Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(1):43-61. doi:10.7150/thno.10350 This issue Cite
Research Paper
Theranostic Nanoparticles Carrying Doxorubicin Attenuate Targeting Ligand Specific Antibody Responses Following Systemic Delivery
Emmy Yang1*, Weiping Qian1*, Zehong Cao1, Liya Wang2, Erica N. Bozeman1, Christina Ward1, Bin Yang3, Periasamy Selvaraj4, Malgorzata Lipowska2, Y. Andrew Wang5, Hui Mao2 ![]() , Lily Yang1,2
, Lily Yang1,2 ![]()
1. Departments of Surgery, Emory University School of Medicine, Atlanta, GA 30322;
2. Departments of Radiology and Imaging Sciences, Emory University School of Medicine, Atlanta, GA 30322.
3. Chengdu Women's and Children's Central Hospital, Chengdu, China;
4. Departments of Pathology, Emory University School of Medicine, Atlanta, GA 30322.
5. Ocean Nanotech, LLC, San Diego, CA 92126.
* Authors contributed equally to this study.
Received 2014-8-17; Accepted 2014-9-18; Published 2015-1-1
Abstract

Understanding the effects of immune responses on targeted delivery of nanoparticles is important for clinical translations of new cancer imaging and therapeutic nanoparticles. In this study, we found that repeated administrations of magnetic iron oxide nanoparticles (IONPs) conjugated with mouse or human derived targeting ligands induced high levels of ligand specific antibody responses in normal and tumor bearing mice while injections of unconjugated mouse ligands were weakly immunogenic and induced a very low level of antibody response in mice. Mice that received intravenous injections of targeted and polyethylene glycol (PEG)-coated IONPs further increased the ligand specific antibody production due to differential uptake of PEG-coated nanoparticles by macrophages and dendritic cells. However, the production of ligand specific antibodies was markedly inhibited following systemic delivery of theranostic nanoparticles carrying a chemotherapy drug, doxorubicin. Targeted imaging and histological analysis revealed that lack of the ligand specific antibodies led to an increase in intratumoral delivery of targeted nanoparticles. Results of this study support the potential of further development of targeted theranostic nanoparticles for the treatment of human cancers.
Keywords: Targeting ligands, nanoparticles, antibody, immune response, tumor imaging, nanoparticle delivery.
Introduction
Multifunctional nanoparticles have been developed for in vivo biomedical applications, particularly biomarker targeted molecular imaging and drug delivery [1-7]. Various targeting ligands, including antibodies, antibody fragments, phage-displayed peptides, and natural ligands for cellular receptors, have been used for functionalizing nanoparticles [3, 8-13]. Preclinical studies in animal models and on-going clinical trials addressing the safety and efficacy are critical for clinical translations of targeted imaging and therapeutic nanoparticles [1-3, 5, 6, 14]. One of the important issues is to determine if repeated administrations of the nanoparticles to patients activate the immune system to produce ligand-specific antibodies that can potentially block the binding of targeted nanoparticles to the intended cell surface receptors and thereby reduce the efficacy of delivery of nanoparticles and their payload drugs into tumors [15].
Antibodies against cell surface biomarkers are the commonly used ligands for the development of targeted nanoparticles [8, 11, 16-20]. Although mouse monoclonal antibodies have been used for making targeted nanoparticles, strong cross-species immune responses limit their potential for future clinical translation. Currently only a few types of humanized monoclonal antibodies, such as HER-2 antibody (Herceptin), are available for the production of targeted nanoparticles [21]. Alternatively, high affinity recombinant antibody fragments have been developed as targeting ligands [22-25]. For example, a human single chain antibody against the epidermal growth factor receptor (ScFvEGFR) that is highly expressed in the majority of epithelial tumors was conjugated to different types of nanoparticles. Specificity of tumor imaging and targeted therapeutic effects of these nanoparticles have been demonstrated in several animal tumor models [8, 18, 19, 26].
The major advantages of using natural ligands for tumor targeting are their high binding affinity, specificity, and most importantly, low immunogenicity. The amino-terminal fragment (ATF) of the receptor binding domain of urokinase plasminogen activator (uPA) has been used for the production of nanoparticles targeting the uPA receptor (uPAR), which is a cellular receptor overexpressed in cancer cells and tumor associated stromal cells in many types of tumor tissues [27, 28]. Our previous studies showed that systemic delivery of ATF-targeted magnetic iron oxide nanoparticles (IONPs) enabled optical imaging and magnetic resonance imaging (MRI) of tumors in mouse mammary and human breast and pancreatic tumor xenograft models in mice [13, 29, 30]. Targeted therapeutic efficacy of theranostic ATF-IONPs carrying a chemotherapy drug, gemcitabine, was also demonstrated in an orthotopic human pancreatic cancer xenograft model [6]. Effects of targeted optical imaging and photodynamic therapy using ATF-human albumin fusion proteins as drug carriers have been demonstrated in a mouse hepatocellular carcinoma model [31].
Mononuclear phagocytes have been shown to efficiently take up nanoparticles [32]. Uptake of antigen-conjugated nanoparticles by macrophages and dendritic cells enhances antigen presentation and stimulates both B and T cell responses [33-38]. Increasing evidence has shown that nanoparticles enhance immune responses to their conjugated protein antigens. Many groups used nanoparticle carriers as immune adjuvant agents for the development of viral, bacterial and tumor vaccines through subcutaneous, mucosal and intranasal administrations [36, 37, 39, 40]. Therefore, for future human applications of targeting ligand conjugated nanoparticles, the potential effects of the activation of immune response following administrations of the nanoparticles on targeted tumor imaging and drug delivery have been a concern in the nanomedicine field. At present, systemic immune responses toward various forms of targeting ligands used to produce targeted nanoparticles are largely unclear. Although targeting ligands derived from the same species, such as human protein based ligands for human use, are preferred choices for the development of targeted nanoparticles or theranostic nanoparticles, the questions concerning whether targeting ligand specific antibodies against those weakly immunogenic ligands can be activated by nanoparticles and the impact of the immune response induced antibody production on targeted delivery of nanoparticles remain to be answered.
Surface modification is a common approach to functionalize targeted nanoparticles and to optimize biodistribution of nanoparticles in vivo after systemic delivery [41-44]. Polyethylene glycol (PEG) is a polymer widely used to stabilize the nanoparticles and modify surface properties to reduce non-specific uptake of nanoparticles by macrophages in the reticuloendothelial system (RES) to improve targeted delivery of the nanoparticles [5, 14, 45, 46]. However, several studies have shown that administrations of PEG-coated nanoparticles stimulated the production of PEG-specific natural IgM antibody and promoted a fast clearance of PEGylated nanoparticles from the blood through antibody-enhanced phagocytic activity [47]. PEG IgM antibody also initiated the activation of complement pathways that may cause side effects in clinical applications [48]. Complement activation was detected in patients following systemic delivery of a PEG-coated liposomal form of doxorubicin (Doxil) [49]. Currently, the effect of PEG-modification on the immune response to targeting ligands conjugated to nanoparticles has yet to be determined.
In this study, we used targeting ligands derived from different species, such as mouse and human ATF peptide, and human single chain anti-EGFR antibody, to conjugate to magnetic IONPs for investigation of the effects of the ligand conjugated nanoparticles, with or without encapsulation of chemotherapy drugs, on the immune response and antibody production in immune competent mice. A mouse mammary tumor model was used to determine the effect of the production of ligand specific antibody on targeted delivery nanoparticles into tumors.
Materials and Methods
Production of recombinant targeting ligands
We used recombinant mouse and human proteins that represent weakly and highly immunogenic proteins relative to the mouse host in this study. Mouse ATF (mATF) is a recombinant protein with 135 amino acids (aa) of the receptor-binding domain of mouse uPA and an additional 14 aa peptide of paramyxovirus of simian virus 5 (V5) and six-histidine (His) tags. It has 87% homology with the natural mouse ATF peptide and is considered weakly immunogenic. Since there is species specificity in the binding of ATF peptides to uPAR, we also produced human ATF (hATF) for targeting human tumor cells. His-tagged hATF peptide has 71% of homology with the natural mouse ATF peptide. A single chain antibody to EGFR (ScFvEGFR) that was derived from the human immunoglobulin (Ig) only has 27% homology with a mouse Ig kappa chain variable region and is highly immunogenic to mice. Mouse serum albumin (MSA) was used as a control for a non-targeted ligand and low immunogenic protein. In total, four peptides and proteins described above were used to conjugate to nanoparticles with or without a near infrared dye (NIR-830) labeling for optical imaging.
Mouse or human amino terminal fragment of uPA: The cDNA fragments of the N-terminal 1 to 135 aa of mouse or human uPA were cloned into ET101/D-TOPO (mATF) or pET20b(+) (hATF) expression vector (Invitrogen, Carlsbad, CA). ATF peptide was expressed in E. coli BL21 as a 17 kDa recombinant peptide and purified by a Ni2+ NTA-agarose column (Qiagen, Valencia, CA) using an established protocol in our laboratory [13]. mATF expressed from this plasmid also contains a RNA polymerase alpha subunit of simian virus 5 epitope (V5, 14 aa) tag and a six-histidine (his) tag (6 aa) at the C-terminal for identification and purification of the recombinant protein. The hATF construct did not contain a V5 tag and only has a six-His tag.
Single-chain Fv epidermal growth factor receptor antibody (ScFvEGFR): Human EGFR specific scFv B10 was isolated from the YUAN-FCCC human naive phage display library using a solid phase biopanning methods [22, 23]. This bacteria expression plasmid was provided by Dr. Gregory Adams at Fox Chase Cancer Center, Philadelphia, PA. Recombinant ScFvEGFR protein (25 kDa) was obtained from the bacterial lysate of scFv B10 transformed TG1 competent cells after Ni2+ NTA-agarose column separation under native conditions [19].
Mouse serum albumin (MSA) was purchased from Sigma-Aldrich, St Louis, MO. This protein was purified from Swiss Webster strain mice.
Production of targeting ligand conjugated nanoparticles
10 nm core size magnetic IONPs were produced by Ocean Nanotech, LLC using an established protocol (San Diego, CA) [50]. IONPs were functionalized with an amphiphilic copolymer layer containing active carboxyl groups [51]. Amine PEG carboxyl (MW2000, Biomatrik, Zhejiang, China) was conjugated to the surface carboxyl groups of the amphiphilic polymer coated IONP to generate PEG-modified IONPs with surface carboxyl groups. mATF, hATF, and human ScFvEGFR as well as control MSA proteins were conjugated to IONPs by cross-linking carboxyl to amino groups of the targeting ligands mediated by 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDAC). About 10 to 15 of targeting ligands were conjugated to each nanoparticle as determined by the Bradford protein assay (BIO-RAD, Hercules, CA). Hydrodynamic sizes of various nanoparticles were determined using Zetasizer Nano (Malvern Instruments Inc., Southborough, MA). The hydrodynamic size ranges for ligand conjugated IONPs were 25 to 27 nm in diameters, depending on the ligands conjugated [19, 29].
