Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results and Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(11):1780-1791. doi:10.7150/thno.14280 This issue Cite
Research Paper
Active-target T1-weighted MR Imaging of Tiny Hepatic Tumor via RGD Modified Ultra-small Fe3O4 Nanoprobes
Zhengyang Jia1,*, Lina Song1,*, Fengchao Zang2, Jiacheng Song3, Wei Zhang1, Changzhi Yan1, Jun Xie1, Zhanlong Ma3, Ming Ma1, Gaojun Teng2, Ning Gu1, ![]() , Yu Zhang1,
, Yu Zhang1, ![]()
1. State Key Laboratory of Bioelectronics, Jiangsu Key Laboratory for Biomaterials and Devices, School of Biological Science and Medical Engineering & Collaborative Innovation Center of Suzhou Nano Science and Technology, Southeast University, Nanjing 210096, China
2. Jiangsu Key Laboratory of Molecular and Functional Imaging, Medical School, Southeast University, Nanjing 210009, China
3. Department of Radiology, The First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, China
* These authors contributed equally to this work.
Received 2015-11-1; Accepted 2016-5-31; Published 2016-7-15
Abstract

Developing ultrasensitive contrast agents for the early detection of malignant tumors in liver is highly demanded. Constructing hepatic tumors specific targeting probes could provide more sensitive imaging information but still faces great challenges. Here we report a novel approach for the synthesis of ultra-small Fe3O4 nanoparticles conjugated with c(RGDyK) and their applications as active-target T1-weighted magnetic resonance imaging (MRI) contrast agent (T1-Fe3O4) for imaging tiny hepatic tumors in vivo. RGD-modified T1-Fe3O4 nanoprobes exhibited high r1 of 7.74 mM-1s-1 and ultralow r2/r1 of 2.8 at 3 T, reflecting their excellent T1 contrast effect at clinically relevant magnetic field. High targeting specificity together with favorable biocompatibility and strong ability to resist against non-specific uptake were evaluated through in vitro studies. Owing to the outstanding properties of tumor angiogenesis targeting with little phagocytosis in liver parenchyma, hepatic tumor as small as 2.2 mm was successfully detected via the T1 contrast enhancement of RGD-modified T1-Fe3O4. It is emphasized that this is the first report on active-target T1 imaging of hepatic tumors, which could not only significantly improve diagnostic sensitivity, but also provide post therapeutic assessments for patients with liver cancer.
Keywords: T1-Fe3O4 nanoprobes, RGD-modified, active-target, tiny hepatic tumor imaging, high diagnostic sensitivity
Introduction
Liver is a frequent region of tumorigenesis and metastases owing to its well-vascularized tissue and accessible capillaries with low blood flow shear rates.[1] Across all cancer types, liver cancer was one of the most frequently diagnosed cancers among men, and it was the second leading cause of cancer deaths worldwide in 2012.[2] The use of early-stage diagnosis, which has implications for patients treatment and prognosis, is an effective strategy to prevent the substantial portion of liver cancer deaths.[2, 3] Therefore, developing ultrasensitive imaging for the accurate detection of tiny malignant hepatic tumors, defined herein as tumors in a size range of 1~4 mm refer to the previous report,[4] is highly demanded in clinic.
During the past decades, Magnetite nanoparticles (Fe3O4 NPs) served as T2-weighted magnetic resonance imaging (MRI) contrast agents (named as T2-Fe3O4 here), such as Feridex and Resovist were widely applied in clinic to improve the detectability of hepatic tumors. Due to the large accumulation of T2-Fe3O4 in liver parenchyma where contains abundant activated macrophages, hepatic tumors would be detected through pseudo-positive contrast as compared with surrounding normal liver tissue.[5, 6] However, sometimes this was lack of diagnostic sensitivity because it was difficult to differentiate solid hepatic tumors from benign lesions (e.g., hemangiomas, cysts) from the enhanced T2-weighted MR images.[3] Recently, large numbers of efforts had been devoted to optimize the parameters (e.g., sizes, morphologies or surface physiochemical properties) of T2-Fe3O4 to achieve higher transversal relaxivity (r2) of NPs and to induce remarkable signal attenuation in normal liver tissue by using a lower dosage.[7, 8] Regardless of this, the hepatobiliary system would be still a major clearance route to degrade Fe3O4 NPs into iron ions, which might lead to an increased risk of toxicity.[9] So, it is desirable to construct imaging probes specifically targeting to hepatic tumors instead of liver parenchyma for the early liver cancer diagnosis. However, this progress faces many challenges. Unlike other solid tumors, nearly no specific targets exist for the detection of hepatic tumors. In addition, permeation of probes through the endothelial fenestrations to hepatic tumor cells with little capture by macrophages residing within and near the liver vasculatures is another great obstacle.[10] Consequently, probes realizing active-target imaging of hepatic tumors have been rarely reported so far.
