Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Former DNA nanostructures
DNA origami
DNA-based drug delivery
Issue of Concerns
Concluding remarks and outlook
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(5):710-725. doi:10.7150/thno.14203 This issue Cite
Review
DNA Nanotechnology for Cancer Therapy
Vinit Kumar1*, Stefano Palazzolo1,2*, Samer Bayda1,2*, Giuseppe Corona1, Giuseppe Toffoli1, Flavio Rizzolio1 ![]()
1. Experimental and Clinical Pharmacology, Department of Translational Research, National Cancer Institute and Center for Molecular Biomedicine, CRO Aviano (PN), Via Franco Gallini, 2, Aviano 33081 - PN - Italy;
2. Doctoral School in Nanotechnology, University of Trieste, Italy.
* Authors equal contribution
Received 2015-10-22; Accepted 2016-1-27; Published 2016-3-20
Abstract

DNA nanotechnology is an emerging and exciting field, and represents a forefront frontier for the biomedical field. The specificity of the interactions between complementary base pairs makes DNA an incredible building material for programmable and very versatile two- and three-dimensional nanostructures called DNA origami. Here, we analyze the DNA origami and DNA-based nanostructures as a drug delivery system. Besides their physical-chemical nature, we dissect the critical factors such as stability, loading capability, release and immunocompatibility, which mainly limit in vivo applications. Special attention was dedicated to highlighting the boundaries to be overcome to bring DNA nanostructures closer to the bedside of patients.
Keywords: Nanomedicine, Nanotechnology, Cancer, Drug delivery, Doxorubicin, Self-assembly, DNA, Origami.
Introduction
The delivery of drug molecules specifically to the tumor site is an exigent requirement to avoid side effects during cancer therapy. However, this requirement is still clinically unmet, mainly due to the lack of effective drug delivery systems (DDSs) for single or combinatorial chemotherapy. In the last two decades, various organic and/or inorganic nanomaterials have been used to create nanostructures that facilitate anticancer drug delivery [1]. In particular, nanoparticle-based DDSs have been shown to enhance permeability and retention (EPR) effects, leading to passive tumor drug accumulation [1]. However, in some cases the observed toxicity versus normal tissues raised concerns over real applications in humans [2-4]. Recently, many research groups have focused their attention on DNA material for the construction of artificial nanostructures not present in biological systems for bio-applications [5-8]. DNA, being a genetic material, possesses high biocompatibility and low cytotoxicity ideal for applications in the biomedical field [5,9]. Its remarkable molecular recognition properties, and complementary base pairing, along with its stability, mechanical rigidity, nano-dimensions of the repeating unit, easily custom synthesis with manipulative length of strands allow the formation of most every shape of nanostructures. Moreover, high drug loading efficiency and effective cellular internalization further support DNA origami as a programmable “smart” building block for the construction and development of versatile highly nontoxic drug nanocarriers [5-7,9,10]. The highly-controllable DNA origami nanostructures with addressable modification sites, simple loading methods for biomolecules, biocompatibility and biodegradability, offer many advantages to be ideal targeted drug delivery comparing to the delivery systems such as liposomes and polymeric nanoparticles which have already been approved for clinical use [11-15].
Due to the aforementioned properties, DNA nanostructures have been synthesized and engineered for various applications, such as: i) scaffolds or templates to arrange organic, inorganic and biomolecules into defined morphology; ii) molecular transporters; iii) highly sensitive molecular- and bio-detector; iv) single molecule spectroscopy; v) protein structure determination, and vi) vehicles for in vitro and in vivo drug delivery [16].
In the present review, we will briefly introduce the properties of DNA and its structure in various self-assembled arrangements to guide readers through the topic of the review focused mainly on the recent advances on the biomedical applications of DNA nanostructures, particularly, their potential as nanocarriers of drug and other molecules across the cellular membrane for specific and effective drug delivery to cancerous cells.
Former DNA nanostructures
The physical and chemical properties of DNA nanostructures are reported in excellent recently published reviews [17-19]. Here, we describe the basic elements that are useful to understand the self-assembled DNA nanostructure as drug nanocarriers. The pyrimidine and purine bases that constitute the nucleotides in single-stranded DNA are linked to pentose sugar and this latter associated unit is called a nucleoside, which is connected to another nucleoside through the phosphodiester bond. The asymmetric ends of DNA strands are called the 5' and 3' ends depending on whether the terminal group is a phosphate group or a free hydroxyl group, respectively. The purine bases are classified into two types: Adenine (A) and Guanine (G). They have a structure derived from the fusion of five- and six-membered heterocyclic structure, while the pyrimidines are Cytosine (C) and Thymine (T) and are six-membered ring (Figure 1A).
A) Adenine (A), Thymine (T), Cytosine (C) and Guanine (G) of DNA responsible for the robust complementary base pair interactions between DNA strands. B) Key features of DNA structures. C) Chemical structure of DNA stabilized by hydrogen bonds between the bases A-T and G-C. Copyright 2002, Pearson Education, Inc. Publishing as Benjamin Cummings.
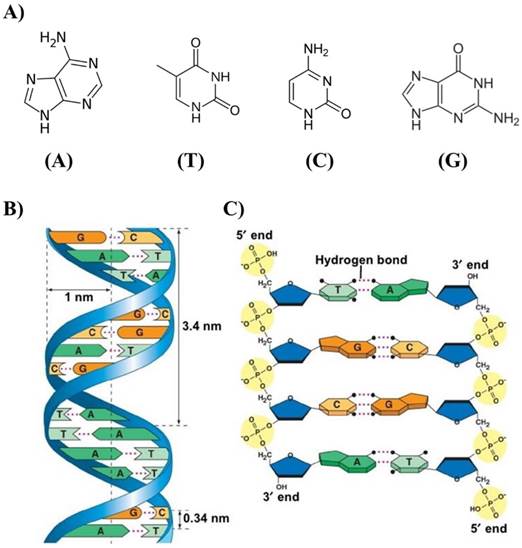
In B-form dsDNA, the most common form of double helix, two nucleotide nanowires are twisted around each other with a replicate unit every 3.4 nm while maintaining a distance of 3.4 Å between the consecutive base pairs in a double helix with a diameter of 2 nm (Figure 1B). The twist angle between base pairs in solution is ~34.38º with C2'-endo sugar pucker. The persistence length of dsDNA, which is a measure of stiffness, is very close to 50 nm if the concentration of NaCl exceeds 10 mM or the concentration of MgCl2 exceeds 0.5 mM. It was found that the dsDNA persistence length in a solution containing 10 mM MgCl2 is equal to 47±1 nm.
Many theoretical and experimental studies were dedicated to the dependence of persistence length on ionic conditions, and it seems that the persistence length is practically independent of ionic concentrations if the concentration of NaCl > 10mM or the concentration of MgCl2 > 0.5 mM [20,21]. Two factors are mainly responsible for the stability of the geometry of the DNA double helix: the presence of hydrogen bonds between complementary bases of the strands, and aromatic π-π stacking between adjacent bases. Two hydrogen bonds are formed between the bases A and T and three between G and C attached to the two strands (Figure 1C). While each hydrogen bond is weak compared to a covalent bond, a large number of hydrogen bonds together represent a strong force that keeps the two strands bound. In addition, other polar groups of the base rings can form external hydrogen bonds with surrounding water that give to the molecule extra stability. The hydrogen bond is not the only force that gives stability to the dsDNA structure. The negatively charge of the phosphate group can interact with positively charged atoms with electrostatic forces [22]. The free energy contribution (-ΔG/kcal mol-1) of the formation of A-T and G-C base pairs due to hydrogen bonds is ~1.34 and 2.17, respectively [23].
A DNA nanostructure is a bottom-up assembly of multiple ssDNA that have to hybridize to other segments or to a scaffold. In 1964, Robin Holliday illustrated a four-armed DNA branched junction, which later became known as the Holliday junction, in which four DNA strands are linked together to form four double helical arms flanking a branch point. A Holliday junction occurs commonly in nature, as it is being the process of genetic recombination called crossing-over involving in passing genetic diversity to the next generation. This specific genetic structure became the foundation of DNA nanotechnology, in which by connecting several Holliday junctions in the form of tiles and yielded DNA lattice in two and three dimensions [24]. The DNA double crossover (DX) molecule is characterized by the crossover of a DNA strand that exchanges between two strands of opposite polarity [25,26]. This enables robust connections among several DNA helices. A DX DNA tile is a DNA nanostructure that has a number of sticky ends on its sides, which are termed pads. A sticky end is a short ssDNA or hang protruding strand from the end of a DNA helix. A DNA lattice is a DNA nanostructure composed of a group of DNA tiles that are assembled together via hybridization of their pads.
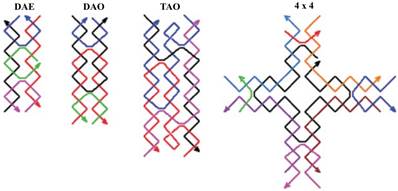
One more modification is the three-domain DNA triple crossover (TX) complex that connects three DNA helix axes by one strand in one plane [16]. Using rigid DX or TX tiles, regular two-dimensional arrays could be created by sticky-ended cohesion of several molecules (Figure 2) [27]. Multiple crossovers could also be created which leads to the formation of more sophisticated structures.
Schematic drawings of four DNA tiles are shown. Colored lines represent different oligonucleotide strands with arrowheads marking the 3 ends. DAE and DAO are double crossover complexes (also known as DX), TAO is an example of a triple crossover (or TX) tile, and the 4 × 4 tile is composed of four arms each of which contains a four-arm junction. Reproduced with permission from reference [27].
DNA origami
DNA nanotechnology uses the molecular recognition properties of DNA to create artificial DNA structures for technological purposes. It holds great promise for a vast range of applications in different fields such as biology, medicine and material science. In 1982, Nadrian Seeman laid the theoretical framework for the use of DNA as a nanoscale building material. This is due to the DNA's capacity for programmable self-assembly and its high stability [24,28].