To produce targeted IONPs with an optical imaging ability, a NIR 830-maleimide dye was conjugated to the free thiol group on cysteine of the peptides or proteins using a standard protocol prior to conjugation onto the IONPs [29,52]. NIR-830-maleimide dye was synthesized from a commercially available cyanine dye, IR-783, by our group [52]. Excitation wavelength of NIR-830 dye labeled onto targeting ligands is 800 nm and emission wavelength is 825 nm. The final targeting ligand-nanoparticle conjugates were purified using a Nanosep 100k OMEGA filter column (Pall Corp, Ann Arbor, MI).
Encapsulation of doxorubicin into targeted IONPs
Doxorubicin HCl (Polymed Therapeutics, Houston, TX) was dissolved in methanol and then added to the targeting ligand conjugated IONPs at a ratio of 1 mg Dox to 2 mg of iron equivalent IONPs in H2O, pH 8.5. After incubating at room temperature for 4 hours, free Dox was separated from the encapsulated Dox using Nanosep 100k column filtration. The amount of Dox in each IONP was determined using our established protocol based on the three standard curves of Dox [53].
Mouse models and nanoparticle delivery protocols
6 to 8 week-old female Balb/c mice or SCID mice were purchased from Harlan Laboratories (Indianapolis, IN). Both normal and tumor bearing mice were used for this study. The mice bearing 4T1 mouse mammary tumors were produced by directly injecting 2x 106 of 4T1 mouse mammary tumor cells into the mammary fat pad. The 4T1 cell line was kindly provided by Dr. Fred R. Miller (Barbara Ann Karmanos Cancer Institute, Detroit, MI).
For systemic delivery, 100 to 300 pmol of various targeting ligand conjugated-IONPs or PEG-IONP, with or without encapsulated Dox, were injected into the tail vein of the mice once per week for two to four injections. Those dosages have been used previously in our group for in vivo tumor imaging and targeted therapy. The number of injections in the tumor bearing mice was not the same between the studies due to the collection of mouse serum samples during different in vivo animal studies to examine the effects of targeted IONPs on tumor targeting and therapy. However, we only compare the levels of antibody production using the same experimental conditions. Mouse serum samples were collected from the mice 5 to 7 days following the last nanoparticle administration for Enzyme-linked immunosorbent (ELISA) assay.
For subcutaneous (s.c.) delivery, nanoparticles were injected into the flank region of the abdominal wall once per week for 2 weeks. Whole body optical imaging of the mice was taken at various time points following each injection using the Kodak FX in vivo imaging system (Carestream Health, Inc., Rochester, NY). At the end of 2 weeks, mouse serum samples were collected for ELISA. Mouse tissues from the last injected tissue sites, sentinel lymph nodes, and normal organs were collected after sacrificing the mice. The tissues were fixed in 10% buffered formalin and embedded in paraffin for histological analysis.
Enzyme-linked immunosorbent assay (ELISA)
96-well microtiter plates were coated with 10 µg/ml of mATF, hATF, ScFvEGFR, or MSA in 0.5 M carbonate-bicarbonate buffer, pH 9.6, for overnight. Serial dilutions of mouse serum samples in PBS were then added into the microtiter plate coated with the same ligand used for injecting the mice. A standard ELISA protocol was followed to detect the antibody level [54]. Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgM or rabbit anti-mouse IgG antibody was used as the secondary antibody. TMB substrate solution (Thermo Fisher Scientific, Waltham, MA) was used to detect HRP enzyme activity, which reflected the level of antibody. The plate was measured using SpectroMax microplate reader (Molecular Devices, LLC, Sunnyvale, CA) using O.D. 450 nm. The OD value was used to represent the level of the antibody in a given serum dilution.
Enzyme-linked immunosorbent spot (ELISPOT) assay
The mice bearing 4T1 mammary tumors received tail vein injections of various ScFvEGFR conjugated nanoparticles once per week for three weeks. Mice were sacrificed and the spleens were removed. Spleen cells were isolated using a standard protocol and red blood cells were lysed using red blood cell lysis buffer. After washing, 3x105 of the viable splenocytes were placed on ELISPOT plates (BD Sciences, Sparks, MD) that were pre-coated with 10 µg/mL of ScFvEGFR for 24 hrs using the manufacturer's protocol. To determine the total IgG antibody producing B cells in the splenocyte fraction, another ELISPOT plate was pre-coated with 10 µg/mL of goat anti-mouse IgG. Then 1x105 splenocytes were added to the plates. The plates were then cultured in RPMI-1640 medium with 10% fetal bovine serum overnight. Unbound cells were washed off using PBS with 0.1% Tween-20. HRP-labeled goat anti-mouse IgM or rabbit anti-IgG was diluted in PBS with 1% BSA and then added to the plates for 1 hour. The plates were then washed and a 3,3'-diaminobenzidine (DAB) substrate kit (Vector laboratories, Burlingame, CA) was used to detect the HRP-antibody labeled B cells. The plates were analyzed using an ELISPOT counter (Cellular Technology Limited, Shaker Heights, OH).
Prussian blue staining
The presence and the level of IONPs in the cells and tissue sections were determined using a Prussian blue staining for iron. Fixed cells or 5 µm thick tissue sections were incubated with Prussian blue staining solution containing equal parts of 20% of hydrochloric acid and 10% potassium ferrocyanide for 2 to 4 hrs. For tissue sections, nuclear fast red was used for counterstaining. Iron-containing cells have a bright blue color while other cells or tissues have red background staining.
Cellular assays to determine uptake of nanoparticles by macrophages and dendritic cells
The mouse macrophage cell line, RAW 264.7, was obtained from American Type Culture Collection (ATCC, Manassas, VA) and maintained in DMEM culture medium with 10% fetal bovine serum. Primary mouse dendritic cells were isolated from the femur bones of normal Balb/c mice. The bones were then sterilized on ice in 70% ethanol for 5 minutes. Marrow was then flushed out of the bones with RPMI-1640 medium using a sterile syringe. Red blood cells were depleted by hypotonic lysis. The remaining cells were then counted and plated in RPMI-1640 medium supplemented with 20 ng/ml mouse recombinant granulocyte-macrophage colony-stimulating factor. After 3 days of culture, 75% of non-adherent cells and media were removed and fresh media was added. Cells were maintained in culture for additional 3 days to obtain immature dendritic cells for future in vitro studies. The nanoparticle uptake study was performed on day 9 to 11 of the cultured cells, when the majority of the cells were dendritic cells [55]. Primary mouse macrophages were isolated from femur bones of normal CD1 mice. The above described isolation protocol was used. Cells were then cultured in RPMI-1640 medium supplemented with 20 ng/ml mouse recombinant macrophage colony-stimulating factor for 5 days before conducting the study.
To determine the efficiency of nanoparticle uptake by different cell types, 5 x104 of cells were seeded on 24-well culture plates for 2 days. Various nanoparticles were then added into the wells and incubated in the tissue culture incubator overnight. Unbound nanoparticles were washed off with PBS and the cells were fixed with 4% formaldehyde in PBS. Prussian blue staining was then performed on the plates. After staining, the plates were observed under an inverted microscope and bright field images of the representative areas were taken. Cells were then lysed and collected for determination of the levels of Prussian blue staining, which reflected IONP uptake, in the cells incubated with nanoparticles using SpectroMax microplate reader at O.D. 700 nm.
Cell Proliferation Assay
4x103 of RAW 264.7 mouse macrophage cell line or mouse primary dendritic cells were plated in 96-well culture plates for overnight. Culture medium was then replaced with the medium containing 50 nM of Dox or various IONPs at 50 nM Dox equivalent concentration of the nanoparticles. Cells were incubated with the above mentioned agents for 48 hours at 37 °C in 5% CO2 tissue culture incubator. Cells were then examined using an inverted fluorescence microscope to determine the presence and intensity of Dox fluorescence in the cells. Percentage of cell growth inhibition was determined by a Crystal Violet Cell Proliferation assay. Briefly, the cells were fixed in the 96-well plate using 4% formaldehyde in PBS for 20 min and then washed with PBS. 0.5% crystal violet in H2O was then added to the wells for 20 min and unstained dye was washed away with H2O. After air-dry, 100 µl of Sorenson's solution containing 30 mmol/L sodium citrate, 0.02 mol/L HCl, and 50% ethanol at room temperature for 20 min was added to the wells to elute the dye. The optical density was read at 590 nm using SpectroMax microplate reader. Absorbance value was normalized to the value of the control cell group without treatment to obtain the percentage of viable cells. Each treatment group was performed in triplicate.
Immunohistochemical analysis
5 µm of paraffin tissue sections were deparaffinized and rehydrated. Slides were then incubated with anti-CD68 antibody (macrophages and dendritic cells) or anti-CD83 antibody (mature dendritic cells) [56, 57] for 2 hours followed by HRP-labeled-secondary antibody for 1 hour (Santa Cruz Biotechnology, Inc., Dallas, Texas). Slides were then incubated with 3, 3'-diaminobenzidine (DAB) substrate using a DAB substrate kit (Vector Laboratories, INC, Burlingame, CA). After hematoxylin background staining, slides were examined under the Leica microscope.
Near infrared optical and MR imaging
Near infrared optical imaging: Tumor bearing mice received a tail vein injection of 100 pmol of NIR-830-dye-mATF-IONP, NIR-830-dye-ScFvEGFR-IONP, or non-targeted control NIR-830-dye-MSA-IONPs. Optical imaging was performed 48 hours after the injection using the Kodak FX In Vivo imaging system. Excitation filter of 800 nm and emission filter of 830 nm were used for optical imaging. Ex vivo tumor imaging was conducted using the above imaging conditions.
MRI: Mice bearing 4T1 mammary tumors received tail vein injections of 200 pmol of different IONPs, respectively, using a dose schedule of once per week for three weeks. MRI was performed on mice 48 hours following the third injection using a 4.7T animal scanner (Varian Unity, Agilent, CA). T2-weighted fast spin echo imaging sequence was used to acquire MR images. MRI contrast in the tumor was quantitatively analyzed using the region of interest (ROI) method and Image J software (National Institutes of Health, Bethesda, MD). Averaged signal intensities of the ROI were obtained from all tumor areas of each MR image and a muscle area as a control. MRI signal intensity in the tumor was normalized with the signal of muscle as the intensity of tumor signal/muscle signal. The mean signal intensity of each tumor was calculated from normalized signal intensities from all slices containing tumor section. The percentage of mean MR signal change as the result of the targeted accumulation of IONPs was calculated from comparing the mean MRI signal of the tumors in the mice that received the same dose and schedule delivery of non-targeted IONP or IONP-PEG.
