Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2016; 6(10):1542-1556. doi:10.7150/thno.15297 This issue Cite
Research Paper
Ultrasound/Magnetic Targeting with SPIO-DOX-Microbubble Complex for Image-Guided Drug Delivery in Brain Tumors
Ching-Hsiang Fan1, Yu-Hang Cheng1, Chien-Yu Ting1, Yi-Ju Ho1, Po-Hung Hsu2, Hao-Li Liu2,3, ![]() , Chih-Kuang Yeh1,
, Chih-Kuang Yeh1, ![]()
1. Department of Biomedical Engineering and Environmental Sciences, National Tsing Hua University, No. 101, Section 2, Kuang-Fu Road, Hsinchu 30013, Taiwan.
2. Department of Electrical Engineering, Chang-Gung University, 259 Wen-Hwa 1st Road, Kuei-Shan, Tao-Yuan 33302, Taiwan.
3. Medical Imaging Research Center, Institute for Radiological Research, Chang Gung University and Chang Gung Memorial Hospital, 259 Wen-Hwa 1st Road, Kuei-Shan, Tao-Yuan 33302, Taiwan.
Received 2016-2-16; Accepted 2016-4-27; Published 2016-6-18
Abstract

One of the greatest challenges in the deployment of chemotherapeutic drugs against brain tumors is ensuring that sufficient drug concentrations reach the tumor, while minimizing drug accumulation at undesired sites. Recently, injection of therapeutic agents following blood-brain barrier (BBB) opening by focused ultrasound (FUS) with microbubbles (MBs) has been shown to enhance drug delivery in targeted brain regions. Nevertheless, the distribution and quantitative deposition of agents delivered to the brain are still hard to estimate. Based on our previous work on superparamagnetic iron oxide (SPIO)-loaded MBs, we present a novel theranostic complex of SPIO-Doxorubicin (DOX)-conjugated MB (SD-MB) for drug delivery to the brain. Magnetic labeling of the drug enables direct visualization via magnetic resonance imaging, and also facilitates magnetic targeting (MT) to actively enhance targeted deposition of the drug. In a rat glioma model, we demonstrated that FUS sonication can be used with SD-MBs to simultaneously facilitate BBB opening and allow dual ultrasound/magnetic targeting of chemotherapeutic agent (DOX) delivery. The accumulation of SD complex within brain tumors can be significantly enhanced by MT (25.7 fold of DOX, 7.6 fold of SPIO). The change in relaxation rate R2 (1/T2) within tumors was highly correlated with SD deposition as quantified by high performance liquid chromatography (R2 = 0.93) and inductively coupled plasma-atomic emission spectroscopy (R2 = 0.94), demonstrating real-time monitoring of DOX distribution. Our results suggest that SD-MBs can serve as multifunction agents to achieve advanced molecular theranostics.
Keywords: superparamagnetic iron oxide, blood-brain barrier, microbubbles, focused ultrasound, brain tumor.
Introduction
Current clinical chemotherapeutic agent treatments for malignant brain tumors such as glioblastoma multiforme (GBM) still show very limited gains in therapeutic efficacy (1,2). The main obstacles to improved performance include structural limitations imposed by the blood-brain barrier (BBB) and blood-tumor barrier (BTB) structure, which prevent drugs from entering tumors, and serious side effects arising from high doses of toxic antineoplastic agents necessary for complete tumor control (3). In addition, the hampering brain tumor treatment effect by BBB/BTB might lead to a high rate of brain-tumor recurrence. Thus, one of the essential issues for improving brain tumor chemotherapeutic agent delivery is to overcome the BBB/BTB.
Transcranial focused ultrasound (FUS) exposure with microbubbles (MBs) can be used to temporarily open the BBB or permeate the BTB, and thus offers great potential in the delivery of therapeutic agents for brain tumor treatment (4). Injection of chemotherapeutic agents followed by FUS-induced BBB opening has been reported to successfully enhance local drug accumulation in the targeted brain regions (5). Since MBs serve as a key catalyst to induce the BBB-opening effect, MBs have attracted considerable attention as drug carriers (6-8). One key advantage in encapsulating unstable agents in MBs is to prevent fast degradation, thus improving treatment efficacy, while reducing the required dose. Also, the encapsulated agents can be control-released during the FUS-triggered MB destruction process, reducing the off-target dose (9,10). However, few studies have explored the use of MBs as carriers or for real-time quantitative or semi-quantitative monitoring of drug distribution, thus limiting the application in clinical practice.
For imaging drug-loaded carriers, some previous studies have attempted to use drug-loaded magnetic nanoparticles or superparamagnetic iron oxide (SPIO) nanoparticles for MRI-T2* imaging (8,11,12). However, T2* imaging only shows the qualitative contrast change, so the actual dynamics, distribution, and deposition of therapeutic agents cannot be truly reflected. Previously, our group demonstrated a SPIO-labeled phospholipid-based bubble structure to allow conjugation of therapeutic agents to concurrently disrupt BBB and perform targeted drug delivery (9). Although the study showed the potential use of MBs as a theranostic tool to perform image-guided drug delivery, a spatial and quantitative correlation between SPIO and chemotherapeutic agents was lacking since the DOX and SPIO have distinct pharmacodynamic behaviors. T2* imaging-based monitoring was used, so only qualitative information was presented without detailed pharmacodynamic behaviors and drug accumulation information.
Another concern is that, despite the success of the targeted opening of the BBB, drug delivery in such cases is still conducted passively, relying on the free diffusion of agents across the barrier. Magnetic nanoparticles could be magnetized and become physically sensitive to external magnetic fields. Magnetic targeting (MT) can then be applied to actively enhance drug deposition at the target site, thus potentially increasing the therapeutic dose delivered far beyond that obtainable by passive diffusion. We thus hypothesized that introducing drug-loaded magnetic nanoparticles into MBs would allow for more direct drug monitoring in vivo. Directly labeling of therapeutic agents with SPIO allows for direct measurement and monitoring of drug delivery, and the MT could serve as an active targeting tool to further enhance targeted drug delivery at the BBB-opening site.
Here, we report the fabrication of a therapeutic complex (SPIO-DOX; SD) in which SPIO nanoparticles are directly conjugated with DOX and are embedded in lipid MBs (SD-MBs). By concurrently triggering FUS exposure and MT, we demonstrate that SD-MBs can simultaneously trigger BBB opening and allow drug penetration into the brain tumor in an actively targeted fashion. We also show that the release and biodistribution of SD complexes can be monitored in real time via the use of MRI relaxometry.
Materials and methods
Fabrication of SPIO-DOX (SD) complex
The SD complexes were freshly prepared prior to use. Superparamagnetic iron oxide (SPIO) nanoparticles modified with surface functional amino group (mean diameter = 35.7 ± 9.23 nm, concentration = 8.13 mg-Fe/mL) were purchased from MagQu Co., Ltd (New Taipei city, Taiwan). Doxorubicin was purchased from Seedchem Company PTY LTD (Vic Melbourne, Australia). The SPIO solution was replaced with deionized distilled water (DDW), and was well mixed with DOX solution under sonication (Model 2510, Branson, NY, USA). The SPIO particles and DOX were conjugated via natural reactions between the amino group and carbonyl group in a 37°C bath for a 4-h incubation period, followed by slow addition of phosphate-buffered saline (PBS) and SD complex collection using magnetic precipitation. The collected SD solution was centrifuged at 11,000 g for 3 min to separate the well-conjugated SD complex from unconjugated DOX molecular, and was re-suspended with PBS. The SD complex conjugation efficiency (i.e., binding of DOX on SPIO) was evaluated by quantifying DOX molecules and SPIO particles of SD via inductive coupled plasma-atomic emission spectroscopy (ICP-AES, JY 2000, JobinYvon, Longjumeau, France) and a plate reader system (Infinite® 200PRO series, Tecan, AG, Switzerland), individually. SD complex samples were treated with the addition of an equal volume of 70% nitric acid under 60°C for 2 h to completely dissolve the iron oxide particles. DDW was mixed with the products after nitrification to a final 35-fold dilution, and the amount of SPIO was finally measured by ICP-AES. DOX concentration on SD complex was measured from the fluorescence intensity of DOX. To avoid the interference of SPIO with the absorption value of DOX, 35% HCl was added before measurement.
