Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Experimental section
Results and discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(3):318-327. doi:10.7150/thno.13533 This issue Cite
Research Paper
Double-stem Hairpin Probe and Ultrasensitive Colorimetric Detection of Cancer-related Nucleic Acids
Jianguo Xu, Hongling Li, Zai-Sheng Wu ![]() , Jun Qian, Chang Xue, Lee Jia
, Jun Qian, Chang Xue, Lee Jia ![]()
Cancer Metastasis Alert and Prevention Center, and Pharmaceutical Photocatalysis of State Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou 350002, China.
Received 2015-8-12; Accepted 2015-10-1; Published 2016-1-1
Abstract

The development of a versatile biosensing platform to screen specific DNA sequences is still an essential issue of molecular biology research and clinic diagnosis of genetic disease. In this work, we for the first time reported a double-stem hairpin probe (DHP) that was simultaneously engineered to incorporate a DNAzyme, DNAzyme's complementary fragment and nicking enzyme recognition site. The important aspect of this hairpin probe is that, although it is designed to have a long ds DNA fragment, no intermolecular interaction occurs, circumventing the sticky-end pairing-determined disadvantages encountered by classic molecular beacon. For the DHP-based colorimetric sensing system, as a model analyte, cancer-related DNA sequence can trigger a cascade polymerization/nicking cycle on only one oligonucleotide probe. This led to the dramatic accumulation of G-quadruplexes directly responsible for colorimetric signal conversion without any loss. As a result, the target DNA is capable of being detected to 1 fM (six to eight orders of magnitude lower than that of catalytic molecular beacons) and point mutations are distinguished by the naked eye. The described DHP as a-proof-of-concept would not only promote the design of colorimetric biosensors but also open a good way to promote the diagnosis and treatment of genetic diseases.
Keywords: double-stem hairpin probe (DHP), DNAzyme, colorimetric sensing, p53 gene
Introduction
With advances in molecular biology, nucleic acids (DNA and RNA) detection has been routinely identified as a highly-valued tool for disease monitoring and genetic disorders screening,[1,2] which can provide valuable information on biological research, medical science and clinical diagnosis. Especially in the diagnosis and treatment of malignant tumors, oncogene and tumor suppressor gene closely associated with tumor progression are often present at a very low concentration, and their expression levels may provide useful clues for clinical application as well as anticancer drug development.[3] Moreover, gene mutations and change in gene expressing levels often are the early molecular events, and the tumor therapy at the early stage is successful in more than 95 percent of cases. Accordingly, nucleic acid detection has been a topic of significant interest, where various amplification strategies, including polymerase chain reaction (PCR), were employed to interrogate nucleic acid levels.[4-11] However, each sensing method has its own advantages and disadvantages with respect to simplicity, sensitivity, operational simplicity, design flexibility and cost. For example, the inherent drawbacks of PCR, such as expensive instruments, complicated procedure, false positive results, and skilled technician, hamper its practical applications in some extent.[12] Further efforts are still needed to address the serious clinical challenge. Additionally, to perform the public and periodic disease screening, it is equally important to simplify the signaling probe design, to reduce the dependence on the medical instruments and to decrease the assay cost, because these improvements can make the sensing platform more readily available and affordable to the people especially in the underdeveloped areas. Thus, development of reliable, convenient and cost-effective screening tools (e.g., visual inspection without any instrument) for nucleic acids detection still remains imperative.
Out of the various sensing DNA probes, molecular beacon (MB) is the most attractive probe for DNA detection due to its operation convenience without separation and better specificity than linear DNA probe originating from its hairpin structure.[13] Conceptually, fluorescent MB is a single-strand nucleic acid probe with the terminally-attached fluorophore and quencher, where the loop portion contains the sequence complementary to target DNA (usually 15-30 bases) and the stem consists of two self-complementary fragments (5-7 bases).[14] In the absence of the target, a stem-loop structure holds the terminal fluorophore/quencher pair in proximity and the fluorescence is quenched. The hybridization of target DNA to loop portion can force the fluorophore and quencher moieties to separate, inducing the fluorescence restoration and producing the detectable signal. In essence, one target DNA only causes the opening of one MB.[15] Namely, the target DNA is detected in a one-to-one manner, and the detection sensitivity of MB is only partially successful.[16,17] Therefore, to improve further the assay performance, MB, as well as the analogous hairpin probe, was combined with various amplification techniques, for example, hybridization chain reaction,[18] nicking enzyme signal amplification,[19] exonuclease-aided target recycling,[20] strand displacement amplification (SDA),[21,22] and rolling circle amplification (RCA).[23] Although considerable research efforts on the innovation and potential application of MBs have been made to implement the amplification detection of nucleic acids, MB-based sensing techniques still suffer from some inherent limitations in some aspects. For example, DNA sticky-end pairing between molecular beacons sometimes takes place especially when long self-complementary base fragments are involved.[24,25] In this case, not only is no fluorescence signal detected even if increasing the target DNA concentration but also non-fluorescent hairpin probe-based sensing systems are unsuccessfully constructed.[25] More stable intramolecular hybridization of hairpin probes is expected to improve the detection specificity and endows the probe with the addition design flexibility, but intermolecular sticky-end pairing is more likely to occur. Therefore, up to now, no MB or analogous hairpin probe with a long stem has been reported. This seems to be a challenging yet promising stepping stone toward achieving synergetic effect of hairpin probes and efficient signal amplification strategies for a variety of new diagnostic screening and biomarker discovery applications.