Statistical analysis
Student's t-test was used for the determination of statistically significant differences between experimental groups. A p value <0.05 is considered statistically significant.
Results
Selective delivery of targeting ligand conjugated IONPs into mammary tumors in mice following systemic delivery
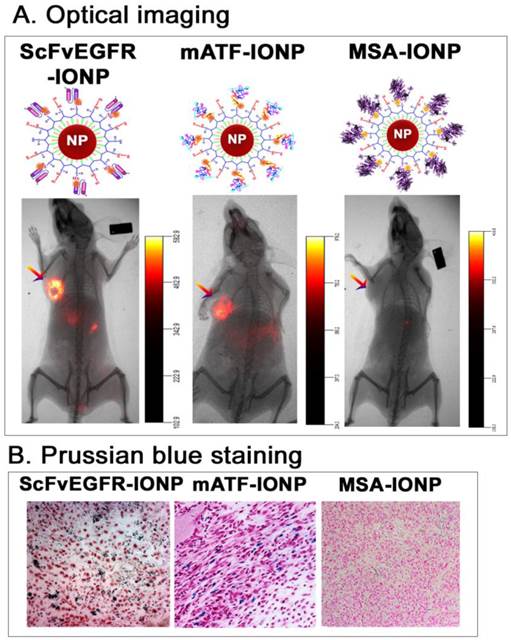
First, we determined the ability of targeted delivery of the ligand conjugated IONPs in the 4T1 mouse mammary tumors growing in the mammary fat pad of immune competent Balb/c mice. An intravenous (i.v.) injection of 100 picomolar (pmol) (IONP equivalent) of near infrared NIR-830 dye-labeled mouse ATF (mATF) or human single chain EGFR antibody (ScFvEGFR) conjugated IONPs led to the accumulation of the IONPs in the tumors and produced strong optical signals that were detectable by NIR optical imaging (Figure 1A). Histological analysis using Prussian blue staining for IONPs showed the presence of cells containing IONPs (blue) in the tumor tissue sections (Figure 1B). However, the mice that received non-targeted NIR-830-mouse serum albumin (MSA)-IONPs did not have detectable optical signals in the tumor and the tumor tissue sections lacked IONP staining (Figure 1).
Targeted delivery of nanoparticles into mouse mammary tumors using targeting ligand conjugated IONPs. A. NIR 830 dye labeled ATF peptides, single chain antibody ScFvEGFR, or non-targeted control MSA were conjugated to amphiphilic polymer coated IONPs, resulting in dual optical and MR imaging modality nanoparticles. Near infrared optical imaging was performed 48 hours following the tail vein injection of the nanoparticles using the Kodak FX in vivo imaging system. Arrows: orthotopic 4T1 mouse mammary tumors. B. Prussian blue staining. Frozen tumor tissue sections were used. Blue: IONP positive cells; Red: nuclear fast red background staining.
Targeting ligand specific antibodies were detected in the mouse serum following systemic administrations of targeted IONPs
To achieve a strong therapeutic effect, anticancer agents are often administrated multiple times to improve the delivery into heterogeneous tumors. We examined antibody responses against highly immunogenic human ScFvEGFR conjugated IONPs after repeated i.v. injections in mice bearing mammary tumors. A high level of anti-ScFvEGFR antibody was detected in the serially diluted serum samples of the mice that received two injections of 200 pmol of ScFvEGFR-IONPs using ELISA. Compared to the serum samples obtained from mice that received i.v. injections of the equal amount of free ScFvEGFR, the level of the antibody in the mice treated with nanoparticle-conjugated ScFvEGFR was three-fold higher (Figure 2A). Conjugation of ScFvEGFR to nanoparticles significantly increased the ligand specific antibody production (p<0.05, 1/200 and 1/2000 dilutions). Anti-ligand specific antibodies were not detected in control serum samples obtained from the mice injected with nanoparticles without targeting ligands (Figure 2B).
Detection of anti-ScFvEGFR antibody in mouse serum samples using ELISA. A. The level of anti-ScFvEGFR antibody in the serially diluted serum samples of tumor bearing mice following two i.v. deliveries of 200 pmol of ScFvEGFR-IONPs or equivalent unconjugated ScFvEGFR (50 µg). The level of mouse IgG antibody was measured. B. ELISA of mouse serum samples obtained from negative control IONP or PEG-IONP injected tumor bearing mice. C. Detection of the production of ligand specific IgG or IgM antibodies. Three injections of ScFvEGFR-IONPs activated high levels of both IgG and IgM ligand specific antibodies. Mice injected with PEG modified ScFvEGFR-PEG-IONPs had significantly higher level of IgG ligand specific antibody, but not IgM antibody, than ScFvEGFR-IONP injected mice. n=3 mice for all bar figures.
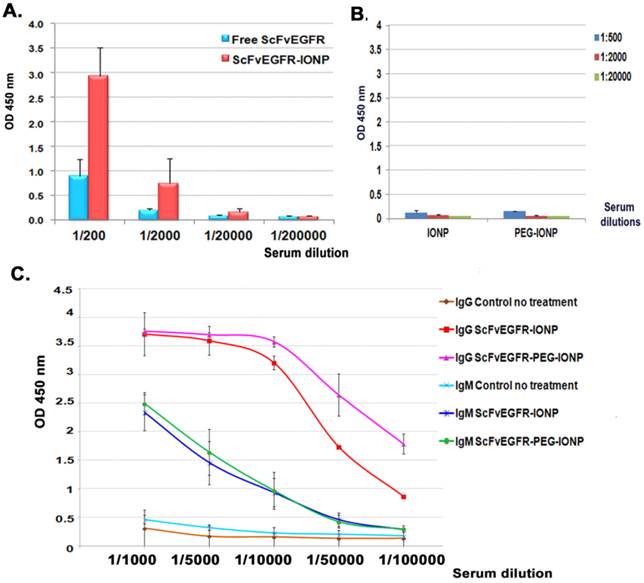
The effect of PEG-modification of the nanoparticles on antibody production was examined. 200 pmol of ScFvEGFR-IONPs coated with an amphiphilic copolymer layer or with an additional PEG modification were injected into the tumor-bearing mice weekly for three weeks. Using secondary antibodies specific for mouse IgG or IgM, we found that both anti-ScFvEGFR IgG and IgM antibodies were induced in the mouse serum (Figure 2C). Results of ELISA also revealed significantly higher levels of anti-ScFvEGFR IgG antibodies in the mice that received PEG modified ScFvEGFR-PEG-IONPs relative to mice injected with ScFvEGFR-IONPs (Figure 2C, p<0.05, 1/50,000 and 1/100,000 dilutions). However, PEG-modification of ScFvEGFR-IONPs did not further enhance the production of anti-ScFvEGFR IgM antibody in the mice (p>0.05).
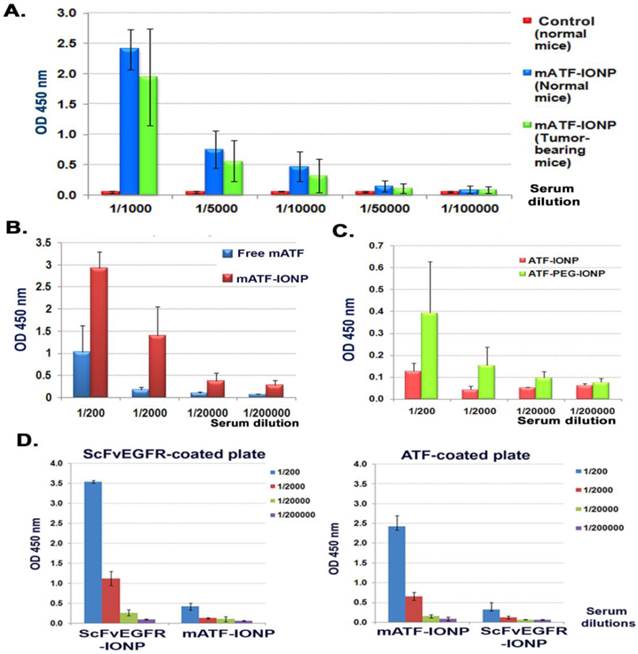
Next, we examined the antibody responses against weakly immunogenic targeting ligands derived from the mice in a mouse tumor model, which represented the immune response to a targeting ligand derived from the same species. Mouse ATF conjugated nanoparticles (mATF-IONP) at a dose of 200 pmol of IONPs were injected via the tail vein into the mice bearing 4T1 mammary tumors once every 5 days for 4 injections. Meanwhile, normal Balb/c mice also received the same dose and schedule of mATF-IONPs. ELISA analysis of mouse serum samples revealed high levels of mouse ATF specific antibody in both normal and tumor bearing mice (Figure 3A). The level of anti-mATF antibody was slightly elevated in normal mice compared with tumor bearing mice but the difference was not statistically significant (p>0.05). The tumor bearing mice that received mATF-conjugated IONPs had 4 to 10 fold higher levels of anti-mATF antibody compared with the mice injected with the equivalent amount of mATF peptides only (Fig. 3b, p<0.001, 1/200 and 1/2,000 dilutions). Additionally, we observed three fold increases in the levels of anti-mATF antibody in the serum of the mice that received two intravenous injections of mATF-PEG-IONPs compared with mATF-IONP injected mice (Figure 3C).
Detection of anti-mATF antibody in mouse serum samples using ELISA. Anti-mATF antibodies were induced in both tumor-bearing and normal Balb/c mice after receiving four i.v. delivery of 200 pmol of mATF-IONPs. B and C. Activation of anti-mATF antibody in tumor-bearing mice that received 50 µg of mATF peptides, 200 pmol of mATF-IONP, or 200 pmol of mATF-PEG-IONP. D. Specific activation of anti-targeting ligand antibodies. Serum samples obtained from ScFvEGFR-IONP or mATF-IONP injected mice were added to mATF and ScFvEGFR coated plates. ELISA results showed that there was no cross reactivity between anti-mATF antibody and ScFvEGFR antibody produced in the mice. The level of mouse IgG antibody was measured. The bar figures show the mean O.D. 450 nm values of each serial dilution of the mouse serum (n=3 mice).