Preparation of SD complex-loaded microbubbles (SD-MBs)
The lipid shell of SD-MBs was composed of 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC, Avanti Polar Lipids, AL, USA), 1,2-Distearoyl-sn-glycero-3-phospho-rac-glycerol sodium salt (DSPG, Avanti Polar Lipids) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(poly(ethyleneglycol))-2000] (DSPE-PEG2000, Avanti Polar Lipids) at a molar ratio of 21:21:1, and was homogeneously dissolved with chloroform. The chloroform was then removed via an evaporator (R‐210, Büchi Labortechnik AG, Flawil, Switzerland). The glycerol PBS (5 wt%) and SD complexes (1-4 mg) were mixed with the dried lipid film. The solution was degassed. Subsequently, the samples were then refilled with perfluoropropane (C3F8). After intensive shaking via an agitator for 45 s, SD-MBs were formed. The products were finally placed on ice for 30 min to stabilize the MB structure before use. Pure lipid MBs were fabricated by a similar protocol to that for SD-MBs, but without mixing SD complexes.
Characterization of SD-MBs
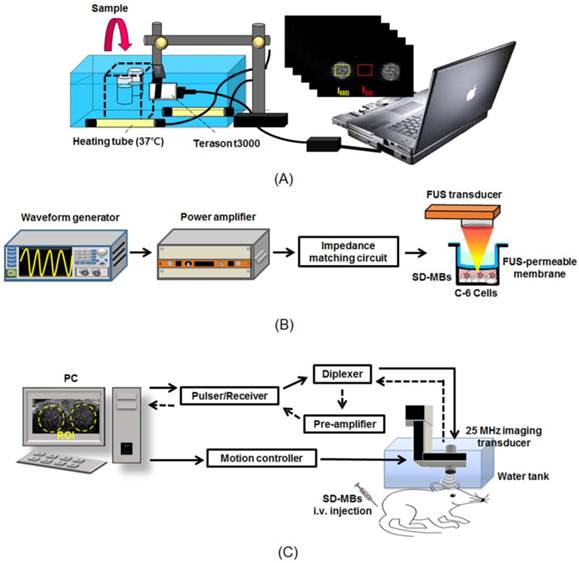
The concentration size and distribution of SD-MBs were analyzed using a coulter counter (Multisizer 3, Beckman Coulter, FL, USA). The structure and DOX localization of SD-MBs were visualized by a fluorescence microscope (IX-71, Olympus, NY, USA) (excitation: 480 nm; emission: 570 nm). To verify the loading of SD complexes on the MBs, the SD-MBs were observed with a transmission electron microscope (TEM, Hitachi H-7100, Tokyo, Japan). The in vitro acoustic stability of SD-MBs was determined by a commercial 7.5-MHz ultrasound imaging system (model t3000, Terason, MA, USA) (Fig. 1A). SD-MBs were loaded into 2% agarose phantom and were diluted 100 times with normal saline under 37°C for 1 h. B-mode images were acquired periodically with a time interval of 10 min. The contrast-to-noise ratio (CNR) was used to quantify the contrast enhancement of SD-MBs as follows:
Experimental setup of (A) in vitro acoustic stability of SD-MBs, (B) in vitro antitumor effect of SD-MBs with FUS and (C) in vivo acoustic stability of SD-MBs.
,
where IMB is denoted as the backscatter intensity of SD-MBs and IBK is denoted as the backscatter intensity of water sample.
The R2 relaxivities of the SD-MBs and SD complexes were measured using a 7-Tesla MRI system (ClinScan, Bruker, Ettlingen, Germany). Samples were loaded in 96-well plates and diluted 50 to 400 times with a 2:1 (v/v) mixture of PBS and ultrasound coupling gel (Aquasonic 100, Parker laboratories, NJ, USA). Standard measurements were performed using a multi-echo spin echo sequence with the following parameters: matrix size = 192 × 384; slice thickness = 1.2 mm; echo time (TE) = 7.1, 14, 21, 28, 35, 45, 112 ms; repetition time (TR) = 2000 ms; resolution = 4.4 × 4.4; and field of view (FOV) = 44 × 88 mm. The acquired images were processed with MATLAB. Spin-spin relaxivity (R2), which is the inverted value of T2, was calculated and a calibration curve was established by correlating R2 values with SPIO concentrations. The real-time monitoring of drug release in vivo was also quantified using this calibration curve. The superparamagnetic properties of SD complexes and SD-MBs suspension were confirmed by a magnetometer (Model 1660, ADE-DMS Co. Ltd., CA, USA).
The payload of SD complex on SD-MBs was also estimated. The supernatant from the SD-MBs suspension was collected as the unloaded SD complex (free SD). The SD-MBs were resuspended with PBS, subjected to sonication for 5 min to completely destruct MBs, and centrifuged at 11,000 g for 2 min. the final precipitate was resuspended with DDW as the loaded SD complex. Subsequently, a nitrification reaction was exerted on both free SD and loaded SD complex followed by ICP-AES estimation. The loading efficiency of SD on MBs was calculated as:
,
where Wloaded SD is the amount of SD complex loaded on SD-MBs, and Wfree SD is the amount of SD complex that was not encapsulated in SD-MBs.
The leakage of SD complex from SD-MBs was observed to assess the drug retention ability of MB carriers. Freshly prepared SD-MBs were diluted 100 times with PBS under 37°C, and the amount of SD complex encapsulated in the lipid shell of SD-MBs at each time point (Wencapsulated) was quantified by analyzing fluorescent intensity of SD complexes. The drug retention rate was calculated as the ratio of Wencapsulated to initial SD complex loading amount.
In vitro studies on C6 glioma cells
In vitro FUS sonication setup
Figure 1B shows the in vitro experimental setup of the FUS sonication system. A 1-MHz FUS transducer (V302, Panametrics, MA, USA) was driven by a function generator (WW2571, Tabor Electronics, Haifa, Israel) and amplified by a power amplifier (Model 150A100B, Amplifier Research, Hazerswoude-Dorp, Netherlands) for transmitting treatment pulses (9). In order to avoid cell contamination, an ultrasound transmittable membrane was placed between the cell dish and the water cone of the transducer. The focal zone had a length and diameter of 26 and 3 mm, respectively. The acoustic pressures used in this study were measured with attenuation by a rat skull (thickness = 0.9 mm) with a polyvinylidene difluoride-type hydrophone (model HGL-0085, ONDA, Sunnyvale, CA, USA) in distilled and degassed water at 25°C. The intact skull was degassed before experiment to avoid any cavitation effects at the skull level that may have affected propagation of the beam of FUS.
In vitro FUS-triggered drug release and cytotoxicity measurements
C-6 glioma cells were cultured as in a previous study (9). Before starting the experiment, 104 of cells were placed in each well of a 96-well plate and incubated in 37°C with 5% CO2 and for 24 h.
Normal saline, DOX, SD complexes, and SD-MBs (with the same DOX concentration of 22 ppm) were co-cultured with the cells for 2 h with or without the process of MT with a 0.48 T permanent magnet (Little Giant International Co., Ltd, Taipei, Taiwan) under the cell dish for 10 min, and with or without MB administration followed by FUS sonication. FUS irradiation was applied with an acoustic pressure of 0.3 MPa (equivalent to mechanical index (MI) value of 0.3), 1,000 cycles, and 1 Hz of pulse repetition frequency (PRF), 1 min of sonication duration, and a total of 4 sonication points. Note that the focus of the transducer was in the cell layer. After a 20-min incubation period, the medium (containing sample) of the cells was moved and refilled with fresh culture medium. The cell viability and proliferation were measured by the Alamar Blue (AbDSerotec, Oxford, UK).
In vivo studies on glioma model
Animal preparation
The animal studies procedures in this study followed the guidelines of the National Tsing-Hua University Institutional Animal Care and Use Committee (IACUC approval number: NTHU10156). Before the experiment, rats (male Sprague-Dawley, 200-250 g) were anesthetized I.P. by chloral hydrate (400 mg/kg). The drugs, dyes and MBs were I.V. administrated by placing a PE50 catheter (IntramedicTM, Clay Adams Inc., NJ, USA) in the jugular vein of the rats.
The rat brain tumor model was constructed by injecting 7 × 105 cells of C-6 glioma cell suspension into the brain of rats (6). Animals were treated 10 days after tumor implantation (tumor volume of 22.9 ± 9.4 mm3 measured by the 7-T MRI).
