Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2016; 6(8):1190-1204. doi:10.7150/thno.15226 This issue Cite
Research Paper
Improvement of Prostate Cancer Diagnosis by Detecting PSA Glycosylation-Specific Changes
Esther Llop1,4*, Montserrat Ferrer-Batallé1,4*, Sílvia Barrabés1,4, Pedro Enrique Guerrero1,4, Manel Ramírez2,4, Radka Saldova5, Pauline M. Rudd5, Rosa N. Aleixandre2,4, Josep Comet3,4, Rafael de Llorens1,4, Rosa Peracaula1,4, ![]()
1. Biochemistry and Molecular Biology Unit. Department of Biology, University of Girona, Campus Montilivi, 17071, Girona, Spain.
2. Clinic Laboratory, Dr. J. Trueta University Hospital, 17007, Girona, Spain.
3. Urology Unit, Dr. J. Trueta University Hospital, 17007, Girona, Spain.
4. Biomedical Research Institute of Girona (IdIBGi). Dr. J. Trueta University Hospital, 17007, Girona, Spain.
5. NIBRT GlycoScience Group, National Institute for Bioprocessing Research and Training, Dublin, Ireland.
*Both authors contributed equally to this work.
Received 2016-2-8; Accepted 2016-4-20; Published 2016-5-24
Abstract

New markers based on PSA isoforms have recently been developed to improve prostate cancer (PCa) diagnosis. However, novel approaches are still required to differentiate aggressive from non-aggressive PCa to improve decision making for patients.
PSA glycoforms have been shown to be differentially expressed in PCa. In particular, changes in the extent of core fucosylation and sialylation of PSA N-glycans in PCa patients compared to healthy controls or BPH patients have been reported. The objective of this study was to determine these specific glycan structures in serum PSA to analyze their potential value as markers for discriminating between BPH and PCa of different aggressiveness.
In the present work, we have established two methodologies to analyze the core fucosylation and the sialic acid linkage of PSA N-glycans in serum samples from BPH (29) and PCa (44) patients with different degrees of aggressiveness. We detected a significant decrease in the core fucose and an increase in the α2,3-sialic acid percentage of PSA in high-risk PCa that differentiated BPH and low-risk PCa from high-risk PCa patients. In particular, a cut-off value of 0.86 of the PSA core fucose ratio, could distinguish high-risk PCa patients from BPH with 90% sensitivity and 95% specificity, with an AUC of 0.94. In the case of the α2,3-sialic acid percentage of PSA, the cut-off value of 30% discriminated between high-risk PCa and the group of BPH, low-, and intermediate-risk PCa with a sensitivity and specificity of 85.7% and 95.5%, respectively, with an AUC of 0.97. The latter marker exhibited high performance in differentiating between aggressive and non-aggressive PCa and has the potential for translational application in the clinic.
Keywords: benign prostatic hyperplasia (BPH), glycosylation, Pholiota squarrosa lectin (PhoSL), prostate cancer (PCa), prostate specific antigen (PSA), Sambucus nigra agglutinin (SNA).
Introduction
Prostate cancer (PCa) represents the highest incidence among carcinomas and is the second most frequent cause of cancer deaths in men in Western countries [1]. PCa is usually suspected on the basis of DRE and/or serum Prostate Specific Antigen (PSA) levels. Definitive diagnosis depends on histopathological verification of adenocarcinoma in prostate biopsy cores or specimens from transurethral resection of the prostate or prostatectomy for benign prostatic enlargement. Men who could develop PCa later in life have increased total PSA levels years or decades before the cancer is diagnosed [2]. PSA is a strong prognostic marker for long-term risk of clinically relevant prostate cancer [3]. These high serum PSA levels may reflect the long duration of prostate carcinogenesis and/or could reflect a causal role of total PSA in PCa development and/or progression [4].
The wide availability of PSA assay revolutionized PCa screening and resulted in a decrease of 29% in the risk of deaths from PCa [5]. However, the ubiquitous application of PSA screening has also led to over-detection and over-treatment resulting in unnecessary biopsies and treatments for low-risk or indolent cancers thus adding to morbidity without prolonging life [3, 6]. The clinical focus must be redirected toward patients who are more likely to develop clinically significant tumors and would benefit from treatment. This goal can only be accomplished through the development of tests that can identify men at risk of developing clinically significant PCa [7-9].
PSA, a glycoprotein secreted by normal prostate epithelial cells and a member of the human kallikrein gene family of serine-proteases [10], is currently used for the detection and monitoring of prostate pathologies. Increased serum PSA level is mostly due to the disruption of the prostate basement membrane in the affected prostate cells. Most of the prostate epithelial cells, including normal, hyperplastic, or cancerous, synthesize PSA. Neoplastic cells produce somewhat lower and varying tissue levels of PSA compared to benign epithelial cells, although both conditions can cause total PSA elevation in the blood. Furthermore, some aggressive prostate cancers do not produce PSA [3]. Thus, serum PSA levels cannot differentiate between changes caused by cancer, benign prostatic hyperplasia (BPH), prostatitis, and manipulations (bicycling, DRE, catheterization) of the prostate since all these situations can cause elevation of the PSA serum concentrations.
PSA serum marker assay has been shown to lack specificity and has a high false positive rate, especially when the serum PSA levels fall in the “grey zone” (4.0-10.0 ng/ml). Several authors have extended this concentration range to 2-20 ng/ml or 1-27 ng/ml [11, 12]. In addition, high-grade cancer can occur in men with low total PSA levels [13]. There is not a fixed PSA serum levels cut off point that could distinguish the indolent from the aggressive PCa. Currently, the accepted cut-off is 2 ng/ml [6] because, as Thompson et al. stated, 15% men with a PSA <4 ng/ml were diagnosed with intermediate to high-risk PCa [14]. The large biological variations in the measured PSA concentrations with up to 20% are an additional problem. PSA test has led to a drastic increase in the detection of PCa, rendering this biomarker the gateway for the diagnosis of prostatic neoplasms. However, the increase in incidence, as detected by the PSA test, has not been mirrored by a similar reduction in mortality [15]. In brief, PSA test has a high rate of false positives resulting in repeated biopsies and unnecessary surgeries [6]. Thus, there is an urgent need for new biomarkers, especially to detect clinically significant and aggressive PCa [6, 16]. Aggressive cancer comprises those tumours destined to recur locally or systemically despite optimal local therapy. Currently, no classification scheme exists that enables outcomes for patients with high-risk PCa to be determined reliably and consistently. Variation in the diagnostic methods to classify patients and in the treatment itself contributes to variations of the specific outcomes between studies. By current estimates, high-risk disease accounts for 15% of all PCa diagnoses and the range of outcomes was highlighted in an analysis showing 5-year relapse-free survival probabilities ranging between 49-80% for the same patient population using different published schemas [8].
Different approaches have been proposed to improve the specificity of the PSA test. The most common approach is to measure the ratio of free PSA (fPSA)/total PSA (tPSA), which is lower in PCa, although it still gives false positives. Other alternatives are PSA density, PSA velocity, and different PSA molecular forms. PSA values and PSA kinetics are useful indicators for prostate biopsy [6]. However, none of these approaches has shown substantial improvements in the ability to distinguish between BPH and PCa [16].
Recently, the introduction of new molecular markers such as Prostate Cancer Antigen 3 (PCA3) and Prostate Health Index (PHI), and sophisticated imaging systems has partially improved PCa diagnosis. PCA3 appears to be useful for detection of cancer, but less useful for differentiation between the low-risk, intermediate-risk, and high-risk disease. PHI includes the determination of [-2] pro-PSA in combination with free and total PSA levels, improves the specificity, and limits the number of unnecessary biopsies. It has been recently approved by FDA and in Europe for PCa diagnosis [17]. Although PHI has better performance than tPSA or %fPSA in distinguishing low-risk from high-risk PCa [18], its specificity is too low to be reliably used for this purpose [19].
It is well known that the oncogenic process results in significant alterations of the cellular glycosylation pattern. Glycoproteins secreted by tumors may reflect the altered glycosylation machinery of cancer cells, and are good candidates as new tumor markers [20]. PSA is a glycoprotein containing 8% of carbohydrates, with a single N-oligosaccharide chain attached to Asn-45 [10, 20, 21]. In previous studies, we analyzed and compared the glycosylation pattern of PSA purified from the seminal fluid of healthy donors with that produced by a human prostate tumor cell line and from prostate cancer patient sera. In comparison with seminal plasma PSA, the fucose content of PSA N-glycans from sera of PCa patients was significantly lower [10, 20-23]. The degree of sialylation, as evaluated by changes in the proportion of the PSA isoforms separated by 2-DE, indicated a decrease in sialylation of PSA in PCa patients. These results suggested that determining the glycosylation changes in serum PSA is a possible way to discriminate between BPH and PCa [10, 20, 21]. In the present study, using lectins as analytes, we have developed two new methodologies to analyze the core fucose and α2,3-linked sialic acid of serum PSA to distinguish between patients with BPH and PCa with different degrees of aggressiveness.
