafficking, tumor-stroma interactions or the regulatory role of scaffold proteins. Although a large variety of inhibitors and molecular pathway regulator are available to date, their application is almost limited to their assessment for clinical purposes rather than to identify their mechanisms of actions. For instance, the observations that mutations in key mediators of growth factor activated pathways are rare in GEP-NENs, is an important fact for novel therapeutic strategies and, especially in the case of Raf, fundamental to define a certain outcome. Without understanding the underlying mechanism, interpretation of unexpected study results will remain unsatisfying. The importance of molecular markers and genetic data to predict therapy outcome before starting cost-intensive clinical trials has been increased dramatically due to the enormous rise of available substances in the context of growth factor signaling. Significant mechanistic knowledge, deducted from generally valid cancerogenetic contexts, needs to be analyzed for its validity in GEP-NENs and thus to furnish stronger rationales for distinct target directed and subtype specific therapeutic trials. Therefore the identification and assessment of new markers and their evolution throughout neuroendocrine cancerogenesis is a promising field of research. Detailed pathway analyses are essential to understand the limitations of the current therapies and to elucidate new possibilities of molecular imaging and targeting in this heterogeneous cancer entity.
Supplementary Material
Suppl. 1-7.
Acknowledgements
The support of the German Wilhelm und Ingeburg Dinse Gedächtnis-Stiftung and of the Sonnenfeld Stiftung Berlin is gratefully acknowledged.
The authors would like to thank Liliana H. Mochmann for critically reading the manuscript, the colleagues of the Theranostics Research Network, especially Prof. Dieter Hörsch and Dr. Daniel Kämmerer, for providing specialist advice and support, and Florentine Lewens and Helma Freitag for technical assistance and support.
Competing Interests
The group of Dr. Patricia Grabowski cooperates with Ipsen Pharma, Novartis and Pfizer Inc. in the context of preclinical trials and receives financial support from Ipsen Pharma and Novartis.
References
1. Schimmack S, Svejda B, Lawrence B, Kidd M, Modlin IM. The diversity and commonalities of gastroenteropancreatic neuroendocrine tumors. Langenbecks Arch Surg. 2011;396:273-98 doi:10.1007/s00423-011-0739-1
2. Capdevila J, Salazar R, Halperin I, Abad A, Yao JC. Innovations therapy: mammalian target of rapamycin (mTOR) inhibitors for the treatment of neuroendocrine tumors. Cancer Metastasis Rev. 2011;30(Suppl 1):27-34 doi:10.1007/s10555-011-9290-3
3. Capdevila J, Tabernero J. A shining light in the darkness for the treatment of pancreatic neuroendocrine tumors. Cancer Discov. 2011;1:213-21 doi:10.1158/2159-8290.cd-11-0151
4. Hilfenhaus G, Gohrig A, Pape UF, Neumann T, Jann H, Zdunek D. et al. Placental growth factor supports neuroendocrine tumor growth and predicts disease prognosis in patients. Endocr Relat Cancer. 2013;20:305-19 doi:10.1530/erc-12-0223
5. Oberstein PE, Saif MW. Update on prognostic and predictive biomarkers for pancreatic neuroendocrine tumors. JOP. 2012;13:368-71 doi:10.6092/1590-8577/965
6. Shah T, Hochhauser D, Frow R, Quaglia A, Dhillon AP, Caplin ME. Epidermal growth factor receptor expression and activation in neuroendocrine tumours. J Neuroendocrinol. 2006;18:355-60 doi:10.1111/j.1365-2826.2006.01425.x
7. Christofori G, Naik P, Hanahan D. Vascular endothelial growth factor and its receptors, flt-1 and flk-1, are expressed in normal pancreatic islets and throughout islet cell tumorigenesis. Mol Endocrinol. 1995;9:1760-70
8. La Rosa S, Chiaravalli AM, Capella C, Uccella S, Sessa F. Immunohistochemical localization of acidic fibroblast growth factor in normal human enterochromaffin cells and related gastrointestinal tumours. Virchows Arch. 1997;430:117-24
9. Nilsson O, Wangberg B, Kolby L, Schultz GS, Ahlman H. Expression of transforming growth factor alpha and its receptor in human neuroendocrine tumours. Int J Cancer. 1995;60:645-51
10. Chaudhry A, Oberg K, Gobl A, Heldin CH, Funa K. Expression of transforming growth factors beta 1, beta 2, beta 3 in neuroendocrine tumors of the digestive system. Anticancer Res. 1994;14:2085-91
11. Krishnamurthy S, Dayal Y. Immunohistochemical expression of transforming growth factor alpha and epidermal growth factor receptor in gastrointestinal carcinoids. Am J Surg Pathol. 1997;21:327-33
12. Chaudhry A, Papanicolaou V, Oberg K, Heldin CH, Funa K. Expression of platelet-derived growth factor and its receptors in neuroendocrine tumors of the digestive system. Cancer Res. 1992;52:1006-12
13. Van Gompel JJ, Chen H. Insulin-like growth factor 1 signaling in human gastrointestinal carcinoid tumor cells. Surgery. 2004;136:1297-302 doi:10.1016/j.surg.2004.06.061
14. Hopfner M, Baradari V, Huether A, Schofl C, Scherubl H. The insulin-like growth factor receptor 1 is a promising target for novel treatment approaches in neuroendocrine gastrointestinal tumours. Endocr Relat Cancer. 2006;13:135-49 doi:10.1677/erc.1.01090
15. La Rosa S, Uccella S, Finzi G, Albarello L, Sessa F, Capella C. Localization of vascular endothelial growth factor and its receptors in digestive endocrine tumors: correlation with microvessel density and clinicopathologic features. Hum Pathol. 2003;34:18-27 doi:10.1053/hupa.2003.56
16. Gross DJ, Munter G, Bitan M, Siegal T, Gabizon A, Weitzen R. et al. The role of imatinib mesylate (Glivec) for treatment of patients with malignant endocrine tumors positive for c-kit or PDGF-R. Endocr Relat Cancer. 2006;13:535-40 doi:10.1677/erc.1.01124
17. Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28:20-47 doi:10.1210/er.2006-0001
18. Furukawa M, Raffeld M, Mateo C, Sakamoto A, Moody TW, Ito T. et al. Increased expression of insulin-like growth factor I and/or its receptor in gastrinomas is associated with low curability, increased growth, and development of metastases. Clin Cancer Res. 2005;11:3233-42 doi:10.1158/1078-0432.ccr-04-1915
19. Vitale L, Lenzi L, Huntsman SA, Canaider S, Frabetti F, Casadei R. et al. Differential expression of alternatively spliced mRNA forms of the insulin-like growth factor 1 receptor in human neuroendocrine tumors. Oncol Rep. 2006;15:1249-56
20. von Wichert G, Jehle PM, Hoeflich A, Koschnick S, Dralle H, Wolf E. et al. Insulin-like growth factor-I is an autocrine regulator of chromogranin A secretion and growth in human neuroendocrine tumor cells. Cancer Res. 2000;60:4573-81
21. Gilbert JA, Adhikari LJ, Lloyd RV, Halfdanarson TR, Muders MH, Ames MM. Molecular markers for novel therapeutic strategies in pancreatic endocrine tumors. Pancreas. 2013;42:411-21 doi:10.1097/MPA.0b013e31826cb243
22. Wulbrand U, Wied M, Zofel P, Goke B, Arnold R, Fehmann H. Growth factor receptor expression in human gastroenteropancreatic neuroendocrine tumours. Eur J Clin Invest. 1998;28:1038-49
23. Nilsson O, Wangberg B, McRae A, Dahlstrom A, Ahlman H. Growth factors and carcinoid tumours. Acta Oncol. 1993;32:115-24
24. Nilsson O, Wangberg B, Theodorsson E, Skottner A, Ahlman H. Presence of IGF-I in human midgut carcinoid tumours--an autocrine regulator of carcinoid tumour growth? Int J Cancer. 1992;51:195-203
25. Gloesenkamp C, Nitzsche B, Lim AR, Normant E, Vosburgh E, Schrader M. et al. Heat shock protein 90 is a promising target for effective growth inhibition of gastrointestinal neuroendocrine tumors. Int J Oncol. 2012;40:1659-67 doi:10.3892/ijo.2012.1328
26. Richardson PG, Mitsiades CS, Laubach JP, Lonial S, Chanan-Khan AA, Anderson KC. Inhibition of heat shock protein 90 (HSP90) as a therapeutic strategy for the treatment of myeloma and other cancers. Br J Haematol. 2011;152:367-79 doi:10.1111/j.1365-2141.2010.08360.x
27. Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16:3-34
28. Hansel DE, Rahman A, House M, Ashfaq R, Berg K, Yeo CJ. et al. Met proto-oncogene and insulin-like growth factor binding protein 3 overexpression correlates with metastatic ability in well-differentiated pancreatic endocrine neoplasms. Clin Cancer Res. 2004;10:6152-8 doi:10.1158/1078-0432.ccr-04-0285
29. Maitra A, Hansel DE, Argani P, Ashfaq R, Rahman A, Naji A. et al. Global expression analysis of well-differentiated pancreatic endocrine neoplasms using oligonucleotide microarrays. Clin Cancer Res. 2003;9:5988-95
30. Papouchado B, Erickson LA, Rohlinger AL, Hobday TJ, Erlichman C, Ames MM. et al. Epidermal growth factor receptor and activated epidermal growth factor receptor expression in gastrointestinal carcinoids and pancreatic endocrine carcinomas. Mod Pathol. 2005;18:1329-35 doi:10.1038/modpathol.3800427
31. Srivastava A, Alexander J, Lomakin I, Dayal Y. Immunohistochemical expression of transforming growth factor alpha and epidermal growth factor receptor in pancreatic endocrine tumors. Hum Pathol. 2001;32:1184-9 doi:10.1053/hupa.2001.28959
32. Di Florio A, Sancho V, Moreno P, Delle Fave G, Jensen RT. Gastrointestinal hormones stimulate growth of Foregut Neuroendocrine Tumors by transactivating the EGF receptor. Biochim Biophys Acta. 2013;1833:573-82 doi:10.1016/j.bbamcr.2012.11.021
33. Missiaglia E, Dalai I, Barbi S, Beghelli S, Falconi M, della Peruta M. et al. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol. 2010;28:245-55 doi:10.1200/jco.2008.21.5988
34. Wu QF, Yang L, Li S, Wang Q, Yuan XB, Gao X. et al. Fibroblast growth factor 13 is a microtubule-stabilizing protein regulating neuronal polarization and migration. Cell. 2012;149:1549-64 doi:10.1016/j.cell.2012.04.046
35. Ono K, Suzuki T, Miki Y, Taniyama Y, Nakamura Y, Noda Y. et al. Somatostatin receptor subtypes in human non-functioning neuroendocrine tumors and effects of somatostatin analogue SOM230 on cell proliferation in cell line NCI-H727. Anticancer Res. 2007;27:2231-9
36. Taniyama Y, Suzuki T, Mikami Y, Moriya T, Satomi S, Sasano H. Systemic distribution of somatostatin receptor subtypes in human: an immunohistochemical study. Endocr J. 2005;52:605-11
37. Pawlikowski M, Pisarek H, Winczyk K. Immunohistochemical detection of dopamine D2 receptors in neuroendocrine tumours. Endokrynol Pol. 2011;62:388-91
38. Diakatou E, Kaltsas G, Tzivras M, Kanakis G, Papaliodi E, Kontogeorgos G. Somatostatin and dopamine receptor profile of gastroenteropancreatic neuroendocrine tumors: an immunohistochemical study. Endocr Pathol. 2011;22:24-30 doi:10.1007/s12022-011-9149-8
39. Patel YC. Somatostatin and its receptor family. Front Neuroendocrinol. 1999;20:157-98 doi:10.1006/frne.1999.0183
40. Sellers LA, Alderton F, Carruthers AM, Schindler M, Humphrey PP. Receptor isoforms mediate opposing proliferative effects through gbetagamma-activated p38 or Akt pathways. Mol Cell Biol. 2000;20:5974-85
41. Rosskopf D, Schurks M, Manthey I, Joisten M, Busch S, Siffert W. Signal transduction of somatostatin in human B lymphoblasts. Am J Physiol Cell Physiol. 2003;284:C179-90 doi:10.1152/ajpcell.00160.2001
42. Meyerhof W. The elucidation of somatostatin receptor functions: a current view. Rev Physiol Biochem Pharmacol. 1998;133:55-108
43. Bruns C, Lewis I, Briner U, Meno-Tetang G, Weckbecker G. SOM230: a novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur J Endocrinol. 2002;146:707-16
44. Appetecchia M, Baldelli R. Somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine tumours, current aspects and new perspectives. J Exp Clin Cancer Res. 2010;29:19. doi:10.1186/1756-9966-29-19
45. Weckbecker G, Lewis I, Albert R, Schmid HA, Hoyer D, Bruns C. Opportunities in somatostatin research: biological, chemical and therapeutic aspects. Nat Rev Drug Discov. 2003;2:999-1017 doi:10.1038/nrd1255
46. Moller LN, Stidsen CE, Hartmann B, Holst JJ. Somatostatin receptors. Biochim Biophys Acta. 2003;1616:1-84
47. Florio T. Somatostatin/somatostatin receptor signalling: phosphotyrosine phosphatases. Mol Cell Endocrinol. 2008;286:40-8 doi:10.1016/j.mce.2007.08.012
48. Reichlin S. Somatostatin. N Engl J Med. 1983;309:1495-501 doi:10.1056/nejm198312153092406
49. Scherubl H, Hescheler J, Riecken EO. Molecular mechanisms of somatostatin's inhibition of hormone release: participation of voltage-gated calcium channels and G-proteins. Horm Metab Res Suppl. 1993;27:1-4
50. Zapata PD, Ropero RM, Valencia AM, Buscail L, Lopez JI, Martin-Orozco RM. et al. Autocrine regulation of human prostate carcinoma cell proliferation by somatostatin through the modulation of the SH2 domain containing protein tyrosine phosphatase (SHP)-1. J Clin Endocrinol Metab. 2002;87:915-26
51. Thangaraju M, Sharma K, Liu D, Shen SH, Srikant CB. Interdependent regulation of intracellular acidification and SHP-1 in apoptosis. Cancer Res. 1999;59:1649-54
52. Florio T, Morini M, Villa V, Arena S, Corsaro A, Thellung S. et al. Somatostatin inhibits tumor angiogenesis and growth via somatostatin receptor-3-mediated regulation of endothelial nitric oxide synthase and mitogen-activated protein kinase activities. Endocrinology. 2003;144:1574-84
53. Florio T, Thellung S, Arena S, Corsaro A, Bajetto A, Schettini G. et al. Somatostatin receptor 1 (SSTR1)-mediated inhibition of cell proliferation correlates with the activation of the MAP kinase cascade: role of the phosphotyrosine phosphatase SHP-2. J Physiol Paris. 2000;94:239-50
54. Held-Feindt J, Forstreuter F, Pufe T, Mentlein R. Influence of the somatostatin receptor sst2 on growth factor signal cascades in human glioma cells. Brain Res Mol Brain Res. 2001;87:12-21
55. Dent P, Wang Y, Gu YZ, Wood SL, Reardon DB, Mangues R. et al. S49 cells endogenously express subtype 2 somatostatin receptors which couple to increase protein tyrosine phosphatase activity in membranes and down-regulate Raf-1 activity in situ. Cell Signal. 1997;9:539-49
56. Reardon DB, Dent P, Wood SL, Kong T, Sturgill TW. Activation in vitro of somatostatin receptor subtypes 2, 3, or 4 stimulates protein tyrosine phosphatase activity in membranes from transfected Ras-transformed NIH 3T3 cells: coexpression with catalytically inactive SHP-2 blocks responsiveness. Mol Endocrinol. 1997;11:1062-9
57. Theodoropoulou M, Zhang J, Laupheimer S, Paez-Pereda M, Erneux C, Florio T. et al. Octreotide, a somatostatin analogue, mediates its antiproliferative action in pituitary tumor cells by altering phosphatidylinositol 3-kinase signaling and inducing Zac1 expression. Cancer Res. 2006;66:1576-82 doi:10.1158/0008-5472.can-05-1189
58. Bousquet C, Delesque N, Lopez F, Saint-Laurent N, Esteve JP, Bedecs K. et al. sst2 somatostatin receptor mediates negative regulation of insulin receptor signaling through the tyrosine phosphatase SHP-1. J Biol Chem. 1998;273:7099-106