In vivo contrast-enhancement capability and stability of SD-MBs
The in vivo acoustic contrast capability and stability of SD-MBs were estimated by evaluating their backscatter intensity in the rat brains (N = 3) with ultrasound B-mode images. In order to permit the delivery of the ultrasound imaging beam into the brain, a craniotomy (approximately 1 × 0.5 cm2) was performed before imaging.
Ultrasound imaging was obtained with a homemade 25-MHz ultrasonic imaging system with MI value of 0.048 (Fig. 1C) (6). A serial B-mode images were acquired prior to and following the SD-MBs bolus administration (1.0 mL, 2 times diluted with 0.9% normal saline). A time-intensity curve (TIC) within the cerebral cortex and dorsal sagittal sinus were created by calculating the intensities of the regions of interest (ROI) of these images. The lifetime of SD-MBs was then estimated by inspecting the clearance time of MBs. The data analyses were applied using MATLAB (MathWorks, MA, USA) offline.
In vivo experimental setup
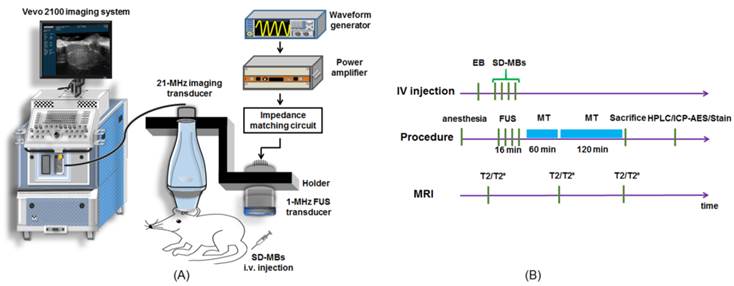
The integrated sonication platform illustrated in Fig. 2A consisted of a 1-MHz FUS transducer utilized for delivering treatment pulses to open the BBB and concurrently elicit the release of SD complexes from SD-MBs, and a commercial ultrasonic imaging system (Vevo2100, VisualSonics, Toronto, Canada) (13). The two transducers were arranged in parallel, separated by approximately 49 mm using a homemade holder that ensured that the foci of the FUS beam and the imaging beam were fixed at the same depth.
(A) Experimental setup of in vivo FUS-triggered SD-MBs releasing SD complexes. (B) Experimental flowchart.
The fur on the top of the tumor-bearing animal's head was shaved before it was placed prone on the stereotaxic apparatus. After guiding by high frequency ultrasound imaging, FUS was transcranially sonicated to the tumor location (PRF: 1 Hz, acoustic pressure: 0.3 MPa, cycle number: 5000, sonication duration: 4 min, and sonication site: 4, MI: 0.3).
The experimental flowchart is presented in Fig. 2B. Tumor-bearing rats were randomly divided into four groups: 1) administration of SD complexes and MT procedure following BBB-opening by pure-lipid MBs and FUS (SD + MBs + FUS + MT group, N = 10); 2) administration of only SD-MBs (SD-MBs, N = 10); 3) FUS sonication following injection of SD-MBs (SD-MBs + FUS group, N = 10); and 4) MT procedure after treating with SD-MBs and FUS (SD-MBs + FUS + MT group, N = 10). The dose of SD-MBs and injection volume for each group were adjusted to 4 mg and 1 mL per animal, individually. It is noteworthy that in the SD + pure-lipid MBs + FUS + MT, SD-MB + FUS and SD-MBs + FUS + MT group, SD-MBs (2.5×1010/mL, total volume: 1 mL) were administrated on a fractionated basis 4 times, together with 4 sonication sites to cover the whole tumor (i.e., 0.25 mL of the MBs/SD-MBs were injected, followed by 4 min of sonication). For animals conducting MT procedure, animals were anesthetized with isoflurane and a 0.48 T permanent magnet was placed tightly to the scalp of the animal's left head for 3 h.
Real-time monitoring of drug release using magnetic resonance imaging (MRI)
The release and tumor morphology of the SD complexes were monitored by MRI R2 map and T2-weighted (T2W) images before treatment, and 1 h and 3 h after treatment. The distribution and concentration of SD complexes delivered into the brain tissues were evaluated in real time by analyzing SPIO signals using the MRI R2 map. Local accumulation of SPIO resulted in an additional loss of phase coherence of the spins, thereby decreasing regional transverse relaxation times and signal intensity (14). Also, the spin-spin relaxivity of SPIO is proportional to the R2 relaxation rate. Therefore, the distribution and concentration of SD complexes delivered into the brain tissues could be evaluated in real time by analyzing the SPIO signals using MRI R2 map.
T2W images were used to identify tumor morphological images with the following parameters: echo time (TE) = 41 ms; TR = 2540 ms; field of view (FOV) = 34 × 40 mm; matrix size = 272 × 320; resolution = 8 × 8 mm; slice thickness = 0.6 mm. R2 maps were generated from serial T2W images using a multi-echo time sequence with the following parameters: TE = 7.1, 14, 21, 28, 35, 45, 112 ms; TR = 2000 ms; matrix size = 320 × 512; slice thickness = 0.7 mm; FOV = 37 × 60 mm; and resolution = 8.6 × 8.5 mm. The obtained MRI images were processed by MATLAB to calculate the R2 value (1/T2) by fitting an exponential curve of the signal intensities as a function of echo time for each pixel. ROIs were set at the tumor site in coronary slices to calculate of average tumor R2 value. Subsequently, color-coded R2 maps were generated.
In vivo quantification of SD complex release
Following MRI imaging, the rats (SD-MBs, SD + pure-lipid MBs + FUS + MT, SD-MBs + FUS, and SD-MBs + FUS + MT group, N=4 for each group) were sacrificed and perfused with 0.9 % normal saline to flush blood and resident SD complexes from the cerebral vessels. Tumor tissues were collected and weighted. The DOX was extracted with the addition of 2 mL of HCl (2 M, at 4 °C). The extracted solution was centrifuged (15,000 g, 15 min, SIGMA 3-30K, Heraeus Co., Germany). The supernatant of the sample was then collected, filtered through a 0.22 μm filter, diluted with mobile phase solution and analyzed using high performance liquid chromatography (HPLC) with a UV detector (S1125, Sykam GmbH, Germany).
The mobile phase solution consisted of 50 vol% DDW diluted with HPLC-grade methanol in DDW. A column packed with RP-18 (Alltima C-18 3u, Alltech, IL, USA) was used with a detection wavelength of 256 nm with a flow rate of 1.0 mL/min. The amount of DOX was analyzed to determine the area under its peak at 3.6 ± 0.2 min retention time. DOX concentration was expressed per gram of tissue.
In vivo quantification of SPIO accumulation
The amount of SPIO accumulation in the tumors (SD-MBs, SD+pure-lipid MBs+FUS+MT, SD-MBs+FUS, and SD-MBs+FUS+MT group, N=4 for each group) was analyzed ICP-AES. Similar volumes of tumor tissues and contralateral brain tissues were collected. These samples were digested in 70 vol% nitric acid for 2 h at 60°C. The digested aqueous samples were measured by ICP-AES (RF power: 1500 W; carrier gas flow: 0.98 L/min; plasma gas flow: 15 L/min; auxiliary gas flow: 0.9 L/min). The concentration of SPIO was expressed per gram of tissue.
BBB opening confirmation and Histological analysis
Successful FUS-induced BBB opening was verified via Evans blue (EB, 100 mg/kg) staining with acoustic pressure of 0.1-0.5 MPa (Sigma-Aldrich, MO, USA) (SD-MB+FUS group, N = 6). The removed brains were embedded in optimal-cutting-temperature compound (Tissue-Tek, Sakura, CA, USA) and frozen at -50°C. Tissues were sliced into coronal sections. The occurrence of erythrocyte extravasation was confirmed by hematoxylin and eosin (H&E) staining. Prussian Blue staining was performed using ferric hexacyanoferrate and hydrochloric acid (Sigma-Aldrich) to verify the accumulation of iron. Slides were immersed in a staining solution consisting of 50 vol% 2% potassium ferrocyanide mixed with 2% hydrochloric acid for 30 min at room temperature, and were rinsed in DDW. Histological confirmations were applied using light microscopy. The distribution of DOX were assessed by red fluorescent microscopic imaging.
Statistical analysis
All data are expressed as mean ± standard error of mean (SEM) of at least three independent samples. All statistical evaluations were carried out with one-way ANOVA. A p-value of less than 0.05 was considered significant. A linear regression analysis with the least square fitting was performed to assess the correlations between samples, and an R square value closer to 1 was considered to indicate the credibility of the selected fitting curve.
