Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results and discussion
Conclusion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(6):808-816. doi:10.7150/thno.13826 This issue Cite
Research Paper
Synthesis and Preclinical Evaluation of a Highly Improved Anticancer Prodrug Activated by Histone Deacetylases and Cathepsin L
Nobuhide Ueki1, ![]() , Wei Wang2, Cooper Swenson1, Caroline McNaughton1, Nicole S. Sampson2, Michael J. Hayman1
, Wei Wang2, Cooper Swenson1, Caroline McNaughton1, Nicole S. Sampson2, Michael J. Hayman1
1. Department of Molecular Genetics & Microbiology, Stony Brook University, Stony Brook, New York, USA;
2. Department of Chemistry, Stony Brook University, Stony Brook, New York, USA.
Received 2015-9-12; Accepted 2016-2-24; Published 2016-3-28
Abstract

Lack of absolute selectivity against cancer cells is a major limitation for current cancer therapies. In the previous study, we developed a prodrug strategy for selective cancer therapy using a masked cytotoxic agent puromycin [Boc-Lys(Ac)-Puromycin], which can be sequentially activated by histone deacetylases (HDACs) and cathepsin L (CTSL) to kill cancer cells expressing high levels of both enzymes. Despite the promise as a selective cancer therapy, its requirement of relatively high dosage could be a potential issue in the clinical setting. To address this issue, we aimed to further improve the overall efficacy of our prodrug strategy. Since the proteolytic cleavage by CTSL is the rate-limiting step for the drug activation, we sought to improve the substrate structure for CTSL activity by modifying the α-amino protecting group of lysine. Here we show that protection with Fmoc [Fmoc-Lys(Ac)-Puromycin] exhibits a marked improvement in overall anticancer efficacy compared to the original Boc-Lys(Ac)-Puromycin and this is mainly due to the highly efficient cellular uptake besides its improved substrate structure. Furthermore, to address a concern that the improved drug efficacy might direct high toxicity to the normal cells, we confirmed that Fmoc-Lys(Ac)-Puromycin still retains excellent cancer selectivity in vitro and no obvious systemic off-target toxicity in vivo. Thus our preclinical evaluation data presented here demonstrate that the Fmoc-Lys(Ac)-Puromycin exhibits substantially improved anticancer efficacy, further supporting our approach for the selective cancer therapy.
Keywords: targeted therapy, prodrug, histone deacetylase, cathepsin L, drug development.
Introduction
A major limitation of current cancer therapies is their low selectivity against tumor cells. Most chemotherapeutic agents affect functions that are common to both tumor and normal cells, resulting in limited therapeutic efficacy due to their adverse effects. Despite recent advances in immunotherapy and targeted therapy [1], highly selective drugs are still urgently needed to eliminate cancer cells while sparing healthy tissue.
During cancer development and progression, tumor cells undergo abnormal changes and acquire abilities to grow and survive by taking advantage of dysregulated cellular systems recognized as phenotypic hallmarks of cancer [2-4]. Extensive research efforts have sought cancer specific target molecules that are more essential for the growth and survival of cancer cells than normal cells. Among those potential target molecules, we have been focusing on histone deacetylases (HDACs) and cathepsin L (CTSL), both of which are considered as valid targets for cancer therapy [5]. HDACs are key enzymes involved in the regulation of histone and non-histone proteins [6]. Increased levels of HDACs in tumor cells are known to be closely associated with tumor initiation, progression and metastasis [7-9]. The therapeutic potential of small molecule HDAC inhibitors (HDACi) has been established as a new class of anticancer drug [10-12], showing potent and highly selective anticancer effects [12-14]. Similarly the lysosomal cysteine protease CTSL plays key roles at multiple stages of tumor progression and metastasis [15-17]. Evidence indicates that upregulation of CTSL is a hallmark of metastatic cancers [15-18]. CTSL acts as an endopeptidase, which can modify and modulate many intracellular and extracellular proteins by proteolytic cleavage in the tumor microenvironment [19]. A number of studies using CTSL inhibitors show a decrease in invasiveness and metastasis in various types of cancer, demonstrating their therapeutic potential [20, 21].
Although many protease-activated prodrugs have been developed [22], stability and selectivity are key factors limiting their clinical efficacy. In the previous study, we developed a prodrug strategy for selective cancer therapy using a masked cytotoxic agent (puromycin, which can be sequentially activated by HDACs and CTSL) to target cancer cells expressing high levels of both enzymes [5]. By introducing an ε-acetylated lysine as the first requirement, we demonstrated that the prodrug is well-protected from proteolytic cleavage until it is deacetylated by the intracellular HDACs, and thus enables highly selective activation in tumor tissues. In the present study, we describe the modification of the masking group for the overall improvement of the prodrug and its preclinical evaluation including assessment of anticancer efficacy and safety.
