Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Materials and methods
3. Results and discussion
4. Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(4):522-532. doi:10.7150/thno.14391 This issue Cite
Research Paper
Development of a Loop Mediated Isothermal Amplification (LAMP) - Surface Enhanced Raman spectroscopy (SERS) Assay for the Detection of Salmonella Enterica Serotype Enteritidis
Mohamed Shehata Draz1,2, Xiaonan Lu1 ![]()
1. Food, Nutrition and Health Program, Faculty of Land and Food Systems, The University of British Columbia, Vancouver, V6T 1Z4, Canada.
2. Botany Department, Faculty of Science, Tanta University, Tanta 31527, Egypt.
Received 2015-11-11; Accepted 2015-12-23; Published 2016-2-13
Abstract

As a major foodborne pathogen, Salmonella enterica serotype Enteritidis is increasingly rising as a global health concern. Here, we developed an integrated assay that combines loop mediated isothermal amplification (LAMP) and surface enhanced Raman spectroscopy (SERS) for DNA detection of S. Enteritidis using specifically designed Raman active Au-nanoprobes. The target DNA was amplified by LAMP and then labeled with Au-nanoprobes comprised of gold nanoparticle-modified with specific cy5/DNA probes to allow the detection by SERS. The sensitivity of the developed LAMP-SERS detection assay (66 CFU/mL) was ~100-fold higher than the conventional polymerase chain reaction (PCR) method. Significantly, this technique allowed highly specific detection of the target DNA of S. Enteritidis and could differentiate it from the DNA of closely related bacterial species or non-specific contamination, making it more accurate and reliable than the standard LAMP technique. The applicability of detection of S. Enteritidis in milk samples using LAMP-SERS assay was validated as well. In sum, the developed LAMP-SERS assay is highly specific and sensitive, and has the potential to be applied for rapid detection of different foodborne pathogens and other microbial contaminants.
Keywords: LAMP, SERS, Raman spectroscopy, nanoprobe, bacterial infections, clinical diagnosis, food safety.
1. Introduction
Salmonella are one of the most common enteric pathogens that widely dispersed in the environment and can be transferred to humans through many agri-food products, such as meat, poultry, fresh produce, and dairy products [1, 2]. Salmonella cause over 80.3 million cases of infections and 155,000 deaths each year worldwide [3, 4]. The majority of Salmonella infections are usually non-symptomatic or associated with mild symptoms ranging from self-limited diarrhea to fever, and the afflicted people can recover within a few days without specific treatment or hospitalization. However, Salmonella may invade into vascular structures, bones, or other localized sites, and subsequently develop potentially life-threatening bacteremia [5].
Traditional methods for the detection of Salmonella include various cultural, biochemical, and immunological methods, but they are labor intensive and time consuming [6-8]. The application of molecular techniques [e.g. polymerase chain reaction (PCR) and real-time PCR] allows faster, highly specific, and sensitive detection of Salmonella in various environmental, clinical, and agri-food samples [8, 9]. However, due to the need for expensive thermal cycling equipments and highly trained personnel, these techniques remain inappropriate for high-throughput screening of a large number of samples as well as in-field application.
The recent development of alternative isothermal amplification techniques for nucleic acid amplification without the use of dedicated equipment has stimulated extensive research interest and applications. Loop mediated isothermal amplification (LAMP) is one representative isothermal amplification technique with wide and potential applications in molecular diagnosis. For example, LAMP has been applied to detect several pathogenic bacteria, such as Bacillus anthracis [10], Escherichia coli O157:H7 [11-13], Listeria monocytogenes [14], Pseudomonas aeruginosa [15], Salmonella [16-18], Staphylococcus aureus [19], and Vibrio parahaemolyticus [20-22]. LAMP is primarily based upon autocycling strand-displacement DNA synthesis using the Bst DNA polymerase enzyme in the presence of 4 to 6 specific primers and dNTPs in an isothermal condition for 60 min [23]. Final amplification products are directly detected by gel electrophoresis or indirectly quantified either by measuring the turbidity derived from magnesium pyrophosphate formed during LAMP amplification or by using a fluorescent DNA-intercalating dye in a way similar to real-time PCR. Although these methods are technically feasible and reliable, it could not differentiate non-specific amplification, thus leading to false positive results and greatly compromising the potential of LAMP in real field applications [24-26].
Surface enhanced Raman spectroscopy (SERS) is rapidly emerging as a promising tool in biological and chemical analysis because it is rapid and requires little or no sample preparation. In the meanwhile, it can offer non-destructive and ultra-sensitive detection to single molecular level [27-29]. One recent approach to SERS biosensing is the development of specific biomolecular probes that contain metal nanostructures for SERS signal enhancement, organic Raman tags as quantitative reporters for the presence of the probes, and capturing molecules (e.g. antibodies, single strand DNA and RNA aptamers, etc.) to interact with target compounds [30-33].
In the current study, we for the first time developed a DNA detection assay that integrates LAMP with SERS by using specific Raman active Au-nanoprobes. This assay leverages the inherent nucleic acid amplification properties of LAMP with the ultrasensitive analytical power of SERS, thus allowing simple and sensitive detection of target DNA. In addition, the postamplification-labeling step with Au-nanoprobes improves the specificity and reliability of detection by limiting the false positive results. We evaluated our developed assay and compared its performance to the conventional PCR and LAMP methods for the specific detection of Salmonella enterica serotype Enteritidis (S. Enteritidis) as a model bacterium in both its pure culture and artificially contaminated food samples. Mainly identified from poultry and dairy food products, S. Enteritidis is one of the most common serotypes of Salmonella reported worldwide and has emerged as the leading cause of human illness in North America. Altogether, our proposed LAMP-SERS assay exhibits excellent analytical performance and is relatively simple, faster, and much more sensitive than the conventional PCR method, validating its potential as a promising technique for high-throughput screening and in-field diagnostic applications.