Material and Methods
Serum samples
Serum samples were obtained from 44 PCa patients (age range 47-83 years) and 29 BPH patients (age range 44-76 years) of the Hospital Universitari Dr. J. Trueta (Girona, Spain), following the standard operating procedures of its Ethics Committee. Serum samples were collected before any diagnostic procedure was started and stored at -80ºC until analysis. The diagnosis was performed by the Urology and Pathology units using Transrectal Ultrasound-guided biopsy and/or adenomectomy/prostatectomy followed by pathological analysis. The follow-up of the BPH patients was made for a minimum of 2 years and 25 out of 29 BPH patients had, at least, two negative biopsies and none of them had evidence of high-grade PIN. The 4 BPH left were subjected to prostate surgery (adenomectomy or prostate transurethral resection) and confirmed by the Pathology Unit not to have prostate cancer. BPH patients had tPSA serum levels ranging from 5.5-18.2 ng/ml (average of 9.8 ng/ml).
PCa patients were classified according to the progression of the disease and the Tumor-Node-Metastasis (TNM) classification following the general guidelines of the European Association of Urology. High-risk PCa patients include patients with Gleason scores equal or above 8 (4+4) and/or patients with metastasis. Low-risk prostate cancers were patients with Gleason scores of ≤6 (3+3), tPSA levels below 10 ng/ml and clinical stage≤pT2a. Intermediate-risk PCa patients were patients that did not meet the above requirements. They had Gleason score 7 (3+4 or 4+3) and 6 (3+3). A patient with focal Gleason 8, tPSA levels below 10 ng/ml and pT2a, was also included in the intermediate-risk group considering his 10-year relapse-free survival. Thus, out of the 44 PCa patients, 22 were classified as high-risk with an average tPSA serum level of 22.9 ng/ml (4.1-109.7 ng/ml), 17 as intermediate-risk with an average tPSA serum level of 8.8 ng/mL (5.1-17.6 ng/ml), and 5 as low-risk with an average tPSA serum level of 6.9 ng/ml (5.2-8.3 ng/ml).
Quantification of tPSA and fPSA
Total and free PSA concentrations were measured using commercially available ELECSYS®1 assays in the Modular Analytics E170 (Roche Diagnostics) as described previously [24]. Elecsys® free or total PSA assay is an electro-chemiluminescence immunoassay (ECLIA) and comprises the biotinylated mouse monoclonal antibody, anti-free PSA M-30, to measure free PSA and a biotinylated mouse monoclonal antibody, anti-total PSA M-36, to measure total PSA [25]. M-30 and M-36 antibodies bind to epitopes 1 and 6b of the PSA molecule, respectively [26]. The N-glycan chain linked to Asn-45 is in proximity to epitope 2 of PSA and the two sialic acids of the N-glycan chain could affect the binding of the antibodies that recognize epitope 2 [27]. This is not the case of the antibodies against PSA used in this study.
Cleavage of PSA- α1-antichymotrypsin (ACT) in PCa and BPH Patients' Sera
Serum PSA complexed to ACT was released using a previously published method with modifications [22, 28]. 200 μl 5 M ethanolamine (Sigma-Aldrich, Germany) was added to 750 μl of serum (final pH of 10.3) and incubated at 25°C for 72h. Subsequently, samples were neutralized to pH 7.8 with 5 M hydrochloric acid.
Isolation of total PSA by immunopurification (total PSA IP)
After PSA release from ACT, total serum PSA was isolated using the Access Hybritech PSA assay Kit (Beckman Coulter, Brea, CA, USA). Briefly, a suspension of 100 μl of paramagnetic particles coated with mouse monoclonal anti-PSA (Access Hybritech PSA Assay, Beckman Coulter, Brea, CA, USA) was washed with 500 μl of incubation buffer (50 mM Tris, 150 mM NaCl, pH 7.4, 0.1% Tween-20, 1% BSA) using magnetic separation. The beads were then incubated with 1 ml of neutralized treated serum (containing 750 μl of original serum) for 1h at room temperature (rt) with shaking. Next, the beads were washed with 500 μl of washing buffer (50 mM Tris, 150 mM NaCl, pH 7.4, 1% Triton X-100) using magnetic separation. The immunoadsorbed PSA was eluted in three steps using 100 μl of Gentle Ag/Ab Elution Buffer, pH 6.6 (Thermo Scientific, Rockford, IL, USA) for 30min each. The final elution volume was of 300 μl. Samples were desalted and concentrated to a final volume of 40 μl using Amicon Ultra-0.5 3K Centrifugal Filter Devices (Millipore, Cork, IRL), which had been previously pre-treated with 5% Brij-35 (Sigma-Aldrich, Germany) and used for sample desalting according to the manufacturer's instructions.
Free PSA immunopurification (free PSA IP)
Free PSA was immunoprecipitated with magnetic beads coated with anti-free PSA M-30 antibody as described previously [22] with some modifications. Briefly, 400 µl of streptavidin coated magnetic beads (0.72 mg/ml) were washed three times with washing buffer (50 mM Tris, 150 mM NaCl, pH 7.4, 1% Triton X-100) and incubated with biotinylated anti-free PSA M-30 antibody for 30min at rt in incubation buffer (50 mM Tris, 150 mM NaCl, pH 7.4, 0.1% Tween-20, 1% BSA) under shaking. After washing three times with washing buffer, samples containing PSA were incubated in a final volume of 500 µl diluted in TBS (10 mM Tris, 100 mM NaCl, pH 7.4) for 1h at rt under shaking. Subsequently, beads were washed three times and free PSA was released twice by the addition of Gentle Ag/Ab Elution Buffer. Finally, the sample was desalted and concentrated as described above.
Western blotting and detection of core fucosylation with PhoSL lectin
Samples of immunoprecipitated PSA (either total PSA IP or both total and free PSA IP) and standard PSA (BBI Solutions, UK) were prepared in Laemmli sample buffer with 1.25% ß-mercaptoethanol and boiled for 5min at 95ºC. Samples were run on a 12% SDS-PAGE and transferred to a PVDF membrane for 3h at 100V in Towbin buffer (25 mM Tris, 192 mM Glycine, 20% v/v methanol, pH 8.3). Following a washing step, membranes were blocked with 2% w/v polyvinylpyrrolidone (PVP) in TBS (10 mM Tris, 100 mM NaCl, 0.05% v/v Tween-20, pH 7.4) for 1h at rt with shaking. Membranes were then washed and incubated with 4 µg/ml of Pholiota squarrosa lectin (PhoSL) labelled with fluorescein isothiocyanate (FITC) in the PhoSL buffer (100 mM Tris pH 7.4, 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2) for 2h at rt. Finally, membranes were washed and incubated with an anti-FITC antibody horseradish peroxidase conjugated (Roche Diagnostics, Basel, Switzerland) diluted 1:7500 in incubation buffer (TBS, 1% w/v BSA, 0.05% v/v Tween-20). After washing out the secondary antibody, the membrane was incubated for 5min with Immobilon Western horseradish peroxidase substrate solution (Millipore, Billerica, MA). Chemiluminescence was visualized using the imaging system Fluorochem SP (AlphaInnotech, San Leandro, CA) under non-saturating conditions.
Enzyme-linked lectin assay (ELLA) to determine PSA core fucosylation
Mouse monoclonal antibodies M-30 against fPSA from Roche Diagnostics were diluted at 8 μg/ml in phosphate buffered saline (PBS) and bound to black maxisorp microplates (FluoroNunc/LumiNunc Maxisorp Surface) overnight at rt. Plates were washed 3 times with saline-T (0.9% NaCl solution, 0.05% v/v Tween-20) and were incubated for 1h at rt with blocking solution (2% w/v PVP in PBS). After washing with saline-T, samples after total PSA immunoprecipitation (total PSA IP) were added in a final volume of 100 μl in PBS - 0.1% Tween-20 (PBST) and allowed to stand for 2h at rt. The plates were washed with saline and the biotinylated PhoSL [29] diluted at 1 μg/ml in the PhoSL buffer (100 mM Tris pH 7.4, 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2) was added and allowed to stand for 2h at rt. Plates were then washed with saline-T and Streptavidin-horseradish peroxidase conjugate (GE-Healthcare, UK) [diluted 1:5000 in PBS] was added and allowed to stand for 1h at rt. Finally, plates were washed with PBS and detection was performed with 100 µl/well of QuantaRed Enhanced Chemifluorescent HRP Substrate (Thermo Scientific, USA). The reaction was stopped with 10 µl/well of QuantaRed Stop Solution (Thermo Scientific, USA) and the fluorescence was read at 585nm after excitation at 530nm in an automated microplate reader (BIO-TEK, USA). Wells containing PBST were used as blanks and wells with pooled female sera (with no endogenous PSA but processed in parallel with samples) were used as controls to standardize the background signal.
