Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Whole Cell as Drug Carrier
Cell Membrane Coated Particles...
Future Prospects
Acknowledgements
References
AUTHOR BIOGRAPHY
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(8):863-881. doi:10.7150/thno.11852 This issue Cite
Review
Cell or Cell Membrane-Based Drug Delivery Systems
Songwei Tan1,2,3, Tingting Wu1, Dan Zhang1, Zhiping Zhang1,2,3, ![]()
1. Tongji School of Pharmacy;
2. National Engineering Research Center for Nanomedicine;
3. Hubei Engineering Research Center for Novel DDS, Huazhong University of Science and Technology, Wuhan 430030, P R China
Received 2015-2-11; Accepted 2015-2-18; Published 2015-4-27
Abstract

Natural cells have been explored as drug carriers for a long period. They have received growing interest as a promising drug delivery system (DDS) until recently along with the development of biology and medical science. The synthetic materials, either organic or inorganic, are found to be with more or less immunogenicity and/or toxicity. The cells and extracellular vesicles (EVs), are endogenous and thought to be much safer and friendlier. Furthermore, in view of their host attributes, they may achieve different biological effects and/or targeting specificity, which can meet the needs of personalized medicine as the next generation of DDS. In this review, we summarized the recent progress in cell or cell membrane-based DDS and their fabrication processes, unique properties and applications, including the whole cells, EVs and cell membrane coated nanoparticles. We expect the continuing development of this cell or cell membrane-based DDS will promote their clinic applications.
Keywords: drug delivery system, nanoparticle, cell membrane, extracellular vesicle, tumor
Introduction
How to design a drug carrier that can specifically deliver drug to the site of disease is always the main focus of current pharmaceutical research[1, 2]. Thanks to recent advances in material science and nanotechnology, many nanoformulations, such as liposomes, micelles and nanoparticles (NPs), have been developed and some of them have been used in the clinic or approaching the clinical translation[3-6]. Among them, liposomes are considered as an effective drug delivery system (DDS) and more than ten types of liposomal drugs have been already approved and commercialized[7-9]. Liposomes possess many advantages in drug delivery applications, such as high biocompatibility, biodegradability and capacity for size and surface manipulations[7]. They have been widely used to entrap hydrophilic and/or hydrophobic agents to increase their in vivo stability during the circulation. More importantly, both the chemical compositions (phospholipids) and the lipid bilayer structures of liposomes are extremely close to the biological membranes, making them highly compatible with the biological milieu. After being modified with targeting groups, their drug delivery efficiency could be further improved[10, 11].
As a kind of biomimetic products, liposome was used as a model of biological membranes to investigate the biologic functions of living cells[12-15]. However, due to the relatively simple structure, it is hard for liposome to mimic the complexity of cell membranes. Recent studies have been focusing on the possibility of using natural cell or cell derived vesicles as drug carriers, including the whole cell, extracellular vesicles (EVs) and cell membrane coated particles[9, 16-21]. For artificial DDS, its “non-self” property may lead to some adverse effects. In contrast, the autologous cells based carriers with the similar membrane structure of body cells are considered as the “self” and thus demonstrate much better biocompatibility and lower toxicity. This cell or cell membrane-based DDS can be produced in a simple way with minimum membrane protein loss. The retained membrane structure consequently endows the carriers various bio-functions and/or targeting specificities as their parent cells without further modifications. For example, carrier erythrocytes (red blood cells, RBCs) were widely used to encapsulate or bind small-molecule agents, nucleic acids, proteins, and NPs to treat systemic disease owing to their long lifespan and high biocompatibility[16, 22, 23]. Stem cells (SCs) could transport therapeutic cargoes to tumor microenvironment via their intrinsic tumor-tropic properties[24]. Cell membrane-derived microparticles (MPs) from apoptotic tumor cells and the mesenchymal stem cell nanoghosts (MSC NGs), can package and deliver therapeutic agents to tumor with enhanced stability and anti-tumor efficiency[25, 26]. Recently, novel cell membrane-coated particles, were developed to combine advantages of natural cells and synthetic polymers with various applications such as drug delivery, toxin absorption and cancer vaccines[27-34].
Herein, we reviewed recent progress made in cell or cell membrane-based DDS and presented their fabrication processes, unique properties and applications (Table 1). According to their structure, this review mainly focused on three main kinds of these systems: whole cells, EVs and cell membrane coated particles. In addition, some future prospects were prudentially addressed. We expect the continuing development of this cell or cell membrane-based DDS will promote the efficiency and safety in the treatment of diseases.
Cell or cell membrane-based drug delivery system
| Platform | Source | Payload | Remark | Ref |
|---|---|---|---|---|
| Carrier RBC | RBCs | antiretroviral agents, cardiovascular drugs, antiparasitics, antibiotics, therapeutic enzymes | biocompatibility, biodegradation, long blood circulation, sustained drug release, RES targeting | 61-66 |
| PS NPs | lung accumulation | 50 | ||
| Au NPs, 5(6)-CF, Rh-dextran | near-IR laser triggered release | 73 | ||
| DOX, IONP-Ce6-PEG | Magnetic targeting; synergistic effect of photodynamic & chemotherapy | 48 | ||
| TAT, OVA | antigen delivery | 58, 59, 75 | ||
| MSC, NSC | SCs | DOX-loaded silica nanorattle, DTX-loaded NPs, photodynamic agents-loaded NPs, Au NPs or AuNRs | tumor homing efficiency | 83-89 |
| Whole blood cells | mRNA | liver and spleen accumulation | 41 | |
| RBC MVs | RBCs | USPIO NPs | label and trace | 118 |
| ICG, Fasudil | long circulation time, increased cell phagocytosis efficiency | 112, 113 | ||
| Tumor cell-derived MPs | Tumor cells (H22, A2780) | MTX, DOX, PTX, cisplatin | “domino-like” tumor killing behavior | 25 |
| MSC NG, MSC MV | MSC | sTRAIL, PTX | hypoimmunogenicity, specific tumor targeting | 26, 119 |
| HUVEC EVs | HUVEC | IONPs, QDs, Au NPs | natural fabrication, easy purification | 120 |
| OMVs | Gram-negative bacteria | siRNA | low endotoxicity, tumor targeting, bioengineered | 123 |
| Exosomes | DCs | OVA, pcDNAgp120, tumor-derived antigen | high antigen presenting efficiency, minimum side effects | 108, 114, 129-131 |
| Curcumin, JSI124, siRNA, miRNA, DOX | passive targeting via the EPR effect specific tissue/cell homing | 109, 110, 132, 134, 135 | ||
| Exosome-mimetic vesicles | embryonic SCs, human U937 monocytic cells | DOX, 5-Fluorouracil, gemcitabine, carboplatin | high yield | 137-139 |
| RBCm coated NPs | RBC | PLGA NPs, DOX-loaded PLA NPs, Au NPs or Au nanocages | increased stability, long blood circulation, sustained drug release | 27-30, 140, 142 |
| toxin | absorbed toxins locking, high safety, strong antibody response | 31, 32 | ||
| Cancer cell membrane-coated NPs | B16-F10, MDA-MB-435 cancer cells | PLGA NPs | specific anti-tumor immune response, homotypic binding capability | 34 |
| Eukaryocyte membranes-coated NPs | Eukaryocyte | Si particle | preventing the internalization/uptake by macrophages, phagocytic cells or vein endothelial cells, binding to and transporting through inflamed endothelium | 145 |
Whole Cell as Drug Carrier
The human body contains various cells with different physiological functions, including long circulation in blood, site-specific migration, physical barriers crossing, and so on. It's worth choosing some types of cells to deliver drugs with retained cell structure and function. Recently, the whole cell-based drug carriers have been emerging as a hot topic, such as RBCs, SCs and immunological cells [23, 24, 35-37].
RBC as Drug Carrier
Fabrication of Carrier RBC
RBCs are the most frequently used whole cells as drug carrier for a variety of bioactive agents, which are known as 'carrier erythrocytes or 'carrier RBCs'. They have gained remarkable interest during the past decades and some of them have undergone clinical tests[23, 37]. Compared with artificial DDS, the 'carrier RBC' shows many advantages. They are intrinsically biocompatible, biodegradable and non-immunogenic. The natural surface can protect encapsulated cargos from inactivation with a remarkably prolonged and controllable lifespan in comparison to the synthetic carriers. These make them a valuable systemic drug release platform[38]. Several approaches have been developed to load agents in RBCs or attach them onto the outer surfaces by either chemical or physical methods (Figure 1).
Schematic illustration of drug loading methods for RBC. (a) electroporation, (b) osmosis-based methods, (c) co-incubation, (d) bio-bridge methods and (e) CPP-mediation method.
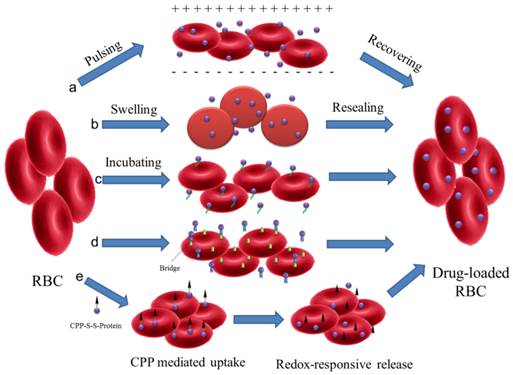
Electroporation: Exposing the RBCs to a strong external electrical field will induce pores in the RBC membrane[39]. The agents can then diffuse into the cells. This method has been used in the encapsulation of enzymes, nucleic acid and anionic drugs (such as diclofenac sodium)[40-42]. However, this method may disrupt the cell membrane and result in a partial and irreversible deterioration of the structural integrity[43]. Phosphatidylserine (PS), a signal to mononuclear-phagocyte system for uptake, was found to be externalized after electroporation treatment[41]. This would result in the reticuloendothelial system (RES) phagocytosis and greatly shorten the blood circulation time.
Osmosis-based method: It's a common strategy used to load therapeutic agents into RBCs. According to the fabrication procedures, the methods are divided into hypotonic dilution, hypotonic pre-swelling, osmotic pulse, hypotonic hemolysis and the most often used hypotonic dialysis[22, 23]. Although the details of the methods may vary, they are all based on the principle that RBCs swell in hypotonic solution followed by the formation of pores with diameter range from 10 to 500 nm. The increase in membrane permeability allows soluble agents to diffuse into cells driven by the concentration gradient. Then the pores are resealed under an isotonic condition to form drug-loaded RBCs. Small molecular drugs, peptides, protein/enzymes, even NPs, such as Au and magnetic NPs, can be encapsulated in this way[44-48]. Hemoglobin-free red blood cells can also be prepared by this method[49]. Incubation time, pH value, temperature and cargo concentration could all affect the encapsulation efficiency (EE) in RBCs[22]. For Au NPs, the EE was found to be more closely related to the surface modification[45]. Compared with negatively charged groups (citrate, thioglycolic acid and 4-mercaptobenzoic acid), amine groups (6-thioguanine), 2-mercaptoethanol or 1-propanthiol surface modifications are more effective to induce the incorporation of Au NPs into the RBCs. Similar to electroporation, osmosis-based methods may also cause the disruption of cell membrane with some unrecoverable structure damage in different degrees. The loss of important cellular constituents (such as hemoglobin and cytoskeleton) via the pore is another fact that cannot be ignored as the physiological functions and the structural integrity of RBCs may have been impaired. This may increase the opportunity of being recognized and cleared by the human body[43].
