Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(10):1083-1097. doi:10.7150/thno.11711 This issue Cite
Research Paper
Superior Performance of Aptamer in Tumor Penetration over Antibody: Implication of Aptamer-Based Theranostics in Solid Tumors
Dongxi Xiang1, Conglong Zheng2, Shu-Feng Zhou3, Shuxi Qiao4, Phuong Ha-Lien Tran1, Chunwen Pu5, Yong Li6, Lingxue Kong7, Abbas Z. Kouzani8, Jia Lin9, Ke Liu10, Lianhong Li11, Sarah Shigdar1 ![]() , Wei Duan1
, Wei Duan1 ![]()
1. School of Medicine, Deakin University, Pigdons Road, Waurn Ponds, Victoria 3216, Australia
2. Department of Biology, Medical College, Dalian University, Liaoning, People's Republic of China
3. Department of Pharmaceutical Sciences, College of Pharmacy, University of South Florida, Tampa, FL 33612, USA
4. Department of Pharmacology and Toxicology, College of Pharmacy and Arizona Cancer Center, University of Arizona, Tucson, AZ 85724, USA
5. Dalian 6th People's Hospital, 269 Huibai Road, Lugang, Ganjingzi District, Dalian, Liaoning 116031, The People's Republic of China
6. Cancer Care Centre, St George Hospital and St George Clinical School, University of New South Wales (UNSW), Kensington, NSW2052, Australia
7. Institute for Frontier Materials, Deakin University, 75 Pigdons Road, Waurn Ponds, Victoria, 3216, Australia
8. School of Engineering, Deakin University, 75 Pigdons Road, Waurn Ponds, Victoria 3216, Australia
9. Department of Biochemistry and Molecular Biology, West China School of Preclinical and Forensic Medicine, Sichuan University, Chengdu, P. R. China, 610041
10. College of Life Sciences, Sichuan University, Chengdu, P. R. China, 610041
11. Liaoning Key Laboratory of Cancer Stem Cell Research, Dalian Medical University, Dalian, China, 116044
Received 2015-1-27; Accepted 2015-4-23; Published 2015-7-2
Abstract

Insufficient penetration of therapeutic agents into tumor tissues results in inadequate drug distribution and lower intracellular concentration of drugs, leading to the increase of drug resistance and resultant failure of cancer treatment. Targeted drug delivery to solid tumors followed by complete drug penetration and durable retention will significantly improve clinical outcomes of cancer therapy. Monoclonal antibodies have been commonly used in clinic for cancer treatment, but their limitation of penetrating into tumor tissues still remains because of their large size. Aptamers, as “chemical antibodies”, are 15-20 times smaller than antibodies. To explore whether aptamers are superior to antibodies in terms of tumor penetration, we carried out the first comprehensive study to compare the performance of an EpCAM aptamer with an EpCAM antibody in theranostic applications. Penetration and retention were studied in in vitro three-dimensional tumorspheres, in vivo live animal imaging and mouse colorectal cancer xenograft model. We found that the EpCAM aptamer can not only effectively penetrate into the tumorsphere cores but can also be retained by tumor sphere cells for at least 24 h, while limited tumor penetration by EpCAM antibody was observed after 4 h incubation. As observed from in vivo live animal imaging, EpCAM aptamers displayed a maximum tumor uptake at around 10 min followed by a rapid clearance after 80 min, while the signal of peak uptake and disappearance of antibody appeared at 3 h and 6 h after intravenous injection, respectively. The signal of PEGylated EpCAM aptamers in xenograft tumors was sustained for 26 h, which was 4.3-fold longer than that of the EpCAM antibody. Consistently, there were 1.67-fold and 6.6-fold higher accumulation of PEGylated aptamer in xenograft tumors than that of antibody, at 3 h and 24 h after intravenous administration, respectively. In addition, the aptamer achieved at least a 4-time better tumor penetration in xenograft tumors than that of the antibody at a 200 μm distances from the blood vessels 3 h after intravenous injection. Taken together, these data indicate that aptmers are superior to antibodies in cancer theranostics due to their better tumor penetration, more homogeneous distribution and longer retention in tumor sites. Thus, aptamers are promising agents for targeted tumor therapeutics and molecular imaging.
Keywords: aptamer, targeted tumor therapeutics, tumor penetration
Introduction
In order to achieve a curable outcome of cancer treatment, anticancer agents should effectively penetrate the extravascular space and gain access to all viable cancer cells with a sufficient concentration [1-4]. However, the properties of abnormal tumor niche, including disorganized vascular system, dysfunctional lymphatics, increased interstitial fluid pressure, the presence of extracellular matrix, and resultant hypoxia with insufficient oxygen and nutrients, limit the penetration and diffusion of molecular medicines into tumor cells [2, 3, 5-10]. This insufficient and heterogeneous drug distribution can significantly reduce therapeutic efficacy by leaving a portion of viable cancer cells behind, in turn leading to the increase of systematic drug resistance and the failure of cancer treatment [11]. Therefore, there is an urgent need to develop effective anticancer therapeutics that could effectively penetrate tumor tissues and accumulate inside tumor cells.
Even though therapeutic antibodies have been routinely used for targeting cancers to overcome the insufficient specificity of traditional anticancer drugs [12-14], their efficacy for in vivo anticancer treatment is restricted due to 1) immunogenicity, 2) limited tumor penetration and distribution, and 3) less accessibility for chemical modification [11, 15-18]. To overcome these challenges, new therapeutics that are tumor-specific, non-immunogenic and with ease of conjugation with various agents to improve tumor penetration are urgently needed. Aptamers are small single-stranded DNA or RNA oligonucleotides that bind to their targets with high affinity and specificity [19]. The limitations of nucleic acid aptamers used in vivo are their shorter circulatory half-life and nucleic acid degradation, which can be alleviated by the conjugation of polyethylene glycol (PEG) to alter the pharmacokinetic profile, and by introducing site-specific chemical modification to minimize the susceptibility to attack from endonucleases and exonucleases [19-21]. Aptamers can be modified and conjugated with functional molecules for cancer diagnostic or therapeutic purposes [19, 22, 23]. In contrast to protein antibodies, aptamers possess little to no immunogenicity and low systemic toxicity in vivo [19, 24, 25].
Because aptamers have a size between antibodies (150 kDa) and small peptides (1-5 kDa) [20], we hypothesized that aptamers with a smaller size and suitable affinity might be advantageous over antibodies in not only effectively penetrating into tumors but also maintaining a durable retention in tumor tissues. The in vitro multicellular tumorsphere possesses several properties of the solid tumor niche in vivo [5, 26-28] and is a simple and practicable method for evaluating drug penetration. To evaluate the tumor penetration performance of aptamers, we systematically evaluated the tumor penetration and retention by aptamers and antibodies in 3D tumorsphere model in vitro and tumor tissues in vivo.
Materials and Methods
Cell culture
HT-29 (human colorectal adenocarcinoma, ATCC® HTB38™) cell line and HEK-293T (human embryonic kidney, ATCC ® CRL-11268™) cell line were purchased from American Type Culture Collection (ATCC, Manassas, VA). Huh-7 and PLC/PRF/7 (human hepatocellular carcinoma) cell lines were kindly provided by Dr. Liang Qiao, Sydney University. All the above cells were cultured in DMEM (Life Technologies, Australia) medium supplemented with 10% fetal bovine serum (FBS, Hyclone, Canada), penicillin (50 U/mL,), and streptomycin (50 µg/mL, Invitrogen, Australia) and 1× Glutamax (Life Technologies, Australia) in a humidified atmosphere containing 5% CO2 at 37 °C.
Animals used in this study
Animal study in this research was approved by Deakin University Animal Welfare Committee. All animals were purchased from The Animal Resources Centre (Perth, Australia). Six to eight weeks old NOD-SCID female mice were used for HT29 tumor xenograft establishment. The mice were housed in TECNIPLAST SealsafeTM Individually Ventilated Cages, which were placed in a temperature-controlled room (25 ± 1 °C) with a 12-h light-dark cycle. Mice were fed ad libitum with a standard diet. Beddings, cages and water were autoclaved at 121 °C for 30 min while the fodder was sterilised by ultraviolet irradiation before use. Male Sprague-Dawley rats (200 to 250 g) were housed in a temperature-controlled room (25 ± 1 °C) with a 12-h light-dark cycle. Rats were fed ad libitum with a standard diet and were fasted overnight before treatments administration.
Antibody and aptamers used in this study
FITC conjugated EpCAM antibody was purchased from Fitzgerald (Cat #10R-2376). Aptamers were synthesized by IBA GmbH (Rudolf-Wissell-Straße 28, 37079 Göttingen, Germany) followed by HPLC purification.