In 2006, Paul Rothemund at the California Institute of Technology introduced the term “scaffolded DNA origami”, which has revolutionized the field of structural DNA nanotechnology by enhancing the complexity and size of self-assembled DNA nanostructures in a simple “one-pot” reaction (Figure 3A) [29].
In DNA origami: A) a long circular single-strand DNA scaffold is folded into a desired shape with the aid of hundreds of short staple strands. In the above example, circular single-stranded genome of M13 bacteriophage (M13mp18) is folded with the assistance of about 200 staple strands into a DNA nanocube with dimension of 35x36x42 nm during a thermal annealing process. Adapted with permission from reference [29]. B) 3D structure with complex curvatures. Adapted with permission from reference [33]. C) DNA gridiron nanostructures. Reproduced with permission from references [35,38]. D) Design of 2D and 3D DNA canvas using single-stranded tiles/bricks. Adapted with permission from references [36,37].
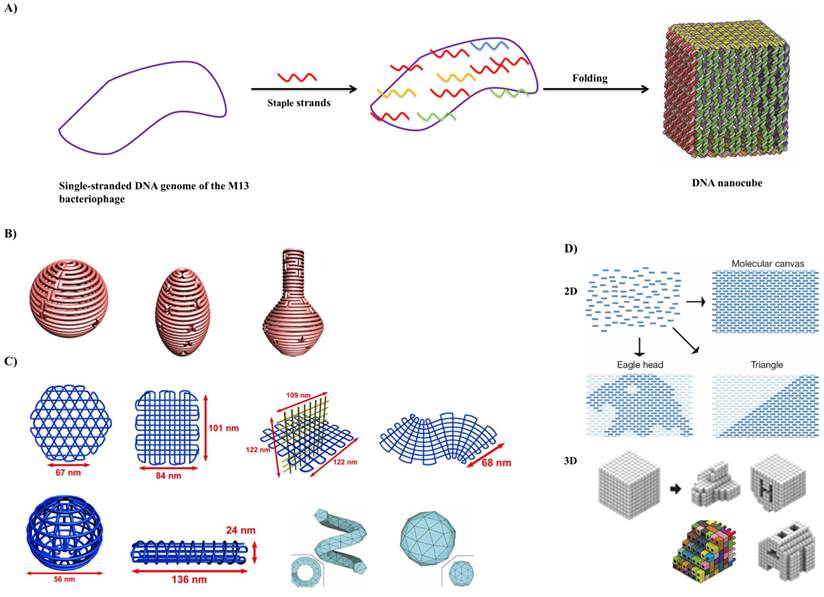
Scaffolded DNA origami involves the folding of a long circular single-strand-scaffold viral DNA derived from the bacteriophage M13mp18 composed of 7249 nucleotide sequences with hundreds of short staple strands or helper strands of DNA into the desired shape. The DNA origami technique has been successfully used for the preparation of different 2D and 3D nanostructures and for the nanopatterning of nanoparticles, proteins and other functional molecular components into well-defined arrangements (Figure 4) [30]. Chemically modified staples can be inserted at a predefined position in the DNA nanostructure, which could be used to impart several additional functionalities in the designed DNA nanostructures. The design of a DNA origami is then fed into computer software called “caDNAno” which calculates the placement of individual staple strands [31]. Each staple binds to a specific region of the DNA template due to Watson-Crick base pairing rules. The scaffold strand and the staple strands are mixed in a one-pot reaction and rapid heating followed by slow cooling allows the various staple strands to pull the long scaffold strand into the desired shape. More recently, many groups invented a versatile strategy that could reliably manipulate the DNA conformation and shape of the crossover networks to design 2D and 3D nanostructures twisted and curved, for example, spherical shells, ellipsoid shells and nanoflask (Figure 3B) [32-34]. Further, they utilized a series of four-arm junctions to create gridiron-like DNA nanostructures, which involves stacking multiple layers of 2D gridiron lattices to form multilayer 3D nanostructures and curved objects (Figure 3C) [35]. Wei et al. and Ke et al. developed a new strategy, which combines the advantages of tile and origami assembly utilizing single-stranded tiles/bricks with concatenated sticky ends as building blocks to form 2D and 3D DNA canvas (Figure 3D) [36,37]. In a different line, Benson et al. developed a method highly automated by using a routeing algorithm based on graph theory and relaxation simulation that traces scaffold strands through the target structures. These structures have one helix per edge and are stable under the ionic condition of biological assays (Figure 3C) [38].
The first examples of the versatile DNA Origami technique. Top row, folding paths. A, square; B, rectangle; C, star; D, disk with three holes; E, triangle with rectangular domains; F, sharp triangle with trapezoidal domains and bridges between them (red lines in inset). Dangling curves and loops represent unfolded sequence. The lower panels contain the resulting DNA structures as imaged by AFM. All images and panels without scale bars are the same size, 165 nm x165 nm. Scale bars for lower AFM images: B, 1 µm; C-F, 100 nm. Reproduced with permission from reference [30].

The design concept of DNA origami, their shape and assembly principles was discussed by Linko and Dietz [39] and Castro et al. and it is illustrated in Figure 5 [40]. The double helix structure of DNA is represented as double helix domains (cylindrical representation) for designing purposes (Figure 5A). Double helix domains are connected to adjacent double helix domains by multiple interhelix connections consisting of immobilized Holliday junctions [25]. As shown in Figure 5B, the interhelix connections are formed by antiparallel crossovers of either the staple or scaffold strand.
Scaffold DNA origami design concept: A) Schematic representation of DNA double helices. B) Two double helices are connected by interhelix crossovers. C) Scaffold strand routing to form three different DNA origami objects. D) For the same three DNA nanostructures, staples are highlighted with different colors to form the structures. E) Cylindrical representation of the three DNA nanostructures. Reproduced with permission from reference [40].
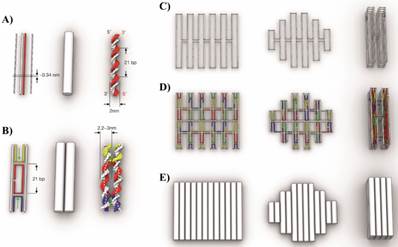
A step-by-step guide to building DNA origami objects is shown in Figure 6 [40].
Step-by-step guide of molecular self-assembly with scaffolded DNA origami. Reproduced with permission from reference [40].
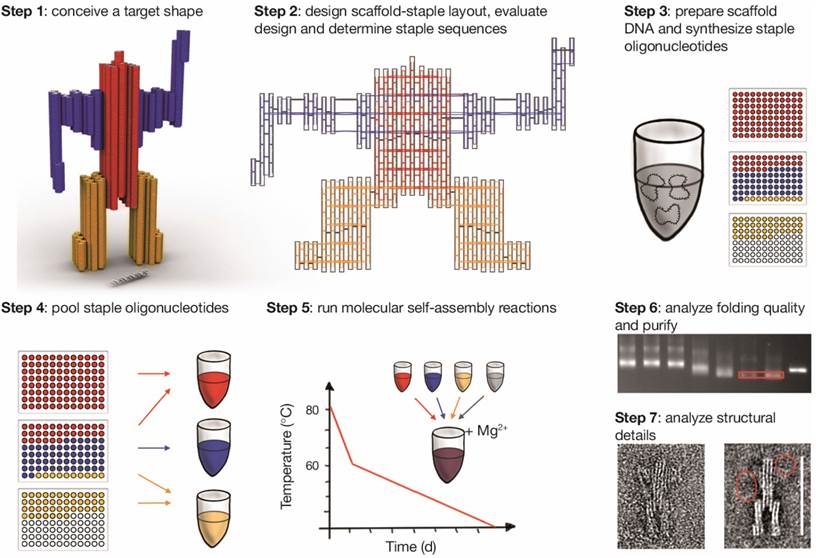
Step 1: Conceive target shape. The work starts with the conception of a target shape with specific functional requirements. Based on the application, it is important to decide on a single-layer or multilayer structure using square lattice or honeycomb lattice. In Figure 6, Step 1, a 72 nm-tall sculpture of a robot is considered as the target shape using a multilayer honeycomb lattice packing.
Step 2: Design layout, evaluate design and determine staple sequences. The designing of the internal layout of the DNA origami object can be accomplished with many computational tools (Figure 6, Step 2). Based on crossover spacing rules, the staple sequence can be determined. Certain applications require site-directed attachment of nanoparticles, proteins or fluorescent dyes. Such attachments need to be considered in this scaffold-staple layout.
After designing the layout, it is very important to validate the designed DNA nanostructure in order to realize their mechanical flexibility and topology by simulating their twists, bends, curvatures and fluctuations and rigidity in a solution using a computational tool named computer-aided engineering for DNA origami (CanDo) [40-42].
Step 3: Prepare scaffold DNA and synthesize staples. The quality of DNA origami folding might depend on the scaffold sequence and the particular cyclic permutation, which means the repeating units of the targeted shape. The single-stranded M13mp18 bacteriophage genome is used as long scaffold strand, which acts as a template for scaffolded DNA origami. This template is commercially available. The scaffold strand could also be prepared by an enzymatic [43] or by a denaturing dialysis method to separate a dsDNA (derived from M13mp18) into two ssDNA scaffolds [44]. The staple sequences, which are generated while designing the DNA origami, are utilized to synthetize the desired oligonucleotides.
Step 4: Pool subsets of concentration-normalized oligonucleotides. This step is important in deciding the right concentration ratio of scaffold strand to staple molecules. For optimal results, this ratio is usually set at 1:5.
Step 5: Run molecular self-assembly reactions. One theory is that the scaffold-staple layout requires a structural solution for the correct mixture of scaffold DNA and staple molecules that minimizes energy through Watson-Crick base pairing. The targeted shape corresponds to a global energy minimum of the system and depends on the solvent and design conditions. The goal of the self-assembly reaction is to reach a minimum energy state in conditions where the targeted structure is folded. The best conditions are identified as a function of the salt concentration and cyclic temperatures.
