Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2015; 5(1):71-85. doi:10.7150/thno.10117 This issue Cite
Research Paper
Copy Number Variation Analysis by Ligation-Dependent PCR Based on Magnetic Nanoparticles and Chemiluminescence
Ming Liu1, Ping Hu2, Gen Zhang1,3, Yu Zeng2, Haowen Yang1, Jing Fan1, Lian Jin1, Hongna Liu1, Yan Deng1,4, Song Li1,4, Xin Zeng1,2 ![]() , Sauli Elingarami1, Nongyue He1
, Sauli Elingarami1, Nongyue He1 ![]()
1. The State Key Laboratory of Bioelectronics, Department of Biological Science and Medical Engineering, Southeast University, Nanjing 210096, China;
2. Nanjing Maternity and Child Health Care Hospital, Nanjing 210029, China;
3. Nanjing Medical University, Nanjing 210029, China;
4. Hunan Key Laboratory of Green Packaging and Application of Biological Nanotechnology, Hunan University of Technology, Zhuzhou 412007, P. R. China.
Received 2014-7-16; Accepted 2014-9-21; Published 2015-1-1
Abstract

A novel system for copy number variation (CNV) analysis was developed in the present study using a combination of magnetic separation and chemiluminescence (CL) detection technique. The amino-modified probes were firstly immobilized onto carboxylated magnetic nanoparticles (MNPs) and then hybridized with biotin-dUTP products, followed by amplification with ligation-dependent polymerase chain reaction (PCR). After streptavidin-modified alkaline phosphatase (STV-AP) bonding and magnetic separation, the CL signals were then detected. Results showed that the quantification of PCR products could be reflected by CL signal values. Under optimum conditions, the CL system was characterized for quantitative analysis and the CL intensity exhibited a linear correlation with logarithm of the target concentration. To validate the methodology, copy numbers of six genes from the human genome were detected. To compare the detection accuracy, multiplex ligation-dependent probe amplification (MLPA) and MNPs-CL detection were performed. Overall, there were two discrepancies by MLPA analysis, while only one by MNPs-CL detection. This research demonstrated that the novel MNPs-CL system is a useful analytical tool which shows simple, sensitive, and specific characters which are suitable for CNV analysis. Moreover, this system should be improved further and its application in the genome variation detection of various diseases is currently under further investigation.
Keywords: Copy number variation, Ligation-dependent PCR, Magnetic nanoparticles, Chemiluminescence.
Introduction
Nucleic acid is the basic genetic material for organisms as it plays a decisive role in life phenomena, including embryonic development, genetic variation, disease occurrence, drug susceptibility, and so on. In the early years, the genomic variation analysis was confined to nucleic acid quantification, for example, simple sequence repeats (SSRs) [1-3], single nucleotide polymorphism (SNP) [4-6], and so forth. The origination of nucleic acid manipulation and analysis was for content determination; for example, the physiological and pathological variations of organisms could be determined by gene dosage, especially the virulence genes. Variation in the gene dosage can cause quantitative change of translated proteins, resulting into pathological alterations. Sonny et al [7] demonstrated that the gene dosage of the common risk variant 9p21 predicted the severity of coronary atheromatous burden and it was useful in determining risk for disease development and risk stratification among patients with documented coronary artery disease. Via amplification of α-synuclein gene (SNCA) exons by quantitative real-time PCR, Singleton et al [8] examined the copy number variant of SNCA in a large family with autosomal dominant Parkinson disease (PD) and yielded results consistent with whole gene triplication, which indicated that the gene dosage effect was one of the causes of Parkinson disease. This current research emphasized on a new method for quantitative analysis of copy number variation (CNV). [9-11].
A series of quantitative technologies have been developed to date, such as molecular spectroanalysis [12] and electrochemical analysis [13]. However, these methods were unable to analyze trace samples. Moreover, due to poor accuracy, traditional technologies have not well adapted the contemporary development of structural molecular biology, genomics and proteomics. In recent years, some methods with high sensitivity, throughput and automation capability have been rapidly developed, for example, real time quantitative PCR [14, 15], serial analysis of gene expression (SAGE) [16,17], microarrays [18,19], and so forth. Besides, there are two techniques, multiplex amplifiable probe hybridization (MAPH) [20] and multiplex ligation-dependent probe amplification (MLPA) [21], which have been continuously developed and updated for measurement of alterations in gene dosage, including CNVs. Both MAPH and MLPA have relied on comparative quantification of specifically bound probes that were amplified by PCR using universal primers. All these methods have been used in diagnosis and gene expression analysis. MAPH has been reported for detection of deletions in Duchenne Muscular Dystrophy (DMD) gene, subtelomeric deletions and Chronic Myeloid Leukemia (CML) tumor typing [22-25]. The application of MLPA has been much more than MAPH, as it has been reported for gene dosage determination in genes such as Human MutS homolog 2 (MSH2), MutL homolog 1 (MLH1), DMD, breast cancer 1 (BRCA1), and so on [26-29]. So far, over 300 MLPA probe sets for a very large range of common and rare genetic disorders have been made commercially available from MRC Holland [21] and the MLPA assay has been widely used in laboratories for molecular diagnosis of several genetic diseases in a few years.
With the rapid development of nanotechnology in life science, nanomaterials, especially magnetic nanoparticles (MNPs), have been widely used in biological fields, such as drug delivery [30-32], magnetic resonance imaging (MRI) [33-35], immunoassays [36-38], purification of nucleic acids and proteins [39-41], owing to their excellent separation and adsorption capacity, high dispersion in aqueous solution and easy operation in automated work-stations [42]. Ranzoni et al [43] described a one-step homogeneous assay based on antibody-coated MNPs which were spiked in very small amount directly into blood plasma to perform protein biomarker detection. This assay could be widely applied in quantitative biology and clinical diagnostics. Zhang et al [44] reported the development of a sensitive colorimetric (MNP)-enzyme-based DNA sandwich assay that was suitable for simultaneous, label-free quantitation of two DNA targets, which could also effectively discriminate SNPs in genes associated with human cancers.
In addition, chemiluminescence (CL) detection is regarded as an effective method for biological detection because of its extremely high sensitivity and wide linear range [45]. CL detection also has other obvious advantages, namely rapidity, low-cost and high-throughput, since there is no additional requirement for excited light and scanning processes [46]. Nelson et al [47] described a rapid in-solution method for detection of all 12 single-base mismatches based on the hybridization protection assay, utilizing oligonucleotide probes labeled with a highly chemiluminescent acridinium ester. This method was simple, sensitive and applicable to both amplified and non-amplified targets. Another sensitive nucleic acid detection method for HBV based on MNPs and chemiluminescence was reported by He et al [48]. The biotinylated DNA was captured by MNPs and different concentrations of HBV-DNA were detected by the chemiluminescence system, which provided a novel tool for the HBV-DNA quantification.
Herein, we report a new method for DNA quantification and CNV analysis, combining the MNPs separation and CL detection by ligation-dependent PCR (Fig. 1). The DNA target was amplified with the base insertion of biotin-dUTP in ligation-dependent PCR and then captured by amino-modified probes immobilized on carboxyl MNPs. The CL signals were detected after streptavidin-modified alkaline phosphatase (STV-AP) bonding and magnetic separation. Gradient concentrations of target DNA were detected under the optimized conditions and relevant CL intensity was obtained. The CL intensity exhibited a linear correlation with the logarithm of the original target concentration. The results from the MNPs-CL quantitative analysis of target genes were compared with results from the MLPA analysis, and it was demonstrated that the MNPs-CL system as a novel and useful analytical tool with simple, sensitive, and specific characters is useful for CNVs analysis. This method displayed potential for applications in the clinical diagnosis of genome variants for various diseases.
Schematic of ligation-dependent PCR and CL detection. Step 1, unique probes (hybridization sequences with universal primers at the end) competitively hybridized to the target sequence. Step 2, DNA ligase-65 enzyme was used to join the probe pairs. Step 3, the ligation products were amplified by PCR with universal primers and dUTP, and extended PCR product oligonucleotides were represented by a broken line. Step 4, the amplification product was hybridized with MNPs-probe and separated specifically for CL detection.