Results
Characterization of SD-MBs
Figure 3A illustrates the fabricated SD complexes. The co-localization of the SD complex in the bright field images and the fluorescent image indicated a good conjunction of SPIO nanoparticles and DOX. The TEM images confirmed that the size of SD complexes was below 50 nm. The conjugation efficiency between DOX and SPIO was measured by calculating the ratio of bound DOX on 0.4 mg SPIO and initial DOX amount (Fig. 3B). The DOX conjugation efficiency declined with increased mixing of DOX into SPIO (0.4 mg: 99.1% ± 1.6% vs. 2 mg: 65.1 ± 2.5%). The DOX binding efficiency to SPIO was optimized at 1 mg of DOX during mixing. Further increasing DOX amount would decrease the conjugation efficiency. The ratio of DOX and SPIO performed in the following experiments was given to be 1:0.4.
Structure and property of SD complex. (A) Microscope bright-field image, fluorescent image and TEM image of the SD complex. (B) Conjugation efficiency of SPIO (0.4 mg) and DOX (0.4-2 mg).
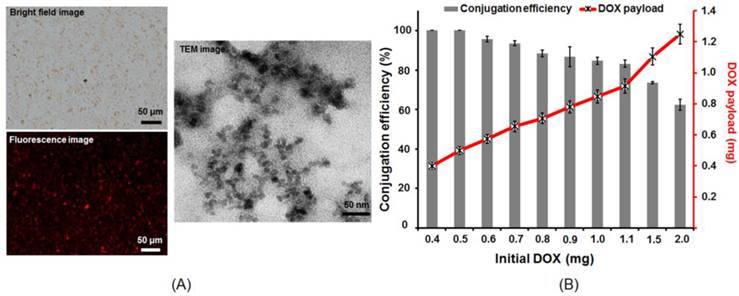
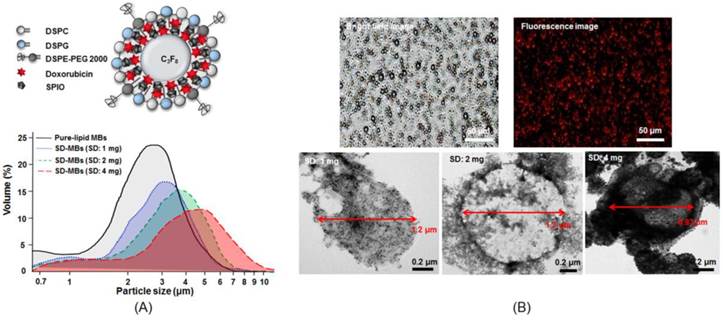
Figure 4A shows the size distribution of SD-MBs and pure-lipid MBs. The mean size and mean concentration of pure-lipid MBs were 2.8 ± 0.9 μm and (9.8 ± 0.2) × 108 MB/mL, respectively. SD complexes (1 mg) loaded into MBs increased the size (3.1 ± 0.2 μm), but decreased the concentration ((6.8 ± 0.3) × 108 MB/mL) of SD-MBs. Further increasing the loaded SD complexes up to 4 mg slightly increased the size (5.4 ± 1.1 μm), but clearly reduced the concentration ((5.1 ± 0.4) × 108 MB/mL) of SD-MBs. Bright-field images illustrated the uniform size distribution of the SD-MBs (Fig. 4B). The conjugated SD complex onto MBs were confirmed by fluorescent image. TEM images showed the SD complexes (black spot) located on the lipid shell (gray layer) of the MBs, suggesting SD complexes were embedded on the shell of the SD-MBs. Also, the embedded SD complexes on the SD-MBs clearly could be regulated by the amount of SD complexes added.
Size distribution and structure of SD-MBs. (A) upper: illustration of SD-MBs structure; lower: Size distribution of pure lipid MBs and SD-MBs with different SD complex payload measured by coulter counter. (B) Microscope bright-field, fluorescent and TEM images of SD-MBs.
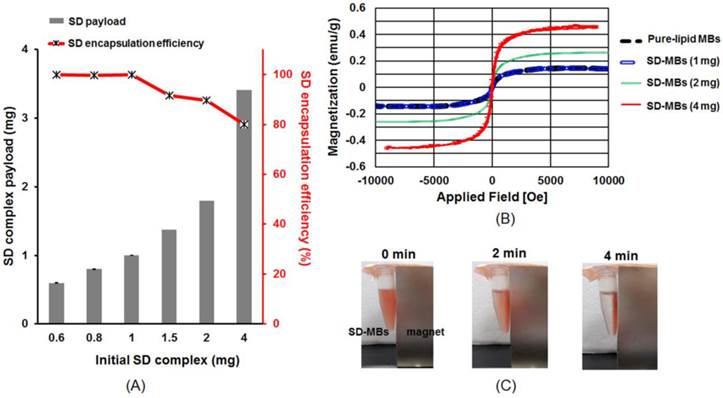
After mixing 0.6-1 mg SD complexes into MBs, the MBs demonstrated a significantly higher SD complex loading capability (nearly 100%) (Fig. 5A). When more than 1.5 mg SD complexes was added into the MBs, the SD complex loading efficiency started to decline. However, 80% of the SD complexes were still found on the lipid shell when 4 mg SD complexes were added to the MBs. To determine how to maximize the theranostic ability of SD-MBs, we conducted the following cell and animal experiments using SD-MBs with 4 mg of SD complexes (the actual SD complexes payloads was 3.4 ± 0.3 mg). The magnetization-magnetic field strength curves (M-H curves) using a showed that SD-MBs achieved a stable state under the applied magnetic fields and presented an overlapping loop without remnant magnetization as well as hysteresis (Fig. 5B), suggesting that the saturation magnetization of SD-MBs also could be enhanced by increasing the loading of SD complexes into MBs (1 mg: 0.1 emu/g vs. 4 mg: 0.5 emu/g). In addition, under a magnetic field for 4 min, the suspended SD-MBs were attracted to the permanent magnet, suggesting the high magnetization capacity of SD-MBs (Fig. 5C).
(A) Encapsulation efficiency of SD complexes onto MBs. (B) Saturation magnetization of SD-MBs with different SD complex payloads (1-4 mg). (C) Magnetization property of SD-MBs.
The acoustic stability of SD-MBs was investigated by time-lapse ultrasound imaging with phantom (Fig. 6A). The SD-MBs showed a high degree of stability under 37⁰C in a 1-h period. Increasing the SD complexes in the MBs up to 4 mg slightly reduced the contrast enhancement ability of the SD-MBs (15.1 ± 0.2 dB to 12.8 ± 1.1 dB). The SD complex retention rate of the SD-MBs was also investigated at 37⁰C over a 24-h period (Fig. 6B). The encapsulated drug started to leak at 14-24 h with a loading of 1-2 mg SD complexes in the MBs (1 mg: 90.1% to 86.6%; 2 mg: 85.4% to 68.6%). Mixing 4 mg of SD complexes into MBs resulted in a reduced drug retention rate (14 h: 64.1%; 24 h: 40.5%). Although SD-MBs with 4 mg of SD complexes had higher drug leakage than other groups, we still used these SD-MBs to perform the cell and animal experiments because the experiments would be complete within 1 h following SD-MBs fabrication.
(A) In vitro acoustic stability of SD-MBs. (B) Drug stability of SD-MBs. (C) MRI contrast-enhanced ability of SD complex, SD-MBs and SD complex released from SD-MBs. (D) Correlation between R2 value and concentration of SD-MBs and SD complex released from SD-MBs.
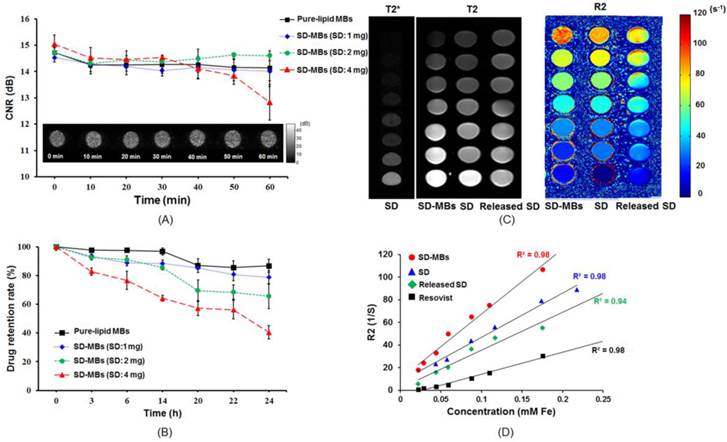
The T2, T2* and R2 maps of the SD complexes (4 mg) and SD-MBs (4 mg) on the MRI images are shown in Fig. 6C. Both the SD complexes and SD-MBs demonstrated highly concentration-dependent contrast enhancement of MRI. The R2 relaxivity of SD complexes (399.6 mM-1s-1) and SD-MBs (600.3 mM-1s-1) were showed by the slope between concentration and R2 (Fig. 6D). The R2 relaxivity of the SD-MBs and SD complexes was 2.9 times and 1.6 times higher, respectively, than clinically used SPIO particles (ResovistR) (12).