In comparison with T2-Fe3O4, Fe3O4 served as T1-weighted MRI contrast agents (named as T1-Fe3O4) with precise particle engineering and rational probe decoration are ideal candidates when combined with specific ligands for hepatic tumors active targeting. It is noteworthy that T1-Fe3O4 commonly possess ultra-small particle size, which not only provides optimal bright T1 contrast effect, but also ensures that T1-Fe3O4 preferentially accumulate in tumors with little liver uptake.[9, 11-13] As a result, it could produce positive T1-weighted MRI contrast enhancement in hepatic tumor sites rather than the normal liver tissue as reported before,[9, 14, 15] which could offer more accurate and sensitive diagnostic information during the early-stage detection of liver cancer. Unfortunately, there are many strict requirements in the process of constructing T1-Fe3O4 based active-target probes. Synthesis of Fe3O4 NPs around core size of 5 nm (generally with hydrodynamic size of 10~20 nm), which has been proved to own reasonable renal excretion rate and favorable longitudinal relaxivity (r1) is the primary request.[4, 15, 16] Secondly, for active-target T1 imaging of tumors, the bioconjugation reaction between specific ligands and T1-Fe3O4 must be carried out in a well-controlled manner to prevent the formation of NPs aggregates as far as possible because the aggregates usually have large magnetic moment, strong T2 effect and could be easily recognized by macrophages and accumulated in liver parenchyma.[8, 17-19] Moreover, it is incontestable that the introduction of targeting moieties will lead to an increase of serum protein adsorption and then a rapid immune clearance.[20] Because of this, dense biocompatible surface modification is additionally important to resist against the non-specific uptake by mononuclear phagocyte system (MPS; e.g., liver and spleen) in living subjects. To fulfill the request aforementioned, herein we synthesized hepatic tumors targeting probes by conjugating c(RGDyK) to PEGylated T1-Fe3O4. RGD peptide could efficiently target to integrin αvβ3 overexpressed endothelium cells on angiogenic tumor vessels, which are regarded as the homogeneous and common hallmarks among different kinds of solid tumors including hepatic tumors.[21, 22] Our work demonstrated that RGD-modified T1-Fe3O4 possessed optimal relaxation properties, remarkable biocompatibility and significant ability to resist against non-specific uptake. Most importantly, Excellent active-target T1 imaging results were validated on the early liver cancer models. To the best of our knowledge, this is the first report on Fe3O4 based contrast agents that realize active-target T1 imaging of orthotopic hepatic tumors.
Materials and methods
Materials
Iron(III) acetylacetonate (Fe(acac)3, 98%), oleylamine (80~90%), benzyl ether (97%) and hexane (98%) were purchased from Aladdin Industrial Co. 1-Ethyl-(3-3-dimethylaminopropyl) carbodiimide hydrochloride (EDC-HCl), N,N′-dicyclohexylcarbodiimide (DCC), N-hydroxysuccinimide (NHS) and a,ω-bis{2-[(3-carboxy-1-oxopropyl)amino]ethyl} poly-(ethylene glycol) (carboxylated PEG, M=2000) were purchased from Sigma Aldrich. Dopamine hydrochloride was purchased from J&K Chemical. Sodium carbonate and other organic solvents used in the syntheses were purchased from Sinopharm Chemical Reagent Co. c(RGDyK) peptide (M=620) were from GLS Biochem Ltd. All the buffers and media were from Sinopharm Chemical Reagent Co. Water was purified by Millipore Water Purification System. All chemicals were used as received without further purification. All cell lines applied in this work were originally obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and cultured under recommended conditions.