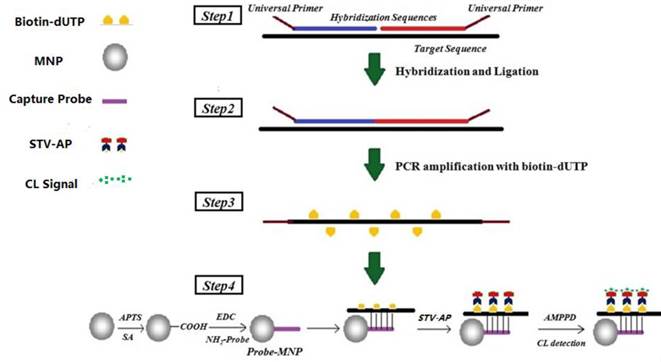
Materials and Methods
Materials
MNPs (SiO2/Fe3O4) were self-made with minor modifications [49]. Succinic anhydride (SA), dimethylfomamide (DMF), 2-(N-morpholino) ethane sulfonic acid (MES), 3-aminopropyl triethoxysilane (APTS), 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide (EDC), acetonitrile, acetic anhydride, phosphate buffered saline (PBS), bovine serum albumin (BSA), Tris, Tween-20, sodium lauryl sulphate (SDS), and salt-sodium citrate (SSC) were purchased from Shanghai Chemical Reagent Co. Ltd (Shanghai, China). MLPA reagents (including MLPA buffer, ligase-65 buffer and ligase-65 enzyme) for MLPA reactions were purchased from MRC-Holland b.v. (Amsterdam, The Netherlands). 3-(2'-spiroadamantane)-4-methoxy-4-(3'-phosphoryloxy) phenyl-1, 2-dioxetane (AMPPD) was purchased from Sichuan Biochem-ZX Research Co. Ltd (Chengdu, China). Streptavidin-modified alkaline phosphatase (STV-AP) was purchased from Vector Laboratories Inc. (Bulingame, USA). All oligonucleotides (including universal primers, hybridization sequences, amino-probe and artificial template) used in this work were synthesized by Shanghai Sangon Biologic Engineering Technology and Service Co. Ltd. (Shanghai, China).
Methods
Preparation of probe-MNPs
MNPs, approximately 400 nm in diameter, were prepared with minor modification of previous reports [49] and characterized by transmission electron microscopy (TEM, JEM2100, JEOL, JPN) and scanning electron microscope (SEM, S100, Hitachi, JPN) (Fig. 2). Briefly, the MNPs were modified with APTS for 7 h and then modified with SA in gradient concentrations (0.1, 0.01, 0.001, 0.0001 and 0.00001M) for optimization. The obtained carboxylated MNPs were blocked by incubating with a solution of 10% (v/v) acetic anhydride in acetonitrile for 1 h and re-suspended in PBS at the concentration of 10 mg/mL. The carboxylated MNPs and amino-probes were incubated in the MES solution (25 mM, pH 6.0) with EDC as a catalytic agent for 10 h at 4℃. The resultant probe-MNPs was then washed for four times with Tris (50 mM, pH 7.4) and finally stored in PBS-T buffer (0.3M PBS buffer containing 0.05% Tween-20, pH 7.0).
The characterization of the MNPs prepared via the combination of self-assembly. (a) The SEM result of the Fe3O4 MNPs; (b) The TEM result of the Fe3O4@SiO2 MNPs. The diameters of the MNPs were about 400 nm with a good dispersion, and the SiO2 shell with about 30 - 60 nm was wrapped on the MNPs surface.

Ligation-dependent PCR
The hybridization master mix contained 1.5 μL MLPA buffer, 5 μL artificial template (glyceraldehyde-3-phosphate dehydrogenase, GAPDH, 0.2 nM), and 1.5 μL ligation-sequence-mix (1 nM). The mixture was incubated at 95℃ for 5 min and subsequently at 60℃ for 16 h. Afterwards, 32 μL ligase master mix containing 3 μL ligase-65 buffer A, 3 μL ligase-65 buffer B, 1 μL ligase-65, and 25 μL ddH2O were added into the hybridization master mix for ligation reaction (Fig. 1, step 1). The ligation conditions are described as follows: incubation at 54℃ for 15 min, followed by 5 min at 98℃ for the heat inactivation of ligase-65 enzyme (Fig. 1, step 2). Finally, 5 μL ligation products were mixed with 20 μL PCR reaction buffer containing 2.5 μL 10×PCR buffer, 2.5 μL Mg2+ (25 mM), 2 μL dNTPs (1 mM, contained biotin-dUTP), 2 μL primermix (10 μM), 0.2 μL Taq DNA polymerase and 10.8 μL ddH2O. The thermal circling conditions were as follows: 95℃ for 5 min, 95℃ for 30 sec, 58℃ for 30 sec, and 72℃ for 40 sec for 35 cycles (Fig. 1, step 3).
Hybridization and CL detection
All the obtained PCR products were mixed with 30 μL probe-MNPs (10 mg/mL) and 10 μL hybridization buffer. The hybridization procedure was as follows: 95℃ for 10 min, 60℃ for 1 h, and the MNPs were shaken well by pipetting up and down every 10 min (Fig. 1, step 3). The MNPs were thereafter washed for three times with 2×SSC containing 10% SDS, 0.2×SSC containing 10% SDS and washing buffer (50 mM Tris, pH 7.5, 0.15 M NaCl), respectively. The MNPs were then re-suspended in blocking buffer (washing buffer containing 0.25% BSA) for 30 min. 30 μL STV-AP (diluted to 1:1000 into blocking buffer) was added into the MNPs, followed by incubation for 30 min at room temperature with gentle shaking. After magnetic separation, followed by transfer into a clean tube, the mixture was washed for three times with washing buffer to remove excess STV-AP. Finally, 300 μL of AMPPD solution (0.25 mM) was added into the above mixture, followed by well mixing. The mixture was then divided into three tubes for independent detection (Fig. 1, step 4). The CL intensity was then measured by EnSpireTM 2300 Multilabel Reader (PerkinElmer Co. Inc., Waltham, USA) and the data recorded every 5 min.
Optimization of CL system
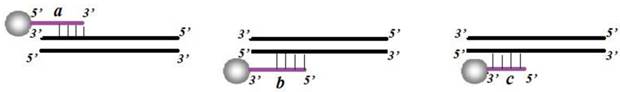
The optimization of CL system consisted of probe optimization, PCR cycle number, and hybridization temperature. Three probes immobilized onto the carboxylated MNPs were designed into following groups: 5'NH2-(T)15-probe (Fig. 3a), 3'NH2-(T)15-probe (Fig. 3b) and 3'NH2-probe (Fig. 3c). The amino ends were immobilized at the surface of the carboxylated MNPs. After ligation, the products were amplified by PCR with 20, 25, 30, 35 and 40 cycles, respectively. The PCR products were then hybridized with probe-MNPs at following gradient temperatures: 50℃, 52℃, 54℃, 56℃, 58℃, 60℃. After hybridization and CL detection, the most suitable and efficient probe, PCR cycle number and hybridization temperature were determined.
Diagrammatic sketch of corresponding positions for three hybridization probes: (a) 5'NH2-(T)15-probe; (b) 3'NH2-(T)15-probe; (c) 3'NH2-probe.
Quantification of nucleic acid
The artificial template was diluted into the following gradient concentrations: 0.002 pM, 0.02 pM, 0.2 pM, 2 pM, 20 pM, and 200 pM. After ligation-dependent PCR, six groups of PCR products were captured by probe-MNPs and detected by CL. According to the relative CL intensity, the dose-response curve was drawn for quantitative analysis.
Samples and DNA extraction
A total of 40 blood samples (including male, female, normal and trisomy 21) were collected from Nanjing Maternity and Child Health Care Hospital after approval by the ethical committee of the hospital for only laboratory study. Genomic DNAs were extracted from peripheral blood samples using ReliaPrepTM Blood gDNA Miniprep System (Promage Co. Ltd., Madison, USA) and quantified by OD-1000 spectrophotometer (OneDrop Co. Ltd., Nanjing, China). DNA samples were diluted at 50 ng/μL with TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA).
Probes and primers design
In addition to the reference gene GAPDH, six genes were selected from the chromosome X, Y and 21, respectively. A series of oligonucleotide probe pairs (Table 1) were designed according to the guidelines from MRC-Holland (www.mlpa.com), such that each pair would locate directly at specific loci within these target genes. Probe sequences were checked for non-specific annealing sites using Basic Local Alignment Search Tool (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Each pair of probes consisted of two parts. The longer part of the probe contained a tag sequence which was complementary with the probe-MNPs and allowed amplification products binding onto them. All probes were diluted to a final concentration of 5 nM in TE buffer.
