Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2016; 6(2):243-253. doi:10.7150/thno.14322 This issue Cite
Research Paper
Chemical Conjugation of Evans Blue Derivative: A Strategy to Develop Long-Acting Therapeutics through Albumin Binding
Haojun Chen1, 2, Guohao Wang3, Lixin Lang2, Orit Jacobson2, Dale O. Kiesewetter2, Yi Liu2, Ying Ma2, Xianzhong Zhang2 ![]() , Hua Wu1
, Hua Wu1 ![]() , Lei Zhu3
, Lei Zhu3 ![]() , Gang Niu2
, Gang Niu2 ![]() , Xiaoyuan Chen2
, Xiaoyuan Chen2 ![]()
1. Department of Nuclear Medicine, Xiamen Cancer Center, the First Affiliated Hospital of Xiamen University, Xiamen, China
2. Laboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda, Maryland, USA
3. State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics & Center for Molecular Imaging and Translational Medicine, School of Public Health, Xiamen University, Xiamen 361005, China
Received 2015-11-5; Accepted 2015-11-12; Published 2016-1-1
Abstract

The efficacy of therapeutic drugs is highly dependent on their optimal in vivo pharmacokinetics. Albumin conjugation is considered to be one of the most effective means of protracting the short lifespan of peptides and proteins. In this study, we proposed a novel platform for developing long lasting therapeutics by conjugating a small molecular albumin binding moiety, truncated Evans blue, to either peptides or proteins. Using the anti-diabetic peptide drug Exendin-4 as a model peptide, we synthesized a new long-acting Exendin-4 derivative (denoted as Abextide). Through complexation with albumin in situ, the biological half-life of Abextide was significantly extended. The hypoglycemic effect of Abextide was also improved remarkably over Exendin-4. Thus, Abextide has considerable potential to treat type 2 diabetes. This strategy as a general technology platform can be applied to other small molecules and biologics for the development of long-acting therapeutic drugs.
Keywords: Exendin-4, Evans blue, Albumin-binding, PET, Abextide
Introduction
The therapeutic efficacy of drugs depends heavily on their intrinsic pharmacokinetics [1]. The efficacies of peptides and proteins are often restricted by their rapid clearance from the body, which means they must be frequently administered to maintain the therapeutic level. The circulating half-lives (t1/2) of most peptides and protein-based drugs range from a few minutes to at most two hours. These short in vivo lifespans of therapeutic peptides and proteins are mainly due to renal filtration or proteolysis [2]. In many cases, sufficiently long half-life is desired to exert expected effect on the patient. Consequently, various strategies have been utilized to increase the half-life of peptide drugs in vivo. One common method is to reduce the rate of clearance, which can be achieved either through direct modification of the drug, or by adding other agents which act on the clearance pathways [3]. Reduction of clearance is particularly desired for protein drugs, as they are highly vulnerable to degradation by proteases.
Renal-urinary tract is one of the major routes for drug clearance and the kidneys generally filter out molecules below 60 kDa [4]. Therefore, enlargement of the molecular size of protein drugs, by protein fusions, glycosylation or PEGylation, reduces the renal clearance and further extends in vivo exposure of the polypeptide or protein therapeutic [5-9]. However, the large size of PEG chain inevitably comprises the biological activity of the peptide [10, 11].
Due to their inherently long blood circulation, immunoglobulins and human serum albumin (HSA) have been used as carriers to protect drugs from enzymatic degradation and increase the half-lives [12, 13]. Due to the high abundance in the blood circulation (35-50 mg/ml), albumin, with a molecular weight of 66.5 kDa, has advantages over immunoglobulins as a drug carrier [14-18]. Two principle albumin-based technologies have been developed in the past 15-20 years. One is to pass lipophilic drugs and HSA under high pressure through a jet to form albumin-drug nanoparticles [19]. In the second approach, albumin-binding peptides or prodrugs are administered intravenously and they bind in situ to circulating albumin, either physically or covalently [20, 21].
For example, fatty acid-conjugation causes hydrophilic peptides to bind to albumin via approximately five binding positions for long chain fatty acids [22]. Several drugs have been enhanced by conjugating fatty acid to increase their circulation time for therapy [23, 24]. However, the biodistribution of the fatty acid conjugates is still not optimal since a high percentage of administered drug molecules will be accumulated in the liver and be degraded. Besides, the lipophilicity also increases the difficulty of chemical reaction and production of these drugs.
Evans blue (EB) dye has been an important tool in studying physiology and malignancy because of its high affinity for serum albumin [25, 26]. Recently, we developed a novel blood pool imaging agent, based on the structure of Evan blue (EB) dye in which one of the 1-amino-naphthol-2,4-disulfonic acid moieties is replaced with a NOTA chelator for radiometal complexation. We refer to these analogs as truncated Evans blue (tEB) [27-29]. In this study, we expanded this strategy with the development of tEB derivatives that can be conjugated to therapeutic drugs and result in enhanced bioavailabilty. Using the tEB derivative as described, many small molecules and biologics can be easily modified with a one-step reaction. Due to the relatively strong binding of tEB moiety with albumin, the number of tEB moieties and linkers per peptide or protein can be controlled to adjust the biodistribution of the conjugates in vivo. By adopting this strategy, complicated in vitro protein labeling procedures and the multiple steps of purifying fusion proteins can be avoided.
The proof-of-concept was demonstrated by using exendin-4, a 39-amino acid peptide that is a potent agonist of mammalian glucagon-like peptide-1 receptor (GLP-1R) that stimulates insulin release and β cell proliferation, while suppressing glucagon secretion and delaying gastric emptying [30-32]. Exendin-4 (Exenatide, Byetta®) has shown antidiabetic effect in patients with type 2 diabetes whose glycemic control is inadequate with metformin alone, sulphonylurea alone, or metformin in combination with sulphonylurea [33-35]. However, because of its short in vivo half-life, Exenatide must be injected by diabetic patients at least twice daily [36], which markedly reduces patient compliance.