Taken together, our results suggest that conjugation of a weakly immunogenic ligand to nanoparticles could increase its immunogenicity and enhance the antibody response in mice (Figure 3). However, the level of anti-ligand antibody in the mice induced by strong immunogenic ligand human ScFvEGFR conjugated IONPs is still higher than that detected in the mice that received the weakly immunogenic ligand mouse ATF-conjugated IONPs. For example, anti-ScFvEGFR antibody level at a given serum dilution (1/5000) in the mice that received three i.v. injections of ScFvEGFR-IONPs was 7 folds higher than the anti-mATF antibody response in the mice treated with four i.v. injections of mATF-IONPs (Figure 2B and 3A). Similar to the observations from the experiments with i.v. injection, ELISA results of the serum samples obtained from normal mice that received two s.c. injections of 100 pmol of ScFvEGFR-IONPs or mATF-IONPs showed high levels of anti-human ScFvEGFR antibody and a relatively low level of anti-mATF antibody (Supplementary Material: Figure S1). On the other hand, MSA protein collected from Swiss Webster mouse strain was not immunogenic in Balb/c mice since a very low level of anti-MSA antibody was detected in the mouse serum sample following s.c. injection of unconjugated MSA. However, conjugation of MSA to IONPs led to 14 fold increases in the level of anti-MSA antibody than that of the mice injected with unconjugated MSA (Supplementary Material: Figure S1). Under the same experimental conditions, the level of anti-MSA antibody was 8 fold higher than the anti-mATF antibody and about the same level as anti-ScFvEGFR antibody after s.c. injection.
We further validated the specificity of ELISA in detecting the anti-ligand antibodies using ELISA plates coated with different ligands. We confirmed that the antibody responses were targeting ligand specific since a high level of anti-mATF antibody was only detected in the serum samples of mATF-IONP injected mice, while anti-ScFvEGFR antibody was detected in the serum of ScFvEGFR-IONP injected mice (Figure 3D).
PEG modifications of nanoparticle coating affected uptake of the nanoparticles by different types of phagocytes
Antibody responses are activated by interactions of antigen-presenting cells with T and B lymphocytes [33, 34]. Efficient uptake of nanoparticles by antigen-presenting macrophages, dendritic cells, and B lymphocytes is thought to contribute to the activation of an antibody response. To interrogate the role of phagocytes in activation of the antibody responses, we examined uptake of the nanoparticles by different types of antigen-presenting phagocytes in vitro and in vivo. First, we evaluated the ability and efficiency of uptake of the targeting ligand conjugated IONPs or PEG-IONPs by macrophages using the RAW 264.7. mouse macrophage cell line. After incubating cultured macrophages with different IONP agents, we found that mouse macrophages efficiently engulfed all types of IONPs tested. Differences in the conjugated targeting ligands did not affect the level of nanoparticle uptake by macrophages under the experimental conditions used (Figure 4A). As expected, macrophages exhibited lower levels of uptake of PEG-modified nanoparticles (PEG-IONP, ScFvEGFR-PEG-IONP or ATF-PEG-IONPs) compared to those without the PEG-coating (Figure 4A). Quantitative analysis of the cell lysates revealed that the amounts of the PEG-modified nanoparticles in the macrophages were 15 to 30% of non-PEG-nanoparticles taken up by the macrophages (Figure 4A). Interestingly, conjugation of MSA to PEG-coated IONPs could reduce the inhibitory effect of PEG modification on nanoparticle uptake in macrophages (Figure 4A). These results were further confirmed using primary mouse macrophage cultures. A marked decrease in nanoparticle uptake was found in cells treated with different ligand conjugated PEG-IONPs for 24 hours compared to the IONPs without PEG modification (Figure 4A).
Uptake of targeting ligand conjugated IONPs without or with PEG modification. A. Differential uptake of targeting ligands conjugated IONP or PEG-IONPs by mouse macrophages detected by Prussian blue staining. The RAW 264.7 mouse macrophage cells or primary mouse macrophages with the nanoparticles were stained blue. Cells were lysed and the amount of iron was measured using a spectrometer at O.D. 700 nm. B. Uptake of the targeting ligand-conjugated IONP or PEG-IONP by mouse dendritic cells. The same experimental conditions were used as the macrophage study.
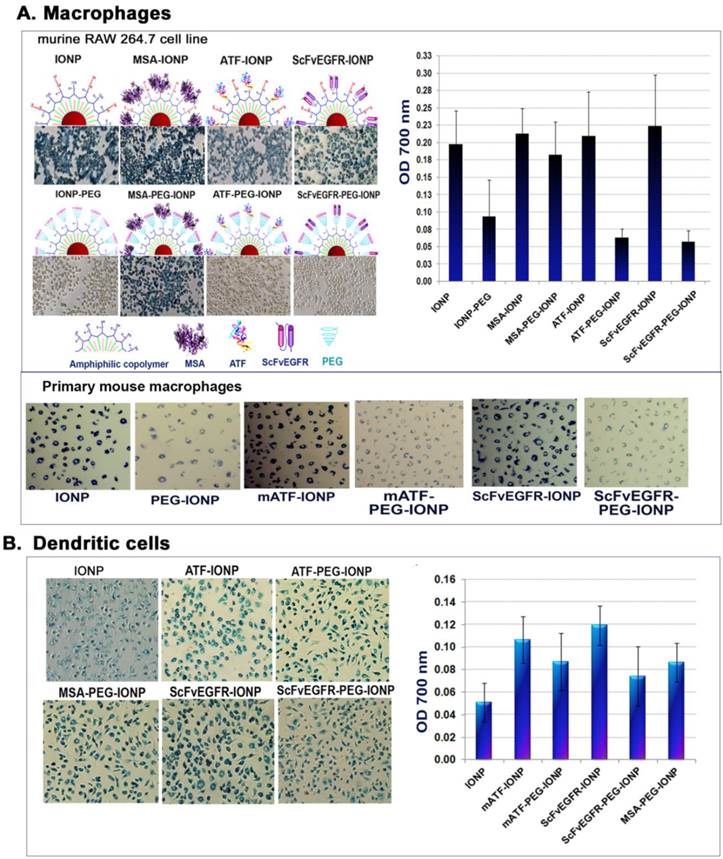
Reduced macrophage uptake of PEG-modified IONPs suggested further investigation of mechanisms of activation of anti-ligand antibody responses. Since dendritic cells are the most potent antigen-presenting cells [58], we then performed a similar uptake study using primary dendritic cells derived from mouse bone marrow progenitor cells. Both targeting ligand conjugated IONPs and PEG-IONPs were taken up by dendritic cells. The levels of ligand-PEG-IONPs in those cells were 62% (ScFvEGFR-PEG-IONP) and 82% (mATF-PEG-IONP) of the uptake of corresponding ligand-IONPs without the PEG modification (Figure 4B).
Production of targeting ligand specific antibodies was not enhanced by subcutaneous injections of ligand conjugated PEG-IONPs
We used s.c. injection of the nanoparticles to determine the difference in uptake of IONPs and PEG-IONPs by phagocytes in vivo and its effect on antibody production. First, we used NIR-830 dye labeled mouse or human ATF conjugated IONPs that represented the same targeting ligands but were derived from different species. Normal Balb/c mice received s.c. injections of 100 pmol of the mATF- or hATF-conjugated nanoparticles, with or without PEG modification, once per week for two injections. ELISA results showed low to intermediate levels of anti-mATF or anti-hATF antibodies in the mouse serum samples (Figure 5A). Unlike the antibody response after i.v. delivery of targeted and PEG-modified IONPs, we found that s.c. delivery of PEG-modified either mATF-PEG-IONPs or hATF-PEG-IONPs did not show an increase in the production of ligand specific antibodies compared to that of mATF-IONP or hATF-IONP injected mice (Figure 5A). It is interesting that human ATF conjugated IONPs induced weaker antibody responses compared to mouse ATF conjugated IONPs in mice. It is possible that the recombinant V5 tag in a mouse ATF peptide was more immunogenic than a hATF peptide that had 71% amino acid sequence homology with a mouse natural ATF. Contribution of the V5 tag to the activation of anti-ligand antibody was demonstrated by comparing antibody levels in mouse serum samples obtained from mice that received s.c. injections of mATF-V5-IONPs using ELISA plates coated with mATF peptides containing the V5 tag or natural mATF peptides without the V5 tag. The level of antibody detected using mATF-V5 peptide coated plate was 2.5 fold higher than mATF peptide without V5 tag coated plate (Supplementary Material: Figure S2).
Comparison of antibody responses, nanoparticle location, and phagocyte uptake of targeting ligand conjugated IONPs and PEG-IONPs following s.c. injections. NIR-830 dye labeled mATF or hATF peptides were conjugated to IONPs or PEG-IONPs and then injected s.c. into the front flank area of normal Balb/c mice. A. Levels of anti-ATF antibodies in the serum of the mice received mATF or hATF conjugated IONPs, without or with PEG-modification. 1 and 2 weeks following IONP injections, mouse serum samples were collected and ELISA assay was used for the detection of antibody responses. n=3 mice/group. B. Optical imaging detected location and migration of the IONPs without or with PEG modification. Optical imaging was performed 1, 2 and 7 days following the first injection using the Kodak FX In Vivo imaging system. Optical imaging signals at the injection site (pink arrows). Diffused signals in the draining lymph node areas of the mice injected with NIR-830-mATF-PEG-IONPs (blue arrows). Numbers shown are the mean signal intensities of the injection site. C. Prussian blue staining showed the presence of cells with IONPs in the injection sites, the draining lymph nodes and the spleen obtained from the mice 7 days following the last nanoparticle injection (red arrows). D. Immunohistochemical staining. Tissue sections of the injection site were stained with antibodies to CD68 (macrophages and dendritic cells) and CD83 (mature dendritic cells). CD68 and CD83 positive cells were found in the injection site (red arrows). A higher level of CD83 positive cells was detected in the tissue sections of the mice injected with PEG modified-ATF-IONPs compared with ATF-IONP injection site. Green arrows indicated the areas of CD83 positive Langerhans cells, which are skin dendritic cells.