RNA EpCAM aptamer: 5'- (DY647) - A (2'-F-C) G (2'-F-U) A (2'-F-U) (2'-F-C) (2'-F-C) (2'-F-C) (2'-F-U) (2'-F-U) (2'-F-U) (2'-F-U) (2'-F-C) G (2'-F-C) G (2'-F-U) -3'
Negative control RNA EpCAM aptamer: 5'- (DY647) - A (2'-O-Me-C) G (2'-O-Me-U) A (2'-O-Me-U) (2'-O-Me-C) (2'-O-Me-C) (2'-O-Me-C) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-C) G (2'-O-Me-C) G (2'-O-Me-U) -3'
Hybrid DNA-RNA EpCAM aptamer: 5'- (DY647) - c g c g c g c c g c A (2'-F-C) G (2'-F-U) A (2'-F-U) (2'-F-C) (2'-F-C) (2'-F-C) (2'-F-U) (2'-F-U) (2'-F-U) (2'-F-U) (2'-F-C) G (2'-F-C) G (2'-F-U) c g g c g c g c g -3'
Negative control Hybrid DNA-RNA EpCAM aptamer: 5'- (DY647) - c g c g c g c c g c A (2'-O-Me-C) G (2'-O-Me-U) A (2'-O-Me-U) (2'-O-Me-C) (2'-O-Me-C) (2'-O-Me-C) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-C) G (2'-O-Me-C) G (2'-O-Me-U) c g g c g c g c g -3'
PEGylated RNA EpCAM aptamer: 5'-(20 kDa PEG-FITC)- c g c g c g c c g c A (2'-F-C) G (2'-F-U) A (2'-F-U) (2'-F-C) (2'-F-C) (2'-F-C) (2'-F-U) (2'-F-U) (2'-F-U) (2'-F-U) (2'-F-C) G (2'-F-C) G (2'-F-U) c g g c g c g c g - (Biotin or DY647) -3'
Negative control PEGylated RNA EpCAM aptamer: 5'-(20 kDa PEG-FITC)- c g c g c g c c g c A (2'-O-Me-C) G (2'-O-Me-U) A (2'-O-Me-U) (2'-O-Me-C) (2'-O-Me-C) (2'-O-Me-C) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-U) (2'-O-Me-C) G (2'-O-Me-C) G (2'-O-Me-U) c g g c g c g c g - (Biotin or DY647) -3'
In the above sequences, 2'-F represents 2'-fluoropyrimidine, 2'-O-Me indicates 2'-O-methyl modification. Lowercase letters indicate DNAs which are modified with 5'-methyl-deoxycytidine (5-Methyl-dC). The negative control aptamer is an aptamer of the same sequence as the EpCAM targeting aptamer but with a different side-chain modification that could affect the 3-dimensional structure of aptamer [29]. As a result, this control aptamer is not able to bind to EpCAM and does not target the cancer cells overexpressing EpCAM. For engineering an effective DOX loading segment for future therapeutic applications (described in a separate manuscript) 5'-methyl-deoxycytidine (dC) was deployed in the newly engineered DNA stem as 5-Methyl dC when substituted for dC will increase the Tm by as much as 0.5°C per insertion. In addition, the presence of 5'-Methyl dC in CpG motifs can prevent or limit unwanted immune responses (Fig. 5-2a) [30-32]. Prior to conducting all the experiments using aptamers, the aptamers are prepared in PBS containing 5 mM MgCl2, and then folded by denaturation at 85˚C for 5 min, followed by 10 min incubation at room temperature and refolding at 37˚C for at least 15 min.
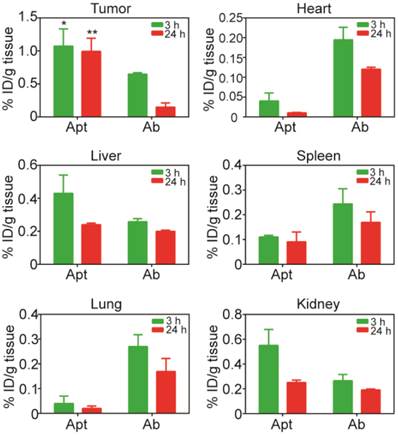
Biodistribution of PEGylated aptamer and antibody in mice bearing xenograft colorectal tumors. NOD/SCID mice bearing HT29 xenograft tumors (~150 mm3) received a single i.v. injection of 1 nmol/mouse of PEGylated aptamer or antibody. The concentration of aptamer or antibody, expressed as % of injected dose (ID) per g of tissue, in tissues iundicated was determined at 3 h and 24 h after the agent administration using ELISA. Data are means ± SEM (n = 4). *, P < 0.05; **, P < 0.01 compared to antibody.
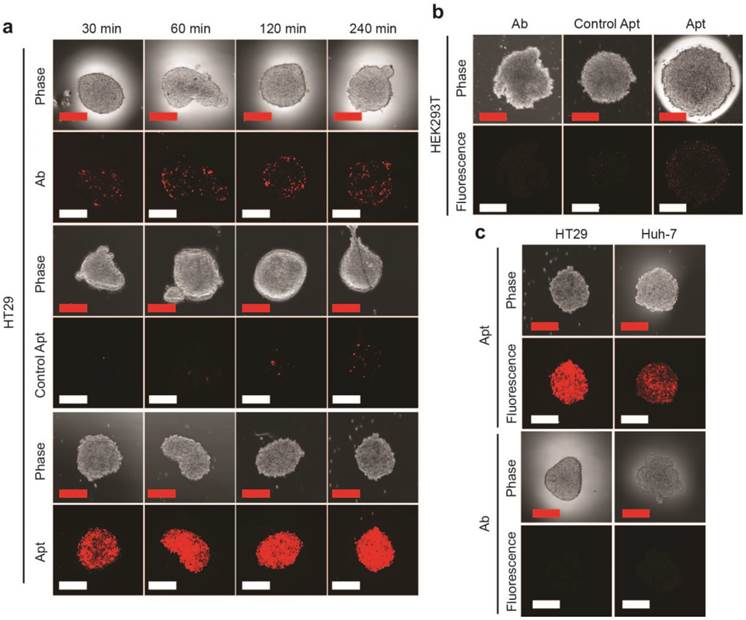
EpCAM aptamer penetrates tumorsphere more effectively than EpCAM antibody. EpCAM aptamer, control aptamer, or EpCAM antibody of the same concentration (100 nM) were incubated with HT29 tumorsphere for up to 240 min at 37 °C. The tumorspheres were then washed three times in PBS and imaged using laser scanning confocal microscopy. (a) Aptamer and antibody staining of HT29 tumorspheres. (b) Aptamer and antibody staining of HEK293T tumorspheres after 240 min incubation. (c) Following a 4 h incubation, the HT-29 and Huh-7 tumorspheres were washed with PBS and incubated in the absence of EpCAM aptamer and EpCAM antibody. Cells incubated with aptamers were imaged using laser scanning confocal microscopy 24 h later; those with antibody imaged 4 h alter. Scale bar = 200 µm.
Determination of particle size of aptamer and antibody
A 2 μL aliquot of EpCAM aptamer or EpCAM antibody (FITC) (Fitzgerald, Cat #10R-2376) (10 nM) was diluted in 998 μL PBS and mixed gently. The vesicle size was measured using a Zetasizer Nano ZS Particle Characterization System (Malvern, UK) following the manufacturer's instruction.
Determination of binding affinity
The equilibrium dissociation constant (K'd) of 2'-F RNA aptamer species to EpCAM proteins expressed on the cell surface was determined using flow cytometry. HT29 or HEK293T cells (5 × 105) were first incubated with blocking buffer (PBS supplemented with 5 mM MgCl2, 0.1 mg/mL tRNA, 0.1 mg⁄mL salmon sperm DNA, and 5% FBS) for 20 min at room temperature followed by two washes with PBS prior to incubation at serial concentrations (0 nM, 10 nM, 20 nM, 40 nM, 60 nM, 80 nM, 100 nM, 150 nM and 200 nM) of DY647-labelled EpCAM aptamer, control aptamer or FITC-labelled EpCAM antibody in a 100 μL blocking buffer at 37 °C for 30 min. The cells were washed three times, resuspended in 150 μL assay buffer containing 5 mM MgCl2 and subjected to flow cytometric analyses. The binding affinity was calculated after subtracting the mean fluorescence intensity (MFI) obtained from target cells to that of negative control cells according to a method described by Ellington and colleagues [33]. Fluorescent histograms were recorded by BD FACS-CantoTM II flow cytometer and analysed using BD FACSDiva software (v6.0).
Confocal microscopy analysis of tumorsphere preparation of aptamer and antibody
Two thousand HT29, Huh-7 and HEK293T cells were plated out in ultralow attachment wells and allowed to form spheres for 3-5 days in DMEM/F12 media (Invitrogen Life Technologies) supplemented with B27 (100 units/mL), Insulin (10 µg/mL), EGF (20 ng/mL) and bFGF (20 ng/mL). The spheres were washed three times in PBS containing 5 mM MgCl2 and blocked for 20 min using blocking buffer. The spheres were then incubated with 100 nM of aptamer or antibody for 30 min, 60 min, 120 min, or 240 min. Following each time point, the spheres were washed three times with PBS prior to visualization using the FluoView FV10i confocal microscope. To determine the retention of aptamers within tumor spheres, HT29 tumor spheres were incubated with EpCAM aptamers or EpCAM antibody (FITC) for a total of 4 h, washed three times in PBS, followed by incubation in sphere medium for a further 24 h before being imaged.