Materials and methods
Materials
Boc-Lys-AMC, Boc-Lys(Ac)-AMC were purchased from Bachem. Z-Lys-AMC, Fmoc-Lys-AMC, Ac-Lys-AMC, Fmoc-Lys(Ac)-AMC were purchased from China Peptide (Shanghai, China). TSA (Trichostatin A), Kollisolve PEG E 400 (PEG400) and Tween 80 were purchased from Sigma-Aldrich. Z-FY-CHO (Cathepsin L Inhibitor II) was purchased from Calbiochem.
Cell culture
Colon cancer (Caco-2, HT29, HCT116) and normal colon CCD841-CoN cells were maintained in Iscove's Modified Dulbecco's Medium (IMDM, Invitrogen) supplemented with 10% fetal bovine serum and 100 U/mL penicillin/streptomycin at 37 °C with 5 % CO2 atmosphere as described previously [5]. All cell lines were routinely tested for mycoplasma contamination.
Enzymatic assays
Live cell CTSL assay was performed as follows. The medium of the overnight culture seeded from 6 x 104 cells in 100 μL growth medium on 96-well tissue culture plate was replaced by the fresh culture medium containing either 25 μM fluorescent substrate Boc-Lys-AMC, Z-Lys-AMC, Fmoc-Lys-AMC or Ac-Lys-AMC in the presence of DMSO or 100 μM Z-FY-CHO, followed by 20 h incubation at 37 °C with 5 % CO2 atmosphere. Then the fluorescent signal of AMC was measured by SpectraMax M5 microplate reader (Molecular Devices) at Ex/Em=355nm/460nm. The reaction time period (20 h) was within a range of linear progression (Fig. S1). Data were obtained from duplicate measurements. Experiments were repeated at least three times. Live cell enzymatic assay using the substrate Fmoc-Lys(Ac)-AMC was performed as follows. The medium of the overnight culture was replaced by the fresh growth medium containing 25 μM Fmoc-Lys(Ac)-AMC and 0.05 % PEG400/Tween80 either in the presence of DMSO, 1 μM TSA or 100 μM Z-FY-CHO, followed by 20 h incubation at 37 °C with 5 % CO2 atmosphere. Then the fluorescent signal of AMC was measured. Data were obtained from duplicate measurements. Experiments were repeated at least three times.
Synthesis of Fmoc-Lys(Ac)-Puromycin
Synthesis of Fmoc-Lys(Ac)-Puromycin is described in Supplementary material.
Cell viability assays
Cell viability was determined by the crystal violet assay. Either 1 μL of vehicle (0.1 % PEG400/Tween80 in saline) containing equivalent amount of DMSO or variable concentrations of Fmoc-Lys(Ac)-Puromycin were added to the overnight culture seeded at 5 x 104 cells per well in 100 μL growth medium on 96-well tissue culture plate, followed by 48 h incubation at 37 °C with 5 % CO2 atmosphere. After the treatment, cells were fixed by 4 % paraformaldehyde solution in PBS. Then crystal violet solution (0.5 % crystal violet in 20 % methanol/water) was added into each well followed by 15-m incubation. The excess crystal violet solution was washed away with distilled water and the remaining crystals were dissolved in Sorenson's buffer (0.1 M sodium citrate in 50 % ethanol/water pH 4.2). Viability was determined by absorbance at 540 nm wavelength using SpectraMax M5 microplate reader. Percent cell viability was expressed relative to the wells containing cells treated with vehicle alone. Data were obtained from triplicate measurements. The IC50 values (the concentration resulting in 50 % inhibition) of Fmoc-Lys(Ac)-Puromycin were determined by dose-response curve analysis (GraphPad Prism software). Experiments were repeated at least three times.
Immunological detection of puromycin-labled proteins
Cells were treated with either vehicle control, Fmoc-Lys(Ac)-Puromycin (0.58 μM), or puromycin (1.1 μM) for 20 h. Cultured cells were sonicated in RIPA lysis buffer (1 % NP-40, 0.1 % SDS, 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5 % sodium deoxycholate, 1 mM EDTA) supplemented with PMSF and protease inhibitor cocktail (Roche) on ice followed by clarification. Protein lysates were separated on sodium dodecyl sulfate-polyacrylamide gels and transferred onto nitrocellulose membranes (Whatman). The primary antibodies used were anti-puromycin (1:5000 dilution, 3RH11, KeraFAST) and anti-α-tubulin (1:4000 dilution, Sigma-Aldrich). Proteins of interest were detected with anti-mouse secondary antibody by chemiluminescence (ECL Kit, GE Healthcare Life Sciences). For flow cytometry, HCT116 cells were untreated or treated with either Boc-Lys(Ac)-Puromycin (54 μM), Fmoc-Lys(Ac)-Puromycin (0.58 μM), or puromycin (1.1 μM) for 20 h, then lifted with Accutase (Sigma-Aldrich), stained with either Alexa Fluor® 647-conjugated anti-puromycin antibody (12D10, EMD Millipore) or appropriate isotype control antibody and analyzed using FACSCalibur with CellQuest software (BD Bioscienses).