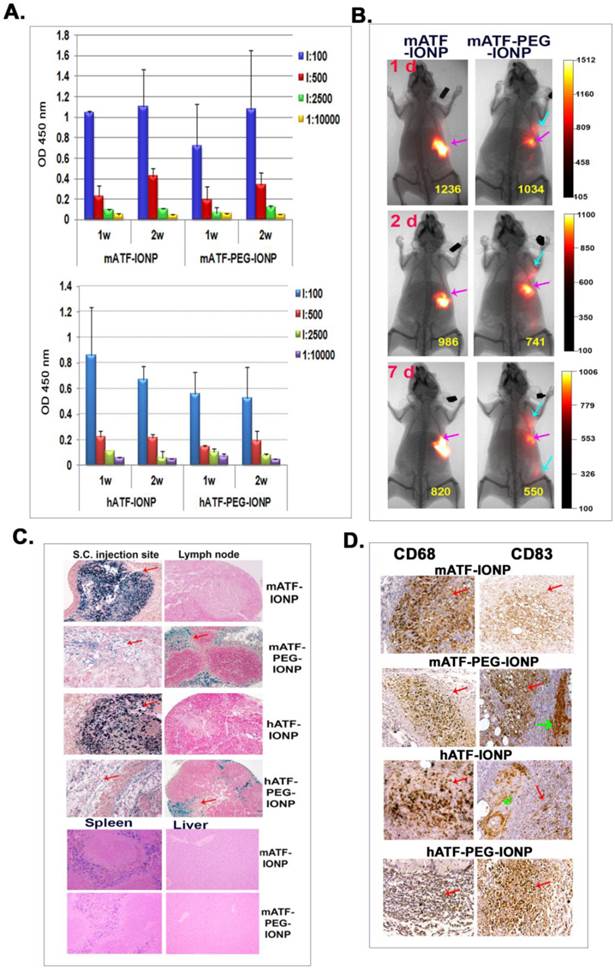
To compare dynamic changes of accumulation of nanoparticles with or without PEG modification in the location and amount of injected nanoparticles, optical imaging was conducted at 1, 2 and 7 days following s.c. injection of different IONPs. Strong and localized optical signals were detected in the injection sites in the mice that received mATF-conjugated IONPs at all time points (Figure 5B). Although the mice that received mATF-PEG-IONPs had lower levels of optical signals at the injection site compared to mATF-IONP injected mice, optical signals were detected in the draining lymph node areas in those mice (Figure 5B). Histological analysis of skin tissues obtained from the injection sites and the draining lymph nodes using Prussian blue staining for IONPs showed that extensive iron positive cells retained in the injection site of the mice that received mATF or hATF conjugated-IONPs for 7 days but not in the lymph nodes (Figure 5B). In contrast, lower levels of the iron positive cells were found in the injection site of the mice that received mATF or hATF conjugated PEG-IONPs (Figure 5C). However, a high level of iron staining cells was detected in the cortex of the draining lymph nodes in those mice (Figure 5C). IONPs were also detected in the spleen of the mice that received s.c. injections of ATF-IONPs, but the level of IONP positive cells in the spleen of the mice injected s.c. with ATF-PEG-IONPs was relatively low (Figure 5C). IONPs were not detected in the liver of the mice that received s.c. injections of either ATF-IONPs or ATF-PEG-IONPs (Figure 5C).
Immunohistochemical staining using antibodies against macrophage (CD68) and dendritic cells (CD68 and CD83) revealed the presence of high levels of CD68 positive cells, and intermediate levels of CD83 positive cells in the subcutaneous areas of the skin tissue sections of the mice that received s.c. injections of mATF-IONPs or hATF-IONPs (Figure 5D). However, strong CD83 positive cells were detected at the injection site of the tissue sections of the mice that received the ligand conjugated PEG-IONPs (Figure 5D).
Production of anti-targeting ligand antibodies was attenuated following systemic administrations of targeted theranostic nanoparticles carrying doxorubicin
To determine the effect of repeated administrations of targeted theranostic nanoparticles on ligand specific antibody production, we examined the level of anti-targeting ligand antibodies following i.v. injections of targeting ligand conjugated IONPs with or without carrying a commonly used chemotherapy drug, doxorubicin (Dox), in the 4T1 mouse mammary tumor model. We found that while systemic delivery of mATF-IONPs or ScFvEGFR-IONPs induced the production of the high levels of anti-mATF or anti-ScFvEGFR antibodies, the mice that received the same amount and dose schedule of mATF-IONP-Dox and ScFvEGFR-IONP-Dox had significantly lower levels of the anti-targeting ligand antibodies (Figure 6A). There was a significant difference in the antibody levels between the mice injected with targeting ligand conjugated IONPs with or without Dox (ATF-IONP vs. ATF-IONP-Dox: p<0.0001; ScFvEGFR-IONP vs. ScFvEGFR-IONP-Dox: p<0.0005). Although ScFvEGFR-PEG-IONP injected mice induced an increased level of anti-ScFvEGFR antibody, the presence of Dox also inhibited the production of the ligand specific antibody (Figure 6A, p<0.01). This finding suggested that the enhanced antibody response against targeting ligands by nanoparticles should not be an issue for the development of targeted theranostic nanoparticles carrying Dox for cancer therapy in human patients.
Inhibition of the production of targeting ligand specific antibodies after systemic delivery of theranostic IONPs carrying doxorubicin. A. ELISA analysis of mouse serum samples obtained from 4T1 tumor bearing mice after systemic delivery of 200 pmol of mATF-IONPs or mATF-IONP-Dox nanoparticles (left) via tail or 200 pmol of ScFvEGFR-IONPs, ScFvEGFR-IONP-Dox, ScFvEGFR-PEG-IONPs, ScFvEGFR-PEG-IONP-Dox nanoparticles (right). Mouse IgG antibody was measured. n=3 mice/group. B. Cell Proliferation assay of the RAW 264.7 mouse macrophage cell line or primary dendritic cells following treatment with 50 nM of Dox or Dox equivalent of IONPs for 48 hours. Crystal Violet Cell Proliferation assay was used for determining the percentage of cell growth inhibition. Fluorescence microscopy detected a higher level of Dox fluorescence (red) in the cells treated with 100 nM of ATF-IONP-Dox for 24 hours than that of the equal concentration of free Dox treatment. Prussian blue staining was used to determine IONP uptake as well as the cell density after the treatment.
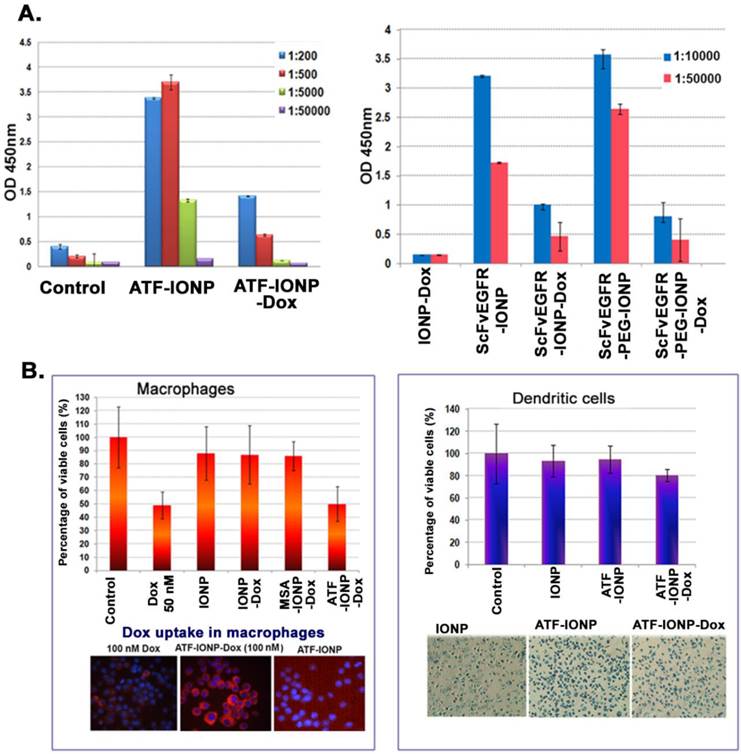
To understand the mechanism of inhibition of the antibody production by chemotherapy drug, we examined the potential cytotoxic effect of Dox on antigen-presenting macrophages and dendritic cells. We found that cultured mouse macrophage RAW 264.7 cells were sensitive to free Dox treatment and 50 nM Dox in the culture medium inhibited cell growth by 50% (Figure 6B). The growth of the cells was also inhibited slightly (~10%) after treatment with non-targeted IONP-Dox or non-targeted MSA-IONP-Dox. However, uPAR-targeted mATF-IONP-Dox treatment led to 50% inhibition of macrophage growth. Since uPAR is highly expressed in activated macrophages, cytotoxic effects detected in mATF-IONP-Dox treated cells may affect their antigen presentation function, and thereby inhibiting the production of the ATF ligand specific antibody. Enhanced uptake of ATF-IONP-Dox was also detected in macrophages by examination of fluorescence intensity in cells after culturing with 100 nM of free Dox and drug equivalent concentration of mATF-IONP-Dox (Figure 6B). We further examined the effect of the theranostic-IONP-Dox on dendritic cells. ATF-IONP-Dox treatment led to 20% growth inhibition of primary mouse dendritic cells (Figure 6B). Similarly, the effects of ScFvEGFR-conjugated IONP or PEG-IONP carrying Dox on the viability of macrophages and dendritic cells were examined. We found strong cytotoxic effects of ScFvEGFR-IONP-Dox or ScFvEGFR-PEG-IONP-Dox on both macrophages and dendritic cells (Supplementary Material: Figure S3).