Synthesis of hydrophobic T1-Fe3O4
Uniform T1-Fe3O4 were synthesized via the thermal decomposition of Fe(acac)3 in the presence of oleylamine and benzyl ether. Specifically, Fe(acac)3 (1.05 g, 3 mmol), oleylamine (12 mL, 37 mmol) and benzyl ether (8 mL, 42 mmol) were mixed together and placed into a 100 mL three-neck round-bottom flask. The mixture was stirred under a flow of N2 for 10 min to remove any oxygen at room temperature. Then, the mixture was heated to 220 °C at a constant heating rate of 3.3 °C/min and refluxed at this temperature for 1 h. Next, the mixture was heated to 290 °C at the same uniform heating rate and lastly maintained at this temperature for 10 min. As the reaction proceeded, the color of the solution changed from the initial transparent red to reddish-brown and finally opaque black. After the reaction, the mixture was rapidly cooled to room temperature. The products were separated by an external magnet, then washed three times using ethanol, and finally dissolved in 20 mL hexane.
PEGylation of hydrophobic T1-Fe3O4
PEG-derivatized dopamine (DA-PEG-COOH) was first synthesized. In detail, carboxylated PEG (60 mg, 0.03 mmol), NHS (6 mg, 0.05 mmol), DCC (9 mg, 0.04 mmol), dopamine hydrochloride (3.9 mg, 0.02 mmol) and anhydrous Na2CO3 (30 mg, 0.28 mmol) were dissolved in a mixture solvent containing CHCl3 (6 mL) and DMF (3 mL). This mixed solution was stirred (500 rpm) for 2 h at 35 °C to obtain DA-PEG-COOH. Next, 15 mg (Fe element mass in Fe3O4 NPs) hydrophobic T1-Fe3O4 were added and the mixed solution was continually stirred overnight (200 rpm) at the same temperature. After the reaction, hexane was added to precipitate the PEGylated T1-Fe3O4, which were collected by an external permanent magnet and dried under N2. Lastly, PEGylated T1-Fe3O4 could be redispersed in distilled water. The extra surfactants and other salts were removed by dialysis using a dialysis bag (MWCO=8000~12000) in distilled water for 48 h. Any precipitation was removed by a 220 nm syringe filter. The final products were stored at 4 °C for next uses.
Conjugation of c(RGDyK) peptide on PEGylated T1-Fe3O4
The immobilization of c(RGDyK) peptide (M=620) on PEGylated T1-Fe3O4 was according to the amide condensation reaction. EDC (35 mg, 0.183 mmol), NHS (20 mg, 0.174 mmol) and 15 mg (Fe element mass in Fe3O4 NPs) above-obtained PEGylated T1-Fe3O4 were dissolved in the MES buffer (pH=5.5). The mixed solution was oscillated (120 rpm) on a shaker for 20 min at 20 °C. Then, the mixture was centrifuged at 4000 g in a ultrafiltration tube (MWCO=30000) to remove the extra surfactants and other salts. Subsequently, above reactants were redispersed in the BB buffer (pH=8.2) containing c(RGDyK) peptide (8 mg, 0.013 mmol) and then oscillated (120 rpm) overnight at the same temperature. After the reaction, free peptide and extra salts were discarded by dialysis using the similar manner aforementioned. Any precipitation was removed by a 220 nm syringe filter. Finally, the purified RGD-modified T1-Fe3O4 were stored at 4 °C for long-term experiments.
Characterizations
The morphology of the nanoparticles was imaged using transmission electron microscopy (TEM) and high resolution TEM (HRTEM) (JEOL, Japan). The iron concentration of Fe3O4 NPs was measured with a classical C-A (absorbance versus Fe concentration) calibration curve on a UV-visible spectrophotometer (UV-3600, Shimadzu, Japan). Magnetism of T1-Fe3O4 was conducted on a vibrating sample magnetometer (VSM, Lakeshore 7407, USA) at room temperature. Hydrodynamic diameter (DH) and Zeta potential measurements were performed on a particle size analyzer (Malvern Zetasizer, UK). The functional groups present in the powder samples were identified using Infrared (IR) analyses on a fourier transform infrared spectrometer (FTIR, Nicolet Nexus 870, USA). The thermal behavior of the powders was studied by thermal gravimetric analyses (TGA) using a Thermo Gravimetric Analyzer (Pyris 1 DSC, USA). Iron content in different tissue samples was quantified by inductively coupled plasma atomic emission spectrometer (ICP-AES, Optima 5300DV, USA).
