Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(2):219-230. doi:10.7150/thno.13178 This issue Cite
Research Paper
BET Bromodomain Inhibition as a Therapeutic Strategy in Ovarian Cancer by Downregulating FoxM1
Zhenfeng Zhang1,2,*, Pengfei Ma1, *, Ying Jing1, *, Ying Yan3, Mei-Chun Cai4, Meiying Zhang5,6, Shengzhe Zhang7, Huixin Peng1, Zhi-Liang Ji4, Wen Di5,6, Zhenyu Gu3, Wei-Qiang Gao1, 7, Guanglei Zhuang1,6 ![]()
1. State Key Laboratory of Oncogenes and Related Genes, Renji-Med X Clinical Stem Cell Research Center, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
2. State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
3. GenenDesign Co., Ltd, Shanghai, China
4. State Key Laboratory of Cellular Stress Biology, School of Life Sciences, Xiamen University, Xiamen, Fujian, China
5. Department of Obstetrics and Gynecology, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
6. Shanghai Key Laboratory of Gynecologic Oncology, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
7. School of Biomedical Engineering & Med-X Research Institute, Shanghai Jiao Tong University, Shanghai, China
*These authors contributed equally to this work.
Received 2015-7-7; Accepted 2015-11-7; Published 2016-1-1
Abstract

Ovarian cancer is responsible for the highest mortality among all gynecologic malignancies, and novel therapies are urgently needed to improve patient outcome. Here we performed an integrative genomic analysis and identified the bromodomain and extraterminal domain (BET) protein BRD4 as a potential therapeutic target in ovarian cancer. Suppression of BRD4 using small-molecule BET inhibitors JQ1 and I-BET151, or dual kinase-bromodomain inhibitor volasertib, led to robust and broad antitumor effects across all subclasses of ovarian cancer. In contrast to many other cancers which are susceptible to BET inhibition due to downregulation of super-enhancer-dependent MYC transcript, we discovered that JQ1-sensitive ovarian cancer cells exhibited marked disruption of Forkhead box protein M1 (FoxM1) pathway, a key driver of ovarian carcinoma. These in vitro findings were further supported by in vivo efficacies of JQ1 targeting both cell line-based and patient-derived xenograft models. Our data establish a new treatment strategy against ovarian cancer by employing epigenetic vulnerabilities, and provide a mechanistic rationale for the clinical investigation of BET bromodomain inhibitors in this deadly disease.
Keywords: ovarian cancer, BET inhibitors, BRD4, FoxM1
Introduction
Epithelial ovarian cancer (EOC) is the fifth most common cancer type in women and remains a significant cause of gynecological cancer mortality, with 140,200 deaths per year globally [1, 2]. The standard treatment is debulking surgery followed by taxane-platinum chemotherapy. Despite initial high response rate, most patients will relapse and when this occurs, ovarian cancer is currently incurable. Therefore, there is an urgent need for new treatment options to improve the therapeutic index [3, 4].
Ovarian cancer is a diverse and genomically complex disease. On the basis of histological characteristics, ovarian tumors of epithelial origin can be categorized into at least five histotypes including high-grade serous, low-grade serous, clear cell, endometrioid and mucinous [1, 5, 6]. Recent genomic and molecular studies have complemented the conventional classification of EOC, revealing heterogeneous genomic and epigenomic abnormalities underlying tumor pathophysiology [7-9]. Importantly, this emerging knowledge base enables integrated analyses to uncover the biological drivers of ovarian cancer. For example, The Cancer Genome Atlas (TCGA) project has reported that the FoxM1 transcription factor network is significantly altered in 87% of high-grade serous ovarian cancers (HGS-OvCa), indicative of tumor dependency [7]. However, these cancer-associated pathways are often undruggable and can not be immediately served as therapeutic targets. As a result, with only several exceptions such as PARP inhibitors being tested in patients with BRCA germline mutations [10-12], molecular targeted strategies against ovarian cancer are largely elusive.
Epigenetic regulators have recently emerged as a new class of therapeutic targets in cancer treatment [13, 14]. In particular, specific inhibitors of the bromodomain and extraterminal domain (BET) proteins have been developed. The BET family proteins, composed of BRD2, BRD3, BRD4 and BRDT, contain two conserved tandem bromodomains and are known as epigenetic “readers” that recognize the acetylated lysine residues on histone tails [15-17]. Small-molecule BET inhibitors such as JQ1 and I-BET mimic the acetyl moiety, occlude the bromodomain's acetyllysine-binding pocket and displace BET proteins from chromatin [18, 19]. BET inhibitors have been extensively evaluated and proven effective in alleviating a growing list of cancers including NUT midline carcinoma, multiple myeloma, leukemia, lymphoma, lung adenocarcinoma, neuroblastoma, medulloblastoma, glioblastoma and prostate cancer [18, 20-27]. The efficacy of BET inhibitors was initially attributed mainly to their ability to suppress MYC, an oncogene marked by BRD4-loaded super-enhancers [20, 28, 29], although recent studies have proposed different modes of action [21, 23]. Nevertheless, the potential activity of BET inhibitors and the central BET-dependent transcriptional program in ovarian cancer have been largely unexplored.
In an effort to identify novel therapeutic targets in ovarian cancer, we performed an integrative genomic analysis and discovered that BRD4 was frequently amplified and correlated with poor prognosis in HGS-OvCa patients. Pharmacological inhibition of BRD4 using JQ1 or I-BET151 substantially abrogated both in vitro growth and in vivo tumorigenesis of ovarian cancer. Unexpectedly, transcriptome profiling revealed that JQ1 selectively downregulated the oncogenic transcription factor FoxM1 and its downstream targets instead of MYC transcriptional machinery. These findings indicate that BET bromodomain inhibition is a promising epigenetic-based treatment avenue to target ovarian cancer, with mechanism of action uniquely reliant on FoxM1 downregulation.