The experimental conditions for the assembly of various DNA nanostructures such as scaffold to staples ratio, buffer, salt concentrations and annealing temperature are of paramount importance for their correct folding, structural integrity and yield. A 1:5 to 1:10 ratio of scaffold to staples is enough for the self-assembly reaction. For most of the folding reaction a buffer containing 10 to 25 mM of Mg2+ in 1xTE, depending of the layers, length and shape of the DNA-origami structure, is adequate. It has been shown that a monovalent ion (Na+) could also be used in place of divalent ion (Mg2+) for DNA-origami preparation, however a very high concentration of Na+ is required [45]. For annealing, the mixture of scaffold and staples in folding buffer is heated to 90 °C and then slowly cool down to 4 °C and this step usually takes up to a week. Dietz and coworker showed that folding occurs in a very narrow temperature range of 60 °C - 45 °C, which has drastically slashed the annealing to few hours [46].
Step 6: Analyze folding quality and purify the object. The quality assessment of DNA origami folding and purification could be accomplished by gel electrophoresis. The gel has to contain magnesium during running. The reactions could be optimized by searching for the best conditions, which give a high yield of nanostructures and good gel separation from the staples. The reactions are purified from the gel slabs by excising the desired bands followed by the DNA electro-elution method [47]. Another method used to purify and concentrate DNA developed by Stahl et al. is based on polyethylene glycol (PEG) precipitation with high molecular weight. This method was efficient and used to reduce the elution volumes and achieve high recovery yields of up to 97% [48]. Ultrafiltration method consists in a repetitive dilution-concentration process across a regenerated cellulose membrane, which retains the large DNA origami structures while the small contaminants flow through [49]. Gel filtration utilizes various dextran or agarose-based size exclusion resins in spin columns to retain contaminants while DNA origami structures flow through during centrifugation [50]. The purification could be also achieved using rate-zonal ultracentrifugation on glycerol density gradient where DNA origami are separated by density. This method allowing for larger quantities of DNA nanostructures to be purified, with less time and effort. The recovery yield is between 40-80% [51].
Due to the difficulties in purifying functionalized DNA origami, in particular with proteins, Shaw et al. investigated the efficiency of different methods. The authors adapted two methods: magnetic bead captures and fast protein liquid chromatography (FPLC). FPLC is commonly used to purify DNA or proteins on a Superose column. This technique is indicated for origami purification since the reaction can be performed in a variety of physiological buffers. The recovery yield is around 70% [49].
Step 7: Single-particle based structural analysis. Single molecule microscopy techniques play an important role in the investigation of advanced DNA origami. They can be imaged by negative-stain transmission electron microscopy (TEM) and with atomic force microscopy (AFM) [30,32]. Alloyeau et al. showed that this is also possible for direct imaging along with chemical analysis of unstained DNA origami with transmission electron microscopy [52].
DNA-based drug delivery
Several DNA-based nanostructures, namely tetrahedral, icosahedral [53], nanotube [54-57], square and triangle [30] have been developed recently for in vivo and in vitro drug delivery applications. In contrast to dsDNA, DNA nanostructures could be internalized within the cells without any aid from transfection agents [58] and, when densely packaged, could be effectively used for the drug delivery purposes [59]. The cellular localization of DNA origami could be detected by fluorescence-based assays, which have the disadvantage of utilizing fluorescent labels. As alternative approach, Okholm et al. use quantitative real time polymerase chain reaction (qPCR) of M13 amplicons to quantify the cellular uptake of DNA origami structures [60]. In order to enhance and improve the cellular uptake/cell transfection of DNA-based systems, Mikkilä et al. demonstrated the possibility of coating DNA origami with virus capsid proteins, which can bind and self-assemble on the surface of origami through electrostatic interactions and pack the DNA nanostructures inside the capsid [61]. More recently, Brglez et al. designed an intercalator based on acridine derivatives that modify the surface properties of DNA nanostructures and increased the cell uptake compared to unmodified origami [62].
In Table 1, an overview of different applications of DNA nanostructures utilized for drug delivery is provided. Starting from DNA tiles, tetrahedral (N1) [63] and icosahedral (N2) [64] nanostructures were demonstrated to be effective for doxorubicin (doxo) delivery to breast cancer cells. In particular, the tetrahedral structure was found to be effective on drug-resistant cells. The icosahedral structure was able to efficiently deliver the doxorubicin in a targeted way only after functionalization with aptamer sequences against the tumor surface marker mucin 1 (MUC1). DNA tetrahedral structures have also been shown to protect single-strand sequences against nuclease degradation; in particular, this kind of structure has been employed to increase the in vivo circulation half-time of siRNA from 6 to 24 minutes (N3) [65] and deliver Cytosine-phosphate-Guanosine (CpG) to elicit an immunoresponse (N4) [66]. Finally, highly biocompatible aptamer-tethered DNA nanotrain (N5) against folic acid receptor exhibited high antitumor efficacy and reduced the side effects of doxo in a mouse xenograft tumor model [67]. Very recently, our group has demonstrated that a half-icosahedral nanostructure (N6) can efficiently delivery doxorubicin to breast and hepatic cancer cells. The study demonstrated the importance of the shape and structure of DNA nanostructures for biomedical applications [68].
Overview of different DNA nanostructures and their applications.
| Structure Characterization | Biological activity | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N° | DNA Nanostructure | Characterization | Size (nm) | Cargo (Loading) | Stability | Targeting | Loading efficiency | Release | Internalization | In vitro tests | In vivo tests | Biodistribution | Ref | ||
| Cells | Activity | Cells | Activity | ||||||||||||
| 1 | Tetrahedral | AFM DLS PAGE (6%) | AFM: height 2-3 nm DLS: 9.08 nm ± 3nm | Doxo | > 4 < 23 hrs Solution: 10% human serum Temp: 37°C | N/A | 26 molecules/structure Time: 1 hr Temp: N/A | 100% in 10hrs. <10% in 3hrs. Solution: PBS pH 7.4 Temp: 37°C 60% in 10 hrs Solution: PBS pH 5 Temp: 37°C | Macropinocytosis and caveolae-mediated endocytosis pathways | Breast cancer MCF7 (drug sensitive) and MCF7-ADR (Doxo resistant). | Doxo-tetrahedral is more effective on cell viability of MCF7-ADR than free doxo. Same efficacy on MCF7 cells. | N/A | N/A | N/A | [63] |
| 2 | Icosahedral: five- (120 bp) and six- (144 bp) (aptamer) point-star structure | AGE (2%) DLS TEM | DLS: - Five stars (28.2 nm ± 3nm) - Six stars (28.6 nm ± 5nm). TEM: 25 nm | Doxo | >30min Solution: cell culture medium Temp: 37°C | Mucin 1 (MUC1). Tumor surface marker | 1200 molecules/structure Time: 1h incubation Temp: room temperature | N/A | Dynamin-dependent and clathrin-mediated endocytosis. Degradation in lysosomes | Breast cancer MCF7 (MUC1+), CHO-K1 (MUC1-) | Doxo-Aptamer six-point-star structure is more effective on cell viability of MUC1+ cell than free doxo. Same efficacy on MUC- cells. | N/A | N/A | N/A | [64] |
| 3 | Tetrahedral (30bp/edge, Total 6 edges) | AFM DLS PAGE (5%) | AFM: height 7.5 nm. DLS: 28.6 nm ± 2.38 nm | siRNA against GFP gene. DNA structure conjugated with folic acid. | N/A | Folic acid receptor (FAR) | 1-6 siRNA per tetrahedral (1 siRNa/edge) | N/A | N/A | HeLa cells (LUC+). KB cells (HeLa cell contaminant overexpressing folate receptor) | HeLa: LUC expression < 50%. KB: GFP expression <40% | KB xenograft tumours | siRNA no effect on LUC. siRNA-DNA- tetrahedral: IC 50 LUC expression: 1.8 mg/kg. Inj: intratumor or tail-vein Stability: siRNA 6 min; siRNA-DNA tetrahedral 24 min. | Tumor, Kidney Time: 12 hours post injection | [65] |
| 4 | Tetrahedral | PAGE (3.5%); | N/A | Straptavidin (STV) and CpGs | 5hrs Solution: FBS 50% Temp: room temperature | N/A | N/A | N/A | Endocytosis. Antigen localized in lysosomes after 2 hours | RAW264.7 (macrophage, Abelson murine leukemia virus transformed). | Increased internalization of the complex tetrahedron-STV-CpG by APC cells | BALB/c immunocompetent mice | Mice immunized with the complex tetrahedron-STV-CpG developed a stronger and a longer immunitary response. | N/A | [66] |
| 5 | Aptamer-tethered DNA nanotrain | AFM AGE (3%) TEM | AFM: length 100 nm. | Doxo, epirubicin, daunorubicin | >45 hrs Solution: PBS + 5mM Mg2+ Temp: N/A | Protein Tyrosin Kinase 7 (PTK7) | N/A | N/A | Endocytosis | CEM cells (human T-cell acute lymphocytic leukemia PTK7+) and Ramos (human B lymphocyte Burkitt's lymphoma PTK7-) | Drug-Aptamer-DNA-drug is more cytotoxic on PTK7+ cells than free drug. Same efficacy on PTK7- cells. | CEM (PTK7+) xenograft mouse model | Increased antitumor efficacy and reduced side effects of doxo delivered via Aptamer-DNA nanotrain. Inj: I.V. | [67] | |
