Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2015; 5(7):656-666. doi:10.7150/thno.10844 This issue Cite
Research Paper
CYLD Regulates Noscapine Activity in Acute Lymphoblastic Leukemia via a Microtubule-Dependent Mechanism
Yunfan Yang1*, Jie Ran1*, Lei Sun1*, Xiaodong Sun2*, Youguang Luo1, Bing Yan1, Tala1, Min Liu1, Dengwen Li1, Lei Zhang3, Gang Bao2 ![]() , Jun Zhou1,2
, Jun Zhou1,2 ![]()
1. State Key Laboratory of Medicinal Chemical Biology, College of Life Sciences, Nankai University, Tianjin 300071, China.
2. Department of Biomedical Engineering, Georgia Institute of Technology & Emory University, Atlanta, GA 30332, USA.
3. State Key Laboratory of Experimental Hematology, Institute of Hematology and Blood Disease Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin 300020, China.
*These authors contributed equally to this work.
Received 2014-10-17; Accepted 2015-1-29; Published 2015-3-2
Abstract

Noscapine is an orally administrable drug used worldwide for cough suppression and has recently been demonstrated to disrupt microtubule dynamics and possess anticancer activity. However, the molecular mechanisms regulating noscapine activity remain poorly defined. Here we demonstrate that cylindromatosis (CYLD), a microtubule-associated tumor suppressor protein, modulates the activity of noscapine both in cell lines and in primary cells of acute lymphoblastic leukemia (ALL). Flow cytometry and immunofluorescence microscopy reveal that CYLD increases the ability of noscapine to induce mitotic arrest and apoptosis. Examination of cellular microtubules as well as in vitro assembled microtubules shows that CYLD enhances the effect of noscapine on microtubule polymerization. Microtubule cosedimentation and fluorescence titration assays further reveal that CYLD interacts with microtubule outer surface and promotes noscapine binding to microtubules. These findings thus demonstrate CYLD as a critical regulator of noscapine activity and have important implications for ALL treatment.
Keywords: leukemia, noscapine, microtubule, mitotic arrest, apoptosis.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common malignancy in children. Although the survival rate of pediatric ALL has been greatly improved over the past decade, the prognosis can vary greatly among patients. Adult ALL is relatively rare but has a strikingly poor prognosis1. Given the body-wide distribution of leukemia cells, chemotherapy is one of the best choices for ALL treatment. However, due to the complex biological nature of leukemia cells, the effectiveness of chemotherapeutic agents currently used in the clinic has been seriously limited. Searching for new chemotherapeutic agents and exploring the molecular mechanisms regulating the sensitivity of leukemia cells would greatly benefit patients suffering from ALL.
Noscapine, a phthalideisoquinoline alkaloid from opium, is a recently discovered anticancer drug and is currently under investigation in phase-I/II clinical trials for the treatment of leukemia and lymphoma2,3. Noscapine is water-soluble and can be absorbed after oral administration4. In addition, this agent causes few or no side effects5,6, and has been widely used as a cough suppressant in developing countries. Due to the great potential of noscapine for cancer chemotherapy, much effort has been put into the development of its analogs with more potent activity7-9. Noscapine has been demonstrated to interact with microtubules2. Interestingly, unlike many other microtubule-targeting agents such as paclitaxel and nocodazole, noscapine does not obviously affect the total amount of microtubule polymers in cells; instead, it significantly increases the time microtubules spend in the pause state10. The alteration of microtubule dynamics then activates the spindle checkpoint and arrests cell cycle progression at mitosis, leading to apoptotic cell death2,10,11. Noscapine has been shown to efficiently inhibit the growth of cancer cells of different types both in vitro and in mouse models2,5,6,12. However, the molecular mechanisms underlying the control of noscapine activity remain largely unknown.
Cylindromatosis (CYLD) is a tumor suppressor protein mutated in familial cylindromatosis and multiple familial trichoepithelioma, genetic conditions associated with the development of skin-appendage tumors13,14. The loss of CYLD has also been implicated in several other malignancies, such as colon and hepatocellular carcinomas, multiple myeloma, melanoma, and leukemia14-16. CYLD interacts with microtubules primarily through its extreme N-terminal cytoskeleton associated protein glycine-rich (CAP-Gly) domain17-19. This domain is also critical for the role of CYLD in regulating the dynamic properties of microtubule assembly17-19. Interestingly, CYLD is highly expressed in mitosis and is critically involved in cell cycle progression19,20. These findings prompted us to explore the potential role of CYLD in regulating the sensitivity of leukemia cells to microtubule-targeting drugs.
Materials and Methods
Materials
Noscapine, paclitaxel, vinblastine, 4',6-diamidino-2-phenylindole (DAPI), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich. Propidium iodide (PI) and Alexa Fluor 488-conjugated annexin V were purchased from Life Technologies. α-Tubulin antibody was obtained from Abcam. CYLD, β-actin, and glutathione S-transferase (GST) antibodies and horseradish peroxidase-conjugated secondary antibody were from Santa Cruz Biotechnology. Rhodamine-conjugated secondary antibody was from Jackson ImmunoResearch Laboratories.
Plasmids, proteins, and siRNAs
Mammalian expression plasmids for GFP-CYLD, GFP-CYLD-ΔCG1 (deletion of amino acids 115-209), GFP-CYLD-C/S (mutation of cysteine 601 to serine), GST-CYLD, and GST-CYLD-ΔCG1 were described previously17,19,21,22. GST, GST-CYLD, and GST-CYLD-ΔCG1 were purified from 293T cells by using glutathione-Sepharose 4B beads according to the manufacturer's instruction (Promega). Purified tubulin and rhodamine-labeled tubulin were purchased from Cytoskeleton. Control siRNA (5'-CGUACGCGGAAUACUUCGA-3') and CYLD siRNAs (#1: 5'-CGAAGAGGCUGAAUCAUAA-3'; #2: 5'-CUGCAAUAGUGGUCAGAAA-3') were synthesized by Dharmacon.