Tumor implantation and evaluation
To establish xenograft tumors, single suspension HT29 cells were harvested after trypsinization. The cells (1 × 105) were mixed and resuspended with DMEM (serum free) and Matrigel (50:50 = V:V) followed by transplantion into the flank of each mouse with a 0.5 mL syringe and 26-gauge needle. Once tumors arose, mice were randomized into treatment groups of 4 mice per group. Treatment was initiated when the tumor volume reached 150 mm3. Tumor fragments were archived in 10% neutral buffered formalin for further analysis. Slides of sections were processed for Immunohistochemistry analysis (double staining of CD31 and aptamer or antibody).
Bio-distribution assay
For determination of PEGylated aptamer and antibody distribution in vivo, HT29 tumor bearing NOD/SCID mice were randomly divided into two groups (4 mice per group, termed 3 h and 24 h time point) once tumors reached an average volume of 150 mm3. Agents were delivered via tail vein injection of 1 nmol aptamer or antibody per mouse. Organs including heart, liver, spleen, kidney, lung and tumor were collected 3 h and 24 h after a single intravenous (i.v.) injection of 1 nmol/mouse of agents and then lightly washed in cold physiological saline to remove any excess blood, blot-dried using filter paper and thoroughly homogenized in PBS (tissue: PBS weight ratio 1:3) using the FastPrep®-24 tissue and cell homogenizer. The tissue homogenate was centrifuged at 21,000 × g for 10 min at 4 °C. The supernatant of homogenate were collected and the aptamer or antibody concentration was quantified by ELISA.
ELISA
Fifty microliters of the 10 µg/mL monoclonal anti-FITC antibody (Sigma, Cat # F5636) in washing buffer (PBS containing 0.1 mg/mL tRNA and 1 mg/mL BSA) was added to goat anti-mouse IgG pre-coated wells (Sapphire Bioscience, Cat #600-11050). After 1 h incubation at room temperature, anti-FITC antibody was removed followed by thorough washes with PBS. The treated wells were blocked with 50 µL 1× SuperBlock Blocking buffer (Thermo Scientific) at room temperature for 1 h, followed by 3 washes with PBS, 3 min per time. Tissue or serum samples containing biotin-labeled aptamer or FITC-labeled antibody (Fitzgerald, Cat #10R-2376) (100 µL/well) were added and incubated for 1 h at room temperature. After extensive washing, 50 µL of 1:5000 diluted Pierce High Sensitivity Streptavidin HRP conjugate (Thermo Scientific, Cat #21140) was added to each well to bind biotin-conjugated aptamer. For detection of bound antibody, HRP conjugated goat-anti-mouse IgG (Pierce, Cat #31430) was added. After 1 h incubation at room temperature and extensive washing, the bound aptamer was detected with a Quanta Blu fluorogenic Peroxidase substrate system (Thermo Scientific, Cat #15169) and measured at a wavelength of 325/420 nm using the VICTOR TM X5 Plate Reader (PerkinElmer Life Sciences).
In vivo imaging
Non-PEGylated aptamer, PEGylated aptamer and antibody were administrated into mice-bearing HT29 xenograft tumors via i.v. injection at a dose of 0.75 nmol/mouse when the tumor volume reached around 150 mm3. The live animal imaging was conducted at 5 min interval using a Xenogen IVIS Lumina II imaging system (PerkinElmer, USA). A circular region of interest (ROI) around the tumor site of each mouse was made and the total flux in this region was quantified using Living Image Software V2.50 (PerkinElmer, USA) with the units of photons/s/cm2/sr. The data were used for semi-quantification of fluorescence signal in tumors and other tissues.
Immunohistochemistry (double staining of CD31 and aptamer or antibody)
The tumor tissues dissected from tumor-bearing mice were fixed at 10% neutral buffered formalin, processed and embedded in paraffin in order to retain their shape and architecture, as well as for their long term storage. Paraffin embedded sections were deparaffinized with Histoclear and rehydrated through graded ethanol. Heat induced antigen retrieval was performed in a microwave oven using sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) for 20 min and the slides were allowed to cool prior to blocking with endogenous peroxidase using 0.3% hydrogen peroxide in PBST (PBS containing 0.1% Tween 20) for 20 minutes at room temperature. Additional blocking with 0.1 mg/mL tRNA, and 0.1 mg/mL bovine serum albumin (BSA) or 10% goat serum in phosphate buffered saline (PBS) for 20 min was carried out before avidin-biotin blocking. Following serial incubation with 100 µL avidin (0.1 mg/mL) and 100 µL biotin (0.5 mg/mL) per slide for 15 min each, the slide was washed with PBST twice and incubated with 1:100 dilution of rabbit anti-CD31/PECAM-1 (platelet endothelial cell adhesion molecule-1) conjugated with biotin (Bioss, Cat #0195R) for 2 h at 37°C. The chromogenic alkaline phosphatase solution (Vector Labs, Cat #SK-5100) was prepared according to the manufacturer's instruction and added to the slides following 1 h staining of secondary anti-biotin antibody (Vector Labs, Cat #SP-3020) (1: 250 dilution). Upon completion of the staining of blood vessels with CD31 antibody and washing with PBST twice for 5 min each. For the detection of FITC-labeled antibody or FITC-labeled-aptamer, the sections were incubated with anti-FITC secondary antibody (Abcam, Cat #AB6656, 1: 100 dilution) for 2 h at room temperature. The slides were then washed in PBST for 5 min for three times prior to be treated with 100 µL DAB peroxidase substrate solution (Vector Laboratories, Cat #SK-4105) for 5-10 min at room temperature for color development, followed by a single wash under running tap water. Counterstaining of tissue sections was performed by immersion in haematoxylin solution for 5 min followed by washing under running tap water for 3-5 min. Slides were then differentiated in 1% acid alcohol for 30 sec and washed under running water for 1 min prior to bluing with Scott's solution for 1 min. Slides were washed under running water for 5 min and then dehydrated with 95% and 100 % alcohol serially. Finally, slides were cleared using two changes of histolene for 5 min each, and then mounted using DPX (Sigma, Cat #317616). Stained sections were examined under a light microscope equipped with an Olympus SC20 camera (Victoria, Australia).
To investigate the distribution of aptamer or antibody in tumor sections, their pixel intensity in relation to distance from the blood vessel within the selected region of interest was quantified using Image Pro software, according to the protocol reported by Lee and Tannock [34]. Briefly, images displaying anti-CD31 staining (black) and aptamer or antibody staining (brown) were converted to black and white binary images: each image was overlaid with the corresponding field of view displaying the intensity of the agent of interest, resulting in an 8-bit black and white image with blood vessels identified by an intensity of 255 (gray scale) and agent intensity ranging from 0-254 (gray scale). The intensity of aptamer/antibody signal was represented as mean ± SEM for all pixels at a given perpendicular distance (20-200 μm) to the nearest vessel and plotted as a function of that distance.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 6.0 (San Diego, CA). An unpaired t test was used for comparisons between two experimental groups, and ANOVA was used for comparisons of more than two groups. Unless otherwise indicated, all results were averaged from biological triplicates and values are reported as means ± SEM. P < 0.05 was considered statistically significant.
Results
EpCAM aptamers internalize into target cells more efficiently than antibody
Efficient delivery of a therapeutic ligand to a tumor requires a sufficient amount of the agent to reach tumors, associated with accompanied rapid elimination from healthy tissues. Such effective tumor delivery is dictated by a number of factors including a suitable binding affinity of the ligand to its targets, the expression of the target antigen at the tumor and the size of the therapeutic ligand [14, 16, 35]. The EpCAM antibody used in this study has high molecular weight (150 kDa) with a size of around 15 nm, while the EpCAM aptamer has a smaller size of around 2.09 nm (Fig. 1a). The equilibrium dissociation constant of EpCAM aptamer or the EpCAM antibody to HT29 cells was determined to be 39.42 nM and 5.18 nM, respectively (Fig. 1b). Adams et al. reported that for a tumor-targeting ligand, the K'd value lower than 1 nM does not necessarily improve tumor penetration but rather limits their further transport and perfusion in a tumor [16, 35]. This phenomena was explained by the “binding site barrier” model which postulated that monoclonal antibodies with very high affinity (K'd < 1 nM) stably bind to the first interacted tumor antigens with a slow rate of dissociation, resulting in a reduction of further diffusion and thus poor penetration into tumors [36, 37]. The K'ds for both the EpCAM aptamer and the antibody used in this study are well above the 1 nM threshold for “binding site barrier”. Therefore, it is extremely unlikely that the K'd of either the aptamer or the antibody used will have a profound negative influence on tumor penetration. Here, we sought to investigate the tumor penetration and retention of an aptamer with a 7-fold lower binding affinity than its antibody counterpart.