The discovery of the horseradish peroxidase (HRP) mimicking hemin/G-quadruplex DNAzyme and related studies provide a good chance for developing a powerful biosensor.[26,27] The hemin/G-quadruplex complex, as one fascinating DNAzyme, possesses several attractive features superior to protein enzymes, such as high thermal stability, simple synthesis, low cost and easy modification. Significantly, DNAzyme sequence can be delicately programmed to implement the unique function, for example, being woven into the common nucleic acid sequence to construct a catalytic DNA biosensing probe [28] or DNA biosensing nanowires [29,30] and being split into two or more separate molecules to achieved the expected objectives.[29,31] Thus, the catalytic utility of G-quadruplex DNAzyme can be compatible with different amplification techniques in a controlled and predictive fashion, leading to several impressive biosensing systems on the basis of nicking enzyme-assisted DNA replication,[12] RCA,[32,33] and PCR.[34]
In this contribution, using p53 cancer suppressor gene as a model analyte, a novel double-stem-contained hairpin probe (DHP) (essentially label-free MB containing very stable stems) was developed for the ultrasensitive colorimetric detection of DNA hybridization. Into the DHP, we simultaneously introduced a binding site of nicking enzyme and two stems, one of which consisted of long G-rich fragment and its complementary fragment. Contrary to the conventional view that the strong base pairing of hairpin probes readily causes the formation of intermolecular hybridization-based DNA duplexes,[24,25] no intermolecular interaction among DHPs occurs. Thus, although only two types of label-free oligonucleotides are involved in the colorimetric sensing system, target binding can easily induce cascade amplification via forcing the DHP to undergo a large-amplitude conformational transition and generates a copious amount of horseradish peroxidase (HRP)-mimicking DNAzyme. This enzyme can catalyze the oxidation of ABTS by H2O2 to a green colored product ABTS•+, producing an exponentially amplified colorimetric signal and improving the intrinsic assay specificity of hairpin probes. As a result, as low as 1 fM p53 gene can be detected and point mutations are easily identified by the naked eye. As a proof-of-concept study, the advanced capabilities of DHP together with the intriguing properties, such as cost-saving and easy construction, are expected to promote the application of hairpin probes in basic research and medical diagnosis.
Experimental section
Materials and reagents
Nt. BbvCI endonuclease, Klenow Fragment (3'-5' exo-) polymerase and low DNA ladder were ordered from New England Biolabs (USA) Ltd, while deoxynucleotide triphosphates (dNTPs) were purchased from Dingguo Changsheng Biotechnology Co., Ltd (Beijing, China). The 25 mM tris-buffer (100 mM NaCl, 50 mM KAc, 10 mM MgAc2 and 1 mM DTT, pH 8.2) was used as the reaction solution.[35] Hemin and ABTS were supplied by Sigma-Aldrich Co. LLC, and H2O2 was from Xilong Chemical Co., Ltd. All chemicals were of analytical reagent grade and used as received unless otherwise stated. Double-distilled water, purified by a Kerton lab MINI water purification system (UK) with electrical resistivity of 18.25 MΩ, was used throughout this study.
Oligonucleotide strands designed in this study were synthesized and PAGE-purified by Invitrogen Bio Inc. (Shanghai, China). Their sequences are listed in Table 1, and the secondary structure of DHP (seen in Figure S1) was determined by the online “mfold” program (http://mfold.rna.albany.edu/). The stock solution of oligonucleotides was prepared in 1× TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) and stored at 4 ℃ before usage.
Oligonucleotide sequences designed in the current study.
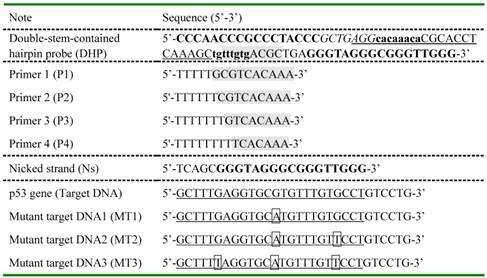
For DHP, two bold base fragments indicated by lower case letter and capital letter cooperatively promote the formation of hairpin structure via self-hybridization and are named as stem I and stem II, respectively. The G-rich fragment known as the aptamer of hemin was added at the 3' end of DHP. The underlined part and segment with gray background are designed to recognize selectively target DNA and polymerization primer, respectively. Nicked strand was synthesized to have the same base sequence as the nicked/displaced oligonucleotide strand generated during DNA detection. A half of recognition site for Nt.BbvCI nickase was designed into DHP and highlighted in italic. Except for the mutant points shown in box, the MTs have the same sequences as the completely complementary target DNA.
Instruments
Absorbance measurements were taken using a UV-2700 spectrophotometer (Shimadzu, Japan) in the wavelength range of 380-500 nm at room temperature, and the absorption value at 417 nm was recorded to evaluate the proposed assay performance and to quantify target molecules in samples. pH value was determined by a pHS-3C digital pH meter (Ohaus Instrument Plant, Shanghai, China), and the reaction temperature was controlled by a TU-200 Block Heater (Yiheng Co. Ltd., Shanghai, China). Canon D3200 was used to visualize the resulting solution.
DHP-based sensing procedure
Prior to experiments, DHP was heated to 90 ℃ for 5 min and allowed to cool down to room temperature slowly. Subsequently, 16 μL of tris-buffer, 3 μ of 5 μM DHP, 3 μL of 5 μM P2 (the primer optimization is shown in Figure S2), and 3 μL of p53 gene at varied concentrations were thoroughly mixed and the hybridization was allowed to proceed at room temperature for 1 h. Then 1 µL of 10 mM dNTPs, 0.5 µL of 5 U/µL Klenow Fragment (3'-5' exo-) polymerase, and 0.5 µL of 10 U/µL Nt. BbvCI endonuclease were consecutively added to the resulting mixture to launch the amplification reaction at 37 ℃ for 40 min with gentle vortex. Followed by incubation at 80 ℃ for 20 min to terminate the activity of enzymes as recommended by the manufacturer, a droplet of 20 μM hemin (3 µL, dissolved by DMSO) was injected and incubated at 37 ℃ for another 1 h to form the DNAzymes. Finally, 82.5 µL of 2× HEPES buffer (50 mM HEPES, 20 mM KCl, 0.4 M NaCl, 2% DMSO, 0.1% Triton X-100, pH =5.2), 5 µL of 18 mM ABTS, and 2.5 µL of 4 mM H2O2 were added to the reaction solution to initiate the catalytic reaction. Notably, the volume of the isothermal amplification reaction and the final oxidation reaction are 30 μL and 120 μL, respectively. We calculated the concentration of DHP, P2, target DNA, and hemin from the 30-µL solution, while the concentration of ABTS and H2O2 mentioned in the text was that in 120-µL solution. After 30-min incubation, the absorption spectra of resulting samples were collected.