In vivo studies
All in vivo experiments were conducted in accordance with the guidelines and protocol approved by the Institutional Animal Care and Use Committee (IACUC) of Stony Brook University. For tumor xenograft study, athymic mice (Foxn1nu, female, age 7 weeks, Harlan) were subcutaneously injected with 5 x 105 cells (HCT116) into the lower flank, then treatment was initiated when small palpable tumors had developed (> 3 mm in diameter). Fmoc-KAc-Puro was first dissolved in DMSO as stock solution (20 mg/ml) and left at room temperature for 5 days. This incubation allows Fmoc-KAc-Puro to completely dissolve in the aqueous vehicle (0.1 % PEG400/Tween80 in saline). The stability and integrity of the prodrug was verified by TLC and LCMS (Supplementary material). Either Fmoc-KAc-Puro or vehicle control containing equivalent amount of DMSO was daily administered intraperitoneally for 12 d. Tumor volumes and body weight were monitored daily. Tumor volume was estimated by the equation vol = (a x b2)/2, where vol, a, and b represent volume (mm3), the length of the major axis (mm), and the length of the minor axis (mm), respectively. Data were expressed as mean ± s.d. For drug toxicity study, Swiss-Webster mice (female, age 7 weeks, Taconic) were treated daily for 10 d with either Fmoc-KAc-Puro or vehicle control (0.1 % PEG400/Tween80 in saline containing equivalent amount of DMSO) via intraperitoneal route. Then blood samples were collected and analyzed for blood chemistry and complete blood count test (ANTECH DIAGNOSTICS, Lake Success, NY).
Statistical analyses
For the in vivo experiments, a two-tailed student's t test was used to calculate statistical significance of the observed differences (GraphPad Prism software). P < 0.05 was considered statistically significant.
Results and discussion
Modification of the masking group for the improved prodrug
Although the original prodrug Boc-Lys(Ac)-Puromycin show promise as a selective cancer therapy [5], its requirement of relatively high dosage could be a potential issue for the clinical applications. To address this issue and move our concept forward, we aimed to improve the rate-limiting step of prodrug activation by CTSL (Fig. 1). The kinetics of proteolytic cleavage of fluorogenic substrate Boc-Lys-AMC by CTSL is very poor [5], thus there is still much room for improvement. Substrate recognition and kinetic parameters of cleavage by structurally similar lysosomal proteases are known to be affected by α-amino protecting groups [23, 24]. Therefore we explored to modify the α-amino protecting group of lysine for the improvement of proteolytic cleavage. Modified substrates Z-Lys-AMC, Fmoc-Lys-AMC and Ac-Lys-AMC, where Z (benzyloxycarbonyl), Fmoc (9-fluorenylmethoxycarbonyl), and Ac (acetyl) represent α-amino protecting groups, were evaluated using Boc-Lys-AMC as a standard. We compared cleavage of the substrates by CTSL in colon cancer HCT116 and normal colon CCD841-CoN cells. The result clearly showed different levels of activation (Fig. 2A). The conjugation of Fmoc moderately increased the activity (up to 70 %) compared to Boc-Lys-AMC, whereas Ac completely disabled the cleavage. Modification to Z showed intermediate behavior between Fmoc and Boc. Only the cancer cell line HCT116 selectively expressed protease activity efficiently cleaving these substrates. Addition of the CTSL inhibitor Z-FY-CHO confirmed the specificity that the major protease responsible was CTSL. Both Fmoc and Z modifications fairly maintained the specificity against CTSL. There was no apparent cytotoxicity observed from the released by-products including Fmoc-Lys-OH as judged by the cell morphology (Fig. S2). Our results clearly indicate that CTSL prefers bulky aromatic structures at the α-amino protecting group of lysine (Fmoc>Z>Boc). Interestingly, this is consistent with the reported substrate specificity profile of human CTSL, which showed a preference for bulky aromatic residues (phenylalanine, tryptophan, tyrosine) over small aliphatic amino acids (valine, leucine) at the P2 position [25]. Further extensive chemical screening could find more optimized structures for the α-amino protecting group of lysine recognized by CTSL.
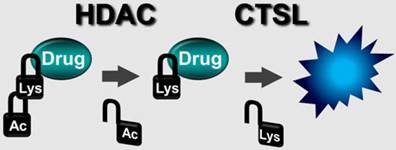
Two-step prodrug activation by HDAC and CTSL for selective cancer targeting.
Effect of the α-amino protecting group modification on Lys-AMC for the proteolytic cleavage by CTSL. (A) Comparative CTSL assay using indicated Lys-AMC substrates either with vehicle control DMSO or CTSL inhibitor Z-FY-CHO (100 μM). (B) Verification of the substrate Fmoc-Lys(Ac)-AMC for the enzyme dependency on HDACs and CTSL and cancer selectivity. The live cell enzymatic assays were performed using HCT116, HT26, CCD841-CoN and Caco-2 cells either in the presence of vehicle control DMSO, HDAC inhibitor TSA (1 μM) or Z-FY-CHO (100 μM).