Materials and methods
Cell culture and reagents
Tumor cell lines were obtained from ATCC and were cultured in RPMI1640 (Invitrogen) supplemented with 10% fetal bovine serum (Millipore). Retroviral vector (pBABE) which contains FoxM1 ORF (FoxM1 1b; NM_021953.3) was transfected into HEK293T cells with packaging mixtures. Virus was collected, filtered and then incubated with target cells in growth medium containing 8μg/ml polybrene (Millipore). Infected cells were selected with 5μg/ml puromycin. For FoxM1 knockdown, siRNA sequences (Dharmacon) were transfected with Lipofectamine RNAiMAX Reagent (Invitrogen). JQ1 and (-)-JQ1 were purchased from Millipore. I-BET151 was purchased from Selleck Chemicals. All inhibitors were reconstituted in DMSO (Sigma-Aldrich) at a stock concentration of 10 mM.
Cell line screening
Cell line screening was performed in a 96-well format. Cells were seeded at optimal density and treated with the indicated inhibitors. Seven concentrations of compounds were applied at a stepwise 3-fold dilution series. Fresh medium and drugs were changed every three days. After six days of drug exposure, cell viability was evaluated using CellTiter-Glo reagent according to the manufacturer's instructions (Promega). Estimates of IC50 were derived from the 7 dose-response curves plotted by GraphPad Prism 6 (GraphPad Software, Inc.).
Cell cycle analysis
Cell cycle analysis was performed 24 hours after JQ1 treatment. Cells were fixed in cold ethanol and resuspended in Propidium Iodide (PI)/RNase Staining Solution (Cell Signaling Technology). After incubation for 15 minutes at room temperature in the dark, flow cytometric analysis was performed on a FACS AriaII cytometer (BD Biosciences). Flow cytometry data was analyzed by using FlowJo software and the cell cycle was plotted as histogram after excluding doublets.
Western blot
Cells were lysed in RIPA buffer (Tris pH 7.4 50mM, NaCl 150mM, NP-40 1%, SDS 0.1%, EDTA 2μM) containing proteinase inhibitors (Roche) and phosphatase inhibitors (Roche). The cell lysates (20μg protein) were subjected to SDS-PAGE and Western blot. Antibodies against the following proteins were used: BRD2, BRD3, BRD4 (Abcam); c-MYC, Hsp90, FoxM1, AURKB, survivin, cyclinB, PLK1, H3, Actin (Cell Signaling Technology).
Microarray analysis and quantitative PCR
RNA was prepared with RNeasy plus mini kit (Qiagen) according to the manufacturer's protocol. Total RNA was subjected to microarray analysis using Affymetrix human genome U133 Plus 2.0. Three biological replicates per treatment group were included for statistical analyses. Affymetrix microarray probe-level data were normalized by Robust Multi-array Average (RMA) procedure. Differential gene expression was analyzed with linear models for microarray data (Limma). TaqMan gene expression assays (Applied Biosystems) were performed to verify the microarray results. Relative expression levels of each gene were normalized to human beta-actin. At least three biological replicates were included for each condition. TissueScan cDNA arrays were purchased from OriGene Technologies.
Chromatin precipitation
Chromatin precipitation was performed as previously described [30]. Briefly, cells were cross-linked with 1% formaldehyde and quenched with glycine. Cell pellets were lysed and sonicated using Covaris M220 Ultrasonicator. Sonicated lysates were cleared and incubated overnight with magnetic beads bound with IgG, BRD4 (Abcam) or FoxM1 (Cell Signaling Technology) antibodies to enrich for DNA fragments associated with the indicated protein. Precipitated complexes were washed. Cross-links were reversed overnight. RNA and protein were digested using RNase A and Proteinase K, respectively. DNA was purified with Qiaquick PCR purification kit. Primer sequences used for qPCR are as follows:
5′- CTGTGATCCAGCCACT -3′ (FoxM1-1 forward)
5′- GTCCCATTTCTGTTCC -3′ (FoxM1-1 reverse)
5′- CCAGCCTTAATTTCCT -3′ (FoxM1-2 forward)
5′- GAGTTTGAGACCAGCC -3′ (FoxM1-2 reverse)
5′- CTGAGCACAGTGGGAGG -3′ (FoxM1-3 forward)
5′- AAGCAGGGCATTCACC -3′ (FoxM1-3 reverse)
5′- ACACCCACTTCCCTCC -3′ (FoxM1-4 forward)
5′- GGCACTACCGCTTCAC -3′ (FoxM1-4 reverse)
5′- GCTGAGGTGGGTGAAT -3′ (BIRC5 forward)
5′- GAGTCTTGCTCTGTGGC -3′ (BIRC5 reverse)
5′- TTGACAAGGATGGGAATA -3′ (PLK1 forward)
5′- CAGCACTTAGGGAGGC -3′ (PLK1 reverse)
5′- GCGGTGGCTCTGGTGAA -3′ (CCNB1 forward)
5′- AGGCTGGTCTCAAACTCC -3′ (CCNB1 reverse)
5′- GGGCGATAGAGCGAGTC -3′ (AURKB forward)
5′- CAGTATTCCTTCCACCT -3′ (AURKB reverse)
Luciferase reporter assay
The promoter segment of human FoxM1 was cloned into a mammalian expression vector with luciferase reporter system (GeneCopoeia). HEK293T cells were transfected in 6-well plates using Lipofectamine 2000 (Invitrogen). Forty-eight hours after transfection, luciferase assays were performed using Secrete-Pair Dual Luminescence Assay Kit (GeneCopoeia) according to the manufacturer's instructions. Gaussia luciferase activity was first normalized to secreted alkaline phosphatase expression control. The normalized value for was then normalized to the value obtained from a control promoter construct. Mean values, standard deviations and Student's t-test were calculated from four independent transfections for each condition.
Tumor xenograft and PDX models
Tumor cells (1×106) were mixed with Matrigel (BD Biosciences) and subcutaneously implanted in the dorsal flank of BALB/c Nude mice. When tumor sizes reached approximately 150 mm3, mice were randomized into 2 groups of 10 mice each. One group of mice was treated with vehicle control (5% DMSO in 10% 2-hydroxypropyl-β-cyclodextrin), and the other group was treated with JQ1 50 mg/kg/day. Tumor volumes (10 animals per group) were measured with digital caliper and calculated as length×width2×0.5. All animal protocols were approved by the institute animal care and use committee.