59. Bousquet C, Guillermet-Guibert J, Saint-Laurent N, Archer-Lahlou E, Lopez F, Fanjul M. et al. Direct binding of p85 to sst2 somatostatin receptor reveals a novel mechanism for inhibiting PI3K pathway. EMBO J. 2006;25:3943-54 doi:10.1038/sj.emboj.7601279
60. Pages P, Benali N, Saint-Laurent N, Esteve JP, Schally AV, Tkaczuk J. et al. sst2 somatostatin receptor mediates cell cycle arrest and induction of p27(Kip1). Evidence for the role of SHP-1. J Biol Chem. 1999;274:15186-93
61. Sharma K, Srikant CB. Induction of wild-type p53, Bax, and acidic endonuclease during somatostatin-signaled apoptosis in MCF-7 human breast cancer cells. Int J Cancer. 1998;76:259-66
62. Ostman A, Hellberg C, Bohmer FD. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer. 2006;6:307-20 doi:10.1038/nrc1837
63. Iuliano R, Trapasso F, Le Pera I, Schepis F, Sama I, Clodomiro A. et al. An adenovirus carrying the rat protein tyrosine phosphatase eta suppresses the growth of human thyroid carcinoma cell lines in vitro and in vivo. Cancer Res. 2003;63:882-6
64. Keane MM, Lowrey GA, Ettenberg SA, Dayton MA, Lipkowitz S. The protein tyrosine phosphatase DEP-1 is induced during differentiation and inhibits growth of breast cancer cells. Cancer Res. 1996;56:4236-43
65. Pera IL, Iuliano R, Florio T, Susini C, Trapasso F, Santoro M. et al. The rat tyrosine phosphatase eta increases cell adhesion by activating c-Src through dephosphorylation of its inhibitory phosphotyrosine residue. Oncogene. 2005;24:3187-95 doi:10.1038/sj.onc.1208510
66. Martelli ML, Trapasso F, Bruni P, Berlingieri MT, Battaglia C, Vento MT. et al. Protein tyrosine phosphatase-eta expression is upregulated by the PKA-dependent and is downregulated by the PKC-dependent pathways in thyroid cells. Exp Cell Res. 1998;245:195-202 doi:10.1006/excr.1998.4257
67. Arena S, Pattarozzi A, Massa A, Esteve JP, Iuliano R, Fusco A. et al. An intracellular multi-effector complex mediates somatostatin receptor 1 activation of phospho-tyrosine phosphatase eta. Mol Endocrinol. 2007;21:229-46 doi:10.1210/me.2006-0081
68. Ferjoux G, Lopez F, Esteve JP, Ferrand A, Vivier E, Vely F. et al. Critical role of Src and SHP-2 in sst2 somatostatin receptor-mediated activation of SHP-1 and inhibition of cell proliferation. Mol Biol Cell. 2003;14:3911-28 doi:10.1091/mbc.E03-02-0069
69. Kaemmerer D, Peter L, Lupp A, Schulz S, Sanger J, Baum RP. et al. Comparing of IRS and Her2 as immunohistochemical scoring schemes in gastroenteropancreatic neuroendocrine tumors. Int J Clin Exp Pathol. 2012;5:187-94
70. Reubi JC, Waser B. Concomitant expression of several peptide receptors in neuroendocrine tumours: molecular basis for in vivo multireceptor tumour targeting. Eur J Nucl Med Mol Imaging. 2003;30:781-93 doi:10.1007/s00259-003-1184-3
71. Reubi JC. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr Rev. 2003;24:389-427
72. Reubi JC. Somatostatin and other Peptide receptors as tools for tumor diagnosis and treatment. Neuroendocrinology. 2004;80(Suppl 1):51-6 doi:10.1159/000080742
73. Papotti M, Bongiovanni M, Volante M, Allia E, Landolfi S, Helboe L. et al. Expression of somatostatin receptor types 1-5 in 81 cases of gastrointestinal and pancreatic endocrine tumors. A correlative immunohistochemical and reverse-transcriptase polymerase chain reaction analysis. Virchows Arch. 2002;440:461-75 doi:10.1007/s00428-002-0609-x
74. Schmid HA, Lambertini C, van Vugt HH, Barzaghi-Rinaudo P, Schafer J, Hillenbrand R. et al. Monoclonal antibodies against the human somatostatin receptor subtypes 1-5: development and immunohistochemical application in neuroendocrine tumors. Neuroendocrinology. 2012;95:232-47 doi:10.1159/000330616
75. Corleto VD, Falconi M, Panzuto F, Milione M, De Luca O, Perri P. et al. Somatostatin receptor subtypes 2 and 5 are associated with better survival in well-differentiated endocrine carcinomas. Neuroendocrinology. 2009;89:223-30 doi:10.1159/000167796
76. Kim HS, Lee HS, Kim WH. Clinical significance of protein expression of cyclooxygenase-2 and somatostatin receptors in gastroenteropancreatic neuroendocrine tumors. Cancer Res Treat. 2011;43:181-8 doi:10.4143/crt.2011.43.3.181
77. Srirajaskanthan R, Watkins J, Marelli L, Khan K, Caplin ME. Expression of somatostatin and dopamine 2 receptors in neuroendocrine tumours and the potential role for new biotherapies. Neuroendocrinology. 2009;89:308-14 doi:10.1159/000179899
78. Zamora V, Cabanne A, Salanova R, Bestani C, Domenichini E, Marmissolle F. et al. Immunohistochemical expression of somatostatin receptors in digestive endocrine tumours. Dig Liver Dis. 2010;42:220-5 doi:10.1016/j.dld.2009.07.018
79. Reubi JC, Kvols LK, Waser B, Nagorney DM, Heitz PU, Charboneau JW. et al. Detection of somatostatin receptors in surgical and percutaneous needle biopsy samples of carcinoids and islet cell carcinomas. Cancer Res. 1990;50:5969-77
80. Oconnor JM, Belli S, Pesce V, Bestani C, Dominichini E, Mendez G. et al. Somatostatin receptor (sstr) expression and proliferative index (ki 67) in 100 patients (pts) with gastroenteropancreatic neuroendocrine tumors (gep-nets). Clinical-pathological correlation. 37th ESMO Conference. Vienna. 2012
81. O'Toole D, Saveanu A, Couvelard A, Gunz G, Enjalbert A, Jaquet P. et al. The analysis of quantitative expression of somatostatin and dopamine receptors in gastro-entero-pancreatic tumours opens new therapeutic strategies. Eur J Endocrinol. 2006;155:849-57 doi:10.1530/eje.1.02307
82. Rocheville M, Lange DC, Kumar U, Patel SC, Patel RC, Patel YC. Receptors for dopamine and somatostatin: formation of hetero-oligomers with enhanced functional activity. Science. 2000;288:154-7
83. Kulaksiz H, Eissele R, Rossler D, Schulz S, Hollt V, Cetin Y. et al. Identification of somatostatin receptor subtypes 1, 2A, 3, and 5 in neuroendocrine tumours with subtype specific antibodies. Gut. 2002;50:52-60
84. Imam H, Eriksson B, Lukinius A, Janson ET, Lindgren PG, Wilander E. et al. Induction of apoptosis in neuroendocrine tumors of the digestive system during treatment with somatostatin analogs. Acta Oncol. 1997;36:607-14
85. Kaemmerer D, Lupp A, Peter L, Fischer E, Schulz S, Kloppel G. et al. Correlation of monoclonal and polyclonal somatostatin receptor 5 antibodies in pancreatic neuroendocrine tumors. Int J Clin Exp Pathol. 2013;6:49-54
86. Lupp A, Hunder A, Petrich A, Nagel F, Doll C, Schulz S. Reassessment of sst(5) somatostatin receptor expression in normal and neoplastic human tissues using the novel rabbit monoclonal antibody UMB-4. Neuroendocrinology. 2011;94:255-64 doi:10.1159/000329876
87. Lupp A, Nagel F, Doll C, Rocken C, Evert M, Mawrin C. et al. Reassessment of sst3 somatostatin receptor expression in human normal and neoplastic tissues using the novel rabbit monoclonal antibody UMB-5. Neuroendocrinology. 2012;96:301-10 doi:10.1159/000337659
88. Fischer T, Doll C, Jacobs S, Kolodziej A, Stumm R, Schulz S. Reassessment of sst2 somatostatin receptor expression in human normal and neoplastic tissues using the novel rabbit monoclonal antibody UMB-1. J Clin Endocrinol Metab. 2008;93:4519-24 doi:10.1210/jc.2008-1063
89. Kao YJ, Ghosh M, Schonbrunn A. Ligand-dependent mechanisms of sst2A receptor trafficking: role of site-specific phosphorylation and receptor activation in the actions of biased somatostatin agonists. Mol Endocrinol. 2011;25:1040-54 doi:10.1210/me.2010-0398
90. Petrich A, Mann A, Kliewer A, Nagel F, Strigli A, Martens JC. et al. Phosphorylation of threonine 333 regulates trafficking of the human sst5 somatostatin receptor. Mol Endocrinol. 2013;27:671-82 doi:10.1210/me.2012-1329
91. Janson ET, Westlin JE, Ohrvall U, Oberg K, Lukinius A. Nuclear localization of 111In after intravenous injection of [111In-DTPA-D-Phe1]-octreotide in patients with neuroendocrine tumors. J Nucl Med. 2000;41:1514-8
92. Jacobs S, Schulz S. Intracellular trafficking of somatostatin receptors. Mol Cell Endocrinol. 2008;286:58-62 doi:10.1016/j.mce.2007.10.005
93. Poll F, Lehmann D, Illing S, Ginj M, Jacobs S, Lupp A. et al. Pasireotide and octreotide stimulate distinct patterns of sst2A somatostatin receptor phosphorylation. Mol Endocrinol. 2010;24:436-46 doi:10.1210/me.2009-0315
94. Csaba Z, Peineau S, Dournaud P. Molecular mechanisms of somatostatin receptor trafficking. J Mol Endocrinol. 2012;48:R1-12 doi:10.1530/jme-11-0121
95. Barbieri F, Bajetto A, Pattarozzi A, Gatti M, Wurth R, Thellung S. et al. Peptide receptor targeting in cancer: the somatostatin paradigm. Int J Pept. 2013;2013:926295. doi:10.1155/2013/926295
96. Bodei L, Cremonesi M, Grana CM, Chinol M, Baio SM, Severi S. et al. Yttrium-labelled peptides for therapy of NET. Eur J Nucl Med Mol Imaging. 2012;39(Suppl 1):S93-102 doi:10.1007/s00259-011-2002-y
97. Bodei L, Ferone D, Grana CM, Cremonesi M, Signore A, Dierckx RA. et al. Peptide receptor therapies in neuroendocrine tumors. J Endocrinol Invest. 2009;32:360-9
98. Giovacchini G, Nicolas G, Forrer F. Peptide receptor radionuclide therapy with somatostatin analogues in neuroendocrine tumors. Anticancer Agents Med Chem. 2012;12:526-42
99. Kam BL, Teunissen JJ, Krenning EP, de Herder WW, Khan S, van Vliet EI. et al. Lutetium-labelled peptides for therapy of neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2012;39(Suppl 1):S103-12 doi:10.1007/s00259-011-2039-y
100. Kwekkeboom DJ, de Herder WW, Krenning EP. Somatostatin receptor-targeted radionuclide therapy in patients with gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin North Am. 2011;40:173-85 ix. doi:10.1016/j.ecl.2010.12.003
101. Horsch D, Bert T, Schrader J, Hommann M, Kaemmerer D, Petrovitch A. et al. Pancreatic neuroendocrine neoplasms. Minerva Gastroenterol Dietol. 2012;58:401-26
102. Horsch D, Ezziddin S, Haug A, Gratz KF, Dunkelmann S, Krause BJ. et al. Peptide receptor radionuclide therapy for neuroendocrine tumors in Germany: first results of a multi-institutional cancer registry. Recent Results Cancer Res. 2013;194:457-65 doi:10.1007/978-3-642-27994-2_25
103. Horsch D, Grabowski P, Schneider CP, Petrovitch A, Kaemmerer D, Hommann M. et al. Current treatment options for neuroendocrine tumors. Drugs Today (Barc). 2011;47:773-86 doi:10.1358/dot.2011.47.10.1673555
104. Sundin A. Radiological and nuclear medicine imaging of gastroenteropancreatic neuroendocrine tumours. Best Pract Res Clin Gastroenterol. 2012;26:803-18 doi:10.1016/j.bpg.2012.12.004
105. Modlin IM, Pavel M, Kidd M, Gustafsson BI. Review article: somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine (carcinoid) tumours. Aliment Pharmacol Ther. 2010;31:169-88 doi:10.1111/j.1365-2036.2009.04174.x
106. Culler MD, Oberg K, Arnold R, Krenning EP, Sevilla I, Diaz JA. Somatostatin analogs for the treatment of neuroendocrine tumors. Cancer Metastasis Rev. 2011;30(Suppl 1):9-17 doi:10.1007/s10555-011-9293-0
107. Arnold R, Benning R, Neuhaus C, Rolwage M, Trautmann ME. Gastroenteropancreatic endocrine tumors: effect of Sandostatin on tumor growth. The German Sandostatin Study Group. Metabolism. 1992;41:116-8
108. Arnold R, Trautmann ME, Creutzfeldt W, Benning R, Benning M, Neuhaus C. et al. Somatostatin analogue octreotide and inhibition of tumour growth in metastatic endocrine gastroenteropancreatic tumours. Gut. 1996;38:430-8
109. Boden G, Ryan IG, Eisenschmid BL, Shelmet JJ, Owen OE. Treatment of inoperable glucagonoma with the long-acting somatostatin analogue SMS 201-995. N Engl J Med. 1986;314:1686-9 doi:10.1056/nejm198606263142606
110. di Bartolomeo M, Bajetta E, Buzzoni R, Mariani L, Carnaghi C, Somma L. et al. Clinical efficacy of octreotide in the treatment of metastatic neuroendocrine tumors. A study by the Italian Trials in Medical Oncology Group. Cancer. 1996;77:402-8 doi:10.1002/(sici)1097-0142(19960115)77:2<402::aid-cncr25>3.0.co;2-4
111. Kvols LK, Moertel CG, O'Connell MJ, Schutt AJ, Rubin J, Hahn RG. Treatment of the malignant carcinoid syndrome. Evaluation of a long-acting somatostatin analogue. N Engl J Med. 1986;315:663-6 doi:10.1056/nejm198609113151102
112. Saltz L, Trochanowski B, Buckley M, Heffernan B, Niedzwiecki D, Tao Y. et al. Octreotide as an antineoplastic agent in the treatment of functional and nonfunctional neuroendocrine tumors. Cancer. 1993;72:244-8
113. Strosberg J, Gardner N, Kvols L. Survival and prognostic factor analysis of 146 metastatic neuroendocrine tumors of the mid-gut. Neuroendocrinology. 2009;89:471-6 doi:10.1159/000197899
114. Yao JC, Phan AT, Chang DZ, Wolff RA, Hess K, Gupta S. et al. Efficacy of RAD001 (everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: results of a phase II study. J Clin Oncol. 2008;26:4311-8 doi:10.1200/jco.2008.16.7858
115. Jann H, Denecke T, Koch M, Pape UF, Wiedenmann B, Pavel M. Impact of Octreotide LAR on Tumour Growth Control as First-Line Treatment in Neuroendocrine Tumours of Pancreatic Origin. Neuroendocrinology. 2013 doi:10.1159/000353785
116. Rinke A, Muller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M. et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27:4656-63 doi:10.1200/jco.2009.22.8510
117. Plockinger U, Wiedenmann B. Treatment of gastroenteropancreatic neuroendocrine tumors. Virchows Arch. 2007;451(Suppl 1):S71-80 doi:10.1007/s00428-007-0446-z
118. Wolin EM, Hu K, Hughes G, Bouillaud E, Giannone V, Resendiz KH. Safety, tolerability, pharmacokinetics, and pharmacodynamics of a long-acting release (LAR) formulation of pasireotide (SOM230) in patients with gastroenteropancreatic neuroendocrine tumors: results from a randomized, multicenter, open-label, phase I study. Cancer Chemother Pharmacol. 2013 doi:10.1007/s00280-013-2202-1
119. Kvols LK, Oberg KE, O'Dorisio TM, Mohideen P, de Herder WW, Arnold R. et al. Pasireotide (SOM230) shows efficacy and tolerability in the treatment of patients with advanced neuroendocrine tumors refractory or resistant to octreotide LAR: results from a phase II study. Endocr Relat Cancer. 2012;19:657-66 doi:10.1530/erc-11-0367
120. Jaquet P, Gunz G, Saveanu A, Barlier A, Dufour H, Taylor J. et al. BIM-23A760, a chimeric molecule directed towards somatostatin and dopamine receptors, vs universal somatostatin receptors ligands in GH-secreting pituitary adenomas partial responders to octreotide. J Endocrinol Invest. 2005;28:21-7
121. Jaquet P, Gunz G, Saveanu A, Dufour H, Taylor J, Dong J. et al. Efficacy of chimeric molecules directed towards multiple somatostatin and dopamine receptors on inhibition of GH and prolactin secretion from GH-secreting pituitary adenomas classified as partially responsive to somatostatin analog therapy. Eur J Endocrinol. 2005;153:135-41 doi:10.1530/eje.1.01950