SNA lectin immunoaffinity column to detect PSA α2,3-linked sialic acid level
Lectin columns (one per sample) were prepared by packing 250 µl of agarose Sambucus nigra (SNA) lectin (Vector Laboratories, INC.) into a spin-X centrifuge tube (0.22 µm cellulose acetate) from Corning, Costar (NY, USA). Lectin columns were washed thoroughly with 4 ml of lectin buffer (LB) [10 mM Hepes (pH 7.6), 150 mM NaCl, 1 mM CaCl2]. PSA from serum samples was purified using the total PSA IP protocol described above. Next, samples were diluted in LB to a final volume of 405 µl and incubated with SNA lectin for 16h at 4ºC. The columns were centrifuged for 2min at 2000 rpm at 4ºC and washed twice (5 min each) with 500 µl of LB containing 1% BSA (unbound fraction; UB). The bound fraction was eluted by incubating the lectin twice with 0.5 M lactose in 1% BSA LB, for 20 min at rt (B1 fraction). The second elution (B2) was performed with 0.5 M lactose in 1% BSA LB, 0.2 M acetic acid. The bound fractions (B) were collected by centrifugation for 2 min at 2000 rpm. In the acidic elution, before the centrifugation, 150 µl of Tris 1 M pH 9.0 was added to the collecting tube in order to neutralize the sample. Total PSA and free PSA levels loaded into column and eluted in each fraction were quantified by ELECSYS assay.
N-glycan sequencing of PSA glycans
N-glycans were extracted from the gel pieces of PSA bands according to the procedure described by Royle et al. [30]. Briefly, the gel pieces were washed and treated with PNGase F to release the N-linked glycans. Subsequently, N-glycans were fluorescently labelled with 2-aminobenzamide (2AB) by reductive amination using a Ludger Tag 2AB labelling kit. The excess 2AB reagent was removed by ascending chromatography on Whatman 3MM paper in acetonitrile.
The 2AB-labelled glycans were digested in 10 µl of 50 mM sodium acetate buffer, pH 5.5 for 18h at 37ºC, using 0.5 U/ml of Arthrobacter ureafaciens sialidase (ABS) (EC 3.2.1.18) and 1.7 U/ml of Streptococcus pneumonia sialidase (NAN1) (EC 3.2.1.23). After incubation, enzymes were removed by filtration through a protein binding EZ filters (Millipore) and N-glycans were then analyzed by hydrophilic interaction liquid chromatography (HILIC).
2AB derivatized N-glycans were separated by ultra-performance liquid chromatography (UPLC) with fluorescence detection on a Waters Acquity UPLC instrument consisting of a binary solvent manager, sample manager and fluorescence detector under the control of Empower 2 chromatography workstation software (Waters, Milford, MA, USA). The hydrophilic interaction chromatography (HILIC) separations were performed using BEH Glycan column 2.1 x 150 mm, 1.7 µm BEH particles. Solvent A was 50 mM formic acid adjusted to pH 4.4 with ammonia solution and solvent B was acetonitrile. The column temperature was set to 40ºC. A 30min method was used with a linear gradient 70-53% acetonitrile at 0.56 ml/min. An injection volume of 20 µl sample prepared in 70% v/v acetonitrile was used throughout. The fluorescence detection excitation/emission wavelengths were λex=330nm and λem=420nm, respectively. Retention times were converted into glucose unit (GU) values by time-based standardization against a dextran hydrolyzed ladder.
Statistics
The patients were divided into four groups; BPH, low-risk PCa, intermediate-risk PCa and high-risk PCa.
Means comparison and correlation analysis of the variables were carried out using IBM SPSS Statistics 19 for Windows. Data with inter-assay CV>15% were not included in the statistical analyses. The variables were tested for normality and homoscedasticity using the Shapiro-Wilk and Levene's tests. When these criteria were accomplished (% α2,3-sialic acid (SA) study), intergroup differences were further analyzed using one-way ANOVA with post-hoc Bonferroni's test. For non-normally distributed groups (core fucosylation study), Mann-Whitney U test was performed. The graphics were generated with SPSS software and GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA).
Receiver operating characteristics (ROC) curve analyses were generated with SPSS software. In ROC analyses, the highest the ROC AUC is over 0.5, the better the variable is for distinguishing between high-risk PCa from indolent PCa and BPH. For α2,3-SA content, the top criterion value corresponding to the highest average of sensitivity and specificity was calculated with SPSS Statistics software.
Bivariate regression (Pearson correlation) was performed to assess the diagnostic value of the α2,3-SA content. In all statistical analyses, the significance level was set at p<0.05. Results are shown as means (95% confidence interval, CI).
Results
Clinical characteristics of the study population
Seventy three serum samples were used in the study comprising 44 from PCa patients that included 22 high-risk, 17 intermediate-risk, and five low-risk PCa samples, as well as 29 BPH samples. The 44 PCa patients were classified as described in Table 1 according to the International Union Against Cancer (IUAC) staging. The five low-risk PCa patients had Gleason scores of 6 and tPSA levels below 10 ng/ml. From the 17 intermediate-risk PCa patients, eight had Gleason score of 7, one presented a focal Gleason score of 8 and the other eight had Gleason score 6 with clinical stage>pT2a. From the 22 high-risk PCa patients, 17 were patients with Gleason scores equal or above 8, three presented a Gleason score of 7 with metastasis. The other two high-risk PCa patients presented metastasis but the Gleason score data of the primary tumor was not determined. The age and the average range of tPSA and fPSA levels of the groups are shown in Table 1.
Clinical and pathological characteristics of the patients
| Pathology | Cases | N | Gleason Score | N | Age Average | ±SD | Range | tPSA ng/ml | ±SD | Range | fPSA ng/ml | ±SD | Range |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BPH | 29 | 29 | 63.24 | 8.08 | 44-76 | 9.76 | 3.05 | 5.53-18.20 | 1.72 | 0.75 | 0.31-3.06 | ||
| PCa N=44 | Low risk | 5 | Gleason 6 | 5 | 67 | 5.34 | 61-74 | 6.91 | 1.10 | 5.22-8.29 | 1.16 | 0.23 | 0.96-1.50 |
| Intermediate risk | 17 | Gleason 6 | 8 | 59 | 9.49 | 47-75 | 7.79 | 3.76 | 5.10-16.40 | 0.92 | 0.50 | 0.27-1.70 | |
| Gleason 7 | 8 | 66.5 | 4.44 | 58-73 | 9.79 | 3.77 | 6.96-17.63 | 0.93 | 0.49 | 0.36-1.98 | |||
| Gleason 8 focal | 1 | 70 | 8.55 | 2.17 | |||||||||
| High risk | 22 | Gleason 7 / metastasis | 3 | 78.3 | 8.10 | 69-83 | 15.13 | 4.39 | 12.42-20.2 | 2.72 | 1.50 | 1.00-3.73 | |
| Gleason 8 | 8 | 61.6 | 10.89 | 51-81 | 24.42 | 15.38 | 4.14-54.12 | 2.17 | 1.95 | 1.04-6.81 | |||
| Gleason 9 | 8 | 71 | 9.77 | 51-79 | 28.55 | 33.35 | 6.48-109.7 | 4.56 | 4.93 | 0.19-15.20 | |||
| Gleason 10 | 1 | 49 | 15.07 | 3.39 | |||||||||
| Gleason ND* / metastasis | 2 | 75 | 11.31 | 67-83 | 10.03 | 1.85 | 8.72-11.33 | 1.84 | 1.13 | 1.04-2.64 |
*ND: non determined
Clinical outcome data of the patients in the high-risk group showed 54.5% of recurrence of PCa one year after treatment while recurrence was 5.9% and 0% in the intermediate group and in the low-risk group, respectively. As for the 5-year relapse-free survival, the data was available for 15 of the 22 high-risk patients and 15 of the 17 intermediate-risk patients. From these data, the 5-year relapse-free survival was 46.7% for the high-risk, 93.3% for the intermediate-risk, and 100% for the low-risk patients.
Sample preparation
Serum samples were first treated with ethanolamine to release the PSA complexed to α1-antichymotrypsin (ACT). To obtain the maximum yield without compromising protein integrity, incubation of serum PSA from a BPH patient with different ethanolamine concentrations and at several incubation times was assessed (Table 2). The cleavage percentage of the PSA-ACT complex was dependent on the ethanolamine concentration. Increasing the ethanolamine molar concentration from 0.1 to 2.5 raised the %fPSA from 32.9 to 57.7%. However, results showed that with higher ethanolamine concentration, the total PSA recovery was accordingly lower. Thus, in order to preserve the PSA integrity, different incubation times and temperatures were assayed with 1 M ethanolamine concentration (Table 2).
Ethanolamine treatment conditions to release PSA from PSA-ACT complex.
| Ethanolamine [M] | Incubation time (h) | Temperature ºC | % fPSA after ethanolamine treatment | % tPSA after ethanolamine treatment |
|---|---|---|---|---|
| 0.1 | 24 h | 25 ºC | 32.88 % | 87.5 % |
| 0.5 | 24 h | 25 ºC | 48.10 % | 89.2 % |
| 1 | 24 h | 25 ºC | 51.63 % | 85.5 % |
| 2 | 24 h | 25 ºC | 54.93 % | 77.3 % |
| 2.5 | 24 h | 25 ºC | 57.74 % | 73.1 % |
| 1 | 24 h | 25 ºC | 50.87 % | 77.0% |
| 1 | 48 h | 25 ºC | 60.06 % | 78.7% |
| 1 | 72 h | 25 ºC | 60.00 % | 81.1% |
| 1 | 24 h | 37ºC | 64.78 % | 66.8 % |
| 1 | 48 h | 37ºC | 71.32 % | 61.9 % |
| 1 | 72 h | 37ºC | 67.10 % | 53.6 % |
The best conditions to release PSA were 1 M ethanolamine for 72 hours. Using this protocol, the percentage of free PSA increased from 19% (untreated serum) to 60% (ethanolamine treated serum). The values of free PSA from the 73 serum samples in the study after ethanolamine treatment ranged from 50-80% of the total PSA values.