Co-incubation: Some membrane active drugs, such as primaquine, hydrocortisone, vinblastine and chlorpromazine, can be encapsulated in RBCs by incubation directly[22]. For NPs, they can attach to the surface of RBCs via electrostatic interaction, van der Waals and/or hydrophobic interactions. This cellular hitchhiking strategy significantly improved the blood pharmacokinetics of the NPs and opened a new horizon for applications[50-53]. Recently, a simple membrane painting method was used to load targeting or therapeutic ligands onto the RBC surface[54-58]. As a model, immunoglobulin (IgG) was first chemically conjugated with DSPE-PEG and then incubated with RBC suspensions. DSPE-PEG-IgG would insert into the RBC membrane through lipid transfer. As a result, IgG was retained in RBCs with maximum membrane retention and long-circulation property in vivo. The method may be further used to bind other smaller ligands, such as Fab fragment, single-chain antibody or small peptide, for in vivo applications.
Bio-bridge method: In general, this method contains a series of strategies to load cargoes by physical or chemical approaches. Avidin-biotin bridge is a common way to conjugate bioactive agents on the surface of RBCs[16, 23, 48, 59]. For example, biotin N-hydrosuccinimide ester (NHS-biotin) was first reacted with the amino groups on RBC membrane. Subsequently, the bioactive agents were conjugated with avidin following by incubation with biotin modified RBCs. Biopharmaceuticals, like fibrinolytic agents, HIV-1 TAT protein, bovine serum albumin, even organic/inorganic hybrid NPs could all be conjugated to RBC surfaces via the biotin-avidin bridge. This procedure could attach approximately 1000 biotin molecules per cell (mouse RBCs) with up to 90% cell recovery but did not affect the cell's 24 h survival in circulation[56].
Some antibodies and peptides were also used as the bridge to accomplish the attachment of targeting antigens or immune antigens to RBC surface[57, 58]. The bispecific antibodies, which were composed of targeting antigens (human IgG or dinitrophenylated bovine gamma globulin) and human RBC complement receptor, were used to rapidly and stably modify RBC for removing pathogenic antigens associated with infectious disease[58]. ERY1 peptide (specifically bind to mouse glycophorin-A) conjugated ovalbumin (OVA) could directly and specifically bind with mouse RBC in vivo after iv injection. The captured OVA amount was up to 1.174 ±0.005 ng of OVA per 106 RBCs with dissociation constant (Kd) of 6.2 ±1.3 nM[58]. This approach provided an effective biomolecular modification to RBCs with minimum damage from any chemical reaction or organic solvent. Shi et al developed another efficient method to engineer RBC with functional probes through sortase-catalyzed reaction under native conditions[60]. The modified RBCs remain detectable in the bloodstream for more than 28 days and could be applied to precisely deliver cargoes to an organ, tissue or some special location in the body.
CPP-mediation method: In the case of loading agents into RBCs, most of the methods discussed above showed limitations due to the possibility of disruption to membrane structure. A new encapsulating method to load proteins into RBCs was presented by Yang' group without altering the structure and/or function[43]. L-asparaginase was conjugated with a low molecular weight protamine based cell penetrating peptide (CPP) via disulfide bond. The CPP-protein conjugation can be easily uptaken by the cells and then release the protein by redox-responsive cleavage of disulfide bond. This method could be also applied to load liposomes, NPs, or micelles into intact RBCs for the treatment of various systemic diseases.
Carrier RBC for Drug Delivery
The most attractive property of RBC-based DDS is its long life-span. Due to the semipermeable membrane nature, carrier RBC allows the exchange of small molecules between intracellular environment and circulation by passive diffusion. Many cargos, such as antiparasitics, antiretroviral agents, antibiotics and cardiovascular drugs, were loaded into the RBCs to avoid their rapid clearance and achieve the sustained release and improved biodistribution[16, 22, 23, 61-65]. Protein-loaded RBCs, known as 'bioreactors', were also developed by taking advantage of the semipermeability[66]. The RBC membrane could avoid unfavorable immune activation or rapid clearance of therapeutic enzymes, but not block the access of enzymatic substrates. This may provide a new strategy for the clinical application of enzymes which cannot be injected directly. RBC was also applied as the carrier of contrast agent or indicator for diagnosis, such as indocyanine green (ICG), fluorescein isothiocyanate (FITC), Au NPs, superparamagnetic iron oxides NPs (SPIONs) or other paramagnetic complexes[45-47, 49, 53, 67]. Compared with free agents, these systems demonstrated enhanced efficiency with reduced adverse effect.
The ultimate fate of carrier RBC is thought to be captured by the RES system, typically, the liver and spleen, just as physiologically processing the “old” RBCs. Compared with natural ones, the carrier RBCs were more likely to be trapped in RES after injection due to the loss of natural integrity during the drug-loading process[43, 68, 69]. In this context, we can treat some RES-related diseases via this strategy, such as hepatoma and RES parasitic diseases[16]. Some more destructive approaches on cell membrane, including cross-link, thermal shock and oxidation, have been reported to achieve a more rapid and efficient RES localization[66, 70, 71]. After coated by some antibodies, the targeting organ or cell for carrier RBC could be further adjusted, such as anti-Rh for RES macrophages, IgG for spleen accumulation and IgM for liver targeting[72].
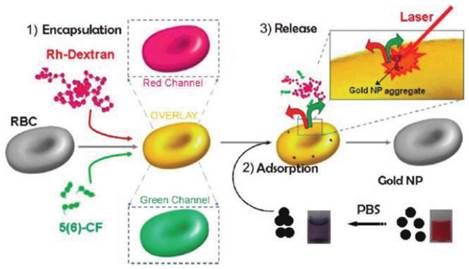
Besides the sustained release in circulation and RES targeting, recent advances of carrier RBC focused on the non-RES targeting. Anselmo et al attached carboxylated polystyrene (PS) NPs to RBCs through non-covalent interaction[50]. This cellular hitchhiking strategy prolonged the circulation time of the NPs in blood and increased the accumulation in lung with diminished uptake by liver and spleen. The lung/liver and lung/spleen NPs accumulation were improved by over 15-fold and 10-fold, respectively. The shear stress derived from RBC-endothelial contact, as well as the large endothelial surface and cardiac blood output in lung vasculature were supposed to be the reason for this special accumulation. However, limited by the large size, it's not easy for carrier RBC to deliver drugs to the desired site. To solve this problem, a thoughtful work has been reported by Delcea et al (Figure 2)[73]. Two model drugs with different molecular weight, 5(6)-carboxyfluorescein (5(6)-CF, 376 Da) and rhodaminelabeled dextran (Rh-dextran, 7500 Da), were co-encapsulated into RBCs by hypotonic-osmotic method. Then the carrier RBC was incubated with highly concentrated Au NPs to achieve the NPs adsorption and aggregation onto the surface of RBC. When the carrier RBC with Au NPs was exposed in a near-IR laser, the local heat from Au NPs would raise the permeability of the membrane and induce the formation of some hydrophilic pores, which led to the fast release of encapsulated model drugs. This light-triggered release strategy could realize site-specific release of encapsulated agents with controlled dose, which may greatly broaden the application of RBC-based DDS. Magnetic targeting is another way to fulfill the designed release of carrier RBC[16]. With the help of external magnetic field (MF), the RBC accumulated in desired organ/tissue and sustainably released the loaded drugs. Recently, another interesting work with remarkable improvement was reported by means of this principle. PEG-chlorine e6 (Ce6) coated iron oxide NPs (IONPs) (IONP-Ce6-PEG) were conjugated onto the surface of doxorubicin (DOX)-loaded RBC[48]. The high tumor tissue accumulation of this novel carrier RBC was observed with the help of MF after iv injection and resulted in a site-specific and continuous DOX and Ce6 delivery. Furthermore, due to the synergistic effect of photodynamic & chemotherapy, a high tumor treating efficiency could be achieved with relatively low therapeutic dosages.
Schematic of the fabrication and controlled release of carrier RBCs: (1) encapsulation: two molecules (Rh-dextran (in red) and 5(6)-CF (in green)) are simultaneously encapsulated. Red, green, and overlay fluorescence channels are shown in the schematics; (2) adsorption of nanoparticle aggregates onto the surface of loaded RBCs (cuvettes demonstrating the behavior of Au NPs in water (right) and after transfer in PBS (left)); and (3) release of both molecules by a near-IR laser. Reproduced with permission[73]. Copyright 2014, ACS.
Carrier RBC for Immunotherapy
Aged and damaged RBCs are removed daily from the blood primarily by phagocytes such as macrophages and DCs. MHC-associated antigens will be then processed and presented to the T cells[37, 74]. This property of RBCs makes them a useful antigen delivery system. In a pioneer work, HIV regulatory protein, TAT was coated on the RBC membrane through avidin-biotin method[59]. Compared with soluble TAT, RBC-TAT was much more effective in delivering antigen to DC and inducing specific CD4+ and CD8+ T-cell responses in vitro. The dose of RBC-TAT was 1250-fold less to induce a similar T-cell response. Another RBC-based antigen delivery system, OVA loaded RBCs were reported to induce specific immune tolerance after being treated with calcium ionophore or BS3[75]. Compared with untreated RBC-OVA, which only induced humoral response, these treated RBCs were capable of delivering a competent amount of antigen to the immune system and then inducing strong and long lasting inhibition of antigen-specific humoral and cellular immune.
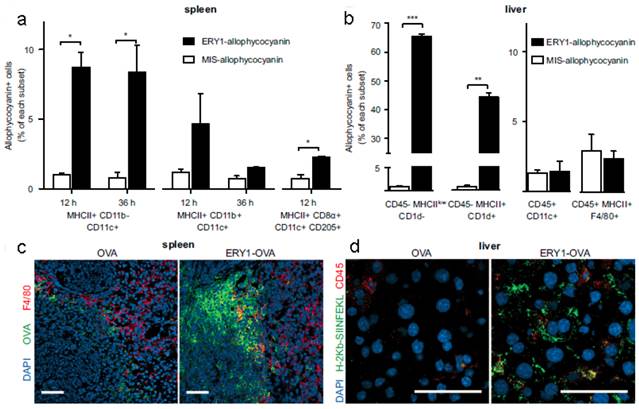
The apoptosis-like programmed cell death of RBCs was also used to induce antigen-specific T-cell deletion[58]. Antigens (such as OVA and peptide islet β-cell autoantigen) were anchored onto the RBCs surface via binding peptide or fusion antibody fragment (against RBC-specific cell surface marker glycophorin A). Compared with mismatch scrambled peptide, OVA coated RBCs were phagocytosed more efficiently by splenic MHC II+ CD11b- CD11c+ DCs, hepatocytes (CD45-MHC IIlow CD1d-) and hepatic stellate cells (CD45- MHC II+ CD1d+), which triggered CD8+ T-cell deletional tolerance, but not by the liver DCs (CD45+ CD11c+) or Kupffer cells (CD45+ MHC II+ F4/80+) (Figure 3). This would induce PD-1 signal related deletional proliferation of CD4+ and CD8+ T cells. Furthermore, this technology was exploited to treat autoimmune type 1 diabetes in a transgenic islet β cell-reactive CD4+ T-cell adoptive transfer model. The activated pathogenic islet-specific CD4+ T cells were successfully deleted with zero morbidity of diabetes for 62 days following adoptive transfer.
RBC-bound allophycocyanin uptake by splenic DC subsets and nonprofessional APCs in the liver. (a) Increased cellular uptake of ERY1-allophycocyanin by MHC II+ CD11b- CD11c+ and MHC II+ CD8α+ CD11c+ CD205+ splenic DCs at 12 and 36 h postinjection, compared with MIS-allophycocyanin. (b) Increased cellular uptake of ERY1-allophycocyanin in the liver by hepatocytes (CD45- MHC IIlow CD1d-) and hepatic stellate cells (CD45- MHC II+ CD1d+) but not by liver DCs (CD45+ CD11c+) or Kupffer cells (CD45+ MHC II+ F4/80+), compared with MIS-allophycocyanin, 36 h following i.v. administration (n = 2). *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. Data represent mean ± SE. (c) Spleen microscopy images of mice 24 h following administration of 10 μg OVA(Left) or ERY1-OVA(Right), stained for OVA (green), F4/80 (red), and DAPI nuclear staining (blue). (Scale bar = 50 μm.) (d) Liver microscopy images of mice 24 h following administration of 10 μg OVA (Left) or ERY1-OVA (Right), stained for MHC I H-2Kb-SIINFEKL (green), CD45 (red), and DAPI for nuclear staining (blue). (Scale bar = 50 μm.) Reproduced with permission[58]. Copyright 2014, ACS.