Cells
Leukemia cell lines CCRF-CEM, HL60, K562, Molt4, RPMI-8226, and SR were obtained from the American Type Culture Collection. Primary ALL cells were obtained from patients who underwent chemotherapy at the Blood Disease Hospital of Chinese Academy of Medical Sciences & Peking Union Medical College. The use of patient samples in this study was approved by our Institutional Medical Ethics Committee. All cells were cultured in RPMI 1640 medium supplemented with 2 mM L-glutamine and 10% fetal calf serum at 37 oC in a humidified atmosphere with 5% CO2. Plasmids were transfected into cells with polyethylenimine (Sigma-Aldrich), and siRNAs were transfected with Lipofectamine 2000 (Invitrogen).
Immunoblot analysis
Proteins were resolved by SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Millipore). The membranes were blocked in Tris-buffered saline containing 0.1% Tween 20 and 5% fat-free dry milk and incubated first with primary antibody and then with horseradish peroxidase-conjugated secondary antibody. Specific proteins were visualized with the enhanced chemiluminescence detection reagent (Pierce Biotechnology).
Quantitative real-time RT-PCR
Total cellular RNA was isolated by using the TRIzol reagent (Invitrogen). Quantitative real-time RT-PCR was performed by using the SYBR Premix Ex Taq (Perfect Real Time) reagent (Takara) as described previously23. β-Actin was used as a control to normalize the reading for CYLD in each sample.
Cytotoxicity assay
Cells were treated with gradient concentrations of drugs for 48 hours in 96-well plates. MTT (5 mg/mL) was then added into each well and incubated for 4 hours. The plates were centrifuged at 1,000 rpm for 10 minutes, and the medium was then removed. DMSO was added into each well to resolve the crystal. Absorbance was then measured by a plate reader at the wavelength of 562 nm. The percentage of cell survival as a function of drug concentration was plotted to determine the IC50 value, which stands for the drug concentration needed to kill cells by 50%.
Fluorescence microscopy
Cells grown on glass coverslips were fixed with cold (-20 oC) methanol for 5 minutes and then washed with phosphate-buffered saline (PBS) for 5 minutes. Non-specific sites were blocked by incubation with 2% bovine serum albumin in PBS for 15 minutes. For annexin V staining, cells were incubated with Alexa Fluor 488-conjugated annexin V for 2 hours. For other experiments, cells were incubated with primary antibody for 2 hours and then rhodamine-conjugated secondary antibody for 1 hour followed by staining with DAPI for 5 minutes. Coverslips were mounted with 90% glycerol in PBS and examined with a TCS SP5 confocal microscope (Leica) as described previously24.
Flow cytometry
Cells were centrifuged, washed twice with ice-cold PBS, and fixed in 70% ethanol. Tubes containing the cell pellets were stored at -20 oC for 4 hours. After this, the cells were centrifuged at 1000 g for 10 minutes, and the pellets were resuspended in phosphate/citrate buffer. Cells were then washed with PBS and incubated with PI and/or Alexa Fluor 488-conjugated annexin V for 30 minutes. Samples were analyzed on a Coulter Elite flow cytometer (BD).
Microtubule assembly in vitro
Purified tubulin (5 μM) was mixed with GST, GST-CYLD, or GST-CYLD-ΔCG1 (20 μM) and noscapine (10 μM) in microtubule assembly buffer (100 mM PIPES, 1 mM EGTA, 1 mM MgSO4, 1 mM GTP, pH 6.8) and loaded in a temperature-controlled spectrophotometer (Amersham Biosciences). The temperature was shifted to 37 oC, and microtubule assembly was monitored by measuring the optical absorbance at 350 nm. To observe the in vitro assembled microtubules, 5 μM purified tubulin was mixed with 10% (wt/wt) rhodamine-labeled tubulin and 20 μM purified GST, GST-CYLD, or GST-CYLD-ΔCG1 and 10 μM noscapine in microtubule assembly buffer. The mixture was incubated at 37 °C for 20 minutes, and the assembled microtubules were then examined with a Zeiss fluorescence microscope.
Microtubule cosedimentation assay
Purified GST, GST-CYLD, or GST-CYLD-ΔCG1 was incubated with untreated or subtilisin-digested microtubules in a buffer containing 100 mM NaCl. The samples were gently mixed and centrifuged at 244,000 g for 10 minutes at 25 °C. Pellets and supernatant fractions were collected and analyzed by immunoblotting.
Fluorescence titration assay
Paclitaxel, vinblastine, or noscapine was incubated with microtubules at 37 oC, and the intrinsic fluorescence intensity of microtubules was monitored with a spectrofluorometer (JASCO) at an excitation wavelength of 295 nm. The dissociation constant (Kd) between each drug and microtubules was then determined by fluorescence titration as described previously7.
Statistics
Analysis of statistical significance was performed by the Student t test capability in Microsoft Excel.
Results
CYLD expression correlates with noscapine activity both in cell lines and in primary cells of leukemia
To test the potential role of CYLD in regulating the sensitivity of microtubule-targeting drugs in leukemia cells, we obtained primary leukemia cells from seven patients suffering from ALL. We examined CYLD protein level in these cells by immunoblotting and CYLD mRNA level by quantitative real-time PCR. We found that CYLD protein and mRNA levels varied significantly among patients (Figures 1A and 1B). The efficacy of noscapine to induce apoptosis in these cells was then analyzed by nuclear staining. We found significant differences between the patients with a high or low expression of CYLD in the apoptotic efficacy induced by noscapine; leukemia cells isolated from the patient (#3) who had the highest CYLD expression showed the highest apoptosis in response to noscapine treatment (Figure 1C).
CYLD expression correlates with noscapine activity both in cell lines and in primary cells of leukemia. (A) Immunoblots showing CYLD and β-actin expression in primary leukemia cells from 7 ALL patients. (B) Quantitative real-time RT-PCR analysis of CYLD mRNA expression in primary leukemia cells. (C) Primary leukemia cells were treated with 2 μM noscapine for 48 hours, and the percentage of apoptotic cells were quantified by nuclear staining. (D-F) Correlation analysis between the IC50 value of noscapine (D), paclitaxel (E), or vinblastine (F) and CYLD protein or mRNA level in primary leukemia cells. CYLD protein level was determined by densitometric analysis of the blots shown in A. (G) Immunoblots for CYLD and β-actin expression in 6 different leukemia cell lines. (H) Correlation analysis between the IC50 value of noscapine and CYLD protein level in 6 leukemia cell lines. (I) Leukemia cell lines were treated with 2 μM noscapine for 48 hours, and the percentage of apoptotic cells were quantified by nuclear staining.