Core fucose determination of serum PSA
PSA fucosylation has been described to occur mainly in the core glycan structure in which fucose is linked α1,6 to N-acetylglucosamine [10, 21, 22, 31-35]. Two lectins were used in this work to asses PSA fucosylation levels. One was the Auleria aurantia lectin (AAL), which binds preferentially to fucose linked α1,6 to N-acetylglucosamine (core fucose) or to fucose linked α1,3 to N-acetyllactosamine related structures, and the other was the Pholiota squarrosa lectin (PhoSL), which recognizes only core fucose [29]. The low concentration of PSA in serum (2-20 ng/ml), which accounts only for a 2.5x10-6 - 2.5x10-5 % of total protein in serum, together with the fact that major serum proteins are glycoproteins with core fucosylation, represents an important challenge for developing an assay to detect specifically PSA core fucosylation with a fucose-recognizing lectin. The method needs to include a PSA immunopurification step to remove most of the glycosylated serum proteins, in particular those that can be core fucosylated, such as the immunoglobulins, in order that the signal generated by the fucose-recognizing lectin is attributed to the PSA molecule.
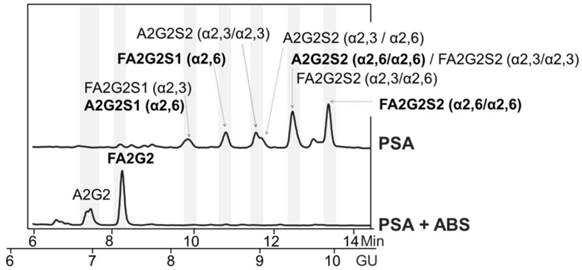
For that, we first checked whether there were remaining core fucosylated glycoproteins other than PSA in the process of immunopurification of the serum PSA with specific antibodies against PSA. These antibodies bind to the PSA epitopes that are not close to the Ans-45 N-glycosylation site and therefore the glycan chain of the PSA would not affect the recognition of these antibodies to PSA [26, 27]. We spiked pooled female sera (without endogenous PSA) with standard PSA of characterized glycosylation. The core fucosylated N-glycans of standard PSA accounted for the 77% of total PSA N-glycans as shown in its N-glycan sequencing analysis (Figure 1). N-glycans from standard PSA before and after sialidase digestion with ABS, which digests all sialic acids, were assigned on the basis of their glucose units (GU) [36] and their profiles after exoglycosidase digestion. The two N-glycan peaks after ABS digestion corresponded to a neutral biantennary complex structure with core fucose (FA2G2) and without core fucose (A2G2) in a proportion of 77% and 33%, respectively.
Glycosylation analysis of standard (Std) PSA. HILIC-UPLC chromatograms of N-glycans released from the Std PSA. N-glycan profiles from the undigested sample (top) and after sialidase digestion with ABS (digests all sialic acids) (bottom). Profiles are standardized against a dextran hydrolysate (GU). N-glycans structures are abbreviated as follows: all N-glycans have two core N-Acetylglucosamines (GlcNAc) and a trimannosyl core; F at the start of the abbreviation indicates a core fucose; A represents the number of antenna; G represents the galactoses linked β1-4 on antenna; S represents the sialic acids linked to the galactose. In peaks containing more than one glycan structure [36], the major one is marked in bold.
Standard PSA spiked in control female sera was immunopurified using antibodies against total PSA-covalently coupled to magnetic beads (total PSA IP) and also using biotinylated M-30 antibodies against fPSA (free PSA IP), which were coupled to streptavidin beads.
The immunoadsorbed PSA in both IPs was eluted by a high saline buffer and further desalted and concentrated using centricon filters. Protein and PSA quantification of the immunoadsorbed PSA indicated that 99% of serum proteins had been removed and PSA recovery was around 40%.
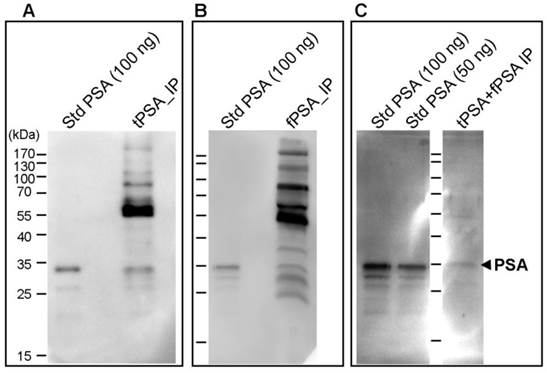
The immunoprecipitated PSA obtained by both IPs was evaluated by Western blot analysis with both lectins, AAL and PhoSL. Major protein bands of higher molecular mass than PSA were detected by both lectins as shown by the data from PhosL displayed in figures 2A and B. These remaining serum glycoproteins could interfere with the fucosylation analysis of PSA. We therefore performed double immunoprecipitation by subjecting immunoprecipitated total PSA to another round of free PSA immunoprecipitation. When the double immunoprecipitated PSA was subjected to Western blotting using both lectins, AAL still detected other remaining serum proteins (data not shown) while PhoSL detected the PSA band but no other core fucosylated glycoproteins (Figure 2C). Subsequently, we chose PhoSL to determine the core fucosylation levels of PSA following two immunopurification steps.
Immunodetection of core fucosylated glycoproteins by Western blot with PhoSL lectin following PSA immunoprecipitation (IP). A) Total PSA IP with Hybritech antibody, B) Free PSA IP with M30 antibody from Roche and C) double IP procedure firstly with Hybritech antibody followed by IP with M30 antibody from Roche. IP starting material consists of 500 µl of pooled female sera spiked with 100 ng (panels A and B) and 200 ng (panel C) of standard (Std) PSA. As a reference, 50 ng and/or 100 ng of Std PSA were loaded in each gel, as well as the molecular markers (identified in panel A). The arrow at the right side indicates the band corresponding to PSA.
We developed an Enzyme-Linked Lectin Assay (ELLA) using PhoSL to determine the core fucose levels in serum PSA. As described above, after the first IP step, the ethanolamine-treated and immunopurified total PSA was captured by the second anti-PSA antibody (M-30), which was bound to the ELISA plate, constituting the second immunopurification step. Although the immunocapture on a microwell plate and the immunoprecipitation procedure with suspended beads cannot be directly compared, this method ensured removal of most fucosylated serum proteins which would otherwise hamper the analysis of core fucosylation of PSA. Subsequently, after a blocking step, incubation with biotinylated labeled PhoSL was used to detect the core fucosylation on the captured PSA. Control pooled female serum sample spiked with standard PSA was also used. To assess the sensitivity of the ELLA, several streptavidin-peroxidase conjugate reagents and colorimetric and chemifluorescent peroxidase substrates were evaluated. The best results were obtained with a chemifluorescent peroxidase substrate achieving a limit of detection (LOD) of 0.56 ng of standard PSA/well and a limit of quantification (LOQ) of 0.71 ng of standard PSA/well. The calibration curve ranged between 0.8-13 ng of standard PSA/well.
The ELLA assay was used to analyze the PSA core fucosylation of the cohort of sera consisting of 20 BPH samples (tPSA from 7.82 to 18.20 ng/ml) and 20 high-risk PCa samples (12 with a tPSA from 8.72 to 19.53 ng/ml and 8 with a tPSA over 20 ng/ml). For each sample, two dilutions per duplicate were used that could be interpolated in the calibration curve. To perform the corresponding replicates, 1.5 ml of sera for each sample, with tPSA values over 8 ng/ml, was required. Only samples with CV below 15% were used. The interassay CV of the ELLA was determined with a high-risk PCa sample of 12.4 ng/ml of tPSA in five runs and was found to be below 10%.
The PSA core fucosylation value obtained from ELLA was divided by the free PSA value of the sample calculated using the ELECSYS platform. The resulting quotient was considered the core fucosylation ratio of the samples, plotted against ng of total serum PSA (Figure 3A), and grouped in the two pathologies of BPH and high-risk PCa patients (Figure 3B).
Representation of the core fucosylation ratio of PSA against either tPSA serum levels (panel A) or the pathology (panel B). Serum samples from BPH are indicated with white circles and high risk PCa serum samples in black circles. In Scatter dot plot, dotted line indicates the cut-off value for discriminating the two pathologies, the center line in the box represents the median, and the top (Q3) and bottom (Q1), the 75th and 25th percentiles, respectively. (C) Comparison of ROC curves for core fucosylation ratio, tPSA and %fPSA. The core fucosylation ratio gave the highest AUC value (AUC=0.94). The cut off value of 0.86 gave the highest sensitivity (90%) and specificity (95%).