Stem Cells-based Drug Carrier
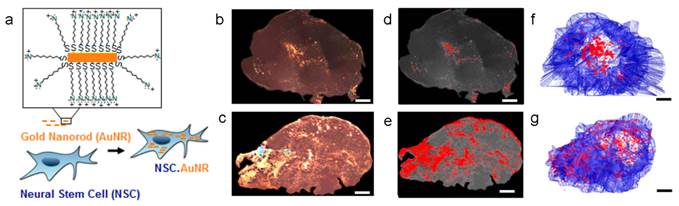
Many types of SCs, including MSCs and neural stem cells (NSCs), have been proved with the ability of migrating towards tumor microenvironment so they are applied widely in tumor-specific drug delivery[24]. For example, SCs with genetic modification could secret therapeutic agents in tumor tissue, such as tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)[76, 77], interferon-β (IFN-β)[78, 79], IL12/18[80-82], to inhibit the growth of the tumor. Recently, a new development was to use SCs as the NPs carriers[83-89]. The NPs-loaded SCs would migrate towards to tumor tissue and thus caused the accumulation of NPs. This “Trojan horse” method seemed to be effective and had been implemented in various cases. Two approaches were reported to achieve the tumor treatment. One is attaching the NPs on the surface of SCs. DOX loaded anti-CD73 or anti-CD90 conjugated silica nanorattle was anchored to MSCs through specific antibody-antigen interaction. These modified MSCs can target the U251 glioma tumor tissue, induce enhanced cell apoptosis and inhibit the tumor metastasis[83]. In another work, docetaxel (DTX)-loaded pH sensitive NPs were coupled onto the surface of NSCs via acid cleavage sialic acid moieties-hydrazone-biotin-avidin linker[84]. These NSC-NP conjugates could overcome the high interstitial pressure in tumor and improve the distribution and retention of the NPs. The other one is encapsulating NPs into SCs. Therapeutic nanoparticle, meso-tetrakis (4-sulfonatophenyl) porphyrin loaded polymeric NPs or purpurin-18 loaded porous hollow silica NPs, could be entrapped in MSCs, which accumulated in tumor and delayed the tumor growth by photodynamic therapy[85]. Au NPs or Au nanorods (AuNRs) were phagocytosed by NSCs with more wide tumor distribution compared to free Au NPs/NRs thus achieving a better photothermal therapy efficiency (Figure 4)[86, 87]. The MSCs containing multifunctional silica NPs were applied in the tumor multimodality imaging, including optical, PET and MR imaging[88]. Despite these progresses, some concerns pointed out that SCs may promote the tumor growth or even differentiate into tumor[24, 90-92]. It's necessary to carefully choose SC type in designing the SC-based delivery system.
Neural stem cell-mediated intratumoral delivery of AuNRs. (a) Schematic depicting AuNR uptake by NSCs. (b-g) Comparison of free AuNR and NSC.AuNR distribution after intratumoral injection. Three days after AuNR injection, tumors were sectioned. Every 150 μm, sections were imaged using dark-field microscopy. (b, c) Tiled, flattened, dark-field micrographs of entire cross sections of tumors injected with free AuNRs (d) or NSC.AuNRs (e). AuNRs are visible as dense, bright gold signals. (c, d) Mapped cross sections of tumors injected with free AuNRs (d) or NSC.AuNRs (e). (f, g) 3D projection of all mapped AuNR (red) and tumor (blue) traces generated using Reconstruct software in tumors that received free AuNR (f) or NSC.AuNR (g). Scale bar = 1 mm and applies to all images. Reproduced with permission[87]. Copyright 2014, ACS.
Other Cell-based Drug Carrier
Tumor tissue is some kind of inflammatory site so that immune cells, such as macrophages, neutrophils, lymphocytes, can be recruited and accumulated into tumor[19, 93-95]. The unique property of these cells was used as 'Trojan horse' delivery systems to tumor tissue[96]. Considering macrophages usually capture foreign materials, it's easily loading therapeutic agents via ex vivo way. RNA loaded liposomes, magnetic NPs, Au nanoshell, imaging agents loaded NPs (zirconium-89 or quantum dot) and drug loaded NPs were able to be phagocytosed by macrophages and accumulate in tumor tissue or even migrate through the blood-brain barrier (BBB) into brain tumors[97-103].
Antigen-encoding mRNA shows many advantages as a source of antigen but its applications are limited by the instability in vivo[104-106]. To prevent the antigen-encoding mRNA from nuclease degradation, the whole blood cells were chosen to load the mRNA (300 ng RNA in 5 × 107 blood cells). This mRNA loaded system could induce anti-tumor immune responses via activated B- and T-cells[41]. This system can not only deliver mRNA but also present the antigen. The blood cells could reach and accumulate in the liver and spleen within 2 h, induce immune responses rapidly and finally achieve a comparable therapeutic efficacy of mRNA-loaded DCs in the B16 melanoma model.
Extracellular Vesicle-Based DDS
EVs are small vesicles secreted by virtually all types of eukaryotic cells for intercellular communication[13, 17, 18, 20, 21, 107, 108]. EVs can deliver biologicals, including lipids, proteins and RNA, so as to affect the function of the receiving cells. Moreover, EVs released from different types of cells may show a high targeting specificity. For example, EVs released from T cells were preferentially taken up by CD11bGr1 cells from inflammatory tissues[109, 110]. MPs from tumor cells exhibited the tumor tropic property[25]. Therefore, EVs have received more attention recently and showed great potential as a novel strategy for drug delivery.
According to the structural, biochemical properties and intracellular site of origin, EVs can be divided into various types: microvesicles (MVs), membrane particles, exosome-like particles, apoptotic vesicles and exosomes[13, 107]. In fact, it is still a challenge to tell apart one EV from another because of their overlapping biophysical characteristics although some discriminating markers have been reported[107, 111]. In this part, we discussed the EVs in two parts, shedding vesicles (or ectosomes) and exosomes, by simply considering the intracellular origin: shedding vesicles are directly derived from the plasma membrane while exosomes are originated from late endosomes and secreted endogenously from multivesicular bodies (Figure 5a)[18, 20]. Except for the two, there is another kind of nano-sized cell membrane vesicles which are prepared artificially[26, 112, 113]. They were also discussed here along with the EVs.
Fabrication and characterization of shedding vesicles and exosomes. (a) Intracellular origin of EVs. Shedding vesicles derive directly from the cell membrane. Exosomes originate from the cell membrane through the endosomal pathway and form via inward budding of the limiting membrane of the multivesicular body, a late endosomal compartment. Exosomes are secreted via fusion of multivesicular bodies with the plasma membrane. (b) TEM imaging, (c) purification procedure and (d) membrane protein analysis of exosomes. Reproduced and modified with permission[18, 114, 115]. Copyright 2014, Elsevier, Nature.
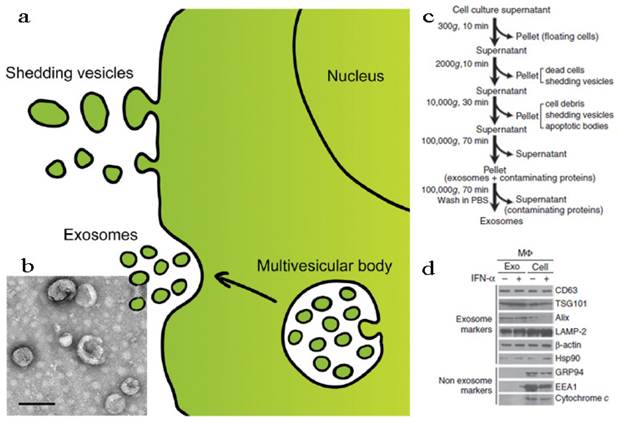
Shedding Vesicles
RBC-derived Microvesicles
It has been established that nano-sized vesicles with denatured hemoglobin, membrane proteins and/or toxicant would be generated in RBC aging processes or by factitious factors such as in vitro osmotic and oxidative stress[116-118]. The RBC aging is a form of apoptosis so the resultant RBC MVs are likely to be phagocytosed by RES. The advantage of RBC MVs is their nano-size, making them able to penetrate certain tissue and achieve the intracellular drug delivery. In one work, ultrasmall superparamagnetic iron oxide (USPIO) particles were loaded into the RBC MVs via incubation method[118]. The USPIO-loaded RBC MVs can be efficiently uptaken by MSCs and hence used to label and trace MSCs by MRI both in vitro and in vivo with minimum adverse effect.
The RBC MVs can also be obtained as the following steps[112, 113]. The RBC membrane was first collected via hypotonic treatment as discussed above, followed by series of membrane extrusion. Two approaches were developed to load cargoes into the RBC MVs. One was co-incubating the payloads with the final MVs directly. The other was loading the drug into RBC during the hypotonic treatment. The latter was thought to be with a higher loading efficiency because the pores on cell membrane allowed the drug to enter the cells more easily. The maximum loading efficiency of these two approaches was 7% and 48%, respectively, when using fasudil as the model drug[113]. ICG, an FDA-approved NIR-transducing molecule, could be also loaded into the RBC MVs via co-incubation to integrate optical imaging and photothermal destruction of tumors[112]. Compared with free ICG, the circulation time and cell phagocytosis efficiency of ICG-loaded RBC MVs had been increased. Accordingly, the photothermal destruction ability was improved by three times, indicating the great potential in further theranostic application.
Tumor Cell-derived Microparticles
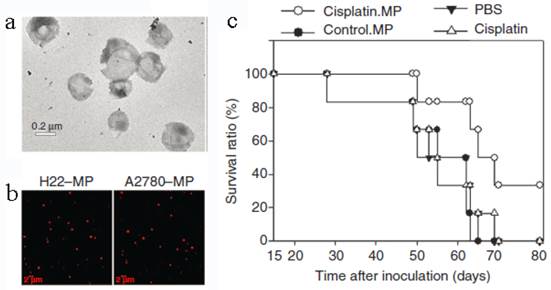
Tumor cell-derived MPs were recently developed to deliver chemotherapeutic drugs[25]. Typically, mouse hepatocarcinoma tumor cell line H22 (5×106) was incubated with methotrexate (MTX, 10 mg/mL) and then exposed to ultraviolet irradiation (UBV, 300 J/m2) for 1 h to induce the apoptosis. After 12 h, the drug-loaded MPs (3×105) with the diameter of 100-1000 nm were collected by multistep centrifugation. The cell line A2780 and the drug cisplatin, paclitaxel (PTX) or DOX can be used to generate MPs in the similar way (Figure 6). The resultant MPs contained a lot of natural biologicals, such as cytochrome c, caspases 3/9, histones and DNA pieces. They also exhibited higher tumor treatment efficacy and much lower side effects than free chemotherapeutic drugs. The mechanism might be that the MPs could be efficiently uptaken by tumor cells and thus directly deliver drugs into the cells with a relatively high concentration. An interesting phenomenon was that the tumor cells killed by MPs can produce a new generation of drug-loaded MPs. These second-generation of MPs were also toxic to tumor cell and then induced the third-generation of MPs. This “domino-like” tumor killing behavior of MPs could improve the anti-tumor efficiency. This study may propose a new way for chemotherapeutic drug delivery.
Tumor cell-derived MPs for anti-cancer drug delivery. (a) TEM image of MPs. (b) Tumor cells treated with chemotherapeutic drugs released drug-packaging MPs. H22 cells or A2780 cells were incubated with 100 μg/mL doxorubicin and irradiated with UBV. MPs were isolated and observed under two-photon laser scanning fluorescence microscope. DOX was shown as the red color. (c) Cisplatin-packaging MPs inhibited ovarian cancer growth in SCID mice. Survival observation A2780 cells were i.p. injected to SCID mice (n=6 per group). From the next day, mouse was administered with MPs or with single cisplatin (2 μg/g) or PBS by i.p. injection once per day for continuous 5 days. Reproduced with permission[25]. Copyright 2014, Nature.