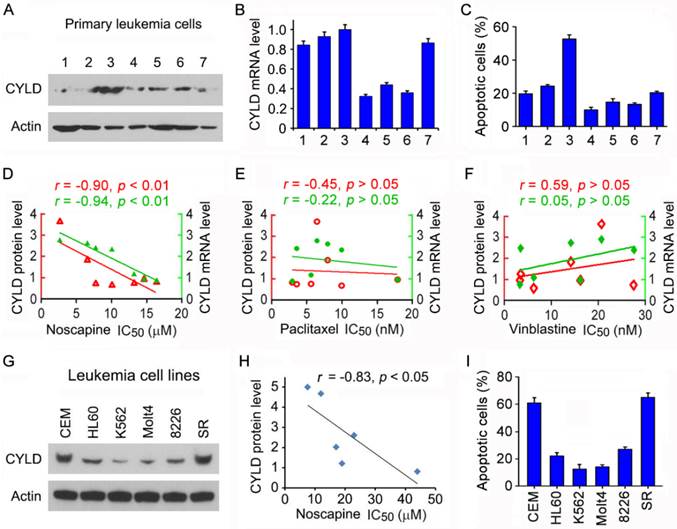
We then performed cytotoxicity assays to evaluate the sensitivity of these cells to noscapine as well as paclitaxel and vinblastine, other two microtubule-targeting drugs, and determined the IC50 values of these drugs, which stand for drug concentrations needed for killing cells by 50%. A negative correlation was found between the IC50 values of noscapine and the protein and mRNA levels of CYLD (Figure 1D). By contrast, no significant correlation was observed between CYLD expression and the IC50 values of paclitaxel or vinblastine (Figures 1E and 1F). These results suggest that CYLD specifically regulates noscapine activity in ALL cells.
To further investigate the relationship between CYLD expression and noscapine activity, we examined CYLD protein levels and noscapine IC50 values in six different leukemia cell lines (two ALL cell lines: CCRF-CEM and Molt4; one promyelocytic leukemia cell line: HL60; one chronic myeloid leukemia cell line: K562; one myelomonocytic leukemia cell line: RPMI-8226; and one large cell lymphoblastic leukemia cell line: SR). Consistent with the data obtained in primary leukemia cells, CYLD protein level in the six leukemia cell lines correlated with cell sensitivity to noscapine treatment (Figures 1G and 1H). In addition, there were significant differences between these cell lines in the apoptotic efficacy induced by noscapine; CCRF-CEM and SR cells, which had the highest CYLD expression, showed the highest apoptosis in response to noscapine treatment, whereas K562 and Molt4 cells, which had the lowest CYLD expression, showed the lowest apoptosis (Figure 1I).
CYLD depletion attenuates noscapine-induced apoptosis in primary leukemia cells
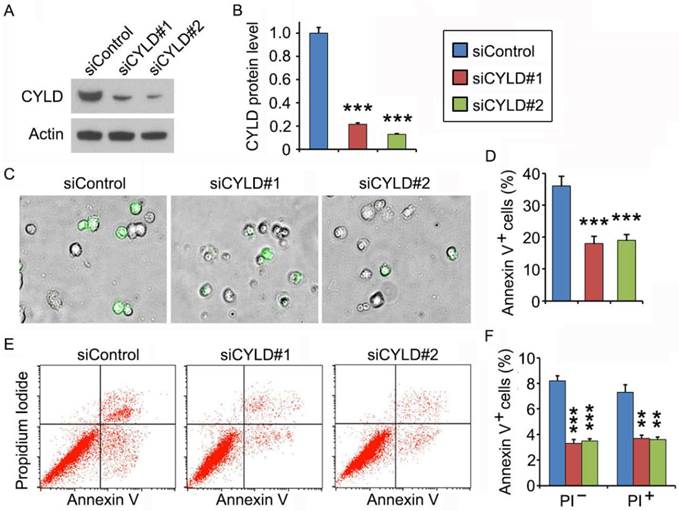
To gain mechanistic insight into the role of CYLD in regulating noscapine activity, we studied the effect of CYLD on downstream cellular events triggered by noscapine. Primary leukemia cells, from #3 ALL patient shown in Figure 1A, were transfected with control or CYLD siRNAs and then treated with noscapine. We found that CYLD expression was dramatically knocked down by the siRNAs (Figures 2A and 2B). Fluorescence microscopy showed that depletion of CYLD remarkably reduced the accumulation of annexin V-positive cells induced by noscapine (Figures 2C and 2D). Flow cytometric analysis of control or CYLD siRNA-transfected primary leukemia cells further demonstrated that both early (annexin V-positive and PI-negative) and late (annexin V-positive and PI-positive) apoptosis induced by noscapine were attenuated by CYLD depletion (Figures 2E and 2F). These data indicate that CYLD is important for the ability of noscapine to induce apoptosis in ALL cells.
CYLD depletion attenuates noscapine-induced apoptosis in primary leukemia cells. (A) Immunoblots for CYLD and β-actin expression in control and CYLD siRNA-treated primary leukemia cells. (B) Experiments were performed as in A, and CYLD protein level was determined by densitometric analysis of the blots. (C) Fluorescence/phase-contrast images of primary leukemia cells transfected with control or CYLD siRNAs followed by treatment with 2 μM noscapine for 36 hours. Cells were stained with Alexa Fluor 488-conjugated annexin V (green). (D) Experiments were performed as in C, and the percentage of annexin V-positive cells was quantified. (E) Primary leukemia cells were transfected with control or CYLD siRNAs followed by treatment with 2 μM noscapine for 24 hours. Cells were then stained with PI and Alexa Fluor 488-conjugated annexin V, and analyzed by flow cytometry. (F) Experiments were performed as in E, and the percentage of annexin V-positive cells was quantified. **, p < 0.01; ***, p < 0.001.