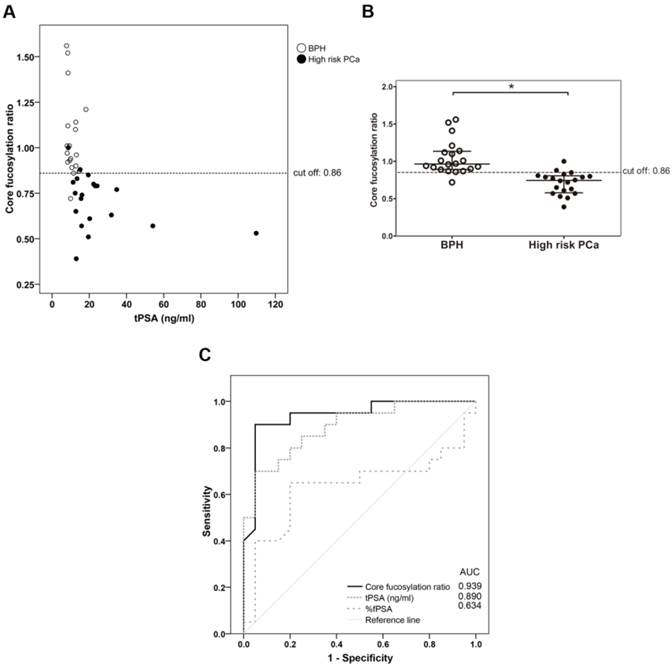
As shown in Figure 3, the BPH group showed significantly higher PSA fucosylation ratio (0.72-1.56) than high-risk PCa patients (0.53-1.00) (p<0.001). PSA fucosylation ratios in each of the BPH and PCa sample groups did not show significant correlation with their tPSA levels. PSA fucosylation ratio of the samples was, therefore, independent of their tPSA levels in each group of patients. ROC analysis of the PSA fucose ratio was also plotted and compared with that of tPSA and the %fPSA. As displayed in figure 3C, ROC assay performed with the 40 serum patient samples showed that high-risk PCa patients separate from BPH with 90% sensitivity and 95% specificity with a cut off of fucose ratio=0.86 (AUC of 0.94).
α2,3-linked sialic acid analysis of serum PSA
The methodology to determine α2,3-sialic acid levels of PSA was based on lectin chromatography using Sambucus nigra agglutinin (SNA)-agarose columns. SNA has been described to bind to α2,6-sialylated glycoconjugates.
The specificity of the SNA chromatography was first validated using standard PSA spiked in the pooled female sera. We first established the proportion of SNA-agarose/serum as 10 µl of serum in 500 µl of agarose-bound SNA. This ratio was sufficient to avoid saturation of the lectin column. To increase the quantity of serum while avoiding saturation of the column due to other sialylated serum proteins, the serum sample containing standard PSA was first immunoadsorbed using monoclonal antibodies against total PSA covalently linked to magnetic beads as described above. This step allowed us to load 0.75 ml of serum. After optimizing the protocol, the selectivity of the SNA lectin column for α2,6-sialic acid was evaluated using standard PSA. The standard PSA was chromatographed and the retained fraction was eluted using 0.5 M lactose, 0.001% BSA. A second bound fraction was eluted with 0.5 M lactose in 0.2 M acetic acid and 0.001% BSA. Quantification of the free PSA of the unbound (α2,3-sialylated PSA) and the two bound fractions (α2,6-sialylated PSA) was performed using the ELECSYS platform (see Material and Methods) and their relative percentages of PSA in these fractions were calculated. In agreement with previously published data [10, 20, 21], we determined that 25% of the standard PSA contained α2,3-sialylated N-glycans.
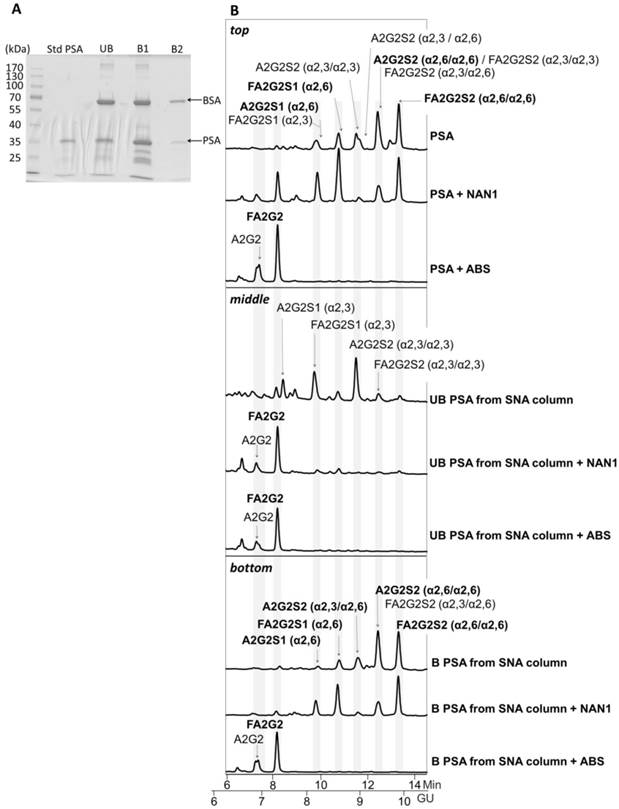
The PSA from the unbound and the two bound fractions was isolated using SDS-PAGE and Coomassie staining (Figure 4A). Densitometry analysis of gel bands revealed a 25.7% of PSA in the unbound (UB) fraction versus 74.3% in bound fractions (B1 + B2) that was consistent with the % α2,3/α2,6-sialic acid obtained in our previous analyses [21]. Protein band excision and N-glycan release with PNGase was performed as described previously [22]. N-Glycan sequencing of the released PSA N-glycans showed that the unbound fraction contained mainly monosialylated glycans and a few disialylated glycans that were digested to neutral structures with a specific α2,3-sialidase (NAN1) treatment (Figure 4B, middle panel). Therefore, the unbound fraction contained only α2,3-sialic acids that were not retained in the SNA column. Bound fractions contained α2,6-sialylated N-glycans since all were digested with ABS, but not with NANI, to neutral glycans (Figure 4B, bottom panel). The proportion of some of the N-glycans from the bound fractions was changed after NANI digestion compared with the undigested sample. This indicated that α2,3 sialic acid was present on the antenna of some of the structures only when α2,6 sialic acid was present on the other antenna in the same structure.
Glycosylation analysis of Std PSA fractionated with SNA chromatography. (A) Coomassie staining of a 10% SDS-PAGE gel containing the molecular weight marker (Mw), Std PSA and the unbound (UB), bound (B) B1, and B2 elution fractions from the SNA column. PSA is indicated with an arrow. Bovine Serum Albumin (BSA) bands arise from the elution buffer (see Materials and Methods for details). (B) HILIC-UPLC chromatograms of N-glycans released from the Std PSA (top panel), UB fraction (middle panel) and B fraction (bottom panel). N-glycan profiles after sialidase digestions with NAN1 (specific for α2,3-sialic acid) and ABS (digests all sialic acids) are depicted under undigested chromatograms. Profiles are standardized against a dextran hydrolysate (GU). N-glycans structures are abbreviated as indicated in Figure 1 legend.
This methodology was then used to analyze the percentage of α2,3-sialic acid of PSA in the cohort of sera consisting of 22 BPH samples (tPSA from 5.5 to 14.7 ng/ml), 5 low-risk samples (tPSA from 5.2 to 8.3 ng/ml), 17 intermediate-risk (tPSA from 5.1 to 17.6 ng/ml), and 21 high-risk PCa samples (13 with a tPSA from 4.1 to 19.5 ng/ml and 8 with a tPSA over 20 ng/ml). For this analysis, 0.75 ml of each serum sample was treated with ethanolamine, immunoprecipitated (total PSA IP) and chromatographed using the SNA column. Quantification of free PSA of the unbound and bound fractions was used to calculate the relative percentages of PSA in the unbound (α2,3-sialylated PSA) and bound fractions (α2,6-sialylated PSA). The inter-assay variation of the method was determined using a standard PCa serum sample (tPSA of 31.89 ng/ml) in 10-runs, and was found to be below 5%.
The plot of the percentage of unbound PSA of the samples, which corresponds to the α2,3-sialylated PSA, is shown against the concentration of total serum PSA of each sample (Figure 5A) and in the four groups (Figure 5B). There was a significant increase (p<0.001) of α2,3-sialylated PSA in the group of high-risk PCa patients (26.5-61.4%) compared with the other three groups, intermediate-risk PCa (12.7-35.5%); low-risk PCa (12.5-29.9%); BPH (13.4-33.5%). No significant differences were found between BPH and low and intermediate-risk PCa patients. Interestingly, α2,3-sialylated PSA values in each of the BPH and PCa sample groups did not show significant correlation with their tPSA levels. Therefore, they were independent variables, and a high percentage of α2,3-sialylated PSA could be found in sera with either low or high tPSA levels and vice versa for each group of patients (Figure 5A).