Unless otherwise specified, the experiments were carried out under identical conditions except for only one issue of interest when investigating the factors that influence the assay performance of the present colorimetric sensing system.
Asymmetric PCR amplifications
Genomic DNA was extracted from the clinical revelant human lung cancer cell line A549 by the universal Genomic DNA exaction kit (from Takara Biotechnology Co. Ltd.) when the cells were cultured to desired density. Asymmetric PCR adopted frequently to generate the single-stranded target strand besides the double-stranded DNA was performed to amplify the codon 273 of p53 gene. The following primers designed with the help of Primer Premier 5.0 software were used: Sense primer 1: 5'-GCTTTGAGGTGCGTGTTT-3'; Anti-sense primer 1: 5'-GTGAGGCTCCCCTTTCTT-3'. Briefly, asymmetric amplification was achieved by thermal cycling for 30 cycles in a total volume of 50 μL of 1× PCR buffer containing 1 μM Forward primer, 0.2 μM Reverse primer, 5 U of Taq polymerase, 0.15 μM dNTP mixture, and 3.4 μg of genomic DNA. Each PCR cycle was initiated by 5 min of denaturation at 94 ℃, followed by 30 s at 94℃, 30 s at 55℃, 55 s at 72 ℃, and then a final extension at 72 ℃ within a time-period of 7 min. Finally, 12% native-PAGE gel electrophoresis was used to verify the amplified products.
For control asymmetric PCR amplification, another pair of primers (Sense primer 2: 5'-GAGGTAAGCAAGCAGGACA-3'; Anti-sense primer 2: 5'-GCAAGGAAAGGTGATAAAAGT-3') were used. The amplification experiment was conducted under identical conditions as described above.
Results and discussion
The design of DHP and working principle
In the present study, to execute the multifunctional signal amplification, the DHP is designed to contain four regions: G-quadruplex sequence, nicking endonuclease-binding site, polymerization primer-binding fragment and target DNA recognition segment. The base sequence of each region of DHP is illustrated in Table 1, while its secondary structure is shown in Figure S1 (mentioned above). DHP is abbreviated from double-stem-contained hairpin probe, containing stem I and stem II as its name implies. One can see that, for DHP, polymerization primer-binding site is locked by stem I, G-quadruplex fragment is locked by its complementary fragment (forming the stem II), and only a half of nicking endonuclease-binding site is involved. Thus, the DHP-based sensing system does not exhibit obvious absorption peak in the UV-vis spectrum. Note that the proposed DHP does not form the sticky-end pairing-based dimer but folds into a unique hairpin structure with two stems containing 25 base pairs. This is an important achievement that can be met without resort (supporting data seen below), establishing the molecular foundation of the present colorimetric assay strategy.
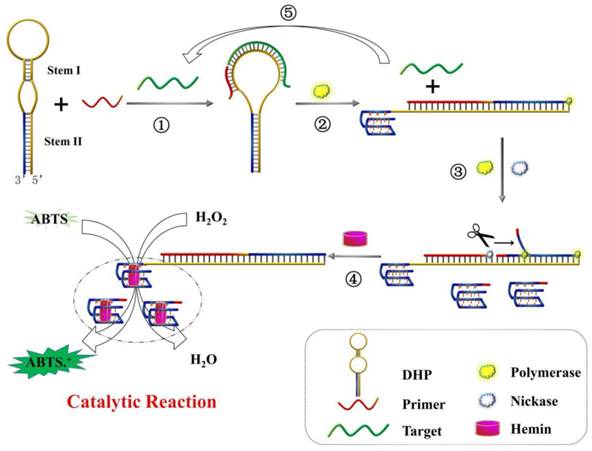
The working principle of DHP for the colorimetric detection of DNA hybridization is illustrated in Scheme 1. In the absence of target, because the DHP were tightly locked by stem I and stem II, the primer could not bind to DHP and G-quadruplex structure is incapable of being formed. Thus, the subsequent enzymatic reaction and signaling do not occur. In contrast, when target gene hybridizes with DHP (step ①), the rigid duplex helix gives rise to a configuration change of DHP and open the hairpin structure of stem I, facilitating the binding of DHP to P2. With the aid of polymerase (step ②), the 3' end of P2 is allowed to extend on DHP and the stem II is completely unfolded. Thus, besides the hybridized target is displaced, the G-rich fragment is released and folds into the G-quadruplex structure, and perfect recognition site of Nt. BbvCI is formed, executing a large-amplitude conformational transition. In the step ③, nicking reaction leads to the generation of new primer and induces the subsequent polymerization, as well as the displacement of pre-replicated G-rich fragment. The nicking, polymerization and displacement can be repeated continuously, resulting in the accumulation of G-quadruplex molecules. Upon addition of hemin, the G-quadruplex can bind to hemin and forms the DNAzyme that has the catalytic activity similar to horseradish peroxidase, catalyzing the oxidation of ABTS by H2O2 and producing the amplified colorimetric signal (step ④). Moreover, the displaced target DNA obtained from the step ② can in turn induce the next round of hybridization, polymerization, nicking and signal conversion, amplifying further the target DNA hybridization event (step ⑤). As a result, an exponentially amplified colorimetric signal is expected to be achieved, enabling the reliable detection of trace target elements.
Schematic representation of DHP design and signaling principle for p53 gene colorimetric detection: ① recognition of p53 gene; ② polymerization to trigger target recycling and G-quadruplex release; ③ accumulation of G-quadruplex induced by the polymerization/nicking cycle; ④ catalytic reaction for signal output; ⑤ displaced-target DNA induces the next round of hybridization and signal transduction.