CYLD siRNAs inhibit the ability of noscapine to induce mitotic arrest and apoptosis in leukemia cell lines
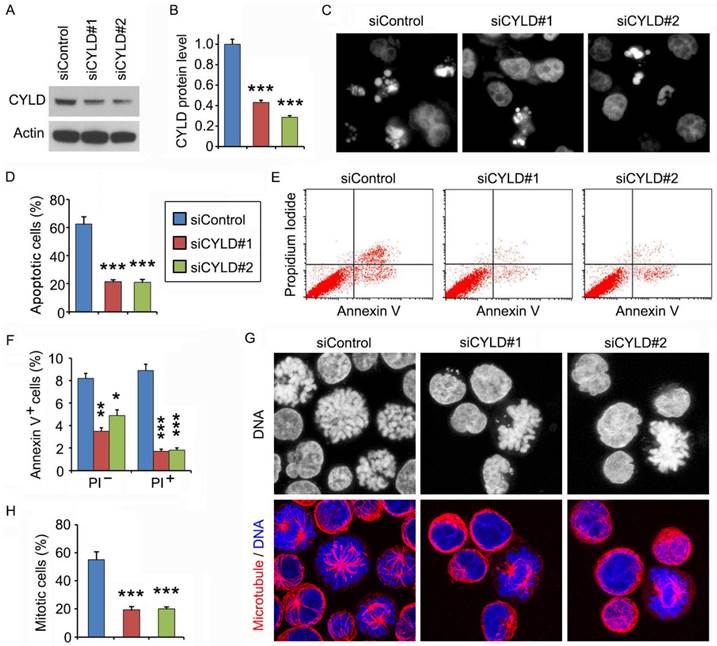
We then used the CCRF-CEM leukemia cell line to further explore the mechanism of how CYLD regulates noscapine activity. Similar to the results obtained in primary leukemia cells, siRNA-mediated knockdown of CYLD expression in CCRF-CEM cells dramatically decreased noscapine-induced apoptosis, as evidenced by the reduction of cells with apoptotic bodies (Figures 3A-3D). Flow cytometry further revealed that CYLD siRNAs inhibited both early and late apoptosis induced by noscapine in CCRF-CEM cells (Figures 3E and 3F). Noscapine is known to arrest cells at mitosis before cells undergo apoptosis2,10,11. Thus, we examined whether CYLD modulates the ability of noscapine to cause mitotic arrest. By staining spindle microtubules and chromosomes, we found that CYLD siRNAs significantly reduced the percentage of cells arrested at mitosis by noscapine; these mitotically arrested cells had chromosome rosettes and either normal or abnormal mitotic spindles (Figures 3G and 3H). Together, these findings suggest that CYLD regulates noscapine activity to induce mitotic arrest and apoptosis in leukemia cells.
CYLD siRNAs inhibit the ability of noscapine to induce mitotic arrest and apoptosis in leukemia cell lines. (A) Immunoblots for CYLD and β-actin expression in control and CYLD siRNA-treated CCRF-CEM cells. (B) Experiments were performed as in A, and CYLD protein level was determined by densitometric analysis of the blots. (C) Fluorescence images of CCRF-CEM cells transfected with control or CYLD siRNAs and treated with 2 μM noscapine for 48 hours, followed by staining with DAPI. (D) Experiments were performed as in C, and the percentage of apoptotic cells (with apoptotic bodies) was quantified. (E) CCRF-CEM cells were transfected with control or CYLD siRNAs followed by treatment with 2 μM noscapine for 24 hours. Cells were then stained with PI and Alexa Fluor 488-conjugated annexin V, and analyzed by flow cytometry. (F) Experiments were performed as in E, and the percentage of annexin V-positive cells was quantified. (G) CCRF-CEM cells were transfected with control or CYLD siRNAs followed by treatment with 2 μM noscapine for 24 hours. Cells were then stained with anti-α-tubulin antibody (red) and DAPI (blue). (H) Experiments were performed as in G, and the percentage of mitotic cells (with chromosome rosettes and either normal or abnormal mitotic spindles) was quantified. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
CYLD enhances noscapine activity to induce mitotic arrest and apoptosis in a microtubule-dependent manner
To understand how CYLD modulates noscapine activity in leukemia cells, we transfected cells with GFP, GFP-CYLD, GFP-CYLD-ΔCG1, in which the extreme N-terminal CAP-Gly domain of CYLD was deleted, or GFP-CYLD-C/S, in which cysteine 601 was mutated to serine to disrupt the deubiquitinase activity of CYLD. Immunofluorescence microscopy revealed that overexpression of CYLD or its C/S mutant greatly enhanced the accumulation of apoptotic cells; by contrast, its ΔCG1 mutant failed to increase the activity of noscapine to induce apoptosis (Figures 4A and 4B). Immunofluorescence microscopy also showed that CYLD and its C/S mutant, but not its ΔCG1 mutant, increased noscapine activity to cause mitotic arrest (Figures 4C and 4D).
CYLD enhances noscapine activity to induce mitotic arrest and apoptosis in a microtubule-dependent manner. (A) Molt4 cells were transfected with GFP, GFP-CYLD, GFP-CYLD-ΔCG1, or GFP-CYLD-C/S and treated with 2 μM noscapine for 36 hours, followed by staining with PI (red). (B) Experiments were performed as in A, and the percentage of apoptotic cells (with apoptotic bodies) was quantified. (C) Molt4 cells were transfected with GFP, GFP-CYLD, GFP-CYLD-ΔCG1, or GFP-CYLD-C/S and treated with 2 μM noscapine for 24 hours. Cells were then stained with PI (red). (D) Experiments were performed as in C, and the percentage of mitotic cells (with chromosome rosettes) was quantified. (E) Molt4 cells were transfected with GFP, GFP-CYLD, GFP-CYLD-ΔCG1, or GFP-CYLD-C/S and treated with 2 μM noscapine for 36 hours. Cells were then stained with PI and Alexa Fluor 488-conjugated annexin V, and analyzed by flow cytometry. (F) Experiments were performed as in E, and the percentage of annexin V-positive cells was quantified. (G) Molt4 cells were transfected with GFP, GFP-CYLD, GFP-CYLD-ΔCG1, or GFP-CYLD-C/S and treated with 2 μM noscapine for 24 hours. Cells were then stained with PI and analyzed by flow cytometry. (H) Experiments were performed as in G, and the percentage of cells in sub-G1, G1/S, and G2/M phases were determined. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant.