Representation of percentage of α2,3-sialic acid (SA) PSA against either tPSA serum levels up to 40 ng/ml (panel A) or the pathology (panel B). Serum samples from BPH are indicated with white circles, low-risk PCa with a cross, intermediate-risk PCa with a black triangle and high-risk PCa with a black circle. In Scatter dot plot, dotted line indicates the cut off value for discriminating the high-risk PCa from the other three pathologies, the center line in the box represents the median, and the top (Q3) and bottom (Q1), the 75th and 25th percentiles, respectively. (C) Comparison of ROC curves for %α2,3-SA, tPSA, and %fPSA. The %α2,3-SA gave the highest AUC value (AUC=0.97) and the cut-off value 30.05 showed the highest sensitivity (85.7%) and specificity (95.5%).
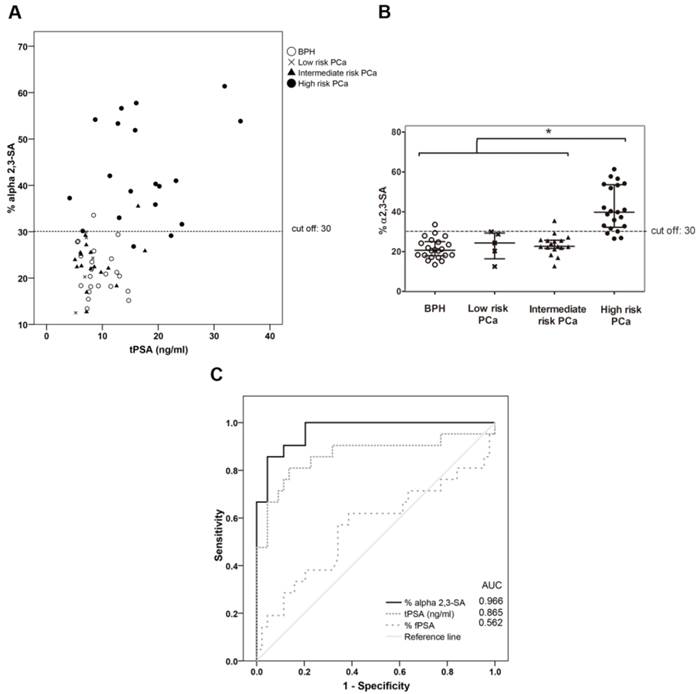
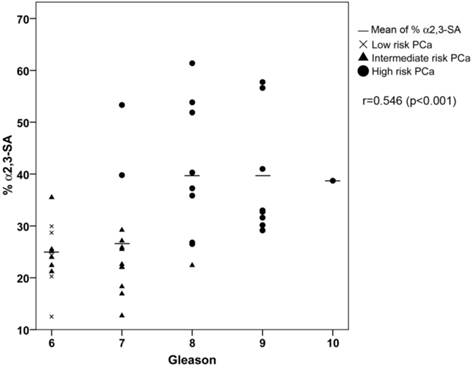
ROC analysis of the PSA α2,3-sialic acid percentage was plotted and compared with that of tPSA and the %fPSA (Figure 5C). The ROC assay performed with the 65 serum samples showed that high-risk PCa patients separate from BPH, low, or intermediate-risk prostate cancers with 85.7% sensitivity and 95.5% specificity with the α2,3-sialic acid percentage cut off of 30%. The value of the area under the curve (AUC = 0.97) was calculated as an indication of the accuracy of α2,3-sialic acid prediction. Percentage of α2,3-sialic acid in PSA correlated significantly (p<0.001) with Gleason score of the tumor (correlation coefficient 0.546) (Figure 6). Thus, this relationship may help clinicians to improve risk evaluation using a serum parameter leading to reduction in unnecessary prostate biopsies.
Correlation plot of percentage of α2,3-sialic acid (%α2,3-SA) in PSA from the PCa serum samples with their Gleason score. Serum samples from low-risk PCa are indicated with a cross, intermediate-risk PCa with a black triangle and high-risk PCa with a black circle. The mean %α2,3-SA for each Gleason score is indicated with a horizontal line.
Discussion
Altered glycosylation of PSA in PCa has already been described [37, 38]. However, this knowledge has still not been translated into clinic. The concentration of serum PSA is very low in patients' sera making the PSA N-glycan analysis very challenging and has therefore limited the development of reliable assays for its detection in large cohorts of patients.
Several approaches for evaluating PSA glycosylation from sera of BPH and PCa patients using lectins have been described [39-44]. Some of these studies have used lectin immunoaffinity columns, but they have not reported the validation of the lectin column specificity and reproducibility. The conditions reported here for the SNA immunoaffinity column have shown to segregate PSA glycoforms according to their type of sialic acid linkage, as confirmed by N-glycan sequencing of the unbound and bound fractions, in a reproducible way with less than 5% interassay variation. Our assay, performed in a large cohort of patients (N=65), showed that high-risk PCa patients can be separated from BPH, low-risk or intermediate-risk prostate cancers with 85.7% sensitivity and 95.5% specificity with a α2,3-sialic acid percentage cut off of 30%, achieving a high performance (AUC of 0.97) not reported so far by other methodologies. In addition, serum α2,3-sialic acid percentage of PSA exhibited a significant positive correlation with Gleason score (r=0.546, p< 0001) and may be used to improve PCa risk classification. This has not been accomplished so far with serum tPSA or %fPSA levels. Furthermore, the non-invasive, glycosylation-specific serum biomarker could be routinely monitored in patients in the diagnostic grey zone to identify high-risk PCa patients and may help to reduce the number of prostate biopsies, one of the main problems of current PCa screening.
In agreement with our results, other authors have also reported similar changes in sialic acid in serum PSA in smaller cohorts. Ohyama et al. [39] focused on distinguishing BPH (N=15) and PCa (N=15) and included patients with different stages in the PCa cohort. They reported an increase of α2,3-sialic acid in serum fPSA in PCa using Maackia amurensis agglutinin (MAA) and SNA binding lectins, but no sensitivity or specificity values were reported. Recently, Yoneyama et al. [45] developed a magnetic microbead-based immunoassay for the analysis of α2,3-sialic acid in fPSA using a monoclonal antibody. The results showed significantly higher serum α2,3-sialic acid levels in PCa than in non-PCa patients, which is consistent with our findings. Tajiri et al. [33] showed by MS that there was an increase in α2,3-sialic acid in the sera of two advanced PCa patients. Similarly, in previous studies we described that PSA from an advanced PCa patient serum showed an increase in α2,3-sialic acid (53%) in comparison with seminal plasma PSA from a healthy donor (22%) by N-glycan sequencing [22]. Also, Kosanovich and Jankovic [40] reported that SNA reactivity of serum PSA was lower in PCa patients with metastatic tumours compared to those with localized tumours, suggesting that the progression of PCa could be linked to an increase in the α2,3-sialic acid content of PSA.
Other lectin-based methodologies have used immune-sandwich assays with an anti-PSA capture antibody and a labelled lectin to detect the specific glycans on the PSA molecule [41, 43]. We used an immunoadsorption step to first remove the major glycosylated serum proteins that would otherwise interfere with the serum PSA analysis. With this prior immunoadsorption step followed by the anti-PSA antibody bound to the ELISA plate to capture the PSA, we could quantify the core fucosylation level of serum PSA using PhoSL. A significant decrease in core fucosylation levels in PSA from high-risk PCa (0.53 - 1.00) compared to BPHs (0.72 - 1.56) was detected. In particular, the sensitivity and the specificity of the core fucosylation PSA ratio were 90% and 95% respectively, giving an AUC of 0.94, with a similar performance as the α2,3-sialic acid percentage. This is consistent with the low core fucosylated N-glycan structures described by N-glycan sequencing in advanced PCa patients with high tPSA levels [21, 22]. These core fucosylation results observed in serum PSA are in agreement with the reported decrease of core fucosylation of proteins, including PSA, from prostate fluids in PCa [34, 46]. This decrease was reported to be more pronounced in aggressive tumors [47]. However, Li et al. recently reported an increase in PSA fucosylation in PCa serum using AAL [48], which detects several types of fucose linkages including core fucosylation. N-glycan structural analysis by MS and N-glycan sequencing have not shown significant amounts of other fucoses than the core fucose in PSA N-glycans [10, 21, 22, 35, 49]; hence the fucosylation of PSA could be attributed basically to core fucosylation. In our hands, AAL detected serum proteins other than PSA, even after two immune absorbing steps, and could not be used in our ELLA assay (data not shown).
The underlying molecular mechanisms that lead to aberrant glycosylation of cancer glycoproteins remain still unclear. However, it is well known that these changes involve altered gene expression and enzymatic activity of glycosyltransferases as well as availability of donor substrates. Changes in the enzymatic activities and gene expression levels of some glycosyltransferases responsible for the assembly of O- and N-glycans have been characterized in several PCa cell lines [50, 51] and in PCa tissues [52, 53]. Recently, the role of androgen receptor, which is an important driver of PCa, in glycogene regulation has also been described [54, 55]. In the case of core fucosylation, the FUT8 gene, which encodes for the enzyme that yields α1,6-fucose linked to the inner core GlcNAc, had not been found to be significantly altered in PCa tissues [52, 53]. However, the FUT8 enzyme has recently been described to be significantly increased in PCa tissues [51] but the expression of PCa core fucosylated proteins was not reported in this study. Mechanisms other than the FUT8 expression, such as substrate availability, the GDP-fucose transporter, and GDP-fucose synthetic enzymes, could also be involved in the synthesis of core fucosylated proteins and remain to be addressed. With respect to α2,3-sialic acid expression, several α2,3-sialyltransferases, ST3Gal I-VI, capable of catalyzing the transfer of sialic acid residues from the donor substrate CMP-sialic acid to the end of the glycoconjugate to form α2,3-sialic acid, have been identified [56]. However, ST3GAL1 and ST3GAL2, which were investigated in PCa tissues, were not found significantly altered [53].