MRI phantom studies
To measure the r1 and r2 of PEGylated and RGD-modified T1-Fe3O4, all samples with different iron concentrations were obtained. T1- and T2-weighted MR images were acquired on a clinic 3.0 T MR scanner (Verio, Siemens, Germany) with a head coil. Modified Look-Locker Inversion Recovery (MOLLI) sequence was used to measure the absolute T1 values of phantoms containing different Fe contents from the quantitative T1 maps. Imaging parameters were as follows: flip angle=35, TR=284.38 ms, TE=1.1 ms, TI=88, 188, 2088, 2204, 4104, 4221 and 6121 ms, field of view (FOV)=340×273, matrix=144×224, slice thickness/gap=5.0 mm/1.0 mm, and NEX=8. True Fast Image with Steady-state Precession (TrueFISP) sequence was used to measure the absolute T2 values of phantoms from the quantitative T2 maps and the following parameters were adopted as flip angle=160, TR=13200 ms, TE=84, 176 and 268 ms, FOV=334×334, matrix=320×320, slice thickness/gap=5.0 mm/1.0 mm and NEX=1.
Non-specific phagocytosis and cytotoxicity experiments
RAW 264.7 cells (Murine macrophage cells) were separately incubated with 3 mL culture medium containing PEGylated, RGD-modified or DMSA-modified T1-Fe3O4 for 24 h. After removing the culture medium, the fixed cells were incubated with Prussian blue solution containing 2% hydrochloric acid aqueous solution and 2% potassium ferrocyanide (II) trihydrate for 30 min to stain the intracellular Fe3O4 NPs. Fixed cells were counterstained with nuclear fast red, and placed under a microscope for the observation of intracellular Fe3O4 NPs after rinse with PBS for 3 times. RAW 264.7 cells, 4T1 cells and H1975 cells were used to evaluate the cytotoxicity of the RGD-modified T1-Fe3O4. For Methyl thiazolyl tetrazolium (MTT) assay, three kinds of cells were incubated with RGD-modified T1-Fe3O4 with a series of concentrations (0~100 μg Fe/mL) for 24 h, respectively. After this, the cells were incubated in the media with 0.5 mg/mL of MTT for 4 h. Precipitated violet crystals were dissolved in 150 mL of Dimethyl sulfoxide (DMSO) and the absorbance was measured at 490 nm on a microplate reader. Cell viability was expressed as the percentage of viable cells compared with the control groups (cells incubated with PBS).
Cellular uptake by HUVECs
To evaluate the targeting specificity of RGD-modified T1-Fe3O4 in vitro, human umbilical vein endothelial cells (HUVECs) that overexpress integrin αvβ3 were chosen and separately incubated with 3 mL culture medium containing PEGylated or RGD-modified T1-Fe3O4 with a concentration of 100 μg Fe/mL for 12 h. Competition experiments were conducted by adding free c(RGDyK) peptide to RGD-modified T1-Fe3O4 group in a ratio of 100:1 before NPs incubation. Culture medium was removed after incubation. The adherent cells were rinsed using PBS for 3 times. Prussian blue and nuclear fast red staining were acquired in the similar manner aforementioned before the observation of intracellular Fe3O4 NPs.
In vivo MRI experiments
Animal experiments were executed according to the protocol approved by Institutional Animal Care and Use Committee of Southeast University. Orthotopic hepatic tumor models were established by inoculation of small subcutaneous HepG2 tumor fragments into the livers of male nude mice (5-week-old, purchased from the Model Animal Research Center of Southeast University). Specifically, 5×106 HepG2 cells in 100 μL cell culture medium were subcutaneously injected into the right leg of nude mice. After implantation for 2 weeks, the subcutaneous tumor tissue was excised and cut into small cubes (size: 1 mm×1 mm×1 mm). Next, the tumor cubes were transplanted into the livers (the upper lobes) of nude mice by using microsurgical operation. 2-week inoculation later, the mice bearing orthotopic hepatic tumor in a size range of 2~3 mm were subjected into 2 groups (one active targeting group and one non-active targeting group, 3 tumor-bearing mice per group) for the following MRI studies. After intravenous injection of RGD-modified T1-Fe3O4 or PEGylated T1-Fe3O4 with a dose of 5.0 mg Fe/kg, T1-weighted MR images across the tumor in transverse plane were acquired by using Multi Slice Multi Echo (MSME) sequence (Respiratory gating contained) on a 7.0 T Micro-MR scanner (PharmaScan, Brukers, Germany) with the following parameters: flip angle=180, TR=498.3 ms, TE=14.0 ms, FOV=3×3, matrix=256×256, SI=1.0 mm/1.0 mm, averages=3, slices=16, NEX=1. Subsequently, T2-weighted images were acquired by using Rapid Acquisition Relaxation Enhanced (RARE) sequence (Respiratory gating contained) under the following parameters: flip angle=180, TR=2500.0 ms, TE=33.0 ms, FOV=3×3, matrix=256×256, SI=1.0 mm/1.0 mm, averages=3, slices=16, NEX=1. MR images were obtained before and at differential time points of 10, 20, 30, 60, 120 and 180 min post administration. Signal-to-noise ratio (SNR) was calculated for each mouse to qualify the signal enhancement in the region of interest (ROI) by the following equation: SNR=SI/SDnoise, where SI stands for signal intensity in ROIs and SD stands for standard deviation analyzed from the MR images. The values of SNRpost/SNRpre were calculated to represent the signal changes. Tumor-to-liver contrast-to-noise ratio (CNR) here was introduced for each mouse to qualify the efficacy of contrast enhancement in hepatic tumors comparing with the surrounding normal liver tissue, where CNR=(SNRtumor-SNRliver)/SNRtumor. The values of CNRpost/CNRpre were calculated to represent the signal changes.
