Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(5):515-529. doi:10.7150/thno.10319 This issue Cite
Research Paper
Molecular Magnetic Resonance Imaging of Angiogenesis In Vivo using Polyvalent Cyclic RGD-Iron Oxide Microparticle Conjugates
Stavros Melemenidis2, Andrew Jefferson1, Neil Ruparelia1, Asim M Akhtar1, Jin Xie3, Danny Allen2, Alastair Hamilton2, James R Larkin2, Francisco Perez-Balderas2, Sean C Smart2, Ruth J Muschel2, Xiaoyuan Chen3, Nicola R Sibson2*, Robin P Choudhury1 ![]() *
*
1. Division of Cardiovascular Medicine, Radcliffe Department of Medicine, University of Oxford, John Radcliffe Hospital, Oxford, OX3 9DU, United Kingdom.
2. Cancer Research UK and Medical Research Council Oxford Institute for Radiation Oncology, Department of Oncology, University of Oxford, Oxford, UK.
3. Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH), Bethesda, Maryland 20892, United States.
*joint senior authors.
Received 2014-8-12; Accepted 2015-1-12; Published 2015-2-15
Abstract

Angiogenesis is an essential component of tumour growth and, consequently, an important target both therapeutically and diagnostically. The cell adhesion molecule αvβ3 integrin is a specific marker of angiogenic vessels and the most prevalent vascular integrin that binds the amino acid sequence arginine-glycine-aspartic acid (RGD). Previous studies using RGD-targeted nanoparticles (20-50 nm diameter) of iron oxide (NPIO) for magnetic resonance imaging (MRI) of tumour angiogenesis, have identified a number of limitations, including non-specific extravasation, long blood half-life (reducing specific contrast) and low targeting valency. The aim of this study, therefore, was to determine whether conjugation of a cyclic RGD variant [c(RGDyK)], with enhanced affinity for αvβ3, to microparticles of iron oxide (MPIO) would provide a more sensitive contrast agent for imaging of angiogenic tumour vessels. Cyclic RGD [c(RGDyK)] and RAD [c(RADyK)] based peptides were coupled to 2.8 μm MPIO, and binding efficacy tested both in vitro and in vivo. Significantly greater specific binding of c(RGDyK)-MPIO to S-nitroso-n-acetylpenicillamine (SNAP)-stimulated human umbilical vein endothelial cells in vitro than PBS-treated cells was demonstrated under both static (14-fold increase; P < 0.001) and flow (44-fold increase; P < 0.001) conditions. Subsequently, mice bearing subcutaneous colorectal (MC38) or melanoma (B16F10) derived tumours underwent in vivo MRI pre- and post-intravenous administration of c(RGDyK)-MPIO or c(RADyK)-MPIO. A significantly greater volume of MPIO-induced hypointensities were found in c(RGDyK)-MPIO injected compared to c(RADyK)-MPIO injected mice, in both tumour models (P < 0.05). Similarly, administration of c(RGDyK)-MPIO induced a greater reduction in mean tumour T2* relaxation times than the control agent in both tumour models (melanoma P < 0.001; colorectal P < 0.0001). Correspondingly, MPIO density per tumour volume assessed immunohistochemically was significantly greater for c(RGDyK)-MPIO than c(RADyK)-MPIO injected animals, in both melanoma (P < 0.05) and colorectal (P < 0.0005) tumours. In both cases, binding of c(RGDyK)-MPIO co-localised with αvβ3 expression. Comparison of RGD-targeted and dynamic contrast enhanced (DCE) MRI assessment of tumour perfusion indicated sensitivity to different vascular features. This study demonstrates specific binding of c(RGDyK)-MPIO to αvβ3 expressing neo-vessels, with marked and quantifiable contrast and rapid clearance of unbound particles from the blood circulation compared to NPIO. Combination of this molecular MRI approach with conventional DCE MRI will enable integrated molecular, anatomical and perfusion tumour imaging.
Keywords: Tumour, angiogenesis, cRGDyK, microparticles, MRI, DCE.
Introduction
Angiogenesis, the formation of new blood vessels from pre-existing vessels, is required for tumour growth, development and metastasis [1-3] and, consequently, is a target for cancer therapy [4]. Therefore, the ability to localise and quantify angiogenesis could have importance for directing treatment selection and monitoring response to therapy [5].
Expression of the integrin αvβ3, also known as vitronectin receptor [6], is characteristic of angiogenic vessels, in which it plays a key role in endothelial cell proliferation, adhesion, and survival [7]. Whilst expression of αvβ3 is particularly high on activated endothelial cells of angiogenic blood vessels, it is low on normal, non-angiogenic blood vessels [8]. Moreover, clinical studies have identified αvβ3 as a biomarker of malignancy and its expression correlates with tumour grade [9, 10]. Accordingly, αvβ3 has become an important target for molecular imaging of angiogenesis [11, 12].
Physiologically, αvβ3 mediates cell adhesion to extracellular matrix proteins, such as fibronectin and collagen, by binding to the conserved amino acid sequence arginine-glycine-aspartic acid (RGD) [13, 14]. The RGD-binding site is located at the interface between the β-propeller domain and the β I-like domain, where amino-acid residues from the two domains interact directly with the RGD peptide [15]. Initial reports of radiolabelled RGD probes for clinical and preclinical PET or SPECT imaging of αvβ3 expression were published over a decade ago [16, 17]. Despite this, progress has been relatively slow, most likely as a consequence of the complex multi-step radiochemistry required for many such agents. At the same time, the relatively poor spatial resolution of PET/SPECT imaging remains a hurdle, particularly in assessing tumour vasculature heterogeneity. In contrast, MRI yields high spatial resolution and excellent soft tissue contrast without the need for ionising radiation, and allows for concomitant angiography and perfusion acquisitions. Paramagnetic contrast agents (e.g. containing gadolinium) targeted to endothelial αvβ3 via monoclonal antibodies [11] or peptidomimetics [18] have been used previously to image tumour angiogenesis [19], as have gadolinium-based lipidic nanoparticles encapsulating fluorescent dyes using RGD as a ligand [5, 20]. However, the MRI contrast effect conferred by gadolinium-containing contrast agents is relatively low. Consequently, interest has also extended to ultrasmall superparamagnetic particles of iron oxide (USPIO; diameter < 100 nm), which yield considerably greater contrast effects than gadolinium-based agents. More recently, USPIO conjugated with RGD peptides have been used to target αvβ3 integrin expression on tumour cells or on the tumour vasculature [21-23].
Despite the improved sensitivity of nanoparticles of iron oxides (NPIO < 500 nm) several issues remain including the possibility of non-specific extravasation [21-23], long blood half-life (reducing specific contrast) and low targeting valency. In contrast, microparticles of iron oxide (MPIO; diameter > 1 μm) have a short half-life in the circulation (< 5 min) [24] together with considerably greater surface area, which can increase binding affinities through multivalency effects [25]. Moreover, the higher iron content of MPIO, compared to NPIO, yields a substantially higher contrast-to-noise ratio per particle, with a dephasing effect approximately 50 times their physical diameter [26]. In addition, whilst NPIO can extravasate from leaky angiogenic vessels, potentially compromising molecular specificity through passive tissue retention, the relatively large MPIO are obligate intravascular agents [27, 28]. Indeed, the potential of MPIO for molecular imaging has been demonstrated in numerous animal models of human disease [27-36].
The aim of the current study was to determine the efficacy of RGD-targeted MPIO for detection of angiogenic tumour vessels. Whilst the relatively large size of such microparticles brings many positive characteristics for imaging purposes (above), their buoyancy in flowing blood means that to achieve effective retention at sites of interest, the target-ligand interaction must be, not only specific, but also of high affinity [37]. Therefore, to increase the effective affinity of the targeting RGD peptide, we have generated a cyclic RGD variant [c(RGDyK)] with high affinity for αvβ3, and have conjugated this polyvalently to microparticles of iron oxide. We have tested the binding characteristics of c(RGDyK)-MPIO to αvβ3-expressing endothelial cells under flow conditions in vitro, and subsequently quantified the MRI contrast effects of c(RGDyK)-MPIO in vivo in animal models of colon carcinoma and melanoma.
Materials and Methods
RGD-MPIO Synthesis
Cyclic RGD and RAD based peptides [c(RGDyK) / c(RADyK)] (CS Bio, Menlo Park, CA) were coupled to 2.8 μm Dynabeads (M-270 amine, Invitrogen), and used throughout for the in vivo work. Dynabeads (100 µL) were first suspended in phosphate buffered saline (PBS) (pH 7.4). A heterodimer crosslinker, 4-maleimidobutyric acid N-hydroxysuccinimide ester (75 µg), was added to the solution. The mixture was incubated for 30 min at room temperature with slow tilt rotation. After incubation, a magnet was placed to the tube to collect the beads, and the supernatant was removed. The beads were washed twice with PBS and resuspended in 100 µL of PBS. c(RGDyK) / c(RADyK) peptides were thiolated as described previously [38], and the thiolated c(RGDyK) / c(RADyK) (200 µg) was added to the Dynabead solution. The mixture was incubated for 1 h at room temperature with slow tilt rotation. After 1 h, cysteine was added to a final concentration of 5 mM, and the mixture was incubated for another 15 min at room temperature to quench non-reacted groups. The beads were washed twice with PBS and the products were resuspended in PBS.