MSC-based Microvesicles
MSCs containing therapeutic cargos could treat the diseases efficiently, but the missing cells might cause some adverse effects and even genetic risks. To overcome these shortcomings, a recent work attempted to remove the nucleic matter and homogenize the cells to form MSC NGs (Figure 7)[26].
MSC NGs can specifically target tumor. (a) Representative Cryo-TEM images (n > 3) of hMSC-NGs (b) Binding of NGs (white arrows) to PC3 cells (NGs, red (DiI); cell, green (GFP); nucleolus, blue (DAPI)) evaluated using confocal microscopy. (c) In vivo prostate tumor targeting, and biodistribution of hMSC-NGs. Harvested tumors were dissociated into single cells and analyzed by flow cytometry for human CD90 as an indicator of NG fusion. Positive expression is calculated in the designated markers normalized to the untreated control group (black curves) based on the test events following ip (blue curves) or iv (purple curves) administration. Reproduced with permission[26]. Copyright 2014, ACS.
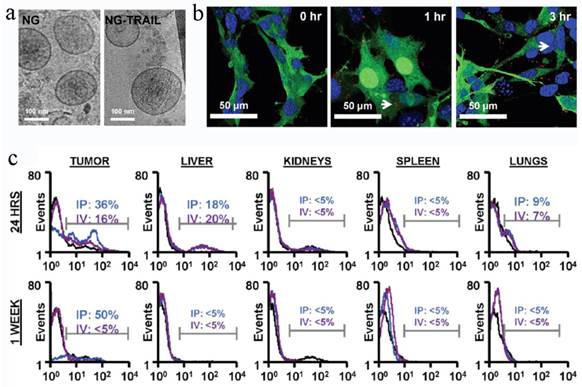
To prepare the NGs, the cells were homogenized after being harvested and re-suspended in cold Tris-Magnesium buffer (TM-buffer, pH 7.4), followed by series of steps of hypotonic treatment, centrifugation and washing. The pellet was then re-suspended, extruded 11 times through 0.4 μm polycarbonate membranes, ultra-centrifuged and re-suspended in TM-buffer (pH 8.6). The final NGs was ~180 nm and retained all the MSC surface markers (>50%), such as CD29, CD44, CD90, CD105. The sTRAIL was loaded into the NGs during the extrusion step with EE of 30%. Blank MSC NGs were non-cytotoxic against human prostate cancer cell-line PC3 cells while exhibiting time-dependent cytotoxicity after sTRAIL loading. In vivo test revealed a remarkable NGs accumulation in tumors after ip or iv administration for 24 h and the targeting effect lasted for more than a week in ip-treated mice. Accordingly, the sTRAIL-loaded NGs caused significant tumor cell apoptosis with a percentage of 89% and inhibited tumor growth by more than 70% after single ip administration compared with free sTRAIL. More importantly, the treatment was tumor-specific, but not species-specific. The NGs derived from either human or rats were found to be with similar treatment efficiency. Recently, Pascucci et al reported PTX loaded MSC MVs by directly incubating MSCs with PTX which exhibited a strong anti-tumor activity on CFPAC-1 cell line[119]. Considering the MSC-based vesicle platform was hypoimmunogenic and specific tumor targeting, it's worth continuing these studies and investigating the precise targeting mechanism to fulfill medical application in future.
Other Extracellular Vesicles
Besides the aforementioned EVs, vesicles generated from other cells, such as vascular endothelial cells and bacteria, were investigated as the drug carrier[19, 120-122].
EV-based nanoplatform encapsulating a set of iron oxide NPs (IONPs) with/without quantum dots (QDs) or Au NPs, had been prepared by Silva et al[120]. The model cells, human umbilical vascular endothelial cells (HUVEC) were cultured with the NPs and then starved in serum-free medium for 2 days. The NP-loaded EVs were finally purified via magnetic sorting. The benefits of this biogenic platform were that we can choose proper parent cell with certain surface features to target desired tissue and then achieve the MRI and/or fluorescence imaging, as well as thermotherapy of certain disease.
Bacterial outer membrane vesicles (OMVs) were another important MVs which were derived from Gram-negative bacteria with a similar structure of EVs, such as nano-size (20-250 nm diameter), lipid bilayer, periplasm and contained membrane protein[19]. These OMVs were usually pathogenic due to the toxins and lipopolysaccharides (LPS) on the surface, which greatly limited their clinic application. However, the OMVs come from attenuated bacterial strains showed no significant pathogenicity or toxicity. They were employed as the vaccines or therapeutic agents carrier considering their various endogenous antigens and the natural self-adjuvanticity[19, 121, 122]. A pioneer work developed OMVs using bioengineered bacteria (Escherichia coli) with an anti-HER2 affibody (AffiHER2OMVs) on the membrane surface[123]. OMVs with low immunogenicity could target and kill cancer cells in a cell-specific manner by delivering the siRNA against kinesin spindle protein (KSP-siRNA). The electroporation method was applied to load the siRNA into AffiHER2OMVs (AffiHER2OMVsiRNA) with an EE around 15%. Even so, the amount of siRNA in the AffiHER2OMVs was sufficient to exert cytotoxic effects against the Her2-positive tumor cells because AffiHER2OMVsiRNA were found to selectively accumulate in the tumor tissue after iv administration. Compared with free siRNA and nontargeted OMVsiRNA, the tumor growth inhibition in AffiHER2OMVsiRNA group was more arrestive accompanying with a significant reduction in KSP protein levels. This genetically engineered OMV was low endotoxic, inflammatory and immunological and thus considered as a safe platform for cancer therapy.
Exosomes
Exosomes are naturally occurring cup-shaped membrane vesicles with a diameter less than 100 nm (Figure 5b)[17, 18, 107, 108]. They can be collected via multi-steps centrifugal separation (Figure 5c). The canonical exosomes are intracellular origin, rich in cholesterol, sphingomyelin, ceramide, and with the protein markers of tetraspanins (CD63, CD9) and TSG101 (Figure 5d). The function of exosomes is mainly to mediate biological immune response, as well as to endow recipient cells some new functional properties[107, 108]. Considering exosomes are natural vehicles of protein and genetic materials (e.g. miRNA) with targeting properties, these vesicles could be excellent candidates for delivery of exogenous therapeutic agents, especially when genetic engineering technology was induced into the modification of exosomes. In the following sections, these two main applications of exosomes, immunomotherapy and drug delivery, were discussed.
Exosomes for Immunotherapy
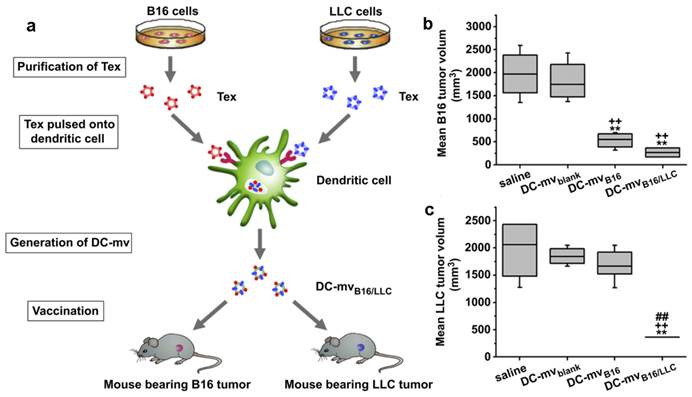
Exosomes come from different cells may play a different role in immunomodulation [107, 108]. Usually, exosomes released by DCs, B cells or macrophages carried both antigenic material and peptide-MHC complexes so they can present exogenous antigens to antigen presenting cells (APCs) for triggering immune responses. Tumor specific peptides pulsed DCs can produce exosomes which can present these peptides to the immune system and induce specific cytotoxic T lymphocyte (CTL)-like responses to inhibit tumor growth[124-128]. Even in AIDS patients with DC dysfunction and CD4+ T cell deficiency, OVA and pcDNAgp120 engineered DC-derived exosomes (Dex) can still induce OVA or pcDNAgp120 specific CTL responses, and cause long-term immunity against OVA-expressing BL6-10(OVA) or Gp120-expressing B16 melanoma[129]. Compared with other DDS, Dex can be used in anti-tumor immunity with minimum side effects[108, 114]. This strategy has been raising the possibility of curing different types of cancers via the patient's immune system. It was supported by some clinical trials, such as Dex cancer vaccines for metastatic melanoma and non-small cell lung cancer treatment[130, 131]. However, the Dex discussed above was site-to-site. That is, one Dex was developed to treat a special cancer. To expand the application of these therapeutic Dex vaccines, a new DC-mvB16/LLC vaccine was developed by pulsing DCs with two kinds of tumor-derived exosomes (Tex) from melanoma (B16) and Lewis lung carcinoma (LLC) simultaneously (Figure 8)[132]. The DC-mvB16/LLC vaccine showed higher efficacy on tumor treatment than single-antigen containing exosomes (DC-mvB16 or DC-mvLLC). This synergistic effect might result from the cross-protection initiated by the shared and specific tumor antigens from B16 and LLC tumor cells.
DC-mvB16/LLC vaccine induced strong lymphocytotoxicity against both B16 and LLC tumor. (a) Schematic diagram of methods used to generate DC-mvB16/LLC dual vaccine and assess anti-tumor effect. Tumor sizes of (b) B16 or (c) LLC at day 30 after tumor cell challenge. Mice were vaccinated twice at week intervals. Mice were then injected with tumor cells 7 days after the last vaccination. Tumor volume was determined at the end of the study (day 30). Significant difference vs. saline group (**p<0.05). Significant difference vs. DC-mvblank group (++p<0.05). Significant difference vs. DC-mvB16 group (##p<0.05). Reproduced with permission[114]. Copyright 2014, Elsevier.
Exosomes for Drug Delivery
Exosomes are with a nano-size structure which are stable in the circulation and can thus achieve a passive targeting via the enhanced permeation retention (EPR) effect[107, 110, 133]. Additionally, exosomes secreted by different cells are able to target specific tissue/cell. The exploratory work about exosomal DDS was reported by Zhang's group[110]. Curcumin was loaded into the EL-4 (mouse lymphoma cell line) derived exosomes by a direct mixing method. The curcumin loaded exosomes can target CD11b+Gr-1+ cells and thus promote the cell apoptosis, resulting in an enhanced anti-inflammatory activity. They further took advantage of this exosomal DDS to deliver anti-inflammatory drugs (curcumin or JSI124) to brain. These drug loaded exosomes could rapidly come across the BBB through a non-invasive intranasal route, target the microglial cells and finally achieve great therapeutic effects[109].
To improve their targeting ability, the exosomes were modified artificially in many ways[132, 134, 135]. A conceptual work was conducted by Alvarez-Erviti et al (Figure 9)[134]. Exosomes generated from immature DCs were hypotoxic and hypo-immunogenic since the low expression of immunostimulatory markers, such as MHC II, CD80 and CD86 on the surface. A peptide derived from the Rabies Virus Glycoprotein (RVG) was fused to Lamp2b (an exosomal membrane protein). These RVG-exosomes could bind to nicotinic acetylcholine receptor expressed on the BBB so as to increase the brain targeting ability. Through this carrier, siRNA was specifically delivered to the neurons, microglia, and oligodendrocytes in the brain with ~ 60% decreased expression of target gene. A subsequent study by Ohno et al developed EGF or GE11 peptide fused exosomes to deliver microRNA (miRNA) to epidermal growth factor receptor (EGFR)-expressing breast cancer cells[135]. As expected, the miRNA was successfully delivered to the xenograft cancer tissue via the exosomes after iv administration. Besides, chemotherapeutic drug, DOX, was also loaded into bioengineered iRGD-Lamp2b exosomes via electroporation (EE, ~ 20%) to target αν integrin-positive cancer cells[114]. This modified exosome showed specific tumor targeting and a powerful tumor growth inhibition.