Sequence of oligonucleotides probes and primers used.
| Probes | Sizea | Oligonucleotide Sequencesb | |
|---|---|---|---|
| Reference gene | GAPDH-R | 122 nt | 5'-gggttccctaagggttggaCAGGAGCGAACGAGATACTCCTGAGGTGGCATAGTGGGGTGGTGAATAC-3' |
| GAPDH-L | 5'-PHO-CATGTACAAAGCTTGTGCCCAGACTGTGAATtctagattggatcttgctggcac-3' | ||
| X- chromosome | RSK4-R | 112 nt | 5'-gggttccctaagggttggaCCAAGCGAATGCAACGTATCTTTACCCCGACAGCATTCCAATGA-3' |
| RSK4-L | 5'-PHO-GTTGGTTAAAGTATGACCACTCACAGtctagattggatcttgctggcac-3' | ||
| PCDH19-R | 114 nt | 5'-gggttccctaagggttggaTAGTCCGGACCTGCTGTTATGTAGAGAACCTCTTGCACAGAAAGAC-3' | |
| PCDH19-L | 5'-PHO-CAGTCCTAACCTTTCAAATGATGCCTtctagattggatcttgctggcac-3' | ||
| Y- chromosome | SRY-R | 110 nt | 5'-gggttccctaagggttggaCCAAGCGAATGCAACGTATCCTGGTAGAAGTGAGTTTTGGATAGTAAAATAAGTTTC-3' |
| SRY-L | 5'-PHO-GAACTCTGGCACCTTTCAATTTTGTCGtctagattggatcttgctggcac-3' | ||
| DAZ1-R | 125 nt | 5'-gggttccctaagggttggaTAGTCCGGACCTGCTGTTATTCCACGCCCAACCATAGCACTTGACC-3' | |
| DAZ1-L | 5'-PHO-GAGAATAATACGTTCTTCATAAATCAGTCAGTCACTTtctagattggatcttgctggcac-3' | ||
| 21- chromosome | DSCR3-R | 111 nt | 5'-gggttccctaagggttggaATACCCAGAACGACTATGCGCTGGTGGCAAAAGCAGGTAGTTTC-3' |
| DSCR3-L | 5'-GAGAGATGAGCACAACCCAGAGAAGtctagattggatcttgctggcac-3' | ||
| DSCR4-R | 112 nt | 5'-gggttccctaagggttggaCTACAGCACCTCAGGAGAGTGCCTTCCAGGTAGCTGCTCATAAG-3' | |
| DSCR4-L | 5'-PHO-CCCTTTCTAGAGAGTGATTGGGACTAtctagattggatcttgctggcac-3' | ||
| Universal primers | Forward | 23 nt | 5'-GTGCCAGCAAGATCCAATCTAGA-3' |
| Reverse | 19 nt | 5'-GGGTTCCCTAAGGGTTGGA-3' |
a Amplification products length.
b The lower-case letters indicate PCR primer sequences, the capital letters indicate DNA hybridization sequences, the underlined letters indicate tag sequences; the 5' end of the right probe is phosphorylated (PHO) to allow ligation to the 3' end of the left probe.
Copy numbers detection by MLPA and MNPs-CL system
For each sample, 200 ng of DNA was used in the hybridization and ligation with the probe mix (including probes for target genes and reference gene). The ligation products were amplified with the universal primers. To compare the accuracy of the MNPs-CL detection system, the copy numbers detection for target genes was also performed by the developed MLPA assay [21]. The amplification products were therefore simultaneously separated by capillary electrophoresis and probe-MNPs, and data analysis was done with Applied Biosystems 3100 Genetic Analyzer (Applied Biosystems Co. Inc., Carlsbad, USA) and EnSpireTM 2300 Multilabel Reader (PerkinElmer Co. Inc., Waltham, USA), respectively.
Data analysis
In the MLPA assay, the copy number alteration could be visually inspected by superimposing the peak profile of the test sample with the profile of the control one [50]. In the MNPs-CL system, the copy number alteration could be investigated by comparing the CL intensity at its stable stage between the test values of sample and the control one. A normal female sample with two copies of PCDH19 and RSK4 on the X chromosome was chosen as the reference sample. The ratio for each test sample was calculated relative to a standard reference created from the reference sample. The equation could be represented as follows: R0 = Vt / Vr, where R0 was the relative copy number of the test genes, Vt was the relative value (the peak intensity value for the MLPA assay and the CL intensity value for the MNPs-CL system) of the test gene within the test sample and Vr was the relative value of the test gene within the reference sample. The relative value V was calculated by the ratio of (test gene value) / (reference gene value) in the same test sample. The standard deviations were based on five replicates of the same subject in the same assay.
Results
The working principle and specificity of MNPs-CL detection method
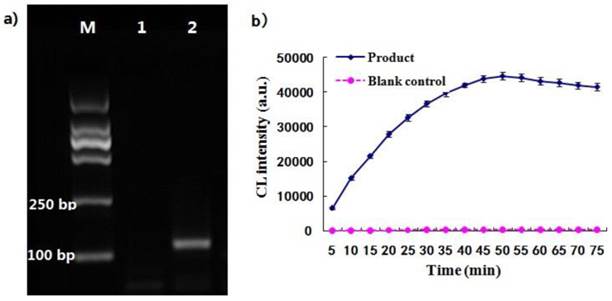
As illustrated in Fig.1, the two oligonucleotides were hybridized to the DNA template and ligated by the ligase enzyme to form a ligated full-length sequence that could be amplified with a pair of universal primers. The PCR product was analyzed with agarose gel electrophoresis. Result showed that there was an amplification target band at the 120 bp and there was no amplification bands in the blank (Fig. 4a), demonstrating that the two probes had been ligated together and amplified with the ends of universal primers. It was thus suggested that the designed ligation probes and universal primers were suitable for the ligation-dependent PCR system due to their good specificity. The CL detection was performed after the dUTP-labeled PCR products were captured by the probe-MNPs. The CL intensity profile was related with the reaction time of STV-AP with AMPPD. As shown in Fig. 4b, the CL signal rose rapidly and became stable at 50 min. The maximum CL value of PCR products was remarkably higher than that of the blank control group. It was therefore suggested that this CL detection system, consisting of ALP and AMPPD, showed a stable light signal and was sensitive to the ligation-dependent PCR product which was labeled with dUTP.
(a) The ligation-dependent PCR products amplified by 35 cycles, and lane 1 represents the blank control and lane 2 represents the product. (b) The curve of CL detection for PCR products, and the blank control group was performed under the same reaction conditions without template.
Optimization of MNPs-CL system
The effects of several reaction parameters were investigated for the detection optimization. The amino groups at surface of the APTS-grafted MNPs were reacted with SA for carboxyl functionalization. The same amount of PCR products from 35 cycles were hybridized with gradiently modified carboxylated probe-MNPs. The values of CL intensity for the MNPs modified with tenfold concentrations of SA (0.1, 0.01, 0.001, 0.0001 and 0.00001M) are shown in Fig. 5a. The CL intensity increased from 0.00001 M to 0.001 M of SA and then decreased under 0.01M SA modification condition. 0.001 M was therefore taken as the optimal concentration for SA modification, and this concentration was chosen for subsequent experiments.
Effect of different parameters on the relative CL intensity for MNPs-CL system. (a) The concentration of SA modified on the carboxyl MNPs impact on the relative CL intensity; (b) Three different probes hybridized with MNPs impact on the relative CL intensity; (c) Hybridization temperature impact on the relative CL intensity; (d) Number of PCR cycles impact on the relative CL intensity.