2. Materials and methods
2.1 Bacterial strains and genomic DNA extraction
Bacterial strains that represent two Salmonella enterica serovars Enteritidis and Typhimurium and four non-Salmonella strains (i.e., E. coli O157:H7, S. aureus, P. aeruginosa, and L. monocytogenes in Table S1) were used to test the specificity and sensitivity of the developed LAMP-SERS assay. All the bacterial strains were cultivated in Tryptic Soy Broth (TSB) for genomic DNA extraction. Briefly, 5 mL of each overnight bacterial culture was harvested by centrifugation and resuspended in 500 μL of TE buffer (10 mM Tris, pH 8.0; 1 mM EDTA; Sigma-Aldrich, St. Louis, MO) and heated at 95°C for 10 min in a dry heating block [16]. The crude cell lysate was centrifuged at 12,000 ×g for 2 min and the supernatant was stored at ˗20°C.
2.2 DNA primers and PCR test
A set of four primers, namely two outer primers (F3 and B3) and two inner primers (FIP and BIP), were selected based upon the sequence of the highly serovar-specific DNA region within the Salmonella gene (GenBank Accession No: AF370707.1) to detect S. Enteritidis DNA using LAMP technique [34]. The sequences of the primers are shown in Table 1. Conventional PCR was carried out to test F1 and B1 using S. Enteritidis ATCC 43353 template DNA. Amplification was conducted in a 25-µL reaction mixture containing 0.5 µL of each primer (0.2 µM), 22 µL of PCR SuperMix (Invitrogen Biotech, Carlsbad, CA), and 2 µL of DNA template. The initial denaturation was carried out at 95°C for 5 min, followed by 30 cycles of denaturation at 94°C for 60 s, annealing at 47°C for 60 s, and extension at 72°C for 60 s. Finally, additional extension was performed at 72°C for 7 min. The amplification product was then analyzed by electrophoresis on 1.7% agarose gel, and visualized under UV light.
List of DNA sequences used in this study.
| Assay type | Oligonucleotide name | Sequence (5'-3') |
|---|---|---|
| PCR | Forward primer (FP) | GGGAGGAGCTTTAGCCAA |
| Reverse primer (RP) | ATGGTGAGCAGACAACAG | |
| LAMP | Forward outer primer (F3) | GGGAGGAGCTTTAGCCAA |
| Backward outer primer (B3) | ATGGTGAGCAGACAACAG | |
| Forward inner primer (FIP) | CATGCTCGCTGCACAAAAGCGAGAGGCGGTTTGATGTG | |
| Backward inner primer (BIP) | CTGGAAAGCCTCTTTATATAGCTCATGATATACTCCCTGAATCTGAGA | |
| LAMP-SERS | Labeling probe | GCCTAAAAAATCAG/iCy5N/GACGA/3ThioMC3-D/ |
| Target fragment | TCGTCACTGATTTTTTAGGC |
2.3 Preparation of Raman active Au-nanoprobes
Au-nanoprobes consist of gold nanoparticles functionalized with Raman dye-labeled oligonucleotides and were synthesized by the following steps. Citrate-capped gold nanoparticles were prepared by sodium citrate-based reduction of chloroauric acid (HAuCl4) solution, and then modified with 5'-thiolated oligonucleotides internally labeled with cy5. Briefly, freshly reduced thiolated-oligonucleotides (50 μM) were added to 1 mL of gold nanoparticles solution and the mixture was incubated at room temperature for 12 h. The solution was then brought to 0.1 M NaCl and allowed to stand for 40 h, followed by centrifugation at 12,000 ×g for 30 min to separate the gold nanoparticles from the excessive reagents [35]. The isolated Au-nanoprobes were finally washed twice with 100 mM phosphate buffer (pH 7.4) and stored at 4°C. The prepared gold nanoparticles and Au-nanoprobes were characterized using UV-vis spectroscopy, transmission electron microscopy, and Raman spectroscopy.
2.4 Synthesis of LAMP products
The isothermal DNA amplification reaction was performed using the loopamp DNA amplification kit, following the protocol from the manufacturer (Eiken Chemical Co., Tokyo, Japan). Each LAMP reaction included the following reagents: 2 µL of genomic DNA, 12.5 µL of reaction mixture, 1 µL of primer mixture (F3, B3, FIP, and BIP), 1 µL of Bst DNA polymerase, and 8.5 µL of molecular grade water. The prepared mixture was then incubated under an isothermal condition (65°C) for 40 min in a real-time PCR machine (Veriti Thermal Cycler, Applied Biosystems). The resultant LAMP products were analyzed by 1.7% gel electrophoresis, visualized with a UV transilluminator, and the images were recorded with a digital gel documentation system (Bio-Rad).
2.5 LAMP-SERS assay
Ten microliters of Au-nanoprobe solution were added to each sample of the amplified DNA products and allowed to anneal by gradually cooling the mixture down from 95°C to 4°C. The hybridization products of Au-nanoprobe and LAMP-amplified DNA were digested with S1 nuclease enzyme at 30°C for 25 min, followed by washing twice with ultrapure water. A confocal micro-Raman spectroscopic system (Renishaw, Gloucestershire, UK) equipped with a Leica microscope (Leica Biosystems, Wetzlar, Germany) was then applied for SERS spectral collection of the resultant hybridization products.
2.6 Specificity and sensitivity of LAMP-SERS assay
DNA templates of different Salmonella and non-Salmonella bacterial strains (Table S1) were prepared by heating at 95°C for 10 min as aforementioned. An aliquot (2 µL) of each template was then tested to determine the specificity of LAMP-SERS assay. The sensitivity of LAMP-SERS assay was determined by using 10-fold serial dilutions of the standard DNA template from S. Enteritidis and an aliquot of each dilution (2 µL) was used as DNA template for LAMP-SERS assay. Both the specificity and sensitivity testing reactions were carried out at least in triplicate.
2.7 Detection of S. Enteritidis in artificially contaminated milk products
Milk products purchased from a local grocery store in Vancouver were used in this study. S. Enteritidis culture was incubated overnight in TSB at 37°C and then used to prepare different concentrations of S. Enteritidis in milk. Bacterial concentration was determined by conventional plating assay (Tryptic Soy Agar, TSA). Simultaneously, 1 mL of each concentration was subjected to DNA extraction using Qiagen QIAamp DNA Mini Kit (Qiagen GmBH, Hilden, Germany). An aliquot (2 µL) of each extracted genomic DNA was then tested using the aforementioned LAMP-SERS assay. Non-inoculated milk sample was used as the negative control and each dilution was tested at least in triplicate.