Targeting peptide expressed with Lamp2b was expressed on the external surface of exosomes. (a) Schematic representation of production, harvest and re-administration of targeted self-exosomes for gene delivery. (b) Size distribution of RVG exosomes as measured by NTA peaking at 88 nm diameter. (c) Electron micrograph of phosphotungstic acid stained RVG exosomes. Reproduced with permission[134]. Copyright 2014, Nature.
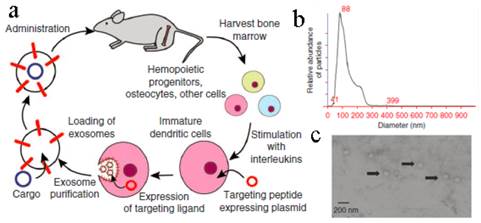
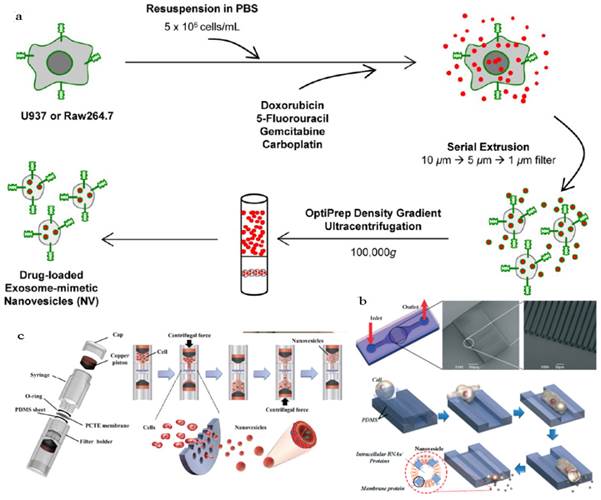
An obstacle for the development of exosome based DDS is that mammalian cells released relatively low quantities of exosomes (e.g., ~ 0.1 μg protein per 106 DCs) and the purification was a little complicated[18, 136]. To improve the yield of exosomes, Jang et al developed exosome-mimetic vesicles (U937NV) to deliver chemotherapeutic drug through a series of extruding steps followed by density gradient ultracentrifugation for collection and purification (Figure 10a)[137]. The yield of DOX-loaded exosome-mimetic vesicles (U937NVDox) was 203 μg of total protein and 210×109 particles per 1×107 U937 cells, which was more than 100-fold of natural produced exosome (U937EXO, only 1.74 μg total protein and 2×109 particles). Moreover, the size, morphology, and protein marker (CD63, Tsg 101 and Moesin) contents of U937NV were almost the same as those of U937EXO. It was also found that U937NVDox had the similar inhibiting ability against tumor growth with U937EXODox. Park's group described the large-scale fabrication methods of NVs via microfluidics or centrifugation device (Figure 10b&c)[138, 139]. The yield of the resulted NVs was 20.18 μg of total protein per 1.5×106 murine embryonic stem (ES) cells and 952 μg of total protein per 1×108 ES cells, respectively. This may promote the comprehensive research on exosomes based DDS.
Generation of exosome-mimetic nanovesicles (NV) in different ways (a) Schematic illustration of the procedure for the generation of chemotherapeutics-loaded NV. (b) Schematic illustration of microfluidic fabrication of NV. (c) Sectional view of centrifuge device and schematic process of NV generation. Reproduced with permission[137-139]. Copyright 2014, ACS, RSC.
Cell Membrane Coated Particles for Drug Delivery
Cell membrane based vesicles are natural sourced drug carriers which have been used in various systems. However, due to their generation method and liposome-like structure, it's hard for them to load hydrophobic agents, co-deliver drugs with different properties or fulfill the well-controlled release. It is well known that polymeric/inorganic NPs are able to achieve aforementioned properties, but they are always cleared by the body quickly owing to their foreign nature. Similar to lipid-enveloped hybrid NPs, researchers have developed a series of cell membrane coated NPs that combine the advantages of the two and realize various functions[5, 9].
RBC Membrane Coated NPs
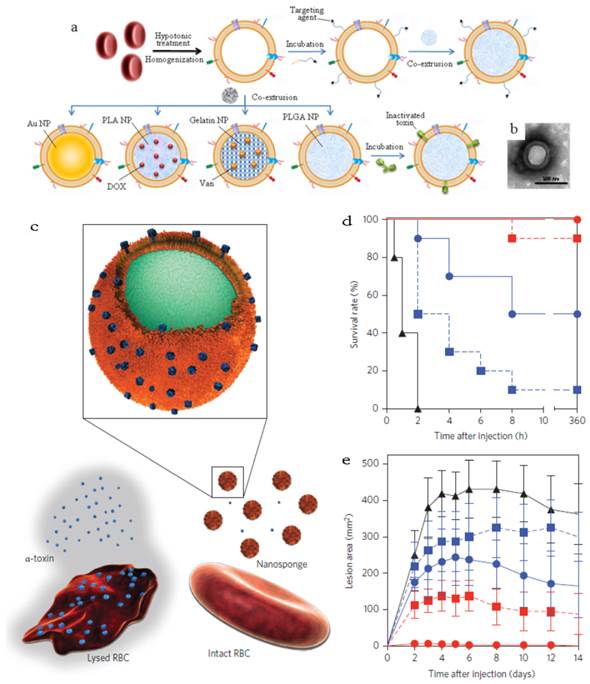
RBC membrane (RBCm) coating technology was developed by Zhang's group since 2011 followed by being applied in many systems with different purposes (Figure 11a)[27-33]. These novel RBC membrane cloaked NPs (RBC-NPs) were fabricated via a three-step method. Simply, RBCs were separated from the whole blood by centrifugation and treated with hypotonic solution to remove the hemoglobin. The RBCm vesicles with the size of about 100 nm could be easily prepared by extrusion through polycarbonate porous membranes (100-400 nm). The last step was co-extruding the RBCm vesicles and NPs several times to fuse the two together. A lipid bilayer shell (7-8 nm) was observed out of the NP core and the zeta potential of the coated NPs was close to the value of RBCm vesicles. Both the results confirmed the successful coating of RBC membrane.
RBCm-cloaking NPs systems. (a) Fabricating illustration and model structure of RBCm-cloaking NPs: RBCm-cloaking Au NPs, DOX-loaded RBCm-cloaking PLA NPs, Van-loaded RBCm-cloaking gelatin NPs, RBCm-cloaking PLGA NPs, Targeting RBCm-cloaking PLGA NPs and toxin-detainment RBCm-cloaking PLGA NPs. (b) TEM image of folic acid modified RBCm-cloaking PLGA NPs (unpublished data). (c) Schematic structure of toxin nanosponges and their mechanism of neutralizing PFTs. The nanosponges consist of substrate-supported RBC bilayer membranes into which PFTs can incorporate. After being absorbed and arrested by the nanosponges, the PFTs are diverted away from their cellular targets, thereby avoiding target cells and preventing toxin-mediated haemolysis. (d) Survival rates of mice over a 15-day period following intravenous injections of 120 μg/kg Hla on day 21 via the tail vein (n=10). Unvaccinated mice were used as negative control and mice vaccinated with heat-treated Hla served as positive controls. Both the prime-only schedule and prime-boost schedule were conducted. (e) Comparison of skin lesion size following subcutaneous injections of 5 μg of Hla on day 21. Lesion size was measured for 14 days following the challenge (n=6). (Unvaccinated mice (black triangles, solid line) and mice vaccinated with heattreated Hla (prime; blue squares, dashed line), nanotoxoid(Hla) (prime; blue circles, solid line), heat-treated Hla (prime+boost; red squares, dashed line) or nanotoxoid(Hla) (prime+boost; red circle, solid line) received intravenous or subcutaneous administration of Hla.) Reproduced with permission[31, 32]. Copyright 2014, Nature.
There are several interfacial aspects which will affect the membrane/particle assembly process, including membrane/polymer ratio, surface charge and NP diameter[28]. For 1 mg PLGA NPs with a diameter of 100 nm, 85 μL of mouse blood was necessary to completely shield the particle surfaces. A recommended ratio by the authors was 100 μL blood/mg polymer. For the cores (ranged from 65 to 340 nm) with negative surface charge (e.g. PLGA NPs), the electrostatic repulsion between the NP cores and asymmetrically charged RBC membrane is not only a key factor in fabricating the RBCm-coated structure, but also a guarantee for the right membrane structure. But if the surface charge of NP cores was positively charged, significant aggregation occurred because the electrostatic interactions would collapse the fluidic lipid bilayer and impede the local arrangement necessary for lipid coverage. The NP cores were effectively coated as proved by TEM. This RBCm coating strategy was also applied in inorganic NP systems. Au NPs or Au nanocages with the diameter of 70 nm were successfully covered by RBCm[30, 140]. Similar to polymeric NPs, increased particle size and decreased zeta potential of the NPs were observed after RBC cloaking. All the results revealed the efficiency and flexibility of this membrane coating technique, as well as the great potential to functionalize various nano-sized system devices.
It was worth noting that this RBC membrane on the NPs was with right-side-out membrane orientation (84%, based on the results of the fluorescence quenching test) and with almost the same protein content and glycan density as natural RBC[27, 28, 30, 141, 142]. The glycans and proteins (such as CD 47) on the surface increased the stability of the NPs in physiological conditions. Glycans are highly hydrophilic and contribute to the steric stabilization of cells while CD 47 is a signal of 'don't eat me'[142]. Both of them leaded to a long circulation property of RBC-NPs in a mouse model (t1/2 39.6 h), which was much higher than that of PEG-based NPs (t1/2 15.8 h)[27]. To evaluate the therapeutic effect of the RBC-NPs, DOX was loaded into the NPs by either chemical conjugation or physical encapsulation[29]. Compared to PEG-PLA NPs, RBC-NPs showed a lower drug release ratio, indicating the diffusion barrier function of RBC membrane. They also exhibited much higher cytotoxicity on an acute myeloid leukemia (AML) Kasumi-1 cell line than free DOX. The folate or aptamer AS1411 were further modified on the RBC-NPs by the bio-bridge method using lipid as the insertion linker[143]. Both of the targeting modified NPs showed enhanced cell uptake against KB or MCF-7 cell lines. Another targeting delivery strategy of this RBCm-cloaking system was developed to take use of the enzyme-responsive property. The bacteria in infection site can secrete gelatinase. Therefore, the vancomycin-loaded RBCm-coated gelatin nanogels can specifically degrade and release the antibiotics in the infection site[144]. The required dose of antibiotics can be subsequently reduced with the minimized side effects.
Other than acting as drug carrier, a recent interesting work afforded the RBC-NPs to the function as vaccine (Figure 11b&c)[31]. Seeing that the particle surface was occupied by RBC membrane, the RBCm-NPs would interact with pore-forming toxins (PFTs) like other cellular membranes and lock the absorbed toxins within the RBCm-coated structure (about 85 a-toxin, 30 streptolysin-O or 850 melittinmonomers per NP) via the anchoring effect of the core-shell structure[31, 32]. These toxin absorbing RBCm-NPs were applied as a safe vaccine to deliver PFTs to the immune system without any denaturation treatment[32]. The virulence of staphylococcal a-haemolysin (Hla), a model toxin, was neutralized after entrapped by the RBCm-NPs without leakage for more than 48 h. Compared with traditional attenuated vaccine (such as heat-treated Hla), the Hla-RBCm-NPs were not only non-toxic, but also with a strong capability of eliciting Hla-specific antibody responses (7- and 15-fold of heat-treated Hla in the prime only and prime-boost vaccinations, respectively) for over 150 days. The protective immunity also proved that the Hla-RBCm-NPs resulted in a much longer survival rate (intravenous injection) and less lesion area (subcutaneous injection) than heat-denatured toxin.