In previous studies, we found that truncated Evans blue maintains albumin binding affinity [27, 28]. In order to conjugate the truncated EB (tEB) to various biomolecules, the compound needs to be functionalized for selective attachment. Therefore, we designed a synthesis route to prepare maleimide-tEB for reaction with a thiol group of Cys40 residue on the Exendin-4 peptide, resulting in an albumin binding drug candidate, denoted as Abextide. The functional group of the tEB can also be amine or carboxyl group as appropriate for conjugation to other therapeutic peptides or proteins.
The main purpose of this study is to determine if EB conjugation would enhance the short in vivo lifespan of therapeutics in the systematic circulation. We demonstrate this approach as a viable route for increasing the half-life of potentially important peptide or protein pharmaceuticals.
Material and methods
General methods
O-tolidine and 1-amino-naphthol-2,4-disulfonic acid monosodium salt were purchased from TCI America (Portland, OR) and NOTA-NHS was obtained from Marcocyclics (Dallas, TX). All other chemicals were purchased from Sigma-Aldrich. Waters 600 high-performance liquid chromatography (HPLC) system with a Waters 996 Photodiode Array Detector (PDA) and an online radioactivity detector (Beckman) using a semi-preparative C18 HPLC column (XTerra Prep RP18, 10 µm, 7.8 x 300 mm, Waters) was used for the purification of products. Varian BOND ELUT C18 column (100 mg) was used for solid phase extraction. A Perkin-Elmer 200 series HPLC pump with a Waters 2487 UV detector and a Bioscan Flow-Count detector using an analytical C18 HPLC column (XTerra 5 µm, 150 x 4.6 mm, Waters) was used for analysis of labeled compounds. HPLC runs a linear gradient starting from 5% A (0.1% TFA in acetonitrile) and 95% B (0.1% TFA in water) for 5 min and increasing to 65% A at 35 min with a flow rate of 5 ml/min for semi-prep HPLC and 1 ml/min for analytical HPLC. Mass spectra were obtained with Waters LC-MS system (Waters, Milford, MA) that includes an Acquity UPLC system coupled to the Waters Q-Tof Premier high-resolution mass spectrometer. The 18F-fluoride was obtained from NIH cyclotron facility.
Preparation of maleimide-tEB (4)
To a 100 ml round bottom flask containing o-tolidine (4.3 g) and methylene chloride (40 ml) was added di-t-butyldicarbonate (4.4 g). The mixture was stirred at room temperature overnight. The reaction was concentrated and the residue was purified by chromatography on silica gel to give 3.2 g of N-Boc-2-tolidine (1). LC-MS: [MH]+ = 313.4135 (m/z), calc: 312.1838. N-Boc-2-tolidine (0.46 g, 1.47 mmol) was dissolved in acetonitrile (10 ml) in a glass vial and cooled to 0 °C, then hydrochloric acid (0.3 M, 15 ml) was added. Cold sodium nitrite solution (0.31 g in 5 ml water) was added dropwise and stirred for 20 min, and the solution turned bright yellow. This solution was added dropwise to another glass vial containing 1-amino-8-naphthol-2,4-disulfonic acid monosodium salt (0.59 g) and sodium bicarbonate (0.49 g) in water (20 ml) at 0 °C. The reaction was deemed complete by LC/MS and the reaction was lyophilized without further purification to provide the Boc-tEB (2) product. [M-H]- = 541.4425, calc: 542.0930.
The Boc-tEB product was added to a solution of 80% TFA, 10% 1,2-ethanedithiol and 10% thioanisole and stirred until the reaction was complete. The mixture was diluted with water (100 ml) and loaded onto a C18 chromatography cartridge (3x15 cm). The column was washed with water and then with 80% ethanol to elute the desired product. After evaporation of the solvent in the eluent, 0.6 g of 80% pure product of tEB (3) was obtained. A small amount of product 3 was further purified by HPLC. LC-MS: [M-H]- = 541.4425, calc: 542.0930.
To a solution of purified tEB (30 mg) in methanol (4 ml), DIPEA (50 μl) and maleic anhydride (160 mg) were added, and the reaction was stirred at room temperature for 2 h. When the reaction to form intermediate was complete as judged by HPLC, the solvent was evaporated, acetic anhydride (1 ml) was added and the reaction was heated at 105 °C for 30 min. When LC/MS showed complete conversion to desired product, the mixture was diluted with water (16 ml) and purified on a Waters Xterra C18 chromatography column running a linear gradient from 5% A (0.1% TFA in acetonitrile) and 95% B (0.1% TFA in water) for 2 min and increasing A to 65% in 30 min. The desired product was collected and lyophilized to give 6.0 mg Maleimide-tEB (4). 1H NMR (MeOD) δ (ppm) 2.17(s, 3H), 2.55 (s, 3H), 6.99 (s, 2H), 7.16 (d, 1H), 7.19 (d, 1H), 7,56 (dd, 1H), 7.63 (m, 3H), 7.97 (d, 1H), 8.00 (d, 1H), 8.70 (s, 1H). LC-MS: [M-H]- = 621.5282, calc: 622.0828.
Preparation of tEB-Exendin-4 (Abextide)
To a solution of cys-40-Exendin-4 (6.3 mg) in 3 mL PBS buffer (pH 7.0) was added 2.0 mg of maleimide-tEB (Figure 1A). The mixture was stirred at room temperature and monitored with HPLC. After the completion of the reaction, the mixture was purified with semi-prep HPLC in 5 injections. The fractions containing the product were collected and lyophilized to give 7.2 mg of desired product, tEB-Exendin-4 (denoted as Abextide). The purity of Abextide was >97% based on the HPLC analysis. LC-MS: [MH]- =4911.00, calc: 4912.32.