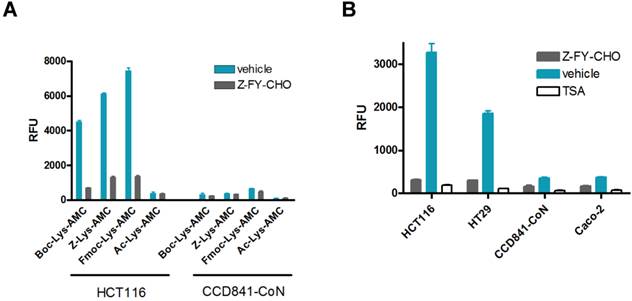
Having found that Fmoc-Lys-AMC works as a better substrate for CTSL, we wanted to test if the Fmoc substitution also works for the two-step activation process mediated by HDACs and CTSL. The fluorogenic substrate Fmoc-Lys(Ac)-AMC was evaluated using HCT116, HT26, CCD841-CoN and Caco-2 cell lines for the cancer selectivity and enzyme specificity. The result confirmed that the substrate was efficiently processed by malignant HCT116 and HT29, but not by normal CCD841-CoN and non-malignant Caco-2 cells showing excellent selectivity against malignant cancer cells (Fig. 2B). Addition of HDAC inhibitor TSA or CTSL inhibitor Z-FY-CHO into HCT116 and HT29 cells efficiently blocked the release of AMC, confirming the enzyme specificity that both HDACs and CTSL activities are required to process this substrate. These data strongly suggest that the Fmoc-Lys(Ac)- masking group can be applied to our prodrug strategy for the improved anticancer efficacy.
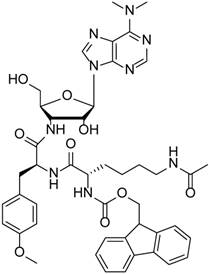
Synthesis and in vitro evaluation of Fmoc-Lys(Ac)-Puromycin
Having chosen the Fmoc-Lys(Ac)- masking group as a lead candidate, we synthesized Fmoc-Lys(Ac)-Puromycin (Fig. 3). To evaluate the anticancer efficacy of Fmoc-Lys(Ac)-Puromycin and confirm its selectivity in vitro, we performed cell viability assays (Fig. 4A). The dose-response curves obtained from the cell lines tested indicate comparable IC50 values for the malignant HCT116 (1.0 μM) and HT29 (1.6 μM) cells. In contrast, the IC50 values of CCD841-CoN and Caco-2 cells were not properly measurable, as sufficient inhibition of cell viability was not achieved. Of note, the IC50 values (1 μM range) of HCT116 and HT29 were obtained from 2 d treatment, compared with previously reported Boc-Lys(Ac)-Puromycin with the IC50 values (10 μM range) of the same cell lines obtained from much longer 5 d treatment [5]. To directly compare the cell killing activity of Fmoc-Lys(Ac)-Puromycin and Boc-Lys(Ac)-Puromycin, cell viability assay was performed using HCT116 cells (Fig. 4B). As early as 2 d, massive cell death was observed in cells treated with Fmoc-Lys(Ac)-Puromycin, whereas no cell death was detected in cells treated with Boc-Lys(Ac)-Puromycin even at much higher (30-fold) drug concentration. Although Boc-Lys(Ac)-Puromycin was able to cause extensive cell death at later time point (4 d), Fmoc-Lys(Ac)-Puromycin exhibited substantially superior cell killing ability with much faster action but still retaining excellent selectivity.
Chemical structure of Fmoc-Lys(Ac)-Puromycin.
In vitro evaluation of Fmoc-Lys(Ac)-Puromycin. (A) Inhibition of cell viability by Fmoc-Lys(Ac)-Puromycin in dose-response curve format for the cell lines as indicated. The cells were treated with DMSO or the indicated doses of agent (0.422, 0.844, 1.69, 3.38, 6.75, or 13.5 μM) for 48 h followed by crystal violet assay. Data represent mean values of triplicate measurements ± s.d. (B) Inhibition of cell viability by Boc-Lys(Ac)-Puromycin (Boc-KAc-Puro) and Fmoc-Lys(Ac)-Puromycin (Fmoc-KAc-Puro). HCT116 cells were treated with vehicle control or the indicated dose of agent for 2 d or 4 d followed by crystal violet assay. Data represent mean values of triplicate measurements ± s.d. (C) Levels of puromycin-incorporated proteins in the cells were monitored by immunoblotting. Indicated cell lines were treated either with vehicle control, Fmoc-Lys(Ac)-Puromycin (Fmoc-KAc-Puro) or puromycin (Puro) for 20 h, followed by preparation of cell lysates. The lower panel (anti-α-Tubulin) serves as a loading control.