By the way, besides mentioned-above exceptional structural features of DHP, no any signal loss occurs for the present colorimetric sensing system compared with traditional nicking/polymerization-based amplification assays.[12,35] Specifically, as illustrated in Scheme S1, the G-quadruplex fragments constantly generated from step ③ are not consumed by hybridizing with DHPs for two reasons: 1) The dual-stem structure makes the DHP stable enough so that it is not easy to open it; 2) even if DHP is opened by displaced G-rich fragments, the G-quadruplex fragment locked by stem II could be released and form an equal amount of DNAzyme. Thus, the likely signal loss is completely circumvented. To demonstrate this important mechanism, Ns with the same base sequence as the nicked/displaced G-quadruplex strand was designed (seen in Table 1), and the corresponding experiments were performed under identical conditions. The measured data were collected and analyzed in Figure S3.
Feasibility of the proposed DHP for target DNA detection
Since the DHP is a novel hairpin probe with very stable stems, the feasibility of colorimetric sensing strategy should be verified at the initial stage. For this purpose, the DNA colorimetric assay and different control experiments were performed, and the measured data are shown in Figure 1A. One can see that there is no obvious difference in absorption spectrum between lines a, b and c, indicating that the DHP can tightly self-lock due to the introduction of its two stems and no free DNA-quadruplex fragment exists even if target DNA was added. In the presence of polymerase, as illustrated in the difference between line d and line e, only a slight absorption peak increase is observed. Excitingly, compared with line f, a dramatic absorption increase is seen in line g where nicking enzyme existed, suggesting that a large number of G-quadruplexes were generated during the autonomous polymerization/scission/displacement cycle and formed the expected catalytic DNAzyme in the presence of hemin. Thus, the optical signal of the proposed colorimetric sensing system can reflect the existence of DNA target.
The feasibility of colorimetric amplification detection of DNA hybridization. (A) Typical UV-vis absorption spectra of the present sensing system in the presence of (a) H2O2+ABTS; (b) DHP+P2+H2O2+ABTS; (c) DHP+P2+p53+H2O2+ABTS; (d) DHP+P2+polymerase+H2O2+ABTS; (e) DHP+P2+p53+polymerase+H2O2+ABTS; (f) DHP+P2+polymerase+nickase+H2O2+ABTS; (g) DHP+P2+p53+polymerase+nickase+H2O2+ABTS; (B) Comparative study of three sensing systems in terms of the capability to signal DNA hybridization. System I: non-amplification system (data obtained from line b and line c); System II: polymerization amplification system (data from line d and line e); System III: polymerization/nicking amplification system (data from line f and line g). A, A0 and As indicate the absorption intensity at 417 nm of target sample, blank and only H2O2-ABTS-contianed sample (namely, sample a), respectively; (C) Representative photographs of different samples described in Figure 1A. Experiments are performed as described in “Experimental section”. [DHP]= 500 nM; [P2]= 500 nM; [p53]= 150 nM; [hemin]= 2 uM; [H2O2]= 83.33 uM; [ABTS]= 0.75 mM.
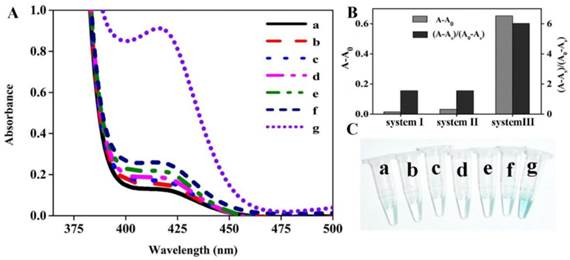
Apparently, three different sensing systems are studied in this section: non-amplification system (system I, line b and c), polymerization amplification system (system II, line d and e), and polymerization/nicking amplification system (system III, line f and g). To obtain the accurate comparative data on the assay performance of sensing systems involved, the optical signals, including the background-subtracted absorption intensity and absorption peak ratio of the target sample to blank, were quantitatively evaluated, and the detailed results are shown in Figure 1B. The system III (namely, the proposed colorimetric assay) exhibits the most desirable sensing performance.
In fact, this optical signal changing trend can be observed by the naked eye. Figure 1C displays the representative photographs corresponding to different samples depicted in Figure 1A. We can see that a remarkable color change occurs in sample g, while all other samples are almost colorless, strongly further convincing that the DHP is rationally designed to execute the amplification detection of target DNA.
In the contribution, we for the first time proposed the DHP with a unique hairpin structure and developed a new amplification sensing system via combining with polymerization and nicking. Although this probe has the self-complementary 25-base fragment, it does not form the sticky-end pairing-based dimer. To confirm the unpredictable molecular structure of DHP and the utility of polymerase and endonuclease, native polyacrylamide gel electrophoresis (PAGE) was used to explore the mechanism of the designed sensing system. The experimental results are represented in Figure S4.
Optimization of experimental conditions
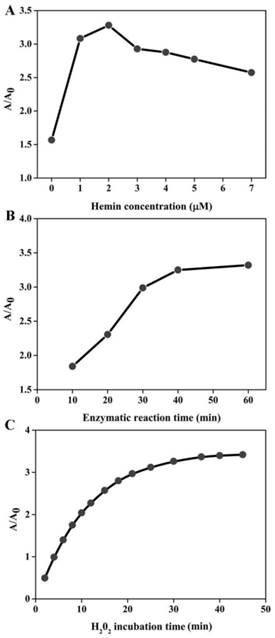
In order to achieve the best sensing performance, the effect of the hemin concentration, enzymatic reaction time and H2O2 incubation time were investigated, respectively. The ration of A/A0 was adopted to evaluate the sensing ability, where A and A0 represent the absorption peak in the presence and absence of target DNA, respectively. As illustrated in Figure 2A, the value of A/A0 first increased with the increase of hemin concentration. The maximal ratio value was achieved when hemin at 2 μM was employed. Then, signal decreased at higher concentration. This is reasonable. In the low hemin concentration range, more hemin molecules can produce more DNAzyme units responsible for the higher signal. However, when the concentration of hemin is more than 2 μM, the background increases and deteriorates the assay performance. Thus, we chose 2 μM as the favorable one for subsequent experiments. The enzymatic reaction time is explored as shown in Figure 2B. The absorption ratio of the solution increases with the increase of reaction time up to 40 min and then levels off. Therefore, the period of 40 min is preferred for the isothermal amplification. The influence of H2O2 incubation time can be seen in Figure 2C, and the A/A0 is found to increase with the incubation time increase and almost reaches equilibrium at 30 min. Thus, we chose 30 min for ABTS oxidation.