3. Results and discussion
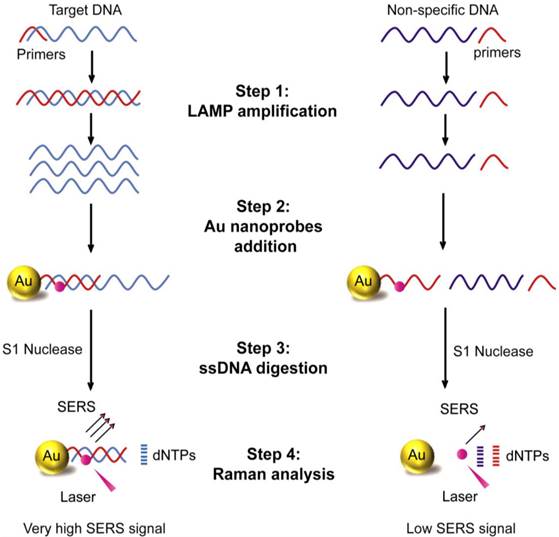
The current study reports the development of a DNA sensing assay that primarily integrates LAMP DNA amplification, hybridization with Raman active Au-nanoprobes, nuclease digestion, and SERS analysis (Figure 1). In this assay, the applied Au-nanoprobes are specifically designed to label the DNA amplification products generated by LAMP, allowing their detection using SERS technique. These probes were mainly comprised of gold nanoparticles conjugated with capturing DNA probes of 20-mer oligonucleotides specifically targeting Sdf I sequence of S. Enteritidis (Figure S1). The capturing DNA oligonucleotides are preferentially modified with a thiol group at 3' end and a Raman active dye of cy5 at the thymine nucleotide exists at position 6 from the same end of DNA to allow controlled and directional coupling of the oligonucleotides to gold nanoparticles. This eventually brings cy5 in the close proximity (< 2 nm) to the surface of gold nanoparticles, allowing a significant enhancement of Raman signal intensity. When these probes are added to the target DNA, and as a result for the specific hybridization of cy5 labeled DNA capturing probes to its complementary sequence, Au nanoprobes-target DNA hybridization complexes are formed and easily detected using SERS based upon the distinctive Raman spectral features of cy5. In addition, we relied on an enzyme digestion step using S1 nuclease to differentiate the presence and absence of target DNA. S1 nuclease is known to specifically degrade single-stranded (ss) DNA into free mononucleotides (dNTPs). In the presence of the target DNA, the probes specifically bind to its complementary sequence and form a double-stranded DNA structure in which the complex cy5-DNAs are protected from nuclease digestion and eventually cy5 remains linked to the surface of gold nanoparticles. In contrast, in the absence of the target DNA, S1 nuclease triggers the release of cy5 from the surface of gold nanoparticles by digesting the surface capturing DNA oligonucleotides of Au-nanoprobes, resulting in a significantly reduced Raman signal intensity.
Schematic illustration of LAMP-SERS assay. The developed LAMP-SERS assay comprises the following steps: 1) LAMP amplification of the target DNA using 4 particular in the presence of Bst enzyme and free dNTPs at 65 C for 40 min; 2) Addition of Au-nanoprobes that preferentially designed of 12-nm gold nanoparticles conjugated to thiolated 20-mer DNA oligonucleotides. In these probes, the surface DNA oligonucleotides act as capturing probes to specifically recognize the target DNA, meanwhile internally labeled with a Raman active dye of cy5 (at position 6 which is < 2 nm from the surface of gold nanoparticles) to allow enhanced Raman detection; 3) DNA digestion with S1 nuclease enzyme that specifically digest the single-stranded DNA into free nucleotides (dNTPs); 4) Raman analysis of the digestion products to detect the signals of cy5. In the presence of the target DNA (left panel), the target DNA is specifically amplified by LAMP reaction generating a huge number of DNA copies, then labeled by Au-nanoprobes forming hybrid DNA duplexes of the probe and LAMP amplicons, which can persist the S1 nuclease activity and maintained a very high Raman signal. In the absence of the target DNA or presence of non-specific DNA (right panel), no amplification takes place and free Au-nanoprobes are readily digested with S1 nuclease and cy5 released from the surface of gold nanoparticles, which results in low Raman signal.
Au-nanoprobes were prepared from citrate-capped gold nanoparticles functionalized with thiolated and cy5 labeled DNA oligonucleotides, following a previously reported thiol-Au coupling protocol (Figure 2) [35]. Transmission electron microscopy (TEM) and the corresponding size distribution histogram indicate that the synthesized gold nanoparticles are spherical in shape with an average diameter of 12.25±2.22 nm (Figure 2a, i and ii). UV-visible spectra of citrate-stabilized gold nanoparticles in aqueous solution showed an intense and narrow surface plasmon absorption peak at 519 nm, confirming the stability and monodispersity of the synthesized nanoparticles (Figure 2c). On the other hand, the conjugation of thiolated DNA oligonucleotides to gold nanoparticles resulted in a slight red-shift of the plasmon band from 519 nm to 523 nm, and the characteristic absorbance peak of cy5 was observed at 650 nm as well (Figure 2c and Supplementary Information, Figure S2). The ratio of cy5 labeled DNA molecules per nanoparticle was estimated from their corresponding absorption values to be 76.3±26.05 cy5-DNA/gold nanoparticle (see Supplementary Methods). This ratio implies ~3% coverage of nanoparticle surface area with cy5-DNA probes and was intended to maintain the colloidal stability of Au-nanoprobes. In addition, it allows efficient hybridization and labeling of the target DNA sequence without the formation of large aggregates of hybridization products, which can limit the efficiency of nuclease digestion and result in a high background signal.