Assessment of peptide-loading
Peptide loading of c(RGDyK)-MPIO and c(RADyK)-MPIO was determined by flow cytometry analysis. The aspartic acid residue of the peptide was fluorescently labelled in a two-step protocol consisting of the activation of the free carboxylic acid by EDC and further reaction of the activated ester with a fluorophore containing a primary amino group (Alexa Fluor 647 cadaverine). When labelled in such way each RGD unit contains one fluorophore. Quifikit calibration beads were used as reference and labelled with a secondary antibody containing 5 fluorophores per protein. Briefly, 10 μg c(RGDyK)- or c(RADyK)-MPIO were diluted in 1 mL of MES buffer 15 mM pH6.0, pelleted on a Dynal magnetic separator (Invitrogen, UK) and redispersed in 200 μL EDC (Sigma Aldrich, UK) solution (10 mg/mL) in MES buffer 15 mM pH 6.0. The sample was shaken at 1000 rpm for 10 min, pelleted, washed with cold water and then redispersed in 200 μL of MES buffer 15 mM pH 6.0. Subsequently, 2 μL of Alexa Fluor 647 cadaverine disodium salt (2 mg/mL; Invitrogen, UK) was added. The sample was shaken for 24 h, pelleted in a magnet, washed 3 times with 1 mL of PBS + 0.1 % Tween-20 and redispersed in 400 μL of PBS. Qifikit calibration beads (Dako, UK), used as a reference, were prepared according to the manufacturer's protocol, but substituting the provided fluorescently-conjugated antibody with Alexa Fluor 647 goat anti-mouse IgG (H+L) (Invitrogen, UK). Flow cytometry experiments were performed on a BD FACScalibur flow cytometer using channel FL4.
RGD-MPIO binding in vitro
In order to assess αvβ3-integrin expression, human umbilical vein endothelial cells (HUVEC)-C were seeded onto glass coverslips in 12-well plates to a final density of 4 x 105 per well, in Media 199 supplemented with 10 % fetal calf serum, penicillin and streptomycin (100 μg/mL). HUVEC-C cells were treated with PBS control or 100 μM S-nitroso-n-acetylpenicillamine (SNAP) (Alexis Corp., San Diego, CA, USA) for 20 h at 37 °C to induce endothelial αvβ3-integrin expression [39]. Cells were then fixed with 4 % PFA, washed, and stored in PBS. Coverslips were blocked with 3 % bovine serum albumin (BSA) in PBS with 0.01 % Tween-20 for 60 min at room temperature. Following blocking, coverslips were incubated with antibodies against one of: either αvβ3-integrin (Clone 21 / CD51, BD Bioscience Oxford, UK); VCAM (Clone 1G11B1, Abcam, Cambridge UK) or PECAM-1 (Clone 9g11, R&D Systems). Following overnight incubation, cells were washed and then incubated with goat anti-mouse AlexaFluor 488 (for VCAM-1 and PECAM-1) or AlexaFluor 594 (for αvβ3-integrin) in the dark for 30 min at 37 °C. Finally, coverslips were washed and counterstained with 4',6-diamidino-2-phenylindole (DAPI) and mounted onto glass slides and stored in the dark at 4 °C. In order to quantify immunofluorescence, automated colour based segmentation was applied to coverslips stained for αvβ3-integrin, using ImagePro Plus (Media Cybernetics, Rockville, Maryland, USA).
To simulate MPIO binding to endothelial cells under physiological conditions, flow chamber experiments were undertaken. HUVEC-C cells were seeded onto 35 mm cell culture dishes (5 x 105 cell per well) and incubated overnight, or until confluent, prior to addition of SNAP or PBS. Further, to assess c(RGDyK)-MPIO specificity, two separate groups of cells were treated with a soluble cyclic RGD-peptide (Cambridge Bioscience, Cambridge, UK) or a control RAD-peptide (Cambridge Bioscience). All flow chamber experiments were conducted on a parallel plate flow chamber (GlycoTech, Gaithersburg, USA) fitted with gasket B (0.25 x 0.025 cm) and connected to a syringe infusion pump (Pump 22; Harvard Apparatus, Cambridge, USA). MPIO stock was diluted in 50 mL PBS (5 x 108 MPIO) and run at estimated shear rates of 0.25, 1.0, and 5.0 dyne cm-2 for 5 min. Subsequent infusion of the flow chamber with PBS removed unbound MPIO. Bound MPIO were counted over 10 fields of view (FOV) by an observer blind to sample identity using a 40X phase contrast objective fitted to an Olympus IX-71 inverted microscope in order to quantify MPIO retention.
Animal tumour models
Female, C57Bl/6 mice (n = 26; Charles River, UK) were housed in ventilated cages with a 12-hour light/dark cycle and controlled temperature (20-22 °C), fed normal chow and given water ad libitum. 7-10 week old mice were anesthetised with 2-3 % isoflurane (IsoFlo) in O2 and injected subcutaneously in the right upper flank with either 5 x 104 B16F10 mouse melanoma cells (n = 10), or 5 x 104 MC-38 mouse colon carcinoma cells (n = 16) in 100 µL sterile PBS. Tumours were allowed to grow for either 21 days to achieve tumour volumes < 400 mm3 (n = 20), or 28 days for tumour volumes > 400 mm3 (n = 6). All experiments were approved by the UK Home Office.
In vivo experimental protocol
For imaging, mice were anaesthetised with 2-3 % isoflurane in room air and a tail vein cannulated for contrast agent administration. Anaesthesia was maintained with 1-2 % isoflurane in room air thereafter. Core temperature was monitored and maintained at 36 ºC with a homeothermic system (Harvard Instruments, Edenbridge, Kent, UK). Respiratory gating was achieved using a pressure balloon, placed in contact with the chest, that was interfaced to a signal processing unit (MP150 unit, Biopac Inc, California, USA) and trigger control unit (DTU200, Biopac Inc).
For each animal, a T2*-weighted 3D dataset was acquired prior to contrast agent administration and again 30 min after injection. After the initial acquisition, the coil was removed from the magnetic field and contrast agent administered intravenously while the mice remained within the coil to enable co-registration of pre- and post-contrast images. Mice were injected with 100 μL of either c(RGDyK)-MPIO or the control c(RADyK)-MPIO at a concentration of 4 mg Fe/kg body weight. Dynamic contrast enhanced (DCE) MRI was performed at the end of each imaging protocol with 30 µL gadolinium-DTPA (Omniscan; 0.5 mmol/mL) injected intravenously during the scan.
In vivo Magnetic Resonance Imaging
All MRI data were acquired using a 4.7 T horizontal bore magnet (Agilent Technologies, Santa Clara, CA, USA) using a 25 mm quadrature birdcage coil (Rapid Biomedical, Wurzburgy, Germany). For detection of MPIO, a respiratory-gated, fat-suppressed, 3D multi-gradient echo, T2*-weighted acquisition was performed with TE = 3.33, 7.82, 12.31 and 16.8 ms; TR = 40 ms; nominal flip angle = 16°; field of view (FOV) = 54 x 27 x 27 mm giving an isotropic resolution of 150 μm. For DCE MRI, a respiratory-gated 3D gradient echo, T1-weighted acquisition was performed with TE = 0.55 ms, TR = 1.1 ms, nominal flip angle = 5°, FOV = 54 x 27 x 27 mm giving an isotropic spatial resolution of ca. 412 μm in a scan time of ca. 10 s/frame. 100 frames were acquired with contrast agent infused (Omniscan, 30 μL) over the duration of frame 11/100. Respiratory gating to eliminate residual motion artefacts, whilst maintaining constant T1-weighting throughout the scans, was achieved by only acquiring data during the respiratory plateau period [40]. B1 inhomogeneity was corrected for using a respiratory-gated implementation of the Actual Flip angle Imaging technique [41], performed with TE = 0.59, TR = 20, 100 ms, nominal flip angle = 60º, covering the same FOV as the DCE scan, but at half the resolution in each of the phase encode directions, to give a spatial resolution of ca. 400 x 800 x 800 μm in a scan time of approximately 4 min. T1 was estimated using the same scan parameters as for the DCE acquisition, but with an array of flip angles (1-6º) in a scan time of approximately 1 min. Both B1 correction and T1 mapping were performed prior to the DCE scan to avoid confounding arising from gadolinium washout.
MRI data analysis
All T2*-weighted datasets were processed with ITK-Snap v2.4.0 (http://www.itksnap.org) [42], tumour volumes were segmented manually and further analysis of datasets was performed using MATLAB v7.11.0.584 (2010b) (Mathworks, Cambridge, UK). T2* relaxation times, for each independent voxel within the tumour volume, were calculated by fitting a T2* curve across the 4 echo times. Values were subsequently averaged across the entire tumour volume to yield a single mean T2* relaxation rate per tumour.