Other Eukaryocyte Membrane Coated Particles
Compared with RBC, the structure of eukaryocyte was more complicated. Considering various physiological functions of eukaryocyte, it's meaningful to develop some biomimetic particles based on the eukaryocyte membrane to realize some different functions[34, 145]. The general preparation step is close to that of RBCm-coated NPs, but a bit more complicated in detail. The eukaryocyte membranes can be collected by ultracentrifugation through a discontinuous sucrose density gradient after hypotonic treatment. Then the membrane-coated structure was fabricated after co-extrusion or incubation with the cores. The components from nucleus, organelle and cytosol were found to be low on the formulated NPs, which may reduce the genetic risk of the technology, especially in the case of taking some pathological cells as the membrane source[34].
To date, two kinds of eukaryocyte membrane coated particles have been investigated, cancer cell membrane-coated PLGA NPs and leukocyte membrane-coated nanoporous silicon particles[34, 145]. Cancer cell membrane-coated NPs were developed to deliver tumor-associated antigens directly to DCs after taking up, processing and presenting to induce specific anti-tumor immune response[34]. With monophosphoryl lipid A (MPLA) entrapment, the NPs would induce the maturation of DCs and subsequent immune activation. Furthermore, this kind of way can realize a new strategy of unique tumor targeting according to the inherent homotypic binding capability among tumor cells.
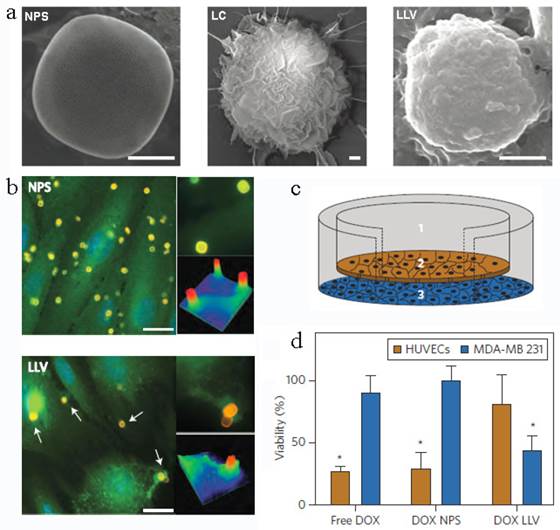
Another leukocyte membrane-coated nanoporous silicon particles were developed to mimic some bio-functions of leukocyte for tumor treatment (Figure 12a)[145]. The membranes were coated onto the Si particles through electrostatic and hydrophobic interactions between the positively charged Si particles and negatively charged cell membranes after incubation overnight at 4 oC. Accordingly, the zeta potential of the particles changed from +7.41 mV to -26 mV. The functional proteins or molecules, such as CD45, CD11a and glycans, were all maintained on the particle surface, which was helpful to prevent the internalization/uptake by macrophages, phagocytic cells or vein endothelial cells but preferentially bind to and transport through inflamed endothelium (Figure 12b). As a result, this leukolike vectors can extend the DOX circulation time, improve the drug accumulation in tumor, and thus enhance the therapeutic efficiency against cancer treatment after DOX-loading. This vector took advantages of the communication of leukocyte with endothelial cells through receptor-ligand interactions (Figure 12c&d). This biomimetic system had paved a new way to transport drugs through vascular barriers.
Leukocyte-like vectors. (a). SEM images of bare NPS, leukocyte (LC) and an NPS camouflaged with leukocyte-derived membranes LLV, Scale bar 1μm. (b) After treatment with NPS and LLV (particles, yellow), ICAM-1 clusters (green, arrows) on the surface of TNF-α stimulated HUVEC cells only in proximity to the LLV. Scale bars, 15 mm. High-magnification images and colorimetric analysis of receptor clustering is shown to the right (particle size, 3 mm). (c) Schematic of a transwell chamber for assaying transport across an endothelial monolayer. Particles were counted in the upper chamber (1, supernatant), endothelial layer (2, intracellular) and lower chamber (3, filtered). (d) Studies of HUVEC (endothelial) and MDA-MB-231 (cancer) cell viability in a transwell system. LLV-loaded doxorubicin (DOX LLV) showed enhanced tumor cell killing and decreased endothelial cell death following 48 h incubation compared with free doxorubicin (free DOX) and NPS-loaded doxorubicin (DOX NPS). Error bars represent standard deviation. *P<0.05. Reproduced with permission[145]. Copyright 2014, Nature.
In a word, these cell membrane coated particles were able to perform the natural properties and even the bio-functions originated from their parent cells. The concept needs further investigations for applying bioactive cellular components of specific functional cells to disease treatment via synthetic and natural processing method.
Future Prospects
Whole cell and EVs have showed many advantages, especially in the concept of biocompatibility and targeting capability. Current synthetic DDS, including inorganic and polymeric NPs, were intrinsically foreign materials and with potential toxicity and immunogenicity although some of them have been approved in clinic. As comparison, whole cell or EVs, which are endogenic, are considered as much more biocompatible with multi-type of bio-functions along with the parent cells. When it comes to the concept 'individual therapy', where the cells are isolated from the patients themselves, the cell or cell membrane-based DDSs are non-toxic and non-immunogenic. For the EVs and cell membrane coated NPs, the retention of the membrane proteins and nano-size structure make them more feasible to achieve site-specific drug delivery than whole cell as drug carrier.
However, there are still some limitations existing in the clinical translation of such research area. As the whole cell delivery system, it's hard for carrier RBC to fulfill the location release except for RES targeting. While for the stem cells, the challenge is still concerned on their genetic risk which may need further investigations. In terms of the EVs and cell membrane coated NPs, the studies on them are far more than sufficient. The potential unwanted side effects of these endogenous carriers are still under investigation. The exorbitant costs may be another problem. It costs about US$93,000 per patient for three doses of DC-based cancer vaccine, Provenge® with modest increase in survival time (4.1 months)[108]. When it comes to EV-based DDS, the fee may be higher than Provenge® due to their low recovery. In view of this, the large scale generation of the EVs is meaningful. Heterogenous EVs from other species with similar bio-functions are also helpful to lower the cost. Another way may be taking use of multifunctional modifications on the EVs and easily available materials, such as RBC MVs or bacterial OMVs. By taking advantage of proteomics analysis, one can identify and purify the key protein and then build some biomimetics, such as the RAFTsome, which can stimulate T cells and prime immune responses like exosomes[15]. It's thus possible to modify natural EVs or create biomimetic NPs in order to achieve some physiological functions. The multidisciplinary investigations with the help of biology, medical science and engineering will be essential to develop a more efficient cell or EVs-based DDS with high safety and therapeutic efficiency.
Acknowledgements
This research is supported by National Basic Research Program of China (2012CB932501), Nature Science Fund of China (21204024 and 81373360), China Postdoctoral Science Foundation funded project (2013T60722), the Fundamental Research Funds for the Central Universities (2014TS091 and 2014QN134) and Chutian Scholar Award.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chertok B, Webber MJ, Succi MD. et al. Drug delivery interfaces in the 21st century: From science fiction ideas to viable technologies. Mol Pharm. 2013;10:3531-43
2. Hubbell JA, Langer R. Translating materials design to the clinic. Nat Mater. 2013;12:963-6
3. Hu C-MJ, Fang RH, Luk BT. et al. Polymeric nanotherapeutics: clinical development and advances in stealth functionalization strategies. Nanoscale. 2013;6:65-75
4. Wicki A, Witzigmann D, Balasubramanian V. et al. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J Control Release. 2015;200:138-157
5. Tan SW, Li X, Guo Y. et al. Lipid-enveloped hybrid nanoparticles for drug delivery. Nanoscale. 2013;5:860-72
6. Bamrungsap S, Zhao ZL, Chen T. et al. Nanotechnology in therapeutics: a focus on nanoparticles as a drug delivery system. Nanomedicine. 2012;7:1253-71
7. Al-Jamal WT, Kostarelos K. Liposomes: from aclinically established drug delivery system to a nanoparticle platform for theranostic nanomedicine. Acc Chem Res. 2011;44:1094-104
8. Muthu MS, Feng S-S. Theranostic liposomes for cancer diagnosis and treatment: current development and pre-clinical success. Expert Opin Drug Del. 2013;10:151-5
9. Gao W, Hu C-MJ, Fang RH. et al. Liposome-like nanostructures for drug delivery. J Mater Chem B. 2013;1:6569-85
10. Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomed. 2006;1:297
11. Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4:145-60
12. Bangham AD. Surrogate cells or Trojan horses. The discovery of liposomes. BioEssays. 1995;17:1081-8
13. van der Meel R, Fens MHAM, Vader P. et al. Extracellular vesicles as drug delivery systems: Lessons from the liposome field. J Control Release. 2014;195:72-85
14. Sicchierolli S, Carmona-Ribeiro A. Incoporation of the cholera toxin receptor in phospholipid-covered polystyrene microspheres. Colloid Surface B. 1995;5:57-61
15. Ding Q, Chen J, Wei X. et al. RAFTsomes containing epitope-MHC-II complexes mediated CD4+ T cell activation and antigen-specific immune responses. Pharm Res. 2013;30:60-9
16. Hamidi M, Zarrin A, Foroozesh M. et al. Applications of carrier erythrocytes in delivery of biopharmaceuticals. J Control Release. 2007;118:145-60
17. Sun D, Zhuang X, Zhang S. et al. Exosomes are endogenous nanoparticles that can deliver biological information between cells. Adv Drug Delivery Rev. 2013;65:342-7
18. van Dommelen SM, Vader P, Lakhal S. et al. Microvesicles and exosomes: Opportunities for cell-derived membrane vesicles in drug delivery. J Control Release. 2012;161:635-44
19. Yoo J-W, Irvine DJ, Discher DE. et al. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat Rev Drug Discov. 2011;10:521-35
20. Choi D-S, Kim D-K, Kim Y-K. et al. Proteomics, transcriptomics and lipidomics of exosomes and ectosomes. Proteomics. 2013;13:1554-71
21. Witwer KW, Buzás EI, Bemis LT. et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracellular Vesicles. 2013;2:20360
22. Millan CG, Marinero MaLS, Castaneda AZ. et al. Drug, enzyme and peptide delivery using erythrocytes as carriers. J Control Release. 2004;95:27-49
23. Hu C-MJ, Fang RH, Zhang L. Erythrocyte-inspired delivery systems. Adv Healthc Mater. 2012;1:537-47
24. Stuckey DW, Shah K. Stem cell-based therapies for cancer treatment: separating hope from hype. Nat Rev Cancer. 2014;14:683-91
25. Tang K, Zhang Y, Zhang H. et al. Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nat Commun. 2012;3:1282
26. Toledano Furman NE, Lupu-Haber Y, Bronshtein T. et al. Reconstructed stem cell nanoghosts: A natural tumor targeting platform. Nano Lett. 2013;13:3248-55
27. Hu C-MJ, Zhang L, Aryal S. et al. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc Natl Acad Sci USA. 2011;108:10980-5
28. Luk BT, Hu C-MJ, Fang RH. et al. Interfacial interactions between natural RBC membranes and synthetic polymeric nanoparticles. Nanoscale. 2014;6:2730-7
29. Aryal S, Hu C-MJ, Fang RH. et al. Erythrocyte membrane-cloaked polymeric nanoparticles for controlled drug loading and release. Nanomedicine. 2013;8:1271-80