| 6 | Open Caged DNA (pyramidal), 408 bp | PAGE (7%) | N/A | Doxo | 35 hrs (half-life) Solution: cell culture medium+10% FBS Temp: 37°C | N/A | 172 molecules doxo/structure (at 15% of loading efficiency) | 50% of Doxo release from py-Doxo in PBS in 5 hrs and 3 hrs in FBS. Free Doxo in 20 minutes | py-Doxo is able to penetrate inside MDA-MB-231, release Doxo in the nucleus | MDA-MB-231, HepG2 | Decrease cell viability compared to free doxo | N/A | N/A | N/A | [68] |
| 7 | Tube with different global twist (Straight (S) 10.5 bp/turn and Twist (T) tube 12 bp/turn) | AGE (2%) TEM | TEM: length: 138 nm, diameter: 13 nm | Doxo | 48hr (T-tube) Solution: 10% FBS Temp: 37°C | N/A | N/A | 80% (T-tube) and 90% (S-tube) in 10 hrs Solution: PBS pH 7.4 Temp: 37°C | N/A | Breast cancer MDA-MB-231; MDA-MB-468; MCF-7 | In all the cell lines tested the T-DNA IC50 is ≈2 times lower than free Doxo. (calculated by our group) | N/A | N/A | N/A | [70] |
| 8 | Tube | AGE (2%) TEM | TEM: length: 80 nm, diameter: ≈20 nm | 62 CpG sequence specific for mouse Toll like receptor 9 | 6 hrs Solution: cell culture medium Temp: 37°C | Toll like receptor 9 (TLR9). | 62 binding sites per tube | N/A | N/A | Splenocytes from female C57BL/6 mice | N/A Immunoresponse through the TLR9. Nontoxic | N/A | N/A | N/A | [71] |
| 9 | Hexagonal barrel with aptamer-based lock (antigen keys). | AFM AGE (2%) DLS TEM | DLS: 90 nm TEM: 35nm x 35 nm x 45 nm | Fab antibody fragments | N/A | CD33, CDw328 | N/A | N/A | N/A | NKL | Increase apoptosis | N/A | N/A | N/A | [72] |
| 10 | Triangle and tube | AFM AGE (1%) | AFM: -Triangle: edge ≈ 150nm -Tube: length ≈ 183 nm | Doxo | N/A | N/A | >200000 molecules (calculated by our group) Time: 24 hrs Temp: room temperature. | ≈15% pH 7.4, 35% pH 5.5 in 48 hrs ≈25% in MCF7 cell lysate >40% with 50U DNAaseI Solution: PBS Temp: 37°C | Endocytosis and localization of origami in lysosomes after 6 hours of treatment. | Breast cancer MCF7 and MCF7 resistant | MCF-7: 2.5µM and MCF-7 resistant >100μM. DNA origami loaded with doxo enhanced the cells death compared to free doxo on MCF7 doxo resistant cells. No differences between free doxo and origami doxo in regular MCF7. | N/A | N/A | N/A | [73] |
| 11 | Tube, triangle and square | AFM AGE (1%) DLS | AFM: -Tube: height: 7 nm, diameter: 380 nm, -Triangle: edge 120 nm, -Square: length*width 90nmx60nm DLS: -Triangle: 59 nm -Square: 80.9 nm -Tube: 98.6 nm. | Doxo | 24 hrs Solution: serum Temp: 37°C | N/A | >200000 molecules (calculated by our group) Time: 24 hrs Temp: room temperature. | ≈20% of Doxo is released in 48 hours at pH 7.4, ≈35% is released at pH 5.5 Solution: PBS Temp: 37°C | N/A | Breast cancer MDA-MB-231 | No significant difference compared to the free doxo. | MDA-MB-231 cells Orthotopic breast cancer model | Increased EPR effect of DNA origami. Significant tumor reduction in mice treated with doxo-origami compared to free doxo Inj: I.V | Tumor: Triangle > tube >square Tumor > liver > kidney > spleen Time: 24 hrs | [74] |
| 12 | Tube (Tile) | AGE (2%) TEM | TEM: Length: 27 nm and diameter: 8 nm | siRNA (GFP) | 8hrs Solution: cell culture medium Temp: 37 °C (Degradation depends on Mg2+ concentration, oligonucleotide sequences, salt concentration, structural extension) | Folic acid receptor (FAR) | N/A | N/A | Endosomal trapping (no release) | N/A | No effects on GFP expression | N/A | NA | NA | [86] |
AGE: Agarose gel electrophoresis. PAGE: Polyacrylamide gel electrophoresis. TEM: Transmission electron microscopy. DLS: Dynamic light scattering. AFM: Atomic force microscopy. N/A: Not Applicable. Temp: Temperature. Inj: Injection. I.V.: Intravenous. Luc: Luciferase
DNA origami, which provides enhanced size, dense packaging of strands and controllable shape, can be used to construct multivalent and multifunctional drug carriers. DNA origami was found to be stable in cell lysates and can be slowly degraded in living cells after 72 hours of treatment, demonstrating its great potential for controlled drug release [69]. This property was demonstrated in an in vitro experiment by Högberg group. The authors were able to control the kinetics of the release of doxo from DNA origami tubes (N7) by regulating the global twist of the structure, showing that the twisted form releases doxo more slowly than the normal structure [70]. The Liedl group constructed a 30-helix DNA origami nanotubes (N8) that were functionalized with CpGs oligonucleotides (up to 62 molecules) and tested for their immunostimulatory efficacy in isolated mouse spleen cells. Splenocytes include a subset of immune cells such as dendritic cells and macrophages that initiate and control the immune response. CpG-DNA nanotubes are internalized better than CpG alone and consequently stimulate a strong immune response [71]. DNA barrels (N9) have been constructed in a structure capable of selectively interfacing with cells to deliver signaling molecules to cell surfaces [72]. The opening lid is based on a DNA aptamer-based lock mechanism, which opens in response to the binding of antigen keys.
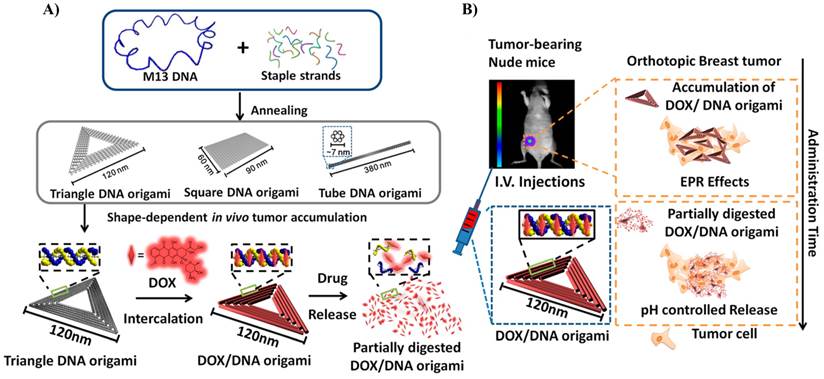
Different DNA nanostructures, namely, triangle, square and tube, were synthesized and tested for drug delivery in in vitro and in vivo experiments (Figure 7) [73,74]. In vivo experiments demonstrated that these structures were able to deliver doxo efficiently to normal and resistant cancer cells [74]. Interestingly, it appears that the triangular structure resided in the tumor for a significantly longer time than the square and tube structures (N10 and N11). This might be attributed to the different shape of DNA structures and was probably due to the enhanced retention time inside the tumor (Figure 7). In most studies, drug loading on DNA nanostructures relies on the intercalation property of doxo molecules with base pairs of DNA duplex [70,73,74]. High loading efficiencies of doxo with different DNA origami structures have been achieved. For instance, in the case of DNA tube, more than 70% loading efficiency was obtained [73].
Schematic design of the DNA carrier-drug complex. A) Long, single-stranded DNA scaffold (M13mp18 phage genomic DNA, blue) hybridizes with rationally designed helper strands to fold into triangular, square, and tube origami shapes. The biodistribution of unstructured M13 DNA and different nanostructures of DNA origami was investigated in subcutaneous breast tumor model. After in vivo biodistribution, the triangle-shaped DNA origami demonstrated optimal tumor accumulation; it was then used for doxorubicin intercalation. The Watson-Crick base pairs in the double helices of DNA origami serve as docking sites for doxorubicin intercalation (DOX/DNA origami, red). B) Tail-injected DOX/DNA origami complexes were delivered via blood circulation, accumulating in the breast tumor of nude mice because of EPR effects. Reproduced with permission from reference [74].
Due to the programmed and well-defined properties of DNA nanostructures, it is possible to precisely control the spatial distribution of cargo molecules over DNA structure. In fact, there is virtually no limit for tethering DNA with various functional molecules through covalent modifications. DNA oligomers with a large variety of functional end groups are commercially available. Amino-functionalized DNA has been employed to bind carboxylic groups, and thiol-modified DNA to maleimide groups [75]. Srinivasan et al. have developed a novel procedure for labeling plasmid DNA using quantum dots (QDs) [75]. This method involves covalent conjugation of plasmid DNA to phospholipid/polyethylene oxide-encapsulated cadmium selenide/zinc sulfide (CdSe/ZnS) core/shell QDs using a peptide nucleic acid-N-succinimidyl-3-(2-pyridylthio) propionate (PNA-SPDP) linker, which facilitates tagging of the plasmid without interfering with its function. QD-tagged DNA can transfect cells with high efficiency and intracellular trafficking can be followed through time. The “tagged” DNA plasmid remains functional and serves as a template for gene transcription upon internalization in the nucleus.
The avidin-biotin system has also been used as a non-covalent receptor-ligand system for the binding of DNA to nanoparticles like gold nanoparticles (AuNPs) or quantum dots (QDs) [76,77]. Other than covalent modifications, DNA is negatively charged and is essentially a polyelectrolyte molecule which could bind through the positive charge of the surface such as gold nanoparticles with quaternary ammonium [78]. Additionally, it has been demonstrated that DNA incubated at high stoichiometric excess over gold nanoparticles shows nonspecific adsorption [79].
Issue of Concerns
Characterization (Table 1, column 3)
The assembly of DNA nanostructures was characterized by multiple techniques, including agarose (AGE) or polyacrylamide gel electrophoresis (PAGE), transmission electron microscopy (TEM), dynamic light scattering (DLS) and atomic force microscopy (AFM).
The AGE/PAGE techniques are performed by applying an electric field allowing DNA migration through an agarose or polyacrylamide matrix. The nanostructures are separated by size (shorter move faster than longer structures) and analyzed under UV-light after staining with ethidium bromide. The nanostructures can be extracted from the gel and dissolved in a suitable buffer. The yields of assembled DNA nanostructures can be estimated by running an agarose or polyacrylamide gel and comparing the intensity of the bands to a standard reference [64,65]. When the structures are loaded with drugs, the DNA exhibits a decrease in folding quality compared to the sharp band before drug loading, as observed with a lower gel mobility, indicating that the drug was intercalated into the DNA [70,73].