Ovarian PDX model OVA9 was established using patient ovarian tumor tissues acquired during surgery resection. Prior written informed consent was obtained from the patient. Experiments were conducted on female BALB/c nude mice aged 6-8 weeks old. The animals were housed in a specific pathogen free (SPF) animal facility in accordance with the Guide for Care and Use of Laboratory Animals and the regulations of the Institutional Animal Care and Use Committee. Freshly collected tumor samples were cut into small pieces and implanted subcutaneously to the flanks of nude mice. Tumors used in this study were over passage-3 with stable tumor growth. Tumor-bearing mice with tumors at 100-250 mm3 range were selected and randomly divided into vehicle (5% DMSO in 10% 2-hydroxypropyl-β-cyclodextrin) or JQ1 treatment groups with 8 animals per group. Animals were dosed by daily intraperitoneal injection (50 mg/kg JQ1 or vehicle). Tumors were measured twice a week in two dimensions with calipers. Tumor volumes were calculated using the following formula: tumor volume = length×width2×0.5.
Statistical analysis
Genomic analysis of BRD4 was performed with TCGA copy number portal (http://www.broadinstitute.org/tcga), UCSC cancer genomics browser [31], cBioPortal for cancer genomics [32, 33] and Project Achilles [34]. Gene set enrichment analysis was performed using the GSEA software and microarray profiles were tested for enrichment based on the hyper-geometric distribution with respect to MSigDB gene sets [35]. Gene sets significantly enriched with FDR q value ≤0.001 were reported. In all experiments, comparisons between two groups were based on two-sided Student's t-test and one-way analysis of variance (ANOVA) was used to test for differences among more groups. P-values of <0.05 were considered statistically significant.
Results
Integrated analyses identify BRD4 as a therapeutic target in ovarian cancer
Large-scale genomic studies have revealed high prevalence of copy number aberrations but low prevalence of somatic mutations in HGS-OvCa, the major subtype of EOC [7]. A search of TCGA copy number portal identified 37 peak regions of amplification and 46 peak regions of deletion among 579 ovarian serous adenocarcinoma samples. We focused on regions that contain putative therapeutic targets and found that BRD4, located at chromatin 19p13, was amplified in a considerable proportion (~20%) of the cases (Figure 1A). We did not observe significant alterations of BRD2 or BRD3, two other members of the BET family (Supplementary Figure 1A). In addition, pan-cancer analysis [32, 33] indicated that ovarian cancer was the most prominent tumor type displaying BRD4 amplification (Supplementary Figure 1B). Importantly, expression of BRD4 correlated with amplification status (Figure 1B). Consistently, quantitative PCR analysis of an ovarian cancer tissue cDNA array containing 192 clinical specimens showed that BRD4 gene expression was significantly higher in tumors compared with nonmalignant control tissues (Figure 1C), whereas BRD2 or BRD3 levels were not statistically different (Supplementary Figure 1C).
Integrated analyses identify BRD4 as a therapeutic target in ovarian cancer. A. Copy number analysis of BRD4 in TCGA ovarian cancer samples. Color scale: amplification in red and deletion in blue. B. BRD4 gene expression in ovarian cancer with different BRD4 copy number alterations. C. Quantitative PCR of BRD4 in an ovarian cancer tissue cDNA array containing 192 clinical samples. D. Results of shRNA lentiviral screen in SNU840 cells were presented in rank order of ascending shRNA scores. The effect of shRNAs targeting BET proteins was highlighted by colored dots. Gray dots represent results for non-BET shRNAs. E. Kaplan-Meier plot of disease free survival in ovarian cancer patients with or without BRD4 gene amplification. F. Kaplan-Meier plot of overall survival in ovarian cancer patients with or without BRD4 gene amplification.
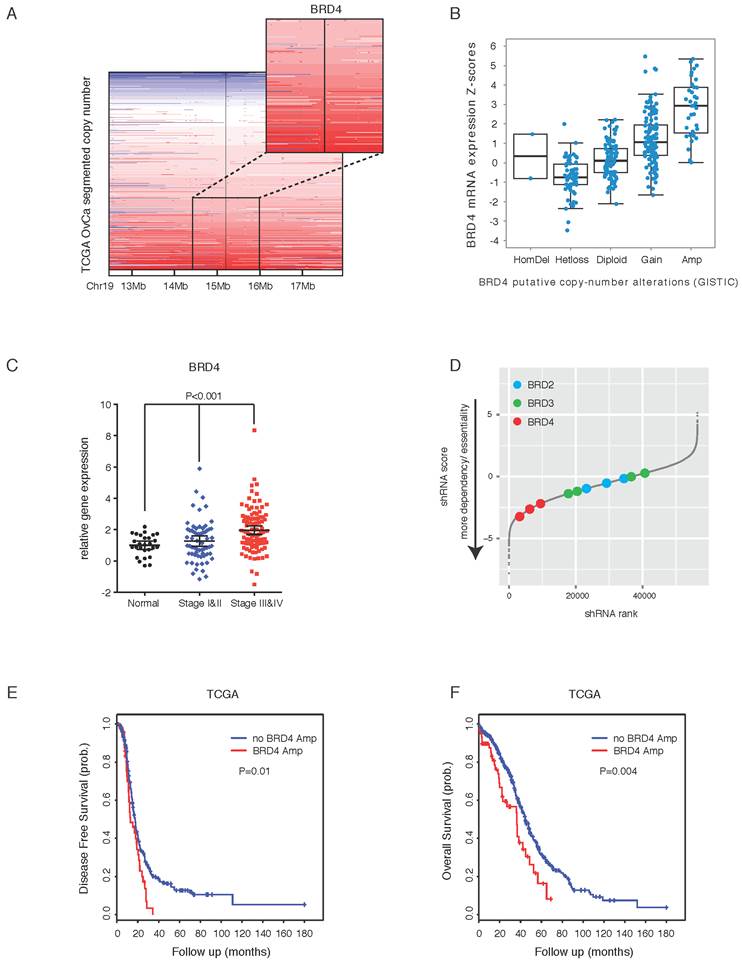
To explore the potential role of BRD4 in ovarian cancer, we first examined the effect of BRD4 short hairpin RNAs (shRNAs) on cell growth in a genome-wide functional screening of cancer cell lines [34]. As illustrated in three ovarian cancer lines (SNU840, TOV-112D, and OVISE), BRD4, but not BRD2 or BRD3, was identified as being essential for cell proliferation (Figure 1D; Supplementary Figure 2A). To investigate the clinical impact of BRD4 on patient prognosis, we performed a meta-analysis using the curatedOvarianData database, which collected standardized gene expression and clinical data for 2970 ovarian cancer patients from 23 studies [36]. BRD4 was a predictor of poor survival in ovarian cancer with mixed stages, grades and histologies (Supplementary Figure 2B; overall hazard ratio=1.08, 95% confidence interval 1.01-1.15). When patients were stratified into two groups based on median BRD4 expression, the BRD4 low group had a significant median overall survival advantage compared to the BRD4 high group (Supplementary Figure 2C). With copy number data, we observed similar results in TCGA cohort, e.g. patients with BRD4 amplification had worse disease-free survival (Figure 1E) and overall survival (Figure 1F). Together, these integrated analyses established a rationale for targeting BRD4 in ovarian cancer.