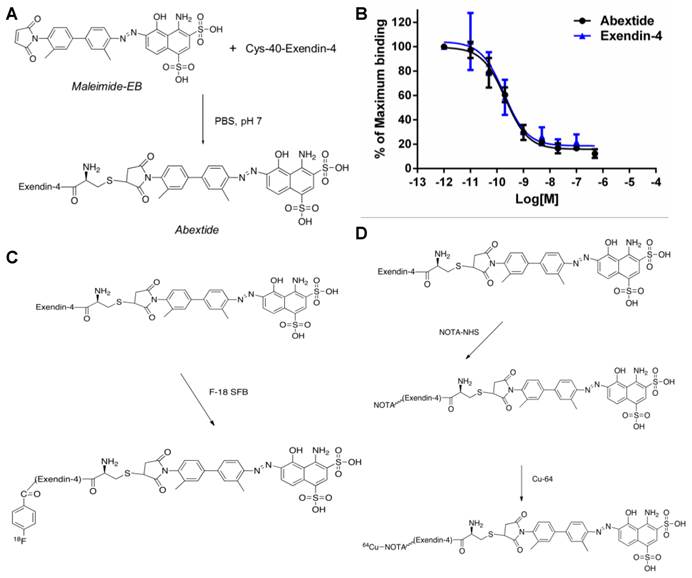
(A) Synthesis of Abextide. (B) Cell binding assay of Exendin-4 and Abextide with INS-1 cells overexpressing glucagon-like peptide-1 receptor (GLP-1R). The IC50 values were determined to be 0.21 ± 0.08 nM and 0.18 ± 0.06 nM for Exendin-4 and Abextide, respectively. (C) Radiolabeling of Abextide with F-18. (D) Radiolabeling Abextide with Cu-64.
Preparation of NOTA-Exendin-4 and NOTA-Abextide
The couplings of NOTA-NHS ester to the amines of Exendin-4 and Abextide were performed with similar procedures using DMSO as the solvent and DIPEA as the base. For a typical run, 2.0 mg of peptide in a 4 mL vial was dissolved in 0.3 mL of DMSO, followed by the addition of 5 μL of DIPEA and 1.3 mg of NOTA-NHS ester (~10 eq.). The reaction mixture was stirred at room temperature and monitored with an analytical HPLC. After all the starting peptide was consumed, the reaction mixture was diluted with 1 mL of water and purified with a semi-preparative HPLC running a linear gradient starting from 5% A (0.1% TFA in acetonitrile) and 95% B (0.1% TFA in water) for 5 min and increasing to 65% A at 35 min with a flow rate of 5 mL/min. The fractions containing the desired product (a mixture di-NOTA and tri-NOTA peptide) were collected and lyophilized to give 1.2 mg powder.
Preparation of NOTA-Albiglutide
Albiglutide (TANZEUM, 30 mg/mL) was washed 4 times with water using a 30 KDa Centricon to remove buffers and small molecules from the original solution to a final concentration of 30 mg/100µL in water. For conjugation reaction with NOTA-NHS ester, only 10 µL of Albiglutide were used. To a solution of Albiglutide were added 20 eq. of NOTA-NHS ester dissolved in 100 µL dimethylsulfoxide and 1 mL of 0.1 M NaHCO3 (pH 8.4). The reaction was left over-night at 4 ºC. Unreacted NOTA was removed by 30 KDa centricon.
Radiolabeling Exendin-4, Albiglutide and Abextide with 18F-SFB
18F-SFB (~5 mCi in 0.5 mL methylene chloride) prepared using an automated synthesizer according to the published procedure [37] was transferred to a 1 ml plastic tube and the solvent was evaporated with an argon flow. 18F-SFB in the tube was re-dissolved in 10 μl of acetonitrile and 0.5 mg of Exendin-4 in 0.3 ml 25 mM phosphate buffer (pH 8.5) was added to the tube. The mixture was injected onto a semi-prep HPLC after 10 min reaction at 37 °C. The radioactive peak (Rt = 23.2 min) was collected and radioactive product was trapped on a 100 mg Varian Bond Elut C18 column and washed with 10 ml water. The radioactivity trapped on the C18 column was eluted off with 0.3 ml of ethanol with 1 mM HCl. For Abextide and Albiglutide labeling, the similar procedure was used except that the Albiglutide product was purified using a PD 10 column.
Radiolabeling Exendin-4, Albiglutide and Abextide with 64Cu
For copper-64 labeling of NOTA-Exendin-4 and NOTA-Abextide, about 0.1 mg of NOTA conjugated peptide in 0.1 mL water were added 5 mCi of 64Cu in 400 mL of 0.4 M ammonium acetate buffer (pH 5.5). The mixture was heated at 80 °C for 10 min and the product was trapped on a 100 mg Varian Bond Elut C18 column and washed with 10 mL water. The product trapped on the C18 column was eluted off with 0.3 mL of ethanol with 1 mM HCl.
For copper-64 labeling of NOTA-Albiglutide, 64CuCl2 (2 mCi) was diluted in 200 μL of 0.4 M ammonium acetate buffer (pH 5.5) and added to 50-100 μg of NOTA conjugate. The reaction mixture was incubated for 1 h at 37 °C. 64Cu-NOTA-Albiglutide was purified by PD-10 column using PBS as the mobile phase. Complexation of 64Cu with the conjugated Albiglutide was monitored by radio-TLC.
Experimental animals
Type 2 diabetic C57BL/6 db/db mice (male, 6-8 weeks old) were obtained from the Nanjing BioMedical Research Institute of Nanjing University (Nanjing, China). Normal BALB/c mice (females, 4-5 weeks old), weighing 18-20 g, were purchased from Harlan Laboratories. All animal studies were conducted in accordance with the principles and procedures outlined in the Guide for the Care and Use of Laboratory Animals [38] and were approved by the Institutional Animal Care and Use Committee of the Clinical Center, National Institutes of Health and Xiamen University. Animals were housed in groups of 5 under a 12-h light/dark cycle (lights on at 6 a.m.), allowed food and water ad libitum, and acclimatized for 2 weeks.
Cell culture and cell binding assay
The INS-1 rat insulinoma cells were purchased from the American Type Culture Collection (ATCC) and grown in RPMI-1640 medium (Life Technologies, Grand Island, NY) which was supplemented with 10% fetal bovine serum, penicillin (100 IU/mL), and streptomycin (100 mg/mL) (Invitrogen, Carlsbad, CA) and in a humidified atmosphere containing 5% CO2 at 37 °C. Cells were passaged three to four times per week.
Incubation was conducted with 96 well MultiScreen filter plates (Millipore, MA). For each well, the reaction volume was 200 µl containing 2 x 105 cells, 200 nCi (7.4 kBq) of 18F-Exendin-4 and 0-500 nM of unlabeled Exendin-4 or Abextide. The plate was incubated for 45 min on a shaker at room temperature. After incubation, cells were washed three times with RPMI medium. Cell bound membranes were dried and isolated. The radioactivity was measured using a gamma counter (1480 Wizard 3, Perkin-Elmer). Binding results were expressed as percent of total counts, IC50 values were calculated using Prism software (GraphPad Software Inc., La Jolla, CA).