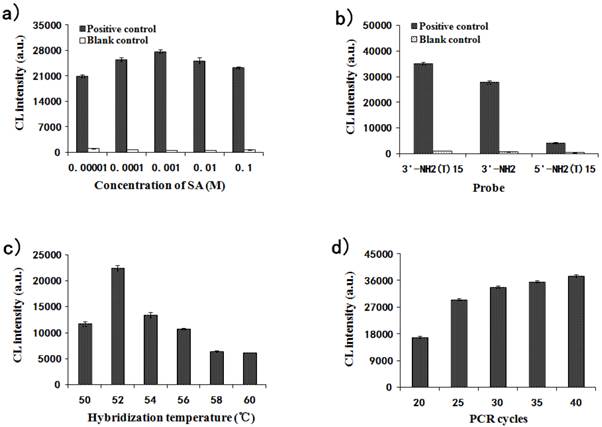
Three probes were designed for immobilization onto the carboxylated MNPs and their effect was investigated by CL detection. The CL intensities for 3'NH2-(T)15-probe, 3'NH2-probe and 5'NH2-(T)15-probe are shown in Fig. 5b. It was demonstrated that the probe with 3'NH2-(T)15 end immobilized onto the MNPs was much more efficient than that without the (T)15 end due to the effect of molecular arms, and it was also more efficient than that with 5'-(T)15 end immobilized onto the MNPs. So the 3'NH2-(T)15-probe was taken as the optimal and chosen for subsequent experiments.
The hybridization temperature was also investigated from 50℃ to 60℃ every 2℃. Fig. 5c illustrates that the effect of hybridization temperature (from 50℃ to 60℃) on CL intensity was great. The CL value reached the peak at 52℃ and decreased from 52℃ to 60℃ sequentially. Therefore, the optimum temperature for hybridization was determined as 52℃.
A PCR cycle gradient (20, 25, 30, 35 and 40) was set up to check its effect on CL intensity. As shown in Fig. 5d, the CL intensity significantly increased with the increase in PCR cycles. The CL values from 25, 30, 35 and 40 cycles were fairly close to each other and the possibility was that the binding capacity of the probe-MNPs tended for saturation. To decrease the methodological error, the binding capacity of the probe-MNPs should be unsaturated. Thus, 25 cycles for PCR amplification were chosen for further studies.
Detection limit of MNPs-CL system
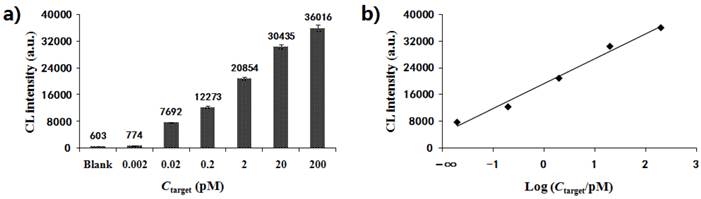
Under the optimum conditions described above, the MNPs-CL system was preliminarily established for quantitative analysis. Fig. 6a depicts the CL signals of the quantitative detection system in response to the artificial template GAPDH at gradient concentrations. The maximum CL values of products amplified from 0.002 pM, 0.02 pM, 0.2 pM, 2 pM, 20 pM, and 200 pM templates were 774, 7692, 12273, 20854, 30435 and 36016, respectively. The CL detection results showed that the CL intensity increased with the concentration of template, ranging from 0.002 pM to 200 pM. As shown in Fig. 6b, the CL intensity exhibited a linear correlation with the logarithm of original concentrations of the targets within the range from 0.02 pM to 200 pM. The linear equation could be represented as y = 7481x + 13973, with a correlation coefficient of R2=0.987, where y was the CL intensity and x the logarithm of target concentration. When the concentration dropped to 0.002 pM, the decrease amplitude of the CL value became much smaller and the signal intensity was close to that of the blank. The detection limit was estimated to be 0.01 pM. The above results showed that this system was feasible and could be used in quantitative analysis for nucleic acids.
(a) Dose-response of artificial template with a gradient concentration (0.002 pM, 0.02 pM, 0.2 pM, 2 pM, 20 pM, and 200 pM) by CL detection; (b) Linear relationship between CL intensity and the logarithm of origination concentration for the template.
Specificity of probes designed for test genes
To validate the methodology for the present new system, six genes were selected for copy number analysis in human genome (Table 1). The amplification products were visualized by agarose gel electrophoresis and the specificity of the designed probes was analyzed (Fig. 7). It was shown that the target bands for six gene loci were yielded at the range of 110 bp to 120 bp, while there were no nonspecific bands in the blank control. As shown in Fig. 7b, the positive bands for SRY and DAZ1 only appeared in male (sample 1) and no corresponding amplification was found in female (sample 2), owing to these two genes only exist on the Y-chromosome, which could also be used for gender determination. These results suggested that the designed probes for target genes were able to amplify all loci presented in the samples, and represented high specificity without cross-reactions. The brightness of the target bands showed the high amplification efficiency of universal primers, laying the foundations for the subsequence experiments of copy number quantification.
The gel electrophoresis results of ligation-dependent PCR for six detected genes. Sample 1 was male, sample 2 was female and CK was the blank control performed under the same reaction compositions without DNA template.

Quantitative analysis of gene copy numbers
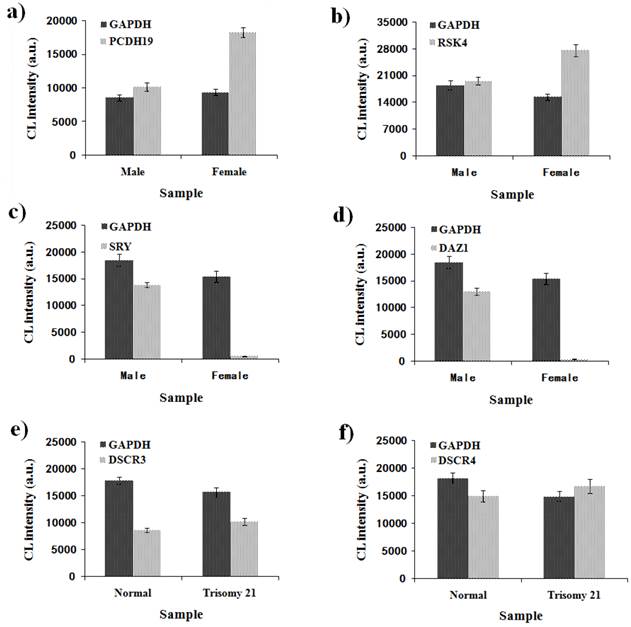
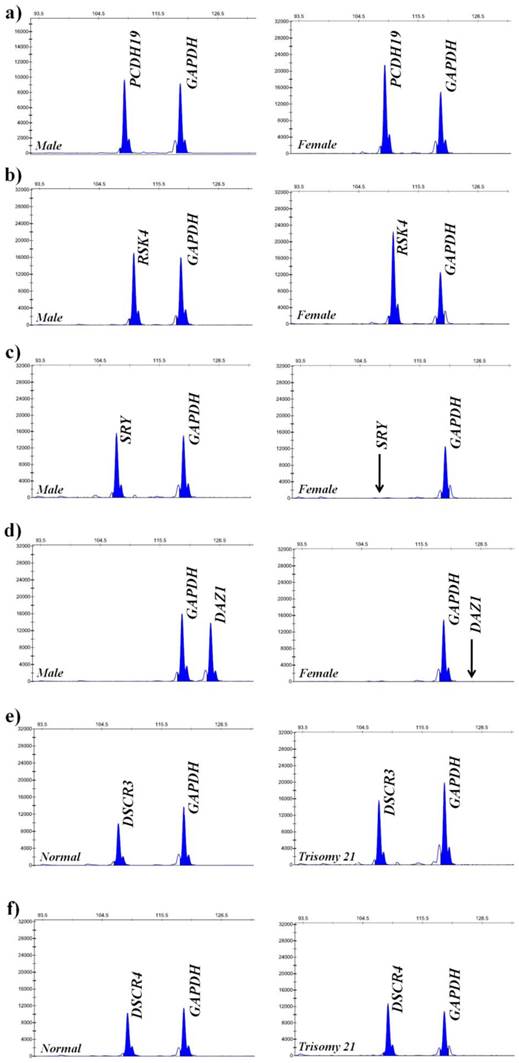
To verify the feasibility and accuracy of the novel MNPs-CL system, the amplification products were quantified by capillary electrophoresis and chemiluminescence simultaneously, and the experimental data from both systems were compared. As illustrated in Fig. 8, the CL intensity of the detected genes on the X-chromosome had obvious multiple relationships between male and female samples and no signals of target genes on the Y-chromosome were detected in the female samples. Although the visual difference of CL intensity was not significant between normal and trisomy-21 samples, the relative quantification was obtained through numerical calculations. These results were consistent with the waveform of MLPA analysis (Fig. 9), and the detailed data are listed in Table 2. Ratios in different ranges correspond to different copy numbers, namely a ratio of 0.3 ~ 0.7 corresponds to 1 copy number, a ratio in the range of 0.7 ~ 1.3 corresponds to 2 copy numbers, a ratio in the range of 1.3 ~ 1.7 corresponds to 3 copy numbers, and a ratio in the range of 1.7 ~ 2.3 corresponds to 4 copy numbers.