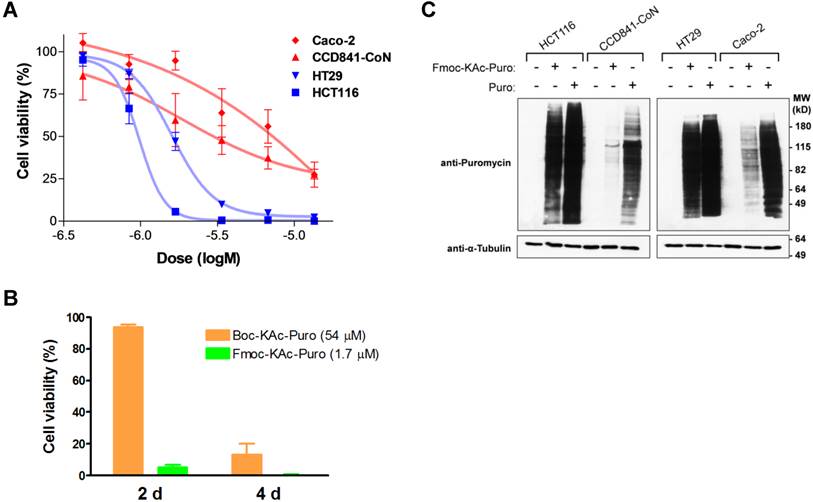
To confirm that the selective cell killing activity of Fmoc-Lys(Ac)-Puromycin is dependent on the release of active puromycin in cancer cells, we examined the cellular uptake of puromycin. As a Tyr-tRNA mimetic, puromycin enters the ribosome A site and blocks peptide chain elongation by covalent incorporation into the C terminus of nascent polypeptide chains [26], whereas Fmoc-Lys(Ac)-Puromycin cannot be incorporated into polypeptides due to the presence of masking group. This allows us to monitor the prodrug activation as well as cellular uptake of active puromycin by detecting puromycin-labeled proteins using anti-puromycin antibody [5, 27]. To confirm that Fmoc-Lys(Ac)-Puromycin can be selectively converted into puromycin in malignant cancer cells, we assessed the level of puromycin incorporation by immunoblotting using cell lysates from cells treated with the prodrug. The intensity of puromycin incorporation was substantially greater in malignant cancer cells (HCT116 and HT29) compared with normal and non-malignant cells (CCD841-CoN and Caco-2) (Fig. 4C). When these cells were treated with parental puromycin, there was no selectivity in terms of puromycin incorporation regardless of malignant or normal. The results provide clear evidence that the Fmoc-Lys(Ac)-Puromycin can be selectively converted into puromycin in malignant cancer cells, leading to cell death. Thus the data demonstrate that the Fmoc modification of the α-amino protecting group of lysine in the masking group has a tremendous impact on the anticancer efficacy of our prodrug strategy.
Highly efficient cellular uptake by the Fmoc modification
Despite the moderate improvement (less than two-fold compared with Boc) by the Fmoc substitution assessed by the AMC substrates (Fig. 2A), Fmoc-Lys(Ac)-Puromycin exhibited dramatic improvement (much faster action and lower concentration) in cancer cell killing ability compared with the original Boc-Lys(Ac)-Puromycin (Fig. 4B). We reasoned that this is mainly due to the difference in cellular uptake of the drugs besides the improvement of substrate structure. As mentioned above, cellular uptake of active puromycin can be monitored by immunological detection of puromycin-labeled proteins using anti-puromycin antibody. The uptake of active puromycin was detected in cancer cells treated with Fmoc-Lys(Ac)-Puromycin at concentration as low as 0.6 μM (Fig. 4C) which is 30-fold lower concentration of previously reported Boc-Lys(Ac)-Puromycin [5]. This appears to correlate well with the improved cell killing ability of Fmoc-Lys(Ac)-Puromycin. Thus we wanted to directly monitor the uptake of active puromycin in live cells treated with the prodrugs.
We took advantage of a flow cytometry method, known as surface sensing of translation (SUnSET), which allows to monitor and quantify the uptake of puromycin in individual mammalian cells [27]. This method is based on the observation that surface detection of puromycin is directly proportional to its incorporation. It utilizes anti-puromycin antibody for the immunological detection of puromycin-labelled peptides and proteins. Free puromycin or its prodrug forms cannot be detected by this method. At high concentration, puromycin acts as an inhibitor of protein synthesis. However at lower concentration, puromycin can serve as an agent for metabolic labelling of cells like radioactive methionine. Similar to amino acids, puromycin (but not its prodrug forms) in the cells can be rapidly incorporated at the carboxyl terminus of newly synthesized proteins, thus all proteins synthesized in the presence of free puromycin are end-labelled with this molecule. In this case using flow cytometry, the levels of puromycin-labelled proteins present on the cell surface of live cancer cells can be measured using anti-puromycin antibody (Fig. 5A).
Monitoring cellular uptake of active puromycin in live cancer cells treated with the prodrug. (A) Illustration of the method to monitor the prodrug activation in live cancer cells using flow cytometry. Cancer cells with increased HDAC and CTSL activities can selectively unmask the prodrug releasing active puromycin which in turn rapidly end-labels newly synthesized proteins, while the prodrug is inactive in normal cells. (B) HCT116 cells were untreated or treated either with Boc-Lys(Ac)-Puromycin (Boc-KAc-Puro), Fmoc-Lys(Ac)-Puromycin (Fmoc-KAc-Puro) or puromycin (Puro) for 20 h, followed by antibody staining and flow cytometry analysis.