Cell binding and internalization of EpCAM aptamer and antibody in vitro. (a) Particle size of EpCAM aptamer and EpCAM antibody as determined by dynamic light scattering. (b) Determination of the equilibrium dissociation constants (K'd) of EpCAM aptamer and EpCAM antibody to HT29 cells using flow cytometry by incubating cells at varying concentrations of aptamer and antibody (1-200 nmol/L). (c) Localization of EpCAM aptamer, control EpCAM aptamer or EpCAM antibody in acidic organelles (late endosome and lysosomes). Following incubation with 100 nM EpCAM aptamer or EpCAM antibody at 37 °C for 15 min and three time washes, HT29 cells were incubated with LysoTracker® Green in the first 90 min of a further 2 h incubation followed by confocal microscopy imaging. (d) Quantification of fluorescence signals from localized aptamer or antibody in acidic organelles (late endosome and lysosomes) as in (c). (e) Specificity of EpCAM aptamer binding and internalization. Three EpCAM-positive cell lines (HT29, Huh-7 and PLC/PRF/5) and the control EpCAM-negative HEK293T cells were incubated with 100 nM EpCAM aptamer or control EpCAM aptamer at 37 °C for 15 min, followed by washing and confocal microscopy imaging. (f) Quantification of fluorescence signals from internalized aptamers in various cell lines as in (e). Ab, antibody; Apt, aptamer, RFI, relative fluorescence intensity; MFI, mean fluorescence intensity. Data are means ± SEM, n=3. Scale bar = 5 μm.
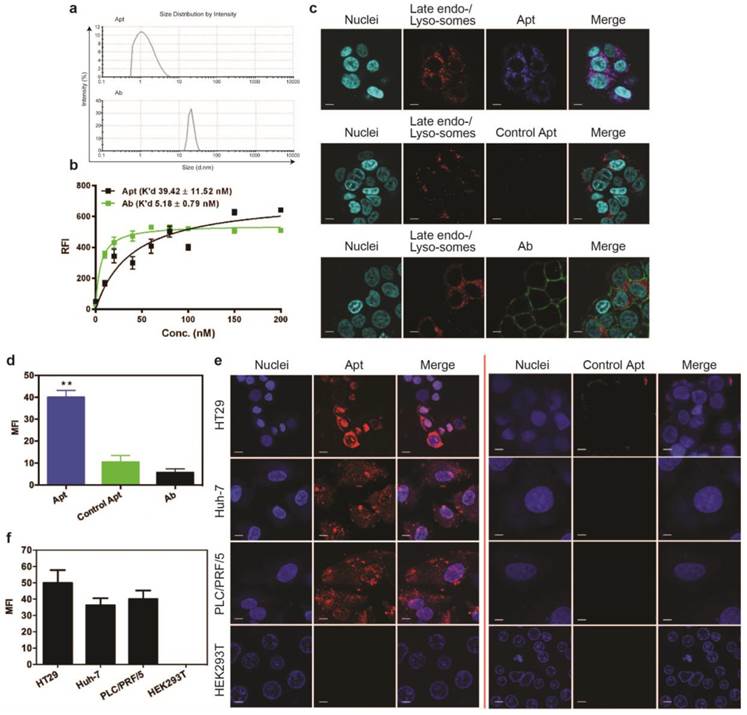
Binding of targeting moieties to the cell surface molecules often results in the uptake and trafficking of the ligand-target complex through endocytic pathways that internalize the targeting molecules into various cellular organelles. To investigate the efficiency in cell internalization and trafficking of the aptamer and antibody after cell binding, we examined their presence in late endosomes and lysosomes using a LysoTracker® Green DND-22 dye that stains acidic compartments in live cells. Confocal imaging of live cells was conducted on HT29 cells incubated with the same concentration of aptamers or antibodies (100 nM). At 15 min the aptamers were transported to and predominantly localized at late endosomes or lysosomes as evident from the colocalization of the blue (for aptamers) and red (for all acidic vesicles) fluorescence (Fig. 1c). In contrast, at least 8-fold less antibodies (green) were observed in these acidic vesicles (red) compared to that of aptamers (Fig. 1d). The ligand (EpCAM)-dependent internalization of the aptamer was assessed by using a control aptamer that has the same nucleic acid sequence as the EpCAM aptamer but with a 2'-O-Me modification of the pyrimidines instead of 2'-fluoro modification thus abolishing its binding to EpCAM proteins. As shown in Fig. 1c and d, very limited negative control aptamers (blue) were found in the late endosome-lysosome compartments; those inside the cells might enter the cells via non receptor-mediated endocytosis.
To be an effective cancer theranostic agent, an aptamer should be ideally internalized following binding to its target expressed on the cell surface of various types of tumors [38-40]. To ascertain if the efficient endocytosis of the aptamer is a general rather than a cell type-specific phenomenon for HT29 cells, two different types of hepatocarcimona cells (Huh-7 and PLC/PRF/5) were employed and incubated with aptamers at 37 °C for 30 min followed by confocal microscopy. The EpCAM aptamer efficiently internalized to these three different EpCAM-positive human cancer cell lines with a specific red fluorescence compared to the control aptamer (Fig. 1e). Such internalization was specific as there was no binding of the aptamer to the negative control HEK293T cells that do not express EpCAM proteins. The cell binding and internalization of the EpCAM aptamer in hepatocarcinoma cells were further quantified (Fig. 1f). After 30 min incubation, there was at least 40-fold enhancement in binding and/or internalization of aptamer to EpCAM-positive cell lines compared with that in the EpCAM-negative cell lines. These results are consistent with our previous observation that the EpCAM aptamer is able to specifically enter a variety of EpCAM-positive tumor cells via receptor-mediated endocytosis [38, 41].
Aptamer is superior to antibody for tumorsphere penetration
Having established that the EpCAM aptamer internalized more efficiently than antibody into monolayer cell culture, we proceeded to investigate the ability of our aptamers to penetrate an in vitro 3D cell structure. For this purpose, we used tumorsphere as a 3D cell model, as it is ideal for in vitro evaluation of penetrating ability of targeting molecules due to the ease of establishment and the similar microenvironment to in vivo tumors such as high inner pressure, low oxygen and low pH situation [42, 43]. We generated both EpCAM-positive (HT29) and EpCAM-negative (HEK293T) tumorspheres in low adherence culture conditions. Upon reaching 200 μm in size, these spheres were incubated with 100 nM EpCAM aptamer or the same concentration of EpCAM antibody for the indicated time, washed 3 times with PBS and subjected to confocal microscopy. The laser scanning confocal microscope recorded images of a serial z-stack optical sections of 500 nm thickness from the periphery to the center of the spheres. The images of the middle of z-stack sections were used for the comparison of tumorsphere penetration (Supplementary Fig. 1). As shown in Fig. 2a, the EpCAM aptamer penetrated into the center of the tumorsphere as early as 30 min, and achieved excellent penetration throughout the center of HT29 tumorspheres between 60 min to 240 min. In sharp contrast, limited tumor penetration into the center of the tumorsphere was observed at 30 min for EpCAM antibody and there was only a patchy presence of antibody in the center of the tumorsphere even after 4 h incubation. The extremely limited penetration by the negative control aptamer throughout entire assay period (up to 6 h) suggests the specific interaction between EpCAM aptamer and its ligand, rather than a passive diffusion of any nucleic acid molecule, is required for the efficient penetration into the center of the tumorsphere. Furthermore, after 240 min incubation, only a weak signal for EpCAM aptamer was discernable with the HEK293T spheres that does not express EpCAM (Fig. 2b).
In order to confirm the observed superior tumorsphere penetration by the EpCAM aptamer was not limited to the 3-D model of HT29 colorectal cancer cells, a liver cancer model, Huh-7 tumorsphere which expresses high level of EpCAM was also included in our studies. As shown in Supplementary Fig. 2, the EpCAM aptamer effectively penetrated into the center of Huh-7 tumorspheres between 30 min and 240 min, while the signal of antibody at the center of the tumorspheres was hardly detectable. Importantly, the fluorescence signals of EpCAM aptamers in both HT29 and Huh-7 tumorspheres were clearly detectable after extensive washing followed by a further incubation in sphere medium for 24 h (Fig. 2c). In contrast, the signals from antibodies in these tumorspheres were hardly detectable after 4 h further incubation. These data indicated that the EpCAM aptamers can not only penetrate into the core of the tumorsphere derived from both colorectal and hepatocellular carcinoma but also were adequately retained by the tumor cells in the center of the tumorsphere for at least 24 h in vitro.