Optimization of experimental conditions for DNA detection. The effect of hemin concentration (A), enzymatic reaction time (B) and H2O2 incubation time (C) on the sensing system. The target detection was conducted according to the method described in “Experimental section”. The concentration of several species involved is detailed below: [DHP]= 500 nM; [P2]= 500 nM; [p53]= 150 nM; [H2O2]= 83.33 uM; [ABTS]= 0.75 mM.
Analytical performance of DHP
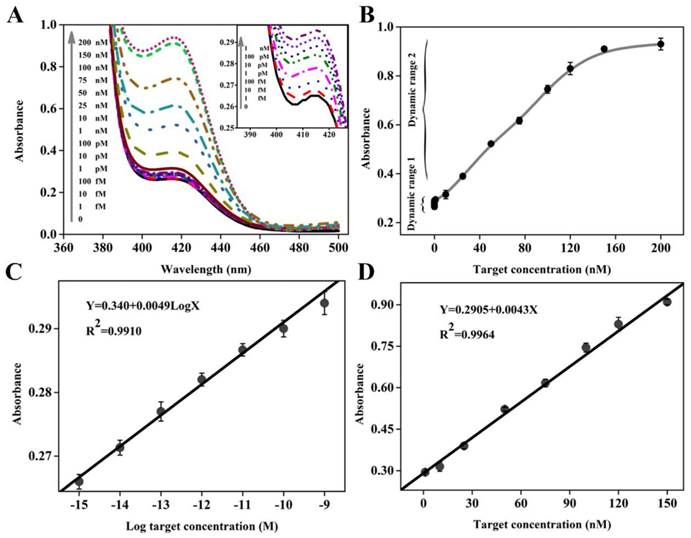
Once possessing the capability to accurately recognize and quantify disease-related genes, especially those associated with cancers, the new biosensing probe would hold considerable promise for early disease diagnosis and monitoring of treatment progress. To evaluate the utility of DHP with unique structural features, different concentrations of target DNA ranging from 0 to 200 nM were prepared and analyzed by this colorimetric sensing system. The UV-vis absorption spectra are collected in Figure 3A, while the corresponding absorption peak is recorded in Figure 3B. It could be seen that an obvious monotonic increase in absorption intensity is observed when increasing target concentration from 0 and 150 nM, and no obvious signal change could be achieved even at the higher target concentration, for example, 200 nM. Excitingly, as low as 1 fM target DNA, equal to the DNA levels in physiological samples, can still induce a detectable signal compared to the blank sample, and thus we define it as the limit of detection (LOD), representing an impressive improvement in term of assay sensitivity. For example, two classical catalytic beacons in the literature reports [28,36] only offer the detection limit of 2 nM and 200 nM, respectively. Namely, the DHP-based colorimetric sensing system can achieve over 6 or 8 orders of magnitude improvement in the detection sensitivity. More details on the sensitivity comparisons with other impressive previously-reported signal amplification strategies, including the methods based on the versatile nanomaterials and electrochemical measurement that is well known to be an extremely sensitive signaling technique, are provided in Table 2. It is clear and unambiguous that the newly-proposed DHP colorimetric assay can offer a very exciting LOD at least 20 times lower than the literature values.[29,37-43] The measured data demonstrates that this homogenous signaling platform is efficient for ultrasensitive colorimetric detection of DNA hybridization.
(A) Absorption spectra collected in the presence of various concentrations of target DNA. The inset shows the absorption spectra in the low target concentration range; (B) Dependence of absorption intensity at 417 nm on target DNA concentration, where two dynamic response ranges are seen. The error bars represent the standard deviation obtained from at least three parallel measurements for each concentration. (C) and (D) depict the linear relationship between absorption peak intensity and the logarithm of the target DNA concentration in the dynamic range I and between absorption peak intensity and the target DNA concentration in the dynamic range II, respectively.
Comparison of detection capability of the present DHP-based sensing system with other optical or electrochemical sensors for nucleic acids detection.
| Method | LOD | Linearity range | Category | Ref |
|---|---|---|---|---|
| Electrochemical interrogation of kinetically-controlled dendritic DNA/PNA assembly | 100 fM | 0.1 pM to 10 nM | Electrochemical assay | 37 |
| Exonuclease III-aided autocatalytic DNA biosensing platform | 0.1 pM | 0.1 pM to 1 nM | Electrochemical assay | 38 |
| Fluorescence Near Gold Nanoparticles | 100 pM | 100 pM to 1000 pM | Fluorescent assay | 39 |
| Pendulum-type optical DNA nanodevice | 0.8 nM | 0.8 nM to 163 nM | Fluorescent assay | 40 |
| Exonuclease-assisted cascade target recycling and DNAzyme amplification | 20 fM | 20 fM to 0.1 nM | Fluorescent assay | 41 |
| Autonomous assembly of Hemin/G-Quadruplex DNAzyme nanowires sensor | 100 fM | 10-13 M to 10-8 M | Colorimetric assay | 29 |
| Exonuclease-assisted cascaded recycling amplification | 0.1 pM | 0.1 pM to 1 μM | Colorimetric assay | 42 |
| G-quadruplex MBzymes mediated label-free amplified detection | 25 fM | 25 fM to 250 nM | Colorimetric assay | 43 |
| DHP-based cascade amplification | 1 fM | 1 fM to 150 nM | Colorimetric assay | Present study |
It is worth pointing out that, compared with these sensing systems,[29,37-43] besides desirable LOD, the dynamic response range is substantially widened, including two linear calibration curves different from each other in the low and high target concentration. Figure 3C displays a good linear relationship between the peak absorption value (Y) and the logarithmic concentration of target DNA (X) in the range from 1.0 fM to 1 nM with a calibration curve fitted to the equation: Y= 0.340+ 0.0049 LogX (R2= 0.9910). Figure 3D presents the linear dynamic response to target DNA in the concentration range from 1 nM to 150 nM, and the regression equation can be expressed as Y= 0.2905+ 0.0043X (R2= 0.9964). It should be easy to understand two different linear relationships. On the whole, two types of reactions can contribute to the detectable signal: primary target DNA hybridization (step ①) and subsequent amplification (steps ②, ③ and ⑤). The former plays a major role in signal conversion in the high target concentration range, while the latter is largely responsible in the low target concentration range. Since there is the difference in signaling process between high and low concentration ranges, two different linear relationships could be obtained.