Copy numbers analysis by MLPA and MNPs-CL system.
| Gene | Meana ± SDb | Expected ratios of copy numbers | ||||
|---|---|---|---|---|---|---|
| Male | Female | Normal | Trisomy 21 | |||
| MLPA | PCDH19 | 0.54 ± 0.06 | 0.99 ± 0.04 | 1:2 | ||
| RSK4 | 0.75 ± 0.05 | 0.95 ± 0.02 | 1:2 | |||
| SRY | 0.48 ± 0.05 | 0 | 1:0 | |||
| DAZ1 | 0.42 ± 0.02 | 0 | 1:0 | |||
| DSCR3 | 0.79 ± 0.04 | 1.81 ± 0.09 | 2:3 | |||
| DSCR4 | 0.94 ± 0.08 | 1.37 ± 0.06 | 2:3 | |||
| MNPs-CL | PCDH19 | 0.60 ± 0.02 | 0.99 ± 0.14 | 1:2 | ||
| RSK4 | 0.58 ± 0.02 | 0.90 ± 0.01 | 1:2 | |||
| SRY | 0.55 ± 0.08 | 0 | 1:0 | |||
| DAZ1 | 0.52 ± 0.03 | 0 | 1:0 | |||
| DSCR3 | 0.96 ± 0.02 | 1.38 ± 0.11 | 2:3 | |||
| DSCR4 | 0.82 ± 0.05 | 1.32 ± 0.02 | 2:3 | |||
a Mean ratio to the reference normalized values with the averages of duplicate in 10 samples.
b Standard Deviation.
Quantitative detection of six genes by MNPs-CL system. (a) CL intensity of GAPDH and PCDH19 in the male and female samples; (b) CL intensity of GAPDH and RSK4 in the male and female samples; (c) CL intensity of GAPDH and SRY in the male and female samples; (d) CL intensity of GAPDH and DAZ1 in the male and female samples; (e) CL intensity of GAPDH and DSCR3 in the normal and trisomy 21 samples; (f) CL intensity of GAPDH and DSCR4 in the normal and trisomy 21 samples.
Quantitative detection of six genes by MLPA. (a) Electropherogram showing peaks of PCDH19 and GAPDH in the male and female samples; (b) Electropherogram showing peaks of RSK4 and GAPDH in the male and female samples; (c) Electropherogram showing peaks of SRY and GAPDH in the male and female samples; (d) Electropherogram showing peaks of DAZ1 and GAPDH in the male and female samples; (e) Electropherogram showing peaks of DSCR3 and GAPDH in the normal and trisomy 21 samples; (f) Electropherogram showing peaks of DSCR4 and GAPDH in the normal and trisomy 21 samples. The horizontal ordinate denotes amplification products length and the vertical ordinate denotes peak values. Black arrows indicate the complete absence of Y-SRY and Y-DAZ1 on the X-chromosome.
The quantitative analysis was performed by both the MLPA and MNPs-CL systems for 40 samples (10 individuals respectively for male, female, normal and trisomy-21). The normal female individuals were detected as the control group. All the samples were successfully genotyped by both platforms, and some of these analyses had mean ratios at unexpected levels. As shown in Table 2, both platforms were able to accurately determine the genes copy number of PCDH19, SRY, DAZ1, and DSCR4. There were two discrepant results for RSK4 and DSCR3 genes quantification by MLPA analysis. The ratio for RSK4 was 0.70-0.80, more than the expected ratio of 0.5 in male subjects, and the ratio of copy number was higher than 0.7, while a ratio of 0.5 was the expected value for males to females. The ratio for DSCR3 was 1.72-1.90, more than the expected ratio of 1.5 for 21-trisomy subjects, and the ratio of copy number was higher than 2.0. A ratio of 1.5 was as expected value for 21-trisomy to normal. In the MNPs-CL system, there was only one discrepant result for DSCR3 copy numbers. The ratio of DSCR3 for 21-trisomy subjects was 1.27-1.49, and it revealed 2-3 copies when 3 copies were expected. By comparison, the data demonstrated that the sensitivity and specificity of the new MNPs-CL system were equivalent to that from the MLPA system, which was acceptable for determining chromosomal DNA copy number changes, and would therefore be suitable for nucleic acid quantitative analysis.
Discussion
Herein, we have successfully designed and tested a new system for quantitative analysis of copy number variants. There were two significant reaction steps in this system. One was ligation reaction; where a pair of probes were hybridized with complementary template and linked together by induction of the DNA ligase to form PCR templates. The other reaction involved PCR amplification of the ligation products by universal primers. Whether these two processes had been accomplished successfully or not was the first key issue for such quantitative analysis. It was important to check whether the obtained PCR products were amplified with the ligation products and universal primers before the subsequent quantitative detection. Amplifications with a universal primer pair laid the foundation for relative quantification and from that the amplification efficiencies among different ligated products could be homogenized [51]. The second key issue from this quantitative analysis was that a cross-reaction appeared between the probes of different target genes [52,53], so probes should be properly optimized. In this work, the gel electrophoresis results showed that the cross-reaction among designed probes had been avoided (Fig. 4 and Fig. 7). Moreover, non-specific amplification might derive from the non-ligated probes, which would produce false positive signals and lower the integral analytical sensitivity [54]. The designed ligation probes should be optimized to reduce or eliminate the non-specific signal. The concentration of ligation probes should also be controlled at an extremely low level in comparison with the universal primers, as this could largely eliminate the non-specific signals. Additionally, adding certain exonucleases to digest unligated oligonucleotides before amplification was also a useful method [55]. The non-specific amplification was checked from the blank control group performed under the same reaction conditions without template.
In the MNPs-CL system, the amino-modified probes were immobilized onto carboxylated MNPs and then hybridized with target DNA for CL detection after magnetic separation, which is currently one of new focuses in biological detection [56-59]. This system had been well developed in many studies, especially those involving original works from our laboratory [48, 60, 61, 62, 63]. Various experimental parameters had been optimized, including the MNPs amounts, carboxyl concentration to amino-MNPs, concentration of specific amino-probes, hybridization temperature and time, and so on. However, some parameters (for example, size, structure, concentration, and so on) tend to vary with detection of different DNA targets. To make our current MNPs-CL system suitable for quantitative detection in this study, we re-optimized some parameters for the functionalization and hybridization of MNPs to capture the target DNA based on the previous work. The density of carboxyl groups on the MNPs surface has a significant effect on the relative CL intensity since it determines the binding capacity of probes [61]. The SA modification with gradient concentration experiment indicated that the appropriate density of carboxyl groups could promote the hybridization efficiency between probe-MNPs and PCR products, leading to the improved CL intensity (Fig. 5a). However, the excess of carboxyl groups hindered the capture ability of probe-MNPs for products. The optimal concentration of SA modification in this study was consistent with the results reported by Wang and Yang et al [61, 62], which was equally to 1 mM or close to 1 mM. This was higher than the reported concentrations (0.1 mM) in the research by He et al [48], probably due to a high amplification and hybridization efficiency of the artificial template. The probe structure also has significant effect on nucleic acid capture efficiency by MNPs. The result from the optimization of hybridization probes suggested that the structure of DNA molecules (probes length, binding site, and so on) tethered to surfaces might significantly have affected the hybridization efficiency. The dynamics and interaction of DNA probes tethered on the silica surface could be investigated by a molecular dynamics simulation, which was helpful to better optimize the DNA probes design [63]. The simulation was performed by use of the CHARMM software package with CHARMM27 force field [64, 65], and the resulting system was subjected to a series of minimizations through the CHARMM program [66]. By calculating the radius of gyration, tilt angles, and distances of the neighboring ssDNAs, the simulation results suggested that the packing density had a significant effect on the change of the molecular orientation and overall structure of surface-tethered ssDNAs. The hybridization temperature is another important factor which greatly affects the hybridization efficiency and final CL intensity (Fig. 5c) [67]. It was also verified by other two genes (GSTT1 and GSTM1), polymorphism of which might be associated with developing gastric cancer [68]. The genes were polymorphic and the null alleles resulted in a lack of corresponding enzyme activities [69]. Null genotypes of both GSTM1 and GSTT1 were found to predispose to stomach cancer [70] and as a result they were selected for the study of CNVs quantification. Likewise, the optimum hybridization temperatures for both two genes were the same as that for GAPDH (Data not shown), which illustrated that different probe sequences had no significant effect on the hybridization temperature. The determination of maximum binding capacity of probe-MNPs was an indispensable step. If the binding capacity of the probe-MNPs was saturated, the excess target DNA fragment would be lost, causing a large deviation from the theoretical value and resulting in non-quantification. This could be investigated in three aspects, the modification concentration of probes, amplification cycles, and quantity of target DNA for hybridization. We chose the PCR cycles for optimization because they are more stable for eliminating the effects of non-specific amplification and adsorption than others.