Transmission electron microscopy (TEM) and atomic force microscopy (AFM) were used to demonstrate the assembly of DNA, the size, the shape and the monodispersity of the nanostructures. These techniques provide direct evidence of the morphology of nanostructures before and after drug loading [64,70,73]. Zhao et al. showed by using TEM images that twisted DNA nanostructures loaded with doxo were more compact, straighter and more elongated than the twisted DNA folded without doxo [70], while Jiang et al. demonstrated with AFM images before and after doxo loading that the morphology of the DNA nanostructure was retained after intercalation of the drug [73], and Douglas et al. utilized TEM to analyze the different states and conformations of DNA nanorobots [72].
The dynamic light scattering (DLS) technique was used to determine the hydrodynamic diameter of the nanostructures. It measures the fluctuations in scattered light intensity due to diffusing particles. When the system is monodisperse, the effective mean diameter of the particles can be determined. This measurement depends on the size of the particle core, the size of surface structures, particle concentration, and the type of ions in the medium. In general, the DLS measurement of the DNA nanostructures has shown a narrow size distribution, representing a uniform and monodisperse hydrodynamic size [63-65,74].
In Table 1, all the nanostructures are characterized by AGE/PAGE taking advantages of the simple and fast procedure to obtain information on the formation of nanostructures. These techniques were equally integrated with AFM or TEM analyses to demonstrate the size and shape of nanostructures. In this regard, there is uniformity and concordance among the papers helping with the interpretation of the results.
Drug loading (Table 1, column 5)
More than half of the studies have utilized doxo as proof-of-concept chemotherapeutic drug. Doxorubicin has the advantage of being widely utilized in cancer therapy, intercalates into DNA and has intrinsic properties such as fluorescence and absorbance that are of help during analysis. Other anthracycline chemotherapeutic drugs (e.g. daunorubicin (DNR), and epirubicin (EPR)) are widely used in therapeutic. They can preferentially intercalate non covalently into double-stranded 5′-GC-3′ or 5′-CG- 3′ [67,80]. The platinum drugs, such as cisplatin, carboplatin, and oxaliplatin, bind to the nitrogen (N) position of the two adjacent guanine (G) bases of DNA, which is responsible for the cytotoxic effect of platinum drugs [81]. Wang et al. designed a cationic core-shell nanoparticles with a hydrophobic core and a cationic shell by self-assembly process from a biodegradable copolymer. A hydrophobic drugs, such as Paclitaxel (PTX), can be incorporated into the core during the self-assembly and the cationic shell of the PTX-loaded nanoparticles bind to the DNA by electrostatic interaction between the negative charge of DNA and the positive charge of the nanoparticle [82]. In addition to the non-covalent interaction, the chemotherapeutic drugs can covalently functionalize the DNA. Each DNA origami staple strand is synthetically made and can be modified in predefined positions and incorporating different functions (e.g. amino groups). For example, the methotrexate (MTX), a chemotherapeutic drug, can be functionalized covalently to the surface of DNA by a chemical reaction between the amino group of DNA and the carboxylic group of the MTX in the presence of EDC (1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide) used as carboxyl activating agent for the coupling of primary amines to form amide bond.
Since the field of DNA nanotechnology is in its infancy, no comparison with other drug delivery systems has been done. Liposomal formulation of doxorubicin (Doxil®), one of a few examples of success in clinic [83-85] represents a gold standard to be compared. Although doxo offers many advantages as cargo, others drugs should be tested to understand whether DNA origami could be applied to different chemotherapeutic treatments. Targeted therapy is now at the forefront of cancer therapy and is based on the application of monoclonal antibodies (e.g. Herceptin, an anti-HER2) or small molecules (e.g. Gifitinib, an anti-EGFR) directed against specific targets. In contrast to doxorubicin, these molecules do not have intercalation properties, thus they should be incorporated on the surface of DNA origami through covalent and non-covalent linkers or be loaded inside a DNA origami cage structure. In the last case, logic gate cages of different shapes and dimension should be designed to entrap the molecules [72]. In this regard, a DNA nanorobot was demonstrated to deliver antibodies against human CD33 and CDw328 to induce growth arrest of natural killer cells as well as antibodies targeting human CD3e and flagellin to induce T cell activation [72]. Other cargos such as siRNAs [65] or specific DNA sequences [66,71] could be loaded to obtain gene-specific downregulation or elicit an immune response, respectively. The data support the DNA nanostructures as multivalent DDS, which could be adapted for any particular requirement.
Stability (Table 1, column 6)
A stability test of the DNA nanostructure used as a drug carrier has been carried out in most the studies. Stability was found to be strictly dependent on the type of solution, that has been utilized and the temperature. Although in most cases the experiments were done at 37 oC corresponding to the physiological temperature of the human body, in a few papers the experiment was carried out at room temperature or the temperature was not reported. With regard to the media, there is less concordance. Cell culture media or fetal bovine serum has been utilized at different concentration or physiological solution such as phosphate buffer (PBS). In this regard, Kocabey et al. reported that degradation depends on Mg2+ concentration, oligonucleotide sequences, salt concentrations and whether structural extensions (e.g. sequences to integrate aptamers) were utilized (N12) [86]. In the light of these observations, a careful analysis should be conducted and actually, it is difficult to draw firm conclusions.
In general, DNA-origami-based structures depict higher stability (more than 24 hours) in physiological buffer as well as in serum. In contrast, non-origami DNA nanostructures exhibit lower stability. The higher density of double helices in DNA-origami nanostructures is likely the main factor contributing to their stability [74]. However, in vivo stability information about DNA nanostructures is still inadequate. Recently, Lee et al. determined that the half-life of siRNAs hybridized to a DNA tetrahedron (1 siRNA/edge) increased the circulation time from 6 to 24 minutes in the blood [65]. These observations showed that the stability of DNA nanostructures is still the critical factor undermining their potential clinical applications. The entrapment of siRNAs inside lipid nanoparticles (LNP) has been useful in overcoming degradation and cell permeability problems related to RNA interference [87]. On the other hand, LNP-siRNA could be entrapped inside the endosomes reducing the efficacy of treatment [88]. Conjugation of siRNAs with polymers could overcome this problem [89]. Nonetheless, the easy custom synthesis of DNA with any arbitrary sequence allows the insertion of modified bases and non-natural chemical modifications at specific positions of the synthetic DNA strands, which could be utilized to tune their in vivo stability along with imparting additional functionalities. The high chemical versatility that allows the easy addition of targeting ligands represents a significant advantage of DNA nanostructures compared to other DDS.
Loading efficiency (Table 1, column 8)
Concerning chemotherapeutic drugs, the loading efficiency is influenced mainly by the DNA origami and drug concentration, the incubation time and the reaction temperature. In a few papers, all these parameters have been reported. We have estimated that the number of doxo molecules per DNA nanostructure was between 26 and more than 200,000 molecules. Although the dimension of the structure and the number of DNA strands are different, the theoretical maximal numbers of doxo molecules that could be intercalated inside the structure does not always correspond to that observed and an overloading of the DNA nanostructure could often be foreseen. Over-saturation of nanostructures is not an ideal condition for in vivo experiments and, may lead to deformation of the nanostructure itself [90], unspecific effects, and altered uptake kinetics and artifacts. Doxo is known to undergo self-association in aqueous solution, which may alter the binding and release properties of doxo from the nanocarrier [91,92].
The loading efficiency was one of the major problems of liposomes. To be used in clinic, the drug/liposome (weight/weight) ratio should be more than 70% to avoid a high concentration of lipids in circulation. An active remote loading was developed that works perfectly with doxo and could be utilized for weak basic or acid amphipathic molecules [93]. Inside the liposome, the high concentration of ammonium sulfate allows the precipitation of doxorubicin. This process produces a stable gradient of doxorubicin between outside and inside the liposome, which allows an efficient drug loading [94]. A hybrid system formed from the encapsulation of DNA nanostructures by a lipid membrane has been suggested to improve the stability of the nanostructures in biological environments, and avoid the activation of inflammatory immune response for the in vivo application. The development of this hybrid system has focused on increasing the bioavailability and targeting of anticancer agents [95].
Release (Table 1, column 9)
Besides physiological stability, another important factor is the control and tuning of the release of the drug cargo from DNA nanostructures. Most of the studies have performed the release tests in PBS at different pH or in cell lysate solutions. It has been observed that there are big differences in the drug release depending on the DNA structures. The DNA tetrahedral, icosahedral and tube release most of the doxorubicin in 10 hours. On the other hand, a DNA triangle, square but also a tube of different sizes (183 nm for N10 and 380 nm for N11) take more than 48 hours to release about only 20% of drug in PBS solutions. In this case, parameters such as different experimental conditions (the release of doxo increased when the pH decreased from 7.4 to 5.5 suggesting that the release of doxo increases when the doxo-intercalated DNA is retained in the acidic tumor region), sequences, salt concentration and structural shape (tetrahedral origami with 9 nm (N1) releases 100% of doxo in 10h, a tube structure of 138 nm in length (N7) releases 80-90% of doxo in 10h and a triangular shape of 150 nm per edge (N10) and (N11) releases 20% of doxo in 48h) and density may influence the results.
Zhao et al. used a very interesting approach to control the release kinetics of doxorubicin from DNA tubes. They designed DNA origami tubes with different global twists through which they were able to tune the encapsulation efficiency and the doxo release rate [70]. They synthesize two types of DNA origami. The first one was a straight nanotube (S-Nano) with 10.5 bases per helical turn of DNA. The second one was a twisted nanotube (T-Nano) with 12 bases per helical turn. The release rate of doxo from the structures was studied by measuring the doxo fluorescence. The T-Nano retains the drug and exhibits a slower release profile (50% of doxo remains bound to the T-Nano after several hours). The encapsulation efficiency of the two structures of DNA showed that the T-Nano encapsulates more doxo per structure than the S-Nano (33% higher for T-Nano than S-Nano). This could be explained by the fact that the T-Nano contains more base pairs per structure (12 bp/turn) and a higher affinity for doxo due to the relaxation of the DNA structure when doxo is intercalated [68].