Preparation and characterization of Au-nanoprobes. (a) Characterization of gold nanoparticles: (i) TEM image; (ii) particle size distribution histogram. (b) Schematic illustration of the preparation of Au-nanoprobes. Citrate capped gold nanoparticles were modified with thiolated DNA oligonucleotides that is 20 mer in length and internally modified with cy5 (icy5). The image shows a slight change in color of gold nanoparticles solution caused by the conjugation of thiolated DNA to the surface of gold nanoparticles. However, no turbidity is visible, indicating that the stability of gold nanoparticles was maintained throughout the preparation reaction of Au-nanoprobes. (c) UV-vis absorption spectra of gold nanoparticles (citrate capped gold nanoparticles, black) and Au-nanoprobes (gold nanoparticles conjugated to DNA, red). (d) Raman spectra of pure cy5 modified DNA oligonucleotides (no gold nanoparticles, black) and Au-nanoprobes (cy5 modified DNA oligonucleotides conjugated gold nanoparticles, red), indicating a strong enhancement of cy5 Raman signal in the presence of gold nanoparticles.
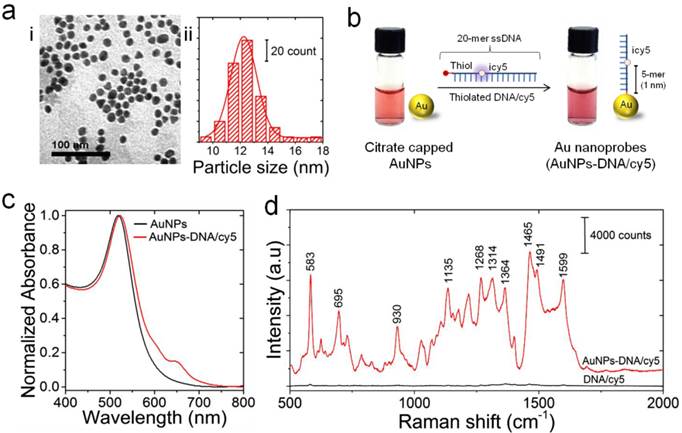
Raman enhancement effect of gold nanoparticles in the prepared Au-nanoprobes is critical to sensitive detection of the LAMP-amplified DNA products. Thus, we investigated Raman enhancement factor by collecting Raman signals of cy5-DNA oligonucleotides before and after conjugation to the surface of gold nanoparticles along with the bare gold nanoparticles. The prepared Au-nanoprobes (cy5-DNA after conjugation to gold nanoparticles) were able to generate a strong SERS signal even at a low concentration of 10 nM, and several intense Raman bands characteristic to cy5 can be observed at 583, 1135, 1465, and 1599 cm-1. The presence of gold nanoparticles resulted in SERS signal enhancement by ~3 orders of magnitudes compared to cy5-DNA (before conjugation to gold nanoparticles) or gold nanoparticles only (Figure 2d and Supplementary Information, Figure S3 & Figure S4). This strong SERS signal enhancement can be attributed to controlling the distance between cy5 and the surface of gold nanoparticles in the prepared probes to be less than 2 nm by using internal cy5-modified oligonucleotides [36]. In addition, this tightly controlled interdistance between cy5 and gold nanoparticles can lead to cy5 fluorescence quenching based on the efficient fluorescence resonance energy that transfers to the surface of particles. Taken together, this greatly minimized the background signal and allowed stable and intense SERS spectral features of Au-nanoprobes [36, 37].
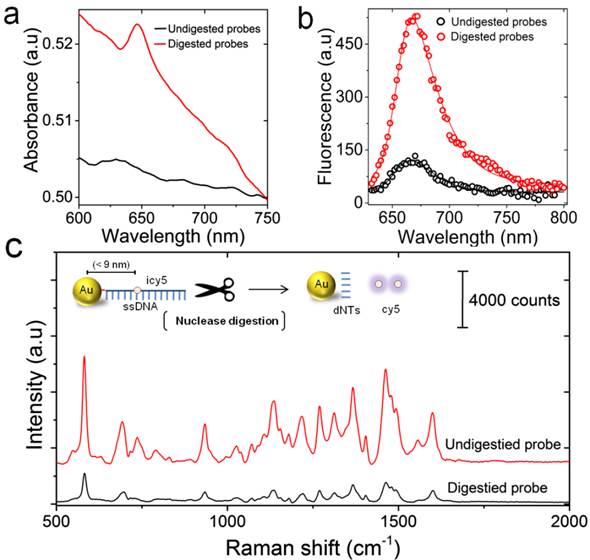
To validate the design of the proposed LAMP-SERS assay, we initially tested the ability of S1 nuclease to selectively digest Au-nanoprobes in the absence of the target DNA. Aliquots of Au-nanoprobe solution were digested with 1 µL of S1 nuclease in 50 µL of 1× S1 nuclease reaction buffer for 20 min. The digested Au-nanoprobe solution was then centrifuged at 14,000 ×g for 20 min and the upper supernatant was tested by UV-vis spectroscopy. The results of UV-vis analysis in Figure 3a showed the characteristic absorbance peak of cy5 at 650 nm. The cy5 dye is indirectly linked to the surface of gold nanoparticles through the conjugated capturing DNA oligonucleotides. Thus, the ability of S1 nuclease to hydrolyze these oligonucleotides liberated cy5 from the surface of gold nanoparticles into the surrounding reaction mixture (not being centrifuged with the particles), allowing its detection by UV-vis spectroscopy. In addition, the fluorescence spectra of Au-nanoprobe solution before and after nuclease digestion demonstrated a substantial increase in the fluorescence signal intensity that is 4-fold over the non-digested Au-nanoprobes (Figure 3b). This additionally confirmed the release of cy5 from the surface of gold nanoparticles by S1 nuclease digestion, as indicated by the relief of cy5 fluorescence quenched by surface plasmon resonance of gold nanoparticles. Furthermore, Raman spectroscopic analysis showed that the signal intensity of cy5 was remarkably decreased (~5-fold, estimated from the peak intensity at 583 cm-1) after nuclease digestion, which is mainly due to the release of cy5 from the surface of gold nanoparticles (Figure 3c).