To calculate the volumes of MPIO-induced hypointensities an automated computational method was used to determine hypointensities based on local contrast levels (Hamilton A., DPhil Thesis 2013, Oxford University). To this end, the signal intensity of each individual voxel was statistically compared with the intensity distribution of all surrounding voxels within a 5 x 5 x 5 (voxel) volume, to determine whether it was significantly lower than the average local signal intensity. To segment true MPIO-induced hypointensities in this way, a threshold value was set at a fraction of the local mean (FLM) signal intensity. The optimal FLM was found to be dependent on tumour type, owing to differences in the global signal intensity distributions; where there are more natural hypointense regions within the tumour (e.g. melanoma) these will be delineated incorrectly with a higher FLM. By assessing a range of FLM values it was possible to determine optimal FLM values that did not return regions of natural hypointenisty (i.e. observed in the pre-injection datasets). On this basis, FLM values of 0.45 for the melanoma model and 0.65 for the colorectal model were found to be optimal. Below these thresholds voxels were designated as MPIO-induced hypointensities. A margin parameter (2 voxel radius excluded in all directions) was applied at the edge of the segmented tumour volumes in order to minimise false counts due to minor segmentation errors. Any independent hypointensity volumes larger than 20 voxels were excluded from the analysis as these reflected pre-existing tumour local inhomogeneities (i.e. present in pre-contrast images), although optimization of the FLM minimised inclusion of such regions in the segmented datasets. 3D reconstructions of MPIO induced hypointensities were created using 3D Slicer 4.3.1 (2013) (http://www.slicer.org). Pairwise statistical comparisons were made between pre- and post-contrast datasets using paired t tests. Unpaired t tests were used to compare between groups.
The DCE datasets were manually segmented for tumour volumes and adequate muscle volume. Areas under the curve of intensity signal over time were estimated from 100 T1-weighted images. Tumour tissue and a sample region of muscle were segmented manually using ITK-Snap [42]. Pharmacokinetic analysis was performed using the reference region model [43-45] implemented in MATLAB v7.11.0.584.
Histological analysis
Immediately after imaging, mice were killed by exsanguination and perfused with 20 mL of sterile heparinised PBS via the right cardiac ventricle within 30 min. Tumours, kidneys and livers were dissected, embedded in optimum cutting temperature compound (OCT-Tissue Tek, VWR International Ltd, Lutterworth, UK) and frozen with isopentane and dry ice. The tissues were stored at -80 ºC until cryosectioning. Tissues were sectioned at 10 µm thickness for further histological assessment.
Excluding the outermost 300 µm of each tumour, series of 30 x 10 µm sections were taken from 3 equidistant representative regions of the tumour. Sections were stained for CD-31 (platelet endothelial cell adhesion molecule-1, anti-PECAM-1, ab28364, Abcam, Cambridge, UK). Frozen sections were thawed for 20 min at room temperature and, subsequently, fixed for 10 min with pre-chilled 100 % ethanol at 4 ºC. Slides were quenched with 1 % v/v of 30 % w/v hydrogen peroxide in methanol (Sigma Aldrich, Gillingham, UK) and blocked with 1 % normal goat serum in PBS (Vector Laboratories) for 1 h. Sections were incubated with primary antibody (ab28364, Abcam) at 4 ºC overnight, then washed with PBS containing 0.01 % Tween-20 (Sigma Aldrich) and incubated with a biotinylated goat polyclonal to chicken IgG secondary antibody (1:1000; ab50579, Abcam) for 1 h. Slides were washed and then incubated in Vectorstain Elite ABC kit (1:1:100; Vector Laboratories) for 45 min. The peroxidase was visualised using 3,3′-diaminobenzidine (DAB; Sigma Aldrich). Sections were counterstained with cresyl violet (Sigma Aldrich).
Slides were further scanned with Leica Biosystem ScanScope CS2 (Aperio, ePathology Solutions, Milton Keynes, UK) from which high magnification images were acquired and further analysed with Image Scope (ePathology Solutions). For each tumour section, all vessels were manually segmented and MPIO, which are visible at high magnification, were manually counted. The vessel densities of the tumours, the MPIO density within each tumour and the adherence of MPIO per unit vessel circumference were calculated. Group-wise differences in these parameters were assessed statistically using unpaired t tests.
Expression of αvβ3 integrin on the tumour vasculature was evaluated by immunofluorescence, and co-localisation of αv and β3 subunits was determined. Frozen sections were thawed for 10 min at room temperature and, subsequently, fixed for 10 min with pre-chilled 100 % ethanol at 4 ºC. Sections were quenched with 1 % v/v of 30 % w/v hydrogen peroxide in PBS, streptavidin- and biotin-blocked (SP-2002, Vector Laboratories) and then incubated with Tris-NaCl blocking buffer (TNB, PerkinElmer, MA, USA). Sections were incubated overnight at 4 ºC with the following primary antibodies: Rabbit anti-Integrin beta3A [CD61] Antibody (EPR2417Y, Milipore, MA, USA) and Rat anti-Mouse Monoclonal anti-ITGAV/Integrin Alpha V/CD51 Antibody (LS-C57970, LifeSpan Biosciences, WA, USA). Sections were washed with PBS and, subsequently, incubated with the appropriate secondary antibody, biotonylated anti-rat (BA-4001, Vector Laboratories) in TNB for 30 min at room temperature. Sections were washed with PBS, incubated with streptavidin-HRP (PerkinElmer, 1:200) in TNB for 30 min, washed and incubated for 8 min in the dark with TSA-biotin (PerkinElmer; 1:100) in amplification buffer (PerkinElmer). Subsequently, slides were washed and incubated with a streptavidin-Alexa Fluor 488 (Invitrogen, Paisley, UK; 1:100) for 30 min. To detect the other fluorophore, a Texas Red-conjugated secondary antibody was added at the same time as streptavidin-Alexa Fluor 488. Slides were cover-slipped using Vectashield mounting medium (Vector Laboratories).
Biological blood clearance protocol
For the iron oxide biological clearance experiments, 7-9 week old female CD1 mice (Charles River, UK) were injected intravenously via a tail vein with 2.8 μm MPIO (n = 10) or ~20 nm nanoparticles (NPIO; n = 15); 4 mg of iron per kg of body mass weight in all cases. Dextran-coated NPIO (20.5 ± 8.9 nm diameter) were synthesised in house, as described in Supplementary Methods (in Supplementary Material). Blood samples from naïve mice were used to assess relaxivity of normal blood (n = 8). For the MPIO experiments, mice were anesthetised with 2-3 % isoflurane in O2 and at different time points after administration of MPIO underwent a thoracotomy, followed by cutting of the inferior vena cava and aorta below the lungs to allow the blood to pool inside the thoracic cavity. Blood was collected with a syringe containing 5 μL of heparin sodium (25,000 I.U./mL, Wockhardt, UK). For early time points, surgery was performed prior to injection of particles, leaving the diaphragm intact. Time points studied were: 10, 20, 30, 40, 50, 60, 90, 120, 180 and 240 s after MPIO injection (n = 1 per time point). For the NPIO experiments, blood was collected by cardiac puncture under terminal anesthesia at different time points after administration of NPIO. A maximum amount of 5 μL heparin sodium was used per sample. Time points studied were: 60, 120, 240, 600 and 1440 min after MPIO injection (n = 3 per time point).
Determination of blood relaxivity
Blood samples were oxygenated by incubation with O2 gas in a sealed tube on a rolling platform for 5 min. Samples were set into agar by mixing blood and 1 % agarose solution in PBS, both at 40.5 °C, at a 1:1 ratio, to a final volume of 300 µL, in a 500 µL microcentrifuge tube. Set samples were stored at 4 °C. T2 relaxivity measurements were made at 4.7 T, within 24 h, using an array of 100 μs hard 90° and 180° pulses, TR = 1000 ms, 2000 complex points and 8 averages. Blood half-life was determined from the fitted one phase decay curves, constrained to plateau at the average naive blood sample values (baseline).
Results
Both c(RGDyK)-MPIO and c(RADyK)-MPIO were successfully synthesised, and peptide loading calculated to be ca. 2,000 ligands per μm2; c(RGDyK)-MPIO 202,632 and c(RADyK)-MPIO 211,407 ligands per MPIO, respectively.
In vitro binding studies
SNAP induced αvβ3-integrin expression on cultured endothelial cells, with αvβ3-integrin largely absent from untreated cells. Expression of αVβ3-integrin was increased 39-fold in SNAP stimulated HUVEC-C cells (5.80 ± 0.60 mm2) compared to cells incubated with PBS (0.15 ± 0.08 mm2, P < 0.001). Both SNAP- and PBS-stimulated cells had similar staining for PECAM-1 (5.10 ± 0.46 mm2 vs. 4.93 ± 0.44 mm2). Under static conditions, in SNAP stimulated cells, c(RGDyK)-MPIO binding was increased 14-fold compared with cells incubated with PBS (P < 0.001; Figure 1A-B). Unconjugated MPIO showed sparse binding to both SNAP-stimulated and PBS-stimulated HUVEC-C (Figure 1C-D).
(A-E). In vitro binding experiments for c(RGDyK)-MPIO. c(RGDyK)-MPIO binding to HUVEC-C following incubation with either SNAP (A) or PBS (C). Binding of RGD-MPIO was sparse to PBS-stimulated cells (B) and unconjugated MPIO binding to SNAP stimulated HUVEC-C (D) was significantly lower than that of c(RGDyK)-MPIO (white arrows). (E) Graph to show binding of c(RGDyK)-MPIO or unconjugated MPIO to HUVEC-C under static conditions , with or without blocking peptide (n = 2, 10 fields of view per coverslip, 40X magnification, P < 0.001). (F) c(RGDyK)-MPIO binding experiments under shear stress conditions. Graph to show comparison of c(RGDyK)-MPIO retention at different shear rates (n = 3 per condition, 10 fields of view, 400X magnification).