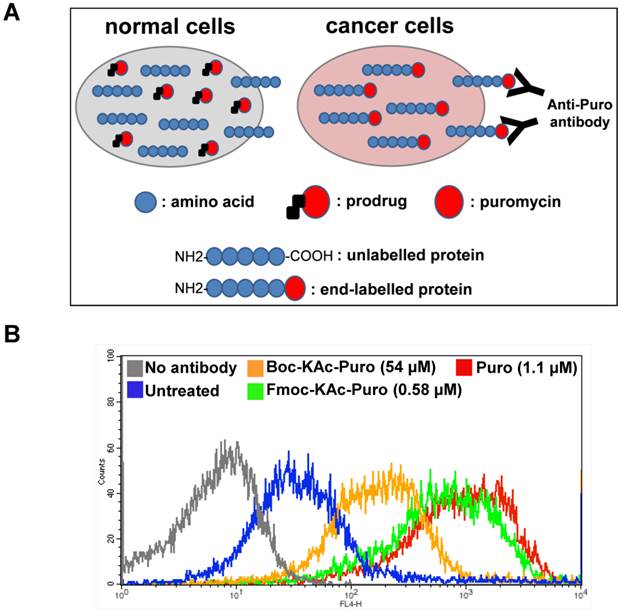
Using Alexa Fluor® 647-conjugated anti-puromycin antibody, we performed flow cytometry analysis to detect cell surface puromycin as an indicative of cellular uptake of active puromycin in live cells (Fig. 5B). The level of fluorescent signal in cells treated with Fmoc-Lys(Ac)-Puromycin is far greater compared to that of cells treated with Boc-Lys(Ac)-Puromycin even at 90-fold lower concentration, reaching close level to that of parental puromycin. These data clearly indicate that the Fmoc modification can dramatically improve the cellular uptake of the prodrug. This is not surprising, since there is a known strategy used successfully to improve cellular uptake of small peptides by replacing the α-amino protecting group at the amino-terminus [28]. Considering that all HDACs are intracellular proteins, the prodrug first needs to cross the cell membrane to be deacetylated. Thus it is possible that the improved substrate cleavage and cellular uptake contribute to a marked synergistic effect on the overall drug efficacy. Of note, in addition to the therapeutic potential, the puromycin-based selective live cancer cell labelling technology established here could have potential to further broaden the utility of our prodrug strategy into the theranostic field including in vivo imaging and diagnostics.
In vivo anticancer efficacy of Fmoc-Lys(Ac)-Puromycin
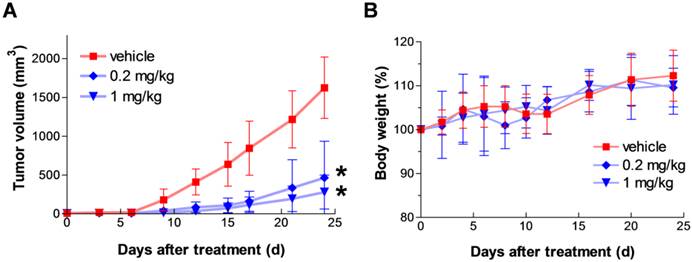
To assess in vivo anticancer efficacy of the agent, we used mouse xenograft models bearing human colon cancer cell line HCT116. First, HCT116 cells were subcutaneously injected into the lower flank of mice, then dosing was initiated when small palpable tumors had developed (> 3 mm in diameter). Fmoc-Lys(Ac)-Puromycin was daily administered intraperitoneally at 0.2 and 1 mg/kg/dose for 12 d. The agent caused a dose-dependent inhibition of tumor growth (Fig. 6A). In this model, animals treated with the prodrug developed significantly smaller tumor mass in comparison to the animals treated with vehicle control. The anticancer effect was observed from as low as 0.2 mg/kg/dose, whereas previously reported Boc-Lys(Ac)-Puromycin required at least 50 mg/kg/dose to achieve similar level of tumor growth inhibition [5]. Furthermore, daily intraperitoneal administration of the prodrug as high as 1 mg/kg/dose for 12 d appeared well-tolerated as judged by weight loss determination (Fig. 6B). These results clearly demonstrate substantially improved in vivo anticancer efficacy of the Fmoc-Lys(Ac)-Puromycin without severe off-target systemic toxicity.
In vivo anticancer efficacy of Fmoc-Lys(Ac)-Puromycin. (A) Tumor growth in HCT116 xenograft model. HCT116 cells were subcutaneously injected into the lower flank of mice, then dosing was initiated when small palpable tumors had developed (> 3 mm in diameter). Animals were treated with either vehicle control or Fmoc-Lys(Ac)-Puromycin (0.2 or 1 mg/kg/dose) daily via intraperitoneal rout. Values are mean ± s.d. (n = 6 mice per group). * P < 0.05 compared to the control group. P = 0.0198 (0.2 mg/kg), P = 0.0186 (1 mg/kg) at 12 d [two-tailed student's t test]. (B) Body weight change during the treatment in A was monitored as a sign of general animal health. Values are mean ± s.d.