Aptamer is superior to antibody for in vivo tumor imaging
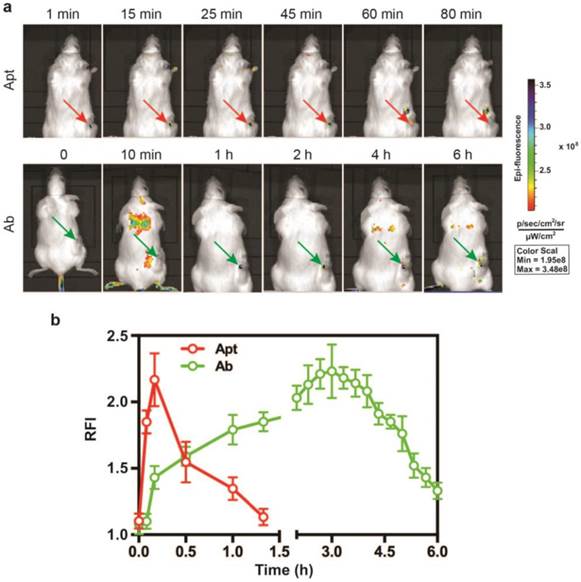
Given that the EpCAM RNA aptamer penetrated the tumorsphere much better than antibody in vitro, we next compared the performance in in vivo imaging between aptamer and antibody. A DY647 fluorophore was conjugated to the 5'-end of the EpCAM aptamer to facilitate molecular imaging. As for the imaging study, NOD/SCID mice-bearing HT29 xenograft tumors with a tumor volume of ~150 mm3 received a bolus i.v. injection of 0.75 nmole of DY647-labelled aptamers or FITC-labeled EpCAM antibodies. The whole body imaging was performed using the IVIS Live Imaging System. Fluorescence signals obtained from aptamers or antibodies at the regions of interest (ROIs) of tumor sites were recorded at a 5 min interval until the signal disappeared. The signal of aptamer on HT29 tumors appeared immediately and the maximum uptake was observed at around 10 min after i.v. injection and lasted for approximately 80 min (Fig. 3a and b). In sharp contrast, the signal for antibody observed in tumor sites had a slow onset, with the maximum uptake reached at 3 h. The signal in the tumor receiving EpCAM antibody lasted for at least 6 h. Thus, the aptamer achieved highest concentration in the tumor within 10 min and the signal in the tumor diminished at least 4.5-times faster than that of the antibody. Such quick onset of the maximum signal at the site of the tumor followed by rapid disappearance confers aptamer with highly desirable characteristics for function as a promising molecular imaging probe which outperform antibodies [44, 45].
Superior performance of EpCAM aptamer to the antibody in in vivo molecular imaging. (a) Representative live animal images of aptamers and antibody. NOD-SCID mice bearing HT29 tumor (150 mm3) received a single i.v. injection of 0.75 nmol of EpCAM aptamer and EpCAM antibody. Log-scale heat map (at the right) of photon flux applies to all panels. p/s/cm2/sr: photons per second per cm2 per steradian. Arrow depicts the position of the subcutaneous HT29 tumor. (b) The fluorescence-time curve of EpCAM aptamer and EpCAM antibody in tumors as indicated in (a) was determined by Living Imaging Software v2.50 (Xenogen) with the units of photons/s/cm2/sr. Data are means ± SEM, n=3.
PEGylated-aptamer is superior to antibody for in vivo drug delivery
Next, the in vivo performance of aptamer and antibody was studied. In order to confer sufficient serum half-life, the EpCAM aptamer was further engineered by conjugating a terminal 20 kDa polyethylene glycol (PEG) (Fig. 4a). PEGylation of the aptamer increased the size of aptamer from 2.09 nm (non-PEGylated version) to approximately 12 nm, exceeding the 10 nm threshold of glomerular filtration (Fig. 4b) [46]. The PEGylation of the aptamer did not introduce adverse effects to the binding affinity. As illustrated in Figure 4c, the K'd of the PEGylated aptamer corresponded well to the reported K'd of the parental EpCAM aptamer against target cell lines [38]. In the case of HT29, the PEGylated EpCAM aptamer had a K'd of 51.79 nM to HT29, compared with that of 39.42 nM for the original EpCAM aptamer (Fig. 4c). In addition, the PEGylated aptamer maintained its binding ability to EpCAM-positive cancer cells (Fig. 4d). To study tumor delivery and retention, 0.75 nmole of DY647-labelled PEGylated aptamers, control DY647-labelled PEGylated aptamers or FITC-labeled EpCAM antibodies was injected i.v. into NOD/SCID mice-bearing HT29 xenograft tumors. The signal from the ROIs of HT29 tumors from mice receiving PEGylated aptamer gradually increased to a plateau at 5 h followed by a slow and prolonged decrease until 26 h or later. However, signals from either control PEGylated aptamer or antibody disappeared at around 6 h (Fig. 4e and f), suggesting that the prolonged tumor residence of PEGylated EpCAM aptamer was not a non-specific accumulation due to EPR effect but rather resulted from the combined EPR effect and specific ligand interaction. The duration of the antibody signal at the tumor, which is indicative of its tumor volume of distribution, was 4.3-fold shorter than the tumor residence time of the PEGylated aptamer. Taken together, the PEGylated aptamers achieved longer tumor retention than the antibody counterpart, paving the way for the development of aptamers as effective targeted drug delivery vehicles.
Superior tumor accumulation and retention of PEGylated aptamer than that of antibody. (a) PEGylated aptamer was developed by attaching a 20 kDa PEG-FITC to the 3'-end and a biotin or a DY647 dye to the 5'- end of the DNA strand. (b) Particle size of PEGylated aptamer and antibody as determined by dynamic light scattering. (c) Determination of the equilibrium dissociation constants (K'd) of PEGylated aptamer to HT29 cells using flow cytometry by incubating cells at varying concentrations (1-200 nmol/L). (d) Binding and internalization of PEGylated aptamer to HT29 cells which were incubated with 100 nM EpCAM aptamer at 37 °C for 30 min, followed by washing and confocal microscopy imaging. Scale bar = 10 μm. (e) Live animal imaging of antibodies and aptamers. NOD-SCID mice bearing HT29 tumor (150 mm3) received a single intravenous injection of 0.75 nmol of control PEGylated aptamer, PEGylated aptamer and antibody followed by live animal imaging at the indicated time points. (f) The fluorescence-time curve of PEGylated aptamer in tumors as in (e) was determined by Living Imaging Software v2.50 (Xenogen) with the units of photons/s/cm2/sr. Log-scale heat map (at the right) of photon flux applies to all panels. Data are means ± SEM, n=3. RFI: relative fluorescence intensity.
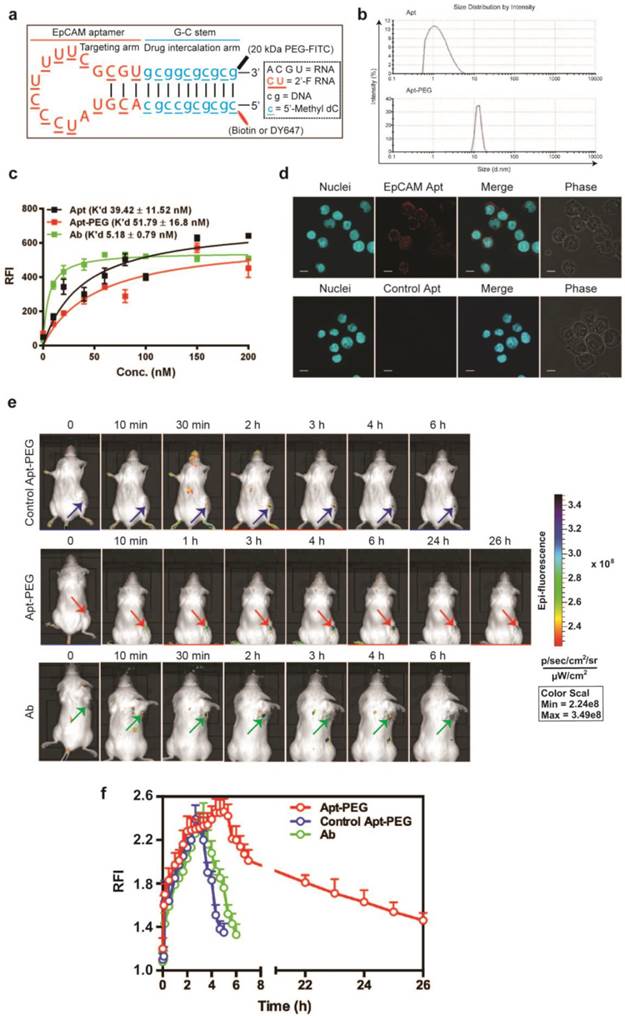
Superior tumor uptake of PEGylated-aptamer over antibody in tumor xenografts
To further evaluate the advantage of PEGylated aptamer over antibody in tumor targeting, the biodistribution of both agents was studied at 3 h and 24 h after a single i.v. injection of these agents into NOD/SCID mice bearing HT29 xenograft tumors (~150 mm3). With regard to the accumulation in tumors, an 1.67-fold and 6.6-fold higher amounts of PEGylated aptamer was found at 3 h and 24 h, respectively, than that of the antibody counterpart (Fig. 5). Furthermore, there was only a 16 % decrease of aptamer accumulation in the tumor xenografts from 3 h to 24 h after in vivo administration, in sharp contrast to a swift decline of 77 % of tumor accumulation of the antibody over the same period. These data are consistent with the sustained retention of aptamer in tumorspheres in vitro, indicating that the PEGylated EpCAM aptamer could also be remained in tumors at a high concentration in vivo for at least 24 h. In the liver and kidney, the aptamer had a higher concentration initially at 3 h compared with that of the antibody, however, at 24 h postinjection, the concentration of both aptamer and antibody were comparable. Whilst in the heart, spleen and lung, the aptamer concentration was much lower than that of the antibody counterpart at both 3 h and 24 h postinjection. Taken together, the PEGylated aptamer exhibited favorable accumulation and retention profile than that of the antibody in xenograft tumors, suggesting that aptamer could be developed as effective modality for targeted cancer therapy.