In vitro activity (Table 1, columns 11 and 12)
All the studies reported the activity of DNA nanostructures on cell culture. Since doxo is the most utilized drug, breast cancer cell lines were chosen. The efficacy of DNA-doxo complex was evaluated in cell viability and cytotoxicity experiments. DNA origami increased the efficacy of doxo and when evaluated for cell internalization, the endocytotic pathway was the principal entry route into the cell.
An in-depth analysis will be necessary to clarify the fate of different DNA nanostructures. Approaches such as RNA interference or chemical inhibitors could be used to assess the mechanism of subcellular redistribution, recycling and excretion of the DNA structure. For example, it has been demonstrated that endocytic recycling restricts siRNA delivery with lipid nanoparticles but little is known about the delivery through DNA nanostructure. Multiple cell signaling effectors are utilized for nanoparticle cellular internalization but gene knockdown effects are almost abrogated by recycling pathways with specific proteins involved in the process [96]. The authors suggested that siRNA delivery efficiency might be improved by designing delivery vehicles that can escape the recycling pathways, an aspect that is not primarily kept in consideration.
In vivo activity (Table 1, columns 13, 14 and 15)
The in vivo application of DNA nanostructures has been limited by the relative structural instability. However, in the few cases tested, DNA nanostructures have been shown to improve the pharmacological effect of their cargo. siRNAs are more stable in the blood ameliorating the knockdown efficiency [65] or CpG DNA sequences can elicit a longer immune response [66]. In regard to chemotherapeutic drugs, DNA tiles and DNA origami have shown increased doxorubicin efficacy in tumor growth assay in xenograft mouse models. Interestingly, all the structures accumulate specifically in the tumor most probably taking advantage of the well-known EPR effect. Among DNA origami, the best activity was obtained by the triangular structure, highlighting that the shape of the structure of DNA origami also has a prominent role in the activity in vivo. Preliminary biodistribution studies have shown that the liver, kidney and spleen are primary organs for DNA nanostructure accumulation [65,74]. Up to now, no studies on pharmacokinetics and pharmacodynamics profiling have been carried out. In our opinion, this point is crucial for understanding the applicability of DNA in the field of therapy. We need to keep in mind that the in vitro efficacy of PEGylated liposomal doxo was two times lower than that of free doxo. The high stability of Doxil® slowly releases the drug in cell culture, which turn out to be an advantage in vivo by increasing the EPR effect in the tumor [97]. Multiple time points after the administration of a drug in animal models allow for complete pharmacokinetic and pharmacodynamics analysis. The parameters typically provided include drug concentration over time, area under the curve (AUC), elimination half-life, clearance, time of maximum concentration, and volume of distribution, which are useful for characterizing the pharmacokinetics properties of the drug associated with the nanocarrier [98]. Moreover, specific drug dosage investigations have to be designed to evaluate the origami effects in vivo over an extended period of time. Dose administration at one or several time points could be used to evaluate overall toxicity in the animal model and the effects on tumor growth.
DNA nanostructures are foreign materials that in theory should be considered as non-self, which could be recognized from the DNA sensing machinery and consequently degraded. DNA is normally confined in the nucleus. When DNA accumulates in the cytoplasm or in endosomes, it is recognized as "anomalous" material. Classical B-form DNA could stimulate immune response through the Toll-like receptor (TLR) pathway. In the endosomes of dendritic cells, TLR9 is activated by anomalous DNA and elicits Type I interferon response. TLR9 preferentially binds unmethylated CpG-rich DNA. These DNA sequences are abundant in numerous pathogen genomes and their binding to the endosomal TLR9 stimulates immune response. In the cytoplasm, cells are embedded with DNases, which degrade foreign DNA [99]. Cyclic GMP-AMP synthase (cGAS) and absent in melanoma 2 (AIM2) are a group of sensors that detect DNA in a sequence-independent manner and are localized in the cytoplasm. There is a third group of DNA sensors localized both in nucleus and cytoplasm including interferon-inducible genes 16 (IFI16), RNA polymerase III and the Mre11-Rad50-Nbs1 (MRN) complex. The first hurdle that DNA meets in the blood stream is a few enzymes capable of degrading it, such as deoxyribonuclease II, phosphodiesterase I, DNA hydrolyzing autoantibodies, and neutral deoxyribonuclease I, which is responsible for more than 90% of deoxyribonuclease activity in blood plasma [100]. DNase I is a secreted protein that is released into the alimentary tract and bloodstream. It acts on single-stranded DNA, double-stranded DNA, and chromatin producing fragments of various lengths [101].
It has been demonstrated that compact structures of DNA present a decreased enzymatic recognition compared to linear DNA. Keum et al. [102] assembled a tetrahedron with edges of about 7 nm containing a centrally located enzyme restriction site, CTNAG (DdeI). It was demonstrated that the tetrahedron structure is less sensitive to DdeI cleavage than the linear DNA presenting the same restriction site. This was probably due to the increased mechanical stability of the ligated DNA. The resistance to non-specific degradation by DNase I was also tested. The tetrahedron structure was digested more slowly than the linear fragment. The explanation of this difference could be attributed to the sensitivity of DNase I activity to local and global helix geometry, which are different between tetrahedral and linear structures. To mimic the physiological condition, the stability of the tetrahedron structure was tested in the presence of 10% FBS. The structure was more resistant to endo- and exo-nuclease cleavage in the serum. The degradation indicated a first-order kinetics and the decay time constant differed by nearly a factor of 50: 0.8 hours for the linear DNA and 42 hours for the tetrahedron. The authors hypothesized that the size is an important feature to confer resistance to the enzymatic degradation, but other types of branched geometries, or curvatures, may also mean protection factors.
To overcome the host immunosurveillance, Perrault et al. developed a new system inspired by the envelope of viral particles. In particular, a DNA octahedron of 50 nm was encapsulated in a lipid bilayer in order to mimic virus like particle. The octahedron structures are composed of bundles of six long double helices (28 nm) engineered with 90° curvatures. The DNA nanostructures could be functionalized by modifying the oligonucleotide structures with high precision. The lipid bilayer was directly assembled around the DNA octahedron, recruited by individual lipid-conjugated oligonucleotides preassembled onto the outer handles. Internal and external diameters were 53 and 76 nm, respectively [95]. In vitro experiments demonstrated that encapsulated DNA drastically decreased the inflammatory response. The production of inflammatory cytokines IL-6 and IL-12 was evident only when the cells were treated with DNA octahedron but not when the structure was encapsulated. These results outlined that a three-stage DDS (Membrane-DNA-Drug) could increase the circulation half-life of the DNA nanostructures in the blood and finely control the drug release. In addition, the outer membrane could be engineered by adding specific molecules such as receptors, ligands or antibodies in order to target specific cells [95].
It will be a key step to understand the interaction between DNA sensing machinery and DNA origami to increase the performance as a delivery system. As for siRNA delivery with lipid nanoparticles [96] in which the recycling pathways are a limiting step for an efficient delivery, DNA immunosurveillance could play a fundamental role in the success of the DNA origami delivery system.
Concluding remarks and outlook
The discipline of DNA nanotechnology is founded on the unique and robust self-assembling properties of DNA through canonical complementary base pair interactions, which allow the programmable design of DNA nanostructures with the required anticipated geometry and functional properties. The modulation of size, shape and charge of DNA nanostructures has been shown to overcome the natural cell membrane barrier allowing the delivery of naked DNA or siRNA that otherwise would not enter the cells. DNA nanostructures could be easily programmed to be a smart container of different cargos of biomedical interest. Many circulating enzymes such as DNase represent a limitation of low-dense DNA nanostructures that especially in tumor cells are overrepresented. Dense packaging of DNA helices within a DNA nanostructure is one strategy that could increase their stability against DNA-degrading enzymes. Another approach could be to encapsulate DNA structures under a sheet of biocompatible materials like membranes, which will protect them from unspecific degradation [95,103]. From a therapeutic point-of-view, considering the elimination and terminal clearance half-life of most chemotherapeutic drugs, the in vivo half-life of DNA-based nanostructures should be higher than 30 hours to enable their feasible clinical applications in drug delivery and formulation. For instance, the terminal clearance half-life of doxo, which is among the most widely used chemotherapeutic agents, is around 30 hours [104].
Another advantage of DNA origami could be represented by the ability to actively release multiple drugs. In the more mature field of liposomes, many research groups are dedicating much effort to loading different drugs inside a single liposome, especially for cancer therapy. As compared to DNA origami, the charge, hydrophobicity and pKa of the drug represent an important limitation for the loading of the liposome. The presence of a huge number of modifiable oligonucleotides ideally represents an advantage and easy way to load multiple drugs. Tweezer-like DNA nanodevices are able to control the activity of an external enzyme and represent an earlier system to be implemented to design a DNA nanomachine with intrinsic ability to actively release drugs [105].
The above studies showed that DNA nanotechnology could provide an excellent designing tool for the construction of novel drug delivery vehicles possessing optimum biocompatibility along with high cellular uptake kinetics of drug-loaded DNA nanostructures. DNA nanotechnology enables tuning and optimization for their best performance in in vivo and in vitro experiments by judiciously selecting the shape, size and functionalities of DNA nanostructures owing to the shape and size-dependent EPR effect. However, before in vivo applications of any perspective DNA nanostructure, a very careful and rigorous in vitro testing is required to avoid any misinterpretation or artifacts in the data [86]. In addition, the cost of production, the yield and purification steps is still an obstacle. Many laboratories are working in this direction and some improvements were recently achieved [46,105,106].
Nevertheless, the research area of DNA nanotechnology for drug delivery is still in the initial stage and a breakthrough is still needed in DNA nanotechnology, that could pave the way for the development of smart functional DNA origami for biomedical applications and drug delivery, and in the long term find a way to be translated into clinics to improve the health of patients.