122. Zitzmann K, Andersen S, Vlotides G, Spottl G, Zhang S, Datta R. et al. The Novel SSTR2/D2R Chimeric Compound BIM-23A758 Decreases the Viability of Human GOT1 Midgut Carcinoid Cells. Neuroendocrinology. 2013 doi:10.1159/000353784
123. Toumpanakis C, Caplin ME. Update on the role of somatostatin analogs for the treatment of patients with gastroenteropancreatic neuroendocrine tumors. Semin Oncol. 2013;40:56-68 doi:10.1053/j.seminoncol.2012.11.006
124. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606-19 doi:10.1038/nrg1879
125. Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655-7 doi:10.1126/science.296.5573.1655
126. Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci. 2001;114:2903-10
127. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB. et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261-9
128. Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A. et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199-203 doi:10.1126/science.1200609
129. Reidy-Lagunes DL, Vakiani E, Segal MF, Hollywood EM, Tang LH, Solit DB. et al. A phase 2 study of the insulin-like growth factor-1 receptor inhibitor MK-0646 in patients with metastatic, well-differentiated neuroendocrine tumors. Cancer. 2012;118:4795-800 doi:10.1002/cncr.27459
130. Couderc C, Poncet G, Villaume K, Blanc M, Gadot N, Walter T. et al. Targeting the PI3K/mTOR pathway in murine endocrine cell lines: in vitro and in vivo effects on tumor cell growth. Am J Pathol. 2011;178:336-44 doi:10.1016/j.ajpath.2010.11.023
131. Pitt SC, Chen H, Kunnimalaiyaan M. Inhibition of phosphatidylinositol 3-kinase/Akt signaling suppresses tumor cell proliferation and neuroendocrine marker expression in GI carcinoid tumors. Ann Surg Oncol. 2009;16:2936-42 doi:10.1245/s10434-009-0591-5
132. Villaume K, Blanc M, Gouysse G, Walter T, Couderc C, Nejjari M. et al. VEGF secretion by neuroendocrine tumor cells is inhibited by octreotide and by inhibitors of the PI3K/AKT/mTOR pathway. Neuroendocrinology. 2010;91:268-78 doi:10.1159/000289569
133. Pfragner R, Behmel A, Hoger H, Beham A, Ingolic E, Stelzer I. et al. Establishment and characterization of three novel cell lines - P-STS, L-STS, H-STS - derived from a human metastatic midgut carcinoid. Anticancer Res. 2009;29:1951-61
134. Svejda B, Kidd M, Kazberouk A, Lawrence B, Pfragner R, Modlin IM. Limitations in small intestinal neuroendocrine tumor therapy by mTor kinase inhibition reflect growth factor-mediated PI3K feedback loop activation via ERK1/2 and AKT. Cancer. 2011;117:4141-54 doi:10.1002/cncr.26011
135. Zitzmann K, Ruden J, Brand S, Goke B, Lichtl J, Spottl G. et al. Compensatory activation of Akt in response to mTOR and Raf inhibitors - a rationale for dual-targeted therapy approaches in neuroendocrine tumor disease. Cancer Lett. 2010;295:100-9 doi:10.1016/j.canlet.2010.02.018
136. von Wichert G, Haeussler U, Greten FR, Kliche S, Dralle H, Bohm BO. et al. Regulation of cyclin D1 expression by autocrine IGF-I in human BON neuroendocrine tumour cells. Oncogene. 2005;24:1284-9 doi:10.1038/sj.onc.1208264
137. Li J, Song J, Cassidy MG, Rychahou P, Starr ME, Liu J. et al. PI3K p110alpha/Akt signaling negatively regulates secretion of the intestinal peptide neurotensin through interference of granule transport. Mol Endocrinol. 2012;26:1380-93 doi:10.1210/me.2012-1024
138. Clement S, Krause U, Desmedt F, Tanti JF, Behrends J, Pesesse X. et al. The lipid phosphatase SHIP2 controls insulin sensitivity. Nature. 2001;409:92-7 doi:10.1038/35051094
139. Hafsi S, Pezzino FM, Candido S, Ligresti G, Spandidos DA, Soua Z. et al. Gene alterations in the PI3K/PTEN/AKT pathway as a mechanism of drug-resistance (review). Int J Oncol. 2012;40:639-44 doi:10.3892/ijo.2011.1312
140. Maehama T, Dixon JE. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. 1999;9:125-8
141. Zhang S, Yu D. PI(3)king apart PTEN's role in cancer. Clin Cancer Res. 2010;16:4325-30 doi:10.1158/1078-0432.ccr-09-2990
142. Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM. et al. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci U S A. 1999;96:2110-5
143. Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA. et al. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A. 1998;95:13513-8
144. Furnari FB, Huang HJ, Cavenee WK. The phosphoinositol phosphatase activity of PTEN mediates a serum-sensitive G1 growth arrest in glioma cells. Cancer Res. 1998;58:5002-8
145. Leslie NR, Foti M. Non-genomic loss of PTEN function in cancer: not in my genes. Trends Pharmacol Sci. 2011;32:131-40 doi:10.1016/j.tips.2010.12.005
146. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627-44 doi:10.1038/nrd2926
147. Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403-14 doi:10.1016/j.cell.2008.04.013
148. Song MS, Carracedo A, Salmena L, Song SJ, Egia A, Malumbres M. et al. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell. 2011;144:187-99 doi:10.1016/j.cell.2010.12.020
149. Lachyankar MB, Sultana N, Schonhoff CM, Mitra P, Poluha W, Lambert S. et al. A role for nuclear PTEN in neuronal differentiation. J Neurosci. 2000;20:1404-13
150. Ginn-Pease ME, Eng C. Increased nuclear phosphatase and tensin homologue deleted on chromosome 10 is associated with G0-G1 in MCF-7 cells. Cancer Res. 2003;63:282-6
151. Gil A, Andres-Pons A, Fernandez E, Valiente M, Torres J, Cervera J. et al. Nuclear localization of PTEN by a Ran-dependent mechanism enhances apoptosis: Involvement of an N-terminal nuclear localization domain and multiple nuclear exclusion motifs. Mol Biol Cell. 2006;17:4002-13 doi:10.1091/mbc.E06-05-0380
152. Chang CJ, Mulholland DJ, Valamehr B, Mosessian S, Sellers WR, Wu H. PTEN nuclear localization is regulated by oxidative stress and mediates p53-dependent tumor suppression. Mol Cell Biol. 2008;28:3281-9 doi:10.1128/mcb.00310-08
153. Gimm O, Attie-Bitach T, Lees JA, Vekemans M, Eng C. Expression of the PTEN tumour suppressor protein during human development. Hum Mol Genet. 2000;9:1633-9
154. Perren A, Komminoth P, Saremaslani P, Matter C, Feurer S, Lees JA. et al. Mutation and expression analyses reveal differential subcellular compartmentalization of PTEN in endocrine pancreatic tumors compared to normal islet cells. Am J Pathol. 2000;157:1097-103 doi:10.1016/s0002-9440(10)64624-x
155. Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H. et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128:141-56 doi:10.1016/j.cell.2006.11.040
156. Tamura M, Gu J, Takino T, Yamada KM. Tumor suppressor PTEN inhibition of cell invasion, migration, and growth: differential involvement of focal adhesion kinase and p130Cas. Cancer Res. 1999;59:442-9
157. Kim JS, Xu X, Li H, Solomon D, Lane WS, Jin T. et al. Mechanistic analysis of a DNA damage-induced, PTEN-dependent size checkpoint in human cells. Mol Cell Biol. 2011;31:2756-71 doi:10.1128/mcb.01323-10
158. Putz U, Howitt J, Doan A, Goh CP, Low LH, Silke J. et al. The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci Signal. 2012;5:ra70. doi:10.1126/scisignal.2003084
159. Speel EJ, Richter J, Moch H, Egenter C, Saremaslani P, Rutimann K. et al. Genetic differences in endocrine pancreatic tumor subtypes detected by comparative genomic hybridization. Am J Pathol. 1999;155:1787-94 doi:10.1016/s0002-9440(10)65495-8
160. Floridia G, Grilli G, Salvatore M, Pescucci C, Moore PS, Scarpa A. et al. Chromosomal alterations detected by comparative genomic hybridization in nonfunctioning endocrine pancreatic tumors. Cancer Genet Cytogenet. 2005;156:23-30 doi:10.1016/j.cancergencyto.2004.04.015
161. Dacic S, Finkelstein SD, Baksh FK, Swalsky PA, Barnes LE, Yousem SA. Small-cell neuroendocrine carcinoma displays unique profiles of tumor-suppressor gene loss in relationship to the primary site of formation. Hum Pathol. 2002;33:927-32
162. Krausch M, Raffel A, Anlauf M, Schott M, Willenberg H, Lehwald N. et al. Loss of PTEN expression in neuroendocrine pancreatic tumors. Horm Metab Res. 2011;43:865-71 doi:10.1055/s-0031-1291333
163. Roldo C, Missiaglia E, Hagan JP, Falconi M, Capelli P, Bersani S. et al. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J Clin Oncol. 2006;24:4677-84 doi:10.1200/jco.2005.05.5194
164. Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647-58 doi:10.1053/j.gastro.2007.05.022
165. Cingarlini S, Bonomi M, Corbo V, Scarpa A, Tortora G. Profiling mTOR pathway in neuroendocrine tumors. Target Oncol. 2012;7:183-8 doi:10.1007/s11523-012-0226-9
166. Han X, Ji Y, Zhao J, Xu X, Lou W. Expression of PTEN and mTOR in pancreatic neuroendocrine tumors. Tumour Biol. 2013 doi:10.1007/s13277-013-0849-1
167. Weisbrod AB, Zhang L, Jain M, Barak S, Quezado MM, Kebebew E. Altered PTEN, ATRX, CHGA, CHGB, and TP53 Expression Are Associated with Aggressive VHL-Associated Pancreatic Neuroendocrine Tumors. Horm Cancer. 2013;4:165-75 doi:10.1007/s12672-013-0134-1
168. O'Toole D, Couvelard A, Rebours V, Zappa M, Hentic O, Hammel P. et al. Molecular markers associated with response to chemotherapy in gastro-entero-pancreatic neuroendocrine tumors. Endocr Relat Cancer. 2010;17:847-56 doi:10.1677/erc-09-0204
169. Gloesenkamp CR, Nitzsche B, Ocker M, Di Fazio P, Quint K, Hoffmann B. et al. AKT inhibition by triciribine alone or as combination therapy for growth control of gastroenteropancreatic neuroendocrine tumors. Int J Oncol. 2012;40:876-88 doi:10.3892/ijo.2011.1256
170. Evers BM, Ishizuka J, Townsend CM Jr, Thompson JC. The human carcinoid cell line, BON. A model system for the study of carcinoid tumors. Ann N Y Acad Sci. 1994;733:393-406
171. Gueli N, Toto A, Palmieri G, Carmenini G, Delpino A, Ferrini U. In vitro growth of a cell line originated from a human insulinoma. J Exp Clin Cancer Res. 1987:281-5
172. Wang L, Ignat A, Axiotis CA. Differential expression of the PTEN tumor suppressor protein in fetal and adult neuroendocrine tissues and tumors: progressive loss of PTEN expression in poorly differentiated neuroendocrine neoplasms. Appl Immunohistochem Mol Morphol. 2002;10:139-46
173. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74 doi:10.1016/j.cell.2011.02.013
174. Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T. et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203-8 doi:10.1101/gad.913901
175. Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB 3rd. et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science. 2001;292:1728-31 doi:10.1126/science.292.5522.1728
176. Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349-52 doi:10.1074/jbc.C100462200
177. Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL. et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest. 2003;112:197-208 doi:10.1172/jci16885
178. Yang ZZ, Tschopp O, Hemmings-Mieszczak M, Feng J, Brodbeck D, Perentes E. et al. Protein kinase B alpha/Akt1 regulates placental development and fetal growth. J Biol Chem. 2003;278:32124-31 doi:10.1074/jbc.M302847200
179. Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P. et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541-51
180. Alessi DR. Discovery of PDK1, one of the missing links in insulin signal transduction. Colworth Medal Lecture. Biochem Soc Trans. 2001;29:1-14
181. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098-101 doi:10.1126/science.1106148
182. Bozulic L, Hemmings BA. PIKKing on PKB: regulation of PKB activity by phosphorylation. Curr Opin Cell Biol. 2009;21:256-61 doi:10.1016/j.ceb.2009.02.002
183. Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J. et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11:859-71 doi:10.1016/j.devcel.2006.10.007
184. Garcia-Martinez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM. et al. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem J. 2009;421:29-42 doi:10.1042/bj20090489
185. Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829-34 doi:10.1101/gad.1110003
186. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS. et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857-68
187. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499-511
188. Vadlakonda L, Pasupuleti M, Pallu R. Role of PI3K-AKT-mTOR and Wnt Signaling Pathways in Transition of G1-S Phase of Cell Cycle in Cancer Cells. Front Oncol. 2013;3:85. doi:10.3389/fonc.2013.00085
189. Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR pathway--beyond rapalogs. Oncotarget. 2010;1:530-43
190. Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY. et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125-37 doi:10.1016/j.cell.2006.08.033
191. Vincent EE, Elder DJ, Thomas EC, Phillips L, Morgan C, Pawade J. et al. Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. Br J Cancer. 2011;104:1755-61 doi:10.1038/bjc.2011.132
192. Ghayouri M, Boulware D, Nasir A, Strosberg J, Kvols L, Coppola D. Activation of the serine/theronine protein kinase Akt in enteropancreatic neuroendocrine tumors. Anticancer Res. 2010;30:5063-7
193. Noh WC, Mondesire WH, Peng J, Jian W, Zhang H, Dong J. et al. Determinants of rapamycin sensitivity in breast cancer cells. Clin Cancer Res. 2004;10:1013-23
194. Breuleux M, Klopfenstein M, Stephan C, Doughty CA, Barys L, Maira SM. et al. Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Mol Cancer Ther. 2009;8:742-53 doi:10.1158/1535-7163.mct-08-0668
195. Meric-Bernstam F, Akcakanat A, Chen H, Do KA, Sangai T, Adkins F. et al. PIK3CA/PTEN mutations and Akt activation as markers of sensitivity to allosteric mTOR inhibitors. Clin Cancer Res. 2012;18:1777-89 doi:10.1158/1078-0432.ccr-11-2123
196. Zitzmann K, Vlotides G, Brand S, Lahm H, Spottl G, Goke B. et al. Perifosine-mediated Akt inhibition in neuroendocrine tumor cells: role of specific Akt isoforms. Endocr Relat Cancer. 2012;19:423-34 doi:10.1530/erc-12-0074
197. Yang Q, Guan KL. Expanding mTOR signaling. Cell Res. 2007;17:666-81 doi:10.1038/cr.2007.64
198. Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S. et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177-89
199. Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H. et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163-75
200. Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E. et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903-15 doi:10.1016/j.molcel.2007.03.003
201. Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM. et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873-86 doi:10.1016/j.cell.2009.03.046
202. Kim DH, Sarbassov DD, Ali SM, Latek RR, Guntur KV, Erdjument-Bromage H. et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. 2003;11:895-904
203. Zeng Z, Sarbassov dos D, Samudio IJ, Yee KW, Munsell MF, Ellen Jackson C. et al. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood. 2007;109:3509-12 doi:10.1182/blood-2006-06-030833
204. O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D. et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500-8 doi:10.1158/0008-5472.can-05-2925