Comparison of penetration of aptamer and antibody in tumors
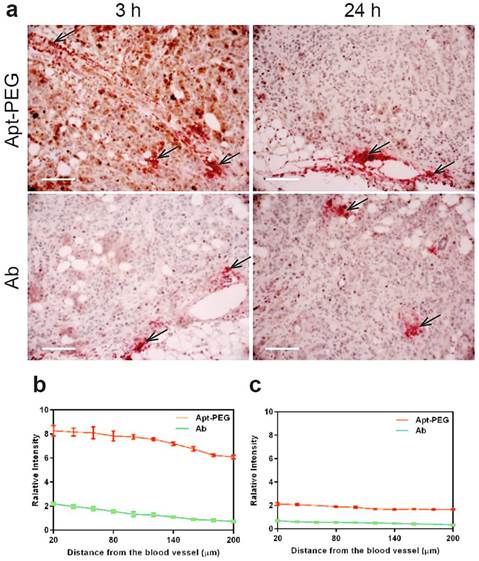
The efficacy of drug transport inside tumors was highly restricted because of limited penetration, due to the abnormality and complication of tumor microenvironment, including poorly organized vascular system, increased interstitial fluid pressure as well as the presence of extracellular matrixes. Thus, the next generation of anticancer therapeutics must penetrate into tumor tissues efficiently and gain access to all cancer cells at a sufficient concentration to eradicate solid tumors [2, 5, 15, 47]. To further determine the penetration ability of aptamer in comparison with antibody in vivo, NOD/SCID mice bearing xenograft colorectal tumous with a tumor volume of 150 mm3 received a bolus i.v. injection of 2 nmol mouse of PEGylated-aptamer or antibody. At 3 h or 24 h post injection, the mice were sacrificed and the formalin fixed paraffin embedded tumor sections were prepared for double staining of blood vessels and aptamer or antibody. To evaluate tumor penetration in vivo, the FITC-labeled aptamer or antibody were detected with an anti-FITC antibody and visualized with the aid of chromogenic substrates 3.3' diaminobenzidine (DAB); while the blood vessels were marked by anti-mouse CD31 antibody and visualized using chromogenic alkaline phosphatase solution. The distribution of aptamer or antibody in tumor sections was assessed by quantification of pixel intensity of aptamer or antibody in relation to distances from the blood vessels within the selected region of interest using Image Pro software. Shown in Fig. 6a are representative images of time-dependent distribution of PEGylated aptamers or antibodies (brown) in relation to blood vessels (red, marked by black arrows) in HT29 xenografts sections. These images show that the majority of brown staining for antibodies was localized near blood vessels, whereas regions away from vessels of the tumors show ample brown staining for aptamers. The quantification of intensity of aptamer or antibody in relation to distances from blood vessels at 3 h after intravenous injection revealed that there were significant differences in the intensity of signals between aptamers and antibodies throughout the entire 200 μm distances from the blood vessels (Fig. 6b). In fact, aptamer achieved at least 4-time better tumor penetration than that of the antibody throughout the tumor spaces studied after 3 h i.v. injection. Importantly, 24 h after administration, intratumoral antibody was hardly discernable even in the close range (20 µm) of the blood vessels. In contrast, the aptamer remained detectable even at regions as far as 200 µm away from the blood vessels (Fig. 6c). These data indicate that aptamers, even after PEGylation, can penetrate tumors much more extensively and efficiently than antibodies. Therefore, there is a great potential for aptamers to become effective modalities for targeted tumor therapeutics and imaging.
Time-dependent penetration of PEGylated aptamer and antibody in relation to blood vessels in HT29 xenograft tumors. (a) Representative images of double staining of aptamer or antibody and blood vessels in tumor sections dissected from treated mice-bearing HT29 xenografts 3 h and 24 h after i.v. administration of aptamer or antibody at a dose of 2 nmol/mouse. Blood vessels were stained by chromogenic alkaline phosphatase (black arrow); while aptamer or antibody were stained using DAB peroxidase substrate (brown). Scale bar: 200 μm. (b-c) Quantitative determination of staining intensity against a given perpendicular distance (20-200 μm) to the blood vessels at 3 h. (b) and 24 h (c) after i.v. injection of aptamer or antibody. Data are means ± SEM (n=8).
Discussion
Recently, increasing data suggest that the size of nanomedicines is critically important for the deep penetration and efficacy of therapeutic agents to be transported into tumor tissues in vivo, which is vital for achieving a curable outcome [48-50]. Therapeutic antibodies are important agents used in clinics, though there have been mixed results in the clinics as tumor immunotherapy [38, 51-53]. Monoclonal antibodies with large molecule weight have been reported to possess limited penetration into solid tumors, as evident from a steep concentration gradient inside a tumor [54, 55]. The mixed outcome of antibody-based therapeutics has been attributed, at least partly, to the large size and binding affinity of the immunoglobulin, as well as immunogenicity [53, 54, 56, 57]. Aptamers, as “chemical antibodies”, have a molecule weight and size approximately 25-fold smaller than that of monoclonal antibodies. And are thus superior to antibodies for in vivo application due to the lack of the Fc fragment and thus do not provide an immune response [10]. Despite 24 years of research into aptamers, however, there is a paucity in our knowledge in terms of difference in the ability of aptamer and antibody in tumor penetration. To our knowledge, this study is the first thorough investigation that compares tumor accumulation and penetration between an aptamer and its antibody counterpart both in vitro and in vivo.
Tumor-targeting efficacy of monoclonal antibodies requires sufficient binding affinity for durable tumor internalization and retention. However, this does not mean that an antibody has to possess extremely high affinity to be therapeutically relevant [35]. Adams et al. revealed that for a tumor-targeting ligand, a K'd value lower than 1 nM has a detrimental effect on its tumor penetration, and in fact limits further transport and perfusion into tumors [16, 35]. This binding site barrier is caused by high-affinity binding of the antibody to the first few layers of tumor cells that the antibodies encountered after extravasation, resulting in a reduction of the amounts of available free antibodies for further penetration into tumors and leaving many viable tumor cells untargeted [1, 11, 25, 36, 37]. Accordingly, targeting ligands with moderate affinity would be desirable to the development of optimal therapeutics as they could effectively perfuse and penetrate into tumors with a durable retention, especially when therapeutic efficacy mostly relies on uniform drug delivery to tumor cells [37, 58]. Of note, the moderate binding affinity of both the EpCAM aptamer (K'd = 39.42 nM) and antibody (K'd = 5.18 nM) used in this study are well above the 1 nM threshold for triggering the “binding site barrier” effect. Therefore, the K'd of the EpCAM aptamer and EpCAM antibody employed in this study are unlikely to impose adverse effects on the diffusion process given the extensive literature on the triggering K'd for binding site barrier being lower than 1 nM. Previous work by others has shown the importance of the size of a molecule in tumor penetration [14, 16, 35]. For example, despite being only 2-fold larger in size than the scFv, Fab fragments display a 16 h delay in moving a distance of 1 mm into a solid tumor [16]. Thus, the observed difference in tumor penetration and retention between the aptamer and the antibody is likely derived from the differences in the molecular weight and size.
We started our comparison of tumor targeting by aptamer and antibody by the examination of cellular internalization upon binding. As shown in Fig. 1c and d, aptamers exhibited at least 8-fold increase in entrance into endosomes and lysosomes than antibodies during a 15 min incubation. The efficient transport into acidic cellular organelles is critically important for a cancer-targeting modality to be used as a theranostic agent as this will facilitate the release of chemotherapy drugs and imaging reporter molecules for Positron emission tomography (PET), single-photon emission computerized tomography (SPECT), computerized tomography (CT) and magnetic resonance imaging (MRI) [59]. The observed marked difference in endocytosis is unlikely the result of the antibody or aptamer engaging different receptor-mediated endocytic pathways in a cell-line specific manner, as similar differences in the efficiency of endocytosis were observed in three different types of cancer cells. However, in the absence of the exact knowledge of the epitope(s) on the EpCAM protein that the aptamer and the antibody binds, one cannot entirely exclude the possibility of the influence from the binding to different epitopes of the target on the different efficiency of endocytosis revealed for the EpCAM antibody and aptamer, although it is unlikely.
The 3D tumorsphere culture model has been proven to possess unique advantages over the monolayer culture system for cancer research [27, 28]. In the sphere model, cells are shaped tightly, which is similar to the condition of tumor tissue in vivo, providing an ideal model for determining the penetration behavior of aptamer and antibody. For example, cells in the outer region of the sphere were exposed to sufficient oxygen and nutrients that corresponded to the tumor sites close to the blood vessel. Moreover, cells located in the inner core were quiescent and more drug resistant due to the decreased supply of oxygen and nutrients, similar to tumor tissues far from blood vessels [15, 28, 60]. Our study in comparing the tumorsphere penetration by aptamer and antibody revealed the aptamer could not only penetrate a tumorsphere efficiently, but could also be retained for at least 24 h (Fig. 2a and Supplementary Fig. 2). In contrast, the EpCAM antibody barely reached the core of tumorsphere after 240 min incubation, and had poor retention as it almost disappeared from the core of the tumorsphere after 4 h (Fig 2a and c). The superior ability of aptamer to antibody in penetrating and being retained in the core of the tumorsphere uncovered in this study highlights an invaluable attribute of aptamers for molecular imaging and targeted therapy.