Abbreviations
AFM: atomic force microscopy; AGE: agarose gel electrophoresis; AuNPs: gold nanoparticles; CdSe/ZnS: cadmium selenide/zinc sulfide; CPG: cytosine-phosphate Guanosine; DDSs: drug delivery systems; DLS: dynamic light scattering; DNA: deoxyribonucleic acid; Doxo, DOX: doxorubicin; dsDNA: double stranded DNA; DX, DAE, DAO: double crossover; FBS: fetal bovine serum; FPLC: fast protein liquid chromatography; LNP: lipid nanoparticles; PAGE: polyacrylamide gel electrophoresis; PBS: phosphate buffer saline; PEG: Polyethylene glycol; PNA-SPDP: peptide nucleic acid-N-succinimidyl-3-(2-pyridythio) propionate; QDs: quantum dots; qPCR: Quantitative PCR; RNA: ribonucleic acid; siRNA: small interfering RNA; ssDNA: single stranded DNA; TEM: transmission electron microscopy; TLR: toll-like receptor; TX, TAO: triple crossover
Acknowledgements
Authors are thankful to My First AIRC (No. 1569), AIRC Special Program Molecular Clinical Oncology, 5x1000, (No. 12214), Italian Ministry of Education MIUR (FIRB prot. RBAP11ETKA) for funding.
Funding Sources
My First AIRC (No. 1569)
AIRC Special Program Molecular Clinical Oncology, 5x1000, (No. 12214)
Italian Ministry of Education MIUR (FIRB prot. RBAP11ETKA)
Competing Interests
The authors have declared that no competing interest exists.
References
1. Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771-82
2. De Jong WH, Borm PJA. Drug delivery and nanoparticles:applications and hazards. Int J Nanomedicine. 2008;3:133-49
3. Buzea C, Pacheco II, Robbie K. Nanomaterials and nanoparticles: sources and toxicity. Biointerphases. 2007;2:MR17-71
4. Kumar V, Toffoli G, Rizzolio F. Fluorescent carbon nanoparticles in medicine for cancer therapy. ACS Med Chem Lett. 2013;4:1012-3
5. Li J, Fan C, Pei H. et al. Smart drug delivery nanocarriers with self-assembled DNA nanostructures. Adv Mater. 2013;25:4386-96
6. Linko V, Ora A, Kostiainen MA. DNA Nanostructures as Smart Drug-Delivery Vehicles and Molecular Devices. Trends Biotechnol. 2015;33:586-94
7. Surana S, Shenoy AR, Krishnan Y. Designing DNA nanodevices for compatibility with the immune system of higher organisms. Nat Nanotechnol. 2015;10:741-7
8. Chen YJ, Groves B, Muscat RA. et al. DNA nanotechnology from the test tube to the cell. Nat Nanotechnol. 2015;10:748-60
9. Chao J, Liu H, Su S. et al. Structural DNA nanotechnology for intelligent drug delivery. Small. 2014;10:4626-35
10. Zhang F, Nangreave J, Liu Y. et al. Structural DNA nanotechnology: state of the art and future perspective. J Am Chem Soc. 2014;136:11198-211
11. Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discov. 2003;2:347-60
12. Tanner P, Baumann P, Enea R. et al. Polymeric vesicles: from drug carriers to nanoreactors and artificial organelles. Acc Chem Res. 2011;44:1039-49
13. Kakizawa Y, Kataoka K. Block copolymer micelles for delivery of gene and related compounds. Adv Drug Deliv Rev. 2002;54:203-22
14. Ganta S, Devalapally H, Shahiwala A. et al. A review of stimuli-responsive nanocarriers for drug and gene delivery. J Control Release. 2008;126:187-204
15. Charoenphol P, Bermudez H. Aptamer-Targeted DNA Nanostructures for Therapeutic Delivery. Mol Pharm. 2014;11:1721-5
16. Reif JH, Chandran H, Gopalkrishnan N. et al. CRCnetBASE - Self-Assembled DNA Nanostructures and DNA Devices. Nanofabrication Handb. 2012
17. Jones MR, Seeman NC, Mirkin CA. Nanomaterials. Programmable materials and the nature of the DNA bond. Science. 2015;347:1260901-11
18. Saccà B, Niemeyer CM. DNA origami: the art of folding DNA. Angew Chem Int Ed Engl. 2012;51:58-66
19. Pinheiro AV, Han D, Shih WM, Yan H. Challenges and opportunities for structural DNA nanotechnology. Nat Nanotechnol. 2011;6:763-72
20. Hagerman PJ. Flexibility of DNA. Annu Rev Biophys Biophys Chem. 1988;17:265-86
21. Vologodskii A. Biophysics of DNA. Cambridge Univ Press. 2015:86-8
22. Fundamentals of Biochemistry: Biochemical Interactions. Voet D, Voet JG, Pratt CW. http://www.amazon.com/Fundamentals-Biochemistry-Biochemical-Interactions-CD-ROM/dp/047132714X
23. Protozanova E, Yakovchuk P, Frank-Kamenetskii MD. Stacked-unstacked equilibrium at the nick site of DNA. J Mol Biol. 2004;342:775-85
24. Seeman NC. DNA in a material world. Nature. 2003;421:427-31
25. Seeman NC. At the Crossroads of Chemistry, Biology, and Materials. Chem Biol. 2003;10:1151-9
26. Seeman NC. An overview of structural DNA nanotechnology. Mol Biotechnol. 2007;37:246-57
27. Gothelf KV, LaBean TH. DNA-programmed assembly of nanostructures. Org Biomol Chem. 2005;3:4023-37
28. Seeman NC. Nucleic acid junctions and lattices. J Theor Biol. 1982;99:237-47
29. Andersen ES, Dong M, Nielsen MM. et al. Self-assembly of a nanoscale DNA box with a controllable lid. Nature. 2009;459:73-6
30. Rothemund PWK. Folding DNA to create nanoscale shapes and patterns. Nature. 2006;440:297-302
31. Douglas SM, Marblestone AH, Teerapittayanon S. et al. Rapid prototyping of 3D DNA-origami shapes with caDNAno. Nucleic Acids Res. 2009;37:5001-6
32. Douglas SM, Dietz H, Liedl T. et al. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature. 2009;459:414-8
33. Han D, Pal S, Nangreave J. et al. DNA origami with complex curvatures in three-dimensional space. Science. 2011;332:342-6
34. Dietz H, Douglas SM, Shih WM. Folding DNA into twisted and curved nanoscale shapes. Science. 2009;325:725-30
35. Han D, Pal S, Yang Y. et al. DNA gridiron nanostructures based on four-arm junctions. Science. 2013;339:1412-5
36. Ke Y, Ong LL, Shih WM. et al. Three-dimensional structures self-assembled from DNA bricks. Science. 2012;338:1177-83
37. Wei B, Dai M, Yin P. Complex shapes self-assembled from single-stranded DNA tiles. Nature. 2012;485:623-6
38. Benson E, Mohammed A, Gardell J. et al. DNA rendering of polyhedral meshes at the nanoscale. Nature. 2015;523:441-4
39. Linko V, Dietz H. The enabled state of DNA nanotechnology. Curr Opin Biotechnol. 2013;24:555-61
40. Castro CE, Kilchherr F, Kim DN. et al. A primer to scaffolded DNA origami. Nat Methods. 2011;8:221-9
41. Pan K, Kim DN, Zhang F. et al. Lattice-free prediction of three-dimensional structure of programmed DNA assemblies. Nat Commun. 2014;5:5578
42. Kim DN, Kilchherr F, Dietz H. et al. Quantitative prediction of 3D solution shape and flexibility of nucleic acid nanostructures. Nucleic Acids Res. 2011;40:2862-8
43. Ducani C, Kaul C, Moche M. et al. Enzymatic production of “monoclonal stoichiometric” single-stranded DNA oligonucleotides. Nat Methods. 2013;10:647-52
44. Yang Y, Han D, Nangreave J. et al. DNA origami with double-stranded DNA as a unified scaffold. ACS Nano. 2012;6:8209-15
45. Martin TG, Dietz H. Magnesium-free self-assembly of multi-layer DNA objects. Nat Commun. 2012;3:1103
46. Sobczak JPJ, Martin TG, Gerling T. et al. Rapid folding of DNA into nanoscale shapes at constant temperature. Science. 2012;338:1458-61
47. Bellot G, McClintock MA, Lin C. et al. Recovery of intact DNA nanostructures after agarose gel-based separation. Nat Methods. 2011;8:192-4
48. Stahl E, Martin TG, Praetorius F. et al. Facile and Scalable Preparation of Pure and Dense DNA Origami Solutions. Angew Chem Int Ed. 2014;53:12735-40
49. Shaw A, Benson E, Högberg B. Purification of functionalized DNA origami nanostructures. ACS Nano. 2015;9:4968-75
50. Wickham SFJ, Endo M, Katsuda Y. et al. Direct observation of stepwise movement of a synthetic molecular transporter. Nat Nanotechnol. 2011;6:166-9
51. Lin C, Perrault SD, Kwak M. et al. Purification of DNA-origami nanostructures by rate-zonal centrifugation. Nucleic Acids Res. 2013;41:e40
52. Alloyeau D, Ding B, Ramasse Q. et al. Direct imaging and chemical analysis of unstained DNA origami performed with a transmission electron microscope. Chem Commun. (Camb). 2011;47:9375-7
53. Zhang C, Su M, He Y. et al. Conformational flexibility facilitates self-assembly of complex DNA nanostructures. Proc Natl Acad Sci. U. S. A. 2008;105:10665-9
54. Yan H, Park SH, Finkelstein G. et al. DNA-templated self-assembly of protein arrays and highly conductive nanowires. Science. 2003;301:1882-4
55. Rothemund PWK, Ekani-Nkodo A, Papadakis N. et al. Design and characterization of programmable DNA nanotubes. J Am Chem Soc. 2004;126:16344-52
56. Liu D, Park SH, Reif JH. et al. DNA nanotubes self-assembled from triple-crossover tiles as templates for conductive nanowires. Proc Natl Acad Sci. U. S. A. 2004;101:717-22