In vitro antitumor cytotoxicity of SD-MBs with MT
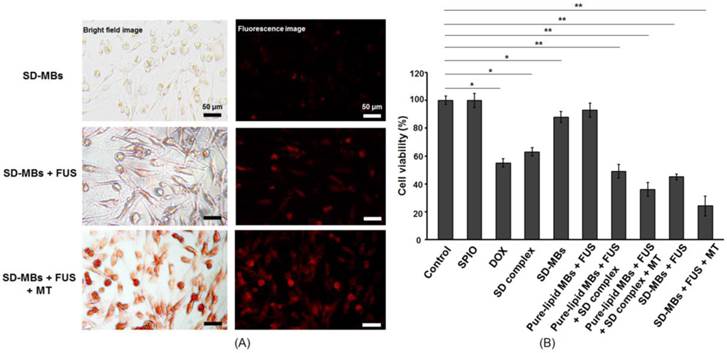
We then studied the antitumor capability of SD-MBs in C-6 cells with FUS and MT processes (Fig. 7A). Fluorescent and bright-field images demonstrated no obvious deposition of SD complexes when applying SD-MBs alone, but in combination with FUS sonication, the delivery of SD complexes could be enhanced. In addition, the SD-MBs + FUS + MT group showed greater delivery efficiency of SD complexes compared with the other groups since the accumulation of released SD complexes within cells could be further increased by the MT process. The in vitro anti-tumor outcome of SD-MBs was also investigated to compare the treatment efficiency of free DOX, free SPIO particles, and pure-lipid MBs, with and without FUS sonication. Cell viability was unchanged when SPIO was applied alone or pure-lipid MBs with FUS sonication were applied separately (Fig. 7B). Administration with SD complexes alone or DOX resulted a lower cell viability (62.9% ± 3.2 % vs. 55.3% ± 3.3 %), suggesting the conjugation of DOX and SPIO did not affect the anti-tumor capability of DOX. Only SD-MBs incubation caused a slight reduction in cell viability (87.9% ± 3.8%), likely due to the natural drug leakage from SD-MBs. It also indicated the toxicity of SD complexes could be limited by the encapsulation of MBs. However, the combined use of SD-MBs, FUS and the MT process produced the most profound anti-tumor ability (24.3% ± 4.7% vs. SD-MBs + FUS: 46.4% ± 1.7%). This finding implies that the SD complexes embedded in SD-MBs could be used for triggered release to kill tumor cells, and this effect can be further enhanced by the MT process.
Antitumor effect of SD-MBs and FUS sonication. (A) Bright-field and dark-field images of C-6 cells treated with the SD-MB, SD-MB + FUS, and SD-MB + FUS + MT. (B) Cell viability after treatment. Single asterisk, p <0.05; double asterisk, p <0.01.
In vivo ultrasound imaging contrast and BBB-opening capability of SD-MBs
The in vivo contrast-enhancement capability of SD-MBs was evaluated by ultrasound imaging (Fig. 8A). The mean intensity was increased to 67.8 ± 9.3 dB after injection of SD-MBs (4 mg). After 18 min of injection, 17.8 ± 6.8 dB mean echo intensity enhancement remained. This long in vivo half-life time of SD-MBs would benefit drug delivery (pure-lipid MBs: 7.6 min; SD-MBs 4 mg: 10.8 min). Moreover, the echogenicity of SD-MBs was higher than that of pure-lipid MBs (pure-lipid MBs: 38.6 ± 2.1 dB; SD-MBs 4 mg: 67.8 ± 9.3 dB) because the embedded SD complexes can increase the impedance mismatch property between the shell of MBs and the interior air gas core.
Acoustic property of SD-MBs. (A) Contrast-enhanced ability and in vivo lifetime of SD-MBs with different SD payloads (1-4 mg). (B) BBB-opening capability with FUS sonication (0.1-0.5 MPa) (Top: brain section; middle: corresponding H&E staining; bottom: 200 × of H&E staining).
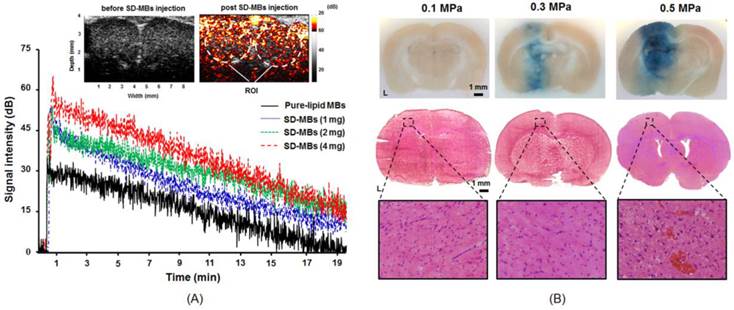
The distribution of BBB openings caused by SD-MBs and FUS was confirmed via EB extravasation (Fig. 8B). Because the acoustic pressure of FUS is a critical issue for brain drug delivery, we thus applied 0.1-0.5 MPa of transcranial FUS sonication after SD-MB (4 mg) injection to determine optimal parameters. There was no EB leakage into the brain tissue following FUS sonication with 0.1 MPa. An apparent BBB-opening effect was observed at the sonication site at 0.3 MPa without visible brain damage. However, serious erythrocyte extravasations occurred in the BBB-opening area at 0.5 MPa from H&E staining probably due to the inertial cavitation effect contributedby FUS and SD-MBs. Brain damage occurring during the FUS-BBB-opening process would reduce drug delivery efficiency (5), so we chose 0.3 MPa for the following in vivo drug delivery experiments. In addition, the delivery of SD complexes into the brain tumor might be improved since the region of BBB-opening was great enough to mantle the whole area of tumor.
Tracing of drug deposition within brain tumor via MRI imaging
The enhancement of SD complex delivery into brain via SD-MBs and FUS was evaluated by MRI. The site of the brain tumor was identified by T2W imaging (indicated by hyperintense edema regions), SD complex deposition was tracked via T2* imaging (indicated by hypointense signal-loss regions), and an R2 map was used to show changes caused by different amounts of SD complexes (represented by a false-color scale) (Fig. 9A-D). IV administration of SD-MBs alone did not provide contrast enhancement within tumors (ΔR2 value 1 h: 1.02 ± 0.1 s-1; 3 h: 1.2 ± 0.8 s-1). Administration of SD-MBs and FUS resulted in minor leakage of SD complexes into the tumor (ΔR2 value at 1 h: 2.9 ± 1.4 s-1; at 3 h: 4.1 ± 1.7 s-1). As the MT procedure was performed following BBB-opening, we detected a gradual enhancement of SD complex accumulation within the tumor location (ΔR2 value at 1 h: 6.3 ± 0.9 s-1; at 3 h: 12.4 ± 1.0 s-1), resulting in an SD complex accumulation of 82.3% as compared to 18.3% without MT. For comparison, injected SD complexes following the BBB-opening procedure also provided high rates of SD deposition in the tumor (ΔR2 value at 1 h: 5.3 ± 2.8 s-1; at 3 h: 9.5 ± 4.3 s-1) (Fig. 9E).
In vivo imaging of SD complex distribution in the brain (Top, before treatment; Middle, 1 h after treatment; Bottom, 3 h after treatment). (A) SD-MB alone. (B) SD + MB + FUS + MT. (C) SD-MB + FUS. (D) SD-MB + FUS + MT. (E) ΔR2 value acquired before and after treatment. Single asterisk, p <0.05; double asterisk, p <0.01.