30. Gao W, Hu C-MJ, Fang RH. et al. Surface functionalization of gold nanoparticles with red blood cell membranes. Adv Mater. 2013;25:3549-53
31. Hu C-MJ, Fang RH, Copp J. et al. A biomimetic nanosponge that absorbs pore-forming toxins. Nat Nanotechnol. 2013;8:336-40
32. Hu C-MJ, Fang RH, Luk BT. et al. Nanoparticle-detained toxins for safe and effective vaccination. Nat Nanotechnol. 2013;8:933-8
33. Copp JA, Fang RH, Luk BT. et al. Clearance of pathological antibodies using biomimetic nanoparticles. Proc Natl Acad Sci USA. 2014;111:13481-6
34. Fang RH, Hu C-MJ, Luk BT. et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14:2181-8
35. Batrakova EV, Kabanov AV. Cell-mediated drug delivery to the brain. J Drug Deliv Sci Tec. 2013;23:419-33
36. Batrakova EV, Gendelman HE, Kabanov AV. Cell-mediated drug delivery. Expert Opin Drug Del. 2011;8:415-33
37. Godfrin Y, Horand F, Franco R. et al. International seminar on the red blood cells as vehicles for drugs. Expert Opin Biol Ther. 2012;12:127-33
38. Kolesnikova TA, Skirtach AG, Möhwald H. Red blood cells and polyelectrolyte multilayer capsules: natural carriers versus polymer-based drug delivery vehicles. Expert Opin Drug Del. 2013;10:47-58
39. Tsong TY. Electroporation of cell membranes. Biophys J. 1991;60:297-306
40. Lizano C, Sanz S, Luque J. et al. In vitro study of alcohol dehydrogenase and acetaldehyde dehydrogenase encapsulated into human erythrocytes by an electroporation procedure. Biochimica et Biophysica Acta-General Subjects. 1998;1425:328-36
41. Phua KK, Boczkowski D, Dannull J. et al. Whole blood cells loaded with messenger RNA as an anti-tumor vaccine. Adv Healthc Mater. 2013:837-42
42. Dong Q, Jin W. Monitoring diclofenac sodium in single human erythrocytes introduced by electroporation using capillary zone electrophoresis with electrochemical detection. Electrophoresis. 2001;22:2786-92
43. He H, Ye J, Wang Y. et al. Cell-penetrating peptides meditated encapsulation of protein therapeutics into intact red blood cells and its application. J Control Release. 2013;176:123-32
44. Biagiotti S, Paoletti MF, Fraternale A. et al. Drug delivery by red blood cells. IUBMB Life. 2011;63:621-31
45. Ahn S, Jung SY, Seo E. et al. Gold nanoparticle-incorporated human red blood cells (RBCs) for X-ray dynamic imaging. Biomaterials. 2011;32:7191-9
46. Antonelli A, Sfara C, Manuali E. et al. Encapsulation of superparamagnetic nanoparticles into red blood cells as new carriers of MRI contrast agents. Nanomedicine. 2011;6:211-23
47. Ferrauto G, Delli Castelli D, Di Gregorio E. et al. Lanthanide-loaded erythrocytes as highly sensitive Chemical Exchange Saturation Transfer (CEST) MRI contrast agents. J Am Chem Soc. 2013;136:638-41
48. Wang C, Sun X, Cheng L. et al. Multifunctional theranostic red blood cells for magnetic-field-enhanced in vivo combination therapy of cancer. Adv Mater. 2014;26:4794-802
49. Flower R, Peiretti E, Magnani M. et al. Observation of erythrocyte dynamics in the retinal capillaries and choriocapillaris using ICG-loaded erythrocyte ghost cells. Invest Ophth Vis Sci. 2008;49:5510-6
50. Anselmo AC, Gupta V, Zern BJ. et al. Delivering nanoparticles to lungs while avoiding liver and spleen through adsorption on red blood cells. ACS nano. 2013;7:11129-37
51. Chambers E, Mitragotri S. Prolonged circulation of large polymeric nanoparticles by non-covalent adsorption on erythrocytes. J Control Release. 2004;100:111-9
52. Chambers E, Mitragotri S. Long circulating nanoparticles via adhesion on red blood cells: mechanism and extended circulation. Exp Biol Med. 2007;232:958-66
53. Mai TD, d'Orlye F, Menager C. et al. Red blood cells decorated with functionalized core-shell magnetic nanoparticles: elucidation of the adsorption mechanism. Chem Commun. 2013;49:5393-5
54. Shi G, Mukthavaram R, Kesari S. et al. Distearoyl anchor-painted erythrocytes with prolonged ligand retention and circulation properties in vivo. Adv Healthc Mater. 2014;3:142-8
55. Mukthavaram R, Shi G, Kesari S. et al. Targeting and depletion of circulating leukocytes and cancer cells by lipophilic antibody-modified erythrocytes. J Control Release. 2014;183:146-53
56. Magnani M, Chiarantini L, Mancini U. Preparation and characterization of biotinylated red blood cells. Biotechnol Appl Biochem. 1994;20:335-45
57. Taylor RP, Sutherland WM, Reist CJ. et al. Use of heteropolymeric monoclonal antibodies to attach antigens to the C3b receptor of human erythrocytes: a potential therapeutic treatment. Proc Natl Acad Sci USA. 1991;88:3305-9
58. Kontos S, Kourtis IC, Dane KY. et al. Engineering antigens for in situ erythrocyte binding induces T-cell deletion. Proc Natl Acad Sci USA. 2013;110:E60-E8
59. Corinti S, Chiarantini L, Dominici S. et al. Erythrocytes deliver Tat to interferon-γ-treated human dendritic cells for efficient initiation of specific type 1 immune responses in vitro. J Leukocyte Biol. 2002;71:652-8
60. Shi J, Kundrat L, Pishesha N. et al. Engineered red blood cells as carriers for systemic delivery of a wide array of functional probes. Proc Natl Acad Sci USA. 2014;111:10131-6
61. Hamidi M, Tajerzadeh H, Dehpour A-R. et al. In vitro characterization of human intact erythrocytes loaded by enalaprilat. Drug Deliv. 2001;8:223-30
62. Talwar N, Jain N. Erythrocytes as carriers of primaquine-preparation: characterization and evaluation. J Control Release. 1992;20:133-41
63. Perno C-F, Santoro N, Balestra E. et al. Red blood cells mediated delivery of 9-(2-phosphonylmethoxyethyl) adenine to primary macrophages: efficiency, metabolism and activity against human immunodeficiency virus or herpes simplex virus. Antivir Res. 1997;33:153-64
64. Fraternale A, Casabianca A, Orlandi C. et al. Macrophage protection by addition of glutathione (GSH)-loaded erythrocytes to AZT and DDI in a murine AIDS model. Antivir Res. 2002;56:263-72
65. Deuticke B, Kim M, Zöllner C. The influence of amphotericin B on the permeability of mammalian erythrocytes to nonelectrolytes, anions and cations. BBA-Biomembranes. 1973;318:345-59
66. Ihler GM, Glew RH, Schnure FW. Enzyme loading of erythrocytes. Proc Natl Acad Sci USA. 1973;70:2663-6
67. Ritter SC, Milanick MA, Meissner KE. Encapsulation of FITC to monitor extracellular pH: a step towards the development of red blood cells as circulating blood analyte biosensors. Biomed Opt Express. 2011;2:2012-21
68. Picas L, Rico F, Deforet M. et al. Structural and mechanical heterogeneity of the erythrocyte membrane reveals hallmarks of membrane stability. ACS nano. 2013;7:1054-63
69. Gutiérrez Millán C, Castañeda AZ, Sayalero Marinero ML. et al. Factors associated with the performance of carrier erythrocytes obtained by hypotonic dialysis. Blood Cells Mol Dis. 2004;33:132-40
70. Jordán JA, Alvarez FJ, Lotero LA. et al. In vitro phagocytosis of carrier mouse red blood cells is increased by Band 3 cross-linking or diamide treatment. Biotechnol Appl Biochem. 2001;34:143-9
71. Jordán JA, Alvarez FJ, Lotero LA. et al. In vivo behaviour of rat band 3 cross-linked carrier erythrocytes. Biochimie. 1998;80:325-32
72. Eichler H, Gasic S, Bauer K. et al. In vivo clearance of antibody-sensitized human drug carrier erythrocytes. Clin Pharmacol Ther. 1986;40:300-3
73. Delcea M, Sternberg N, Yashchenok AM. et al. Nanoplasmonics for dual-molecule release through nanopores in the membrane of red blood cells. ACS nano. 2012;6:4169-80
74. Ing R, Segura M, Thawani N. et al. Interaction of mouse dendritic cells and malaria-infected erythrocytes: uptake, maturation, and antigen presentation. J Immunol. 2006;176:441-50
75. Cremel M, Guérin N, Horand F. et al. Red blood cells as innovative antigen carrier to induce specific immune tolerance. Int J Pharma. 2013;443:39-49
76. Hu Y-L, Huang B, Zhang T-Y. et al. Mesenchymal stem cells as a novel carrier for targeted delivery of gene in cancer therapy based on nonviral transfection. Mol Pharm. 2012;9:2698-709
77. Sasportas LS, Kasmieh R, Wakimoto H. et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc Natl Acad Sci USA. 2009;106:4822-7
78. Ito S, Natsume A, Shimato S. et al. Human neural stem cells transduced with IFN-beta and cytosine deaminase genes intensify bystander effect in experimental glioma. Cancer Gene Ther. 2010;17:299-306
79. Dembinski JL, Wilson SM, Spaeth EL. et al. Tumor stroma engraftment of gene-modified mesenchymal stem cells as anti-tumor therapy against ovarian cancer. Cytotherapy. 2013;15:20-32
80. Seo SH, Kim KS, Park SH. et al. The effects of mesenchymal stem cells injected via different routes on modified IL-12-mediated antitumor activity. Gene Ther. 2011;18:488-95
81. Ryu CH, Park SH, Park SA. et al. Gene therapy of intracranial glioma using interleukin 12-secreting human umbilical cord blood-derived mesenchymal stem cells. Hum Gene Ther. 2011;22:733-43
82. Xu G, Jiang X-D, Xu Y. et al. Adenoviral-mediated interleukin-18 expression in mesenchymal stem cells effectively suppresses the growth of glioma in rats. Cell Biol Int. 2009;33:466-74
83. Li L, Guan Y, Liu H. et al. Silica nanorattle-doxorubicin-anchored mesenchymal stem cells for tumor-tropic therapy. ACS nano. 2011;5:7462-70
84. Mooney R, Weng YM, Garcia E. et al. Conjugation of pH-responsive nanoparticles to neural stem cells improves intratumoral therapy. J Control Release. 2014;191:82-9
85. Cao BR, Yang MY, Zhu Y. et al. Stem cells loaded with nanoparticles as a drug carrier for in vivo breast cancer therapy. Adv Mater. 2014;26:4627-31
86. Schnarr K, Mooney R, Weng YM. et al. Gold nanoparticle-loaded neural stem cells for photothermal ablation of cancer. Adv Healthc Mater. 2013;2:976-82
87. Mooney R, Roma L, Zhao DH. et al. Neural stem cell-mediated intratumoral delivery of gold nanorods improves photothermal therapy. ACS Nano. 2014;8:12450-60
88. Huang X, Zhang F, Wang H. et al. Mesenchymal stem cell-based cell engineering with multifunctional mesoporous silica nanoparticles for tumor delivery. Biomaterials. 2013;34:1772-80
89. Mooney R, Weng Y, Tirughana-Sambandan R. et al. Neural stem cells improve intracranial nanoparticle retention and tumor-selective distribution. Future Oncology. 2014;10:401-15
90. Ljujic B, Milovanovic M, Volarevic V. et al. Human mesenchymal stem cells creating an immunosuppressive environment and promote breast cancer in mice. Scientific Reports. 2013;3:2298
91. Karnoub AE, Dash AB, Vo AP. et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557-63
92. Rosland GV, Svendsen A, Torsvik A. et al. Long-term Cultures of Bone Marrow-Derived Human Mesenchymal Stem Cells Frequently Undergo Spontaneous Malignant Transformation. Cancer Res. 2009;69:5331-9
93. Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104:2224-34
94. Wang HY, Chen L. Tumor microenviroment and hepatocellular carcinoma metastasis. J Gastroen Hepatol. 2013;28:43-8
95. Watanabe MAE, Oda JMM, Amarante MK. et al. Regulatory T cells and breast cancer: implications for immunopathogenesis. Cancer Metast Rev. 2010;29:569-79
96. Moon JJ, Huang B, Irvine DJ. Engineering nano- and microparticles to tune immunity. Adv Mater. 2012;24:3724-46