Detection specificity
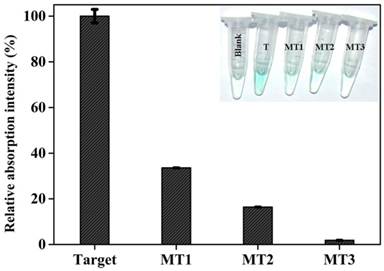
Gene mutation is a very common phenomenon occurred frequently in some biological events.[44] A large number of human diseases have been known to originate from gene mutation, and the information on gene mutation is expected to provide direct insight into molecular mechanisms of some diseases [45] and to promote the targeted therapy. Thus, reliably distinguishing mutant target DNA from the wild type one is of great significance. To evaluate the detection specificity of the current colorimetric sensing system, different target DNAs, including complementary target DNA (T), single-base mutant DNA (MT1), two-bases mutant DNA (MT2), and three-base mutant DNA (MT3), were detected under identical experimental conditions, and the measured data are comparatively depicted in Figure 4. As can be seen, mutations existing in every target DNA induce a substantial decrease in signal intensity. Specifically, if the optical signal triggered by complementary target gene is defined as 100%, the signal intensity corresponding to MT1, MT2, and MT3 are 33.6%, 16.4%, and 1.9%, respectively, reflecting that the newly-proposed DHP possesses an excellent specificity. Clearly, the capability to distinguish the single-base mismatched targets has been fundamentally improved compared with literature values (about 75% and 95% ) [46] offered respectively by common MB and adapted one (LMB).[47] More excitingly, as shown in Figure 4 Inset, all mutant target DNAs can be easily visualized by the naked eye according to the distinct color changes. Note that the MT1 is the mutant type DNA identified as one of the oncogenes,[48] and the reliable identification of MT1 indicates the potential application of DHP in clinical settings to a certain extent.
Relative colorimetric signal induced by 150 nM target DNA and mutant targets. The net signal increase (At-A0) induced by target gene was defined as 100%, while the mutant targets were calculated as (Am-A0)/(A-A0). At, Am, and A0 are the absorption intensity at 417 nm corresponding to target DNA, mutant targets, and blank, respectively. Error bars are the standard deviation for three repetitive experiments. The inset shows the photographs of reaction solutions in the absence and presence of target DNA and mutant targets.
Undoubtedly, the excellent analytical performance achieved should be attributed to the unique DHP and efficient signaling scheme to amplify DNA hybridization event. Several issues should be mentioned: stem I that inhibits the primer binding, stem II that locks the folding of pre-incorporated G-rich fragment into DNAzyme and binding site for nicking endonuclease responsible for the successive generation of new primer. In this case, besides the low background in the absence of target DNA, the synergetic effect of nicking and polymerization causes the target DNA cycle and in turn the exponential accumulation of DNAzyme products, and no any signal loss occurs, enabling the detection of trace targets. Meanwhile, stem I and stem II enhance the stability of DHP hairpin in a cooperative manner, making the detection specificity higher. Essentially, all the distinct advantages are based on the successful design of DHP.
In fact, DHP is a more stable hairpin probe than common MB to some degree, but it is never reported because of the difficulty of obtaining the highly stable intramolecular hairpin. The functional fragments of DHP are shown in Scheme S2. Its part I consisting of loop and stem I is similar to the classical MB, and part II have Stem II and ss (single-stranded) fragment. While the ss fragment severs as the bridge between the stem I and stem II and stabilizes further the part I, the stem can be opened by target DNA hybridization, but stem II is still in the close state (no signal detected in line c of Figure 1A). On the basis of theoretical analysis [24] and our own experience,[25] when each of oligonucleotides contains more than 9 complementary base pairs, the dimer via intermolecular interaction is easily formed. Thus, the construction of two stem-contained DHP with 25 base pairs is a very inspiring achievement in the development of DNA sensing probe. Presumably, the stem II is a unique double-stranded fragment (rather than a common ds DNA) composed of a G-quadruplex sequence and its complementary segment and promotes the intramolecular hybridization. The present study is expected to give invaluable insight into the DNA probe design and clinic genetic diagnosis.
Target DNA detection in human serum samples
To verify the potential application of the proposed DHP to complex biological samples, we conducted the recovery tests by adding varying amounts of target DNA into the sensing system containing 1% human serum (kindly provided by Fuzhou general hospital of Nanjing military command). As demonstrated in Table 3, the recoveries for the spiked target DNA are in the range of 99-106%, suggesting that the developed DHP is a promising tool for possible applications since human serum is one of the most challenging media with a variety of proteins and other serious interference.
Recovery tests for target DNA in human serum.
| Samples | Target DNA added (nM) | Found | RSDa (n=3)b | Recovery (%) | |
|---|---|---|---|---|---|
| 1 | 25 | 25.38 | 1.65% | 101.52 | |
| 2 | 50 | 53.01 | 0.72% | 106.02 | |
| 3 | 100 | 99.68 | 8.31% | 99.68 | |
aRSD is the abbreviation of relative standard deviation. bThe data given above are obtained from three repeated experiments.