BET bromodomain inhibition induces pan-subtype cell-cycle arrest in ovarian cancer
We assembled a panel of 28 ovarian cancer cell lines, representing all major histological subtypes of EOC (16 serous, 5 clear cell, 4 mucinous, 3 endometrioid). BET family members, including BRD2, BRD3 and BRD4, were ubiquitously expressed in various ovarian cancer cell lines (Figure 2A). Interestingly, oncoprotein c-MYC, a previously reported BET target [20, 29], showed a more restricted expression pattern. We conducted a pharmacological screen of 28 cell lines to determine the response to BET bromodomain inhibitors. Both JQ1 and I-BET151 exerted broad inhibition on cell viability, without noticeable discrepancy between serous and non-serous ovarian cancer (Figure 2B; Supplementary Table 1). A subset of highly sensitive cell lines demonstrated half maximal inhibitory concentration (IC50) values of less than 1 μM (Figure 2C). Long-term colony-forming assays on 2D or 3D culture consistently showed that proliferation of sensitive cell lines was severely inhibited (Figure 2D; Supplementary Figure 3A&B). It has been recently reported that certain clinical kinase inhibitors also inhibit BET bromodomains with therapeutically relevant potencies [37]. We tested one such dual kinase-bromodomain inhibitor BI-6727 (volasertib), which targets both PLK1 and BRD4, and found that BI-6727 exhibited comparable efficacy to JQ1 in suppressing ovarian cancer cell viability (Supplementary Figure 4A).
BET bromodomain inhibition induces pan-subtype cell-cycle arrest in ovarian cancer. A. Western blot analysis of BRD2, BRD3, BRD4 and c-MYC in a panel of ovarian cancer cell lines. B. Heatmap of IC50s of the BET inhibitors (7-day treatment) in the indicated panel of ovarian cancer cell lines. Color scale: sensitive in red and resistant in black. C. Cell viability was assayed in cells treated with (-)-JQ1, JQ1 or I-BET151 (n = 6 biological replicates). D. Cells were treated with (-)-JQ1, JQ1 (1 μM) or I-BET151 (1 μM) for 10 days. The remaining cells were stained with crystal violet. E. Cell cycle analysis following 24-hour treatment with (-)-JQ1, JQ1 (1 μM) or I-BET151 (1 μM).
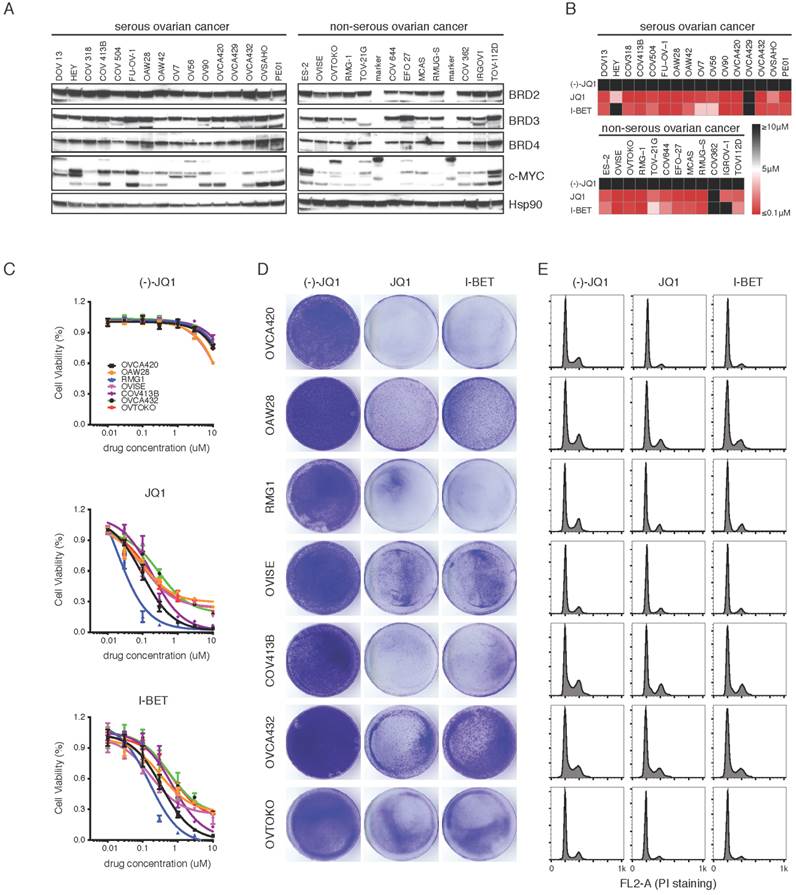
To further characterize the anti-proliferative effects of BET inhibitors, we performed flow cytometry of drug treated cells to determine the consequences on cell-cycle progression and apoptosis. We observed a profound S phase and G2/M peak reduction, indicative of a G0-G1 cell-cycle arrest (Supplementary Figure 4B). Consistent with previous findings in lymphoma [21], BET inhibition did not induce apoptosis in most ovarian cancer cell lines studied, as evidenced by the absence of a sub-G1 fraction (Figure 2E). We conclude that BET bromodomain inhibitors induce cell-cycle arrest and attenuate cell growth in different histological subtypes of ovarian cancer.