Reduction of targeting ligand specific antibody producing B lymphocytes in the spleen after systemic delivery of theranostic IONP-Dox
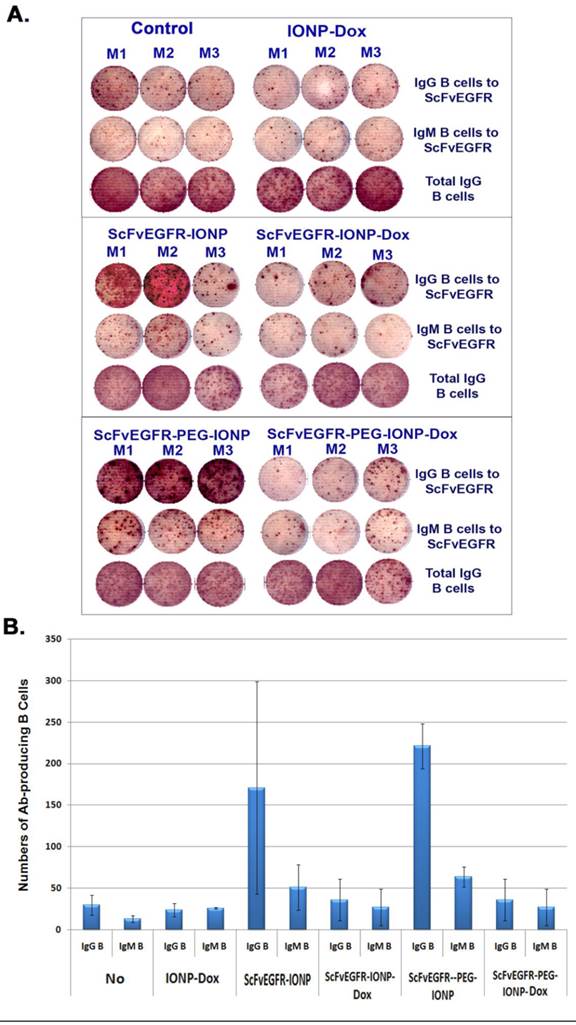
To further determine the mechanisms of action of PEG-modified nanoparticles on enhanced antibody production and inhibition of antibody production by theranostic IONPs carrying Dox, we collected splenocytes from mice that received three systemic deliveries of control IONP, ScFvEGFR-IONP, ScFvEGFR-IONP-Dox, ScFvEGFR-PEG-IONP, or ScFvEGFR-PEG-IONP-Dox. ELISPOT assay was performed in a 96-well plate coated with ScFvEGFR, which allows the capture of B lymphocytes that produce ScFvEGFR specific IgM or IgG antibodies onto the plate. We found ScFvEGFR specific IgM or IgG antibody producing B cells in the spleens of mice that received systemic deliveries of ScFvEGFR-IONPs or ScFvEGFR-PEG-IONPs, but not in mice that received IONPs without any ligands (Figure 7A). Furthermore, the highest level of ScFvEGFR specific IgM or IgG producing B cells was detected in the spleens of mice that received ScFvEGFR-PEG-IONPs (Figure 7A & B). The mice that received ScFvEGFR-IONP-Dox or ScFvEGFR-PEG-IONP-Dox had significantly fewer ScFvEGFR specific IgG or IgM-producing B cells than that of the mice injected with ScFvEGFR-IONPs (p <0.05), or ScFvEGFR-PEG-IONPs (p <0.005). Additionally, we found that decreases in the levels of ScFvEGFR specific IgG or IgM-producing B cells in the mice that received the targeted IONP-Dox were not due to non-specific immune suppression effects or cytotoxic effect of Dox on B lymphocytes since ELISPOT assay revealed that the total numbers of IgG producing B cells were not decreased in the spleen (Figure 7A).
Detection of ligand specific antibody producing B cells in mouse spleens using ELISPOT assay. Balb/c mice bearing 4T1 mouse mammary tumors received tail vein injections of 200 pmol IONP, ScFvEGFR-IONP or ScFvEGFR-PEG-IONP, without or with carrying Dox, once per week for three weeks. The mice were then sacrificed and spleens collected for ELISPOT assay. A. Images of antibody-producing B cell spots (brown) in wells incubated with spleen cells from the mice. An ELISPOT reader was used to take the image. The number of brown colored dots represented the number of antibody producing B cells. M1, M2 and M3 were spleen cells from three mice in each group. B. Quantification of the amount of ScFvEGFR specific IgG or IgM producing B cells in mouse spleen. A total of 3x105 of spleen cells were added in each well for detection of targeting ligand specific antibody. The number was the mean of three mice in each group. Ab: antibody.
Inhibition of targeting ligand specific antibody production increased intratumoral nanoparticle delivery of targeted nanoparticles
Although results of our study demonstrated the activation of anti-targeting ligand antibodies by nanoparticles and PEG-nanoparticles, it was unclear if those antibodies affected targeted delivery of the nanoparticles into tumors. The observed inhibitory effect of theranostic nanoparticles carrying Dox on the antibody production suggested an experimental system for determining whether the anti-targeting ligand antibody decreased the efficiency of intratumoral nanoparticle delivery. The 4T1 mouse mammary tumor model has a low sensitivity to Dox treatment. There was no tumor growth inhibition in the tumor bearing mice after receiving 5 mg/Kg of free Dox or the drug dose equivalent amount of mATF-IONP-Dox for four injections. The tumor growth inhibitory effect was only detected when the mice were treated with 10 mg/Kg of Dox or mATF-IONP-Dox for four times (Supplementary Material: Figure S4). Therefore, we expected that systemic delivery of ATF-IONP-Dox at a 5 mg/Kg Dox dose should not affect the growth and structure of the tumor significantly while allowing determination of the efficiency of targeted nanoparticle delivery after repeated administrations in the absence of a high level of anti-targeting ligand antibody. First, non-invasive MRI was performed to monitor the amount of IONPs delivered into the tumors using T2-weighted MRI. We detected decreased MRI signals in the tumors that received targeting ligand conjugated IONP or PEG-IONPs since the presence of IONPs led to a signal decrease in T2 weighted MRI and reduction of MRI signals dependent on the amount of IONPs in the tumor (Figure 8A). As compared with unconjugated IONPs, tumors in the mice receiving mATF-IONPs or mATF-PEG-IONPs showed 21 or 27% MRI signal decreases, respectively. Repeated injections of mATF-IONP-Dox theranostic nanoparticles led to further signal decrease (43.3%) in the tumor of the mice (Figure 8A). Similarly, the MRI signal of the tumors in ScFvEGFR-IONP injected mice also showed 29.8% decrease (Figure 8A). Histological analysis of tumor tissue sections using Prussian blue staining further confirmed higher levels of iron positive cells in the tumors collected from repeated injections of either mATF-IONP-Dox or ScFvEGFR-IONP-Dox, compared with the tumors receiving the same nanoparticles without Dox (Figure 8B).
Determination of the effects of the production of anti-targeting ligand antibody on efficiency of intratumoral nanoparticle delivery. Mice bearing 4T1 mammary tumors received tail vein deliveries of 200 pmol of different IONPs once per week for three injections (A &B). A. T2-weighted MRI was performed 24 hours after the last injection. Pink-lined areas were mammary tumors. Numbers in the figure were the mean MRI signal intensity of the entire tumor. B. Histological analysis of tumor tissue sections using Prussian blue staining. Tumors were collected 48 hours after the third injection. Paraffin tissue sections were used for Prussian blue staining. Blue: IONP positive cells; Red: nuclear fast red background staining. C. Balb/c or SCID mice bearing 4T1 mouse mammary tumors received tail vein injections of 300 pmol of NIR-830-ScFvEGFR-IONP or NIR-830-ScFvEGFR-PEG-IONP once every 5 days for three injections. 72 hours following the last injection, the mice were sacrificed. Tumors were excised for ex vivo organ optical imaging, which provided more accurate quantification of optical imaging signals that reflected the level of IONP accumulation. Yellow arrows: comparison of optical imaging of tumors in Balb/c or SCID mice treated with NIR-830-ScFvEGFR-PEG-IONP; Pink arrows, optical imaging of tumors of Balb/c or SCID mice that received NIR-830-ScFvEGFR-IONP. Mean signal intensity and standard derivations of each tumor are shown. The levels of anti ScFvEGFR-antibody in mouse serum samples shown are O.D. 450 nm at a mouse serum dilution of 1:5000. Prussian blue staining of frozen tumor sections revealed the accumulation of IONPs in tumor tissues.
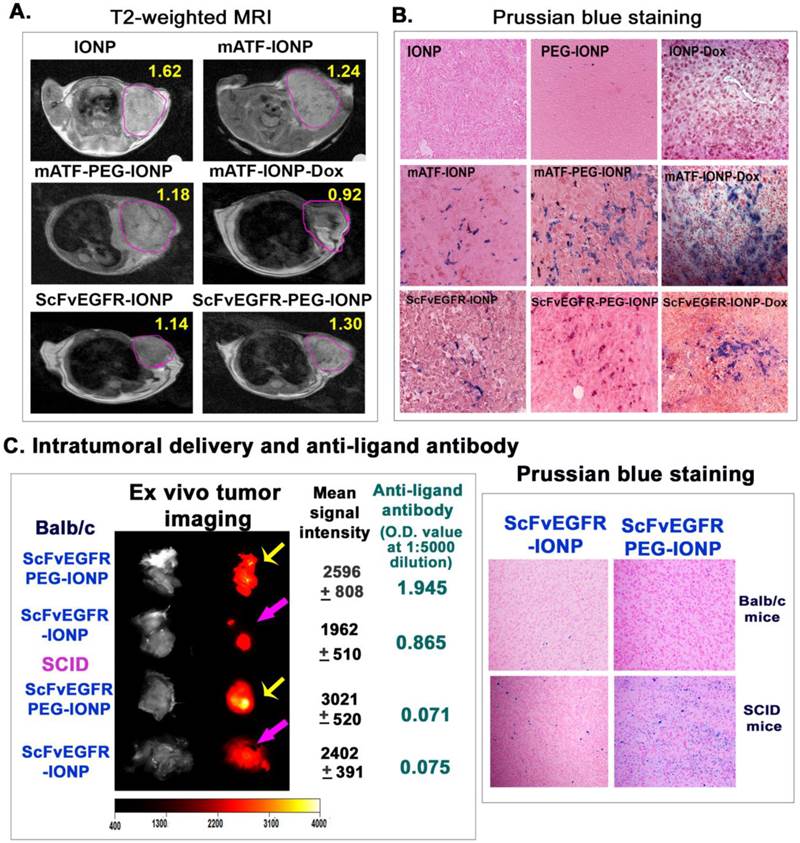
To further elucidate the effect of anti-targeting ligand specific antibody on targeted nanoparticle delivery, we established the 4T1 mouse mammary tumor model in immune competent Balb/c and immunocompromised SCID mice, which lack both T and B cells. 300 pmol of NIR-830-ScFvEGFR-IONP or NIR-830-ScFvEGFR-PEG-IONP were injected i.v. into the tumor bearing mice once every 5 days for three injections. At the end of the study, ex vivo optical imaging of tumors revealed that tumors of either Balb/c or SCID mice that received PEG-modified ScFvEGFR-IONPs had higher signals than tumors obtained from those mice treated with ScFvEGFR-IONPs (Figure 8C), suggesting that PEG-modification improved nanoparticle delivery. Furthermore, the tumors of SCID mice had higher levels of optical signals compared to the same nanoparticle treated tumors of Balb/c mice (Figure 8C). Balb/c mouse received ScFvEGFR-IONPs had the lowest signal in the tumor (Figure 8C). ELISA assay showed that the level of anti-ScFvEGFR antibody in the serum samples collected from the Balb/c mouse treated with ScFvEGFR-PEG-IONPs was 25-fold higher than that in the serum sample of SCID mouse treated with the same nanoparticles (serum dilution, 1:5000, Figure 8C). Histological analysis using Prussian blue staining for iron showed a high level of accumulation of ScFvEGFR-PEG-IONPs in the tumors from SCID mice (Figure 8C). Tumor tissues from ScFvEGFR-IONPs-treated SCID mouse had a medium level of IONPs while a low level of IONPs was detected in tumors from the Balb/c mouse following the above treatment (Figure 8C). Therefore, results of this study demonstrated that the production of targeting ligand specific antibodies reduced efficiency of nanoparticle delivery into tumors. Theranostic IONPs carrying Dox were able to inhibit the antibody responses and achieve higher intratumoral delivery efficiency.