To be clinical useful as in vivo molecular imaging agents in oncology, an imaging probe/ligand should display a rapid uptake in target sites with a maximum tumor uptake at 15 min followed by a rapid blood clearance [20]. Our in vivo imaging data demonstrate that the EpCAM aptamer was taken up by the tumor swiftly, reaching a peak signal at around 10 min followed by a rapid decline of the signal in tumor up to 80 min (Fig. 3a and b). In addition, the aptamer displayed a durable accumulation and retention in the tumor area within the window of imaging as it had approximately 50% of signal retained within 30 min. Taken together, these parameters suggest a potential match with short half-life radiotracer species such as typical isotopes (fluorine-18, carbon-11 and nitrogen-13) used in PET scanning [20, 61]. In contrast, the EpCAM antibody took 3 h to reach a maximum uptake after i.v. injectionand showed a slow blood clearance over a 6 h period. These data suggest that the EpCAM aptamer would produce a much better clinically viable signal-to-background ratio than the EpCAM antibody for molecular imaging applications.
Generally, RNA-based drugs have limited stability due to the susceptibility to endonuclease or exonuclease cleavage, which can be mitigated by the 2′-fluoropyrimidine modifications or terminal modifications such as PEGylation. The PEGylated EpCAM was found to have a serum half-life of approximately 14 hours (Supplementary Fig. 3). In order to facilitate in vivo therapeutic delivery, the EpCAM aptamer conjugated with a 20 kDa PEG was used in our in vivo studies to prolong serum retention and minimize systemic clearance without compromising its binding affinity (Fig. 4b, c and d). In our live animal imaging study for therapeutic application, the signal of PEGylated aptamer in the tumor persisted more than 24 h, which is consistent with the period of aptamer retention in the tumorsphere model. In contrast, the antibody produced only a short-lasting signal for up to 6 h (Fig. 4e and f). The 4.3-fold increase of tumor targeting of aptamer than that of antibody indicates that aptamer is a valuable class of targeting ligand for targeted drug delivery.
The distribution of monoclonal antibody (trastuzumab) in HER2 overexpressing xenografts (MDA-435/LCC6HER2) was reported to display considerable intervessel heterogeneity following a single i.p. injection of 20 mg/kg trastuzumab. The functional distance of trastuzumab, derived from average intensity, from nearest microvessel was up to 150 μm throughout the tumor cord [62]. This is consistent with the distribution profile of the EpCAM antibody we observed within the tumor in that EpCAM antibody was hardly discernable at 150 - 200 μm away from the blood vessel (Fig. 6). In contrast, the EpCAM aptamer displayed at least 4-fold better tumor penetration than that of the antibody at all distances (20 - 200 μm) from the blood vessels studied after 3 h i.v. injection. Work from several independent laboratories have revealed the heterogeneous distribution of monoclonal antibodies at various times after their administration [16, 62, 63]. Dennis et al. used intravital microscopy to detect perivascular localization of FITC-conjugated trastuzumab at 24 hours after i.v. injection of 10 mg/kg (about 0.25 mg/mouse) to mice bearing allograft mammary tumors [17]. Compared to the deep and even penetration of Fab4D5 (a 52 kDa antibody fragment), limited penetration and heterogeneous distribution of trastuzumab (only 4 cell layers) were observed 24 h post-treatment. It has been appreciated that size plays a role in affecting a targeting agent to penetrate tissues. Intact antibodies tend to localize besides to blood vessels and are less uniform in distribution throughout tumors, while smaller agents possess the ability to penetrate depths of 8 to 10 cell layers [55, 64]. This is consistent with our findings that the EpCAM aptamer established a 2-fold higher and more uniform distribution than that of antibody in tumors 24 h after i.v. injection, even though there was less amount of aptamers present at 24 h compared with that at 3 h (Fig. 6). Furthermore, assuming the average size of a cancer cell being 15 µm, at the distance of 200 µm away from the blood vessels, the EpCAM aptamers would have travelled a distance that is well beyond 10 cell layers from the blood vessels at both 3 h and 24 h. Therefore, our results suggests that aptamers have superior ability of tumor penetration and thus possess great potential as a targeting moiety for targeted therapeutics as well as in molecular imaging in which deep and uniform tumor penetration is desirable.
In summary, the EpCAM aptamers studied, as chemical antibodies, possess a number of superior attributes over monoclonal EpCAM antibodies in terms of tumor penetration, accumulation and retention. Consistent with the criteria of an ideal in vivo imaging agent [44, 45], the EpCAM aptamer displays a rapid tumor uptake at around 10 min followed by a rapid clearance. This parameter suggests that the EpCAM aptamers used in this study could be used as an attractive alternative to monoclonal antibodies for in vivo imaging for imaging tumors using radioactive tracers. The EpCAM aptamer can also be chemically modified including PEGylation to achieve a better pharmacokinetic performance for targeted drug delivery. The superior tumor penetration ability demonstrated for the tested EpCAM aptamers compared to the antibody counterpart makes aptamers promising escort modalities for the development of active targeting-based theranostics for both cancer therapy and in vivo imaging without the side effects associated with conventional immunotherapy.
Supplementary Material
Supplementary Figures 1-3.
Acknowledgements
D. Xiang was supported by a Deakin University Postgraduate Research Scholarship. This work was supported by grants from Indo-Australia Science and Technology Fund. (Grant No. ST040007), Victorian Cancer Agency Platform Technology Capacity Building Grant (Grant No. PTCP-02) and CASS Foundation (Australia).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Muchekehu R, Liu D, Horn M. et al. The Effect of Molecular Weight, PK, and Valency on Tumor Biodistribution and Efficacy of Antibody-Based Drugs. Transl Oncol. 2013;6(5):562-72
2. Tunggal JK, Cowan DS, Shaikh H, Tannock IF. Penetration of anticancer drugs through solid tissue: a factor that limits the effectiveness of chemotherapy for solid tumors. Clin Cancer Res. 1999;5(6):1583-6
3. Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF. The distribution of the anticancer drug Doxorubicin in relation to blood vessels in solid tumors. Clin Cancer Res. 2005;11(24 Pt 1):8782-8
4. O'Connor ML, Xiang D, Shigdar S. et al. Cancer stem cells: A contentious hypothesis now moving forward. Cancer Lett. 2014;344(2):180-7
5. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6(8):583-92
6. Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 1998;58(7):1408-16
7. Leu AJ, Berk DA, Lymboussaki A, Alitalo K, Jain RK. Absence of functional lymphatics within a murine sarcoma: a molecular and functional evaluation. Cancer Res. 2000;60(16):4324-7
8. Heldin C-H, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure - an obstacle in cancer therapy. Nat Rev Cancer. 2004;4(10):806-13
9. Brown E, McKee T, diTomaso E. et al. Dynamic imaging of collagen and its modulation in tumors in vivo using second-harmonic generation. Nat Med. 2003;9(6):796-800
10. Zeng Z, Parekh P, Li Z, Shi Z-Z, Tung C-H, Zu Y. Specific and sensitive tumor imaging using biostable oligonucleotide aptamer probes. Theranostics. 2014;4(9):945-52
11. Thurber GM, Schmidt MM, Wittrup KD. Factors determining antibody distribution in tumors. Trends Pharmacol Sci. 2008;29(2):57-61
12. Waldmann TA, Morris JC. Development of antibodies and chimeric molecules for cancer immunotherapy. Adv Immunol. 2006;90:83-131
13. Boyiadzis M, Foon KA. Approved monoclonal antibodies for cancer therapy. Expert Opin Biol Ther. 2008;8(8):1151-8
14. Zahnd C, Kawe M, Stumpp MT. et al. Efficient tumor targeting with high-affinity designed ankyrin repeat proteins: effects of affinity and molecular size. Cancer Res. 2010;70(4):1595-605
15. Huo S, Ma H, Huang K. et al. Superior penetration and retention behavior of 50 nm gold nanoparticles in tumors. Cancer Res. 2013;73(1):319-30
16. Adams GP, Schier R, McCall AM. et al. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001;61(12):4750-5