BET inhibition represses FoxM1 transcriptional program
To gain insights into the molecular mechanisms through which BET inhibitors led to cell-cycle arrest, we performed kinetic transcriptional profiling of JQ1-treated ovarian cancer cells (OVTOKO). At 24 hours following JQ1 treatment, the most differentially expressed genes were associated with pathways that involve in DNA replication and cell cycle progression, consistent with previous reports [20, 21] and aforementioned phenotype in ovarian cancer (Figure 3A). Additionally, it was of great interest to observe that FoxM1 pathway, one of the central networks activated in HGS-OvCa [7], was significantly altered by JQ1 inhibitor. Indeed, gene set enrichment analysis (GSEA) revealed a marked downregulation of cell-cycle and FoxM1 pathway genes, as well as transcripts in FoxM1-related Aurora-B and PLK1 pathways (Figure 3B). In addition, GSEA of common transcription factor binding motifs (C3; MSigDB v4.0) identified that gene sets with E2F binding sites were significantly downregulated upon JQ1 treatment (Supplementary Figure 5A). BET inhibition suppressed FoxM1 and its proliferation-related target genes in a time-dependent manner (Figure 3C). Analysis of CHIP-sequencing data in multiple cancer cell lines [28, 38] revealed that BRD4 was accumulated to the promoter and enhancer regions of FoxM1 gene, implicating BET proteins in modulating FoxM1 transcription (Supplementary Figure 5B). These results were confirmed by CHIP-qPCR analysis showing that BRD4 was bound to the promoters/enhancers of FoxM1 and FoxM1 target genes (Supplementary Figure 5C). However, BRD4 alone, in contrast to FoxM1, was not sufficient to activate FoxM1 gene expression (Supplementary Figure 5D). Interestingly, c-MYC, a reported BET-regulated oncogene, was only transiently and modestly reduced by JQ1 inhibition and became unchanged at later time points, in contrast to the sustained and dramatic downregulation of FoxM1 (Figure 3D).
BET inhibition represses FoxM1 transcriptional program. A. Enriched gene sets of differentially expressed genes in OVTOKO cells following 24-hour of treatment with JQ1 (1 μM) or DMSO. FoxM1 pathway was highlighted in red. B. GSEA plots of indicated functionally defined gene sets in DMSO versus JQ1 treated OVTOKO cells at 24 hour. C. Heatmap of FoxM1 pathway components in DMSO versus JQ1 treated OVTOKO cells. D. Gene expression of c-MYC and FoxM1 in DMSO versus JQ1 treated OVTOKO cells. E. OVTOKO cells were treated with DMSO, JQ1 (1 μM) or I-BET151 (1 μM). FoxM1 and c-MYC were detected by Western blot. F. OVTOKO cells were treated with JQ1 and fractionated. Cell lysates were immunoblotted with indicated antibodies. G. HEK293T cells were transfected with the indicated expression plasmids, and subjected to co-immunoprecipitation using a BRD4 antibody.
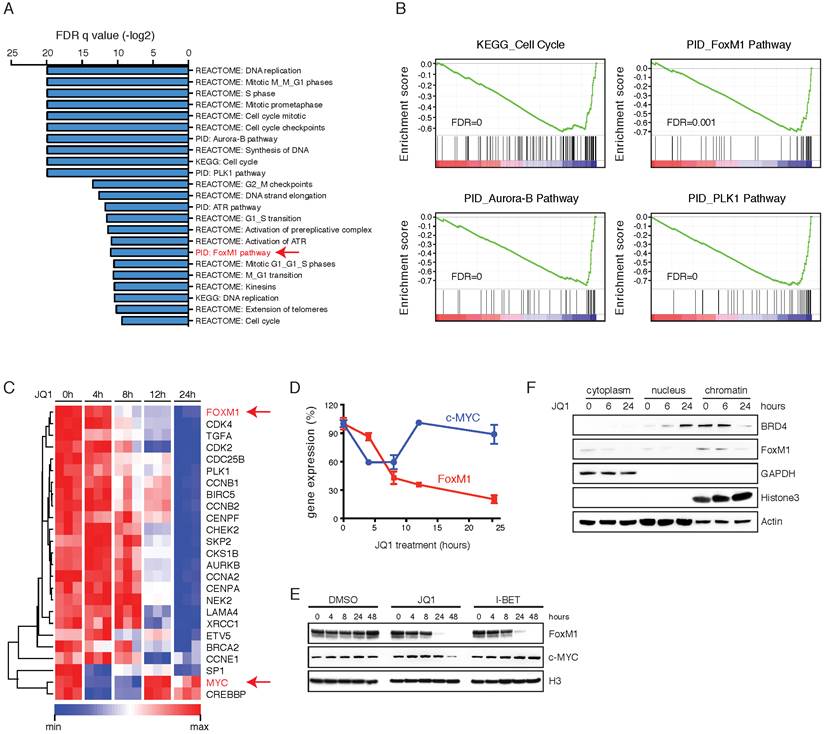
Importantly, BET bromodomain inhibition resulted in a more rapid and dramatic decrease of FoxM1 than c-MYC at the protein level (Figure 3E). Mechanistically, JQ1 displaced BRD4 from chromatin, coupled with reduced FoxM1 protein expression (Figure 3F). Analysis of the TCGA data revealed that FoxM1 expression significantly correlated with BRD4 expression (Supplementary Figure 6A), and that FoxM1 gene amplification was associated with poor prognosis of ovarian cancer patients (Supplementary Figure 6B). Therefore, FoxM1 transcriptional program is more likely the relevant target of BET inhibitors in ovarian cancer.
FoxM1 is a functional target of BET bromodomain inhibitors
We performed quantitative RT-PCR and Western blot analyses of various ovarian cancer cell lines treated with JQ1 or I-BET151, to further validate the inhibition of FoxM1 pathway by BET bromodomain inhibitors. In cell lines that responded to BET bromodomain inhibition, FoxM1, as well as its putative transcriptional targets including AURKB, survivin, cyclinB and PLK1, were significantly downregulated at both mRNA (Figure 4A) and protein (Figure 4B) levels upon JQ1 or I-BET151 treatment.