205. Shepherd PR, Nave BT, O'Rahilly S. The role of phosphoinositide 3-kinase in insulin signalling. J Mol Endocrinol. 1996;17:175-84
206. Peng T, Golub TR, Sabatini DM. The immunosuppressant rapamycin mimics a starvation-like signal distinct from amino acid and glucose deprivation. Mol Cell Biol. 2002;22:5575-84
207. Hannan KM, Brandenburger Y, Jenkins A, Sharkey K, Cavanaugh A, Rothblum L. et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol Cell Biol. 2003;23:8862-77
208. Mayer C, Zhao J, Yuan X, Grummt I. mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev. 2004;18:423-34 doi:10.1101/gad.285504
209. Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K. et al. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003;278:15461-4 doi:10.1074/jbc.C200665200
210. Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5:671-88 doi:10.1038/nrd2062
211. Dennis PB, Pullen N, Kozma SC, Thomas G. The principal rapamycin-sensitive p70(s6k) phosphorylation sites, T-229 and T-389, are differentially regulated by rapamycin-insensitive kinase kinases. Mol Cell Biol. 1996;16:6242-51
212. Pause A, Belsham GJ, Gingras AC, Donze O, Lin TA, Lawrence JC Jr. et al. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5'-cap function. Nature. 1994;371:762-7 doi:10.1038/371762a0
213. Redpath NT, Foulstone EJ, Proud CG. Regulation of translation elongation factor-2 by insulin via a rapamycin-sensitive signalling pathway. EMBO J. 1996;15:2291-7
214. Seidel ER, Ragan VL. Inhibition by rapamycin of ornithine decarboxylase and epithelial cell proliferation in intestinal IEC-6 cells in culture. Br J Pharmacol. 1997;120:571-4 doi:10.1038/sj.bjp.0700936
215. Azpiazu I, Saltiel AR, DePaoli-Roach AA, Lawrence JC. Regulation of both glycogen synthase and PHAS-I by insulin in rat skeletal muscle involves mitogen-activated protein kinase-independent and rapamycin-sensitive pathways. J Biol Chem. 1996;271:5033-9
216. Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F. et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol. 2002;22:7004-14
217. Huffman TA, Mothe-Satney I, Lawrence JC Jr. Insulin-stimulated phosphorylation of lipin mediated by the mammalian target of rapamycin. Proc Natl Acad Sci U S A. 2002;99:1047-52 doi:10.1073/pnas.022634399
218. Parekh D, Ziegler W, Yonezawa K, Hara K, Parker PJ. Mammalian TOR controls one of two kinase pathways acting upon nPKCdelta and nPKCepsilon. J Biol Chem. 1999;274:34758-64
219. Peterson RT, Desai BN, Hardwick JS, Schreiber SL. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycinassociated protein. Proc Natl Acad Sci U S A. 1999;96:4438-42
220. Huang S, Liu LN, Hosoi H, Dilling MB, Shikata T, Houghton PJ. p53/p21(CIP1) cooperate in enforcing rapamycin-induced G(1) arrest and determine the cellular response to rapamycin. Cancer Res. 2001;61:3373-81
221. Nourse J, Firpo E, Flanagan WM, Coats S, Polyak K, Lee MH. et al. Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature. 1994;372:570-3 doi:10.1038/372570a0
222. Usui I, Haruta T, Iwata M, Takano A, Uno T, Kawahara J. et al. Retinoblastoma protein phosphorylation via PI 3-kinase and mTOR pathway regulates adipocyte differentiation. Biochem Biophys Res Commun. 2000;275:115-20 doi:10.1006/bbrc.2000.3201
223. Yokogami K, Wakisaka S, Avruch J, Reeves SA. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol. 2000;10:47-50
224. Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA. et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol. 2006;16:1865-70 doi:10.1016/j.cub.2006.08.001
225. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H. et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296-302 doi:10.1016/j.cub.2004.06.054
226. Pearce LR, Huang X, Boudeau J, Pawlowski R, Wullschleger S, Deak M. et al. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem J. 2007;405:513-22 doi:10.1042/bj20070540
227. Martin J, Masri J, Bernath A, Nishimura RN, Gera J. Hsp70 associates with Rictor and is required for mTORC2 formation and activity. Biochem Biophys Res Commun. 2008;372:578-83 doi:10.1016/j.bbrc.2008.05.086
228. Zierhut B, Mechtler K, Gartner W, Daneva T, Base W, Weissel M. et al. Heat shock protein 70 (Hsp70) subtype expression in neuroendocrine tissue and identification of a neuroendocrine tumour-specific Hsp70 truncation. Endocr Relat Cancer. 2004;11:377-89
229. Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem. 2005;280:40406-16 doi:10.1074/jbc.M508361200
230. Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF. et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159-68 doi:10.1016/j.molcel.2006.03.029
231. Gulhati P, Bowen KA, Liu J, Stevens PD, Rychahou PG, Chen M. et al. mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res. 2011;71:3246-56 doi:10.1158/0008-5472.can-10-4058
232. Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H. et al. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052-8 doi:10.1158/0008-5472.can-05-0917
233. Shi Y, Yan H, Frost P, Gera J, Lichtenstein A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol Cancer Ther. 2005;4:1533-40 doi:10.1158/1535-7163.mct-05-0068
234. Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, Poulikakos PI, Scaltriti M, Moskatel E. et al. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011;1:248-59 doi:10.1158/2159-8290.cd-11-0085
235. Catena L, Bajetta E, Milione M, Ducceschi M, Valente M, Dominoni F. et al. Mammalian target of rapamycin expression in poorly differentiated endocrine carcinoma: clinical and therapeutic future challenges. Target Oncol. 2011;6:65-8 doi:10.1007/s11523-011-0171-z
236. Shida T, Kishimoto T, Furuya M, Nikaido T, Koda K, Takano S. et al. Expression of an activated mammalian target of rapamycin (mTOR) in gastroenteropancreatic neuroendocrine tumors. Cancer Chemother Pharmacol. 2010;65:889-93 doi:10.1007/s00280-009-1094-6
237. Kasajima A, Pavel M, Darb-Esfahani S, Noske A, Stenzinger A, Sasano H. et al. mTOR expression and activity patterns in gastroenteropancreatic neuroendocrine tumours. Endocr Relat Cancer. 2011;18:181-92 doi:10.1677/erc-10-0126
238. Larson AM, Hedgire SS, Deshpande V, Stemmer-Rachamimov AO, Harisinghani MG, Ferrone CR. et al. Pancreatic neuroendocrine tumors in patients with tuberous sclerosis complex. Clin Genet. 2012;82:558-63 doi:10.1111/j.1399-0004.2011.01805.x
239. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36:320-8 doi:10.1016/j.tibs.2011.03.006
240. Zitzmann K, De Toni EN, Brand S, Goke B, Meinecke J, Spottl G. et al. The novel mTOR inhibitor RAD001 (everolimus) induces antiproliferative effects in human pancreatic neuroendocrine tumor cells. Neuroendocrinology. 2007;85:54-60 doi:10.1159/000100057
241. Chiu CW, Nozawa H, Hanahan D. Survival benefit with proapoptotic molecular and pathologic responses from dual targeting of mammalian target of rapamycin and epidermal growth factor receptor in a preclinical model of pancreatic neuroendocrine carcinogenesis. J Clin Oncol. 2010;28:4425-33 doi:10.1200/jco.2010.28.0198
242. Bollard J, Couderc C, Blanc M, Poncet G, Lepinasse F, Hervieu V. et al. Antitumor effect of everolimus in preclinical models of high-grade gastroenteropancreatic neuroendocrine carcinomas. Neuroendocrinology. 2013;97:331-40 doi:10.1159/000347063
243. Yao JC, Lombard-Bohas C, Baudin E, Kvols LK, Rougier P, Ruszniewski P. et al. Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: a phase II trial. J Clin Oncol. 2010;28:69-76 doi:10.1200/jco.2009.24.2669
244. Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E. et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514-23 doi:10.1056/NEJMoa1009290
245. Yao JC, Phan AT, Jehl V, Shah G, Meric-Bernstam F. Everolimus in advanced pancreatic neuroendocrine tumors: the clinical experience. Cancer Res. 2013;73:1449-53 doi:10.1158/0008-5472.can-12-3923
246. Azzoni C, Bottarelli L, Cecchini S, Lagrasta C, Pizzi S, D'Adda T. et al. Involvement of HER-2/neu and metastasis-related proteins in the development of ileal neuroendocrine tumors. Virchows Arch. 2011;458:525-36 doi:10.1007/s00428-011-1069-y
247. Cuevas BD, Abell AN, Johnson GL. Role of mitogen-activated protein kinase kinase kinases in signal integration. Oncogene. 2007;26:3159-71 doi:10.1038/sj.onc.1210409
248. Chong H, Vikis HG, Guan KL. Mechanisms of regulating the Raf kinase family. Cell Signal. 2003;15:463-9
249. Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299-304 doi:10.1126/science.1062023
250. Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding Ras: 'it ain't over 'til it's over'. Trends Cell Biol. 2000;10:147-54
251. Kawesha A, Ghaneh P, Andren-Sandberg A, Ograed D, Skar R, Dawiskiba S. et al. K-ras oncogene subtype mutations are associated with survival but not expression of p53, p16(INK4A), p21(WAF-1), cyclin D1, erbB-2 and erbB-3 in resected pancreatic ductal adenocarcinoma. Int J Cancer. 2000;89:469-74
252. Moskaluk CA, Hruban RH, Kern SE. p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997;57:2140-3
253. Kimura W, Zhao B, Futakawa N, Muto T, Makuuchi M. Significance of K-ras codon 12 point mutation in pancreatic juice in the diagnosis of carcinoma of the pancreas. Hepatogastroenterology. 1999;46:532-9
254. Jancik S, Drabek J, Radzioch D, Hajduch M. Clinical relevance of KRAS in human cancers. J Biomed Biotechnol. 2010;2010:150960. doi:10.1155/2010/150960
255. Ellenbroek SI, Collard JG. Rho GTPases: functions and association with cancer. Clin Exp Metastasis. 2007;24:657-72 doi:10.1007/s10585-007-9119-1
256. Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10:842-57 doi:10.1038/nrc2960
257. Vigil D, Martin TD, Williams F, Yeh JJ, Campbell SL, Der CJ. Aberrant overexpression of the Rgl2 Ral small GTPase-specific guanine nucleotide exchange factor promotes pancreatic cancer growth through Ral-dependent and Ral-independent mechanisms. J Biol Chem. 2010;285:34729-40 doi:10.1074/jbc.M110.116756
258. Prenen H, Tejpar S, Van Cutsem E. New strategies for treatment of KRAS mutant metastatic colorectal cancer. Clin Cancer Res. 2010;16:2921-6 doi:10.1158/1078-0432.ccr-09-2029
259. Salhia B, Tran NL, Chan A, Wolf A, Nakada M, Rutka F. et al. The guanine nucleotide exchange factors trio, Ect2, and Vav3 mediate the invasive behavior of glioblastoma. Am J Pathol. 2008;173:1828-38 doi:10.2353/ajpath.2008.080043
260. Patel V, Rosenfeldt HM, Lyons R, Servitja JM, Bustelo XR, Siroff M. et al. Persistent activation of Rac1 in squamous carcinomas of the head and neck: evidence for an EGFR/Vav2 signaling axis involved in cell invasion. Carcinogenesis. 2007;28:1145-52 doi:10.1093/carcin/bgm008
261. Qin J, Xie Y, Wang B, Hoshino M, Wolff DW, Zhao J. et al. Upregulation of PIP3-dependent Rac exchanger 1 (P-Rex1) promotes prostate cancer metastasis. Oncogene. 2009;28:1853-63 doi:10.1038/onc.2009.30
262. Lambert JM, Lambert QT, Reuther GW, Malliri A, Siderovski DP, Sondek J. et al. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat Cell Biol. 2002;4:621-5 doi:10.1038/ncb833
263. Tannapfel A, Vomschloss S, Karhoff D, Markwarth A, Hengge UR, Wittekind C. et al. BRAF gene mutations are rare events in gastroenteropancreatic neuroendocrine tumors. Am J Clin Pathol. 2005;123:256-60
264. Paraskevakou H, Saetta A, Skandalis K, Tseleni S, Athanassiadis A, Davaris PS. Morphological-histochemical study of intestinal carcinoids and K-ras mutation analysis in appendiceal carcinoids. Pathol Oncol Res. 1999;5:205-10
265. Dammann R, Schagdarsurengin U, Liu L, Otto N, Gimm O, Dralle H. et al. Frequent RASSF1A promoter hypermethylation and K-ras mutations in pancreatic carcinoma. Oncogene. 2003;22:3806-12 doi:10.1038/sj.onc.1206582
266. Liedke M, Karnbach C, Kalinin V, Herbst B, Frilling A, Broelsch CE. [Detection of H-ras and K-ras in tumors of gastrointestinal-pancreatic system]. Langenbecks Arch Chir Suppl Kongressbd. 1998;115:255-9
267. Morrison DK, Cutler RE. The complexity of Raf-1 regulation. Curr Opin Cell Biol. 1997;9:174-9
268. Wittinghofer A, Nassar N. How Ras-related proteins talk to their effectors. Trends Biochem Sci. 1996;21:488-91
269. Mason CS, Springer CJ, Cooper RG, Superti-Furga G, Marshall CJ, Marais R. Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J. 1999;18:2137-48 doi:10.1093/emboj/18.8.2137
270. Zhang BH, Guan KL. Activation of B-Raf kinase requires phosphorylation of the conserved residues Thr598 and Ser601. EMBO J. 2000;19:5429-39 doi:10.1093/emboj/19.20.5429
271. Emuss V, Garnett M, Mason C, Marais R. Mutations of C-RAF are rare in human cancer because C-RAF has a low basal kinase activity compared with B-RAF. Cancer Res. 2005;65:9719-26 doi:10.1158/0008-5472.can-05-1683
272. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S. et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54 doi:10.1038/nature00766
273. Kumar SM, Yu H, Edwards R, Chen L, Kazianis S, Brafford P. et al. Mutant V600E BRAF increases hypoxia inducible factor-1alpha expression in melanoma. Cancer Res. 2007;67:3177-84 doi:10.1158/0008-5472.can-06-3312
274. Sharma A, Tran MA, Liang S, Sharma AK, Amin S, Smith CD. et al. Targeting mitogen-activated protein kinase/extracellular signal-regulated kinase kinase in the mutant (V600E) B-Raf signaling cascade effectively inhibits melanoma lung metastases. Cancer Res. 2006;66:8200-9 doi:10.1158/0008-5472.can-06-0809
275. Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP. Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Res. 2005;65:2412-21 doi:10.1158/0008-5472.can-04-2423
276. Balmanno K, Cook SJ. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009;16:368-77 doi:10.1038/cdd.2008.148
277. Lehmann K, Janda E, Pierreux CE, Rytomaa M, Schulze A, McMahon M. et al. Raf induces TGFbeta production while blocking its apoptotic but not invasive responses: a mechanism leading to increased malignancy in epithelial cells. Genes Dev. 2000;14:2610-22
278. Sobczak I, Galabova-Kovacs G, Sadzak I, Kren A, Christofori G, Baccarini M. B-Raf is required for ERK activation and tumor progression in a mouse model of pancreatic beta-cell carcinogenesis. Oncogene. 2008;27:4779-87 doi:10.1038/onc.2008.128
279. Riesco-Eizaguirre G, Rodriguez I, De la Vieja A, Costamagna E, Carrasco N, Nistal M. et al. The BRAFV600E oncogene induces transforming growth factor beta secretion leading to sodium iodide symporter repression and increased malignancy in thyroid cancer. Cancer Res. 2009;69:8317-25 doi:10.1158/0008-5472.can-09-1248
280. Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J. et al. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol. 2002;156:299-313 doi:10.1083/jcb.200109037
281. Pritchard CA, Hayes L, Wojnowski L, Zimmer A, Marais RM, Norman JC. B-Raf acts via the ROCKII/LIMK/cofilin pathway to maintain actin stress fibers in fibroblasts. Mol Cell Biol. 2004;24:5937-52 doi:10.1128/mcb.24.13.5937-5952.2004