To evaluate the feasibility for quantitative analysis, we investigated the linear range of MNPs-CL system by detecting the artificial DNA sequence at varying concentrations (Fig. 6). The CL results showed a good linear relationship in the concentration range from 0.02 pM to 200 pM. The sensitivity of this system was compared with several other DNA assay techniques based on MNPs. As shown in Table 3, the sensitivity of the MNPs-CL system in this work is equal to or better than most other DNA assay techniques, and even better than the same MNPs-CL assay for amplified DNA sequence [62]. It was therefore indicated that this system provided good specificity and sensitivity for DNA detection. This might be attributed to highly efficient signal amplification by ligation-dependent PCR with biotin-labeled dUTP and magnetic separation. However, the background value was high and it did not perform well as expected, possibly due to the target sequences binding to the probe-MNPs nonspecifically. Non-specific binding might result from the special structure of molecules immobilized on the MNPs surface. Optimizing non-specific binding to reduce the background is a prerequisite for high-sensitive detection. Therefore, the CL system is for further optimization, both on the concentration of probes modified onto MNPs and concentration of STV-AP which could bind with AMPPD to generate the CL signals. The system would then be more suitable in the analysis of genomic diversity with a better detection limit and wider linear range.
Comparison of sensitivity for different DNA assay methods based on MNPs.
| Analytical method | Label | Detection limit |
|---|---|---|
| Colorimetric detection | Ag/SiO2 | 100 pM [71] |
| Colorimetric detection | Carbon nanotube-HRP | 1 pM [72] |
| Surface plasma resonance (SPR) | Label-free | 10-100 pM [73] |
| Quartz crystal microbalance (QCM) | Gold nanoparticle | 32 pM [74] |
| MNPs-electrochemical | Gold nanoparticle | 6 pM [75] |
| Inductively coupled plasma mass spectrometry (ICPMS) | Gold nanoparticle | 200 fM [76] |
| Magnetic concentration-fluorescence | Quantum dots | 500 fM [77] |
| Fluorescence imaging | Counting silica nanoparticles | 0.8 fM [78] |
| MNPs-CL | Catalytic DNAzyme | 100 pM [56] |
| MNPs-CL | Gold nanoparticle | 1 pM [58] |
| MNPs-CL | Horseradish peroxidase (HRP) | 100 fM [57] |
| MNPs-CL | Alkaline phosphatase | 5 pM [62] |
| MNPs-CL (this work) | Alkaline phosphatase | 10 fM |
To demonstrate the usefulness of such a new system, we adapted it for the copy numbers detection of six selected genes. To obtain reliable results, a stably expressed reference gene (GAPDH) was used for data normalization, so that sample-to-sample variation could be controlled [79]. The ligation probes for both reference and test genes was mixed together to participate in the ligation-dependent PCR reaction within the same sample for statistical validity of relative quantification. In the design of ligation probes, a tag sequence, which could be captured by the relative probe-MNPs to separate multiple targets, was inserted between the universal primers and hybridization sequences (Fig. 1). To develop a high-throughput DNA assay, it is important to reach a high degree of multiplexing. In the RT-PCR assay, the multiplexing required to interrogate a useful number of loci may be difficult to optimize, which is limited by the number of available fluorophores, the detection capabilities of the instrument, and high-budget for fluorescently labeled probes for each intended target. Likewise, the multiplexing of the MLPA assay would be reduced due to limitations on the maximum size of oligonucleotide synthesis [80]. In our assay, the tag sequence was designed in the probes for identifying multiple targets without the limitation of the number. The generation of probes with tag sequences is far simpler than the M13 cloning required for MLPA probes which contain the variable length fragments for the identification of multiple targets [80]. Considering the said advantages, including unlimited number of multiple targets, simplicity and low-cost, tag sequences will have potential for application in the multiplexing of this MNPs-CL system.
The accuracy of the system was validated by analyzing 40 human genome samples in comparison with the results by MLPA assay (Table 2). The MLPA test failed to yield expected results in two target genes, which suggested that the MLPA assay had some variability in results and should probably be performed repeatedly, or some more pairs of probes should be designed for the same loci in order to assure an accurate result [81]. In contrast, only one inconclusive result was reported in the MNPs-CL detection. It was thus inferred that a high background value would decrease the sensitivity of CL system, and the inevitable loss of MNPs in practical operation affected the accuracy of quantification, which produced a significant deviation from the expected values. However, the results from this novel system suggested that such developed system was acceptable to determine chromosomal DNA copy number changes, especially deletions and duplications, and would therefore be suitable for CNV analysis. Further optimization for sensitivity improvement would be required to capture template DNA of low copy numbers and obtain an accurate result, such as use of multiple probes. The CNVs are distributed widely in the human genome [82,83] and the single probe-based hybridization might cause systemic errors unpredictably and unavoidably. Some studies reported that more pairs of hybridization probes produced much better differentiation results than one pair of probes. For example, Guo et al [51] investigated the effect of hybridization probe number on the discrimination power of the MLPA/RT-PCR method, and the results showed that eight pairs of hybridization probes for one chromosome demonstrated substantially larger F value than one pair of probes. Shen et al [50] developed their own MLPA assays for quick validation of array comparative genomic hybridization (CGH) findings using all synthetic MLPA probes, where they designed 14 pairs of probes for 12 exons in Park2 gene, and two pairs of probes for the exons with large size. The MLPA assay confirmed all the duplications and deletions, demonstrating 100% specificity and sensitivity. Therefore, the random errors could largely be eliminated via the use of multiple probes, and that would yield a cumulative signal.
The system developed for CNV analysis in this study required DNA hybridization to probe-MNPs and CL detection after ligation-dependent PCR, which seems more complex than the MLPA assay. However, it is still feasible and useful for DNA quantification and CNV analysis, with advantages such as, low-cost, high accuracy, good specificity and sensitivity. Moreover, a high throughput SNP automatic detection system based on MNPs and dual-color hybridization or single base extension was recently developed at our group [84]. This detection system can realize sample preparation, target sequence amplification, signal detection and data analysis automatically. It is therefore our continuous work to integrate such MNPs-CL assay with the system described above for simplicity, automation and high throughput.
Conclusion
In summary, a novel method based on magnetic nanoparticles, ligation-dependent PCR, and chemiluminescence detection technique for quantitative analysis of copy number polymorphisms has been developed and described herein. The PCR, hybridization and CL detection conditions for the system were optimized and nucleic acid sequences were detected by a CL signal.. To verify the feasibility of the MNPs-CL system, copy number variation analysis for six target genes in genomic DNA was successfully performed using this new system. In comparison with the MLPA assay, the new MNPs-CL system showed high accuracy for detection of copy number changes. This system has the advantages of having low-cost, simplicity, high specificity and sensitivity, which will have great potential for application in analysis of genome diversity. Moreover, the method should further be improved for its stability, repeatability, and application in detecting the variants of genomic DNA in different diseases under further investigation.