Discussions
Current advances in the development of targeted nanoparticle imaging and drug delivery agents have shown promise towards novel approaches for the detection and treatment of human cancer. It is increasingly recognized that the issues concerning the host immune responses to administrated nanoparticles must be addressed in order to translate developed nanomaterials into clinical applications in humans [15]. Although various nanoparticles, especially liposomal or polymeric formulated nanoparticles, have been used as carriers to enhance immune responses to bacterial, viral, or tumor antigens for vaccination, immune responses to targeting moieties conjugated on nanoparticles or theranostic nanoparticles are largely unknown [35, 36, 40]. Therefore, there is a need to investigate the antibody responses to different targeting ligand-conjugated nanoparticles and the effect of ligand specific antibody activation on the efficiency of targeted nanoparticle delivery.
In this study, we showed that ligand specific antibodies could be induced by in vivo administrations of highly, as well as weakly, immunogenic targeting ligand conjugated nanoparticles. Our results demonstrated that a commonly used PEG coating for nanoparticles further enhanced the production of targeting ligand specific antibodies. However, systemic delivery of targeted theranostic nanoparticles carrying Dox inhibited this antibody production. Results of non-invasive imaging along with histological analysis showed that a reduction or the absence of targeting ligand specific antibodies markedly improved the efficiency of nanoparticle delivery into tumors. Results of this study address important concerns on antibody responses and their inhibitory effects on the intratumoral nanoparticle delivery following in vivo administrations of targeted nanoparticles. Our finding that significant inhibition of targeting ligand specific antibody responses following systemic administrations of theranostic nanoparticles carrying chemotherapy drug Dox support the translation potential of this novel class of targeted cancer therapeutics for clinic applications.
While it is expected that highly immunogenic human ScFvEGFR ligands would induce stronger antibody responses in mice than a weakly immunogenic mouse ATF peptide, induction of ligand-specific antibodies after conjugation of relatively weak immunogenic mouse proteins, such as mouse ATF and MSA, to nanoparticles indicates an additional challenge for the development of targeted nanoparticles. It seems that specific properties of targeting ligands affected the nanoparticle-mediated immune responses. For example, a lower level of anti-human ATF antibody was detected in mice that received hATF-IONP or hATF-PEG-IONP compared to the level of anti-mouse ATF antibody detected in mice treated with mATF-IONPs. On the other hand, conjugation of weakly immunogenic MSA to IONPs activated a strong antibody response that was comparable to that induced by highly immunogenic human single chain antibody conjugated IONPs.
Mononuclear phagocytes have been shown to efficiently take up nanoparticles [32]. Uptake of antigen-conjugated nanoparticles by macrophages and dendritic cells enhances antigen presentation and stimulates both B and T cell responses [33-38]. PEG surface coating has been widely used for the development of various nanoparticle imaging probes and drug delivery carriers. Unlike previously reported production of PEG specific natural IgM antibodies [47], our results showed that the enhancement of anti-targeting ligand IgG antibodies by systemic delivery of targeted PEG-IONPs is likely to be mediated by the interaction of antigen presenting cells and T cells, leading to the induction of IgG producing B cells. It is intriguing that PEG-coated nanoparticles, with significantly reduced uptake by macrophages, activated strong IgG antibody responses that relied on the function of antigen presenting cells. This finding of differential uptake of PEG-coated nanoparticles by macrophages and dendritic cells suggested a novel mechanism for the activation of the ligand specific antibody responses by targeted PEG-nanoparticles. It is well known that a high percentage of intravenously delivered nanoparticles without the PEG-coating nonspecifically accumulate in Kupffer cells in the liver [15, 32]. We found a high level of nonspecific uptake of ScFvEGFR-IONPs in the liver following intravenous delivery and an intermediate level of the IONPs in the spleen. However, systemic delivery of ScFvEGFR-PEG-IONPs significantly reduced the level of the IONPs in the liver while slightly lowered the IONP level in the spleen (Supplementary Material: Figure S5). Kupffer cells have much lower efficiency in antigen presentation for activation of antibody responses than macrophages and other antigen presenting cells in the spleen [59]. It is likely that inhibition of nonspecific uptake by Kupffer cells in the liver and macrophages in the spleen using PEG-coated nanoparticles resulted in longer blood circulation time of the nanoparticles and increased availability of the nanoparticles to more efficient antigen presenting cells, such as dendritic cells and B lymphocytes, in the spleen and peripheral lymph nodes to activate the production of targeting ligand specific antibodies.
Our in vivo study results following subcutaneous delivery of targeted IONPs with or without PEG modification further supports the above mechanism. Skin and subcutaneous areas have high numbers of macrophages and dendritic cells with strong antigen presentation ability. Subcutaneous injection of ATF conjugated nanoparticles facilitated differential uptake of nanoparticles with or without PEG-coating by macrophages and dendritic cells in vivo using immunochemical staining with antibodies for macrophage (CD68) and dendritic cell (CD68 and CD83) biomarkers. ATF conjugated IONPs were taken up by both CD68 positive macrophages and CD83 expressing dendritic cells while ATF-PEG-IONPs were detected mostly in CD83 positive dendritic cells. Our results revealed that the differences in nanoparticle uptake also affected the distribution of ATF-IONPs and ATF-PEG-IONPs following subcutaneous delivery. ATF-IONPs taken up by macrophages had prolonged retention time at the injection site. However, uptake of ATF-PEG-IONPs by dendritic cells resulted in the accumulation of ATF-PEG-IONPs in the draining lymph nodes. ATF-IONPs and ATF-PEG-IONPs have a small hydrodynamic size of ~25 nm. They were able to enter the lymphatic vessels and be transported to the lymph nodes and the spleen to stimulate antibody responses. In spite of their differences in uptake by different macrophages and dendritic cells, efficient uptake of both types of targeted nanoparticles by strong antigen presenting cells at the subcutaneous injection site, spleen and lymph nodes led to a similar level of targeting ligand antibody production in the mice that received either ATF-IONPs or ATF-PEG-IONPs. Results of this study also supported the notion that enhanced ligand specific antibody production after intravenous delivery of PEG-coated nanoparticles was not due to an increased immunogenicity of targeting ligands by PEG-coating, rather through reduced uptake of the nanoparticles by Kupffer cells in the liver and increased uptake by professional antigen presenting cells in the spleen and lymph nodes.
Although targeted theranostic nanoparticles enhance intratumoral delivery of chemotherapy agents, effective cancer therapy requires repeated administrations due to low delivery efficiency into the tumor and the presence of heterogeneous tumor cells. Currently, the effect of targeted delivery of chemotherapy drugs encapsulated in nanoparticles on immune responses is largely unknown. Our finding demonstrated that the presence of chemotherapy drug Dox in the nanoparticles attenuated the antibody production and increased targeted delivery of the theranostic IONPs into tumors. After administrations of targeted theranostic IONPs carrying Dox, the levels of anti-targeting ligand antibodies in mouse serum samples significantly decreased. Our results indicated that the direct cytotoxic effects on macrophages and dendritic cells following uptake of the theranostic nanoparticles may inhibit antigen presentation. Additionally, detection of significant decreases in the numbers of the ligand specific IgG producing B cells in the spleen of the mice that received multiple doses of theranostic IONP-Dox in our ELISPOT assay suggests the possibility of selective elimination of targeting ligand epitope specific IgG producing B cells. Further investigations are ongoing to elucidate mechanisms of the action mediating the inhibitory effect of theranostic nanoparticles carrying Dox on antibody responses.
In summary, results of this study provide novel information on immune responses, notably antibody responses, against targeted nanoparticles and PEG-coated nanoparticles following multiple in vivo administrations. Our discoveries of activation of targeting ligand specific antibodies using weakly immunogenic ligands and the enhanced antibody responses by PEG coating raise important issues to be considered for future design and development of targeted nanoparticle imaging probes and drug delivery carriers, and for conducting preclinical animal studies and clinical trials. However, it is likely that nanoparticle imaging probes for cancer detection will be administrated at low concentrations with only one probe administration, whereby the production of ligand specific antibody responses may not be a major concern. Importantly, demonstration of an inhibitory effect on the production of targeting ligand specific antibodies and improved delivery efficiency following systemic administrations of targeted theranostic nanoparticles carrying Dox is significant for the development of novel, targeted drug delivery and imaging platforms for future clinical translations.
Supplementary Material
Figures S1 - S5.