Small animal PET imaging
All the PET scans were conducted with an Inveon small-animal PET scanner (Siemens Preclinical Solution). Mice were anesthetized with mixtures of 1 ml/min O2 and 1.5% isoflurane and kept warm with a heating pad thermostat during the imaging. All data acquisitions were initiated immediately before the tracer injections. For dynamic PET imaging, the duration of a scan was 60 min. A catheter was placed in the tail vein before each scan for tracer administration. About 3.7 MBq (100 μCi) of 18F-Exendin-4, 18F-Albiglutide or 18F-Abextide was injected through the catheter immediately after the scan was started (n = 4/group). The reconstruction frames were 1×5, 1×25, 9×30, 5×120, and 10×240 s. After the 60 min dynamic scan, all mice underwent 10-min static PET scans at 120 min post-injection (p.i.).
For the subcutaneous injection study, 1.5-1.85 MBq (40-50 μCi) of 64Cu-Exendin-4, 64Cu-Albiglutide or 64Cu-Abextide was diluted in 30-50 μl of saline solution, then injected into the loose skin over the left hind limb (n = 4/group). Each injection was given into the fat layer between the skin and muscle. Static PET scans were acquired at 1, 2, 4, 24, 48 and 72 h after tracer injection. The acquisition time was 5 min at 1, 2 and 4 h p.i., and was 10, 15 and 20 min at 24, 48 and 72h p.i.
Image reconstruction and data analysis
The images were reconstructed using a 3-dimensional ordered subset expectation maximum (OSEM) algorithm, and no correction was applied for attenuation or scatter. For each scan, regions of interest (ROIs) were drawn using vendor software (ASI Pro 5.2.4.0; Siemens Medical Solutions) on decay-corrected whole-body coronal images. The radioactivity concentrations (accumulation) within the tumor and heart were obtained from mean pixel values within the multiple ROI volumes and then converted to megabecquerel per milliliter. These values were then divided by the administered activity to obtain (assuming a tissue density of 1 g/ml) an image-ROI-derived percentage injected dose per gram (%ID/g).
Ex vivo biodistribution
Immediately after 64Cu-Exendin-4, 64Cu-Albiglutide or 64Cu-Abextide PET imaging at 72 h p.i., the mice were sacrificed and dissected. Blood, major organs, and tissues were collected and wet-weighed. The radioactivity in the wet whole tissue was measured with a γ-counter (Packard). The results were expressed as percentage of injected dose per gram of tissue (%ID/g) for a group of 4 animals. For each mouse, the radioactivity of the tissue samples was calibrated against a known aliquot of the injected radiotracer and normalized to a body mass of 20 g. Values were expressed as mean ± SD (n = 4/group).
Hypoglycemic efficacy and in vivo pharmacokinetics test of Abextide in db/db mice
Hypoglycemic efficacies of Exendin-4 analogues were investigated using an intraperitoneal glucose tolerance test (IPGTT) in male db/db mice (6-7 weeks old). Under non-fasting conditions with free access to food and water, animals received a single subcutaneous injection of saline, Exendin-4 or Abextide (25 nmol/kg body weight, n = 3/group). Blood glucose levels were then monitored using a convenient blood glucose meter (ACCU-CHEK Sensor, Roche Diagnostics Corp., USA). At predetermined times (0, 1, 2, 4, 6, 8, 12, 24, 48, 72, 96 h), blood samples were collected from tail vein of each animal and blood glucose levels were measured as mentioned above. Hypoglycemic durations to a blood glucose level of <8.35 nM (150 mg/dL) were checked.
The pharmacokinetic profiles of subcutaneously administered Exendin-4 or Abextide were evaluated by using a previously described method [39]. The animals were randomly divided into two groups and Exendin-4 or Abextide (25 nmol/kg) was administered. Blood samples, drawn at predetermined times, were placed in ice-cold polyethylene tubes which contained anticoagulant (heparin solution, 1/100 volume of blood). After that, plasma samples were obtained by centrifugation and stored at -70°C until required for assay. Amount of Exendin-4 or Abextide in plasma was measured by commercial Exendin-4 EIA kits (Phoenix Biotech., USA).
Statistics
Quantitative data were expressed as mean ± SD. Means were compared using one-way analysis of variance and Student's t test. P value of < 0.05 was considered statistically significant.
Results
In vitro cell receptor-binding assay
The GLP-1 receptor-binding affinities of Abextide were compared with Exendin-4 on INS-1 cells. The cell binding affinities of exendin-4 and Abextide for GLP-1R were measured using competitive displacement studies with 18F-Exendin-4 as the radioligand and plotted in sigmoid curves. As shown in Figure 1B, compared with exendin-4, Abextide shows similar high binding affinity with GLP-1R (0.21±0.08 vs 0.18 ± 0.06 nM). The comparable IC50 values between exendin-4 and Abextide suggested that EB conjugation did not comprise the binding affinity of the exendin-4 peptide.
Radiochemistry
The procedure for radiolabeling of Exendin-4, Abextide and Albiglutide with 18F-SFB was indicated in Figure 1C. The radiochemical yield for 18F-exendin-4, 18F-Abextide and 18F-Albiglutide was around 34%, 28% and 70%, respectively, with a total synthesis and work-up time of 2-3 h. A single peak was detected on thin-layer chromatography, and the radiochemical purity was greater than 95% based on HPLC analysis. To facilitate radioactive copper labeling of the three compounds, we synthesized NOTA-Exendin-4, NOTA-Abextide and NOTA-Albiglutide in 2 steps as shown in Figure 1D. Labeling of NOTA-exendin-4, NOTA-Abextide and NOTA-Albiglutide with 64Cu took 20-30 min, with a radiochemical yield of 34%, 71% and 95%, respectively.