Nuclease digestion of the prepared Au-nanoprobes. (a) UV-vis absorption spectra of the centrifugation supernatants for the undigested (without the addition of nuclease, black) and the digested (after incubation with S1 nuclease, black) Au-nanoprobes. The presence of cy5 characteristic peak at 650 nm indicates the release of cy5 into the solution after digestion of Au-nanoprobe solution by S1 nuclease. (b) Fluorescence spectra of Au-nanoprobes before (black) and after (red) nuclease digestion, confirming a remarkable release of cy5 fluorescence after probe digestion by S1 nuclease. (c) Raman spectral features of the digested (black) and undigested (red) Au-nanoprobes. The corresponding schematic illustration shows the detailed structure of Au-nanoprobes and the products by S1 nuclease digestion. The digestion of ssDNA that is linked to the surface of gold nanoparticles by thiol-Au bond (red) results in the release of cy5 and free nucleotides (dNTs) into the reaction mixture. The release of cy5 from the surface of gold nanoparticles can be detected by measuring cy5 fluorescence and Raman signal. Increasing the distance between cy5 and gold nanoparticles results in the recovery of its fluorescence signal quenched by gold nanoparticles, but the effect of gold nanoparticles to enhance Raman signal intensity of cy5 is minimized. All spectra were obtained by averaging at least three independent measurements.
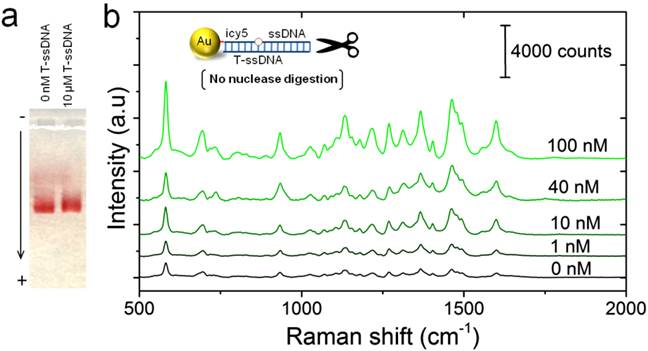
The ability of the target DNA to protect Au-nanoprobes against S1 nuclease activity and maintain cy5 enhanced Raman signals was tested using SERS technique. Mixtures of Au-nanoprobes and the target DNA sequence were gradually cooled down from 95°C to 4°C to ensure a complete hybridization. The formation of specific Au nanoprobes-target DNA hybridization products was first confirmed using gel electrophoresis before they were subjected to digestion with S1 nuclease (Figure 4a). Gel electrophoresis indicated that the formed Au nanoprobes-DNA hybridization products exhibited a slight decrease in the electrophoretic mobility of the nanoprobes, which is due to the increase in their size when they are hybridized with the target DNA oligonucleotides. The formed Au nanoprobe-target DNA complexes were then subjected to S1 nuclease digestion and tested by SERS along with the control sample (without S1 nuclease enzyme digestion). The results indicated that the presence of the target DNA significantly protected the original structure of Au-nanoprobes and the enhanced SERS signal intensity of cy5 was maintained compared to the control (digested solution in the absence of the target DNA). Further, the intensity of the corresponding SERS spectra increased along with the increase in the amount of the target DNA added to the same amount of Au-nanoprobes, indicating the possibility for quantitative detection (Figure 4b). Specifically, the increase in the concentration of the target DNA sequence, which is complementary to the capturing probe on the surface of gold nanoparticles, increases the number of the duplex DNA structures of Au nanoprobes-target DNA hybridization product, leading to an enhanced SERS signal intensity in highly specific and quantitative manner.
Nuclease digestion of Au-nanoprobes in the presence of different concentrations of the target DNA. (a) Agarose gel electrophoresis of Au-nanoprobes (2 nM) after incubation with 50 µL of phosphate buffer (free of DNA) or with 1 µM of the synthetic target DNA (20-mer). (b) SERS detection of the target DNA (concentrations from 1-100 nM) using Au-nanoprobes (0.1 µM). Raman spectroscopic analysis was performed using a 785-nm laser for excitation at 25 mW and 10 sec as integration time. The raw SERS spectra were baseline corrected and the final spectrum was obtained by averaging three different measurements without spectral normalization.
One of the key factors that affect the performance of this LAMP-SERS assay is the stability of Au-nanoprobes under our experimental conditions. The stability was tested using gel electrophoresis and fluorescence spectroscopy. Using gel electrophoresis, both of the treated and untreated nanoprobes showed identical electrophoretic properties, validating the stability of the applied Au-nanoprobes (Figure S5). Similarly, the fluorescence analysis showed a very slight change in the intensity of cy5 emission values at 670 nm measured for Au-nanoprobes solution before and after hybridization (prior to the nuclease digestion step), confirming an acceptable stability of Au-nanoprobes with only minor dissociation of cy5 labeled DNA oligonucleotides from the surface of gold nanoparticles (Figure S6). Furthermore, optimization of the digestion reaction time with the concentration of the probes is crucial to minimize the background noise caused by undigested probes. The effect of the nuclease digestion time on SERS signal of Au-nanoprobes was investigated by monitoring the response of fluorescence intensity at different times. As displayed in Figure S7, the fluorescence intensity increased rapidly along with the increase of reaction time in the range from 10 to 30 min and reached a plateau after 20 min. Therefore, 25 min was selected as the optimum digestion time for our assay.