97. Vinogradov S, Warren G, Wei X. Macrophages associated with tumors as potential targets and therapeutic intermediates. Nanomedicine. 2014;9:695-707
98. Akao Y, Iio A, Itoh T. et al. Microvesicle-mediated RNA molecule delivery system using monocytes/macrophages. Mol Ther. 2011;19:395-9
99. Tao YH, Ning MM, Dou HY. A novel therapeutic system for malignant glioma: nanoformulation, pharmacokinetic, and anticancer properties of cell-nano-drug delivery. Nanomed-Nanotechnol. 2013;9:222-32
100. Choi MR, Stanton-Maxey KJ, Stanley JK. et al. A cellular Trojan horse for delivery of therapeutic nanoparticles into tumors. Nano Lett. 2007;7:3759-65
101. Keliher EJ, Yoo J, Nahrendorf M. et al. 89Zr-labeled dextran nanoparticles allow in vivo macrophage imaging. Bioconjugate Chem. 2011;22:2383-9
102. Alizadeh D, Zhang LY, Hwang J. et al. Tumor-associated macrophages are predominant carriers of cyclodextrin-based nanoparticles into gliomas. Nanomed-Nanotechnol. 2010;6:382-90
103. He CB, Hu YP, Yin LC. et al. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31:3657-66
104. Kreiter S, Selmi A, Diken M. et al. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 2010;70:9031-40
105. Van Lint S, Goyvaerts C, Maenhout S. et al. Preclinical evaluation of TriMix and antigen mRNA-based antitumor therapy. Cancer Res. 2012;72:1661-71
106. Sheikh NA, Petrylak D, Kantoff PW. et al. Sipuleucel-T immune parameters correlate with survival: an analysis of the randomized phase 3 clinical trials in men with castration-resistant prostate cancer. Cancer Immunol Immun. 2013;62:137-47
107. Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581-93
108. Tan A, De La Pena H, Seifalian AM. The application of exosomes as a nanoscale cancer vaccine. Int J Nanomed. 2010;5:889-900
109. Zhuang XY, Xiang XY, Grizzle W. et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol Ther. 2011;19:1769-79
110. Sun D, Zhuang X, Xiang X. et al. A novel nanoparticle drug delivery system: the anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol Ther. 2010;18:1606-14
111. Al-Nedawi K, Meehan B, Kerbel RS. et al. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc Natl Acad Sci USA. 2009;106:3794-9
112. Bahmani B, Bacon D, Anvari B. Erythrocyte-derived photo-theranostic agents: hybrid nano-vesicles containing indocyanine green for near infrared imaging and therapeutic applications. Sci Rep-UK. 2013;3:2180
113. Gupta N, Patel B, Ahsan F. Nano-engineered erythrocyte ghosts as inhalational carriers for delivery of fasudil: Preparation and characterization. Pharm Res. 2014;31:1553-65
114. Tian X, Zhu M, Tian Y. et al. A membrane vesicle-based dual vaccine against melanoma and Lewis lung carcinoma. Biomaterials. 2012;33:6147-54
115. Li JH, Liu KC, Liu Y. et al. Exosomes mediate the cell-to-cell transmission of IFN-alpha-induced antiviral activity. Nat Immunol. 2013;14:793-803
116. Bosman G, Werre J, Willekens F. et al. Erythrocyte ageing in vivo and in vitro: structural aspects and implications for transfusion. Transfusion Med. 2008;18:335-47
117. Rho J, Chung J, Im H. et al. Magnetic nanosensor for detection and profiling of erythrocyte-derived microvesicles. ACS nano. 2013;7:11227-33
118. Chang M, Hsiao J-K, Yao M. et al. Homologous RBC-derived vesicles as ultrasmall carriers of iron oxide for magnetic resonance imaging of stem cells. Nanotechnology. 2010;21:235103
119. Pascucci L, Coccè V, Bonomi A. et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: A new approach for drug delivery. J Control Release. 2014;192:262-70
120. Silva AKA, Di Corato R, Pellegrino T. et al. Cell-derived vesicles as a bioplatform for the encapsulation of theranostic nanomaterials. Nanoscale. 2013;5:11374-84
121. Gaillard ME, Bottero D, Errea A. et al. Acellular pertussis vaccine based on outer membrane vesicles capable of conferring both long-lasting immunity and protection against different strain genotypes. Vaccine. 2014;32:931-7
122. Bottero D, Gaillard M, Errea A. et al. Outer membrane vesicles derived from Bordetella parapertussis as an acellular vaccine against Bordetella parapertussis and Bordetella pertussis infection. Vaccine. 2013;31:5262-8
123. Gujrati V, Kim S, Kim S-H. et al. Bioengineered bacterial outer membrane vesicles as cell-specific drug-delivery vehicles for cancer therapy. ACS nano. 2014;8:1525-37
124. Wolfers J, Lozier A, Raposo G. et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med. 2001;7:297-303
125. Simpson RJ, Jensen SS, Lim JWE. Proteomic profiling of exosomes: Current perspectives. Proteomics. 2008;8:4083-99
126. Zeelenberg IS, Ostrowski M, Krumeich S. et al. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res. 2008;68:1228-35
127. Paulis LE, Mandal S, Kreutz M. et al. Dendritic cell-based nanovaccines for cancer immunotherapy. Curr Opin Immunol. 2013;25:389-95
128. Lakhal S, Wood MJ. Exosome nanotechnology: an emerging paradigm shift in drug delivery: exploitation of exosome nanovesicles for systemic in vivo delivery of RNAi heralds new horizons for drug delivery across biological barriers. BioEssays. 2011;33:737-41
129. Nanjundappa RH, Wang R, Xie Y. et al. GP120-specific exosome-targeted T cell-based vaccine capable of stimulating DC- and CD4(+) T-independent CTL responses. Vaccine. 2011;29:3538-47
130. Escudier B, Dorval T, Chaput N. et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of thefirst phase I clinical trial. J Transl Med. 2005;3:10
131. Morse MA, Garst J, Osada T. et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med. 2005;3:9
132. Tian Y, Li S, Song J. et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35:2383-90
133. Skog J, Würdinger T, van Rijn S. et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470-6
134. Alvarez-Erviti L, Seow Y, Yin H. et al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nature biotechnology. 2011;29:341-5
135. Ohno S-i, Takanashi M, Sudo K. et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 2012;21:185-91
136. Théry C, Amigorena S, Raposo G. et al. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Current Protocols in Cell Biology. John Wiley & Sons, Inc. 2006:3-22
137. Jang SC, Kim OY, Yoon CM. et al. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS nano. 2013;7:7698-710
138. Jo W, Kim J, Yoon J. et al. Large-scale generation of cell-derived nanovesicles. Nanoscale. 2014;6:12056-64
139. Jo W, Jeong D, Kim J. et al. Microfluidic fabrication of cell-derived nanovesicles as endogenous RNA carriers. Lab Chip. 2014;14:1261-9
140. Piao J-G, Wang L, Gao F. et al. Erythrocyte membrane is an alternative coating to polyethylene glycol for prolonging the circulation lifetime of gold nanocages for photothermal therapy. ACS nano. 2014;8:10414-25
141. Cheng H, Fan Z, Zhou H. et al. Structural elucidation of cell membrane-derived nanoparticles using molecular probes. J Mater Chem B. 2014;2:8231-8
142. Hu C-MJ, Fang RH, Luk BT. et al. 'Marker-of-self' functionalization of nanoscale particles through a top-down cellular membrane coating approach. Nanoscale. 2013;5:2664-8
143. Fang RH, Hu C-MJ, Chen KNH. et al. Lipid-insertion enables targeting functionalization of erythrocyte membrane-cloaked nanoparticles. Nanoscale. 2013;5:8884-8
144. Li L-L, Xu J-H, Qi G-B. et al. Core-shell supramolecular gelatin nanoparticles for adaptive and “on-demand” antibiotic delivery. ACS nano. 2014;8:4975-83
145. Parodi A, Quattrocchi N, van de Ven AL. et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat Nanotechnol. 2013;8:61-8
AUTHOR BIOGRAPHY
 Professor Zhiping Zhang earned his Ph.D. degree in 2007 from National University of Singapore, Singapore. During 2007-2010, he was awarded as a research fellow in Dana-Farber Cancer Institute at Harvard Medical School, USA. Now he is a professor of Pharmaceutics in Tongji School of Pharmacy & National Engineering Research Center for Nanomedicine, Huazhong University of Science and Technology. His research interests include Vitamin E TPGS-related Nanomedicine and biomimetic nanoparticles for cancer chemotherapy, immunotherapy and combinational therapy.
Professor Zhiping Zhang earned his Ph.D. degree in 2007 from National University of Singapore, Singapore. During 2007-2010, he was awarded as a research fellow in Dana-Farber Cancer Institute at Harvard Medical School, USA. Now he is a professor of Pharmaceutics in Tongji School of Pharmacy & National Engineering Research Center for Nanomedicine, Huazhong University of Science and Technology. His research interests include Vitamin E TPGS-related Nanomedicine and biomimetic nanoparticles for cancer chemotherapy, immunotherapy and combinational therapy.
 Dr. Songwei Tan received the BS degree in polymer science and engineering in 2005 and a Ph.D. degree in polymer chemistry and physics in 2011 from Zhejiang University, China. From 2011 to 2013 he was a postdoctoral fellow with Prof. Zhiping Zhang at Huazhong University of Science and Technology. Now he is a lecturer in Tongji School of Pharmacy, Huazhong University of Science and Technology, China. His research interest is biomaterials synthesis and smart polymeric drug delivery system.
Dr. Songwei Tan received the BS degree in polymer science and engineering in 2005 and a Ph.D. degree in polymer chemistry and physics in 2011 from Zhejiang University, China. From 2011 to 2013 he was a postdoctoral fellow with Prof. Zhiping Zhang at Huazhong University of Science and Technology. Now he is a lecturer in Tongji School of Pharmacy, Huazhong University of Science and Technology, China. His research interest is biomaterials synthesis and smart polymeric drug delivery system.
 Tingting Wu received the BS degree in pharmaceutics in 2013 from Henan University. Now she is a postgraduate student in Prof. Zhiping Zhang's group in Tongji School of Pharmacy, Huazhong University of Science and Technology, China. Her research interests mainly focus on the construction of biomimetic nano-carriers and combination of chemotherapy and immunotherapy for cancer treatment.
Tingting Wu received the BS degree in pharmaceutics in 2013 from Henan University. Now she is a postgraduate student in Prof. Zhiping Zhang's group in Tongji School of Pharmacy, Huazhong University of Science and Technology, China. Her research interests mainly focus on the construction of biomimetic nano-carriers and combination of chemotherapy and immunotherapy for cancer treatment.
 Dan Zhang received the BS degree in pharmaceutical science in 2014 from Shandong University. Now she is a first-year graduate student in Prof. Zhiping Zhang's group at Huazhong University of Science and Technology. Her current research interests include nanomedicine and the biomedical applications of functional nanomaterials.
Dan Zhang received the BS degree in pharmaceutical science in 2014 from Shandong University. Now she is a first-year graduate student in Prof. Zhiping Zhang's group at Huazhong University of Science and Technology. Her current research interests include nanomedicine and the biomedical applications of functional nanomaterials.
![]() Corresponding author: Dr. Zhiping Zhang, E-mail: zhipingzhangedu.cn
Corresponding author: Dr. Zhiping Zhang, E-mail: zhipingzhangedu.cn