FoxM1 is a functional target of BET bromodomain inhibitors. A. Quantitative PCR of FoxM1 pathway genes in OVTOKO and OVCA420 cell lines treated with DMSO, JQ1 (1 μM) or I-BET151 (1 μM). Each group had three biological replicates. P<0.05, ANOVA followed by Tukey's post-test. B. OVTOKO and OVCA420 cells were treated with DMSO, JQ1 (1 μM) or I-BET151 (1 μM). Cell lysates were immunoblotted with indicated antibodies. C. FoxM1 was knocked down in OVTOKO cells. Western blot demonstrated FoxM1 knockdown. D. OVTOKO cell growth upon FoxM1 knockdown or JQ1 treatment. *P<0.05, ANOVA followed by Tukey's post-test. E. FoxM1 was overexpressed in ES-2 cells. Western blot demonstrated FoxM1 overexpression. F. ES-2 cells were treated with DMSO or JQ1 (1 μM) for 10 days. The remaining cells were stained with crystal violet. G. Relative cell viability of ES-2 cells treated with DMSO or JQ1 (1 μM) in the presence or absence of FoxM1 overexpression.
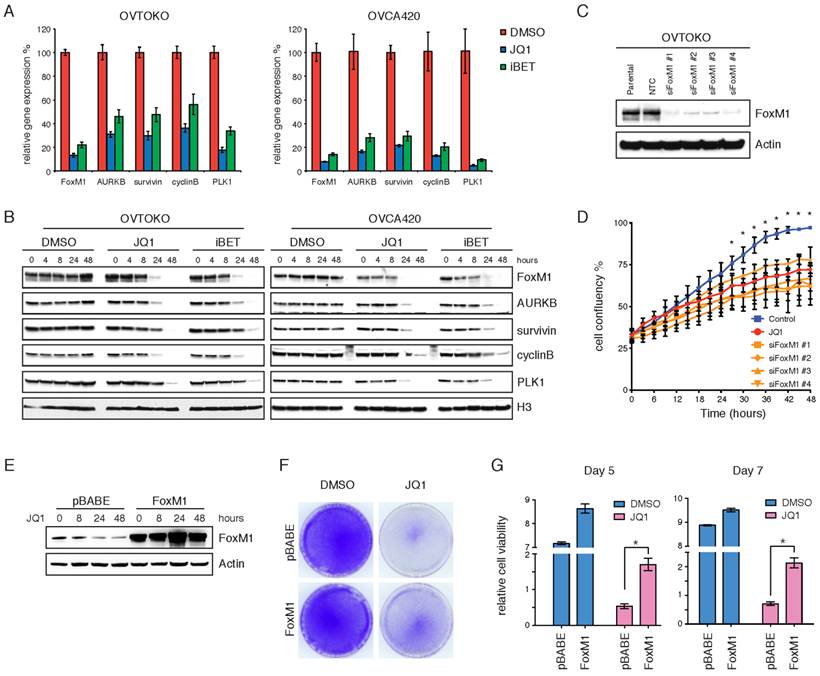
In strikingly contrast, JQ1 or I-BET151 failed to diminish FoxM1 transcriptional program in two cell lines relatively resistant to BET bromodomain inhibition (Supplementary Figure 7A&B). To test whether FoxM1 downregulation was merely a consequence of cell cycle arrest, we treated OVTOKO cells with docetaxel or etoposide and found that both chemo drugs had only moderate effects on FoxM1 protein levels (Supplementary Figure 7C). These results suggest that the biological consequences observed with BET bromodomain inhibitors may be dependent on transcriptional repression of FoxM1. Indeed, knockdown of FoxM1 in OVTOKO cells (Figure 4C) phenocopied the anti-proliferative effects of JQ1 treatment (Figure 4D; Supplementary Figure 7D). Conversely, when FoxM1 was exogenously overexpressed (Figure 4E), ovarian tumor cells could be partially rescued from the growth-inhibitory activity of JQ1 (Figure 4F&G). Taken together, these data suggest that FoxM1 serves as a functional downstream effector, at least partially, mediating the growth inhibition induced by BET bromodomain inhibitors in ovarian cancer.
BET bromodomain inhibition attenuates tumor growth and downregulates FoxM1 in vivo
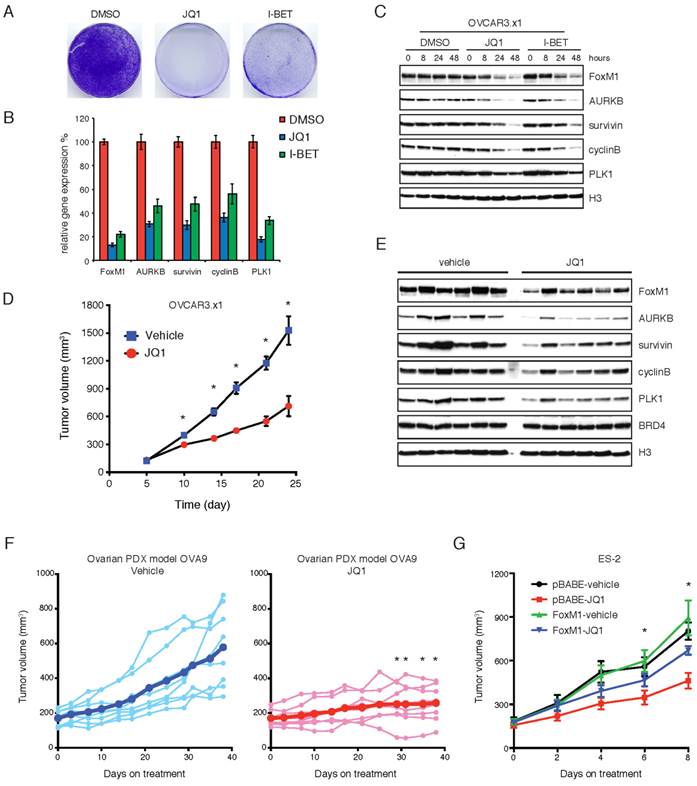
We next investigated the therapeutic potential of BET bromodomain inhibition in ovarian xenograft models. A widely used ovarian cell line OVCAR3 was passaged in immunodeficient mice to further improve its xenotransplantation efficiency. The resulting cell line (termed OVCAR.x1) was sensitive to JQ1 or I-BET151 inhibitors in vitro (Figure 5A). As expected, the FoxM1 pathway was significantly inhibited at both mRNA (Figure 5B) and protein (Figure 5C) levels upon JQ1 or I-BET151 treatment. When OVCAR.x1 was transplanted in vivo, JQ1 significantly suppressed tumor growth by ~60% compared with vehicle-treated animals at day 24 (Figure 5D). In addition, immunoblotting of JQ1-treated xenografts confirmed effective downregulation of FoxM1 transcriptional program in tumors at end point of the study, indicating that the efficacy of JQ1 against FoxM1 signaling was maintained during the course of treatment (Figure 5E). We further evaluated JQ1 antitumor activity in ovarian cancer patient-derived xenograft (PDX), and found that JQ1 significantly abrogated tumor growth of the PDX model (Figure 5F). To verify FoxM1 as a functional target of BET bromodomain inhibitors in vivo, ES-2 cells were transplanted in nude mice and treated with JQ1 or vehicle control. As anticipated, exogenous expression of FoxM1 was able to rescue the tumors from the inhibitory effects of JQ1 (Figure 5G). We conclude that BET bromodomain inhibition is an effective therapeutic approach for ovarian cancer in vivo.