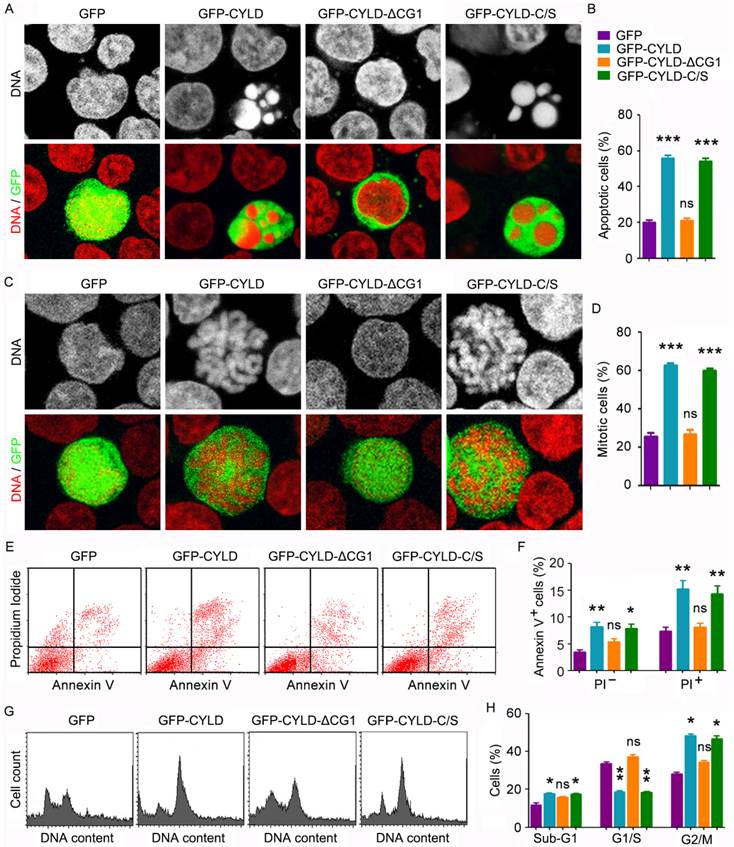
To corroborate the above findings, we examined the effects of CYLD and its mutants on noscapine-induced apoptosis and mitotic arrest by flow cytometry. Consistent with the immunofluorescence results, annexin V/PI staining-based flow cytometry showed that CYLD and its C/S mutant, but not its ΔCG1 mutant, promoted both early and late apoptosis induced by noscapine (Figures 4E and 4F). In addition, flow cytometric analysis of DNA content showed that CYLD and its C/S mutant, but not its ΔCG1 mutant, promoted the ability of noscapine to induce the accumulation of G2/M cells (Figures 4G and 4H). Given that the extreme N-terminal CAP-Gly domain of CYLD is essential for its interaction with microtubules17-19, the above data indicate that the effect of CYLD on noscapine activity is dependent on the binding of CYLD to microtubules and is independent of its deubiquitinase activity.
CYLD enhances the effect of noscapine on microtubule polymerization
To provide additional insight into how CYLD regulates noscapine activity in leukemia cells, we examined the morphology of microtubules in GFP, GFP-CYLD, or GFP-CYLD-ΔCG1-transfected cells treated with noscapine. Immunofluorescence microscopy showed that overexpression of CYLD led to the accumulation of microtubule bundles in both CCRF-CEM and Molt4 cell lines; by contrast, no significant effect was observed for the ΔCG1 mutant (Figures 5A and 5B). These data indicate that CYLD increases the effect of noscapine on microtubule polymerization.
CYLD enhances the effect of noscapine on microtubule polymerization. (A) CCRF-CEM and Molt4 cells were transfected with GFP, GFP-CYLD, or GFP-CYLD-ΔCG1 and treated with 2 μM noscapine for 6 hours. Cells were then stained with anti-α-tubulin antibody (red) and DAPI (blue). (B) Experiments were performed as in A, and the percentage of cells with abnormal microtubules was quantified. (C) Coomassie blue staining of GST, GST-CYLD, and GST-CYLD-ΔCG1 purified from 293T cells. (D) Purified tubulin (5 μM) was mixed with GST, GST-CYLD, or GST-CYLD-ΔCG1 (20 μM) and noscapine (10 μM), and microtubule assembly was monitored by measuring the optical absorbance at 350 nm. (E) Images of microtubules incorporated with rhodamine-labeled tubulin, polymerized in the presence of noscapine (10 μM) and purified GST, GST-CYLD, or GST-CYLD-ΔCG1 (20 μM). (F) Experiments were performed as in E, and microtubule length was measured. (G) Experiments were performed as in E, and the number of microtubules per field was quantified. ***, p < 0.001; ns, not significant.
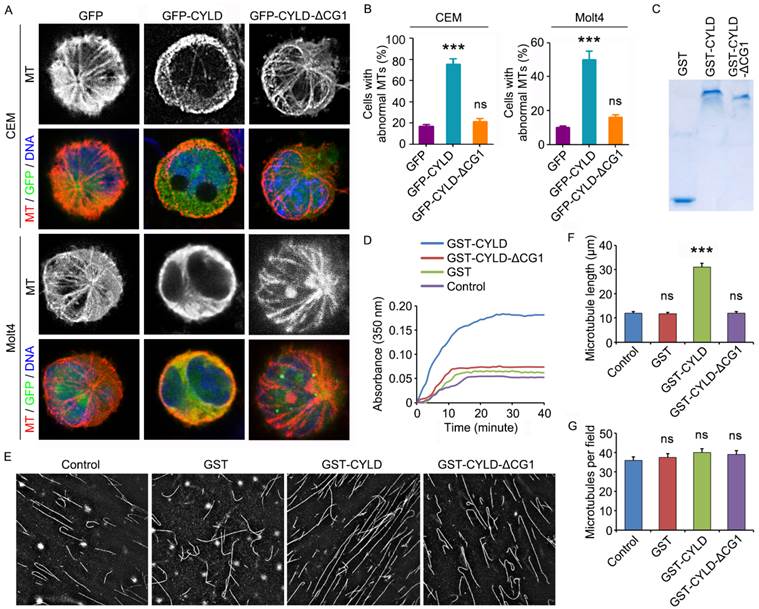
To verify the above finding, we transfected 293T cells with GST, GST-CYLD, and GST-CYLD-ΔCG1 and then purified these proteins (Figure 5C). We then evaluated their effects on microtubule assembly in vitro. Purified tubulin was incubated with noscapine and GST or GST-CYLD fusion proteins at 37 oC, and the polymerization of microtubules was assessed by measuring the optical absorbance at 350 nm of wavelength. We found that CYLD, but not its ΔCG1 mutant, significantly promoted the effect of noscapine on microtubule assembly in vitro (Figure 5D).