In vivo distribution pattern of Abextide through intravenous injection
The in vivo distribution pattern of 18F-Abexide was evaluated with dynamic PET in healthy BALB/C mice (through intravenous injection). For comparison, we also labeled Exendin-4 and Albiglutide, a glucagon-like peptide-1 dimer fused to human albumin with a half-life of four to seven days [40]. As shown in Figure 2A, at early time points (at 1 min p.i.), 18F-exendin-4 presented an initial distribution in the circulation system, indicated by relative high tracer accumulation in heart and liver region. The compound was cleared out very quickly through the urinary tract, demonstrated by the high accumulation of radioactivity in kidneys and bladder. After 60 min, the background signal from other part of the whole body was extremely low.
(A-C) Whole-body PET images (series of maximum-intensity-projection) of healthy BALB/c mice at different time points after tail vein injection of 3.7-7.4 MBq of 18F-Exendin-4 (A), 18F-Albiglutide (B) or 18F-Abextide (C). The highest scale is 20 %ID/g and the lowest scale is 0 %ID/g. (D-F) Time-activity curves of blood (D), liver (E) and kidneys (F) quantified based on PET images (n = 4/group).
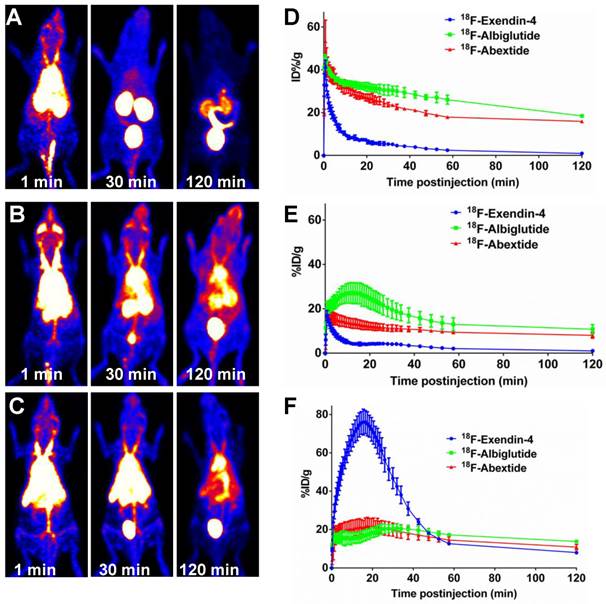
However, the distribution pattern of 18F-Abextide is dramatically different from that of 18F-exendin-4. With the EB moiety, the compound could bind with serum albumin after i.v. injection. Consequently, very strong radioactive signal could be observed over the heart and major vessel regions, even at 2 h post injection (18F-Abextide vs. 18F-Exendin-4, 15.92±0.40 %ID/g vs. 0.94±0.04 %ID/g, P<0.01). The time-activity curves (TACs) showed the fast clearance of 18F-Exendin-4, but not 18F-Abextide, through the kidneys (Figure 2B). Regarding GLP-1-HSA fusion protein Albiglutide, it showed a similar distribution patter as compared to 18F-Abextide (Figure 2C). The blood TACs from 18F-Albiglutide showed slightly less but nonsignificant decline rates as compared to those from 18F-Abextide (-0.14 ± 0.05 vs. -0.15 ± 0.01, P > 0.05) (Figure 2D). Furthermore, we also noticed that 18F-Albiglutide showed higher tracer uptake in the liver but similar uptake in the kidney as compared to 18F-Abextide (Figure 2E-F).
In vivo distribution pattern of Abextide through subcutaneous injection
In the next step, we move forward to evaluate the in vivo distribution pattern of Abexide after subcutaneous administration. In order to cover the whole process of drug release from the injection site, a radionuclide with longer physical half-life (Copper-64, t1/2=12.6 h) was used to labeled all three compounds, which were given into the fat layer over the left hind legs of healthy BALB/c mice. As shown in Figure 3A-C, native Exendin-4 was released from the injection site very fast with most of the activity cleared out within 24 h. Both Abextide and Albiglutide showed much slower migration from the injection site, demonstrated by the gradually reduced radioactivity from 1 h to 72 h. As for quantification, within the first 2 h and between 48 and 72 h, the drug residue at the injection site showed no significant difference between Abextide and Albiglutide, quantified by the percentage of the total injected dose (%ID) (Figure 3D). Albiglutide had somewhat higher residual amount than Abextide at 4 h and 24 h time points.
(A-C) Whole body PET images (series of maximum-intensity-projection) of healthy BALB/c mice at different time points after subcutaneous injection of 1.5-1.8 MBq of 64Cu-Exendin-4 (A), 64Cu-Albiglutide (B) or 64Cu-Abextide (C). The injection was given into the fat layer over the left hind leg. (D) Residence of 64Cu-Exendin-4, 64Cu-Albiglutide or 64Cu-Abextide in the injection site expressed as percentage of injection dose (%ID). (E-F) The distribution of different compounds in blood circulation (E) and kidneys (F) at different time points after subcutaneous injection. The quantification is based on PET images (n = 4/group). (G) Biodistribution of 64Cu-Exendin-4, 64Cu-Albiglutide or 64Cu-Abextide in healthy BALB/C mice right after PET imaging at the 72 h time point after tracer injection (n = 4/group). *, P < 0.05
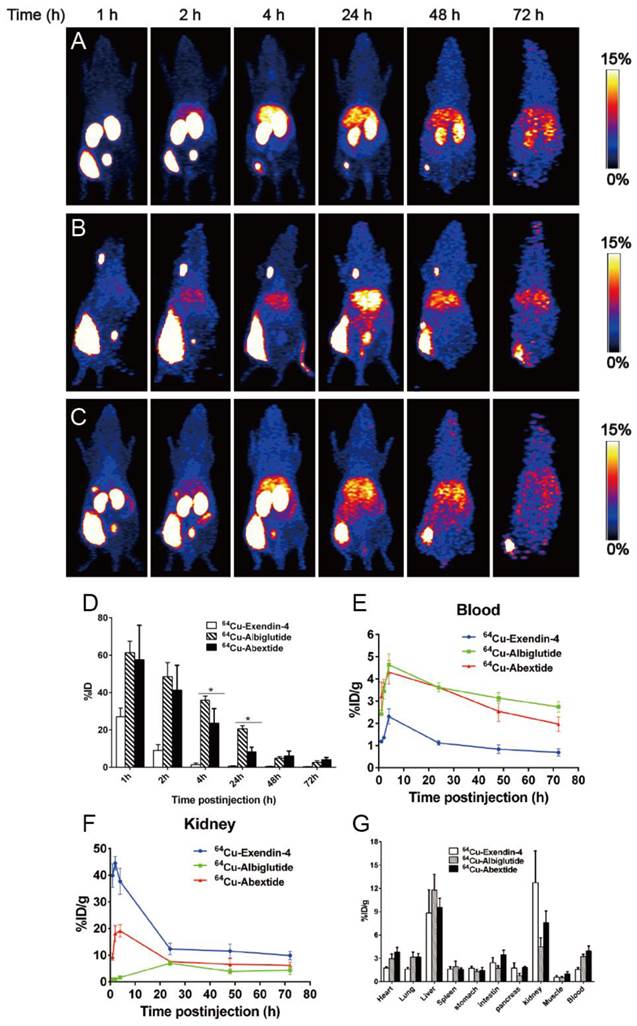
With respect to the in vivo distribution patterns of the three compounds after subcutaneous injection, Abextide and Albiglutide showed similar blood TACs, which peaked at 4 h p.i. and reduced gradually over time. The blood TACs of Exendin-4 also showed a similar trend, but with a much lower magnitude (Figure 3E). The ex vivo biodistribution assay was also performed to confirm the reliability of the imaging quantification (Figure 3G).