BET bromodomain inhibition attenuates tumor growth and downregulates FoxM1 in vivo. A. OVCAR.x1 cells were treated with DMSO, JQ1 (1 μM) or I-BET151 (1 μM) for 10 days. The remaining cells were stained with crystal violet. B. Quantitative PCR of FoxM1 pathway genes in OVCAR.x1 cells treated with DMSO, JQ1 (1 μM) or I-BET151 (1 μM). Each group had three biological replicates. P<0.05, ANOVA followed by Tukey's post-test. C. OVCAR.x1 cells were treated with DMSO, JQ1 (1 μM) or I-BET151 (1 μM). Cell lysates were immunoblotted with indicated antibodies. D. Tumor growth of OVCAR.x1 cells treated with JQ1 (50mg/kg/day) or vehicle control, ten mice per group. *P<0.05, Student's t-test. E. OVCAR.x1 tumor lysates were immunoblotted with indicated antibodies. F. Tumor growth of ovarian PDX model OVA9 treated with JQ1 (50mg/kg/day) or vehicle control, eight mice per group. Each line represents an individual tumor growth curve, and the thick blue/red lines indicate mean tumor volume of the treatment group. Tumor volumes of the JQ1 group were statistically significantly lower than those of the vehicle control after 29 days of treatment (*P<0.05, unpaired Student's t-test). G. Tumor growth of ES-2 xenografts in the presence or absence of FoxM1 overexpression and treated with JQ1 (50mg/kg/day) or vehicle control. *P<0.05, ANOVA followed by Tukey's post-test.
Discussion
Through integrative genomic analyses, we were able to identify BRD4 as a candidate oncogenic driver of epithelial ovarian carcinoma and provide mechanistic evidence supporting the investigation of BET bromodomain inhibitors in different subtypes of ovarian cancer. BET bromodomain inhibition caused a profound G0-G1 cell cycle arrest in a large panel of ovarian cancer cell lines and significantly delayed tumor growth in both cell line-based and patient-derived xenograft models. Gene expression profiling revealed downregulation of the FoxM1 pathway in response to BET bromodomain inhibitors. The pathologic activation of FoxM1 plays a central role in ovarian tumorigenesis, with FoxM1 transcription factor network reported to be altered in 87% of HGS-OvCa patients [7]. Although FoxM1 has been a compelling target in ovarian cancer, inhibiting transcription factor oncoproteins remains challenging for conventional drug development. Here, we showed that JQ1 displaced BET proteins from chromatin and presumably FoxM1 gene locus, leading to reduced FoxM1 expression. Overall, our study has unambiguously established a rationale for targeting FoxM1 by BET bromodomain inhibitors as a therapeutic strategy in diverse human ovarian cancers.
By taking an in vivo functional genomics approach, a recent report by Livingston and colleagues also identified BRD4 as a candidate therapeutic target particularly in high-grade serous ovarian carcinoma [39]. Given that ovarian cancer is a complex disease and contains several histologic subtypes, we have further explored the therapeutic effects of BET bromodomain inhibitors in other histotypes beyond HGS-OvCa such as clear cell, endometrioid and mucinous, and found that BET bromodomain inhibition exerts pan-subtype pharmacologic efficacy in ovarian cancer. These data imply that different subtypes of ovarian cancer might share common transcription regulation that renders tumor cells vulnerable to BET bromodomain inhibitors.
Downstream of BRD4, suppression of c-MYC or MYCN and their target genes was previously proposed as the main mechanism mediating the anti-proliferative effects of BET inhibition in a variety of human cancers [20, 24]. Along this line, Livingston et al. suggested that sensitivity to BRD4 inhibition in HGS-OvCa correlates with either c-MYC or MYCN overexpression [39]. However, we found that c-MYC was only transiently and modestly reduced by JQ1 at the mRNA level and remained largely unchanged at the protein level in OVTOKO cells upon short-term treatment, in contrast to rapid and robust c-MYC downregulation observed in other tumor types [20, 24]. Instead, FoxM1 was identified as a relevant functional target of BET bromodomain inhibitors in multiple ovarian cancer cell lines and tumor xenografts. More importantly, our data indicate that efficient downregulation of FoxM1 and its transcriptional targets is likely required for ovarian cancer to respond to BET bromodomain inhibition. Therefore, at least a subset of ovarian cancer responds to BET inhibitors through FoxM1 rather than c-MYC or MYCN, an important consideration when we develop these drugs in clinical trials involving ovarian cancer patients. Additionally, it will be of interest to determine the potential value of FoxM1 as candidate pharmacodynamic and/or predictive biomarkers of BET bromodomain inhibitors in the ovarian cancer setting.
Consistent with the functional role of FoxM1 as a BET protein target, ectopic overexpression of FoxM1 can partially rescue tumor cells from JQ1 treatment. However, it is noteworthy that tumor cells are still significantly inhibited by JQ1, although to a lesser extent. There are at least two possible reasons. First, reduction of FoxM1 may not be solely responsible for the anti-proliferative effects of BET bromodomain inhibition and BET proteins may regulate other oncogenic transcriptional pathways in addition to FoxM1. For example, our microarray analysis discovered that JQ1 decreased the abundance of gene sets with E2F binding sites, reminiscent to a recent finding in diffuse large B cell lymphoma [21]. Second, BET bromodomain proteins may function as coactivators of FoxM1-dependent gene transcription. Previous studies have highlighted the important role of androgen receptor (AR)-binding BRD4 as a coactivator of AR-mediated transcription [25]. It remains to be determined whether similar coactivator mechanism exists between BRD4 and FoxM1. Nevertheless, our findings that FoxM1 is a functional BET protein target may provide a therapeutic strategy for targeting ovarian tumors, and potentially other cancers dependent on this pathway.