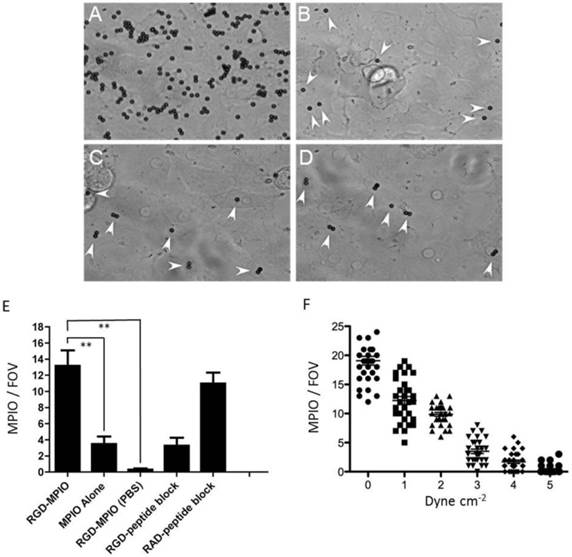
c(RGDyK)-MPIO retention under flow conditions (1 dyne cm-2) in SNAP stimulated cells was increased 44-fold compared with PBS-treated cells (P < 0.001), 3.8-fold compared with unconjugated MPIO in SNAP-stimulated cells (P < 0.001) and 4-fold compared with c(RGDyK)-MPIO retention in cells treated with soluble (blocking) RGD-peptide following SNAP stimulation and prior to MPIO administration (P < 0.001; Figure 1E). c(RGDyK)-MPIO incubation in cells treated with the irrelevant RAD-peptide following SNAP stimulation MPIO was increased 3.3-fold in comparison with cells undergoing similar conditions but treated with soluble RGD-peptide prior to MPIO administration (P < 0.001; Figure 1E). c(RGDyK)-MPIO retention at shear rate of 0.25 dyne cm-2 was increased 1.6-fold vs. c(RGDyK)-MPIO retention at a 1.0 dyne cm-2 (P < 0.001) and 12.3-fold compared with c(RGDyK)-MPIO binding at 5.0 dyne cm-2 in SNAP stimulated cells (P < 0.001; Figure 1F).
In vivo c(RGDyK)-MPIO MRI
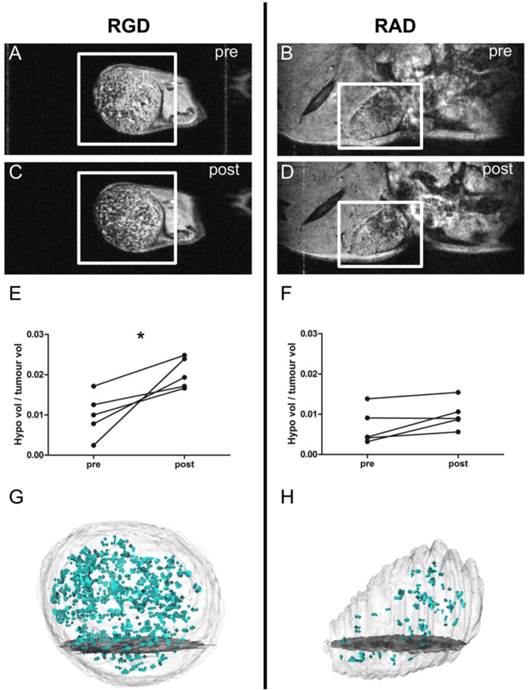
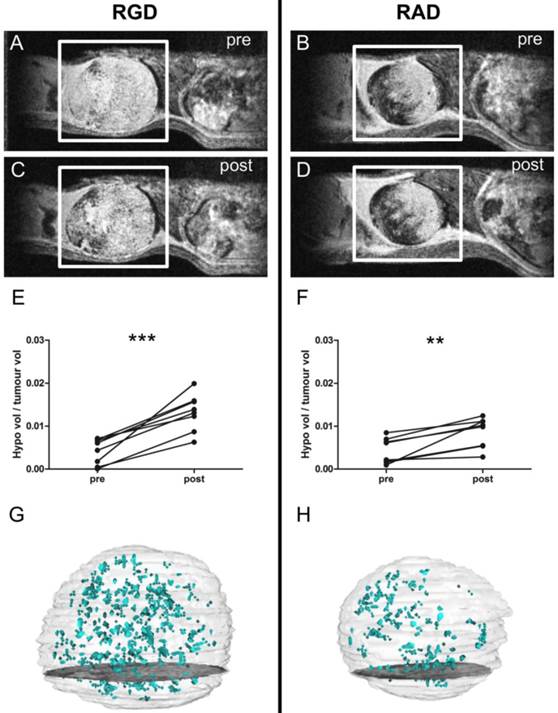
T2*-weighted images acquired post-injection of c(RGDyK)-MPIO showed marked focal hypointensities throughout the tumour in both the B16F10 (melanoma-derived) and MC-38 (colorectal-derived) models, which were not present in the pre-contrast images (Figures 2A,C and 3A,C, respectively). Few such hypointensities were observed on images acquired post-injection of c(RADyK)-MPIO in either model (Figures 2B,D and 3B,D, respectively). Intravenous administration of c(RGDyK)-MPIO to mice bearing either melanoma or colorectal subcutaneous tumours resulted in new signal hypointensities within the tumours on T2*-weighted images compared to pre-contrast images. The ratio of the hypointensity volume to the total tumour volume, showed a significant increase following c(RGDyK)-MPIO injection in both the melanoma (P < 0.05; Figure 2E) and colorectal models (P < 0.001; Figure 3E). No significant increase in hypointense volume was seen following administration of the control c(RADyK)-MPIO to the group of mice with melanoma derived tumours (Figure 2F). In contrast, the group with colorectal-derived tumours had a significant increase in the volume of MPIO-induced hypointensity (as a fraction of total tumour volume) following administration of the control c(RADyK)-MPIO (P < 0.005; Figure 3F), but to a lesser extent than with c(RGDyK)-MPIO. Comparison between the colorectal model injected with c(RGDyK)-MPIO and c(RADyK)-MPIO confirmed the significant difference between groups; ratio of hypointensity volume to total tumour volume (post - pre) = 0.017 ± 0.008 vs. 0.006 ± 0.006 (P < 0.005; data not shown).
(A-D) Selected images from a 3D gradient echo dataset from the B16F10 (melanoma) subcutaneous tumour model, averaged over the 4 echoes. Images acquired pre- (A) and (B) and post- (C) and (D) injection of c(RGDyK)-MPIO or c(RADyK)-MPIO, respectively. (E-F) Graphs showing paired analysis of hypointensity volumes from c(RGDyK)-MPIO (E) and c(RADyK)-MPIO (F) treated animals. Data are expressed as a ratio of the volume of MPIO-induced hypoinintensity vs. the total tumour volume. (G-H) 3D reconstructions of hypointesities induced by c(RGDyK)-MPIO (G) or c(RADyK)-MPIO (H). *P < 0.05.
(A-D) Selected images from a 3D gradient echo dataset from the MC38 (colorectal) subcutaneous tumour model, averaged over the 4 echoes. Images acquired pre- (A) and (B) and post- (C) and (D) injection of c(RGDyK)-MPIO or c(RADyK)-MPIO, respectively. (E-F) Graphs showing paired analysis of hypointensity volumes from c(RGDyK)-MPIO (E) and c(RADyK)-MPIO (F) treated animals. Data are expressed as a ratio of the volume of MPIO-induced hypoinintensity vs. the total tumour volume. (G-H) 3D reconstructions of hypointesities induced by c(RGDyK)-MPIO (G) or c(RADyK)-MPIO (H). **P < 0.005, ***P < 0.0005.
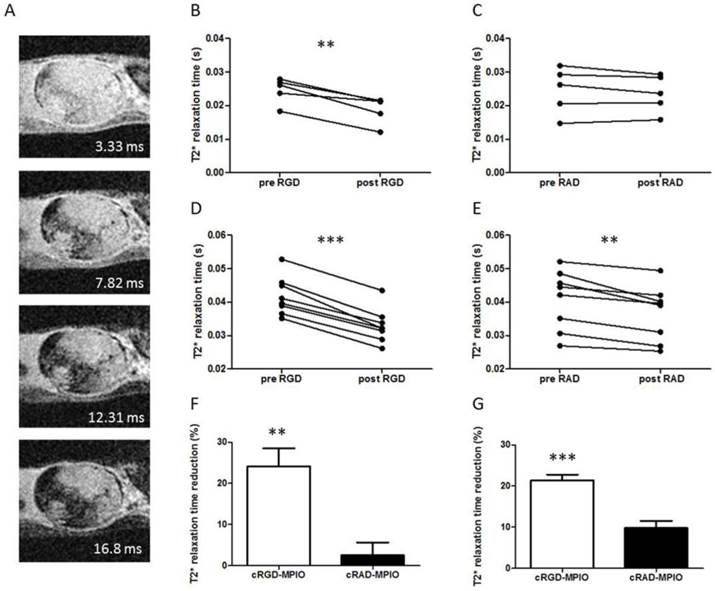
As an additional means of assessing the degree of c(RGDyK)-MPIO binding, mean T2* relaxation rates across the whole tumour were calculated for both the melanoma and colorectal groups (Figure 4). In accord with the analysis of segmented hypointensities (above), administration of c(RGDyK)-MPIO to mice bearing subcutaneous melanoma significantly reduced the mean tumour T2* relaxation time (P < 0.001; Figure 4B). No significant reduction in T2* relaxation times was seen following administration of the control c(RADyK)-MPIO to the group of mice with melanoma derived tumours (Figure 4C). Again, the reduction in T2* relaxation time in the c(RGDyK)-MPIO group was significantly greater than in the c(RADyK)-MPIO group (P < 0.001; Figure 4F). Similarly, in the colorectal tumours mean tumour T2* relaxation time was significantly reduced (P < 0.0001; Figure 4D). The reduction in T2* relaxation times in the c(RGDyK)-MPIO group was significantly greater than in the c(RADyK)-MPIO group (P < 0.0001; Figure 4G). However, the c(RADyK)-MPIO injected tumours showed a significant, but lesser reduction in T2* relaxation time from pre- to post-contrast datasets (P < 0.005; Figure 4E).