Conclusion
In summary, a facile DHP for colorimetric screening of p53 gene has been proposed, into which a G-rich fragment and a cleavable restriction site for Nt. BbvCI are easily introduced. The DHP exclusively folds into an extremely stable hairpin structure consisting of double stems, indicating the exceptional characteristics distinct from classical MB. As a result, the DHP-based colorimetric sensing system can execute the exponential amplification of target DNA hybridization and offers the dramatically-improved detection sensitivity, enabling the ultrasensitive p53 gene detection. Moreover, the unparalleled detection specificity is obtained compared with the traditional MB, and the point mutation occurring in target DNA is able to be visualized by the naked eye. Besides, the DHP has several outstanding advantages of easy synthesis, simple operation and low cost. The impressive research advances, as well as the innovative design of sensing probe as a proof-of-concept, are expected to benefit the development of multifunctional probes for various important purposes and would create a great potential for the clinical diagnosis of genetic diseases.
Supplementary Material
Schemes S1-S2, Figures S1-S5.
Acknowledgements
This work was supported by Ministry of Science and Technology of China (2015CB931804), National Natural Science Foundation of China (NSFC) (grant NO: 21275002 and 81273548), and Independent Research Project of State Key Laboratory of Photocatalysis on Energy and Environment (NO. 2014CO1).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Grompe M. The rapid detection of unknown mutations in nucleic acids. Nat Genet. 1993;5:111-7
2. Várallyay E, Burgyán J, Havelda Z. MicroRNA detection by northern blotting using locked nucleic acid probes. Nat Protoc. 2008;3:190-6
3. Tamura G, Kihana T, Nomura K, Terada M, Sugimura T, Hirohashi S. Detection of frequent p53 gene mutations in primary gastric cancer by cell sorting and polymerase chain reaction single-strand conformation polymorphism analysis. Cancer Res. 1991;51:3056-8
4. Cronin M, Pho M, Dutta D, Stephans JC, Shak S, Kiefer MC. et al. Measurement of gene expression in archival paraffin-embedded tissues: development and performance of a 92-gene reverse transcriptase-polymerase chain reaction assay. Am J Pathol. 2004;164:35-42
5. Li H, Rothberg LJ. Label-free colorimetric detection of specific sequences in genomic DNA amplified by the polymerase chain reaction. J Am Chem Soc. 2004;126:10958-61
6. Gilliland G, Perrin S, Blanchard K, Bunn HF. Analysis of cytokine mRNA and DNA: detection and quantitation by competitive polymerase chain reaction. Proc Natl Acad Sci U.S.A. 1990;87:2725-9
7. Xu J, Wu Z-S, Shen W, Xu H, Li H, Jia L. Cascade DNA nanomachine and exponential amplification biosensing. Biosen Bioelectron. 2015;73:19-25
8. Cui H-F, Xu T-B, Sun Y-L, Zhou A-W, Cui Y-H, Liu W. et al. Hairpin DNA as a Biobarcode Modified on Gold Nanoparticles for Electrochemical DNA Detection. Anal Chem. 2015;87:1358-65
9. Ding C, Zhang W, Wang W, Chen Y, Li X. Amplification strategies using electrochemiluminescence biosensors for the detection of DNA, bioactive molecules and cancer biomarkers. TrAC, Trac-Trend Anal Chem. 2015;65:137-50
10. Wang F, Elbaz J, Teller C, Willner I. Amplified Detection of DNA through an Autocatalytic and Catabolic DNAzyme-Mediated Process. Angew Chem Int Ed. 2011;50:295-9
11. Wang F, Elbaz J, Orbach R, Magen N, Willner I. Amplified analysis of DNA by the autonomous assembly of polymers consisting of DNAzyme wires. J Am Chem Soc. 2011;133:17149-51
12. Weizmann Y, Beissenhirtz MK, Cheglakov Z, Nowarski R, Kotler M, Willner I. A virus spotlighted by an autonomous DNA machine. Angew Chem. 2006;118:7544-8
13. Häner R, Biner SM, Langenegger SM, Meng T, Malinovskii VL. A Highly Sensitive, Excimer-Controlled Molecular Beacon. Angew Chem Int Ed. 2010;49:1227-30
14. Martinez K, Estevez M-C, Wu Y, Phillips JA, Medley CD, Tan W. Locked nucleic acid based beacons for surface interaction studies and biosensor development. Anal Chem. 2009;81:3448-54
15. Tyagi S, Bratu DP, Kramer FR. Multicolor molecular beacons for allele discrimination. Nat Biotechnol. 1998;16:49-53
16. Conlon P, Yang CJ, Wu Y, Chen Y, Martinez K, Kim Y. et al. Pyrene excimer signaling molecular beacons for probing nucleic acids. J Am Chem Soc. 2008;130:336-42
17. Tyagi S, Marras SA, Kramer FR. Wavelength-shifting molecular beacons. Nat Biotechnol. 2000;18:1191-6
18. Huang J, Wu Y, Chen Y, Zhu Z, Yang X, Yang CJ. et al. Pyrene-Excimer Probes Based on the Hybridization Chain Reaction for the Detection of Nucleic Acids in Complex Biological Fluids. Angew Chem Int Ed. 2011;50:401-4
19. Li JJ, Chu Y, Lee BY-H, Xie XS. Enzymatic signal amplification of molecular beacons for sensitive DNA detection. Nucleic Acids Res. 2008;36:e36
20. Zuo X, Xia F, Xiao Y, Plaxco KW. Sensitive and selective amplified fluorescence DNA detection based on exonuclease III-aided target recycling. J Am Chem Soc. 2010;132:1816-8