Acknowledgements
This work was financially supported by National Basic Research Program (973 Program) of China (2010CB933903, 2014CB744501), National Nature Science Foundation of China (NSFC) (61271056, 31201003, 81301305, 21205013, 61471168), funding from the state key laboratory of bioelectronics of southeast university (2012F12, 2012F13) and Natural Science Foundation of Jiangsu Province (BK20130892).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Polymeropoulos MH, Rath DS, Xiao H. et al. A simple sequence repeat polymorphism at the human growth-hormone locus. Nucleic Acids Res. 1991;19:689
2. Cumming AM, Armstrong JG, Pendry K. et al. Polymerase chain-reaction amplification of 2 polymorphic simple repeat sequences within the vonwillebrand-factor gene-application to family studies in vonwillebrand disease. Hum Genet. 1992;89:194-198
3. Subramanian S, Madgula VM, George R. et al. SSRD: simple sequence repeats database of the human genome. Comp Funct Genomics. 2003;4:342-345
4. Qin WJ, Yung L. Nanoparticle-based detection and quantification of DNA with single nucleotide polymorphism (SNP) discrimination selectivity. Nucleic Acids Res. 2007;35:e111
5. Wu JX, Jiang R. Prediction of deleterious nonsynonymous single-nucleotide polymorphism for human diseases. Scientific World Journal. 2013;2013:675851.DOI 10.1155/2013/675851
6. Van den Broeck T, Joniau S, Clinckemalie L. et al. The role of single nucleotide polymorphisms in predicting prostate cancer risk and therapeutic decision making. Biomed Res Int. 2014;2014:627510. DOI: 10.1155/2014/627510
7. Dandona S, Stewart A, Chen L. et al. Gene dosage of the common variant 9p21 predicts severity of coronary artery disease. J Am Coll Cardiol. 2010;56:479-486
8. Singleton AB, Farrer M, Johnson J. et al. Alpha-synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841
9. Shlien A, Malkin D. Copy number variations and cancer. Genome Med. 2009;1:62
10. Sebat J, Lakshmi B, Troge J. et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525-528
11. Stranger BE, Forrest MS, Dunning M. et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848-853
12. Chen C, Sun SR, Gong YP. et al. Quantum dots-based molecular classification of breast cancer by quantitative spectroanalysis of hormone receptors and HER2. Biomaterials. 2011;32:7592-7599
13. Li SJ, Li J, Wang K. et al. A Nanochannel Array-based electrochemical device for quantitative label-free DNA analysis. ACS Nano. 2010;4:6417-6424
14. Shain EB, Clemens JM. A new method for robust quantitative and qualitative analysis of real-time PCR. Nucleic Acids Res. 2008;36:e91
15. Jozefczuk J, Adjaye J. Quantitative real-time pcr-based analysis of gene expression. Methods Enzymol. 2011;500:99-109
16. Horan MP. Application of serial analysis of gene expression to the study of human genetic disease. Hum Genet. 2009;126:605-614
17. Alvarez H, Corvalan A, Roa JC. et al. Serial analysis of gene expression identifies connective tissue growth factor expression as a prognostic biomarker in gallbladder cancer. Clin Cancer Res. 2008;14:2631-2638
18. Suter B, Fontaine JF, Yildirimman R. et al. Development and application of a DNA microarray-based yeast two-hybrid system. Nucleic Acids Res. 2013;41:1496-1507
19. Kim H, Takei H, Yasuda K. Quantitative evaluation of a gold-nanoparticle labeling method for detecting target DNAs on DNA microarrays. Sensor Actuat B-Chem. 2010;144:6-10
20. Armour J, Sismani C, Patsalis PC. et al. Measurement of locus copy number by hybridization with amplifiable probes. Nucleic Acids Res. 2000;28:605-609
21. Schouten JP, McElgunn CJ, Waaijer R. et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57
22. White S, Kalf M, Liu Q. et al. Comprehensive detection of genomic duplications and deletions in the DMD gene, by use of multiplex amplifiable probe hybridization. Am J Hum Genet. 2002;71:365-374
23. Hollox EJ, Atia T, Cross G. et al. High throughput screening of human subtelomeric DNA for copy number changes using multiplex amplifiable probe hybridisation (MAPH). J Med Genet. 2002;39:790-795
24. Reid AG, Tarpey PS, Nacheva EP. High-resolution analysis of acquired genomic imbalances in bone marrow samples from chronic myeloid leukemia patients by use of multiple short DNA probes. Gene Chromosome Canc. 2003;37:282-290
25. Sismani C, Armour J, Flint J. et al. Screening for subtelomeric chromosome abnormalities in children with idiopathic mental retardation using multiprobe telomeric FISH and the new MAPH telomeric assay. Eur J Hum Genet. 2001;9:527-532
26. Taylor CE, Charlton RS, Burn J. et al. Genomic deletions in MSH2 or MLH1 are a frequent cause of hereditary non-polyposis colorectal cancer: Identification of novel and recurrent deletions by MLPA. Hum Mutat. 2003;22:428-433
27. Gille J, Hogervorst F, Pals G. et al. Genomic deletions of MSH2 and MLH1 in colorectal cancer families detected by a novel mutation detection approach. Brit J Cancer. 2002;87:892-897
28. Sansovic I, Barisic I, Dumic K. Improved detection of deletions and duplications in the DMD gene using the Multiplex Ligation-Dependent Probe Amplification (MLPA) method. Biochem Genet. 2013;51:189-201
29. Lips EH, Laddach N, Savola SP. et al. Quantitative copy number analysis by Multiplex Ligation-dependent Probe Amplification (MLPA) of BRCA1-associated breast cancer regions identifies BRCAness. Breast Cancer Res. 2011;13:R107
30. Knezevic NZ, Lin V. A magnetic mesoporous silica nanoparticle-based drug delivery system for photosensitive cooperative treatment of cancer with a mesopore-capping agent and mesopore-loaded drug. Nanoscale. 2013;5:1544-1551
31. Gupta J, Bhargava P, Bahadur D. Methotrexate conjugated magnetic nanoparticle for targeted drug delivery and thermal therapy. J Appl Phys. 2014;115:17B516
32. Ruiz-Hernandez E, Baeza A, Vallet-Regi M. Smart drug delivery through DNA/magnetic nanoparticle gates. ACS Nano. 2011;5:1259-1266
33. Hou Y, Qiao RR, Fang F. et al. NaGdF4 nanoparticle-based molecular probes for magnetic resonance imaging of intraperitoneal tumor xenografts in vivo. ACS Nano. 2013;7:330-338
34. Leung K, Lee SF, Wong CH. et al. Nanoparticle-DNA-polymer composites for hepatocellular carcinoma cell labeling, sensing, and magnetic resonance imaging. Methods. 2013;64:315-321
35. Hayashi K, Nakamura M, Sakamoto W. et al. Superparamagnetic nanoparticle clusters for cancer theranostics combining magnetic resonance imaging and hyperthermia treatment. Theranostics. 2013;3:366-376
36. Nagaoka H, Sato Y, Xie XM. et al. Coupling stimuli-responsive magnetic nanoparticles with antibody-antigen detection in immunoassays. Anal Chem. 2011;83:9197-9200
37. Wang GN, Gao YA, Huang H. et al. Multiplex immunoassays of equine virus based on fluorescent encoded magnetic composite nanoparticles. Anal Bioanal Chem. 2010;398:805-813
38. Han JH, Sudheendra L, Kim HJ. et al. Ultrasensitive on-chip immunoassays with a nanoparticle-assembled photonic crystal. ACS Nano. 2012;6:8570-8582
39. Mohapatra S, Pal D, Ghosh SK. et al. Design of superparamagnetic iron oxide nanoparticle for purification of recombinant proteins. J Nanosci Nanotechnol. 2007;7:3193-3199
40. Wang W, Wang D, Li Z. Facile fabrication of recyclable and active nanobiocatalyst: purification and immobilization of enzyme in one pot with Ni-NTA functionalized magnetic nanoparticle. Chem Commun. 2011;47:8115-8117
41. Nash MA, Yager P, Hoffman AS. et al. Mixed stimuli-responsive magnetic and gold nanoparticle system for rapid purification, enrichment, and detection of biomarkers. Bioconjug Chem. 2010;21:2197-2204