Toxicity assessment
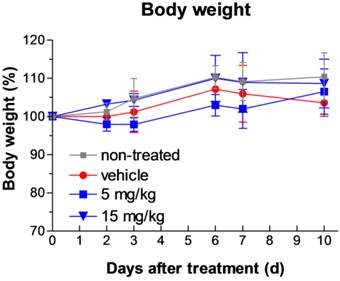
We extended our study to the toxicity assessment of the Fmoc-Lys(Ac)-Puromycin. Acute phase toxic effects can be monitored by measuring the levels of serum enzymes and proteins associated with disease or injury of the liver or kidney. Elevations in the levels of the serum transaminases aspartate aminotransferase (AST) and alanine aminotransferase (ALT) are often indicators of liver disease or injury. Serum total bilirubin is an indicator of liver and biliary function, and albumin, globulin and blood urea nitrogen (BUN) are indicators of renal function. The levels of ALT, AST, total bilirubin, albumin, globulin, and BUN were measured in mice treated with the Fmoc-Lys(Ac)-Puromycin daily for 10 d at two different concentrations (5 and 15 mg/kg/dose). Blood samples were collected and analyzed. The results are shown in Table 1. The levels of these clinical indicators of liver and kidney injury and disease are within normal ranges and are not significantly changed relative to non-treated or vehicle treated animals, demonstrating that the prodrug did not significantly affect renal or hepatic function. Furthermore analysis of blood cell counts also indicated that all cells tested including RBC, WBC, platelets, and neutrophils as well as hemoglobin and hematocrit levels were all within normal and vehicle control levels (Supplementary Table S1 and S2), indicating that there is no obvious indication of acute toxicity. By contrast, the parental puromycin treatment has been reported to cause apparent renal damage resulting in significant elevation of BUN levels in animals as soon as 2 d after the onset of administration [29]. Consistent with these blood lab results, daily intraperitoneal administration of the prodrug as high as 15 mg/kg/dose for 10 d appeared well-tolerated as judged by weight loss determination (Fig. 7).
Blood chemistry after Fmoc-Lys(Ac)-Puromycin treatment.
| Serum indicator | Normal range | No treatment | Vehicle | 5 mg/kg | 15 mg/kg |
|---|---|---|---|---|---|
| BUN (mg/dL) | 15-40 | 22 ± 1.2 | 23 ± 1.2 | 21 ± 3.1 | 17 ± 2.6 |
| Albumin (g/dL) | 2.5-4.0 | 3.4 ± 0.21 | 3.5 ± 0.12 | 3.4 ± 0.44 | 3.3 ± 0.15 |
| Globulin (g/dL) | 1.3-3.0 | 2.3 ± 0.058 | 2.3 ± 0.12 | 2.4 ± 0.31 | 2.2 ± 0.15 |
| Bilirubin (mg/dL) | 0.1-1.0 | 0.23 ± 0.058 | 0.2 ± 0 | 0.2 ± 0.1 | 0.2 ± 0 |
| AST (U/L) | 30-300 | 140 ± 76 | 180 ± 93 | 130 ± 82 | 140 ± 52 |
| ALT (U/L) | 30-200 | 83 ± 82 | 95 ± 48 | 100 ± 57 | 96 ± 46 |
Toxicity assessment of the Fmoc-Lys(Ac)-Puromycin. Body weight change during the treatment was monitored as a sign of general toxicity. Swiss-Webster mice were treated with either none, vehicle control or Fmoc-Lys(Ac)-Puromycin (5 or 15 mg/kg/dose) daily for 10 d via intraperitoneal rout. Values are mean ± s.d. (n = 3 mice per group). Values are mean ± s.d.
Conclusion
Our preclinical evaluation data presented here demonstrate that the Fmoc-Lys(Ac)-Puromycin exhibits substantially improved anticancer efficacy without compromising off-target toxicity, further supporting our approach for the selective cancer therapy. Although more extensive long-term safety studies are warranted, the outcome of this preclinical research provides crucial foundations for our therapeutic approach. Successful development of such agents to selectively kill cancer cells has the potential to impact the mortality and quality of life caused by advanced cancer.
Supplementary Material
Supplementary information, tables and figures.
Abbreviations
HDAC: histone deacetylase; CTSL: cathepsin L; AMC: 7-amino-4-methylcoumarin: Boc: tert-butyloxycarbonyl; Z: benzyloxycarbonyl; Fmoc: 9-fluorenylmethoxycarbonyl; Ac: acetyl; Lys: lysine; TSA: Trichostatin A.
Acknowledgements
We thank the Division of Laboratory Animal Resources (DLAR) at Stony Brook University for support on our animal studies. In particular, we wish to express our appreciation to Tom Zimmerman, Jean Rooney, and other staff in DLAR.
Grant Support
This work was supported by CA170041 (to M.J. Hayman) from the National Cancer Institute and the Fusion Award (to N. Ueki and N.S. Sampson) from The Stony Brook Institute for Clinical and Translational Sciences, School of Medicine, Stony Brook University.
Competing Interests
N.U. and M.J.H. are co-inventors on a pending patent application entitled “Novel prodrugs for selective anticancer therapy” (PCT/US2013/060443). All other authors declare no competing financial interests.