282. Ehrenreiter K, Kern F, Velamoor V, Meissl K, Galabova-Kovacs G, Sibilia M. et al. Raf-1 addiction in Ras-induced skin carcinogenesis. Cancer Cell. 2009;16:149-60 doi:10.1016/j.ccr.2009.06.008
283. Ehrenreiter K, Piazzolla D, Velamoor V, Sobczak I, Small JV, Takeda J. et al. Raf-1 regulates Rho signaling and cell migration. J Cell Biol. 2005;168:955-64 doi:10.1083/jcb.200409162
284. Piazzolla D, Meissl K, Kucerova L, Rubiolo C, Baccarini M. Raf-1 sets the threshold of Fas sensitivity by modulating Rok-alpha signaling. J Cell Biol. 2005;171:1013-22 doi:10.1083/jcb.200504137
285. Schulte TW, Blagosklonny MV, Ingui C, Neckers L. Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J Biol Chem. 1995;270:24585-8
286. Schulte TW, Blagosklonny MV, Romanova L, Mushinski JF, Monia BP, Johnston JF. et al. Destabilization of Raf-1 by geldanamycin leads to disruption of the Raf-1-MEK-mitogen-activated protein kinase signalling pathway. Mol Cell Biol. 1996;16:5839-45
287. Stancato LF, Chow YH, Hutchison KA, Perdew GH, Jove R, Pratt WB. Raf exists in a native heterocomplex with hsp90 and p50 that can be reconstituted in a cell-free system. J Biol Chem. 1993;268:21711-6
288. Kolch W, Heidecker G, Kochs G, Hummel R, Vahidi H, Mischak H. et al. Protein kinase C alpha activates RAF-1 by direct phosphorylation. Nature. 1993;364:249-52 doi:10.1038/364249a0
289. Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science. 1999;286:1741-4
290. Jin S, Zhuo Y, Guo W, Field J. p21-activated Kinase 1 (Pak1)-dependent phosphorylation of Raf-1 regulates its mitochondrial localization, phosphorylation of BAD, and Bcl-2 association. J Biol Chem. 2005;280:24698-705 doi:10.1074/jbc.M413374200
291. Diaz B, Barnard D, Filson A, MacDonald S, King A, Marshall M. Phosphorylation of Raf-1 serine 338-serine 339 is an essential regulatory event for Ras-dependent activation and biological signaling. Mol Cell Biol. 1997;17:4509-16
292. Baumann B, Weber CK, Troppmair J, Whiteside S, Israel A, Rapp UR. et al. Raf induces NF-kappaB by membrane shuttle kinase MEKK1, a signaling pathway critical for transformation. Proc Natl Acad Sci U S A. 2000;97:4615-20 doi:10.1073/pnas.080583397
293. Kinkade R, Dasgupta P, Carie A, Pernazza D, Carless M, Pillai S. et al. A small molecule disruptor of Rb/Raf-1 interaction inhibits cell proliferation, angiogenesis, and growth of human tumor xenografts in nude mice. Cancer Res. 2008;68:3810-8 doi:10.1158/0008-5472.can-07-6672
294. Polzien L, Baljuls A, Rennefahrt UE, Fischer A, Schmitz W, Zahedi RP. et al. Identification of novel in vivo phosphorylation sites of the human proapoptotic protein BAD: pore-forming activity of BAD is regulated by phosphorylation. J Biol Chem. 2009;284:28004-20 doi:10.1074/jbc.M109.010702
295. Eves EM, Shapiro P, Naik K, Klein UR, Trakul N, Rosner MR. Raf kinase inhibitory protein regulates aurora B kinase and the spindle checkpoint. Mol Cell. 2006;23:561-74 doi:10.1016/j.molcel.2006.07.015
296. Alessi DR, Saito Y, Campbell DG, Cohen P, Sithanandam G, Rapp U. et al. Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J. 1994;13:1610-9
297. Zheng CF, Guan KL. Activation of MEK family kinases requires phosphorylation of two conserved Ser/Thr residues. EMBO J. 1994;13:1123-31
298. Ferrell JE Jr. Tripping the switch fantastic: how a protein kinase cascade can convert graded inputs into switch-like outputs. Trends Biochem Sci. 1996;21:460-6
299. Ferrell JE Jr. How responses get more switch-like as you move down a protein kinase cascade. Trends Biochem Sci. 1997;22:288-9
300. Ferrell JE Jr. Building a cellular switch: more lessons from a good egg. Bioessays. 1999;21:866-70 doi:10.1002/(sici)1521-1878(199910)21:10<866::aid-bies9>3.0.co;2-1
301. Ferrell JE Jr, Bhatt RR. Mechanistic studies of the dual phosphorylation of mitogen-activated protein kinase. J Biol Chem. 1997;272:19008-16
302. Huang CY, Ferrell JE Jr. Ultrasensitivity in the mitogen-activated protein kinase cascade. Proc Natl Acad Sci U S A. 1996;93:10078-83
303. Robbins DJ, Cobb MH. Extracellular signal-regulated kinases 2 autophosphorylates on a subset of peptides phosphorylated in intact cells in response to insulin and nerve growth factor: analysis by peptide mapping. Mol Biol Cell. 1992;3:299-308
304. Murugan AK, Dong J, Xie J, Xing M. MEK1 mutations, but not ERK2 mutations, occur in melanomas and colon carcinomas, but none in thyroid carcinomas. Cell Cycle. 2009;8:2122-4
305. Wang GG, Yao JC, Worah S, White JA, Luna R, Wu TT. et al. Comparison of genetic alterations in neuroendocrine tumors: frequent loss of chromosome 18 in ileal carcinoid tumors. Mod Pathol. 2005;18:1079-87 doi:10.1038/modpathol.3800389
306. Rindi G, Candusso ME, Solcia E. Molecular aspects of the endocrine tumours of the pancreas and the gastrointestinal tract. Ital J Gastroenterol Hepatol. 1999;31(Suppl 2):S135-8
307. Perren A, Schmid S, Locher T, Saremaslani P, Bonvin C, Heitz PU. et al. BRAF and endocrine tumors: mutations are frequent in papillary thyroid carcinomas, rare in endocrine tumors of the gastrointestinal tract and not detected in other endocrine tumors. Endocr Relat Cancer. 2004;11:855-60 doi:10.1677/erc.1.00841
308. Karkouche R, Bachet JB, Sandrini J, Mitry E, Penna C, Cote JF. et al. Colorectal neuroendocrine carcinomas and adenocarcinomas share oncogenic pathways. A clinico-pathologic study of 12 cases. Eur J Gastroenterol Hepatol. 2012;24:1430-7 doi:10.1097/MEG.0b013e3283583c87
309. Karhoff D, Sauer S, Schrader J, Arnold R, Fendrich V, Bartsch DK. et al. Rap1/B-Raf signaling is activated in neuroendocrine tumors of the digestive tract and Raf kinase inhibition constitutes a putative therapeutic target. Neuroendocrinology. 2007;85:45-53 doi:10.1159/000100508
310. Mordant P, Loriot Y, Leteur C, Calderaro J, Bourhis J, Wislez M. et al. Dependence on phosphoinositide 3-kinase and RAS-RAF pathways drive the activity of RAF265, a novel RAF/VEGFR2 inhibitor, and RAD001 (Everolimus) in combination. Mol Cancer Ther. 2010;9:358-68 doi:10.1158/1535-7163.mct-09-1014
311. Zitzmann K, de Toni E, von Ruden J, Brand S, Goke B, Laubender RP. et al. The novel Raf inhibitor Raf265 decreases Bcl-2 levels and confers TRAIL-sensitivity to neuroendocrine tumour cells. Endocr Relat Cancer. 2011;18:277-85 doi:10.1530/erc-10-0108
312. Sippel RS, Carpenter JE, Kunnimalaiyaan M, Lagerholm S, Chen H. Raf-1 activation suppresses neuroendocrine marker and hormone levels in human gastrointestinal carcinoid cells. Am J Physiol Gastrointest Liver Physiol. 2003;285:G245-54 doi:10.1152/ajpgi.00420.2002
313. Sippel RS, Chen H. Activation of the ras/raf-1 signal transduction pathway in carcinoid tumor cells results in morphologic transdifferentiation. Surgery. 2002;132:1035-9 discussion 9. doi:10.1067/msy.2002.128877
314. Ning L, Chen H, Kunnimalaiyaan M. Focal adhesion kinase, a downstream mediator of Raf-1 signaling, suppresses cellular adhesion, migration, and neuroendocrine markers in BON carcinoid cells. Mol Cancer Res. 2010;8:775-82 doi:10.1158/1541-7786.mcr-09-0525
315. Georgieva I, Koychev D, Wang Y, Holstein J, Hopfenmuller W, Zeitz M. et al. ZM447439, a novel promising aurora kinase inhibitor, provokes antiproliferative and proapoptotic effects alone and in combination with bio- and chemotherapeutic agents in gastroenteropancreatic neuroendocrine tumor cell lines. Neuroendocrinology. 2010;91:121-30 doi:10.1159/000258705
316. Kanakis G, Kaltsas G. Biochemical markers for gastroenteropancreatic neuroendocrine tumours (GEP-NETs). Best Pract Res Clin Gastroenterol. 2012;26:791-802 doi:10.1016/j.bpg.2012.12.006
317. O'Toole D, Grossman A, Gross D, Delle Fave G, Barkmanova J, O'Connor J. et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: biochemical markers. Neuroendocrinology. 2009;90:194-202 doi:10.1159/000225948
318. Cook MR, Pinchot SN, Jaskula-Sztul R, Luo J, Kunnimalaiyaan M, Chen H. Identification of a novel Raf-1 pathway activator that inhibits gastrointestinal carcinoid cell growth. Mol Cancer Ther. 2010;9:429-37 doi:10.1158/1535-7163.mct-09-0718
319. Pinchot SN, Adler JT, Luo Y, Ju J, Li W, Shen B. et al. Tautomycin suppresses growth and neuroendocrine hormone markers in carcinoid cells through activation of the Raf-1 pathway. Am J Surg. 2009;197:313-9 doi:10.1016/j.amjsurg.2008.10.007
320. Lee JH, Lee JS, Kim SE, Moon BS, Kim YC, Lee SK. et al. Tautomycetin inhibits growth of colorectal cancer cells through p21cip/WAF1 induction via the extracellular signal-regulated kinase pathway. Mol Cancer Ther. 2006;5:3222-31 doi:10.1158/1535-7163.mct-06-0455
321. Van Gompel JJ, Kunnimalaiyaan M, Holen K, Chen H. ZM336372, a Raf-1 activator, suppresses growth and neuroendocrine hormone levels in carcinoid tumor cells. Mol Cancer Ther. 2005;4:910-7 doi:10.1158/1535-7163.mct-04-0334
322. Callahan MK, Rampal R, Harding JJ, Klimek VM, Chung YR, Merghoub T. et al. Progression of RAS-mutant leukemia during RAF inhibitor treatment. N Engl J Med. 2012;367:2316-21 doi:10.1056/NEJMoa1208958
323. Carnahan J, Beltran PJ, Babij C, Le Q, Rose MJ, Vonderfecht S. et al. Selective and potent Raf inhibitors paradoxically stimulate normal cell proliferation and tumor growth. Mol Cancer Ther. 2010;9:2399-410 doi:10.1158/1535-7163.mct-10-0181
324. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427-30 doi:10.1038/nature08902
325. Kaplan FM, Shao Y, Mayberry MM, Aplin AE. Hyperactivation of MEK-ERK1/2 signaling and resistance to apoptosis induced by the oncogenic B-RAF inhibitor, PLX4720, in mutant N-RAS melanoma cells. Oncogene. 2011;30:366-71 doi:10.1038/onc.2010.408
326. Rushworth LK, Hindley AD, O'Neill E, Kolch W. Regulation and role of Raf-1/B-Raf heterodimerization. Mol Cell Biol. 2006;26:2262-72 doi:10.1128/mcb.26.6.2262-2272.2006
327. Cichowski K, Janne PA. Drug discovery: inhibitors that activate. Nature. 2010;464:358-9 doi:10.1038/464358a
328. Ritt DA, Monson DM, Specht SI, Morrison DK. Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol Cell Biol. 2010;30:806-19 doi:10.1128/mcb.00569-09
329. Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R. et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431-5 doi:10.1038/nature08833
330. Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A. et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065-74 doi:10.1172/jci34739
331. Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527-41 doi:10.1038/onc.2008.247
332. Gao J, Wagnon JL, Protacio RM, Glazko GV, Beggs M, Raj V. et al. A stress-activated, p38 MAPK-ATF/CREB pathway regulates post-transcriptional, sequence-dependent decay of target RNAs. Mol Cell Biol. 2013 doi:10.1128/mcb.00349-13
333. Hollenhorst PC. RAS/ERK pathway transcriptional regulation through ETS/AP-1 binding sites. Small GTPases. 2012;3:154-8 doi:10.4161/sgtp.19630
334. Rolli M, Kotlyarov A, Sakamoto KM, Gaestel M, Neininger A. Stress-induced stimulation of early growth response gene-1 by p38/stress-activated protein kinase 2 is mediated by a cAMP-responsive promoter element in a MAPKAP kinase 2-independent manner. J Biol Chem. 1999;274:19559-64
335. Xing J, Kornhauser JM, Xia Z, Thiele EA, Greenberg ME. Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol Cell Biol. 1998;18:1946-55
336. Tan Y, Rouse J, Zhang A, Cariati S, Cohen P, Comb MJ. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J. 1996;15:4629-42
337. Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T. et al. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025-37
338. Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135-48
339. Chen RH, Abate C, Blenis J. Phosphorylation of the c-Fos transrepression domain by mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase. Proc Natl Acad Sci U S A. 1993;90:10952-6
340. Chen RH, Juo PC, Curran T, Blenis J. Phosphorylation of c-Fos at the C-terminus enhances its transforming activity. Oncogene. 1996;12:1493-502
341. O'Donnell A, Odrowaz Z, Sharrocks AD. Immediate-early gene activation by the MAPK pathways: what do and don't we know? Biochem Soc Trans. 2012;40:58-66 doi:10.1042/bst20110636
342. Janknecht R, Ernst WH, Pingoud V, Nordheim A. Activation of ternary complex factor Elk-1 by MAP kinases. EMBO J. 1993;12:5097-104
343. Gille H, Kortenjann M, Thomae O, Moomaw C, Slaughter C, Cobb MH. et al. ERK phosphorylation potentiates Elk-1-mediated ternary complex formation and transactivation. EMBO J. 1995;14:951-62
344. Rowan BG, Weigel NL, O'Malley BW. Phosphorylation of steroid receptor coactivator-1. Identification of the phosphorylation sites and phosphorylation through the mitogen-activated protein kinase pathway. J Biol Chem. 2000;275:4475-83
345. Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B. et al. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403-14
346. Smith CL, Onate SA, Tsai MJ, O'Malley BW. CREB binding protein acts synergistically with steroid receptor coactivator-1 to enhance steroid receptor-dependent transcription. Proc Natl Acad Sci U S A. 1996;93:8884-8
347. Schuringa JJ, Dekker LV, Vellenga E, Kruijer W. Sequential activation of Rac-1, SEK-1/MKK-4, and protein kinase Cdelta is required for interleukin-6-induced STAT3 Ser-727 phosphorylation and transactivation. J Biol Chem. 2001;276:27709-15 doi:10.1074/jbc.M009821200
348. Schuringa JJ, Jonk LJ, Dokter WH, Vellenga E, Kruijer W. Interleukin-6-induced STAT3 transactivation and Ser727 phosphorylation involves Vav, Rac-1 and the kinase SEK-1/MKK-4 as signal transduction components. Biochem J. 2000 347 Pt 1: 89-96
349. Briest F, Berndt A, Clement J, Junker K, Eggeling F, Grimm S. et al. Tumor-stroma interactions in tumorigenesis: lessons from stem cell biology. Front Biosci (Elite Ed). 2012;4:1871-87
350. Zugowski C, Lieder F, Muller A, Gasch J, Corvinus FM, Moriggl R. et al. STAT3 controls matrix metalloproteinase-1 expression in colon carcinoma cells by both direct and AP-1-mediated interaction with the MMP-1 promoter. Biol Chem. 2011;392:449-59 doi:10.1515/bc.2011.038
351. Tsareva SA, Moriggl R, Corvinus FM, Wiederanders B, Schutz A, Kovacic B. et al. Signal transducer and activator of transcription 3 activation promotes invasive growth of colon carcinomas through matrix metalloproteinase induction. Neoplasia. 2007;9:279-91
352. Bode JG, Ehlting C, Haussinger D. The macrophage response towards LPS and its control through the p38(MAPK)-STAT3 axis. Cell Signal. 2012;24:1185-94 doi:10.1016/j.cellsig.2012.01.018
353. Dijkgraaf EM, Welters MJ, Nortier JW, van der Burg SH, Kroep JR. Interleukin-6/interleukin-6 receptor pathway as a new therapy target in epithelial ovarian cancer. Curr Pharm Des. 2012;18:3816-27