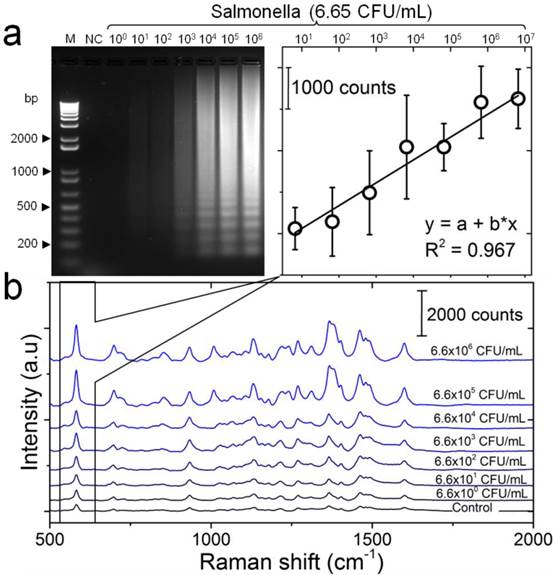
The detection sensitivity of the developed LAMP-SERS assay was tested against different concentrations of diluted S. Enteritidis standard DNA template (6.6- 6.6×106 CFU/mL). The target DNA template was initially amplified by LAMP assay and then detected using SERS. Successful LAMP amplification of target DNA was confirmed with gel electrophoresis. Figure 5a represents the electrophoresis patterns of the amplification products generated by LAMP, confirming the formation of ladder-like patterns that is characteristic of LAMP. Moreover, the amount of the amplification products was proportional to the concentration of target DNA, and 66 CFU/mL was the lowest concentration that could be visually detected on gel. Following up the working protocol depicted in Figure 1, the amplified DNA products were incubated with Au-nanoprobes, digested by S1 nuclease, and then analyzed by SERS. The generated SERS spectra are shown in Figure 5b. All the spectra were reproducibly showing the major Raman bands characteristic of cy5, as previously observed in the spectra of Au-nanoprobes (Figure 2). Most interesting, the SERS signal intensity was directly proportional to the initial concentration of the target DNA, indicating the potential application of the developed assay for quantitative detection even without any process of the collected raw SERS signals. A calibration curve was obtained by plotting the SERS peak intensity at 583 cm-1 as a function of the concentration of S. Enteritidis template DNA in the LAMP reaction (Figure 5b). A linear curve yielded the best fit for the experimental data with a coefficient of determination of 0.967. The limit of detection for LAMP-SERS assay was 66 CFU/mL with Raman intensity of 2385.36, which was 2-fold higher than (S/N > 2) the average signal of control sample with Raman intensity of 1174.48 calculated at 583 cm-1. Compared to the conventional PCR (Figure S8), the detection limit of both LAMP and the developed LAMP-SERS assay is about 100 times higher. In addition, there was no need to process the collected SERS signals because the LAMP assay could facilitate amplifying the amount of DNA from low to a tremendously high level, allowing an efficient and reliable detection.
Detection sensitivity of the developed LAMP-SERS assay. (a) Electrophoresis patterns of LAMP amplification products generated from different concentrations of S. Enteritidis DNA template. LAMP reaction was performed using 10-fold serial dilutions of S. Enteritidis DNA template (6.6×100 CFU/mL - 6.6×106 CFU/mL). M: 1-kb DNA ladder marker; NC: negative control (without target DNA template). (b) SERS spectral features of Au-nanoprobes incubated with LAMP products of different concentrations of S. Enteritidis DNA template. SERS peak intensity centered at 583 cm-1 was used for quantitative analysis of S. Enteritidis. Variations in SERS peak intensity centered at 583 cm-1 were as a function of S. Enteritidis concentration. The inset shows a linear relationship in the concentration range from 6.6×100 CFU/mL - 6.6×106 CFU/mL. Error bars are standard deviations from a total of three independent measurements. Raman spectroscopic analysis was performed using a 785-nm laser for excitation at 25 mW and 10 sec as integration time. The raw SERS spectra were baseline corrected and the final spectrum was obtained by averaging three different measurements without spectral normalization.
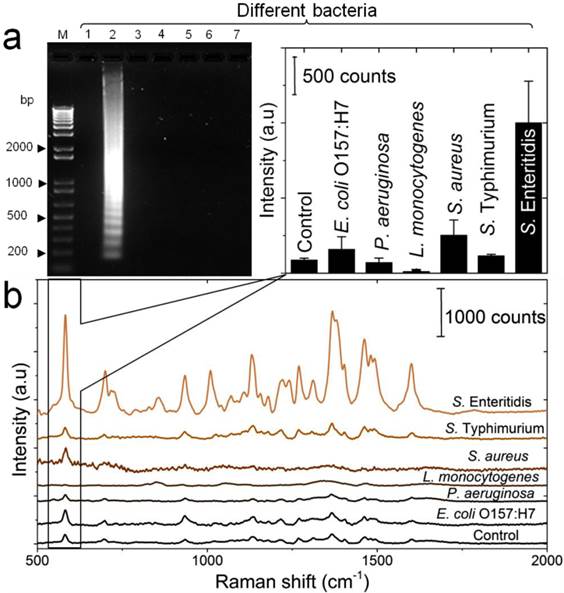
To determine the specificity of the developed LAMP-SERS assay, a series of control experiments between the target S. Enteritidis and other Salmonella and non-Salmonella species were performed. Figure 6a displays the results of the electrophoretic analysis of the LAMP products, confirming that the characteristic ladder bands of LAMP were only observed with the target S. Enteritidis. Figure 6b illustrated the SERS spectra in response to different bacterial strains. An intense SERS signal was observed for the target S. Enteritidis. On the contrary, the same concentration of all other bacterial strains, even the closely related S. Typhimurium, could not induce a significant SERS signal compared to the control. In addition, the corresponding intensity histogram obtained from the peak intensity at 583 cm-1 indicated high SERS intensity ratios (5 to 7-times) between S. Enteritidis and other bacteria. This result confirms that our LAMP-SERS assay exhibits great selectivity and specificity for the detection of S. Enteritidis. There are two main factors contributed to the high specificity of the LAMP SERS assay established in this study: 1) LAMP primers that allow specific amplification of the target S. Enteritidis, 2) Au-nanoprobes that designed to specifically recognize the target amplification products. Due to the increasingly reported susceptibility of LAMP to false positives because of carry-over or cross-contamination, we challenged the specificity of our assay against positive LAMP samples to confirm its reliability (Figure S9). No significant SERS signal was observed either from the negative control (without any DNA) or from the positive control (with non-Salmonella DNA amplification products). This can be attributed to the combined action of Au-nanoprobes post-amplification labeling and nuclease digestion that allow differentiating the specific amplifications form the non-specific amplifications in the final step of SERS detection.