References
1. Dobbelstein M, Moll U. Targeting tumour-supportive cellular machineries in anticancer drug development. Nat Rev Drug Discov. 2014;13:179-96
2. Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823-37
3. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57-70
4. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
5. Ueki N, Lee S, Sampson NS, Hayman MJ. Selective cancer targeting with prodrugs activated by histone deacetylases and a tumour-associated protease. Nat Commun. 2013;4:2735
6. Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009;277:8-21
7. Haberland M, Johnson A, Mokalled MH, Montgomery RL, Olson EN. Genetic dissection of histone deacetylase requirement in tumor cells. Proc Natl Acad Sci U S A. 2009;106:7751-5
8. Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280:168-76
9. Wu MZ, Tsai YP, Yang MH, Huang CH, Chang SY, Chang CC. et al. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol Cell. 2011;43:811-22
10. Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38-51
11. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769-84
12. Marks PA, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem. 2009;107:600-8
13. Qian DZ, Wei YF, Wang X, Kato Y, Cheng L, Pili R. Antitumor activity of the histone deacetylase inhibitor MS-275 in prostate cancer models. Prostate. 2007;67:1182-93
14. Wedel SA, Sparatore A, Soldato PD, Al-Batran SE, Atmaca A, Juengel E. et al. New histone deacetylase inhibitors as potential therapeutic tools for advanced prostate carcinoma. J Cell Mol Med. 2008;12:2457-66
15. Joyce JA, Baruch A, Chehade K, Meyer-Morse N, Giraudo E, Tsai FY. et al. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell. 2004;5:443-53
16. Jedeszko C, Sloane BF. Cysteine cathepsins in human cancer. Biol Chem. 2004;385:1017-27
17. Gonzalez-Suarez I, Redwood AB, Grotsky DA, Neumann MA, Cheng EH, Stewart CL. et al. A new pathway that regulates 53BP1 stability implicates cathepsin L and vitamin D in DNA repair. EMBO J. 2011;30:3383-96
18. Brindle NR, Joyce JA, Rostker F, Lawlor ER, Swigart-Brown L, Evan G. et al. Deficiency for the cysteine protease cathepsin L impairs Myc-induced tumorigenesis in a mouse model of pancreatic neuroendocrine cancer. PLoS One. 2015;10:e0120348
19. Sevenich L, Joyce JA. Pericellular proteolysis in cancer. Genes Dev. 2014;28:2331-47
20. Lankelma JM, Voorend DM, Barwari T, Koetsveld J, Van der Spek AH, De Porto AP. et al. Cathepsin L, target in cancer treatment? Life Sci. 2010;86:225-33
21. Sudhan DR, Siemann DW. Cathepsin L inhibition by the small molecule KGP94 suppresses tumor microenvironment enhanced metastasis associated cell functions of prostate and breast cancer cells. Clin Exp Metastasis. 2013;30:891-902
22. Choi KY, Swierczewska M, Lee S, Chen X. Protease-activated drug development. Theranostics. 2012;2:156-78
23. Dubowchik GM, Firestone RA. Cathepsin B-sensitive dipeptide prodrugs. 1. A model study of structural requirements for efficient release of doxorubicin. Bioorg Med Chem Lett. 1998;8:3341-6
24. Dubowchik GM, Mosure K, Knipe JO, Firestone RA. Cathepsin B-sensitive dipeptide prodrugs. 2. Models of anticancer drugs paclitaxel (Taxol), mitomycin C and doxorubicin. Bioorg Med Chem Lett. 1998;8:3347-52
25. Choe Y, Leonetti F, Greenbaum DC, Lecaille F, Bogyo M, Bromme D. et al. Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J Biol Chem. 2006;281:12824-32
26. Pestka S, Rosenfeld H, Harris R, Hintikka H. Studies on transfer ribonucleic acid-ribosome complexes. XXI. Effect of antibiotics on peptidyl-puromycin synthesis by mammalian polyribosomes. J Biol Chem. 1972;247:6895-900
27. Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6:275-7
28. Tornatore L, Sandomenico A, Raimondo D, Low C, Rocci A, Tralau-Stewart C. et al. Cancer-selective targeting of the NF-kappaB survival pathway with GADD45beta/MKK7 inhibitors. Cancer Cell. 2014;26:495-508
29. Watanabe M, Nomura G, Hirata M, Imai K, Koizumi H. Studies on the validity of urinary enzyme assay in the diagnosis of drug-induced renal lesions in rats. Toxicol Pathol. 1980;8:22-33
Author contact
![]() Corresponding author: Nobuhide Ueki, #234 Life Sciences Building, Department of Molecular Genetics & Microbiology, Stony Brook University, Stony Brook, NY 11794-5222. Phone: 631-632-4314, Fax: 631-632-8891, E-mail: nobuhide.uekiedu
Corresponding author: Nobuhide Ueki, #234 Life Sciences Building, Department of Molecular Genetics & Microbiology, Stony Brook University, Stony Brook, NY 11794-5222. Phone: 631-632-4314, Fax: 631-632-8891, E-mail: nobuhide.uekiedu