A comprehensive study of the overall expression pattern of fucosyl and sialyltransferases and other related glycosyltransferases competing for the same substrates in PCa is of paramount importance to understand changes in the level of a specific glycoepitope. To accomplish this objective, it is also important to have tools to precisely identify the glycoepitope of interest in PCa proteins.
Our results have demonstrated the potential of the α2,3-sialic acid percentage and the core fucosylation ratio in separating high-risk PCa from low-risk PCa or BPH patients. However, validation in larger independent cohorts, including those that allow correlation with prostate cancer-specific mortality, will be required to confirm these data. The highest performance was achieved with the α2,3-sialic acid percentage in an assay that allows measurement of this marker in 0.75 ml of PCa sera with tPSA levels above 4 ng/ml. The ELLA assay requires 1.5 ml of starting serum with total PSA levels above 8 ng/ml to determine PSA core fucosylation. The sensitivity of both detection methods is being improved to be able to analyze samples in the range of 2-4 ng/ml of total PSA. In addition, a methodology to analyze altered PSA glycosylation determinants in a high-throughput fashion amenable for automation for its use in the clinical setting is under development.
There is a growing consensus that men with low-risk cancer do not usually need immediate definitive therapy and can be placed in active surveillance programs. However, some of these cancers may harbor high-grade cancer unrepresented in the biopsy which may lead to progression to aggressive disease [57, 58]. Therefore, new diagnostic tools to monitor them closely and to perform a risk reclassification are required [15]. On the other hand, men with high-risk cancers are less likely to develop metastases or to die of cancer if they are treated with radical prostatectomy or radiation therapy [8, 59]. Our described methodology, based on the determination of α2,3-sialic acid percentage in serum PSA, identifies aggressive prostate cancers with high sensitivity and specificity independent of their tPSA levels. This non-invasive technique has the potential of addressing the clinical problem of under-treatment of aggressive PCa and over-treatment of non-aggressive tumors. Identification of patients with potentially aggressive cancers (α2,3-sialic acid percentage in PSA over 30%) will allow their management accordingly [6, 8]. On the other hand, men with less aggressive cancers, based upon the serum markers, might be under active surveillance and followed up by assessing the changes in the sialylation marker to monitor the PCa evolution. The introduction of this serum marker could help improve the diagnostic pathway of prostate cancers and together with imaging techniques, such as mpMRI, unnecessary biopsies and prostatectomies could be avoided. Further analyses are being performed to validate this novel diagnostic marker in a large panel of samples.
Acknowledgements
We thank Dr. Y. Kobayashi for kindly providing PhoSL, biotinylated PhoSL and FITC-PhoSL lectin. This work was supported by the Spanish Ministerio de Economía y Competitividad (grant BIO 2010-16922 and CDTI grant IDI20130186), by the Generalitat de Catalunya, Spain (grant 2014 SGR 229), and by Roche Diagnostics (Barcelona, Spain). R.S. acknowledges funding from the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement number 260600 (“GlycoHIT”) and funding from Science foundation Ireland Starting Investigator Research grant (SFI SIRG) under grant number 13/SIRG/2164
Competing interests
The authors have filed a Spanish patent: “In vitro method for prostate cancer diagnosis”. P201531608. Date: 6th November 2015.
References
1. Siegel RL, Miller KD, Jemal A. CA Cancer J Clin. 2015. CA: a cancer journal for clinicians. 2015;65:5-29
2. Kirby R. Optimising the management of early prostate cancer. The Practitioner. 2014;258:15-8 2
3. Shariat SF, Semjonow A, Lilja H, Savage C, Vickers AJ, Bjartell A. Tumor markers in prostate cancer I: blood-based markers. Acta Oncol. 2011;50(Suppl 1):61-75
4. Heidenreich A, Bastian PJ, Bellmunt J, Bolla M, Joniau S, van der Kwast T. et al. EAU guidelines on prostate cancer. part 1: screening, diagnosis, and local treatment with curative intent-update 2013. Eur Urol. 2014;65:124-37
5. Schroder FH, Hugosson J, Roobol MJ, Tammela TL, Ciatto S, Nelen V. et al. Prostate-cancer mortality at 11 years of follow-up. N Engl J Med. 2012;366:981-90
6. Sountoulides P, Moutzouris G. Prostate-specific antigen screening, why have the guidelines changed? Expert Rev Anticancer Ther. 2014;14:1277-81
7. Salinas CA, Tsodikov A, Ishak-Howard M, Cooney KA. Prostate cancer in young men: an important clinical entity. Nat Rev Urol. 2014;11:317-23
8. Chang AJ, Autio KA, Roach M 3rd, Scher HI. High-risk prostate cancer-classification and therapy. Nat Rev Clin Oncol. 2014;11:308-23
9. Bjurlin MA, Rosenkrantz AB, Beltran LS, Raad RA, Taneja SS. Imaging and evaluation of patients with high-risk prostate cancer. Nat Rev Urol. 2015;12:617-28
10. Peracaula R, Tabares G, Royle L, Harvey DJ, Dwek RA, Rudd PM. et al. Altered glycosylation pattern allows the distinction between prostate-specific antigen (PSA) from normal and tumor origins. Glycobiology. 2003;13:457-70
11. Jung K, Reiche J, Boehme A, Stephan C, Loening SA, Schnorr D. et al. Analysis of subforms of free prostate-specific antigen in serum by two-dimensional gel electrophoresis: potential to improve diagnosis of prostate cancer. Clin Chem. 2004;50:2292-301
12. Stephan C, Cammann H, Meyer HA, Muller C, Deger S, Lein M. et al. An artificial neural network for five different assay systems of prostate-specific antigen in prostate cancer diagnostics. BJU Int. 2008;102:799-805
13. Michalakis K, Ilias I, Triantafyllou A, Polymeris A, Kastriotis I, Chairakaki AD. et al. Detection of prostate cancer by sialic acid level in patients with non-diagnostic levels of prostate-specific antigen. Maturitas. 2012;73:325-30
14. Thompson IM, Pauler DK, Goodman PJ, Tangen CM, Lucia MS, Parnes HL. et al. Prevalence of prostate cancer among men with a prostate-specific antigen level < or =4.0 ng per milliliter. N Engl J Med. 2004;350:2239-46
15. Polascik TJ, Passoni NM, Villers A, Choyke PL. Modernizing the diagnostic and decision-making pathway for prostate cancer. Clin Cancer Res. 2014;20:6254-7
16. Stephan C, Ralla B, Jung K. Prostate-specific antigen and other serum and urine markers in prostate cancer. Biochim Biophys Acta. 2014;1846:99-112
17. Lazzeri M, Haese A, Abrate A, de la Taille A, Redorta JP, McNicholas T. et al. Clinical performance of serum prostate-specific antigen isoform [-2]proPSA (p2PSA) and its derivatives, %p2PSA and the prostate health index (PHI), in men with a family history of prostate cancer: results from a multicentre European study, the PROMEtheuS project. BJU Int. 2013;112:313-21
18. Boegemann M, Stephan C, Cammann H, Vincendeau S, Houlgatte A, Jung K. et al. The percentage of prostate-specific antigen (PSA) isoform [-2]proPSA and the Prostate Health Index improve the diagnostic accuracy for clinically relevant prostate cancer at initial and repeat biopsy compared with total PSA and percentage free PSA in men aged </=65 years. BJU Int. 2016;117:72-9
19. Wang W, Wang M, Wang L, Adams TS, Tian Y, Xu J. Diagnostic ability of %p2PSA and prostate health index for aggressive prostate cancer: a meta-analysis. Sci Rep. 2014;4:5012
20. Peracaula R, Barrabes S, Sarrats A, Rudd PM, de Llorens R. Altered glycosylation in tumours focused to cancer diagnosis. Dis Markers. 2008;25:207-18
21. Tabares G, Radcliffe CM, Barrabes S, Ramirez M, Aleixandre RN, Hoesel W. et al. Different glycan structures in prostate-specific antigen from prostate cancer sera in relation to seminal plasma PSA. Glycobiology. 2006;16:132-45
22. Sarrats A, Saldova R, Comet J, O'Donoghue N, de Llorens R, Rudd PM. et al. Glycan characterization of PSA 2-DE subforms from serum and seminal plasma. OMICS. 2010;14:465-74