(A) Selected slice from a 3D gradient echo dataset from MC38 (colorectal) subcutaneous tumour model showing all 4 echoes (echo times shown for each image) acquired post-c(RGDyK)-MPIO administration. (B) Graph showing paired analysis of tumour T2* relaxation times between pre- and post-c(RGDyK)-MPIO treatment in the B16F10 model. (C) Graph showing paired analysis of tumour T2* relaxation times between pre- and post-c(RADyK)-MPIO treatment in the B16F10 model. (D) Graph showing paired analysis of tumour T2* relaxation times between pre- and post-c(RGDyK)-MPIO treatment in the MC-38 model. (E) Graph showing paired analysis of tumour T2* relaxation times between pre- and post-c(RADyK)-MPIO treatment in the MC-38 model. (F,G) Graphs showing percentage reduction in T2* relaxation times following administration of either targeted (c(RGDyK)-MPIO) or control (c(RADyK)-MPIO) contrast agent for B16F10 (F) or MC-38 (G) models. Data are presented as Mean + S.E.M. in bar charts. *P < 0.01, **P < 0.005, ***P < 0.0001.
Figures 2G,H and 3G,H show projections of the 3D reconstructions of hypointensities induced by c(RGDyK)-MPIO (G) or c(RADyK)-MPIO (H), in melanoma or colorectal tumour-bearing mice, respectively. From Figure 2G, it is evident that the MPIO-induced hypointensities are present across the majority of the tumour volume in the melanoma-derived tumours, with the exception of the rim of the tumour, which shows little MPIO retention. These findings are supported by immunohistochemical assessment of the vessel distribution and MPIO retention (Supplementary Material: Figure S1A). Similarly, from Figure 3G, it appears that the MPIO-induced hypointensities arise throughout much of the tumour volume excluding a small rim at the outer edges. Again these observations were confirmed immunohistochemically (Figure S1B), and in this case there appeared to be slightly greater heterogeneity in vessel distribution across the tumour, with some edge areas showing high local vascularity.
Comparison of c(RGDyK)-MPIO and DCE MRI
Supplementary Material: Figure S2 shows representative slices across the DCE dataset from the largest (Figure S2A) and the smallest (Figure S2B) colorectal tumours. Gadolinium enhancement over time is evident in both, but is noticeably faster in the smaller tumour (Figure S2B). Correspondingly, linear regression analysis of the ratios of tumour-to-muscle area under the curve (AUC) against tumour volume revealed a significant negative correlation (R2 = 0.6716; P < 0.05; Figure S2C) indicating an inverse relationship between contrast agent uptake and tumour size. A negative correlation was also observed between Ktrans and tumour volume (R2 = 0.8097; P < 0.05; Figure S2D). In contrast, the volumes of hypointensity measured in the cohort of colorectal tumour animals injected with c(RGDyK)-MPIO (see above) showed a strong positive correlation with increasing tumour size (Figure S2E), indicating increased c(RGDyK)-MPIO retention with increasing tumour volume. Interestingly, no significant correlation was found between the percent reduction in T2* relaxation time (from pre- to post-contrast images) and tumour volume in the same cohort of animals (Figure S2F), indicating that the contrast effect induced by c(RGDyK)-MPIO binding is independent of tumour size when measured globally in this way.
Immunohistochemistry and immunofluorescence
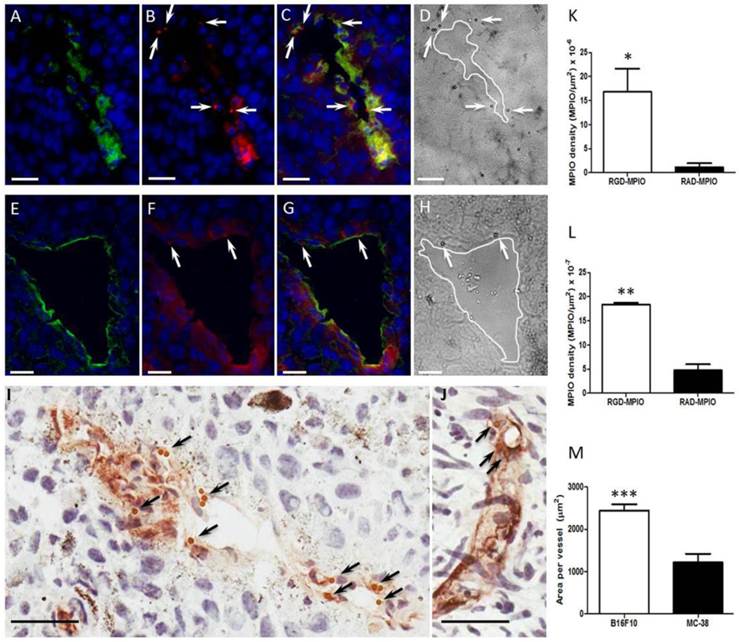
Co-localisation of αV and β3, and specific binding of c(RGDyK)-MPIO to αVβ3 expressing endothelium, was assessed by immunofluorescence microscopy. As shown in Figure 5 for melanoma and colorectal tumours, respectively, expression of αV and β3 co-localised on blood vessels. At the same time, the MPIO were detectable in the red channel owing to autofluorescence at this wavelength and could be seen also to co-localise with regions of αV and β3 expression (Figures 5C and G; arrows). The MPIO were also clearly visualised in the equivalent phase contrast images (Figures 5D and H; arrows). Under bright field microscopy the MPIO appeared brown (Figures 5I and J; arrows). Quantitative analysis of MPIO binding indicated significant differences in MPIO density per μm2 tumour area between c(RGDyK)-MPIO and c(RADyK)-MPIO injected animals, for both melanoma tissue (P < 0.05; Figure 5K) and colorectal tissue (P < 0.001; Figure 5L). Assessment of vessel diameter in the two tumour models revealed a significant difference in mean vessel cross-sectional area (P < 0.0001; Figure 5M). This greater vessel diameter in the melanoma derived tumours was also reflected in the 10-fold higher c(RGDyK)-MPIO retention, despite a lower vessel density (per unit tumour area) than in the colorectal tumours (P < 0.0001; Supplementary Material: Figure S3A). Vessel density analysis within groups showed little variation, and no significant change with tumour volume (Figure S3B and C).
Epifluorescence photomicrographs of (A-C) B16F10 or (E-G) MC-38 tumour tissue stained for αV (green), β3 (red) and nuclei counterstain with DAPI (blue). (A,E) Showing αV expression with nuclei, (B,F) β3 expression with nuclei, and (C,G) merged 3 colour channels for visual co-localisation of αVβ3 expression (yellow). (D,H) Equivalent bright field micrographs in which c(RGDyK)-MPIO are clearly visible (arrowheads). (B,C,F,G,) c(RGDyK)-MPIO may also appear as red under this protocol (arrowheads). (I,J) Immunohistochemical micrographs from B16F10 (I) and MC38 (J) tumours showing vessels (CD31) stained brown; again bound c(RGDyK)-MPIO are clearly visible (arrowheads). (K,L) Quantitation of MPIO density per μm2 tumour area for both c(RGDyK)-MPIO and c(RADyK)-MPIO treated groups in the B16F10 (K) and MC38 (L) models. (M) Graph showing quantitation of total vessel area per tumour. Data are presented as Mean + S.E.M. in bar charts. Scale bars = 20 μm. *P < 0.05; **P < 0.005; ***P < 0.0001.
Clearance
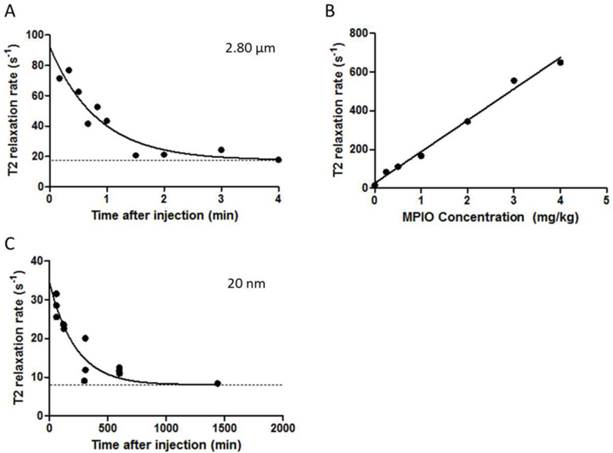
Clearance of iron oxide micro- and nanoparticles from the blood circulation was determined for 2.8 μm and 20 nm iron oxide particles. The initial relaxivity (10 s sample) for blood containing the 2.8 μm MPIO was approximately 2.5 times higher than for the 20 nm NPIO (60 min samples; n=3), 1/T2 = 74.6 vs. 30.3 s-1, respectively (Figure 6A,B). The half-life of the 2.8 μm MPIO was 34.9 s (Figure 6A), which is significantly shorter than the half-life of the 20 nm NPIO (2.6 h; Figure 6B). The MPIO are porous polymers containing evenly distributed iron oxide cores of 6-12 nm, and have been characterised previously [46].