354. Brami-Cherrier K, Roze E, Girault JA, Betuing S, Caboche J. Role of the ERK/MSK1 signalling pathway in chromatin remodelling and brain responses to drugs of abuse. J Neurochem. 2009;108:1323-35 doi:10.1111/j.1471-4159.2009.05879.x
355. Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH, Hazzalin CA. et al. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J. 2003;22:2788-97 doi:10.1093/emboj/cdg273
356. Deak M, Clifton AD, Lucocq LM, Alessi DR. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998;17:4426-41 doi:10.1093/emboj/17.15.4426
357. Shapiro PS, Whalen AM, Tolwinski NS, Wilsbacher J, Froelich-Ammon SJ, Garcia M. et al. Extracellular signal-regulated kinase activates topoisomerase IIalpha through a mechanism independent of phosphorylation. Mol Cell Biol. 1999;19:3551-60
358. Sassone-Corsi P, Mizzen CA, Cheung P, Crosio C, Monaco L, Jacquot S. et al. Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science. 1999;285:886-91
359. Clayton AL, Mahadevan LC. MAP kinase-mediated phosphoacetylation of histone H3 and inducible gene regulation. FEBS Lett. 2003;546:51-8
360. Dunn KL, Espino PS, Drobic B, He S, Davie JR. The Ras-MAPK signal transduction pathway, cancer and chromatin remodeling. Biochem Cell Biol. 2005;83:1-14 doi:10.1139/o04-121
361. Hopfner M, Sutter AP, Gerst B, Zeitz M, Scherubl H. A novel approach in the treatment of neuroendocrine gastrointestinal tumours. Targeting the epidermal growth factor receptor by gefitinib (ZD1839). Br J Cancer. 2003;89:1766-75 doi:10.1038/sj.bjc.6601346
362. Clerk A, Aggeli IK, Stathopoulou K, Sugden PH. Peptide growth factors signal differentially through protein kinase C to extracellular signal-regulated kinases in neonatal cardiomyocytes. Cell Signal. 2006;18:225-35 doi:10.1016/j.cellsig.2005.04.005
363. Weng LP, Smith WM, Brown JL, Eng C. PTEN inhibits insulin-stimulated MEK/MAPK activation and cell growth by blocking IRS-1 phosphorylation and IRS-1/Grb-2/Sos complex formation in a breast cancer model. Hum Mol Genet. 2001;10:605-16
364. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117-34 doi:10.1016/j.cell.2010.06.011
365. Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010;40:310-22 doi:10.1016/j.molcel.2010.09.026
366. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21-35 doi:10.1038/nrm3025
367. Kodaki T, Woscholski R, Hallberg B, Rodriguez-Viciana P, Downward J, Parker PJ. The activation of phosphatidylinositol 3-kinase by Ras. Curr Biol. 1994;4:798-806
368. Suire S, Hawkins P, Stephens L. Activation of phosphoinositide 3-kinase gamma by Ras. Curr Biol. 2002;12:1068-75
369. Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A. 2004;101:13489-94 doi:10.1073/pnas.0405659101
370. McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007;26:3113-21 doi:10.1038/sj.onc.1210394
371. Dougherty MK, Ritt DA, Zhou M, Specht SI, Monson DM, Veenstra TD. et al. KSR2 is a calcineurin substrate that promotes ERK cascade activation in response to calcium signals. Mol Cell. 2009;34:652-62 doi:10.1016/j.molcel.2009.06.001
372. Costanzo-Garvey DL, Pfluger PT, Dougherty MK, Stock JL, Boehm M, Chaika O. et al. KSR2 is an essential regulator of AMP kinase, energy expenditure, and insulin sensitivity. Cell Metab. 2009;10:366-78 doi:10.1016/j.cmet.2009.09.010
373. Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X. et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008;10:138-48 doi:10.1038/ncb1676
374. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261-74 doi:10.1016/j.cell.2007.06.009
375. Tang ED, Nunez G, Barr FG, Guan KL. Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem. 1999;274:16741-6
376. Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410-25 doi:10.1038/sj.onc.1209086
377. Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501-14
378. Zhu J, Blenis J, Yuan J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc Natl Acad Sci U S A. 2008;105:6584-9 doi:10.1073/pnas.0802785105
379. Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9-22 doi:10.1038/nrm2822
380. Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123:569-80 doi:10.1016/j.cell.2005.10.024
381. van Gorp AG, van der Vos KE, Brenkman AB, Bremer A, van den Broek N, Zwartkruis F. et al. AGC kinases regulate phosphorylation and activation of eukaryotic translation initiation factor 4B. Oncogene. 2009;28:95-106 doi:10.1038/onc.2008.367
382. Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M. Regulation of Raf-Akt Cross-talk. J Biol Chem. 2002;277:31099-106 doi:10.1074/jbc.M111974200
383. Reusch HP, Zimmermann S, Schaefer M, Paul M, Moelling K. Regulation of Raf by Akt controls growth and differentiation in vascular smooth muscle cells. J Biol Chem. 2001;276:33630-7 doi:10.1074/jbc.M105322200
384. Rommel C, Clarke BA, Zimmermann S, Nunez L, Rossman R, Reid K. et al. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science. 1999;286:1738-41
385. Wang X, Hawk N, Yue P, Kauh J, Ramalingam SS, Fu H. et al. Overcoming mTOR inhibition-induced paradoxical activation of survival signaling pathways enhances mTOR inhibitors' anticancer efficacy. Cancer Biol Ther. 2008;7:1952-8
386. Mi R, Ma J, Zhang D, Li L, Zhang H. Efficacy of combined inhibition of mTOR and ERK/MAPK pathways in treating a tuberous sclerosis complex cell model. J Genet Genomics. 2009;36:355-61 doi:10.1016/s1673-8527(08)60124-1
387. Pavel M, Baudin E, Couvelard A, Krenning E, Oberg K, Steinmuller T. et al. ENETS Consensus Guidelines for the management of patients with liver and other distant metastases from neuroendocrine neoplasms of foregut, midgut, hindgut, and unknown primary. Neuroendocrinology. 2012;95:157-76 doi:10.1159/000335597
388. Raymond E, Hammel P, Dreyer C, Maatescu C, Hentic O, Ruszniewski P. et al. Sunitinib in pancreatic neuroendocrine tumors. Target Oncol. 2012;7:117-25 doi:10.1007/s11523-012-0220-2
389. Pavel ME, Hainsworth JD, Baudin E, Peeters M, Horsch D, Winkler RE. et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomised, placebo-controlled, phase 3 study. Lancet. 2011;378:2005-12 doi:10.1016/s0140-6736(11)61742-x
390. Strosberg JR, Chan JA, Ryan DP, Meyerhardt JA, Fuchs CS, Abrams T. et al. A multi-institutional, phase II open-label study of ganitumab (AMG 479) in advanced carcinoid and pancreatic neuroendocrine tumors. Endocr Relat Cancer. 2013;20:383-90 doi:10.1530/erc-12-0390
391. Bao XH, Takaoka M, Hao HF, Wang ZG, Fukazawa T, Yamatsuji T. et al. Esophageal cancer exhibits resistance to a novel IGF-1R inhibitor NVP-AEW541 with maintained RAS-MAPK activity. Anticancer Res. 2012;32:2827-34
392. Baumann P, Hagemeier H, Mandl-Weber S, Franke D, Schmidmaier R. Myeloma cell growth inhibition is augmented by synchronous inhibition of the insulin-like growth factor-1 receptor by NVP-AEW541 and inhibition of mammalian target of rapamycin by Rad001. Anticancer Drugs. 2009;20:259-66 doi:10.1097/CAD.0b013e328328d18b
393. Cunningham MP, Thomas H, Marks C, Green M, Fan Z, Modjtahedi H. Co-targeting the EGFR and IGF-IR with anti-EGFR monoclonal antibody ICR62 and the IGF-IR tyrosine kinase inhibitor NVP-AEW541 in colorectal cancer cells. Int J Oncol. 2008;33:1107-13
394. Garcia-Echeverria C, Pearson MA, Marti A, Meyer T, Mestan J, Zimmermann J. et al. In vivo antitumor activity of NVP-AEW541-A novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell. 2004;5:231-9
395. Gariboldi MB, Ravizza R, Monti E. The IGFR1 inhibitor NVP-AEW541 disrupts a pro-survival and pro-angiogenic IGF-STAT3-HIF1 pathway in human glioblastoma cells. Biochem Pharmacol. 2010;80:455-62 doi:10.1016/j.bcp.2010.05.011
396. Hagerstrand D, Lindh MB, Pena C, Garcia-Echeverria C, Nister M, Hofmann F. et al. PI3K/PTEN/Akt pathway status affects the sensitivity of high-grade glioma cell cultures to the insulin-like growth factor-1 receptor inhibitor NVP-AEW541. Neuro Oncol. 2010;12:967-75 doi:10.1093/neuonc/noq029
397. Ioannou N, Seddon AM, Dalgleish A, Mackintosh D, Modjtahedi H. Treatment with a combination of the ErbB (HER) family blocker afatinib and the IGF-IR inhibitor, NVP-AEW541 induces synergistic growth inhibition of human pancreatic cancer cells. BMC Cancer. 2013;13:41. doi:10.1186/1471-2407-13-41
398. Isebaert SF, Swinnen JV, McBride WH, Haustermans KM. Insulin-like growth factor-type 1 receptor inhibitor NVP-AEW541 enhances radiosensitivity of PTEN wild-type but not PTEN-deficient human prostate cancer cells. Int J Radiat Oncol Biol Phys. 2011;81:239-47 doi:10.1016/j.ijrobp.2011.03.030
399. Maiso P, Ocio EM, Garayoa M, Montero JC, Hofmann F, Garcia-Echeverria C. et al. The insulin-like growth factor-I receptor inhibitor NVP-AEW541 provokes cell cycle arrest and apoptosis in multiple myeloma cells. Br J Haematol. 2008;141:470-82 doi:10.1111/j.1365-2141.2008.07049.x
400. Moser C, Schachtschneider P, Lang SA, Gaumann A, Mori A, Zimmermann J. et al. Inhibition of insulin-like growth factor-I receptor (IGF-IR) using NVP-AEW541, a small molecule kinase inhibitor, reduces orthotopic pancreatic cancer growth and angiogenesis. Eur J Cancer. 2008;44:1577-86 doi:10.1016/j.ejca.2008.04.003
401. Mukohara T, Shimada H, Ogasawara N, Wanikawa R, Shimomura M, Nakatsura T. et al. Sensitivity of breast cancer cell lines to the novel insulin-like growth factor-1 receptor (IGF-1R) inhibitor NVP-AEW541 is dependent on the level of IRS-1 expression. Cancer Lett. 2009;282:14-24 doi:10.1016/j.canlet.2009.02.056
402. Premkumar DR, Jane EP, Pollack IF. Co-administration of NVP-AEW541 and dasatinib induces mitochondrial-mediated apoptosis through Bax activation in malignant human glioma cell lines. Int J Oncol. 2010;37:633-43
403. Tanno B, Mancini C, Vitali R, Mancuso M, McDowell HP, Dominici C. et al. Down-regulation of insulin-like growth factor I receptor activity by NVP-AEW541 has an antitumor effect on neuroblastoma cells in vitro and in vivo. Clin Cancer Res. 2006;12:6772-80 doi:10.1158/1078-0432.ccr-06-1479
404. Tazzari PL, Tabellini G, Bortul R, Papa V, Evangelisti C, Grafone T. et al. The insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 induces apoptosis in acute myeloid leukemia cells exhibiting autocrine insulin-like growth factor-I secretion. Leukemia. 2007;21:886-96 doi:10.1038/sj.leu.2404643
405. Zumsteg A, Caviezel C, Pisarsky L, Strittmatter K, Garcia-Echeverria C, Hofmann F. et al. Repression of malignant tumor progression upon pharmacologic IGF1R blockade in a mouse model of insulinoma. Mol Cancer Res. 2012;10:800-9 doi:10.1158/1541-7786.mcr-11-0522
406. He Y, Zhang J, Zheng J, Du W, Xiao H, Liu W. et al. The insulin-like growth factor-1 receptor kinase inhibitor, NVP-ADW742, suppresses survival and resistance to chemotherapy in acute myeloid leukemia cells. Oncol Res. 2010;19:35-43
407. Warshamana-Greene GS, Litz J, Buchdunger E, Garcia-Echeverria C, Hofmann F, Krystal GW. The insulin-like growth factor-I receptor kinase inhibitor, NVP-ADW742, sensitizes small cell lung cancer cell lines to the effects of chemotherapy. Clin Cancer Res. 2005;11:1563-71 doi:10.1158/1078-0432.ccr-04-1544
408. Warshamana-Greene GS, Litz J, Buchdunger E, Hofmann F, Garcia-Echeverria C, Krystal GW. The insulin-like growth factor-I (IGF-I) receptor kinase inhibitor NVP-ADW742, in combination with STI571, delineates a spectrum of dependence of small cell lung cancer on IGF-I and stem cell factor signaling. Mol Cancer Ther. 2004;3:527-35
409. Zhou H, Rao J, Lin J, Yin B, Sheng H, Lin F. et al. The insulin-like growth factor-I receptor kinase inhibitor NVP-ADW742 sensitizes medulloblastoma to the effects of chemotherapy. Oncol Rep. 2011;25:1565-71 doi:10.3892/or.2011.1233
410. Beauchamp MC, Knafo A, Yasmeen A, Carboni JM, Gottardis MM, Pollak MN. et al. BMS-536924 sensitizes human epithelial ovarian cancer cells to the PARP inhibitor, 3-aminobenzamide. Gynecol Oncol. 2009;115:193-8 doi:10.1016/j.ygyno.2009.07.009
411. Dool CJ, Mashhedi H, Zakikhani M, David S, Zhao Y, Birman E. et al. IGF1/insulin receptor kinase inhibition by BMS-536924 is better tolerated than alloxan-induced hypoinsulinemia and more effective than metformin in the treatment of experimental insulin-responsive breast cancer. Endocr Relat Cancer. 2011;18:699-709 doi:10.1530/erc-11-0136
412. Hou X, Huang F, Carboni JM, Flatten K, Asmann YW, Ten Eyck C. et al. Drug efflux by breast cancer resistance protein is a mechanism of resistance to the benzimidazole insulin-like growth factor receptor/insulin receptor inhibitor, BMS-536924. Mol Cancer Ther. 2011;10:117-25 doi:10.1158/1535-7163.mct-10-0438
413. Huang F, Greer A, Hurlburt W, Han X, Hafezi R, Wittenberg GM. et al. The mechanisms of differential sensitivity to an insulin-like growth factor-1 receptor inhibitor (BMS-536924) and rationale for combining with EGFR/HER2 inhibitors. Cancer Res. 2009;69:161-70 doi:10.1158/0008-5472.can-08-0835
414. Wahner Hendrickson AE, Haluska P, Schneider PA, Loegering DA, Peterson KL, Attar R. et al. Expression of insulin receptor isoform A and insulin-like growth factor-1 receptor in human acute myelogenous leukemia: effect of the dual-receptor inhibitor BMS-536924 in vitro. Cancer Res. 2009;69:7635-43 doi:10.1158/0008-5472.can-09-0511
415. Gilbert JA, Adhikari LJ, Lloyd RV, Rubin J, Haluska P, Carboni JM. et al. Molecular markers for novel therapies in neuroendocrine (carcinoid) tumors. Endocr Relat Cancer. 2010;17:623-36 doi:10.1677/erc-09-0318
416. Svejda B, Kidd M, Timberlake A, Harry K, Kazberouk A, Schimmack S. et al. Serotonin and the 5-HT7 receptor: The link between hepatocytes, IGF-1 and small intestinal neuroendocrine tumors. Cancer Sci. 2013 doi:10.1111/cas.12174
417. Gilbert JA, Lloyd RV, Ames MM. Lack of mutations in EGFR in gastroenteropancreatic neuroendocrine tumors. N Engl J Med. 2005;353:209-10 doi:10.1056/nejm200507143530219
418. Bojko A, Reichert K, Adamczyk A, Ligeza J, Ligeza J, Klein A. The effect of tyrphostins AG494 and AG1478 on the autocrine growth regulation of A549 and DU145 cells. Folia Histochem Cytobiol. 2012;50:186-95
419. Yan Y, Ai Z, Wang J, Xu Y, Teng Y. Influence of epidermal growth factor receptor inhibitor AG1478 on epithelial-mesenchymal transition in endometrial carcinoma cells. Int J Gynecol Cancer. 2012;22:1457-62 doi:10.1097/IGC.0b013e31826ed2be
420. Jin X, Jin X, Sohn YW, Yin J, Kim SH, Joshi K. et al. Blockade of EGFR signaling promotes glioma stem-like cell invasiveness by abolishing ID3-mediated inhibition of p27(KIP1) and MMP3 expression. Cancer Lett. 2013;328:235-42 doi:10.1016/j.canlet.2012.09.005
421. Eriksson B, Janson ET, Bax ND, Mignon M, Morant R, Opolon P. et al. The use of new somatostatin analogues, lanreotide and octastatin, in neuroendocrine gastro-intestinal tumours. Digestion. 1996;57(Suppl 1):77-80
422. Cap J, Marekova M, Cerman J, Malirova E, Suba P, Netuka D. et al. Inhibition of hormone secretion in GH-secreting pituitary adenomas by receptor-subtype specific somatostatin analogues in vitro. Gen Physiol Biophys. 2003;22:201-12