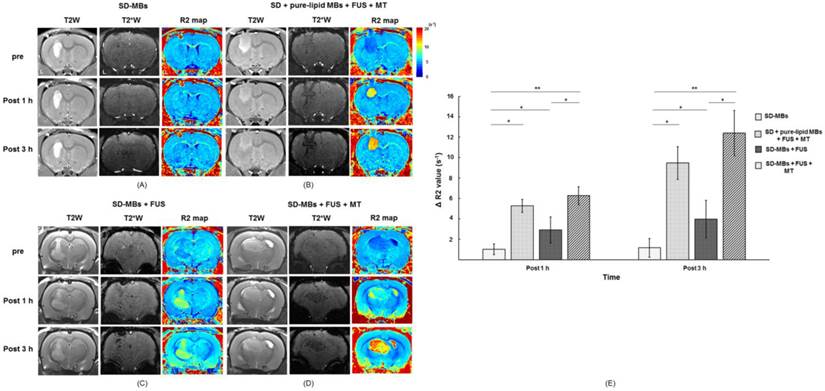
FUS-induced SD release from SD-MBs in the brain tumor
The brain tissues were collected following the treatment, and SPIO and DOX were extracted and individually quantitated with ICP-AES and HPLC systems. ICP-AES results indicated no SPIO accumulation occurred in the tumor after injecting SD-MBs alone. We discovered that the SPIO deposition in the SD-MBs + FUS + MT group was 2.8 fold higher than that of the SD-MBs + FUS group, indicating that SPIO deposition can be enhanced by MT (with MT: 36.1 ± 4.2 μg/g tissue; without MT: 13.3 ± 4.1 μg/g tissue) (Fig. 10A). IV administration of SD complexes with MT after performing BBB-opening also provided good drug accumulation in the tumor site (22.6 ± 4.5 μg/g tissue). DOX delivery into the brain tumors was also evaluated by HPLC (Fig. 10B). Only slight amounts of DOX were measured by applying SD-MBs alone (0.1 ± 0.02 μg/g tissue). Importantly, the deposition of DOX into brain tissues also could be enhanced 2.1-fold via the MT process following administration of FUS and SD-MBs (with MT: 3.6 ± 0.7 μg/g tissue; without MT: 1.7 ± 0.4 μg/g tissue). Delivery of free SD complexes with MT following BBB-opening also increased DOX accumulation in brain tumors (2.9 ± 0.3 μg/g tissue). These results suggest that the combinative effects between MT and FUS sonication improve the delivery of local SD complexes.
SD complex deposition within tumor after treatment. (A) SPIO deposition analyzed by ICP-AES. (B) DOX accumulation measured by HPLC. (C) Correlation between SPIO deposition and ΔR2 value. (D) Correlation between SPIO deposition and DOX accumulation. Single asterisk, p <0.05; double asterisk, p <0.01.
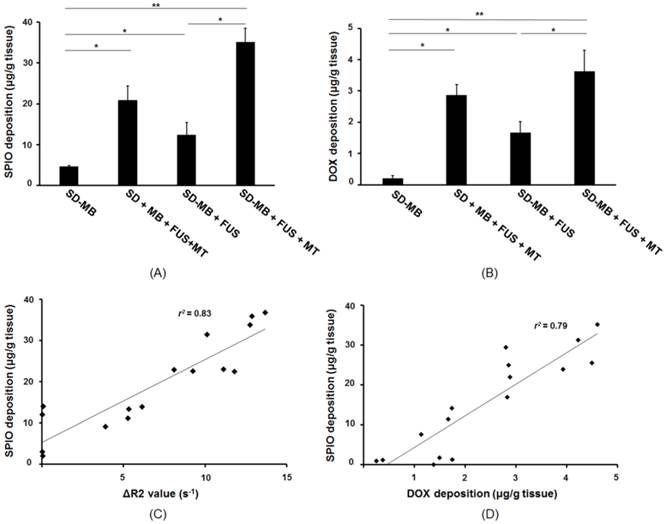
Analysis of the MR R2 value and the SPIO concentration in the tumor regions showed a high correlation between values (r2 = 0.83) (Fig. 10C). Furthermore, the SPIO concentration was highly correlated with DOX deposition (r2 = 0.79) (Fig. 10D). Based on comparisons between the ICP-AES, HPLC and R2 relaxivity results, 1 μg/g of SPIO detected on the R2 maps was equivalent to a DOX concentration of 2.2 μg/g in tissue, or 3.1 μg/g tissue of DOX per change in R2 (s-1) Therefore, the R2 map not only provided good correlations with DOX, but also could be used to quantitatively monitor tumor drug delivery.
Histological distribution of FUS-triggered SD release from SD-MBs in brain tumor
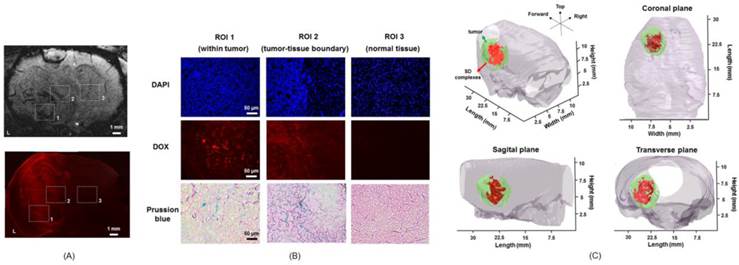
Finally, the distribution of SD complexes in brain tumors was tracked by fluorescent imaging and Prussian blue staining. After SD-MB + FUS + MT treatment, brain tissues were sliced and stained with Prussian blue and DAPI. DAPI staining was used to identify the tumor location. Figure 11A shows the MRI image and the corresponding fluorescent DOX image after treatment. Three ROIs were selected, containing the area of the normal brain tissue near the tumor site (N), the tumor region (T), and the contralateral normal brain tissue (C). Representative images show that obvious SD complexes accumulate at the tumor and tumor-normal tissue boundary (Fig. 11B). However, SD complexes were undetectable in contralateral normal brain tissue (Fig. 11B). We considered that SD complexes indeed specifically enter the tumor site following treatment. Figure 11C shows the 3D reconstruction image of the R2 map after treatment. The high correlation between tumor location and released SD complex distribution (tumor size: 132.1 ± 32.5 mm3; delivered SD complex: 91.3 ± 24.6 mm3) provides the possibility of theranostic brain tumor drug delivery by SD-MBs.
Histological section. (A) MRI image and corresponding fluorescent DOX image. (B) DAPI, DOX and Prussian blue staining images of tumor, tumor-tissue boundary and normal tissue. (C) 3D reconstruction of R2 map for demonstrating SD complex distribution within tumor.
Discussion
We presented the design of a novel MB-based nanomedicine platform to concurrently conjugate SPIOs and chemotherapeutic agents DOX as multi-functional CNS theranostic agents. We demonstrate that, the use of FUS with SD-MBs can successfully open the targeted BBB and trigger targeted release of chemotherapeutic agent DOX into the tumor. With the concurrent application of magnetic targeting, we showed that the SPIO-DOX (SD) complexes could be significantly enhanced. We previously demonstrated this strategy can be implemented via thin film hydration method in synthesizing the novel bubble system (9), and we showed the novel bubble-drug system provided satisfied R2 relaxivity (107.3 mM-1s-1 and similar with the commercialized SPIO agent) to carry sufficient high DOX concentration (0.2 μg/g tissue measured in brain, and can increased to 0.3 μg/g tissue after FUS shown in (9)). In this study, we redesigned the bubble-drug system synthesis by first synthesizing SPIO-DOX complex prior to MBs conjugation, and we successfully shown the significantly improved magnetization property (R2 relaxivity of 600.3 mM-1s-1; i.e., 5.6-fold increase to (9)) and further increasing DOX-carrying capability (brain-measured concentration of 1.7 μg/g tissue and can be increased to 3.6 μg/g tissue after FUS and MT, i.e., about one-order DOX concentration higher than (9)). The improved R2 relaxivity allows the monitoring of the distribution and accumulation of the released SD complexes through MR relaxometry, supporting the theranostic capability of the SD-complexed bubble-drug system.
In in-vitro experiment, we confirmed that SPIO-DOX conjugation did not affect the anti-tumor capability of DOX, and the delivery can be benefit from FUS and MT process and with the in-vitro cytotoxicity regulated by MBs encapsulation On the other hand, the cellular drug uptake mechanism between free DOX and SD complexes might different. Distinct from free DOX that contribute cytotoxicity from DOX diffusion into cytosol and bound to chromosomal DNA, SPIO-DOX complexes induce cytotoxicity majorly contributes to cell endocytosis with DOX accumulated in endosomes, diffusing DOX into the cytosol and then into the nucleus (15).