We then added rhodamine-labeled tubulin into the in vitro microtubule assembly system, to visualize the assembled microtubules. Consistent with the data obtained with the absorbance measurement, CYLD, but not its ΔCG1 mutant, remarkably increased the average length of microtubules assembled in vitro (Figures 5E and 5F). The number of microtubules per field was not affected by CYLD or its ΔCG1 mutant (Figures 5E and 5G). Collectively, these results suggest that CYLD stimulates noscapine activity to promote microtubule polymerization, without affecting microtubule nucleation.
CYLD interacts with microtubule outer surface and promotes noscapine binding to microtubules
We then sought to study how CYLD regulates noscapine effect on microtubule polymerization. To address this question, we incubated the in vitro assembled microtubules with purified GST, GST-CYLD, or GST-CYLD-ΔCG1 and then pelleted microtubules by centrifugation. In agreement with the role of the extreme N-terminal CAP-Gly domain in mediating CYLD interaction with microtubules, immunoblotting revealed that a significant proportion of CYLD, but not its ΔCG1 mutant, was present in the microtubule pellet fraction (Figure 6A). The in vitro assembled microtubules were then treated with subtilisin, which is known to remove the C-terminal tails of α- and β-tubulin exposed to the outer surface of microtubules25. We found that while CYLD resided in the pellet of control microtubules, it was not detectable in the pellet of subtilisin-digested microtubules (Figure 6A). These data indicate that CYLD associates with microtubule outer surface.
CYLD interacts with microtubule outer surface and promotes noscapine binding to microtubules. (A) Purified GST, GST-CYLD, or GST-CYLD-ΔCG1 was incubated with subtilisin-treated or control untreated microtubules. Microtubules were pelleted by centrifugation and proteins present in the pellet (P) and supernatant (S) fractions were detected by immunoblotting with anti-GST antibody. (B) Paclitaxel was incubated with microtubules in the presence of GST or GST-CYLD. The dissociation constant (Kd) between paclitaxel and microtubules was then determined by fluorescence titration assays. (C) Vinblastine was incubated with microtubules in the presence of GST or GST-CYLD. The dissociation constant between vinblastine and microtubules was then determined by fluorescence titration assays. (D) Noscapine was incubated with microtubules in the presence of GST, GST-CYLD, or GST-CYLD-ΔCG1. The dissociation constant between noscapine and microtubules was then determined by fluorescence titration assays. (E) A model illustrating the role of CYLD in promoting noscapine activity in leukemia cells. When CYLD expression in leukemia cells is low, noscapine interacts weakly with microtubules, arrests only a small subset of cells at mitosis (round cells with mitotic spindles and chromosomes), and then induce these mitotically arrested cells to undergo apoptosis (cells with blebs and apoptotic bodies). When CYLD expression is high, noscapine interacts strongly with microtubules, arrests a large subset of cells at mitosis, and then triggers these cells to undergo apoptosis.
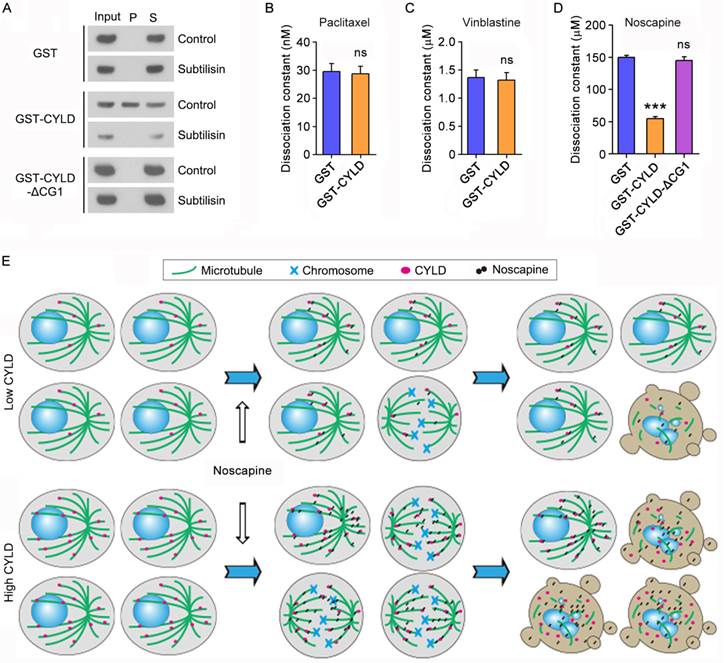
Next, we examined the effect of CYLD on the interaction of paclitaxel, vinblastine, and noscapine with microtubules. Fluorescence titration assays were performed to determine the dissociation constant (Kd) between these drugs and microtubules. We found that CYLD did not significantly affect the Kd values of paclitaxel-microtubule interaction and vinblastine-microtubule interaction (Figures 6B and 6C). By contrast, CYLD, but not its ΔCG1 mutant, remarkably decreased the Kd value of noscapine-microtubule interaction (Figure 6D). This result indicates that CYLD specifically enhances the interaction between noscapine and microtubules, but does not alter the interaction of paclitaxel and vinblastine with microtubules.
Discussion
Microtubule-targeting drugs are widely used in cancer chemotherapy26. These agents arrest cell cycle progression at mitosis by altering microtubule dynamics and activating the spindle checkpoint and subsequently result in cell death. This is believed to be the major mechanism underlying the action of these agents in killing cancer cells26-28. Despite the effectiveness of microtubule-targeting drugs, patients show variable response to them either at the initial treatment or at the recurring stage26. The molecular events regulating the sensitivity of microtubule-targeting drugs remain poorly understood.