Acknowledgements
We thank Dr. Gregory Adams for providing the scFv B10 plasmid, Dr. Fred Millar for the 4T1 cell line, Dr. Minati Satpathy for assistance in Prussian blue staining of tissue sections, and Dr. Chris Ibegbu for using the IMMUNOSPOT equipment. This research project was supported by the following research grants from the National Institute of Health: R01#CA133722 (Yang), U01#CA151810 (Yang and Mao), R01#CA154129A (Yang), SBIR Phase II HHSN261201200029C (Wang and Yang), and R01# CA138993 (Selvaraj). Dr. Yang is supported in part by Nancy Panoz Chair of Surgery in Cancer Research Funds.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Davis ME, Zuckerman JE, Choi CHJ. et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067-70
2. Benezra M, Penate-Medina O, Zanzonico PB. et al. Multimodal silica nanoparticles are effective cancer-targeted probes in a model of human melanoma. J Clin Invest. 2011;121:2768-80
3. Lee JH, Huh YM, Jun Y. et al. Artificially engineered magnetic nanoparticles for ultra-sensitive molecular imaging. Nat Med. 2007;13:95-9
4. Nasongkla N, Bey E, Ren JM. et al. Multifunctional polymeric micelles as cancer-targeted, MRI-ultrasensitive drug delivery systems. Nano Lett. 2006;6:2427-30
5. Hrkach J, Von Hoff D, Mukkaram Ali M. et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4:128ra39
6. Lee GY, Qian WP, Wang L. et al. Theranostic nanoparticles with controlled release of gemcitabine for targeted therapy and MRI of pancreatic cancer. ACS Nano. 2013;7:2078-89
7. Bhirde AA, Liu G, Jin A. et al. Combining portable Raman probes with nanotubes for theranostic applications. Theranostics. 2011;1:310-21
8. Chen Y, Zhu X, Zhang X. et al. Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Mol Ther. 2010;18:1650-6
9. Gao J, Chen K, Miao Z. et al. Affibody-based nanoprobes for HER2-expressing cell and tumor imaging. Biomaterials. 2011;32:2141-8
10. Lee HY, Li Z, Chen K. et al. PET/MRI dual-modality tumor imaging using arginine-glycine-aspartic (RGD)-conjugated radiolabeled iron oxide nanoparticles. J Nucl Med. 2008;49:1371-9
11. Han H, Davis ME. Single-antibody, targeted nanoparticle delivery of camptothecin. Mol Pharm. 2013;10:2558-67
12. Kelly KA, Bardeesy N, Anbazhagan R. et al. Targeted nanoparticles for imaging incipient pancreatic ductal adenocarcinoma. PLoS Med. 2008;5:e85
13. Yang L, Peng XH, Wang YA. et al. Receptor-Targeted Nanoparticles for In vivo Imaging of Breast Cancer. Clin Cancer Res. 2009;15:4722-32
14. Thakor AS, Luong R, Paulmurugan R. et al. The fate and toxicity of Raman-active silica-gold nanoparticles in mice. Sci Transl Med. 2011;3:79ra33
15. Dobrovolskaia MA, Aggarwal P, Hall JB. et al. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol Pharm. 2008;5:487-95
16. Moore A, Medarova Z, Potthast A. et al. In vivo targeting of underglycosylated MUC-1 tumor antigen using a multimodal imaging probe. Cancer Res. 2004;64:1821-7
17. Kievit FM, Stephen ZR, Veiseh O. et al. Targeting of primary breast cancers and metastases in a transgenic mouse model using rationally designed multifunctional SPIONs. ACS Nano. 2012;6:2591-601
18. Zhou Y, Drummond DC, Zou H. et al. Impact of single-chain Fv antibody fragment affinity on nanoparticle targeting of epidermal growth factor receptor-expressing tumor cells. J Mol Biol. 2007;371:934-47
19. Yang L, Mao H, Wang YA. et al. Single Chain Epidermal Growth Factor Receptor Antibody Conjugated Nanoparticles for in vivo Tumor Targeting and Imaging. Small. 2009;5:235-43
20. Sakamoto JH, Smith BR, Xie B. et al. The molecular analysis of breast cancer utilizing targeted nanoparticle based ultrasound contrast agents. Technol Cancer Res Treat. 2005;4:627-36
21. Slamon D, Pegram M. Rationale for trastuzumab (Herceptin) in adjuvant breast cancer trials. Semin Oncol. 2001;28:13-9
22. Schier R, Bye J, Apell G. et al. Isolation of high-affinity monomeric human Anti-c-erbB-2 single chain Fv using affinity-driven selection. J Mol Biol. 1996;255:28-43
23. Adams GP, Schier R. Generating improved single-chain Fv molecules for tumor targeting. J Immunol Methods. 1999;231:249-60
24. Steffen AC, Orlova A, Wikman M. et al. Affibody-mediated tumour targeting of HER-2 expressing xenografts in mice. Eur J Nucl Med Mol Imaging. 2006;33:631-8
25. Kramer-Marek G, Gijsen M, Kiesewetter DO. et al. Potential of PET to predict the response to trastuzumab treatment in an ErbB2-positive human xenograft tumor model. J Nucl Med. 2012;53:629-37
26. Peng XH, Wang Y, Huang D. et al. Targeted delivery of cisplatin to lung cancer using ScFvEGFR-heparin-cisplatin nanoparticles. ACS Nano. 2011;5:9480-93
27. Blasi F, Carmeliet P. uPAR: A versatile signalling orchestrator. Nat Rev Mol Cell Biol. 2002;3:932-43
28. Han B, Nakamura M, Mori I. et al. Urokinase-type plasminogen activator system and breast cancer. Oncol Rep. 2005;14:105-12
29. Yang L, Mao H, Cao ZH. et al. Molecular Imaging of Pancreatic Cancer in an Animal Model Using Targeted Multifunctional Nanoparticles. Gastroenterology. 2009;136:1514-25
30. Yang L, Sajja HK, Cao Z. et al. uPAR-targeted optical imaging contrasts as theranostic agents for tumor margin detection. Theranostics. 2013;4:106-18
31. Li R, Zheng K, Hu P. et al. A novel tumor targeting drug carrier for optical imaging and therapy. Theranostics. 2014;4:642-59
32. Moore A, Weissleder R, Bogdanov A Jr. Uptake of dextran-coated monocrystalline iron oxides in tumor cells and macrophages. J Magn Reson Imaging. 1997;7:1140-5
33. Cruz LJ, Rueda F, Cordobilla B. et al. Targeting nanosystems to human DCs via Fc receptor as an effective strategy to deliver antigen for immunotherapy. Mol Pharm. 2011;8:104-16
34. Lutsiak ME, Kwon GS, Samuel J. Biodegradable nanoparticle delivery of a Th2-biased peptide for induction of Th1 immune responses. J Pharm Pharmacol. 2006;58:739-47
35. Moon JJ, Suh H, Li AV. et al. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc Natl Acad Sci U S A. 2012;109:1080-5
36. Patel J, Galey D, Jones J. et al. HIV-1 Tat-coated nanoparticles result in enhanced humoral immune responses and neutralizing antibodies compared to alum adjuvant. Vaccine. 2006;24:3564-73
37. Sloat BR, Sandoval MA, Hau AM. et al. Strong antibody responses induced by protein antigens conjugated onto the surface of lecithin-based nanoparticles. J Control Release. 2010;141:93-100
38. Stano A, van der Vlies AJ, Martino MM. et al. PPS nanoparticles as versatile delivery system to induce systemic and broad mucosal immunity after intranasal administration. Vaccine. 2011;29:804-12
39. Roy A, Singh MS, Upadhyay P. et al. Combined chemo-immunotherapy as a prospective strategy to combat cancer: a nanoparticle based approach. Mol Pharm. 2010;7:1778-88
40. Wadhwa S, Jain A, Woodward JG. et al. Lipid nanocapsule as vaccine carriers for his-tagged proteins: evaluation of antigen-specific immune responses to HIV I His-Gag p41 and systemic inflammatory responses. Eur J Pharm Biopharm. 2012;80:315-22
41. Chen H, Wang L, Yeh J. et al. Reducing non-specific binding and uptake of nanoparticles and improving cell targeting with an antifouling PEO-b-PgammaMPS copolymer coating. Biomaterials. 2010;31:5397-407
42. Yu T, Hubbard D, Ray A. et al. In vivo biodistribution and pharmacokinetics of silica nanoparticles as a function of geometry, porosity and surface characteristics. J Control Release. 2012;163:46-54
43. Shenoy D, Little S, Langer R. et al. Poly(ethylene oxide)-modified poly(beta-amino ester) nanoparticles as a pH-sensitive system for tumor-targeted delivery of hydrophobic drugs: part 2. In vivo distribution and tumor localization studies. Pharm Res. 2005;22:2107-14
44. Bartneck M, Keul HA, Singh S. et al. Rapid uptake of gold nanorods by primary human blood phagocytes and immunomodulatory effects of surface chemistry. ACS Nano. 2010;4:3073-86
45. Cole AJ, David AE, Wang J. et al. Magnetic brain tumor targeting and biodistribution of long-circulating PEG-modified, cross-linked starch-coated iron oxide nanoparticles. Biomaterials. 2011;32:6291-301
46. Dhar S, Kolishetti N, Lippard SJ. et al. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc Natl Acad Sci U S A. 2011;108:1850-5
47. Ishida T, Kiwada H. Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int J Pharm. 2008;354:56-62
48. Pham CT, Mitchell LM, Huang JL. et al. Variable antibody-dependent activation of complement by functionalized phospholipid nanoparticle surfaces. J Biol Chem. 2011;286:123-30
49. Szebeni J. Complement activation-related pseudoallergy: a new class of drug-induced acute immune toxicity. Toxicology. 2005;216:106-21
50. Yu WW, Falkner JC, Yavuz CT. et al. Synthesis of monodisperse iron oxide nanocrystals by thermal decomposition of iron carboxylate salts. Chem Commun. 2004:2306-7
51. Chen K, Xie J, Xu H. et al. Triblock copolymer coated iron oxide nanoparticle conjugate for tumor integrin targeting. Biomaterials. 2009;30:6912-9
52. Zhou Z, Chen H, Lipowska M. et al. A dual-modal magnetic nanoparticle probe for preoperative and intraoperative mapping of sentinel lymph nodes by magnetic resonance and near infrared fluorescence imaging. J Biomater Appl. 2013;28:100-11
53. Yang L CZ, Sajja HK, Mao H, Wang L, Geng H, Xu H, Jiang T, Wood WC, Nie S, Wang YA. Development of Receptor Targeted Magnetic Iron Oxide Nanoparticles for Efficient Drug Delivery and Tumor Imaging. J Biomed Nanotechnol. 2008;4:1-11
54. Lequin RM. Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA). Clin Chem. 2005;51:2415-8
55. Inaba K, Inaba M, Romani N. et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693-702
56. Wolenski M, Cramer SO, Ehrlich S. et al. Expression of CD83 in the murine immune system. Med Microbiol Immunol. 2003;192:189-92
57. Lechmann M, Shuman N, Wakeham A. et al. The CD83 reporter mouse elucidates the activity of the CD83 promoter in B, T, and dendritic cell populations in vivo. Proc Natl Acad Sci U S A. 2008;105:11887-92
58. Guermonprez P, Valladeau J, Zitvogel L. et al. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20:621-67
59. Roland CR, Walp L, Stack RM. et al. Outcome of Kupffer cell antigen presentation to a cloned murine Th1 lymphocyte depends on the inducibility of nitric oxide synthase by IFN-gamma. J Immunol. 1994;153:5453-64
Author contact
![]() Corresponding authors: Dr. Lily Yang, Department of Surgery, Emory University School of Medicine, Clinic C, Room C-4088, 1365 C Clifton Road, NE, Atlanta, GA 30322. Telephone: 404-778-4269; Fax: 404-778-5530. E-mail address: Lyang02edu Or Dr. Hui Mao, Department of Radiology and Imaging Sciences, Emory University School of Medicine, 1364 Clifton Road, NE, Atlanta, Georgia 30322, USA. Phone: 404-712-0357. Fax: 404-712-5948. E-mail: Hmaoedu.
Corresponding authors: Dr. Lily Yang, Department of Surgery, Emory University School of Medicine, Clinic C, Room C-4088, 1365 C Clifton Road, NE, Atlanta, GA 30322. Telephone: 404-778-4269; Fax: 404-778-5530. E-mail address: Lyang02edu Or Dr. Hui Mao, Department of Radiology and Imaging Sciences, Emory University School of Medicine, 1364 Clifton Road, NE, Atlanta, Georgia 30322, USA. Phone: 404-712-0357. Fax: 404-712-5948. E-mail: Hmaoedu.