423. Duran-Prado M, Saveanu A, Luque RM, Gahete MD, Gracia-Navarro F, Jaquet P. et al. A potential inhibitory role for the new truncated variant of somatostatin receptor 5, sst5TMD4, in pituitary adenomas poorly responsive to somatostatin analogs. J Clin Endocrinol Metab. 2010;95:2497-502 doi:10.1210/jc.2009-2247
424. Saveanu A, Gunz G, Guillen S, Dufour H, Culler MD, Jaquet P. Somatostatin and dopamine-somatostatin multiple ligands directed towards somatostatin and dopamine receptors in pituitary adenomas. Neuroendocrinology. 2006;83:258-63 doi:10.1159/000095536
425. Zatelli MC, Tagliati F, Taylor JE, Rossi R, Culler MD, degli Uberti EC. Somatostatin receptor subtypes 2 and 5 differentially affect proliferation in vitro of the human medullary thyroid carcinoma cell line tt. J Clin Endocrinol Metab. 2001;86:2161-9
426. Bocci G, Culler MD, Fioravanti A, Orlandi P, Fasciani A, Colucci R. et al. In vitro antiangiogenic activity of selective somatostatin subtype-1 receptor agonists. Eur J Clin Invest. 2007;37:700-8 doi:10.1111/j.1365-2362.2007.01848.x
427. Fusco A, Gunz G, Jaquet P, Dufour H, Germanetti AL, Culler MD. et al. Somatostatinergic ligands in dopamine-sensitive and -resistant prolactinomas. Eur J Endocrinol. 2008;158:595-603 doi:10.1530/eje-07-0806
428. Gruszka A, Kunert-Radek J, Radek A, Pisarek H, Taylor J, Dong JZ. et al. The effect of selective sst1, sst2, sst5 somatostatin receptors agonists, a somatostatin/dopamine (SST/DA) chimera and bromocriptine on the "clinically non-functioning" pituitary adenomas in vitro. Life Sci. 2006;78:689-93 doi:10.1016/j.lfs.2005.05.061
429. Ruscica M, Arvigo M, Gatto F, Dozio E, Feltrin D, Culler MD. et al. Regulation of prostate cancer cell proliferation by somatostatin receptor activation. Mol Cell Endocrinol. 2010;315:254-62 doi:10.1016/j.mce.2009.11.006
430. Ludvigsen E, Stridsberg M, Taylor JE, Culler MD, Oberg K, Janson ET. Subtype selective interactions of somatostatin and somatostatin analogs with sst1, sst2, and sst5 in BON-1 cells. Med Oncol. 2004;21:285-95 doi:10.1385/mo:21:3:285
431. Barbieri F, Pattarozzi A, Gatti M, Aiello C, Quintero A, Lunardi G. et al. Differential efficacy of SSTR1, -2, and -5 agonists in the inhibition of C6 glioma growth in nude mice. Am J Physiol Endocrinol Metab. 2009;297:E1078-88 doi:10.1152/ajpendo.00292.2009
432. Barbieri F, Pattarozzi A, Gatti M, Porcile C, Bajetto A, Ferrari A. et al. Somatostatin receptors 1, 2, and 5 cooperate in the somatostatin inhibition of C6 glioma cell proliferation in vitro via a phosphotyrosine phosphatase-eta-dependent inhibition of extracellularly regulated kinase-1/2. Endocrinology. 2008;149:4736-46 doi:10.1210/en.2007-1762
433. Gruszka A, Culler MD, Melmed S. Somatostatin analogs and chimeric somatostatin-dopamine molecules differentially regulate human growth hormone and prolactin gene expression and secretion in vitro. Mol Cell Endocrinol. 2012;362:104-9 doi:10.1016/j.mce.2012.05.020
434. Zatelli MC, Tagliati F, Piccin D, Taylor JE, Culler MD, Bondanelli M. et al. Somatostatin receptor subtype 1-selective activation reduces cell growth and calcitonin secretion in a human medullary thyroid carcinoma cell line. Biochem Biophys Res Commun. 2002;297:828-34
435. Tallent M, Liapakis G, O'Carroll AM, Lolait SJ, Dichter M, Reisine T. Somatostatin receptor subtypes SSTR2 and SSTR5 couple negatively to an L-type Ca2+ current in the pituitary cell line AtT-20. Neuroscience. 1996;71:1073-81
436. Prevost G, Veber N, Viollet C, Roubert V, Roubert P, Benard J. et al. Somatostatin-14 mainly binds the somatostatin receptor subtype 2 in human neuroblastoma tumors. Neuroendocrinology. 1996;63:188-97
437. Cescato R, Loesch KA, Waser B, Macke HR, Rivier JE, Reubi JC. et al. Agonist-biased signaling at the sst2A receptor: the multi-somatostatin analogs KE108 and SOM230 activate and antagonize distinct signaling pathways. Mol Endocrinol. 2010;24:240-9 doi:10.1210/me.2009-0321
438. Cescato R, Schulz S, Waser B, Eltschinger V, Rivier JE, Wester HJ. et al. Internalization of sst2, sst3, and sst5 receptors: effects of somatostatin agonists and antagonists. J Nucl Med. 2006;47:502-11
439. Mentlein R, Eichler O, Forstreuter F, Held-Feindt J. Somatostatin inhibits the production of vascular endothelial growth factor in human glioma cells. Int J Cancer. 2001;92:545-50
440. Stark A, Mentlein R. Somatostatin inhibits glucagon-like peptide-1-induced insulin secretion and proliferation of RINm5F insulinoma cells. Regul Pept. 2002;108:97-102
441. Yang SK, Parkington HC, Blake AD, Keating DJ, Chen C. Somatostatin increases voltage-gated K+ currents in GH3 cells through activation of multiple somatostatin receptors. Endocrinology. 2005;146:4975-84 doi:10.1210/en.2005-0696
442. Arvigo M, Gatto F, Ruscica M, Ameri P, Dozio E, Albertelli M. et al. Somatostatin and dopamine receptor interaction in prostate and lung cancer cell lines. J Endocrinol. 2010;207:309-17 doi:10.1677/joe-10-0342
443. Florio T, Barbieri F, Spaziante R, Zona G, Hofland LJ, van Koetsveld PM. et al. Efficacy of a dopamine-somatostatin chimeric molecule, BIM-23A760, in the control of cell growth from primary cultures of human non-functioning pituitary adenomas: a multi-center study. Endocr Relat Cancer. 2008;15:583-96 doi:10.1677/erc-07-0271
444. Ferone D, Arvigo M, Semino C, Jaquet P, Saveanu A, Taylor JE. et al. Somatostatin and dopamine receptor expression in lung carcinoma cells and effects of chimeric somatostatin-dopamine molecules on cell proliferation. Am J Physiol Endocrinol Metab. 2005;289:E1044-50 doi:10.1152/ajpendo.00209.2005
445. Gatto F, Barbieri F, Gatti M, Wurth R, Schulz S, Ravetti JL. et al. Balance between somatostatin and D2 receptor expression drives TSH-secreting adenoma response to somatostatin analogues and dopastatins. Clin Endocrinol (Oxf). 2012;76:407-14 doi:10.1111/j.1365-2265.2011.04200.x
446. Ren SG, Kim S, Taylor J, Dong J, Moreau JP, Culler MD. et al. Suppression of rat and human growth hormone and prolactin secretion by a novel somatostatin/dopaminergic chimeric ligand. J Clin Endocrinol Metab. 2003;88:5414-21
447. Saveanu A, Lavaque E, Gunz G, Barlier A, Kim S, Taylor JE. et al. Demonstration of enhanced potency of a chimeric somatostatin-dopamine molecule, BIM-23A387, in suppressing growth hormone and prolactin secretion from human pituitary somatotroph adenoma cells. J Clin Endocrinol Metab. 2002;87:5545-52
448. Gruszka A, Ren SG, Dong J, Culler MD, Melmed S. Regulation of growth hormone and prolactin gene expression and secretion by chimeric somatostatin-dopamine molecules. Endocrinology. 2007;148:6107-14 doi:10.1210/en.2007-0378
449. Chen L, Han L, Shi Z, Zhang K, Liu Y, Zheng Y. et al. LY294002 enhances cytotoxicity of temozolomide in glioma by down-regulation of the PI3K/Akt pathway. Mol Med Rep. 2012;5:575-9 doi:10.3892/mmr.2011.674
450. Gong C, Liao H, Wang J, Lin Y, Qi J, Qin L. et al. LY294002 induces G0/G1 cell cycle arrest and apoptosis of cancer stem-like cells from human osteosarcoma via down-regulation of PI3K activity. Asian Pac J Cancer Prev. 2012;13:3103-7
451. Guo RX, Zhang RF, Wang XY, Shi HR, Qiao YH. [Effects of PD98059 and LY294002 on subcutaneous xenograft of human endometrial carcinoma in nude mice]. Zhonghua Fu Chan Ke Za Zhi. 2011;46:446-52
452. Li C, Liu VW, Chan DW, Yao KM, Ngan HY. LY294002 and metformin cooperatively enhance the inhibition of growth and the induction of apoptosis of ovarian cancer cells. Int J Gynecol Cancer. 2012;22:15-22 doi:10.1097/IGC.0b013e3182322834
453. Liu J, Fu XQ, Zhou W, Yu HG, Yu JP, Luo HS. LY294002 potentiates the anti-cancer effect of oxaliplatin for gastric cancer via death receptor pathway. World J Gastroenterol. 2011;17:181-90 doi:10.3748/wjg.v17.i2.181
454. Sun JL, Zhu BS, Gong W, Zhang P, Yu LY, Zhao K. et al. [Effects of combined therapy of LY294002 and SN50 on nude mice model with gastric cancer]. Zhonghua Wei Chang Wai Ke Za Zhi. 2011;14:364-7
455. Wempe SL, Gamarra-Luques CD, Telleria CM. Synergistic lethality of mifepristone and LY294002 in ovarian cancer cells. Cancer Growth Metastasis. 2013;6:1-13 doi:10.4137/cgm.s11124
456. Wu D, Tao J, Xu B, Qing W, Li P, Lu Q. et al. Phosphatidylinositol 3-kinase inhibitor LY294002 suppresses proliferation and sensitizes doxorubicin chemotherapy in bladder cancer cells. Urol Int. 2011;87:105-13 doi:10.1159/000322849
457. Yoshioka T, Yogosawa S, Yamada T, Kitawaki J, Sakai T. Combination of a novel HDAC inhibitor OBP-801/YM753 and a PI3K inhibitor LY294002 synergistically induces apoptosis in human endometrial carcinoma cells due to increase of Bim with accumulation of ROS. Gynecol Oncol. 2013;129:425-32 doi:10.1016/j.ygyno.2013.02.008
458. Al-Rawi MA, Rmali K, Mansel RE, Jiang WG. Interleukin 7 induces the growth of breast cancer cells through a wortmannin-sensitive pathway. Br J Surg. 2004;91:61-8 doi:10.1002/bjs.4449
459. Boehle AS, Kurdow R, Boenicke L, Schniewind B, Faendrich F, Dohrmann P. et al. Wortmannin inhibits growth of human non-small-cell lung cancer in vitro and in vivo. Langenbecks Arch Surg. 2002;387:234-9 doi:10.1007/s00423-002-0314-x
460. Li J, Li F, Wang H, Wang X, Jiang Y, Li D. Wortmannin reduces metastasis and angiogenesis of human breast cancer cells via nuclear factor-kappaB-dependent matrix metalloproteinase-9 and interleukin-8 pathways. J Int Med Res. 2012;40:867-76
461. Teranishi F, Takahashi N, Gao N, Akamo Y, Takeyama H, Manabe T. et al. Phosphoinositide 3-kinase inhibitor (wortmannin) inhibits pancreatic cancer cell motility and migration induced by hyaluronan in vitro and peritoneal metastasis in vivo. Cancer Sci. 2009;100:770-7
462. Yun J, Lv YG, Yao Q, Wang L, Li YP, Yi J. Wortmannin inhibits proliferation and induces apoptosis of MCF-7 breast cancer cells. Eur J Gynaecol Oncol. 2012;33:367-9
463. Zhang F, Zhang T, Jiang T, Zhang R, Teng ZH, Li C. et al. Wortmannin potentiates roscovitine-induced growth inhibition in human solid tumor cells by repressing PI3K/Akt pathway. Cancer Lett. 2009;286:232-9 doi:10.1016/j.canlet.2009.05.039
464. Bagci-Onder T, Wakimoto H, Anderegg M, Cameron C, Shah K. A dual PI3K/mTOR inhibitor, PI-103, cooperates with stem cell-delivered TRAIL in experimental glioma models. Cancer Res. 2011;71:154-63 doi:10.1158/0008-5472.can-10-1601
465. Gedaly R, Angulo P, Hundley J, Daily MF, Chen C, Koch A. et al. PI-103 and sorafenib inhibit hepatocellular carcinoma cell proliferation by blocking Ras/Raf/MAPK and PI3K/AKT/mTOR pathways. Anticancer Res. 2010;30:4951-8
466. Kojima K, Shimanuki M, Shikami M, Samudio IJ, Ruvolo V, Corn P. et al. The dual PI3 kinase/mTOR inhibitor PI-103 prevents p53 induction by Mdm2 inhibition but enhances p53-mediated mitochondrial apoptosis in p53 wild-type AML. Leukemia. 2008;22:1728-36 doi:10.1038/leu.2008.158
467. Lopez-Fauqued M, Gil R, Grueso J, Hernandez-Losa J, Pujol A, Moline T. et al. The dual PI3K/mTOR inhibitor PI-103 promotes immunosuppression, in vivo tumor growth and increases survival of sorafenib-treated melanoma cells. Int J Cancer. 2010;126:1549-61 doi:10.1002/ijc.24926
468. Park S, Chapuis N, Bardet V, Tamburini J, Gallay N, Willems L. et al. PI-103, a dual inhibitor of Class IA phosphatidylinositide 3-kinase and mTOR, has antileukemic activity in AML. Leukemia. 2008;22:1698-706 doi:10.1038/leu.2008.144
469. Zou ZQ, Zhang XH, Wang F, Shen QJ, Xu J, Zhang LN. et al. A novel dual PI3Kalpha/mTOR inhibitor PI-103 with high antitumor activity in non-small cell lung cancer cells. Int J Mol Med. 2009;24:97-101
470. Zhao Y, Duan S, Zeng X, Liu C, Davies NM, Li B. et al. Prodrug strategy for PSMA-targeted delivery of TGX-221 to prostate cancer cells. Mol Pharm. 2012;9:1705-16 doi:10.1021/mp3000309
471. Meuillet EJ, Zuohe S, Lemos R, Ihle N, Kingston J, Watkins R. et al. Molecular pharmacology and antitumor activity of PHT-427, a novel Akt/phosphatidylinositide-dependent protein kinase 1 pleckstrin homology domain inhibitor. Mol Cancer Ther. 2010;9:706-17 doi:10.1158/1535-7163.mct-09-0985
472. Reid JM, Walden CA, Qin R, Ziegler KL, Haslam JL, Rajewski RA. et al. Phase 0 clinical chemoprevention trial of the Akt inhibitor SR13668. Cancer Prev Res (Phila). 2011;4:347-53 doi:10.1158/1940-6207.capr-10-0313
473. Mandal M, Kim S, Younes MN, Jasser SA, El-Naggar AK, Mills GB. et al. The Akt inhibitor KP372-1 suppresses Akt activity and cell proliferation and induces apoptosis in thyroid cancer cells. Br J Cancer. 2005;92:1899-905 doi:10.1038/sj.bjc.6602595
474. Mandal M, Younes M, Swan EA, Jasser SA, Doan D, Yigitbasi O. et al. The Akt inhibitor KP372-1 inhibits proliferation and induces apoptosis and anoikis in squamous cell carcinoma of the head and neck. Oral Oncol. 2006;42:430-9 doi:10.1016/j.oraloncology.2005.09.011
475. Zeng Z, Samudio IJ, Zhang W, Estrov Z, Pelicano H, Harris D. et al. Simultaneous inhibition of PDK1/AKT and Fms-like tyrosine kinase 3 signaling by a small-molecule KP372-1 induces mitochondrial dysfunction and apoptosis in acute myelogenous leukemia. Cancer Res. 2006;66:3737-46 doi:10.1158/0008-5472.can-05-1278
476. de Frias M, Iglesias-Serret D, Cosialls AM, Coll-Mulet L, Santidrian AF, Gonzalez-Girones DM. et al. Akt inhibitors induce apoptosis in chronic lymphocytic leukemia cells. Haematologica. 2009;94:1698-707 doi:10.3324/haematol.2008.004028
477. Gallia GL, Tyler BM, Hann CL, Siu IM, Giranda VL, Vescovi AL. et al. Inhibition of Akt inhibits growth of glioblastoma and glioblastoma stem-like cells. Mol Cancer Ther. 2009;8:386-93 doi:10.1158/1535-7163.mct-08-0680
478. Han EK, Leverson JD, McGonigal T, Shah OJ, Woods KW, Hunter T. et al. Akt inhibitor A-443654 induces rapid Akt Ser-473 phosphorylation independent of mTORC1 inhibition. Oncogene. 2007;26:5655-61 doi:10.1038/sj.onc.1210343
479. Liu X, Shi Y, Woods KW, Hessler P, Kroeger P, Wilsbacher J. et al. Akt inhibitor a-443654 interferes with mitotic progression by regulating aurora a kinase expression. Neoplasia. 2008;10:828-37
480. Somnay Y, Simon K, Harrison AD, Kunnimalaiyaan S, Chen H, Kunnimalaiyaan M. Neuroendocrine phenotype alteration and growth suppression through apoptosis by MK-2206, an allosteric inhibitor of AKT, in carcinoid cell lines in vitro. Anticancer Drugs. 2013;24:66-72 doi:10.1097/CAD.0b013e3283584f75
481. Feldman RI, Wu JM, Polokoff MA, Kochanny MJ, Dinter H, Zhu D. et al. Novel small molecule inhibitors of 3-phosphoinositide-dependent kinase-1. J Biol Chem. 2005;280:19867-74 doi:10.1074/jbc.M501367200
482. Duran I, Kortmansky J, Singh D, Hirte H, Kocha W, Goss G. et al. A phase II clinical and pharmacodynamic study of temsirolimus in advanced neuroendocrine carcinomas. Br J Cancer. 2006;95:1148-54 doi:10.1038/sj.bjc.6603419