In vivo pharmacokinetics and hypoglycemic durations of Abextide in db/db Mice
The time vs. blood concentration curves of exendin-4 and Abextide after subcutaneous injection were plotted in Figure 4A. The pharmacokinetic parameters of extendin-4 and Abextide were summarized in Table 1. As shown in Figure 4A and Table 1, subcutaneously administered Exendin-4 was rapidly removed from the circulation with a t1/2 of 5.16 ± 5.23h, whereas the t1/2 of Abextide was 36.28 ± 7.01 h, resulting in a 7-fold greater time than that of native exendin-4. Furthermore, its AUCinf value (3533.70 ± 236.45 ng/mL*h) was 18 times greater than that of native Exendin-4 (193.21 ± 40.98 ng/mL*h).
Pharmacokinetic parameters of native Extendin-4 and Abextide after subcutaneous administration to SD rats.
| Parameter | Native Extendin-4 | Abextide |
|---|---|---|
| C max (ng/mL) | 42.08 ± 1.85 | 87.29 ± 3.71 |
| T max (h) | 4.0 ± 1.32 | 12.3 ± 3.88 |
| t1/2 (h) | 5.16 ± 5.23 | 36.28 ± 7.01 |
| AUCinf (ng*h/mL) | 193.21±40.98 | 3533.70±236.45 |
(A) Time vs. blood concentration curves of Exendin-4 and Abextide after subcutaneous injection determined by a commercial EIA kit (n=4/group). (B) Hypoglycemic efficacies of Exendin-4 and Abextide (n = 3/group). (C) The cut-off line was set at 8.35 mM (150 mg/dL) since the euglycemic duration under that value is considered a practical indication of the potential for antidiabetic treatment. The time required to rebound to a glucose level of 8.35 mM was 36 h for Abextide as compared to 10 h for exendin-4 treated mice.
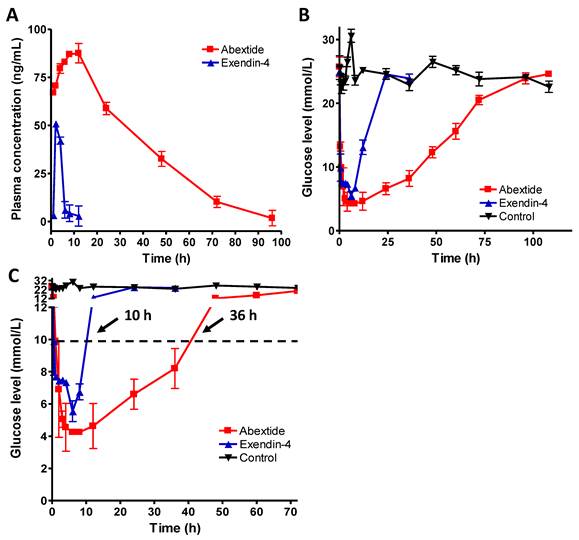
The hypoglycemic effects of exendin-4 and Abextide were examined in db/db mice. At a dose of 25 nmol/kg, as shown in Figure 4B, the lowest glucose level (4.25±0.21 mM) in Abextide treated mice was slightly lower, albeit insignificantly, than in exendin-4 treated mice (5.55±0.64 mM). However, the hypoglycemic duration in Abextide treated mice was much greater, the time required to rebound to a glucose level of 8.35 mM (a blood glucose level below 8.35 mM was regarded as normal) was 36 h as compared to 10 h for Exendin-4 treated mice (Figure 4C).
Discussion
So far, there are several albumin binding moieties available such as albumin binding domain, albumin binding peptide or fatty acids [41]. For example, a recombinant fusion of the albumin binding domain from streptococcal protein G to human complement receptor type 1 increased its half-life 3 times to 5 h in rats [42]. Albumin binding peptide SA21 fused to the anti-tissue factor D3H44 Fab increased its half-life 37-fold to 32.4 h in rabbits and 26-fold to 10.4 h in mice [43]. In another example, when exendin-4 was modified with fatty acids to promote association with albumin, an extended therapeutic effect was observed when injected subcutaneously in mice [24]. However, the big molecular size and/or unfavorable hydrophobicity of these albumin binding moieties will compromise target recognition and therapeutic efficacy of the drugs. As an alternative, we conjugated the biomolecule with our newly developed small molecule, truncated Evans blue (tEB), which was proved to be highly albumin binding [27-29]. The proof-of-concept was demonstrated by using the anti-diabetic drug Exendin-4. The tEB-conjugated drug candidate was denoted as Abextide. Due to the small molecular size of the tEB, it does not inhibit the biological function of the drug molecule. Indeed, in vitro receptor binding assay using INS-1 cells demonstrated that Abextide had similarly high GLP-1R binding affinity as compared to Exendin-4 (0.21 ± 0.08 vs. 0.18 ± 0.06 nM) (Figure 1B).