42. Salgueirino-Maceira V, Correa-Duarte MA. Increasing the complexity of magnetic core/shell structured nanocomposites for biological applications. Adv Mater. 2007;19:4131-4144
43. Ranzoni A, Sabatte G, van IJzendoorn LJ. et al. One-step homogeneous magnetic nanoparticle immunoassay for biomarker detection directly in blood plasma. ACS Nano. 2012;6:3134-3141
44. Zhang Y, Pilapong C, Guo Y. et al. Sensitive, simultaneous quantitation of two unlabeled DNA targets using a magnetic nanoparticle-enzyme sandwich assay. Anal Chem. 2013;85:9238-9244
45. Dodeigne C, Thunus L, Lejeune R. Chemiluminescence as a diagnostic tool. A review. Talanta. 2000;51:415-439
46. Li ZP, Li W, Cheng YQ. et al. Chemiluminescent detection of DNA hybridization and single-nucleotide polymorphisms on a solid surface using target-primed rolling circle amplification. Analyst. 2008;133:1164-1168
47. Nelson NC, Hammond PW, Matsuda E. et al. Detection of all single-base mismatches in solution by chemiluminescence. Nucleic Acids Res. 1996;24:4998-5003
48. He NY, Wang F, Ma C. et al. Chemiluminescence analysis for HBV-DNA hybridization detection with magnetic nanoparticles based DNA extraction from positive whole blood samples. J Biomed Nanotechnol. 2013;9:267-273
49. Jiang HR, Zeng X, Xi ZJ. et al. Improvement on controllable fabrication of streptavidin-modified three-layer core-shell Fe3O4@SiO2@Au magnetic nanocomposites with low fluorescence background. J Biomed Nanotechnol. 2013;9:674-684
50. Shen YP, Wu BL. Designing a simple multiplex ligation-dependent probe amplification (MLPA) assay for rapid detection of copy number variants in the genome. J Genet Genomics. 2009;36:257-265
51. Guo QW, Zhou YL, Wang XB. et al. Simultaneous detection of trisomies 13, 18, and 21 with multiplex ligation-dependent probe amplification-based real-time PCR. Clin Chem. 2010;56:1451-1459
52. Huang CH, Chang YY, Chen CH. et al. Copy number analysis of survival motor neuron genes by multiplex ligation-dependent probe amplification. Genet Med. 2007;9:241-248
53. Arkblad EL, Darin N, Berg K. et al. Multiplex ligation-dependent probe amplification improves diagnostics in spinal muscular atrophy. Neuromuscul Disord. 2006;16:830-838
54. Liao YQ, Wang XB, Sha C. et al. Combination of fluorescence color and melting temperature as a two-dimensional label for homogeneous multiplex PCR detection. Nucleic Acids Res. 2013;41:e76
55. Battistella S, Damin F, Chiari M. et al. Genotyping beta-globin gene mutations on copolymer-coated glass slides with the ligation detection reaction. Clin Chem. 2008;54:1657-1663
56. Cao ZJ, Li ZX, Zhao YJ. et al. Magnetic bead-based chemiluminescence detection of sequence-specific DNA by using catalytic nucleic acid labels. Anal Chim Acta. 2006;557:152-158
57. Li HG, He ZK. Magnetic bead-based DNA hybridization assay with chemiluminescence and chemiluminescent imaging detection. Analyst. 2009;134:800-804
58. Fan AP, Lau CW, Lu JZ. Colloidal gold-polystyrene bead hybrid for chemiluminescent detection of sequence-specific DNA. Analyst. 2008;133:219-225
59. An YQ, Chen M, Xue QJ. et al. Preparation and self-assembly of carboxylic acid-functionalized silica. J Colloid Interf Sci. 2007;311:507-513
60. Li ZY, He L, He NY. et al. Polymerase chain reaction coupling with magnetic nanoparticles-based biotin-avidin system for amplification of chemiluminescent detection signals of nucleic acid. J Nanosci Nanotechnol. 2011;11:1074-1978
61. Wang F, Ma C, Zeng X. et al. Chemiluminescence molecular detection of sequence-specific HBV-DNA using magnetic nanoparticles. J Biomed Nanotechnol. 2012;8:786-790
62. Yang HW, Guo YF, Li S. et al. Magnetic beads-based chemiluminescent assay for ultrasensitive detection of pseudorabies virus. J Nanosci Nanotechnol. 2014;14:3337-3342
63. Wang ZL, Zeng X, Deng Y. et al. Molecular dynamics simulations of end-tethered single-stranded DNA probes on a silica surface. J Nanosci Nanotechnol. 2011;11:8457-8468
64. Foloppe N, MacKerell AD. All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J Comput Chem. 2000;21:86-104
65. MacKerell AD, Banavali NK. All-atom empirical force field for nucleic acids: II. Application to molecular dynamics simulations of DNA and RNA in solution. J Comput Chem. 2000;21:105-120
66. Brooks BR, Brooks CL, Mackerell AD. et al. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30:1545-1614
67. Tang YJ, Zou J, Ma C. et al. Highly sensitive and rapid detection of pseudomonas aeruginosa based on magnetic enrichment and magnetic separation. Theranostics. 2013;3:85-92
68. Tamer L, Ates NA, Ates C. et al. Glutathione S-transferase M1, T1 and P1 genetic polymorphisms, cigarette smoking and gastric cancer risk. Cell Biochem Funct. 2005;23:267-272
69. Hayes JD, Pulford DJ. The glutathione S-Transferase supergene family: Regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol. 1995;30:445-600
70. Saadat I, Saadat M. Glutathione S-transferase M1 and T1 null genotypes and the risk of gastric and colorectal cancers. Cancer Lett. 2001;169:21-26
71. Liu SH, Zhang ZH, Han MY. Gram-scale synthesis and biofunctionalization of silica-coated silver nanoparticles for fast colorimetric DNA detection. Anal Chem. 2005;77:2595-2600
72. Lee AC, Ye JS, Tan SN. et al. Carbon nanotube-based labels for highly sensitive colorimetric and aggregation-based visual detection of nucleic acids. Nanotechnology. 2007;18:455102
73. Lee HJ, Li Y, Wark AW. et al. Enzymatically amplified surface plasmon resonance imaging detection of DNA by exonuclease III digestion of DNA microarrays. Anal Chem. 2005;77:5096-5100
74. Zhou XC, O'Shea SJ, Li SFY. Amplified microgravimetric gene sensor using Au nanoparticle modified oligonucleotides. Chem Commun. 2000;11:953-954
75. Kawde AN, Wang J. Amplified electrical transduction of DNA hybridization based on polymeric beads loaded with multiple gold nanoparticle tages. Electroanalysis. 2004;16:101-107
76. Merkoci A, Aldavert M, Tarrason G. et al. Toward an ICPMS-linked DNA assay based on gold nanoparticles immunoconnected through peptide sequences. Anal Chem. 2005;77:6500-6503
77. Kim YS, Kim BC, Lee JH. et al. Specific detection of DNA using quantum dots and magnetic beads for large volume samples. Biotechnol Bioprocess Eng. 2006;11:449-454
78. Zhao XJ, Tapec-Dytioco R, Tan WH. Ultrasensitive DNA detection using highly fluorescent bioconjugated nanoparticles. J Am Chem Soc. 2003;125:11474-11475
79. Guo Y, Pennell ML, Pearl DK. et al. The choice of reference gene affects statistical efficiency in quantitative PCR data analysis. Biotechniques. 2013;55:207-209
80. Sellner LN, Taylor GR. MLPA and MAPH: New techniques for detection of gene deletions. Hum Mutat. 2004;23:413-419
81. Strom C, Anderson B, Peng M. et al. 1000 sample compasison of MLPA and RT-PCR for carrier detection and diagnostic testing for Spinal Muscular Atrophy Type 1. O J Genet. 2013;3:111-114
82. Wong KK, DeLeeuw RJ, Dosanjh NS. et al. A comprehensive analysis of common copy-number variations in the human genome. Am J Hum Genet. 2007;80:91-104
83. Redon R, Ishikawa S, Fitch KR. et al. Global variation in copy number in the human genome. Nature. 2006;444:444-454
84. Liu B, Jia YY, Ma M. et al. High throughput SNP detection system based on magnetic nanoparticles separation. J Biomed Nanotechnol. 2013;9:247-256
Author contact
![]() Corresponding authors: Tel: 86-25-83790885, Fax: +86-25-83790885, E-mail: august555482com (X. Zeng), nyhe1958com (N. He).
Corresponding authors: Tel: 86-25-83790885, Fax: +86-25-83790885, E-mail: august555482com (X. Zeng), nyhe1958com (N. He).