483. Eum KH, Ahn SK, Kang H, Lee M. Differential inhibitory effects of two Raf-targeting drugs, sorafenib and PLX4720, on the growth of multidrug-resistant cells. Mol Cell Biochem. 2013;372:65-74 doi:10.1007/s11010-012-1446-0
484. Jiang CC, Lai F, Thorne RF, Yang F, Liu H, Hersey P. et al. MEK-independent survival of B-RAFV600E melanoma cells selected for resistance to apoptosis induced by the RAF inhibitor PLX4720. Clin Cancer Res. 2011;17:721-30 doi:10.1158/1078-0432.ccr-10-2225
485. Nehs MA, Nagarkatti S, Nucera C, Hodin RA, Parangi S. Thyroidectomy with neoadjuvant PLX4720 extends survival and decreases tumor burden in an orthotopic mouse model of anaplastic thyroid cancer. Surgery. 2010;148:1154-62 discussion 62. doi:10.1016/j.surg.2010.09.001
486. Nucera C, Nehs MA, Nagarkatti SS, Sadow PM, Mekel M, Fischer AH. et al. Targeting BRAFV600E with PLX4720 displays potent antimigratory and anti-invasive activity in preclinical models of human thyroid cancer. Oncologist. 2011;16:296-309 doi:10.1634/theoncologist.2010-0317
487. Oikonomou E, Koc M, Sourkova V, Andera L, Pintzas A. Selective BRAFV600E inhibitor PLX4720, requires TRAIL assistance to overcome oncogenic PIK3CA resistance. PLoS One. 2011;6:e21632. doi:10.1371/journal.pone.0021632
488. Shao Y, Aplin AE. BH3-only protein silencing contributes to acquired resistance to PLX4720 in human melanoma. Cell Death Differ. 2012;19:2029-39 doi:10.1038/cdd.2012.94
489. Gibney GT, Zager JS. Clinical development of dabrafenib in BRAF mutant melanoma and other malignancies. Expert Opin Drug Metab Toxicol. 2013;9:893-9 doi:10.1517/17425255.2013.794220
490. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M. et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-65 doi:10.1016/s0140-6736(12)60868-x
491. Long GV, Trefzer U, Davies MA, Kefford RF, Ascierto PA, Chapman PB. et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13:1087-95 doi:10.1016/s1470-2045(12)70431-x
492. Lee NV, Lira ME, Pavlicek A, Ye J, Buckman D, Bagrodia S. et al. A novel SND1-BRAF fusion confers resistance to c-Met inhibitor PF-04217903 in GTL16 cells though MAPK activation. PLoS One. 2012;7:e39653. doi:10.1371/journal.pone.0039653
493. Torti VR, Wojciechowicz D, Hu W, John-Baptiste A, Evering W, Troche G. et al. Epithelial tissue hyperplasia induced by the RAF inhibitor PF-04880594 is attenuated by a clinically well-tolerated dose of the mek inhibitor PD-0325901. Mol Cancer Ther. 2012;11:2274-83 doi:10.1158/1535-7163.mct-11-0984
494. Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE. et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853-61 doi:10.1158/0008-5472.can-07-6787
495. Whittaker SR, Theurillat JP, Van Allen E, Wagle N, Hsiao J, Cowley GS. et al. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013;3:350-62 doi:10.1158/2159-8290.cd-12-0470
496. Baines P, Fisher J, Truran L, Davies E, Hallett M, Hoy T. et al. The MEK inhibitor, PD98059, reduces survival but does not block acute myeloid leukemia blast maturation in vitro. Eur J Haematol. 2000;64:211-8
497. Kanda S, Kanetake H, Miyata Y. Long-term exposure of human renal carcinoma cells to PD98059 induces epithelial-mesenchymal transition-like phenotype and enhanced motility. Mol Cell Biochem. 2008;309:69-76 doi:10.1007/s11010-007-9644-x
498. Mandic A, Viktorsson K, Heiden T, Hansson J, Shoshan MC. The MEK1 inhibitor PD98059 sensitizes C8161 melanoma cells to cisplatin-induced apoptosis. Melanoma Res. 2001;11:11-9
499. Xu LL, Mei JH, Chen JX, Xu S, Qin HY, Wang SS. [Study the role of PD98059 in ovarian carcinoma cell line HO-8910]. Zhonghua Bing Li Xue Za Zhi. 2008;37:625-6
500. Yao J, Qian C, Shu T, Zhang X, Zhao Z, Liang Y. Combination treatment of PD98059 and DAPT in gastric cancer through induction of apoptosis and downregulation of WNT/beta-catenin. Cancer Biol Ther. 2013;14(9):833-839
501. Zhang YJ, Fang JY, Sun DF, Zhao SL, Shen GF, Zheng Q. et al. [Synergistic effect of rapamycin (RPM) and PD98059 on cell cycle and mTOR signal transduction in human colorectal cancer cells]. Zhonghua Zhong Liu Za Zhi. 2007;29:889-93
502. Flores LG 2nd, Yeh HH, Soghomonyan S, Young D, Bankson J, Hu Q. et al. Monitoring therapy with MEK inhibitor U0126 in a novel Wilms tumor model in Wt1 knockout Igf2 transgenic mice using 18F-FDG PET with dual-contrast enhanced CT and MRI: early metabolic response without inhibition of tumor growth. Mol Imaging Biol. 2013;15:175-85 doi:10.1007/s11307-012-0588-5
503. Freeman MR, Kim J, Lisanti MP, Di Vizio D. A metabolic perturbation by U0126 identifies a role for glutamine in resveratrol-induced cell death. Cancer Biol Ther. 2011;12:966-77 doi:10.4161/cbt.12.11.18136
504. Ge X, Fu YM, Meadows GG. U0126, a mitogen-activated protein kinase kinase inhibitor, inhibits the invasion of human A375 melanoma cells. Cancer Lett. 2002;179:133-40
505. Georgakis GV, Li Y, Rassidakis GZ, Medeiros LJ, Younes A. The HSP90 inhibitor 17-AAG synergizes with doxorubicin and U0126 in anaplastic large cell lymphoma irrespective of ALK expression. Exp Hematol. 2006;34:1670-9 doi:10.1016/j.exphem.2006.07.002
506. Hong SK, Kim JH, Lin MF, Park JI. The Raf/MEK/extracellular signal-regulated kinase 1/2 pathway can mediate growth inhibitory and differentiation signaling via androgen receptor downregulation in prostate cancer cells. Exp Cell Res. 2011;317:2671-82 doi:10.1016/j.yexcr.2011.08.008
507. Horiuchi H, Kawamata H, Fujimori T, Kuroda Y. A MEK inhibitor (U0126) prolongs survival in nude mice bearing human gallbladder cancer cells with K-ras mutation: analysis in a novel orthotopic inoculation model. Int J Oncol. 2003;23:957-63
508. Horiuchi H, Kawamata H, Furihata T, Omotehara F, Hori H, Shinagawa Y. et al. A MEK inhibitor (U0126) markedly inhibits direct liver invasion of orthotopically inoculated human gallbladder cancer cells in nude mice. J Exp Clin Cancer Res. 2004;23:599-606
509. Ito M, Zhao N, Zeng Z, Chang CC, Zu Y. Synergistic growth inhibition of anaplastic large cell lymphoma cells by combining cellular ALK gene silencing and a low dose of the kinase inhibitor U0126. Cancer Gene Ther. 2010;17:633-44 doi:10.1038/cgt.2010.20
510. James JA, Smith MA, Court EL, Yip C, Ching Y, Willson C. et al. An investigation of the effects of the MEK inhibitor U0126 on apoptosis in acute leukemia. Hematol J. 2003;4:427-32 doi:10.1038/sj.thj.6200327
511. Kerr AH, James JA, Smith MA, Willson C, Court EL, Smith JG. An investigation of the MEK/ERK inhibitor U0126 in acute myeloid leukemia. Ann N Y Acad Sci. 2003;1010:86-9
512. Lind CR, Gray CW, Pearson AG, Cameron RE, O'Carroll SJ, Narayan PJ. et al. The mitogen-activated/extracellular signal-regulated kinase kinase 1/2 inhibitor U0126 induces glial fibrillary acidic protein expression and reduces the proliferation and migration of C6 glioma cells. Neuroscience. 2006;141:1925-33 doi:10.1016/j.neuroscience.2006.05.038
513. Marampon F, Gravina GL, Di Rocco A, Bonfili P, Di Staso M, Fardella C. et al. MEK/ERK inhibitor U0126 increases the radiosensitivity of rhabdomyosarcoma cells in vitro and in vivo by downregulating growth and DNA repair signals. Mol Cancer Ther. 2011;10:159-68 doi:10.1158/1535-7163.mct-10-0631
514. Yip-Schneider MT, Schmidt CM. MEK inhibition of pancreatic carcinoma cells by U0126 and its effect in combination with sulindac. Pancreas. 2003;27:337-44
515. Zou ZQ, Zhang LN, Wang F, Bellenger J, Shen YZ, Zhang XH. The novel dual PI3K/mTOR inhibitor GDC-0941 synergizes with the MEK inhibitor U0126 in non-small cell lung cancer cells. Mol Med Rep. 2012;5:503-8 doi:10.3892/mmr.2011.682
516. Iida S, Miki Y, Ono K, Akahira J, Nakamura Y, Suzuki T. et al. Synergistic anti-tumor effects of RAD001 with MEK inhibitors in neuroendocrine tumors: a potential mechanism of therapeutic limitation of mTOR inhibitor. Mol Cell Endocrinol. 2012;350:99-106 doi:10.1016/j.mce.2011.11.024
517. Kunnimalaiyaan M, Ndiaye M, Chen H. Neuroendocrine tumor cell growth inhibition by ZM336372 through alterations in multiple signaling pathways. Surgery. 2007;142:959-64 discussion -64. doi:10.1016/j.surg.2007.09.020
518. Hail N Jr, Chen P, Bushman LR. Teriflunomide (leflunomide) promotes cytostatic, antioxidant, and apoptotic effects in transformed prostate epithelial cells: evidence supporting a role for teriflunomide in prostate cancer chemoprevention. Neoplasia. 2010;12:464-75
519. Hail N Jr, Chen P, Rower J, Bushman LR. Teriflunomide encourages cytostatic and apoptotic effects in premalignant and malignant cutaneous keratinocytes. Apoptosis. 2010;15:1234-46 doi:10.1007/s10495-010-0518-4
520. Adler JT, Cook M, Luo Y, Pitt SC, Ju J, Li W. et al. Tautomycetin and tautomycin suppress the growth of medullary thyroid cancer cells via inhibition of glycogen synthase kinase-3beta. Mol Cancer Ther. 2009;8:914-20 doi:10.1158/1535-7163.mct-08-0712
521. Magae J, Watanabe C, Osada H, Cheng XC, Isono K. Induction of morphological change of human myeloid leukemia and activation of protein kinase C by a novel antibiotic, tautomycin. J Antibiot (Tokyo). 1988;41:932-7
522. Suganuma M, Okabe S, Sueoka E, Nishiwaki R, Komori A, Uda N. et al. Tautomycin: an inhibitor of protein phosphatases 1 and 2A but not a tumor promoter on mouse skin and in rat glandular stomach. J Cancer Res Clin Oncol. 1995;121:621-7
523. Kappes A, Vaccaro A, Kunnimalaiyaan M, Chen H. ZM336372, a Raf-1 activator, inhibits growth of pheochromocytoma cells. J Surg Res. 2006;133:42-5 doi:10.1016/j.jss.2006.02.002
524. Tatake RJ, O'Neill MM, Kennedy CA, Wayne AL, Jakes S, Wu D. et al. Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem Biophys Res Commun. 2008;377:120-5 doi:10.1016/j.bbrc.2008.09.087
525. Razumovskaya E, Sun J, Ronnstrand L. Inhibition of MEK5 by BIX02188 induces apoptosis in cells expressing the oncogenic mutant FLT3-ITD. Biochem Biophys Res Commun. 2011;412:307-12 doi:10.1016/j.bbrc.2011.07.089
526. Yang Q, Lee JD. Targeting the BMK1 MAP kinase pathway in cancer therapy. Clin Cancer Res. 2011;17:3527-32 doi:10.1158/1078-0432.ccr-10-2504
527. Weekes CD, Von Hoff DD, Adjei AA, Leffingwell DP, Eckhardt SG, Gore L. et al. Multicenter phase I trial of the mitogen-activated protein kinase 1/2 inhibitor BAY 86-9766 in patients with advanced cancer. Clin Cancer Res. 2013;19:1232-43 doi:10.1158/1078-0432.ccr-12-3529
528. Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A. et al. Discovery of a Novel ERK Inhibitor with Activity in Models of Acquired Resistance to BRAF and MEK Inhibitors. Cancer Discov. 2013 doi:10.1158/2159-8290.cd-13-0070
529. Li H, Wang X, Chen T, Qu J. p38 inhibitor SB203580 sensitizes the resveratrol-induced apoptosis in human lung adenocarcinoma (A549) cells. J Biochem Mol Toxicol. 2012;26:251-7 doi:10.1002/jbt.21413
530. Zhao YM, Su B, Yang XJ, Shi JY, Tang L, Zhang J. et al. [Small molecule inhibitor SB203580 enhances the antitumor effect of gefitinib in PC-9 and A549 lung cancer cell lines]. Zhonghua Zhong Liu Za Zhi. 2013;35:103-8
531. He D, Zhao XQ, Chen XG, Fang Y, Singh S, Talele TT. et al. BIRB796, the inhibitor of p38 mitogen-activated protein kinase, enhances the efficacy of chemotherapeutic agents in ABCB1 overexpression cells. PLoS One. 2013;8:e54181. doi:10.1371/journal.pone.0054181
532. Yasui H, Hideshima T, Ikeda H, Jin J, Ocio EM, Kiziltepe T. et al. BIRB 796 enhances cytotoxicity triggered by bortezomib, heat shock protein (Hsp) 90 inhibitor, and dexamethasone via inhibition of p38 mitogen-activated protein kinase/Hsp27 pathway in multiple myeloma cell lines and inhibits paracrine tumour growth. Br J Haematol. 2007;136:414-23 doi:10.1111/j.1365-2141.2006.06443.x
533. Grossi V, Liuzzi M, Murzilli S, Martelli N, Napoli A, Ingravallo G. et al. Sorafenib inhibits p38alpha activity in colorectal cancer cells and synergizes with the DFG-in inhibitor SB202190 to increase apoptotic response. Cancer Biol Ther. 2012;13:1471-81 doi:10.4161/cbt.22254
534. Hirosawa M, Nakahara M, Otosaka R, Imoto A, Okazaki T, Takahashi S. The p38 pathway inhibitor SB202190 activates MEK/MAPK to stimulate the growth of leukemia cells. Leuk Res. 2009;33:693-9 doi:10.1016/j.leukres.2008.09.028
535. Hideshima T, Akiyama M, Hayashi T, Richardson P, Schlossman R, Chauhan D. et al. Targeting p38 MAPK inhibits multiple myeloma cell growth in the bone marrow milieu. Blood. 2003;101:703-5 doi:10.1182/blood-2002-06-1874
536. Jemaa M, Vitale I, Kepp O, Berardinelli F, Galluzzi L, Senovilla L. et al. Selective killing of p53-deficient cancer cells by SP600125. EMBO Mol Med. 2012;4:500-14 doi:10.1002/emmm.201200228
537. Jin HO, Seo SK, Woo SH, Kim ES, Lee HC, Yoo DH. et al. SP600125 negatively regulates the mammalian target of rapamycin via ATF4-induced Redd1 expression. FEBS Lett. 2009;583:123-7 doi:10.1016/j.febslet.2008.11.035
538. Kim JA, Lee J, Margolis RL, Fotedar R. SP600125 suppresses Cdk1 and induces endoreplication directly from G2 phase, independent of JNK inhibition. Oncogene. 2010;29:1702-16 doi:10.1038/onc.2009.464
539. Kim JH, Kim TH, Kang HS, Ro J, Kim HS, Yoon S. SP600125, an inhibitor of Jnk pathway, reduces viability of relatively resistant cancer cells to doxorubicin. Biochem Biophys Res Commun. 2009;387:450-5 doi:10.1016/j.bbrc.2009.07.036
540. Li JY, Huang JY, Xing B, Ren KW, Li M, Wei D. et al. SP600125, a JNK inhibitor, suppresses growth of JNK-inactive glioblastoma cells through cell-cycle G2/M phase arrest. Pharmazie. 2012;67:942-6
541. Moon DO, Choi YH, Kim GY. Role of p21 in SP600125-induced cell cycle arrest, endoreduplication, and apoptosis. Cell Mol Life Sci. 2011;68:3249-60 doi:10.1007/s00018-011-0626-5
542. Moon DO, Kim MO, Choi YH, Kim ND, Chang JH, Kim GY. Bcl-2 overexpression attenuates SP600125-induced apoptosis in human leukemia U937 cells. Cancer Lett. 2008;264:316-25 doi:10.1016/j.canlet.2008.02.011
543. Renlund N, Pieretti-Vanmarcke R, O'Neill FH, Zhang L, Donahoe PK, Teixeira J. c-Jun N-terminal kinase inhibitor II (SP600125) activates Mullerian inhibiting substance type II receptor-mediated signal transduction. Endocrinology. 2008;149:108-15 doi:10.1210/en.2007-0529
544. Wang M, Atayar C, Rosati S, Bosga-Bouwer A, Kluin P, Visser L. JNK is constitutively active in mantle cell lymphoma: cell cycle deregulation and polyploidy by JNK inhibitor SP600125. J Pathol. 2009;218:95-103 doi:10.1002/path.2521
545. Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T. et al. Discovery of potent and selective covalent inhibitors of JNK. Chem Biol. 2012;19:140-54 doi:10.1016/j.chembiol.2011.11.010
Author contact
![]() Corresponding author: Franziska Briest, Phone: +49 30 8445 4579 e-mail: franziska.briestde.
Corresponding author: Franziska Briest, Phone: +49 30 8445 4579 e-mail: franziska.briestde.