Although this study did not directly access anti-tumor efficiency, previous DOX anticancer studies provide comprehensive understanding of the required DOX concentration to reach therapeutic level. Clinically, it has been reported that the intratumoral DOX concentrations reaching 0.82 ± 0.5 μg/g tissue correlated with partial or complete responses in breast cancer patients (16). In preclinical test, Kovacs et al. showed in glioma model that DOX concentration increase from 0.114 to 0.189 μg/g tissue after FUS-BBB opening provided 68% of median survival improve (22 days compared to 37 days) (17). In addition, Aryal et al. showed the use of multiple-secession FUS-BBB opening to enhance liposomal DOX delivery (accumulated DOX concentration increased from 0.8 to 4.8 μg/g tissue) can improve median survival to 75% (20 days compared to 35 days) with partial animals survived exceeding observation period (18, 19). In this study, we showed that the proposed bubble-drug system capable of increasing local DOX concentration up to 3.6 μg/g tissue when synergistically apply FUS and MT at one time, and therefore is believed to provide sufficiently high anti-glioma response with the proposed brain drug delivery system.
The magnetic property of the nanoparticles allowed them to be steered, moved and trapped at a local target location by manipulating the magnetic field gradient, volume of the magnetic core and magnetic permeability of the free space. The use of magnetically targeted delivery of therapeutic agents was first proposed by Widder et al. (20). Magnetically enhanced drug delivery has already been applied in several clinical trials, for conditions such as leukemia and brain neoplasms (21). However, the use of magnetic nanoparticles for MT is still subject to several concerns. First, to ensure the drug can be captured by the magnet in the tumor, the force of the applied magnetic field should be larger than the drag forces associated with the convective blood flow. Second, the efficiency of MT is determined by the characteristics of the magnetic nanoparticles (size, magnetic susceptibility, etc.). Many studies have indicated that the magnetic force and retention time within the tumor depend on the nanoparticle size (22). However, larger nanoparticles increase the reticuloendothelial system capture and shorten the in vivo life-time. Third, although the in vivo stability of the magnetic nanoparticles can be improved by surface modification with organic ligands (23), these surface protectants would decrease the surface magnetic moment of the metal atoms located at the particle surface, thus reducing the magnetic properties of the particles (24).
Although DOX has been shown to have anti-tumor properties in vivo (25), DOX is associated with several complications, such as cardiotoxicity, myelosuppression, and nephrotoxicity (26,27). Shailendra et al. demonstrated that pegylated liposomal DOX was equally active but associated with a significantly lower risk of cardiotoxicity compared with DOX delivery alone (28). Our group has previously demonstrated that the systemic cytotoxic effects of chemotherapeutic drug (BCNU) could be significantly reduced by the encapsulation in MBs (6). Furthermore, the MB shells also prolong the life-time due to pegilated modification of MBs to reduce phagocytes/ RES uptake and delay the systemic immune reaction. The in-vitro experiments in this study also confirmed that the cytotoxicity of the SD complex could be reduced when encapsulated in MBs, suggesting the superiority of using MBs as a drug carrier to in systemic DOX toxicity reduction.
This study successfully demonstrated the feasibility of using SD-MBs as an imaging indicator for monitoring drug distribution and concentrations following FUS treatment. On the other hand, many studies have shown that pure MBs can also serve as a contrast agent for MR imaging due to the gas-liquid interface producing large local magnetic susceptibility differences. The potential of albumin-shell MBs for tumor imaging with MRI was confirmed by Moseley et al. (29). Therefore, it should be possible to achieve MRI theranostic imaging using drug-loaded MBs without carrying SPIO.
An alternative means to fabricate dual modality ultrasound/MR imaging contrast agents is to load MBs with a paramagnetic contrast agent, such as Gd-DTPA. Gd-DTPA enhances the positive signal of blood by shortening both the transverse and longitudinal proton relaxation times (30). In addition, after FUS destruction, the contrast properties of Gd-loaded MBs would increase due to the enhancement of proton exchange between water and the fragments of Gd-bound lipid shell (31). Liao et al. reported the use of Gd-DTPA loading MBs for monitoring FUS-induced BBB opening and for detecting intracerebral hemorrhage in T1-weighted MR imaging and T2-weighted MR images (32). However, since the accumulation of SPIO can be manipulated by MT, we thus chose SPIO to fabricated the SD-MBs.
Previous studies have indicated that FUS and MBs can improve the delivery of nanoparticles across the BBB such as DOX-loaded liposomal nanoparticles or brain-penetrating nanoparticles [18, 33]. However, benefits might be further gained when conjugating nanoparticles with MBs for enhanceing CNS drug delivery as follows: 1) the drug-loaded MBs can simplify IV administration from separated microbubbles/ drugs injection to one; 2) the drug release only occurred at FUS exposure site, thereby reducing off-target toxicity of the drug in CNS [6]; 3) the surface of MBs can be modified with disease-associated molecular markers to increase drug targeting (34); 4) the drug delivered efficiency into cells might superior than free-form drugs since ultrasound-microbbuble interaction can trigger local cavitational effect to trigger phargocytosis and therefore enhance cell uptake of the released drug (35).
Previous studies have pointed out that the synthesis of SPIO-lipid complex could alter the MB physical and chemical properties, including: 1) The imaging contrast or gas diffusion rate change due to the inclusion of SPIO in the MBs (36), 2) increased bubble oscillation nonlinearity due to viscoelastic property change (37), 3) change of tumor-endocytosis efficiency (38), and 4) resonance-frequency shift and half-life change of MBs (39-41). Additional characterization of SD-MBs should be performed to optimize safety and drug release efficiency before clinical use. Compared with our previous study, this study provides a further demonstration of the using SD complex loaded MBs in a catalytic role to induce FUS-induced BBB opening, and also concurrently as dual-imaging agents, therapeutic agents, and active-targeting agents. The SD-MBs could be further adapted towards specific purposes (e.g., for drug carrying or magnetic targeting).
Conclusions
In this study, we successfully fabricated a therapeutic SD complex that can be loaded onto MBs. The SD-MB could be triggered by FUS exposure to targeted release SD complexes. High magnetization of the designed SD complexes was capable of in-vivo monitored via MR relaxometry, and support the feasibility of the theranostic capability of the SD-MBs complex. We also confirmed that the drug delivery efficiency of SD-MB can be significantly improved in this new SD synthesis approach, and also confirmed the DOX targeted delivery can be superior than co-administration of SD complexes and MBs. The results of this study may provide useful information promoting FUS-triggered drug-loaded MB delivery for future brain tumor treatment.