23. Sarrats A, Comet J, Tabares G, Ramirez M, Aleixandre RN, de Llorens R. et al. Differential percentage of serum prostate-specific antigen subforms suggests a new way to improve prostate cancer diagnosis. Prostate. 2010;70:1-9
24. Butch AW, Crary D, Yee M. Analytical performance of the Roche total and free PSA assays on the Elecsys 2010 immunoanalyzer. Clin Biochem. 2002;35:143-5
25. Becker C, Wigheden I, Lilja H. Characterization of epitope structure for 53 monoclonal antibodies against prostate-specific antigen. Tumour Biol. 1999;20(Suppl 1):13-7
26. Paus E, Nustad K, Bormer OP. Epitope mapping and affinity estimation of 83 antibodies against prostate-specific antigen. Tumour Biol. 1999;20(Suppl 1):52-69
27. Stenman UH, Paus E, Allard WJ, Andersson I, Andres C, Barnett TR. et al. Summary report of the TD-3 workshop: characterization of 83 antibodies against prostate-specific antigen. Tumour Biol. 1999;20(Suppl 1):1-12
28. Peter J, Unverzagt C, Krogh TN, Vorm O, Hoesel W. Identification of precursor forms of free prostate-specific antigen in serum of prostate cancer patients by immunosorption and mass spectrometry. Cancer Res. 2001;61:957-62
29. Kobayashi Y, Tateno H, Dohra H, Moriwaki K, Miyoshi E, Hirabayashi J. et al. A novel core fucose-specific lectin from the mushroom Pholiota squarrosa. J Biol Chem. 2012;287:33973-82
30. Royle L, Radcliffe CM, Dwek RA, Rudd PM. Detailed structural analysis of N-glycans released from glycoproteins in SDS-PAGE gel bands using HPLC combined with exoglycosidase array digestions. Methods Mol Biol. 2006;347:125-43
31. Belanger A, van Halbeek H, Graves HC, Grandbois K, Stamey TA, Huang L. et al. Molecular mass and carbohydrate structure of prostate specific antigen: studies for establishment of an international PSA standard. Prostate. 1995;27:187-97
32. Okada T, Sato Y, Kobayashi N, Sumida K, Satomura S, Matsuura S. et al. Structural characteristics of the N-glycans of two isoforms of prostate-specific antigens purified from human seminal fluid. Biochim Biophys Acta. 2001;1525:149-60
33. Tajiri M, Ohyama C, Wada Y. Oligosaccharide profiles of the prostate specific antigen in free and complexed forms from the prostate cancer patient serum and in seminal plasma: a glycopeptide approach. Glycobiology. 2008;18:2-8
34. White KY, Rodemich L, Nyalwidhe JO, Comunale MA, Clements MA, Lance RS. et al. Glycomic characterization of prostate-specific antigen and prostatic acid phosphatase in prostate cancer and benign disease seminal plasma fluids. J Proteome Res. 2009;8:620-30
35. Behnken HN, Ruthenbeck A, Schulz JM, Meyer B. Glycan analysis of Prostate Specific Antigen (PSA) directly from the intact glycoprotein by HR-ESI/TOF-MS. J Proteome Res. 2014;13:997-1001
36. Saldova R, Asadi Shehni A, Haakensen VD, Steinfeld I, Hilliard M, Kifer I. et al. Association of N-glycosylation with breast carcinoma and systemic features using high-resolution quantitative UPLC. J Proteome Res. 2014;13:2314-27
37. Vermassen T, Speeckaert MM, Lumen N, Rottey S, Delanghe JR. Glycosylation of prostate specific antigen and its potential diagnostic applications. Clin. Chim Acta. 2012;413:1500-5
38. Drake RR, Jones EE, Powers TW, Nyalwidhe JO. Altered glycosylation in prostate cancer. Adv Cancer Res. 2015;126:345-82
39. Ohyama C, Hosono M, Nitta K, Oh-eda M, Yoshikawa K, Habuchi T. et al. Carbohydrate structure and differential binding of prostate specific antigen to Maackia amurensis lectin between prostate cancer and benign prostate hypertrophy. Glycobiology. 2004;14:671-9
40. Kosanovic MM, Jankovic MM. Sialylation and fucosylation of cancer-associated prostate specific antigen. J BUON. 2005;10:247-50
41. Meany DL, Zhang Z, Sokoll LJ, Zhang H, Chan DW. Glycoproteomics for prostate cancer detection: changes in serum PSA glycosylation patterns. J Proteome Res. 2009;8:613-9
42. Fukushima K, Satoh T, Baba S, Yamashita K. alpha1,2-Fucosylated and beta-N-acetylgalactosaminylated prostate-specific antigen as an efficient marker of prostatic cancer. Glycobiology. 2010;20:452-60
43. Dwek MV, Jenks A, Leathem AJ. A sensitive assay to measure biomarker glycosylation demonstrates increased fucosylation of prostate specific antigen (PSA) in patients with prostate cancer compared with benign prostatic hyperplasia. Clin Chim Acta. 2010;411:1935-9
44. Li C, Zang T, Wrobel K, Huang JT, Nabi G. Quantitative urinary proteomics using stable isotope labelling by peptide dimethylation in patients with prostate cancer. Anal Bioanal Chem. 2015;407:3393-404
45. Yoneyama T, Ohyama C, Hatakeyama S, Narita S, Habuchi T, Koie T. et al. Measurement of aberrant glycosylation of prostate specific antigen can improve specificity in early detection of prostate cancer. Biochem Biophys Res Commun. 2014;448:390-6
46. Vermassen T, Van Praet C, Vanderschaeghe D, Maenhout T, Lumen N, Callewaert N. et al. Capillary electrophoresis of urinary prostate glycoproteins assists in the diagnosis of prostate cancer. Electrophoresis. 2014;35:1017-24
47. Nyalwidhe JO, Betesh LR, Powers TW, Jones EE, White KY, Burch TC. et al. Increased bisecting N-acetylglucosamine and decreased branched chain glycans of N-linked glycoproteins in expressed prostatic secretions associated with prostate cancer progression. Proteomics Clin Appl. 2013;7:677-89
48. Li QK, Chen L, Ao MH, Chiu JH, Zhang Z, Zhang H. et al. Serum fucosylated prostate-specific antigen (PSA) improves the differentiation of aggressive from non-aggressive prostate cancers. Theranostics. 2015;5:267-76
49. Leymarie N, Griffin PJ, Jonscher K, Kolarich D, Orlando R, McComb M. et al. Interlaboratory study on differential analysis of protein glycosylation by mass spectrometry: the ABRF glycoprotein research multi-institutional study 2012. Mol Cell Proteomics. 2013;12:2935-51
50. Gao Y, Chachadi VB, Cheng PW, Brockhausen I. Glycosylation potential of human prostate cancer cell lines. Glycoconj J. 2012;29:525-37
51. Wang X, Chen J, Li QK, Peskoe SB, Zhang B, Choi C. et al. Overexpression of alpha (1,6) fucosyltransferase associated with aggressive prostate cancer. Glycobiology. 2014;24:935-44
52. Barthel SR, Wiese GK, Cho J, Opperman MJ, Hays DL, Siddiqui J. et al. Alpha 1,3 fucosyltransferases are master regulators of prostate cancer cell trafficking. Proc Natl Acad Sci U S A. 2009;106:19491-6
53. Chen Z, Gulzar ZG, St Hill CA, Walcheck B, Brooks JD. Increased expression of GCNT1 is associated with altered O-glycosylation of PSA, PAP, and MUC1 in human prostate cancers. Prostate. 2014;74:1059-67
54. Itkonen HM, Engedal N, Babaie E, Luhr M, Guldvik IJ, Minner S. et al. UAP1 is overexpressed in prostate cancer and is protective against inhibitors of N-linked glycosylation. Oncogene. 2015;34:3744-50
55. Munkley J, Oltean S, Vodak D, Wilson BT, Livermore KE, Zhou Y. et al. The androgen receptor controls expression of the cancer-associated sTn antigen and cell adhesion through induction of ST6GalNAc1 in prostate cancer. Oncotarget. 2015;6:34358-74
56. Harduin-Lepers A, Vallejo-Ruiz V, Krzewinski-Recchi MA, Samyn-Petit B, Julien S, Delannoy P. The human sialyltransferase family. Biochimie. 2001;83:727-37
57. Klotz L, Emberton M. Management of low risk prostate cancer-active surveillance and focal therapy. Nat Rev Clin Oncol. 2014;11:324-34
58. Lund L, Svolgaard N, Poulsen MH. Prostate cancer: a review of active surveillance. Res Rep Urol. 2014;6:107-12
59. Ehdaie B, Scardino PT. Decade in review-prostate cancer: a decade of progress in detection and treatment. Nat Rev Urol. 2014;11:618-20
Footnotes
ELECSYS® is trademark of Roche.
Author contact
![]() Corresponding author: Dr. Rosa Peracaula, Department of Biology, Faculty of Sciences. University of Girona, Campus Montilivi s/n. 17071 Girona, Spain. Phone: +34 972418370; Fax: +34 972418150, E-mail: rosa.peracaulaedu
Corresponding author: Dr. Rosa Peracaula, Department of Biology, Faculty of Sciences. University of Girona, Campus Montilivi s/n. 17071 Girona, Spain. Phone: +34 972418370; Fax: +34 972418150, E-mail: rosa.peracaulaedu