57. Mathieu F, Liao S, Kopatsch J. et al. Six-helix bundles designed from DNA. Nano Lett. 2005;5:661-5
58. Walsh AS, Yin H, Erben CM. et al. DNA cage delivery to mammalian cells. ACS Nano. 2011;5:5427-32
59. Li J, Pei H, Zhu B. et al. Self-assembled multivalent DNA nanostructures for noninvasive intracellular delivery of immunostimulatory CpG oligonucleotides. ACS Nano. 2011;5:8783-9
60. Okholm AH, Nielsen JS, Vinther M. et al. Quantification of cellular uptake of DNA nanostructures by qPCR. Methods. 2014;67:193-7
61. Mikkilä J, Eskelinen AP, Niemelä EH. et al. Virus-Encapsulated DNA Origami Nanostructures for Cellular Delivery. Nano Lett. 2014;14:2196-200
62. Brglez J, Nikolov P, Angelin A. et al. Designed Intercalators for Modification of DNA Origami Surface Properties. Chem Eur J. 2015;21:9440-6
63. Kim KR, Kim DR, Lee T. et al. Drug delivery by a self-assembled DNA tetrahedron for overcoming drug resistance in breast cancer cells. Chem Commun. (Camb). 2013;49:2010-2
64. Chang M, Yang CS, Huang DM. Aptamer-conjugated DNA icosahedral nanoparticles as a carrier of doxorubicin for cancer therapy. ACS Nano. 2011;5:6156-63
65. Lee H, Lytton-Jean AKR, Chen Y. et al. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat Nanotechnol. 2012;7:389-93
66. Liu X, Xu Y, Yu T. et al. A DNA nanostructure platform for directed assembly of synthetic vaccines. Nano Lett. 2012;12:4254-9
67. Zhu G, Zheng J, Song E. et al. Self-assembled, aptamer-tethered DNA nanotrains for targeted transport of molecular drugs in cancer theranostics. Proc Natl Acad Sci. U. S. A. 2013;110:7998-8003
68. Kumar V, Bayda S, Hadla M. et al. Enhanced Chemotherapeutic Behavior of Open-Caged DNA@Doxorubicin Nanostructures for Cancer Cells. J Cell Physiol. 2016;231:106-10
69. Mei Q, Wei X, Su F. et al. Stability of DNA origami nanoarrays in cell lysate. Nano Lett. 2011;11:1477-82
70. Zhao YX, Shaw A, Zeng X. et al. DNA origami delivery system for cancer therapy with tunable release properties. ACS Nano. 2012;6:8684-91
71. Schüller VJ, Heidegger S, Sandholzer N. et al. Cellular immunostimulation by CpG-sequence-coated DNA origami structures. ACS Nano. 2011;5:9696-702
72. Douglas SM, Bachelet I, Church GM. A logic-gated nanorobot for targeted transport of molecular payloads. Science. 2012;335:831-4
73. Jiang Q, Song C, Nangreave J. et al. DNA origami as a carrier for circumvention of drug resistance. J Am Chem Soc. 2012;134:13396-403
74. Zhang Q, Jiang Q, Li N. et al. DNA origami as an in vivo drug delivery vehicle for cancer therapy. ACS Nano. 2014;8:6633-43
75. Srinivasan C, Lee J, Papadimitrakopoulos F. et al. Labeling and intracellular tracking of functionally active plasmid DNA with semiconductor quantum dots. Mol Ther. 2006;14:192-201
76. Levy M, Cater SF, Ellington AD. Quantum-dot aptamer beacons for the detection of proteins. Chembiochem. 2005;6:2163-6
77. Niemeyer CM, Bürger W, Peplies J. Covalent DNA-Streptavidin Conjugates as Building Blocks for Novel Biometallic Nanostructures. Angew Chem Int Ed. 1998;37:2265-8
78. Wang G, Murray RW. Controlled Assembly of Monolayer-Protected Gold Clusters by Dissolved DNA. Nano Lett. 2004;4:95-101
79. Pellegrino T, Sperling RA, Alivisatos AP. et al. Gel electrophoresis of gold-DNA nanoconjugates. J Biomed Biotechnol. 2007;2007:26796
80. Halley PD, Lucas CR, McWilliams EM. et al. Daunorubicin-Loaded DNA Origami Nanostructures Circumvent Drug-Resistance Mechanisms in a Leukemia Model. Small. 2016;12:308-20
81. N'soukpoé-Kossi CN, Descôteaux C, Asselin E. et al. DNA interaction with novel antitumor estradiol-platinum(II) hybrid molecule: a comparative study with cisplatin drug. DNA Cell Biol. 2008;27:101-7
82. Wang Y, Gao S, Ye WH. et al. Co-delivery of drugs and DNA from cationic core-shell nanoparticles self-assembled from a biodegradable copolymer. Nat Mater. 2006;5:791-6
83. Nellis DF, Giardina SL, Janini GM. et al. Preclinical manufacture of anti-HER2 liposome-inserting, scFv-PEG-lipid conjugate. 2. Conjugate micelle identity, purity, stability, and potency analysis. Biotechnol Prog. 2005;21:221-32
84. Laginha KM, Moase EH, Yu N. et al. Bioavailability and therapeutic efficacy of HER2 scFv-targeted liposomal doxorubicin in a murine model of HER2-overexpressing breast cancer. J Drug Target. 2008;16:605-10
85. Puri A, Loomis K, Smith B. et al. Lipid-based nanoparticles as pharmaceutical drug carriers: from concepts to clinic. Crit Rev Ther Drug Carrier Syst. 2009;26:523-80
86. Kocabey S, Meinl H, MacPherson I. et al. Cellular Uptake of Tile-Assembled DNA Nanotubes. Nanomaterials. 2014;5:47-60
87. Coelho T, Adams D, Silva A. et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369:819-29
88. Gilleron J, Querbes W, Zeigerer A. et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. 2013;31:638-46
89. Wooddell CI, Rozema DB, Hossbach M. et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol Ther. 2013;21:973-85
90. Ke Y, Bellot G, Voigt NV. et al. Two design strategies for enhancement of multilayer-DNA-origami folding: underwinding for specific intercalator rescue and staple-break positioning. Chem Sci. 2012;3:2587-97
91. Agrawal P, Barthwal SK, Barthwal R. Studies on self-aggregation of anthracycline drugs by restrained molecular dynamics approach using nuclear magnetic resonance spectroscopy supported by absorption, fluorescence, diffusion ordered spectroscopy and mass spectrometry. Eur J Med Chem. 2009;44:1437-51
92. Karukstis KK, Thompson EH, Whiles JA. et al. Deciphering the fluorescence signature of daunomycin and doxorubicin. Biophys Chem. 1998;73:249-63
93. Haran G, Cohen R, Bar LK. et al. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151:201-15
94. Fritze A, Hens F, Kimpfler A. et al. Remote loading of doxorubicin into liposomes driven by a transmembrane phosphate gradient. Biochim Biophys Acta. 2006;1758:1633-40
95. Perrault SD, Shih WM. Virus-inspired membrane encapsulation of DNA nanostructures to achieve in vivo stability. ACS Nano. 2014;8:5132-40
96. Sahay G, Querbes W, Alabi C. et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat Biotechnol. 2013;31:653-8
97. Hong RL, Huang CJ, Tseng YL. et al. Direct comparison of liposomal doxorubicin with or without polyethylene glycol coating in C-26 tumor-bearing mice: is surface coating with polyethylene glycol beneficial? Clin Cancer Res. 1999;5:3645-52
98. Marcato PD. Pharmacokinetics and pharmacodynamics of Nanomaterials. Durán N, Guterres SS, Alves OL.(Eds). Nanotoxicology Materials, Methodologies and assessments, New York, USA: Springer New York. 2014:97-110
99. Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013;38:870-80
100. Tamkovich SN, Cherepanova AV, Kolesnikova EV. et al. Circulating DNA and DNase activity in human blood. Ann N Y Acad Sci. 2006;1075:191-6
101. Samejima K, Earnshaw WC. Trashing the genome: the role of nucleases during apoptosis. Nat Rev Mol Cell Biol. 2005;6:677-88
102. Keum JW, Bermudez H. Enhanced resistance of DNA nanostructures to enzymatic digestion. Chem Commun. (Camb). 2009;45:7036-8
103. Toffoli G, Hadla M, Corona G. et al. Exosomal doxorubicin reduces the cardiac toxicity of doxorubicin. Nanomedicine (Lond). 2015 [Epub ahead of print]
104. Greene RF, Collins JM, Jenkins JF. et al. Plasma pharmacokinetics of adriamycin and adriamycinol: implications for the design of in vitro experiments and treatment protocols. Cancer Res. 1983;43:3417-21
105. Liu M, Fu J, Hejesen C. et al. A DNA tweezer-actuated enzyme nanoreactor. Nat Commun. 2013;4:2127
106. Fu Y, Zeng D, Chao J. et al. Single-step rapid assembly of DNA origami nanostructures for addressable nanoscale bioreactors. J Am Chem Soc. 2013;135:696-702
Author contact
![]() Corresponding author: Flavio Rizzolio, PhD. Clinical Pharmacology, Department of Molecular Biology and Translational Research, National Cancer Institute and Center for Molecular Biomedicine, CRO Aviano (PN), Via Franco Gallini, 2, Aviano 33081 - PN - Italy. Phone. +39-0434 659384 Fax. +39-0434 659799 E-mail: frizzolioit.
Corresponding author: Flavio Rizzolio, PhD. Clinical Pharmacology, Department of Molecular Biology and Translational Research, National Cancer Institute and Center for Molecular Biomedicine, CRO Aviano (PN), Via Franco Gallini, 2, Aviano 33081 - PN - Italy. Phone. +39-0434 659384 Fax. +39-0434 659799 E-mail: frizzolioit.