Detection specificity of the developed LAMP-SERS assay. (a) Electrophoresis patterns of LAMP amplification products generated from different bacteria. LAMP reaction was performed using DNA template extracted from different bacterial suspensions (OD600=1). Lane M: 1-kb DNA ladder marker; NC: negative control (without target DNA template); Lane1: S. Enteritidis (target bacterium); Lane 2: S. Typhimurium; Lane 3: S. aureus; Lane 4: L. monocytogenes; Lane 5: P. aeruginosa; Lane 6: E. coli O157:H7. (b) SERS spectral features of Au-nanoprobes incubated with LAMP products of different bacteria. The inset shows the variation in SERS peak intensity centered at 583 cm-1 as a function of different bacteria. Error bars are standard deviations from a total of three independent measurements. Raman spectroscopic analysis was performed using a 785-nm laser for excitation at 25 mW and 10 sec as integration time. The raw SERS spectra were baseline corrected and the final spectrum was obtained by averaging three different measurements without spectral normalization.
To evaluate the potential of the developed LAMP-SERS assay for the detection of real samples, three different concentrations (i.e., 6×103 CFU/mL, 6×104 CFU/mL, and 6×105 CFU/mL) of S. Enteritidis were spiked into milk samples (Figure S10). The total genomic DNA of each sample was directly extracted without any enrichment and then tested using our LAMP-SERS assay. The obtained recoveries of the samples were 80.5%, 112.2%, and 109.1%, respectively. In the meanwhile, no obvious SERS signal was observed from the non-spiked milk samples. These results indicated that the developed LAMP-SERS assay has the potential to be applied to detect S. Enteritidis in the environmental and agri-food products.
This work has demonstrated the development of a LAMP-SERS assay for sensitive and specific detection of S. Enteritidis by taking the advantage of LAMP technique for DNA amplification and the detection sensitivity of SERS. This is the first study of integrating LAMP with SERS for the detection of microorganisms by using specifically designed Au-nanoprobes. We attribute the high sensitivity (100-times higher than PCR) of this LAMP-SERS assay to the elegant design of the used Au nanoprobes, in which the distance between cy5 (Raman tag) is precisely controlled to be less than 2 nm, allowing optimum plasmon-induced electromagnetic field enhancement without interfering the ability of the capturing DNA to recognize and interact with the amplified target DNA. Further, tuning the characteristics of those Au-nanoprobes offers a capability for controlling the limit and range of detection. Using higher concentrations of Au-nanoprobes will result in a wide detection range. In addition, reducing the amount of cy5-DNA molecules on the surface of gold nanoparticles will lead to a higher signal-to-noise ratio, allowing greatly enhanced detection sensitivity. Nuclease digestion was primarily used in this assay to differentiate the presence and absence of the target DNA and contributed significantly to the stability and quality of the collected SERS signal. By removing the non-interacted cy5 from the surface of gold nanoparticles, it allowed a highly quantitative detection even without the need for additional spectral processing steps required in other studies of SERS detection.
Compared to the most commonly used methods for the detection of Salmonella, the developed LAMP-SERS assay was highly sensitive, specific, and cost-effective (Table S2). In addition, it allowed more reliable and accurate detection compared to the standard, turbidity-based, and fluorescence-based LAMP assays that are not able to differentiate non-specific amplifications. The post-amplification labeling with specific probes in our assay combined with nuclease digestion greatly facilitated achieving such improved detection reliability, as the non-specific DNA or any potential contamination were digested, causing minimal interference to the collected SERS signal derived from the target DNA. Although this additional step leads to relative complexity and extra time for operation, our assay requires no enrichment step and this ultimately saves tremendous times.
4. Conclusions
We have demonstrated the feasibility and usefulness of a new LAMP-based nucleic acid detection using Raman spectroscopy. The results in this study indicate that the introduced LAMP-SERS assay, which involves LAMP amplification of the target DNA and SERS detection after labeling with Raman active Au-nanoprobes, greatly simplifies the experimental procedures by excluding the need of thermal cycling or complicated gel documentation systems and could potentially used in assays that require simple and rapid identification of genomic materials. The intrinsic Raman enhancement in the presence of colloidal nanoprobes enables a detection sensitivity that is comparable to that of LAMP and 100-times more sensitive than PCR. In addition, the designed assay shows dynamic range from 6.6×100-6.6×106 CFU/mL of bacterial template DNA with excellent detection specificity and reliability. The proposed assay has a great potential for broad applications in DNA detection in the area of clinical diagnosis, foodborne pathogen detection, and environmental monitoring.
Supplementary Material
Figure S1-S10 and Table S1-S2.
Acknowledgements
This study was supported by funds awarded to X.L. by Natural Sciences and Engineering Research Council of Canada (NSERC RGPIN-2014-05487).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hoelzer K, Switt AM, Wiedmann M. Animal contact as a source of human non-typhoidal salmonellosis. Veterinary Research. 2011;42:41-28
2. Doyle ME, Kaspar C, Archer J, Klos R. White paper on human illness caused by Salmonella from all food and non-food vectors. FRI Briefings. 2009;24:1-81
3. Majowicz SE, Musto J, Scallan E. et al. The global burden of nontyphoidal Salmonella gastroenteritis. Clinical Infectious Diseases. 2010;50:882-889
4. Hendriksen RS, Vieira AR, Karlsmose S. et al. Global monitoring of Salmonella serovar distribution from the World Health Organization Global Foodborne Infections Network Country Data Bank: results of quality assured laboratories from 2001 to 2007. Foodborne Pathogens and Disease. 2011;8:887-900
5. Acheson D, Hohmann EL. Nontyphoidal salmonellosis. Clinical Infectious Diseases. 2001;32:263-269