CYLD dysregulation has been demonstrated previously as one of the essential events driving acute and chronic lymphocytic leukemia. The function of CYLD in preventing lymphocytes from tumorigenesis has been shown to be partially mediated by its role in regulating cell death and necroptosis15,16,29-32. These findings suggest that CYLD may serve as a potential biomarker and therapeutic target in leukemia treatment. In this study, we show that CYLD expression varies among leukemia patients and that the response of leukemia cells to noscapine treatment correlates with CYLD expression. These findings suggest a potential for using CYLD as a biomarker for predicting the efficacy of noscapine in leukemia treatment. Our data also suggest that CYLD might be exploited as a therapeutic target to improve noscapine sensitivity. Interestingly, CYLD down-regulation has been shown to confer glucocorticoid resistance in Jurkat T cells by interfering with drug-induced necroptosis32. Thus, the dysregulation of CYLD might be critical for both tumorigenesis and chemotherapeutic resistance of leukemia, and targeting CYLD may represent an attractive strategy for the development of more efficient treatment for refractory leukemia.
The molecular mechanisms of how CYLD is regulated in leukemia remain elusive. Lymphoid enhancer-binding factor 1, a transcription factor in the Wnt signaling, has been demonstrated to suppress the transcription of CYLD for the dysregulation of tumor necrosis factor α-induced necroptotic signaling in chronic lymphocytic leukemia30. Hes1, another transcription factor, has been shown to function in the Notch signaling to repress CYLD transcription in T-cell leukemia31. In addition, miR-19 has been reported to repress CYLD expression in T-cell ALL16. Interestingly, CYLD has also been shown to be regulated by post-translational modifications. For instance, mucosa-associated lymphoid tissue lymphoma translocation protein 1 promotes the initiation of ALL by cleaving CYLD into a 35 kDa segment29. Thus, the expression of CYLD seems to be regulated at different levels, and further studies are warranted to better understand the molecular events underlying its dysregulation in leukemia.
Based on the data presented in this work, we propose the following model for how CYLD promotes noscapine activity in leukemia cells. CYLD binds to the outer surface of microtubules and promotes noscapine binding to microtubules, which in turn stimulates the ability of noscapine to cause mitotic arrest and apoptosis (Figure 6E). It should be noted, however, that CYLD interacts with several other microtubule-binding proteins, such as histone deacetylase 6 (HDAC6) and end-binding protein 1 (EB1)18,33. Therefore, the interaction of CYLD with HDAC6 or EB1 may also contribute to the effect of CYLD on noscapine activity in leukemia cells. In addition, CYLD is known as a critical regulator of nuclear factor κB, c-Jun N-terminal kinase, and several other signaling pathways involved in the control of cell proliferation and survival34-36. It is possible that CYLD may also exert on these pathways to stimulate noscapine activity.
The present study demonstrates by fluorescence titration assays that CYLD decreases the dissociation constant between noscapine and microtubules, suggesting that CYLD promotes noscapine binding to microtubules. Regarding the underlying molecular mechanism, it is possible that CYLD may increase the interaction between noscapine and microtubules by directly exerting structural or allosteric effects on microtubules, in a pattern similar to other microtubule-binding proteins such as Tau, parkin, EB1, and cytoplasmic linker protein 170, which have been shown to regulate the anticancer activity of paclitaxel25,37-39. Given the complex structural feature of CYLD, it would not be surprising if other mechanisms were discovered in the future mediating the actions of CYLD in regulating noscapine binding to microtubules and noscapine activity in leukemia cells. It will be important to examine in the future whether CYLD affects noscapine activity in animal models of leukemia and whether this action is mediated by the effect of CYLD on noscapine-induced mitotic arrest and apoptosis.
Abbreviations
ALL: acute lymphoblastic leukemia; CYLD: cylindromatosis; CAP-Gly: cytoskeleton associated protein glycine-rich; DAPI: 4´,6-diamidino-2-phenylindole; GST: glutathione S-transferase; MTT: 3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide; DMSO: dimethyl sulfoxide; PI: propidium iodide; PBS: phosphate-buffered saline; HDAC6: histone deacetylase 6; EB1: end-binding protein 1.
Acknowledgements
This work was supported by grants from the National Basic Research Program of China (2012CB945002 to JZ), the National Natural Science Foundation of China (31130015, 31471262, and 91313302 to JZ), and the US National Science Foundation (CBET-0939511 to GB).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet. 2013;381:1943-55
2. Ye K, Ke Y, Keshava N. et al. Opium alkaloid noscapine is an antitumor agent that arrests metaphase and induces apoptosis in dividing cells. Proc Natl Acad Sci U S A. 1998;95:1601-6
3. Joshi HC, Zhou J. Noscapine and analogues as potential chemotherapeutic agents. Drug News Perspect. 2000;13:543-6
4. Haikala V, Sothmann A, Marvola M. Comparative bioavailability and pharmacokinetics of noscapine hydrogen embonate and noscapine hydrochloride. Eur J Clin Pharmacol. 1986;31:367-9
5. Ke Y, Ye K, Grossniklaus HE. et al. Noscapine inhibits tumor growth with little toxicity to normal tissues or inhibition of immune responses. Cancer Immunol Immunother. 2000;49:217-25
6. Aneja R, Zhou J, Vangapandu SN. et al. Drug-resistant T-lymphoid tumors undergo apoptosis selectively in response to an antimicrotubule agent, EM011. Blood. 2006;107:2486-92