The high concentration of albumin in plasma guarantees the complexation of tEB with albumin after intravenous administration of Abextide. We evaluated the in vivo distribution pattern of Abextide using positron emission tomography (PET), a non-invasive function imaging technology that has been widely used to monitor the pharmacokinetics and pharmacodynamics of drugs and biologics [44]. As expected, very strong radioactive signal could be observed over the heart region, even at 2 h post injection. Due to the complexation with serum albumin, Abextide showed a similar distribution patterns as the macromolecular drug Albiglutide. However, Abextide showed advantage over Albiglutide with less liver accumulation, probably due to fact that Abextide did not interrupt the physiological behavior of the endogenous albumin after complexation. Even at 2 h p.i., the 18F-Abextide uptake in blood is still nearly 20%ID/g, confirming the long-lasting bioavailability of this EB conjugate. The results also suggest that functionalized EB can be used as the albumin binding moiety to increase the blood circulation half-life of other drug candidates as well.
In most cases, the anti-diabetic drugs based on stimulation of insulin secretion, GLP-1 and Exendin-4 are administered subcutaneously instead of being intravenously injection [45], so we moved forward to evaluate the pharmacokinetics of Abextide after subcutaneous administration. All of the three compounds were labeled with a radionuclide with longer physical half-life (Copper-64, t1/2=12.6 h) to cover the whole process of drug release from the injection site. Both Abextide and Albiglutide showed much slower migration from the injection site (Figure 3). The underlying mechanism of uptake is that Abextide binds with albumin in the interstitial fluid [28] as does the macromolecule as Albiglutide. However, Albiglutide had somewhat higher residual amount than Abextide at 4 h and 24 h time points and the exact reason will need further investigation.
Besides the non-invasive imaging method, we also evaluated the pharmacokinetic parameters of Abextide and Exendin-4 via traditional sample collection and commercial Exendin-4 EIA kits measurement after subcutaneous administration, which were summarized in Figure 4A and Table 1. Similar to PET quantification, the in vivo pharmacokinetic profile of Abextide after subcutaneous injection was obviously better than that of exendin-4. Furthermore, the concentration of Abextide in the circulation was twice than that of exendin-4. More importantly, the hypoglycemic duration of Abextide in db/db mice was greatly prolonged as compared to native Exendin-4. A blood glucose level below 8.35 mM (150 mg/dL) was regarded as normal, and the euglycemic duration under that value is considered a practical indication of the potential for antidiabetic treatment. The administration of Abextide sustained normal glycemia for 3.6 times longer than native Exendin-4 (Figure 4C). It is well known that drug clearance in humans will be much slower than that in rodents, and thus, the antidiabetic duration of Abextide may be substantially longer than 36 h in human than in db/db mice in the present study. Therefore, Abextide has great potential to be used as a long-lasting anti-diabetic drug.
In conclusion, Abextide can be easily synthesized with high yield and high purity; in addition tEB conjugation does not compromise the binding affinity of Exendin-4. Abextide was found to have greatly improved in vivo distribution pattern, pharmacokinetic and pharmacodynamic characteristics compared to Exendin-4 in healthy BALB/c or diabetic rodent models. This enhanced in vivo performance of Abextide is mostly due to albumin-binding property through tEB conjugation. Based on these results, we believe that Abextide has considerable pharmaceutical potential as a treatment for type 2 diabetes. This strategy of tEB conjugation also holds potential to be applied to other small molecules and biologics for the development of long-acting therapeutic drugs.
Acknowledgements
The authors gratefully acknowledge the National Natural Science Foundation of China (81471684 and 81371596) and the Intramural Research Program, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health. Haojun Chen was partially funded by the China Scholarship Council (CSC).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Caliceti P, Veronese FM. Pharmacokinetic and biodistribution properties of poly(ethylene glycol)-protein conjugates. Adv Drug Deliv Rev. 2003;55:1261-77
2. Shechter Y, Mironchik M, Rubinraut S. et al. Albumin-insulin conjugate releasing insulin slowly under physiological conditions: a new concept for long-acting insulin. Bioconjug Chem. 2005;16:913-20
3. Lin JH, Lu AY. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol Rev. 1997;49:403-49
4. Meibohm B, Zhou H. Characterizing the impact of renal impairment on the clinical pharmacology of biologics. J Clin Pharmacol. 2012;52:54S-62S
5. Keyt BA, Paoni NF, Refino CJ. et al. A faster-acting and more potent form of tissue plasminogen activator. Proc Natl Acad Sci U S A. 1994;91:3670-4
6. Chae SY, Chun YG, Lee S. et al. Pharmacokinetic and pharmacodynamic evaluation of site-specific PEGylated glucagon-like peptide-1 analogs as flexible postprandial-glucose controllers. J Pharm Sci. 2009;98:1556-67
7. Syed S, Schuyler PD, Kulczycky M. et al. Potent antithrombin activity and delayed clearance from the circulation characterize recombinant hirudin genetically fused to albumin. Blood. 1997;89:3243-52
8. Chapman AP. PEGylated antibodies and antibody fragments for improved therapy: a review. Adv Drug Deliv Rev. 2002;54:531-45
9. Veronese FM, Pasut G. PEGylation, successful approach to drug delivery. Drug Discov Today. 2005;10:1451-8