(A-C) Graph demonstrating biological clearance of injected micro- and nanoparticles from circulation, at constant dose of 4 mg of iron per kg of body weight. (A) Graph shows T2 relaxivity values of blood samples at different time points after administration of 2.80 μm MPIO. The clearance half-life was calculated to be 34.91 s (K = 0.019 s-1, Y0 = 92.18 s-1). (B) Scatter plot showing T2 relaxation rates of various concentrations of 2.8 μm MPIO in blood samples. T2 relaxation rate increases linearly with increasing MPIO concentration (R2 = 0.9907). (C) Graph showing T2 relaxivity values of blood samples at different time points after administration of 20 nm NPIO. The clearance half-life for 20 nm NPIO was calculated to be 153.70 min (K = 0.0045 min-1, Y0 = 34.69 s-1). Curves are fitted as one phase exponential decay constrained to plateau at the average naïve blood sample values (baseline; dashed lines).
Discussion
Here, we show that a high affinity cyclic RGD moiety conjugated to microparticles of iron oxide binds to endothelial cells expressing the integrin αvβ3 and demonstrate potent and quantifiable contrast effects in vivo owing to specific retention of c(RGDyK)-MPIO in two different mouse tumour models. Thus, these methods might be useful in detection of αvβ3 integrin in tumour vascular beds and, as such, could provide opportunities both for early tumor detection and for determining the applicability of specific treatments
Targeting the integrin αvβ3 with in vivo imaging agents has been attempted extensively previously. The novelty of the approach described here lies in the delivery of particles that are orders of magnitude larger than the nanoparticles that have usually been used for this application. In an imaging context, the substantially enhanced payload of iron oxide delivers conspicuous, quantifiable contrast effects, which are aided by the rapid clearance of unbound blood phase MPIO, leading to excellent target to background contrast. Clearly, the ability to deliver large cargo loads also opens possibilities for therapeutic purposes.
Micron size range particles of iron oxide have proven successful in imaging molecules differentially expressed on vascular endothelial cells in disease states across a variety of pathologies [27-36]. In earlier work, to enhance target affinity, leukocyte mimetic strategies have been developed, in which dual targeting of antibodies, e.g. recognizing VCAM-1 and P-selectin, markedly improved target-MPIO binding [33, 36]. These molecules are regulated at sites of vascular inflammation, where they mediate the binding of mononuclear leukocytes prior to their migration across the vessel wall. Systematic evaluation of a variety of ligands under variable flow conditions demonstrated that dual antibody targeting provided superior binding compared with either individually [47]. The integrin αvβ3 comprises two subunits, raising the possibility of a similar dual targeting approach to increase MPIO-target affinity. Although antibodies can be generated against each subunit, the spatial relationship of MPIO surface mounted antibodies and cell membrane-associated αvβ3 would not be predictable. In order to enhance binding, we therefore adopted an alternative approach of using the cognate ligand RGD mounted on the surface of MPIO in a polyvalent configuration. The binding affinity of c(RGDyK) to αvβ3 has previously been described in comparison to c(RGDyK)-functionalised quantum dots (QD) [48]. Owing to the polyvalent presentation of the c(RGDyK) on the MPIO, we believe that the IC50 of the functionalised particle will be in the low nanomolar range. Variable shear rate flow chamber experiments demonstrated that c(RGDyK)-MPIO bound at low shears equivalent to what might be expected in small vessels and veins. In vivo administration enabled molecular imaging of integrin αvβ3 on tumour neovasculature. Retention was specific, confirmed by enhanced binding of c(RGDyK)-MPIO compared with c(RADyK)-MPIO.
The contrast effects of MPIO extend for a volume that far exceeds their physical size. However, the contrast effect is 'negative' (i.e. low signal), which can present challenges to distinguish MPIO from features of tissue heterogeneity. In the current study both tumour models showed intrinsic T2* weighted inhomogeneities, although to varying degrees, reflecting differences in tumour growth and cell density. For this reason, it was necessary to obtain pre- and post-contrast MR acquisitions to enable distinction between pre-existing and MPIO-specific hypointensities. In addition to volumetric analysis of MPIO-induced hypointensitites, as described previously, we also calculated pan-tumour T2* relaxivity rates. The aim was to determine whether this analysis approach (which requires considerably less post-processing) yielded comparable and/or complementary information to the more complex volumetric image segmentation approach. Although this approach included the effects of intrinsic tumour inhomogeneities on the global T2* relaxivity, a comparison of pre- and post-contrast values allowed assessment of the reduction in T2* induced by MPIO retention. As described, this approach also revealed the specific retention of c(RGDyK)-MPIO compared to c(RADyK)-MPIO, as demonstrated by a greater shortening of global tumour T2* relaxivity in both models. Interestingly, however, a significant reduction in tumour T2* relaxivity was also found in the c(RADyK)-MPIO injected animals in the colorectal model, but to a lesser extent than in the c(RGDyK)-MPIO injected animals. These data suggest that the relaxivity analysis method may be more sensitive to non-specific retention of MPIO that are too dispersed to detect with the thresholded volumetric analysis. Importantly, both MRI analysis methods correlated well with histological quantification of the c(RGDyK)-MPIO binding, for both tumour models. The two subcutaneous tumour models showed slightly different patterns of c(RGDyK)-MPIO retention, in accord with the spatial distribution of their vasculature. The melanoma-derived tumours, with double the mean vessel area of the colorectal-derived tumours, showed largely constant c(RGDyK)-MPIO induced hypointensities across the tumour volume, with the exception of a small poorly-vascularised rim region that did not show c(RGDyK)-MPIO retention. In contrast, the colorectal-derived tumours tended to show greater heterogeneity in c(RGDyK)-MPIO induced hypointensities across the tumour volume. These findings are in accord with other reports in the literature of the binding distribution of angiogenesis-targeted 19F- and Gd-based nanoparticles in experimental tumour models, in which patterns of particle and vessel distribution vary according to tumour type and site [49-52].
Alternative, widely used MRI contrast agents are based on paramagnetic gadolinium, in which the Gd(III) ion is chelated to low molecular weight ligands, such as diethylenetriaminepentacetic acid (DTPA). However, binding of only a few gadolinium complexes to molecular targets cannot provide a sufficiently high contrast-to-noise ratio for robust detection. In order to enhance the detection sensitivity, Gd(III) complexes are very often loaded onto nanocarriers [11, 53]. For instance, cross-linked liposomes with a high payload of gadolinium (containing 30 % Gd[III] chelate-labelled lipid) and a mean size of 250-350 nm have been conjugated with an anti-αvβ3 antibody or specific peptomimetic [11, 53]. After 2 hours of intravenous administration into rabbits with squamous cell carcinoma, regions of tumour angiogenesis were detectable by MRI. Work in mice-bearing subcutaneous tumours using similar paramagnetic nanocarriers conjugated to anti-integrin αvβ3 peptidomimetic was able to characterise the angiogenic phenotype in early melanoma [18], whilst α5β1(αvβ3)-targeted nanocarriers have enabled 3D maps of angiogenesis in xenographs to be constructed [54]. Again, to enable sufficient contrast accumulation, MRI assessments were performed 2 h post-administration of the contrast agent, owing to the limited binding efficiency at earlier time points. Other endeavours for improving MRI sensitivity for tumour angiogenesis imaging include developing Gd(III)-containing micelles, liposomes, or high-density lipoprotein-like nanoparticles. Work in mice bearing subcutaneous tumours with RGD-liposomes have been able to reduce the time of MRI assessment post-administration to 35 minutes, with sufficient T1-W contrast to characterise angiogenesis [5, 20]. In that work, there was evidence that unbound nanoparticles had extravasated from the vasculature [5, 20], but this was considered insignificant for T1-W imaging of the nanoparticles, owing to the relatively small contrast effect of the Gd. However, extravasation of an equivalent number of USPIO would cause significant non-specific contrast on T2-W imaging. For example, specific targeting of tumour angiogenesis has been demonstrated using RGD-USPIO conjugates (< 100 nm) [23], in squamous cell carcinoma-bearing subcutaneous xenographs. One limitation that was identified, was the tendency for passive extravasation and accumulation, thereby diminishing molecular specificity, as also found in tumour xenograft models implanted with non-small cell lung cancer cells [55]. In contrast, we have previously shown that MPIO are not internalised by either endothelial cells or peripheral immune cells (e.g. macrophages) during the period between injection and imaging [33].