21. Qiu L-P, Wu Z-S, Shen G-L, Yu R-Q. Highly sensitive and selective bifunctional oligonucleotide probe for homogeneous parallel fluorescence detection of protein and nucleotide sequence. Anal Chem. 2011;83:3050-7
22. Connolly AR, Trau M. Isothermal Detection of DNA by Beacon-Assisted Detection Amplification. Angew Chem. 2010;122:2780-3
23. Dahl F, Banér J, Gullberg M, Mendel-Hartvig M, Landegren U, Nilsson M. Circle-to-circle amplification for precise and sensitive DNA analysis. Proc Natl Acad Sci U.S.A. 2004;101:4548-53
24. Li JJ, Tan W. A real-time assay for DNA sticky-end pairing using molecular beacons. Anal Biochem. 2003;312:251-4
25. Wu Z-S, Lu H, Liu X, Hu R, Zhou H, Shen G. et al. Inhibitory effect of target binding on hairpin aptamer sticky-end pairing-induced gold nanoparticle assembly for light-up colorimetric protein assay. Anal Chem. 2010;82:3890-8
26. Rojas AM, Gonzalez PA, Antipov E, Klibanov AM. Specificity of a DNA-based (DNAzyme) peroxidative biocatalyst. Biotechnol Lett. 2007;29:227-32
27. Chan C, Khachigian L. DNAzymes and their therapeutic possibilities. Intern Med J. 2009;39:249-51
28. Xiao Y, Pavlov V, Niazov T, Dishon A, Kotler M, Willner I. Catalytic beacons for the detection of DNA and telomerase activity. J Am Chem Soc. 2004;126:7430-1
29. Shimron S, Wang F, Orbach R, Willner I. Amplified detection of DNA through the enzyme-free autonomous assembly of hemin/G-quadruplex DNAzyme nanowires. Anal Chem. 2011;84:1042-8
30. Yuan Y, Chai Y, Yuan R, Zhuo Y, Gan X. An ultrasensitive electrochemical aptasensor with autonomous assembly of hemin-G-quadruplex DNAzyme nanowires for pseudo triple-enzyme cascade electrocatalytic amplification. Chem Commun. 2013;49:7328-30
31. Lu C-H, Qi X-J, Orbach R, Yang H-H, Mironi-Harpaz I, Seliktar D. et al. Switchable catalytic acrylamide hydrogels cross-linked by Hemin/G-Quadruplexes. Nano lett. 2013;13:1298-302
32. Tian Y, He Y, Mao C. Cascade signal amplification for DNA detection. ChemBioChem. 2006;7:1862-4
33. Cheglakov Z, Weizmann Y, Basnar B, Willner I. Diagnosing viruses by the rolling circle amplified synthesis of DNAzymes. Org Biomol Chem. 2007;5:223-5
34. Cheglakov Z, Weizmann Y, Beissenhirtz MK, Willner I. Ultrasensitive detection of DNA by the PCR-Induced generation of DNAzymes: the DNAzyme primer approach. Chem Commun. 2006:3205-7
35. Xie S-J, Zhou H, Liu D, Shen G-L, Yu R, Wu Z-S. In situ amplification signaling-based autonomous aptameric machine for the sensitive fluorescence detection ofcocaine. Biosen Bioelectron. 2013;44:95-100
36. Stojanovic MN, de Prada P, Landry DW. Catalytic molecular beacons. ChemBioChem. 2001;2:411-5
37. Xuan F, Fan TW, Hsing I-M. Electrochemical Interrogation of Kinetically-Controlled Dendritic DNA/PNA Assembly for Immobilization-Free and Enzyme-Free Nucleic Acids Sensing. ACS nano. 2015;9:5027-33
38. Liu S, Lin Y, Wang L, Liu T, Cheng C, Wei W. et al. Exonuclease III-Aided Autocatalytic DNA Biosensing Platform for Immobilization-Free and Ultrasensitive Electrochemical Detection of Nucleic Acid and Protein. Anal Chem. 2014;86:4008-15
39. Cheng Y, Stakenborg T, Van Dorpe P, Lagae L, Wang M, Chen H. et al. Fluorescence Near Gold Nanoparticles for DNA Sensing. Anal Chem. 2011;83:1307-14
40. Wu Z, Zhou H, Zhang S, Zhang X, Shen G, Yu R. Pendulum-type optical DNA nanodevice. Chem Commun. 2010;46:2232-4
41. Liu S, Cheng C, Liu T, Wang L, Gong H, Li F. Highly sensitive fluorescence detection of target DNA by coupling exonuclease-assisted cascade target recycling and DNAzyme amplification. Biosen Bioelectron. 2015;63:99-104
42. Bi S, Li L, Cui Y. Exonuclease-assisted cascaded recycling amplification for label-free detection of DNA. Chem Commun. 2012;48:1018-20
43. Li H, Wu Z, Qiu L, Liu J, Wang C, Shen G. et al. Ultrasensitive label-free amplified colorimetric detection of p53 based on G-quadruplex MBzymes. Biosen Bioelectron. 2013;50:180-5
44. Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas N. et al. The human gene mutation database: 2008 update. Genome Med. 2009;1:13
45. Stenson PD, Ball EV, Howells K, Phillips AD, Mort M, Cooper DN. The Human Gene Mutation Database: providing a comprehensive central mutation database for molecular diagnostics and personalised genomics. Hum genomics. 2009;4:69
46. Marquette C, Lawrence M, Blum L. DNA covalent immobilization onto screen-printed electrode networks for direct label-free hybridization detection of p53 sequences. Anal Chem. 2006;78:959-64
47. Liu X, Tan W. A fiber-optic evanescent wave DNA biosensor based on novel molecular beacons. Anal Chem. 1999;71:5054-9
48. Eeles R, Warren W, Knee G, Bartek J, Averill D, Stratton M. et al. Constitutional mutation in exon 8 of the p53 gene in a patient with multiple primary tumours: molecular and immunohistochemical findings. Oncogene. 1993;8:1269-76
Author contact
![]() Corresponding authors: Zai-Sheng Wu and Lee Jia, CMAPC, Fuzhou University, Fuzhou, Fujian 350002, China. Phone: 086-591-8357-6912. Email: wuzaishengcom (Z.S. Wu) and pharmlinkcom (L. Jia)
Corresponding authors: Zai-Sheng Wu and Lee Jia, CMAPC, Fuzhou University, Fuzhou, Fujian 350002, China. Phone: 086-591-8357-6912. Email: wuzaishengcom (Z.S. Wu) and pharmlinkcom (L. Jia)