6. Wallace H, June G, Sherrod P, Hammack T, Amaguana R. Salmonella In Food and Drug Administration Bacteriological Analytical Manual. Tomlinson ed, LA Hypertext Source: http://vm.cfsan.fda.gov/~comm/bam-5.html# Intro. 1999
7. Liu T, Liljebjelke K, Bartlett E, Hofacre C, Sanchez S, Maurer JJ. Application of nested polymerase chain reaction to detection of Salmonella in poultry environment. Journal of Food Protection. 2002;65:1227-1232
8. Eriksson E, Aspan A. Comparison of culture, ELISA and PCR techniques for Salmonella detection in faecal samples for cattle, pig and poultry. BMC Veterinary Research. 2007;3:1-19
9. Botteldoorn N, Van Coillie E, Grijspeerdt K. et al. Real-time reverse transcription PCR for the quantification of the mntH expression of Salmonella enterica as a function of growth phase and phagosome-like conditions. Journal of Microbiological Methods. 2006;66:125-135
10. Qiao Y-M, Guo Y-C, Zhang X-E. et al. Loop-mediated isothermal amplification for rapid detection of Bacillus anthracis spores. Biotechnology Letters. 2007;29:1939-1946
11. Maruyama F, Kenzaka T, Yamaguchi N, Tani K, Nasu M. Detection of bacteria carrying the stx2 gene by in situ loop-mediated isothermal amplification. Applied and Environmental Microbiology. 2003;69:5023-5028
12. Ohtsuka K, Tanaka M, Ohtsuka T, Takatori K, Hara-Kudo Y. Comparison of detection methods for Escherichia coli O157 in beef livers and carcasses. Foodborne Pathogens and Disease. 2010;7:1563-1567
13. Zhao X, Li Y, Wang L. et al. Development and application of a loop-mediated isothermal amplification method on rapid detection Escherichia coli O157 strains from food samples. Molecular Biology Reports. 2010;37:2183-2188
14. Wang D, Wang Y, Wang J, Zhang X, Xiao F. Rapid detection of viable Listeria monocytogenes in raw milk using loop-mediated isothermal amplification with the aid of ethidium monoazide. Milchwissenschaft. 2011;66:426-429
15. Zhao X, Wang L, Li Y. et al. Development and application of a loop-mediated isothermal amplification method on rapid detection of Pseudomonas aeruginosa strains. World Journal of Microbiology and Biotechnology. 2011;27:181-184
16. Chen S, Wang F, Beaulieu JC, Stein RE, Ge B. Rapid detection of viable salmonellae in produce by coupling propidium monoazide with loop-mediated isothermal amplification. Applied and Environmental Microbiology. 2011;77:4008-4016
17. Ye Y, Wang B, Huang F. et al. Application of in situ loop-mediated isothermal amplification method for detection of Salmonella in foods. Food Control. 2011;22:438-444
18. Zhao X, Wang L, Chu J. et al. Development and application of a rapid and simple loop-mediated isothermal amplification method for food-borne Salmonella detection. Food Science and Biotechnology. 2010;19:1655-1659
19. Yang H, Ma X, Zhang X, Wang Y, Zhang W. Development and evaluation of a loop-mediated isothermal amplification assay for the rapid detection of Staphylococcus aureus in food. European Food Research and Technology. 2011;232:769-776
20. Nemoto J, Ikedo M, Kojima T, Momoda T, Konuma H, Hara-Kudo Y. Development and evaluation of a loop-mediated isothermal amplification assay for rapid and sensitive detection of Vibrio parahaemolyticus. Journal of Food Protection. 2011;74:1462-1467
21. Yamazaki W, Kumeda Y, Uemura R, Misawa N. Evaluation of a loop-mediated isothermal amplification assay for rapid and simple detection of Vibrio parahaemolyticus in naturally contaminated seafood samples. Food Microbiology. 2011;28:1238-1241
22. Zhao X, Wang L, Chu J. et al. Rapid detection of Vibrio parahaemolyticus strains and virulent factors by loop-mediated isothermal amplification assays. Food Science and Biotechnology. 2010;19:1191-1197
23. Notomi T, Okayama H, Masubuchi H. et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Research. 2000;28:e63
24. Njiru ZK. Loop-mediated isothermal amplification technology: towards point of care diagnostics. PLoS Neglected Tropical Diseases. 2012;6:e1572
25. Karthik K, Rathore R, Thomas P. et al. New closed tube loop mediated isothermal amplification assay for prevention of product cross-contamination. MethodsX. 2014;1:137-143
26. Savan R, Kono T, Itami T, Sakai M. Loop-mediated isothermal amplification: an emerging technology for detection of fish and shellfish pathogens. Journal of Fish Diseases. 2005;28:573-581
27. Li JF, Huang YF, Ding Y. et al. Shell-isolated nanoparticle-enhanced Raman spectroscopy. Nature. 2010;464:392-395
28. Haran G. Single-molecule Raman spectroscopy: a probe of surface dynamics and plasmonic fields. Accounts of Chemical Research. 2010;43:1135-1143
29. Wang Y, Irudayaraj J. Surface-enhanced Raman spectroscopy at single-molecule scale and its implications in biology. Philosophical Transactions of the Royal Society B: Biological Sciences. 2013;368:20120026
30. Bizzarri AR, Cannistraro S. SERS detection of thrombin by protein recognition using functionalized gold nanoparticles. Nanomedicine: Nanotechnology, Biology and Medicine. 2007;3:306-310
31. Drachev VP, Thoreson MD, Khaliullin EN, Davisson VJ, Shalaev VM. Surface-enhanced Raman difference between human insulin and insulin lispro detected with adaptive nanostructures. Journal of Physical Chemistry B. 2004;108:18046-18052
32. Tripp RA, Dluhy RA, Zhao Y. Novel nanostructures for SERS biosensing. Nano Today. 2008;3:31-37
33. Wang Y, Lee K, Irudayaraj J. Silver nanosphere SERS probes for sensitive identification of pathogens. Journal of Physical Chemistry C. 2010;114:16122-16128
34. Yang JL, Ma G-p, Yang R. et al. Simple and rapid detection of Salmonella serovar Enteritidis under field conditions by loop-mediated isothermal amplification. Journal of Applied Microbiology. 2010;109:1715-1723
35. Storhoff JJ, Elghanian R, Mucic RC, Mirkin CA, Letsinger RL. One-pot colorimetric differentiation of polynucleotides with single base imperfections using gold nanoparticle probes. Journal of the American Chemical Society. 1998;120:1959-1964
36. Lakowicz JR, Geddes CD, Gryczynski I. et al. Advances in surface-enhanced fluorescence. Journal of Fluorescence. 2004;14(4):425-441
37. Lakowicz JR. Radiative decay engineering: biophysical and biomedical applications. Analytical Biochemistry. 2001;298:1-24
Author contact
![]() Corresponding author: Prof./Dr. Xiaonan Lu. xiaonan.luca; +1-604-822-2551.
Corresponding author: Prof./Dr. Xiaonan Lu. xiaonan.luca; +1-604-822-2551.