A further limitation of nano-scale particles is their relatively long blood half-life, which increases background contrast effects and greatly reduces the binding-specific contrast at short time frames after administration. As demonstrated here, the half-life of micron-sized particles in the blood is considerably shorter (< 1min). We have previously demonstrated that this clearance occurs primarily through the liver and spleen, with minimal retention in non-target organs (e.g. kidney, lungs, brain), but cannot exclude a role of complement in this process. In common with previously described molecular imaging approaches [27-36], we show empirically that specific MPIO binding to target is sufficient to attain effective contrast. In imaging vascular inflammation using a particle targeting vascular cell adhesion molecule-1 (VCAM-1) we have previously shown that specific binding has occurred within 20 minutes of administration and that this was maintained at a constant level for at least 60 minutes [28]. If reproduced in humans, these characteristics would be ideal for clinical application.
These features are important advantages of MPIO over NPIO and Gd-based nanocarriers for potential clinical application. Towards clinical application we have developed a new class of biodegradable iron oxide microparticle in which 60 nm nanoparticles are linked together to form larger (approx. 600 nm) particles using peptide linkers that are selectively susceptible to cleavage by macrophage enzymes [56].
Finally, it is important to note that the molecular MRI approaches described here, are compatible within the same imaging session with other MRI-based measurement of tumour vascularity and permeability. Interestingly, whilst DCE measures of vascular perfusion and permeability showed a negative relationship with increasing tumour volume, c(RGDyK)-MPIO binding (as determined by hypointense volumes) showed a positive correlation with tumour volume. The former likely reflects slow blood flow through the poorly perfused central tumour regions (as can be seen in the DCE images) which, nevertheless, does not appear to alter the ability of the c(RGDyK)-MPIO to bind effectively within these vessels. No correlation was found between the percent reduction in global tumour T2* relaxation time and increasing tumour volume, indicating that this measure was independent of tumour size. This finding suggests that the average vascularity per unit tumour volume did not change markedly with increasing tumour size (across the range studied), as confirmed histologically. Taken together, the molecular MRI data (volume and T2* relaxation) demonstrate that vascularity increases linearly with tumour growth over the size range studied. It is possible that this relationship will change once substantial necrosis develops within the core of the tumour, leading to either vessel destruction and reduced MPIO access and binding, and/or pooling of contrast agent owing to greater compromise of vascular perfusion through intact vessels. Nevertheless, over the tumour size range studied, our data indicate that despite some reduction in tumour perfusion with tumour growth, the c(RGDyK)-MPIO are able to reach their targets and provide a reliable biomarker of intact tumour vessels. Thus, the molecular approach described here may provide a more accurate measure of vessel number, whilst the complementary DCE data provide information on vascular perfusion and permeability. Together, these methods may enable more effective patient stratification for specific therapies and provide useful biomarkers of efficacy in therapeutic trials.
In conclusion, we present a high affinity c(RGDyK)-MPIO that binds specifically to the integrin αvβ3 in tumour neo-vessels. Unbound MPIO are cleared rapidly from circulation. The result is a marked and quantifiable contrast effect that is evident both on T2* weighted images as intensely low signal volumes and through automated quantitative pan-tumour T2* relaxivity analysis. Recently developed biodegradable MPIO make clinical translation a realistic possibility, thereby enabling integrated molecular, anatomical and perfusion sensitive tumour imaging with MRI.
Supplementary Material
Figures S1 - S3, Supplementary Methods.
Abbreviations
AUC: area under the curve; c(RGDyK): arginine-glycine-aspartic acid-D configuration tyrosine-lysine; c(RADyK): arginine-alanine-aspartic acid-D configuration tyrosine-lysine DCE: dynamic contrast enhanced; DTPA: diethylenetriaminepentacetic; FLM: fraction of local mean; FOV: field of view; Gd: gadolinium; HUVEC: human umbilliant vascular endothelia cells; IsoFlo: isoflurane; MPIO: microparticles of iron oxide; MRI: magnetic resonance imaging; NPIO: nanoparticles of iron oxide; PBS: phosphate buffered saline; PET: positron emission tomography; SNAP: S-nitroso-n-acetylpenicillamine; SPECT: single photon emission computed tomography; T: tesla; TE: echo time; TR: repetition time.
Acknowledgements
This work was funded by Cancer Research UK (grant number C5255/A12678), the CR-UK & EPSRC Oxford Cancer Imaging Center, the Wellcome Trust (Senior Research Fellowship to RC) and British Heart Foundation Oxford Centre for Research Excellence (NR). SM was funded by a CR-UK & EPSRC Oxford Cancer Imaging Centre Studentship. The authors would like to thank Dr Veerle Kersemans, Dr Sébastien Serres and Dr Manuel Sarmiento Soto for technical assistance.
Competing Interests
The authors have declared that no competing interests exist.
References
1. Folkman J. The role of angiogenesis in tumor growth. Semin Cancer Biol. 1992;3(2):65-71
2. Weinstat-Saslow D, Steeg PS. Angiogenesis and colonization in the tumor metastatic process: basic and applied advances. FASEB J. 1994;8(6):401-407
3. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1(1):27-31
4. Kerbel RS. Antiangiogenic therapy: a universal chemosensitization strategy for cancer? Science. 2006;312(5777):1171-1175
5. Mulder WJ, van der Schaft DW, Hautvast PA, Strijkers GJ, Koning GA, Storm G. et al. Early in vivo assessment of angiostatic therapy efficacy by molecular MRI. FASEB J. 2007;21(2):378-383
6. Horton MA. The alpha v beta 3 integrin "vitronectin receptor". Int J Biochem Cell Biol. 1997;29(5):721-725
7. Drake CJ, Cheresh DA, Little CD. An antagonist of integrin alpha v beta 3 prevents maturation of blood vessels during embryonic neovascularization. J Cell Sci. 1995;108(7):2655-2661
8. Griffioen AW, Molema G. Angiogenesis: potentials for pharmacologic intervention in the treatment of cancer, cardiovascular diseases, and chronic inflammation. Pharmacol Rev. 2000;52(2):237-268
9. Brooks PC, Montgomery AM, Rosenfeld M, Reisfeld RA, Hu T, Klier G. et al. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell. 1994;79(7):1157-1164
10. Gladson CL. Expression of integrin αvβ3 in small blood vessels of glioblastoma tumors. Journal of Neuropathology & Experimental Neurology. 1996;55:1143-1149
11. Sipkins DA, Cheresh DA, Kazemi MR, Nevin LM, Bednarski MD, Li KC. Detection of tumor angiogenesis in vivo by alphaVbeta3-targeted magnetic resonance imaging. Nat Med. 1998;4(5):623-626
12. Cai W, Chen X. Multimodality molecular imaging of tumor angiogenesis. J Nucl Med. 2008;49(Suppl 2):113S-28S
13. Buck CA, Horwitz AF. Cell surface receptors for extracellular matrix molecules. Annu Rev Cell Biol. 1987;3:179-205
14. Ruoslahti E, Pierschbacher MD. Arg-Gly-Asp: a versatile cell recognition signal. Cell. 1986;44(4):517-518
15. Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL. et al. Crystal structure of the extracellular segment of integrin alpha Vbeta3 in complex with an Arg-Gly-Asp ligand. Science. 2002;296(5565):151-155
16. Sivolapenko GB, Skarlos D, Pectasides D, Stathopoulou E, Milonakis A, Sirmalis G. et al. Imaging of metastatic melanoma utilising a technetium-99m labelled RGD-containing synthetic peptide. Eur J Nucl Med. 1998;25(10):1383-1389
17. Haubner R, Wester HJ, Weber WA, Mang C, Ziegler SI, Goodman SL. et al. Noninvasive imaging of alpha(v)beta3 integrin expression using 18F-labeled RGD-containing glycopeptide and positron emission tomography. Cancer Res. 2001;61(5):1781-1785
18. Schmieder AH, Winter PM, Caruthers SD, Harris TD, Williams TA, Allen JS. et al. Molecular MR imaging of melanoma angiogenesis with alphanubeta3-targeted paramagnetic nanoparticles. Magn Reson Med. 2005;53(3):621-627
19. Kiessling F, Morgenstern B, Zhang C. Contrast agents and applications to assess tumor angiogenesis in vivo by magnetic resonance imaging. Curr Med Chem. 2007;14(1):77-91