Acknowledgements
The authors gratefully acknowledge the support of the Ministry of Science and Technology, Taiwan under Grant No. 102-2221-E-007-020-MY3 and 104-2221-E-007-076, National Tsing Hua University (Hsinchu, Taiwan) under Grant No. 105N522CE1, and Chang Gung Memorial Hospital (Linkou, Taiwan) under Grant No. CIRPD2E0051.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ricard D, Idbaih A, Ducray F. et al. Delattre. Primary brain tumours in adults. Lancet. 2003;379(9830):1984-96
2. Grossman SA, Batara JF. Current management of glioblastoma multiforme. Semin Oncol. 2004;31(5):635-44
3. Gately MK, Warrier RR, Honasoge S. et al. Administration of recombinant IL-12 to normal mice enhances cytolytic lymphocyte activity and induces production of IFN-gamma in vivo. Int Immunol. 1994;6(1):157-67
4. Hynynen K, McDannold N, Vykhodtseva N. et al. Noninvasive MR imaging-guided focal opening of the blood-brain barrier in rabbits. Radiology. 2001;220(3):640-6
5. Liu HL, Hua MY, Chen PY. et al. Blood-brain barrier disruption with focused ultrasound enhances delivery of chemotherapeutic drugs for glioblastoma treatment. Radiology. 2010;255(2):415-25
6. Ting CY, Fan CH, Liu HL. et al. Concurrent blood-brain barrier opening and local drug delivery using drug-carrying microbubbles and focused ultrasound for brain glioma treatment. Biomaterials. 2012;33(2):704-12
7. Åslund AK, Berg S, Hak S. et al. Nanoparticle delivery to the brain - By focused ultrasound and self-assembled nanoparticle-stabilized microbubbles. J Control Release. 2015;220(Pt A):287-94
8. Lammers T, Koczera P, Fokong S. et al. Theranostic USPIO-Loaded Microbubbles for Mediating and Monitoring Blood-Brain Barrier Permeation. Adv Funct Mater. 2015;25(1):36-43
9. Fan CH, Ting CY, Lin HJ. et al. SPIO-conjugated, doxorubicin-loaded microbubbles for concurrent MRI and focused-ultrasound enhanced brain-tumor drug delivery. Biomaterials. 2013;34(14):3706-15
10. Park J, Zhang Y, Vykhodtseva N. et al. The kinetics of blood brain barrier permeability and targeted doxorubicin delivery into brain induced by focused ultrasound. J Control Release. 2012;162(1):134-42
11. Liu HL, Hua MY, Yang HW. et al. Magnetic resonance monitoring of focused ultrasound/magnetic nanoparticle targeting delivery of therapeutic agents to the brain. Proc Natl Acad Sci U S A. 2010;107(34):15205-10
12. Liu HL, Hsu PH, Chu PC. et al. Magnetic resonance imaging enhanced by superparamagnetic iron oxide particles: usefulness for distinguishing between focused ultrasound-induced blood-brain barrier disruption and brain hemorrhage. J Magn Reson Imaging. 2009;29(1):31-8
13. Fan CH, Lin WH, Ting CY. et al. Contrast-enhanced ultrasound imaging for the detection of focused ultrasound-induced blood-brain barrier opening. Theranostics. 2014;4(10):1014-25
14. Corot C, Robert P, Idée JM. et al. Recent advances in iron oxide nanocrystal technology for medical imaging. Adv Drug Deliv Rev. 2006;58(14):1471-504
15. Yang X, Grailer JJ, Rowland IJ. et al. Multifunctional SPIO/DOX-loaded wormlike polymer vesicles for cancer therapy and MR imaging. Biomaterials. 2010;31(34):9065-73
16. Cummings J, McArdle CS. Studies on the in vivo disposition of adriamycin in human tumours which exhibit different responses to the drug. Br J Cancer. 1986;53(6):835-8
17. Kovacs Z, Werner B, Rassi A. et al. Prolonged survival upon ultrasound-enhanced doxorubicin delivery in two syngenic glioblastoma mouse models. J Control Release. 2014;10(187):74-82
18. Aryal M, Vykhodtseva N, Zhang YZ. et al. Multiple treatments with liposomal doxorubicin and ultrasound-induced disruption of blood-tumor and blood-brain barriers improve outcomes in a rat glioma model. J Control Release. 2013;169(1-2):103-11
19. Aryal M, Vykhodtseva N, Zhang YZ. et al. Multiple sessions of liposomal doxorubicin delivery via focused ultrasound mediated blood-brain barrier disruption: A safety study. J Control Release. 2015Apr28;204:60-9
20. Widder KJ, Senyei AE, Ranney DF. Magnetically responsive microspheres and other carriers for the biophysical targeting of antitumor agents. Adv Pharmacol Chemother. 1979;16:213-71
21. Singh A, Sahoo SK. Magnetic nanoparticles: a novel platform for cancer theranostics. Drug Discov Today. 2014;19(4):474-81
22. Gupta AK, Gupta M. Synthesis and surface engineering of iron oxide nanoparticles for biomedical applications. Biomaterials. 2005;26(18):3995-4021
23. Reddy LH, Arias JL, Nicolas J. et al. Magnetic nanoparticles: design and characterization, toxicity and biocompatibility pharmaceutical and biomedical applications. Chem Rev. 2012;112(11):5818-78
24. Paulus PM, Bonnemann H, an der Kraan AM. et al. Magnetic properties of nanosized transition metal colloids: the influence of noble metal coating. Eur Phy J D. 1999;9:501-504
25. Walter KA, Tamargo RJ, Olivi A. et al. Intratumoral chemotherapy. Neurosurgery. 1995;37(6):1128-1145
26. Carvalho C, Santos RX, Cardoso S. et al. Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem. 2009;16(25):3267-85
27. Pajic M, Iyer JK, Kersbergen A. et al. Moderate increase in Mdr1a/1b expression causes in vivo resistance to doxorubicin in a mouse model for hereditary breast cancer. Cancer Res. 2009;69(16):6396-404
28. Verma S, Dent S, Chow BJ. et al. Metastatic breast cancer: the role of pegylated liposomal doxorubicin after conventional anthracyclines. Cancer Treat Rev. 2008;34(5):391-406
29. Cheung JS, Chow AM, Guo H. et al. Microbubbles as a novel contrast agent for brain MRI. Neuroimage. 2009;46(3):658-64
30. Caravan P, Ellison JJ, McMurry TJ. et al. Gadolinium(III) Chelates as MRI Contrast Agents: Structure, Dynamics, and Applications. Chem Rev. 1999;99(9):2293-352
31. Feshitan JA, Boss MA, Borden MA. Magnetic resonance properties of Gd(III)-bound lipid-coated microbubbles and their cavitation fragments. Langmuir. 2012;28(43):15336-43
32. Liao AH, Liu HL, Su CH. et al. Paramagnetic perfluorocarbon-filled albumin-(Gd-DTPA) microbubbles for the induction of focused-ultrasound-induced blood-brain barrier opening and concurrent MR and ultrasound imaging. Phys Med Biol. 2012;57(9):2787-802
33. Nance E, Timbie K, Miller GW. et al. Non-invasive delivery of stealth, brain-penetrating nanoparticles across the blood-brain barrier using MRI-guided focused ultrasound. J Control Release. 2014;189:123-32
34. Zhou Y, Gu H, Xu Y. et al. Targeted antiangiogenesis gene therapy using targeted cationic microbubbles conjugated with CD105 antibody compared with untargeted cationic and neutral microbubbles. Theranostics. 2015;5(4):399-417
35. De Cock I, Lajoinie G, Versluis M. et al. Sonoprinting and the importance of microbubble loading for the ultrasound mediated cellular delivery of nanoparticles. Biomaterials. 2016;83:294-307
36. Yang F, Gu A, Chen Z. et al. Multiple emulsion microbubbles for ultrasound imaging. Mater Lett. 2008;62(1):121-4
37. Hea W, Yanga F, Wua Y. et al. Microbubbles with surface coated by superparamagnetic iron oxide nanoparticles. Mater Lett. 2012;68(1):64-7
38. Yang F, Li Y, Chen Z. et al. Superparamagnetic iron oxide nanoparticle-embedded encapsulated microbubbles as dual contrast agents of magnetic resonance and ultrasound imaging. Biomaterials. 2009;30(23-24):3882-90
39. Brismar TB, Grishenkov D, Gustafsson B. et al. Magnetite nanoparticles can be coupled to microbubbles to support multimodal imaging. Biomacromolecules. 2012;13(5):1390-9
40. Raisinghani A, DeMaria AN. Physical principles of microbubble ultrasound contrast agents. Am J Cardiol. 2002;90(10A):3J-7J
41. Stride E. The influence of surface adsorption on microbubble dynamics. Philos Trans A Math Phys Eng Sci. 2008;366(1873):2103-15
Author contact
![]() Corresponding authors: Department of Biomedical Engineering and Environmental Sciences, National Tsing Hua University, No. 101, Section 2, Kuang-Fu Road, Hsinchu 30013, Taiwan. Tel: +886-3-571-5131, ext. 34240; Fax: +886-3-571-8649 (C.-K. Yeh). Department of Electrical Engineering, Chang-Gung University, 259 Wen-Hwa 1st Road, Kuei-Shan, Tao-Yuan 33302, Taiwan, R.O.C. Tel: +886-3-211-8800, ext. 5677; Fax: +886-3-211-8026 (H.-L. Liu). E-mail addresses: ckyehnthu.edu.tw (C.-K. Yeh), haoliliucgu.edu.tw (H.-L. Liu).
Corresponding authors: Department of Biomedical Engineering and Environmental Sciences, National Tsing Hua University, No. 101, Section 2, Kuang-Fu Road, Hsinchu 30013, Taiwan. Tel: +886-3-571-5131, ext. 34240; Fax: +886-3-571-8649 (C.-K. Yeh). Department of Electrical Engineering, Chang-Gung University, 259 Wen-Hwa 1st Road, Kuei-Shan, Tao-Yuan 33302, Taiwan, R.O.C. Tel: +886-3-211-8800, ext. 5677; Fax: +886-3-211-8026 (H.-L. Liu). E-mail addresses: ckyehnthu.edu.tw (C.-K. Yeh), haoliliucgu.edu.tw (H.-L. Liu).