7. Zhou J, Gupta K, Aggarwal S. et al. Brominated derivatives of noscapine are potent microtubule-interfering agents that perturb mitosis and inhibit cell proliferation. Mol Pharmacol. 2003;63:799-807
8. Aneja R, Vangapandu SN, Joshi HC. Synthesis and biological evaluation of a cyclic ether fluorinated noscapine analog. Bioorg Med Chem. 2006;14:8352-8
9. Naik PK, Chatterji BP, Vangapandu SN. et al. Rational design, synthesis and biological evaluations of amino-noscapine: a high affinity tubulin-binding noscapinoid. J Comput Aided Mol Des. 2011;25:443-54
10. Zhou J, Panda D, Landen JW. et al. Minor alteration of microtubule dynamics causes loss of tension across kinetochore pairs and activates the spindle checkpoint. J Biol Chem. 2002;277:17200-8
11. Zhou J, Gupta K, Yao J. et al. Paclitaxel-resistant human ovarian cancer cells undergo c-Jun NH2-terminal kinase-mediated apoptosis in response to noscapine. J Biol Chem. 2002;277:39777-85
12. Landen JW, Hau V, Wang M. et al. Noscapine crosses the blood-brain barrier and inhibits glioblastoma growth. Clin Cancer Res. 2004;10:5187-201
13. Masoumi KC, Shaw-Hallgren G, Massoumi R. Tumor Suppressor Function of CYLD in Nonmelanoma Skin Cancer. J Skin Cancer. 2011;2011:614097
14. Massoumi R. CYLD: a deubiquitination enzyme with multiple roles in cancer. Future Oncol. 2011;7:285-97
15. Wu W, Zhu H, Fu Y. et al. Clinical significance of down-regulated cylindromatosis gene in chronic lymphocytic leukemia. Leuk Lymphoma. 2014;55:588-94
16. Ye H, Liu X, Lv M. et al. MicroRNA and transcription factor co-regulatory network analysis reveals miR-19 inhibits CYLD in T-cell acute lymphoblastic leukemia. Nucleic Acids Res. 2012;40:5201-14
17. Gao J, Huo L, Sun X. et al. The tumor suppressor CYLD regulates microtubule dynamics and plays a role in cell migration. J Biol Chem. 2008;283:8802-9
18. Wickstrom SA, Masoumi KC, Khochbin S. et al. CYLD negatively regulates cell-cycle progression by inactivating HDAC6 and increasing the levels of acetylated tubulin. EMBO J. 2010;29:131-44
19. Yang Y, Liu M, Li D. et al. CYLD regulates spindle orientation by stabilizing astral microtubules and promoting dishevelled-NuMA-dynein/dynactin complex formation. Proc Natl Acad Sci U S A. 2014;111:2158-63
20. Stegmeier F, Sowa ME, Nalepa G. et al. The tumor suppressor CYLD regulates entry into mitosis. Proc Natl Acad Sci U S A. 2007;104:8869-74
21. Sun L, Gao J, Huo L. et al. Tumour suppressor CYLD is a negative regulator of the mitotic kinase Aurora-B. J Pathol. 2010;221:425-32
22. Yang Y, Ran J, Liu M. et al. CYLD mediates ciliogenesis in multiple organs by deubiquitinating Cep70 and inactivating HDAC6. Cell Res. 2014;24:1342-53
23. Gao J, Sun L, Huo L. et al. CYLD regulates angiogenesis by mediating vascular endothelial cell migration. Blood. 2010;115:4130-7
24. Tala Xie S, Sun X et al. Microtubule-associated protein mdp3 promotes breast cancer growth and metastasis. Theranostics. 2014;4:1052-61
25. Wang H, Liu B, Zhang C. et al. Parkin regulates paclitaxel sensitivity in breast cancer via a microtubule-dependent mechanism. J Pathol. 2009;218:76-85
26. Zhou J, Giannakakou P. Targeting microtubules for cancer chemotherapy. Curr Med Chem Anticancer Agents. 2005;5:65-71
27. Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253-65
28. Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell. 2005;8:7-12
29. Arora M, Kaul D, Varma N. et al. Cellular proteolytic modification of tumor-suppressor CYLD is critical for the initiation of human T-cell acute lymphoblastic leukemia. Blood Cells Mol Dis. 2015;54:132-8
30. Liu P, Xu B, Shen W. et al. Dysregulation of TNFalpha-induced necroptotic signaling in chronic lymphocytic leukemia: suppression of CYLD gene by LEF1. Leukemia. 2012;26:1293-300
31. Espinosa L, Cathelin S, D'Altri T. et al. The Notch/Hes1 pathway sustains NF-kappaB activation through CYLD repression in T cell leukemia. Cancer Cell. 2010;18:268-81
32. Bonapace L, Bornhauser BC, Schmitz M. et al. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J Clin Invest. 2010;120:1310-23
33. Li D, Gao J, Yang Y. et al. CYLD coordinates with EB1 to regulate microtubule dynamics and cell migration. Cell Cycle. 2014;13:974-83
34. Sun SC. CYLD: a tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ. 2010;17:25-34
35. Reiley W, Zhang M, Sun SC. Negative regulation of JNK signaling by the tumor suppressor CYLD. J Biol Chem. 2004;279:55161-7
36. Yang Y, Sun L, Tala. et al. CYLD regulates RhoA activity by modulating LARG ubiquitination. PLoS One. 2013;8:e55833
37. Rouzier R, Rajan R, Wagner P. et al. Microtubule-associated protein tau: a marker of paclitaxel sensitivity in breast cancer. Proc Natl Acad Sci U S A. 2005;102:8315-20
38. Luo Y, Li D, Ran J. et al. End-binding protein 1 stimulates paclitaxel sensitivity in breast cancer by promoting its actions toward microtubule assembly and stability. Protein Cell. 2014;5:469-79
39. Sun X, Li D, Yang Y. et al. Microtubule-binding protein CLIP-170 is a mediator of paclitaxel sensitivity. J Pathol. 2012;226:666-73
Author contact
![]() Corresponding authors: Jun Zhou, Ph.D., State Key Laboratory of Medicinal Chemical Biology, College of Life Sciences, Nankai University, Tianjin 300071, China. Telephone: +86-22-2350-4946; Fax: +86-22-2350-4946; E-mail: junzhouedu.cn or Gang Bao, Ph.D., Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30332, USA. Telephone: +1-404-385-0373; Fax: +1-404-894-4243; E-mail: gang.baogatech.edu
Corresponding authors: Jun Zhou, Ph.D., State Key Laboratory of Medicinal Chemical Biology, College of Life Sciences, Nankai University, Tianjin 300071, China. Telephone: +86-22-2350-4946; Fax: +86-22-2350-4946; E-mail: junzhouedu.cn or Gang Bao, Ph.D., Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA 30332, USA. Telephone: +1-404-385-0373; Fax: +1-404-894-4243; E-mail: gang.baogatech.edu