17. Dennis MS, Jin H, Dugger D. et al. Imaging tumors with an albumin-binding Fab, a novel tumor-targeting agent. Cancer Res. 2007;67(1):254-61
18. Cobleigh MA, Vogel CL, Tripathy D. et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17(9):2639-48
19. Xiang D, Shigdar S, Qiao G. et al. Nucleic Acid Aptamer-Guided Cancer Therapeutics and Diagnostics: the Next Generation of Cancer Medicine. Theranostics. 2015;5(1):23-42
20. Hicke BJ, Stephens AW, Gould T. et al. Tumor targeting by an aptamer. J Nucl Med. 2006;47(4):668-78
21. Watson SR, Chang YF, O'Connell D, Weigand L, Ringquist S, Parma DH. Anti-L-selectin aptamers: binding characteristics, pharmacokinetic parameters, and activity against an intravascular target in vivo. Antisense Nucleic Acid Drug Dev. 2000;10(2):63-75
22. Shi H, He X, Wang K. et al. Activatable aptamer probe for contrast-enhanced in vivo cancer imaging based on cell membrane protein-triggered conformation alteration. Proc Natl Acad Sci U S A. 2011;108(10):3900-5
23. Shi H, Cui W, He X. et al. Whole cell-SELEX aptamers for highly specific fluorescence molecular imaging of carcinomas in vivo. PLoS One. 2013;8(8):e70476
24. Scaggiante B, Dapas B, Farra R. et al. Aptamers as targeting delivery devices or anti-cancer drugs for fighting tumors. Curr Drug Metab. 2013;14(5):565-82
25. Pei X, Zhang J, Liu J. Clinical applications of nucleic acid aptamers in cancer. Mol Clin Oncol. 2014;2(3):341-8
26. Choi I-K, Strauss R, Richter M, Yun C-O, Lieber A. Strategies to increase drug penetration in solid tumors. Front Oncol. 2013;3:193
27. Pickl M, Ries CH. Comparison of 3D and 2D tumor models reveals enhanced HER2 activation in 3D associated with an increased response to trastuzumab. Oncogene. 2009;28(3):461-8
28. Ong S-M, Zhao Z, Arooz T. et al. Engineering a scaffold-free 3D tumor model for in vitro drug penetration studies. Biomaterials. 2010;31(6):1180-90
29. Chushak Y, Stone MO. In silico selection of RNA aptamers. Nucleic Acids Res. 2009;37(12):e87
30. June CH, Blazar BR, Riley JL. Engineering lymphocyte subsets: tools, trials and tribulations. Nat Rev Immunol. 2009;9(10):704-16
31. Woloszynska-Read A, James SR, Song C, Jin B, Odunsi K, Karpf AR. BORIS/CTCFL expression is insufficient for cancer-germline antigen gene expression and DNA hypomethylation in ovarian cell lines. Cancer Immun. 2010;10:6
32. Wang HH, Xu G, Vonner AJ, Church G. Modified bases enable high-efficiency oligonucleotide-mediated allelic replacement via mismatch repair evasion. Nucleic Acids Res. 2011;39(16):7336-47
33. Li N, Ebright JN, Stovall GM. et al. Technical and biological issues relevant to cell typing with aptamers. J Proteome Res. 2009;8(5):2438-48
34. Lee CM, Tannock IF. The distribution of the therapeutic monoclonal antibodies cetuximab and trastuzumab within solid tumors. BMC Cancer. 2010;10:255
35. Rudnick SI, Lou J, Shaller CC. et al. Influence of affinity and antigen internalization on the uptake and penetration of Anti-HER2 antibodies in solid tumors. Cancer Res. 2011;71(6):2250-9
36. Fujimori K, Covell DG, Fletcher JE, Weinstein JN. Modeling analysis of the global and microscopic distribution of immunoglobulin G, F(ab')2, and Fab in tumors. Cancer Res. 1989;49(20):5656-63
37. Fujimori K, Covell DG, Fletcher JE, Weinstein JN. A modeling analysis of monoclonal antibody percolation through tumors: a binding-site barrier. J Nucl Med. 1990;31(7):1191-8
38. Shigdar S, Lin J, Yu Y, Pastuovic M, Wei M, Duan W. RNA aptamer against a cancer stem cell marker epithelial cell adhesion molecule. Cancer Sci. 2011;102(5):991-8
39. Hussain S, Pluckthun A, Allen TM, Zangemeister-Wittke U. Antitumor activity of an epithelial cell adhesion molecule targeted nanovesicular drug delivery system. Mol Cancer Ther. 2007;6(11):3019-27
40. Zangemeister-Wittke U. Antibodies for targeted cancer therapy -- technical aspects and clinical perspectives. Pathobiology. 2005;72(6):279-86
41. Shigdar S, Qiao L, Zhou S-F. et al. RNA aptamers targeting cancer stem cell marker CD133. Cancer Lett. 2013;330(1):84-95
42. Perche F, Patel NR, Torchilin VP. Accumulation and toxicity of antibody-targeted doxorubicin-loaded PEG-PE micelles in ovarian cancer cell spheroid model. J Control Release. 2012;164(1):95-102
43. Santini MT, Rainaldi G. Three-dimensional spheroid model in tumor biology. Pathobiology. 1999;67(3):148-57
44. Guhlke S, Famulok M, Biersack HJ. Aptamares: a novel class of radiopharmaceutical with diagnostic and therapeutic potential. Eur J Nucl Med Mol Imaging. 2003;30(11):1441-3
45. Zhu H, Li J, Zhang XB, Ye M, Tan W. Nucleic Acid aptamer-mediated drug delivery for targeted cancer therapy. ChemMedChem. 2015;10(1):39-45
46. Choi HS, Liu W, Misra P. et al. Renal clearance of quantum dots. Nat Biotechnol. 2007;25(10):1165-70
47. Jain RK. Barriers to drug delivery in solid tumors. Sci Am. 1994;271(1):58-65
48. Huang K, Ma H, Liu J. et al. Size-dependent localization and penetration of ultrasmall gold nanoparticles in cancer cells, multicellular spheroids, and tumors in vivo. ACS Nano. 2012;6(5):4483-93
49. Tang L, Gabrielson NP, Uckun FM, Fan TM, Cheng J. Size-dependent tumor penetration and in vivo efficacy of monodisperse drug-silica nanoconjugates. Mol Pharm. 2013;10(3):883-92
50. Huo S, Ma H, Huang K. et al. Superior penetration and retention behavior of 50 nm gold nanoparticles in tumors. Cancer Res. 2013;73(1):319-30
51. Das M, Mohanty C, Sahoo SK. Ligand-based targeted therapy for cancer tissue. Expert Opin Drug Deliv. 2009;6(3):285-304
52. White RR, Sullenger BA, Rusconi CP. Developing aptamers into therapeutics. J Clin Invest. 2000;106(8):929-34
53. McLaughlin PM, Kroesen BJ, Harmsen MC, de Leij LF. Cancer immunotherapy: insights from transgenic animal models. Crit Rev Oncol Hematol. 2001;40(1):53-76
54. Oberneder R, Weckermann D, Ebner B. et al. A phase I study with adecatumumab, a human antibody directed against epithelial cell adhesion molecule, in hormone refractory prostate cancer patients. Eur J. Cancer. 2006;42(15):2530-8
55. Sutherland R, Buchegger F, Schreyer M, Vacca A, Mach JP. Penetration and binding of radiolabeled anti-carcinoembryonic antigen monoclonal antibodies and their antigen binding fragments in human colon multicellular tumor spheroids. Cancer Res. 1987;47(6):1627-33
56. Schmidt M, Scheulen ME, Dittrich C. et al. An open-label, randomized phase II study of adecatumumab, a fully human anti-EpCAM antibody, as monotherapy in patients with metastatic breast cancer. Ann Oncol. 2010;21(2):275-82
57. Schwartzberg LS. Clinical experience with edrecolomab: a monoclonal antibody therapy for colorectal carcinoma. Crit Rev Oncol Hematol. 2001;40(1):17-24
58. Juweid M, Neumann R, Paik C. et al. Micropharmacology of monoclonal antibodies in solid tumors: direct experimental evidence for a binding site barrier. Cancer Res. 1992;52(19):5144-53
59. Shigdar S, Macdonald J, O'Connor M. et al. Aptamers as theranostic agents: modifications, serum stability and functionalisation. Sensors (Basel). 2013;13(10):13624-37
60. Fracasso G, Colombatti M. Effect of therapeutic macromolecules in spheroids. Crit Rev Oncol Hematol. 2000;36(2-3):159-78
61. Kinahan PE, Fletcher JW. Positron emission tomography-computed tomography standardized uptake values in clinical practice and assessing response to therapy. Semin Ultrasound CT MR. 2010;31(6):496-505
62. Baker JHE, Lindquist KE, Huxham LA, Kyle AH, Sy JT, Minchinton AI. Direct visualization of heterogeneous extravascular distribution of trastuzumab in human epidermal growth factor receptor type 2 overexpressing xenografts. Clin Cancer Res. 2008;14(7):2171-9
63. Jones PL, Gallagher BM, Sands H. Autoradiographic analysis of monoclonal antibody distribution in human colon and breast tumor xenografts. Cancer Immunol Immunother. 1986;22(2):139-43
64. Yokota T, Milenic DE, Whitlow M, Schlom J. Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. Cancer Res. 1992;52(12):3402-8
Author contact
![]() Corresponding authors: School of Medicine, Deakin University, Waurn Ponds, Victoria 3216, Australia. Tel.: +61 - 3 5227 1149; Fax: +61 - 3 5227 2945; E-mail: sarah.shigdaredu.au (S. Shigdar) and wduanedu.au (W. Duan).
Corresponding authors: School of Medicine, Deakin University, Waurn Ponds, Victoria 3216, Australia. Tel.: +61 - 3 5227 1149; Fax: +61 - 3 5227 2945; E-mail: sarah.shigdaredu.au (S. Shigdar) and wduanedu.au (W. Duan).