10. Gaberc-Porekar V, Zore I, Podobnik B. et al. Obstacles and pitfalls in the PEGylation of therapeutic proteins. Curr Opin Drug Discov Devel. 2008;11:242-50
11. Veronese FM. Peptide and protein PEGylation: a review of problems and solutions. Biomaterials. 2001;22:405-17
12. Chuang VT, Kragh-Hansen U, Otagiri M. Pharmaceutical strategies utilizing recombinant human serum albumin. Pharm Res. 2002;19:569-77
13. Rath T, Baker K, Dumont JA. et al. Fc-fusion proteins and FcRn: structural insights for longer-lasting and more effective therapeutics. Crit Rev Biotechnol. 2015;35:235-54
14. Kratz F. Albumin as a drug carrier: design of prodrugs, drug conjugates and nanoparticles. J Control Release. 2008;132:171-83
15. Doumas BT, Watson WA, Biggs HG. Albumin standards and the measurement of serum albumin with bromcresol green. Clin Chim Acta. 1971;31:87-96
16. Kurtzhals P, Havelund S, Jonassen I. et al. Effect of fatty acids and selected drugs on the albumin binding of a long-acting, acylated insulin analogue. J Pharm Sci. 1997;86:1365-8
17. Sinn H, Schrenk HH, Friedrich EA. et al. Design of compounds having an enhanced tumour uptake, using serum albumin as a carrier. Part I. Int J Rad Appl Instrum B. 1990;17:819-27
18. Franssen EJ, Jansen RW, Vaalburg M. et al. Hepatic and intrahepatic targeting of an anti-inflammatory agent with human serum albumin and neoglycoproteins as carrier molecules. Biochem Pharmacol. 1993;45:1215-26
19. Gradishar WJ. Albumin-bound paclitaxel: a next-generation taxane. Expert Opin Pharmacother. 2006;7:1041-53
20. Havelund S, Plum A, Ribel U. et al. The mechanism of protraction of insulin detemir, a long-acting, acylated analog of human insulin. Pharm Res. 2004;21:1498-504
21. Kratz F, Ehling G, Kauffmann HM. et al. Acute and repeat-dose toxicity studies of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin (DOXO-EMCH), an albumin-binding prodrug of the anticancer agent doxorubicin. Hum Exp Toxicol. 2007;26:19-35
22. Myers SR, Yakubu-Madus FE, Johnson WT. et al. Acylation of human insulin with palmitic acid extends the time action of human insulin in diabetic dogs. Diabetes. 1997;46:637-42
23. Yoon HJ, Kang KW, Chun IK. et al. Correlation of breast cancer subtypes, based on estrogen receptor, progesterone receptor, and HER2, with functional imaging parameters from 68Ga-RGD PET/CT and 18F-FDG PET/CT. Eur J Nucl Med Mol Imaging. 2014;41:1534-43
24. Lee J, Lee C, Kim I. et al. Preparation and evaluation of palmitic acid-conjugated exendin-4 with delayed absorption and prolonged circulation for longer hypoglycemia. Int J Pharm. 2012;424:50-7
25. Spahr PF, Edsall JT. Amino Acid Composition of Human and Bovine Serum Mercaptalbumins. J Biol Chem. 1964;239:850-4
26. Gibson JG, Evans WA. Clinical Studies of the Blood Volume. I. Clinical Application of a Method Employing the Azo Dye "Evans Blue" and the Spectrophotometer. J Clin Invest. 1937;16:301-16
27. Niu G, Lang L, Kiesewetter DO. et al. In Vivo Labeling of Serum Albumin for PET. J Nucl Med. 2014;55:1150-6
28. Wang Y, Lang L, Huang P. et al. In vivo albumin labeling and lymphatic imaging. Proc Natl Acad Sci U S A. 2015;112:208-13
29. Zhang J, Lang L, Li F. et al. Clinical Translation of a Novel Albumin-Binding PET Radiotracer 68Ga-NEB. J Nucl Med. 2015;56:1609-14
30. Edwards CM, Stanley SA, Davis R. et al. Exendin-4 reduces fasting and postprandial glucose and decreases energy intake in healthy volunteers. Am J Physiol Endocrinol Metab. 2001;281:E155-61
31. Xu G, Stoffers DA, Habener JF. et al. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes. 1999;48:2270-6
32. Deacon CF. Therapeutic strategies based on glucagon-like peptide 1. Diabetes. 2004;53:2181-9
33. Buse JB, Henry RR, Han J. et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care. 2004;27:2628-35
34. Kendall DM, Riddle MC, Rosenstock J. et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care. 2005;28:1083-91
35. DeFronzo RA, Ratner RE, Han J. et al. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care. 2005;28:1092-100
36. Davidson MB, Bate G, Kirkpatrick P. Exenatide. Nat Rev Drug Discov. 2005;4:713-4
37. Gao H, Niu G, Yang M. et al. PET of insulinoma using 18F-FBEM-EM3106B, a new GLP-1 analogue. Mol Pharm. 2011;8:1775-82
38. National Research Council. Guide for the Care and Use of Laboratory Animals. Washington (DC). 1996
39. Kim TH, Jiang HH, Lee S. et al. Mono-PEGylated dimeric exendin-4 as high receptor binding and long-acting conjugates for type 2 anti-diabetes therapeutics. Bioconjug Chem. 2011;22:625-32
40. Matthews JE, Stewart MW, De Boever EH. et al. Pharmacodynamics, pharmacokinetics, safety, and tolerability of albiglutide, a long-acting glucagon-like peptide-1 mimetic, in patients with type 2 diabetes. J Clin Endocrinol Metab. 2008;93:4810-7
41. Yang M, Hoppmann S, Chen L. et al. Human serum albumin conjugated biomolecules for cancer molecular imaging. Curr Pharm Design. 2012;18:1023-31
42. Makrides SC, Nygren PA, Andrews B. et al. Extended in vivo half-life of human soluble complement receptor type 1 fused to a serum albumin-binding receptor. J Pharmacol Exp Ther. 1996;277:534-42
43. Dennis MS, Zhang M, Meng YG. et al. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J Biol Chem. 2002;277:35035-43
44. Gupta N, Price PM, Aboagye EO. PET for in vivo pharmacokinetic and pharmacodynamic measurements. Eur J Cancer. 2002;38:2094-107
45. Tahrani AA, Bailey CJ, Del Prato S. et al. Management of type 2 diabetes: new and future developments in treatment. Lancet. 2011;378:182-97
Author contact
![]() Corresponding authors: Xianzhong Zhang (zhangxzedu.cn); Hua Wu (wuhua1025com); Lei Zhu (lei.zhuedu.cn); Gang Niu (niugnih.gov); Xiaoyuan Chen (shawn.chengov)
Corresponding authors: Xianzhong Zhang (zhangxzedu.cn); Hua Wu (wuhua1025com); Lei Zhu (lei.zhuedu.cn); Gang Niu (niugnih.gov); Xiaoyuan Chen (shawn.chengov)