A number of investigational BET bromodomain inhibitors are being evaluated in Phase I clinical trials and it is still too early to completely establish the toxicity profile of this drug family. Recently, a clinical PLK1 inhibitor BI-6727 (volasertib) has been shown to exhibit potent activity on BRD4 [37] and we found that BI-6727 recapitulated the anti-proliferative effects of JQ1 in suppressing ovarian cancer cell viability. Volasertib showed generally manageable safety profile in clinic and has been granted a breakthrough therapy designation by the United States Food and Drug Administration for the treatment of acute myeloid leukemia [40]. Interestingly, early-phase clinical trials of volasertib have revealed signs of antitumor activity in advanced ovarian cancer and it would be important to determine its precise mechanism of action in these patients [41]. Therefore, in addition to specific BET bromodomain inhibitors, dual kinase-bromodomain inhibitors are promising regimens and warrant further investigation in ovarian cancer.
In summary, we have preclinically addressed a major unmet medical need by identifying BET bromodomain inhibitors as a promising therapeutic approach for the treatment of diverse ovarian cancers. We have identified oncoprotein FoxM1 as a key target of BET bromodomain inhibitors, although other complementary mechanisms may exist. Our study defines a general treatment strategy to target ovarian cancer through the direct modulation of epigenetic machinery.
Supplementary Material
Supplementary Figures and Tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81472537 to G Zhuang, 81502597 to Y Jing, 81130038 and 81372189 to WQ Gao), the Chinese Ministry of Science and Technology (2012CB966800 to WQ Gao), the Grants from the State Key Laboratory of Oncogenes and Related Genes (No. 91-14-18 and 91-15-12 to G Zhuang), the Shanghai Institutions of Higher Learning (Eastern Scholar to G Zhuang), the Shanghai Health Bureau Key Discipline and Specialty Foundation and the KC Wong foundation to WQ Gao.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bowtell DD. The genesis and evolution of high-grade serous ovarian cancer. Nat Rev Cancer. 2010;10:803-8
2. Liu J, Matulonis UA. New strategies in ovarian cancer: translating the molecular complexity of ovarian cancer into treatment advances. Clin Cancer Res. 2014;20:5150-6
3. Banerjee S, Kaye SB. New strategies in the treatment of ovarian cancer: current clinical perspectives and future potential. Clin Cancer Res. 2013;19:961-8
4. Bookman MA, Gilks CB, Kohn EC, Kaplan KO, Huntsman D, Aghajanian C. et al. Better therapeutic trials in ovarian cancer. J Natl Cancer Inst. 2014;106:dju029
5. Coleman RL, Monk BJ, Sood AK, Herzog TJ. Latest research and treatment of advanced-stage epithelial ovarian cancer. Nat Rev Clin Oncol. 2013;10:211-24
6. Berns EM, Bowtell DD. The changing view of high-grade serous ovarian cancer. Cancer Res. 2012;72:2701-4
7. Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609-15
8. Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T. et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532-43
9. Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R. et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228-31
10. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G. et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366:1382-92
11. Liu JF, Barry WT, Birrer M, Lee JM, Buckanovich RJ, Fleming GF. et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. Lancet Oncol. 2014;15:1207-14
12. Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RH, Sonke GS. et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol. 2015;16:87-97
13. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12-27
14. Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339:1567-70
15. Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13:337-56
16. Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54:728-36
17. Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D. et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214-31
18. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O. et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067-73
19. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI. et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529-33
20. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM. et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904-17
21. Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J. et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24:777-90
22. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA. et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524-8
23. Lockwood WW, Zejnullahu K, Bradner JE, Varmus H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc Natl Acad Sci U S A. 2012;109:19408-13
24. Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH. et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3:308-23
25. Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R. et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278-82
26. Cheng Z, Gong Y, Ma Y, Lu K, Lu X, Pierce LA. et al. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin Cancer Res. 2013;19:1748-59
27. Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y. et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin Cancer Res. 2014;20:912-25
28. Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR. et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320-34
29. Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA. et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108:16669-74
30. Lee TI, Johnstone SE, Young RA. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat Protoc. 2006;1:729-48
31. Cline MS, Craft B, Swatloski T, Goldman M, Ma S, Haussler D. et al. Exploring TCGA Pan-Cancer data at the UCSC Cancer Genomics Browser. Sci Rep. 2013;3:2652
32. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
33. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
34. Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA. et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci U S A. 2011;108:12372-7
35. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545-50
36. Ganzfried BF, Riester M, Haibe-Kains B, Risch T, Tyekucheva S, Jazic I. et al. curatedOvarianData: clinically annotated data for the ovarian cancer transcriptome. Database (Oxford). 2013;2013:bat013
37. Ciceri P, Muller S, O'Mahony A, Fedorov O, Filippakopoulos P, Hunt JP. et al. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat Chem Biol. 2014;10:305-12
38. De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N. et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014;514:247-51
39. Baratta MG, Schinzel AC, Zwang Y, Bandopadhayay P, Bowman-Colin C, Kutt J. et al. An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc Natl Acad Sci U S A. 2015;112:232-7
40. Gjertsen BT, Schoffski P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia. 2015;29:11-9
41. Schoffski P, Awada A, Dumez H, Gil T, Bartholomeus S, Wolter P. et al. A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. Eur J Cancer. 2012;48:179-86
Author contact
![]() Corresponding author: Guanglei Zhuang, State Key Laboratory of Oncogenes and Related Genes, Renji-Med X Clinical Stem Cell Research Center, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China. Email: zhuangguangleicom
Corresponding author: Guanglei Zhuang, State Key Laboratory of Oncogenes and Related Genes, Renji-Med X Clinical Stem Cell Research Center, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China. Email: zhuangguangleicom