20. Mulder WJ, Strijkers GJ, Habets JW, Bleeker EJ, van der Schaft DW, Storm G. et al. MR molecular imaging and fluorescence microscopy for identification of activated tumor endothelium using a bimodal lipidic nanoparticle. FASEB J. 2005;19(14):2008-2010
21. Montet X, Montet-Abou K, Reynolds F, Weissleder R, Josephson L. Nanoparticle imaging of integrins on tumor cells. Neoplasia. 2006;8(3):214-222
22. Persigehl T, Matuszewski L, Kessler T, Wall A, Meier N, Ebert W. et al. Prediction of antiangiogenic treatment efficacy by iron oxide enhanced parametric magnetic resonance imaging. Invest Radiol. 2007;42(12):791-796
23. Zhang C, Jugold M, Woenne EC, Lammers T, Morgenstern B, Mueller MM. et al. Specific targeting of tumor angiogenesis by RGD-conjugated ultrasmall superparamagnetic iron oxide particles using a clinical 1.5-T magnetic resonance scanner. Cancer Res. 2007;67(4):1555-1562
24. Yang Y, Yang Y, Yanasak N, Schumacher A, Hu TC. Temporal and noninvasive monitoring of inflammatory-cell infiltration to myocardial infarction sites using micrometer-sized iron oxide particles. Magn Reson Med. 2010;63(1):33-40
25. Weissleder R, Kelly K, Sun EY, Shtatland T, Josephson L. Cell-specific targeting of nanoparticles by multivalent attachment of small molecules. Nat Biotechnol. 2005;23(11):1418-1423
26. Shapiro EM, Skrtic S, Sharer K, Hill JM, Dunbar CE, Koretsky AP. MRI detection of single particles for cellular imaging. Proc Natl Acad Sci U S A. 2004;101(30):10901-10906
27. McAteer MA, Sibson NR, von Zur Muhlen C, Schneider JE, Lowe AS, Warrick N. et al. In vivo magnetic resonance imaging of acute brain inflammation using microparticles of iron oxide. Nat Med. 2007;13(10):1253-1258
28. McAteer MA, Mankia K, Ruparelia N, Jefferson A, Nugent HB, Stork LA. et al. A leukocyte-mimetic magnetic resonance imaging contrast agent homes rapidly to activated endothelium and tracks with atherosclerotic lesion macrophage content. Arterioscler Thromb Vasc Biol. 2012;32(6):1427-1435
29. McAteer MA, Schneider JE, Ali ZA, Warrick N, Bursill CA, von zur Muhlen C. et al. Magnetic resonance imaging of endothelial adhesion molecules in mouse atherosclerosis using dual-targeted microparticles of iron oxide. Arterioscler Thromb Vasc Biol. 2008;28(1):77-83
30. von Zur Muhlen C, Sibson NR, Peter K, Campbell SJ, Wilainam P, Grau GE. et al. A contrast agent recognizing activated platelets reveals murine cerebral malaria pathology undetectable by conventional MRI. J Clin Invest. 2008;118(3):1198-1207
31. von zur Muhlen C, von Elverfeldt D, Moeller JA, Choudhury RP, Paul D, Hagemeyer CE. et al. Magnetic resonance imaging contrast agent targeted toward activated platelets allows in vivo detection of thrombosis and monitoring of thrombolysis. Circulation. 2008;118(3):258-267
32. Serres S, Anthony DC, Jiang Y, Broom KA, Campbell SJ, Tyler DJ. et al. Systemic inflammatory response reactivates immune-mediated lesions in rat brain. J Neurosci. 2009;29(15):4820-4828
33. Akhtar AM, Schneider JE, Chapman SJ, Jefferson A, Digby JE, Mankia K. et al. In vivo quantification of VCAM-1 expression in renal ischemia reperfusion injury using non-invasive magnetic resonance molecular imaging. PLoS One. 2010;5(9):e12800
34. Hoyte LC, Brooks KJ, Nagel S, Akhtar A, Chen R, Mardiguian S. et al. Molecular magnetic resonance imaging of acute vascular cell adhesion molecule-1 expression in a mouse model of cerebral ischemia. J Cereb Blood Flow Metab. 2010;30(6):1178-1187
35. Serres S, Mardiguian S, Campbell SJ, McAteer MA, Akhtar A, Krapitchev A. et al. VCAM-1-targeted magnetic resonance imaging reveals subclinical disease in a mouse model of multiple sclerosis. FASEB J. 2011;25(12):4415-4422
36. Serres S, Soto MS, Hamilton A, McAteer MA, Carbonell WS, Robson MD. et al. Molecular MRI enables early and sensitive detection of brain metastases. Proc Natl Acad Sci U S A. 2012;109(17):6674-6679
37. Jefferson A, Wijesurendra RS, McAteer MA, Digby JE, Douglas G, Bannister T. et al. Molecular imaging with optical coherence tomography using ligand-conjugated microparticles that detect activated endothelial cells: rational design through target quantification. Atherosclerosis. 2011;219(2):579-587
38. Cai W, Shin DW, Chen K, Gheysens O, Cao Q, Wang SX. et al. Peptide-labeled near-infrared quantum dots for imaging tumor vasculature in living subjects. Nano Lett. 2006;6(4):669-676
39. Lee PC, Kibbe MR, Schuchert MJ, Stolz DB, Watkins SC, Griffith BP. et al. Nitric oxide induces angiogenesis and upregulates alpha(v)beta(3) integrin expression on endothelial cells. Microvasc Res. 2000;60(3):269-280
40. Cassidy PJ, Schneider JE, Grieve SM, Lygate C, Neubauer S, Clarke K. Assessment of motion gating strategies for mouse magnetic resonance at high magnetic fields. J Magn Reson Imaging. 2004;19(2):229-237
41. Yarnykh VL. Actual flip-angle imaging in the pulsed steady state: a method for rapid three-dimensional mapping of the transmitted radiofrequency field. Magn Reson Med. 2007;57(1):192-200
42. Yushkevich PA, Piven J, Hazlett HC, Smith RG, Ho S, Gee JC. et al. User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage. 2006;31(3):1116-1128
43. Yankeelov TE, Luci JJ, Lepage M, Li R, Debusk L, Lin PC. et al. Quantitative pharmacokinetic analysis of DCE-MRI data without an arterial input function: a reference region model. Magn Reson Imaging. 2005;23(4):519-529
44. Yankeelov TE, DeBusk LM, Billheimer DD, Luci JJ, Lin PC, Price RR. et al. Repeatability of a reference region model for analysis of murine DCE-MRI data at 7T. J Magn Reson Imaging. 2006;24(5):1140-1147
45. Yankeelov TE, Luci JJ, DeBusk LM, Lin PC, Gore JC. Incorporating the effects of transcytolemmal water exchange in a reference region model for DCE-MRI analysis: theory, simulations, and experimental results. Magn Reson Med. 2008;59(2):326-335
46. Fonnum G, Johansson C, Molteberg A, Mørup S, Aksnes E. Characterisation of Dynabeads< sup>® by magnetization measurements and Mössbauer spectroscopy. J Magn Magn Mater. 2005;293(1):41-47
47. Xie J, Chen K, Lee HY, Xu C, Hsu AR, Peng S. et al. Ultrasmall c(RGDyK)-coated Fe3O4 nanoparticles and their specific targeting to integrin alpha(v)beta3-rich tumor cells. J Am Chem Soc. 2008;130(24):7542-7543
48. Cai W, Chen X. Preparation of peptide-conjugated quantum dots for tumor vasculature-targeted imaging. Nat Protoc. 2008;3(1):89-96
49. Huo T, Du X, Zhang S, Liu X, Li X. Gd-EDDA/HYNIC-RGD as an MR molecular probe imaging integrin alphanubeta3 receptor-expressed tumor-MR molecular imaging of angiogenesis. Eur J Radiol. 2010;73(2):420-427
50. Kiessling F, Farhan N, Lichy MP, Vosseler S, Heilmann M, Krix M. et al. Dynamic contrast-enhanced magnetic resonance imaging rapidly indicates vessel regression in human squamous cell carcinomas grown in nude mice caused by VEGF receptor 2 blockade with DC101. Neoplasia. 2004;6(3):213-223
51. Park JA, Lee JJ, Jung JC, Yu DY, Oh C, Ha S. et al. Gd-DOTA conjugate of RGD as a potential tumor-targeting MRI contrast agent. Chembiochem. 2008;9(17):2811-2813
52. Giraudeau C, Geffroy F, Meriaux S, Boumezbeur F, Robert P, Port M. et al. 19F molecular MR imaging for detection of brain tumor angiogenesis: in vivo validation using targeted PFOB nanoparticles. Angiogenesis. 2013;16(1):171-179
53. Winter PM, Caruthers SD, Kassner A, Harris TD, Chinen LK, Allen JS. et al. Molecular imaging of angiogenesis in nascent Vx-2 rabbit tumors using a novel alpha(nu)beta3-targeted nanoparticle and 1.5 tesla magnetic resonance imaging. Cancer Res. 2003;63(18):5838-5843
54. Schmieder AH, Caruthers SD, Zhang H, Williams TA, Robertson JD, Wickline SA. et al. Three-dimensional MR mapping of angiogenesis with alpha5beta1(alpha nu beta3)-targeted theranostic nanoparticles in the MDA-MB-435 xenograft mouse model. FASEB J. 2008;22(12):4179-4189
55. Jiang T, Zhang C, Zheng X, Xu X, Xie X, Liu H. et al. Noninvasively characterizing the different alphavbeta3 expression patterns in lung cancers with RGD-USPIO using a clinical 3.0T MR scanner. Int J Nanomedicine. 2009;4:241-249
56. Perez-Balderas F. Multimeric iron oxide micro particles: Novel high sensitivity and biodegradable mri contrast agents. Intl Soc Mag Reson Med. 2010
Author contact
![]() Corresponding author: Professor Robin P. Choudhury, Division of Cardiovascular Medicine, Level 6, West Wing, John Radcliffe Hospital, Oxford. OX3 9DU. Telephone: +44-1865-234664 Fax: +44-1865-234667 E-mail: robin.choudhuryox.ac.uk.
Corresponding author: Professor Robin P. Choudhury, Division of Cardiovascular Medicine, Level 6, West Wing, John Radcliffe Hospital, Oxford. OX3 9DU. Telephone: +44-1865-234664 Fax: +44-1865-234667 E-mail: robin.choudhuryox.ac.uk.