Histopathological staining
Tumors and livers from 6 experimental mice (3 injected with RGD-modified T1-Fe3O4 and 3 injected with PEGylated T1-Fe3O4) were excised and fixed in 10% neutral buffered formalin for 2 days. Then, the tissues were processed routinely into paraffin and sectioned into 5-micrometer thickness slices. For Prussian blue staining, the sections were put into Prussian blue solution and incubated at room temperature for 30 min. After rinse with PBS, sections were counterstained with nuclear fast red. After rinsing again, sections were dehydrated by 70%, 80% and 100% ethanol, then transferred into xylene, and finally mounted. Sections could be observed under an optical microscopy. An immunohistochemical analysis method was used to further evaluate the specific location of RGD-modified T1-Fe3O4 accumulated in hepatic tumors. Tumor angiogenesis were firstly stained with primary rat anti-mouse CD31 antibody. CD31 staining with brown color could be reached by using glucose oxidase-diaminobenzidine (DAB) method. The procedures for Prussian blue and nuclear fast red staining were virtually the same aforementioned and then the sections could be observed under an optical microscopy.
In vivo blood half-life and biodistribution studies
5-week-old mice were adopted for the blood half-life studies of RGD-modified T1-Fe3O4, which was intravenously injected into the mice (n=6). The dose level was the same as that for imaging experiments. Blood samples were obtained via retro-orbital blood collection at 0 min, 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h postinjection. Then, the samples were digested by aqua regia solution (nitric acid/hydrochloric acid, volume/volume=1 : 3) after the red blood cells were removed by centrifugation at 1000 rpm. The Fe content in different samples was determined by ICP-AES. Tumor-bearing mice were euthanized at 5 h post-injection (3 injected with RGD-modified T1-Fe3O4 and 3 injected with PEGylated T1-Fe3O4, the dose level was the same as that for imaging experiments). The heart, liver, spleen, lung, kidney and tumor were extracted and weighed. The tissues were then cut into pieces and digested by aqua regia solution for 24 h. The Fe content in different tissues was quantified by ICP-AES. For comparison, 3 mice without injection were used as control group.
Statistical analysis
Student's t test statistical analysis was performed to evaluate the significance of the experimental data. A p value of 0.05 was selected as the level of significance. The data was indicated with (*) for p<0.05 and (**) for p<0.01, respectively.
Results and Discussion
Preparation and characterizations of RGD-modified T1-Fe3O4
Uniform T1-Fe3O4 was synthesized via thermal decomposition of Fe(acac)3 as precursors in the presence of oleylamine and benzyl ether. TEM image presented that oleylamine coated T1-Fe3O4 had a relatively narrow size distribution with mean core size of 5.3±0.6 nm (Figure S1). The hydrophobic NPs could be easily transformed to aqueous medium for further applications through a simple and effective ligand exchange reaction with the biocompatible ligands, carboxylated PEG (Figure 1a). As mentioned in Materials and methods section, PEG ligands bearing a dopamine group at one end (denoted as DA-PEG-COOH) were firstly fabricated, and the catechol units of dopamine having strong binding affinity to transition metal were chosen as anchor groups to decorate on the surface of T1-Fe3O4 by replacing the oleylamine molecular. According to the TEM and HRTEM images (Figure 2a and b), PEGylated T1-Fe3O4 were monodispersed in quasi-spherical shapes with good crystalline and negligible surface etching. Dynamic light scattering (DLS) measurements revealed that PEGylated T1-Fe3O4 had ultra-small DH of 10±0.7 nm and homogeneously dispersed in aqueous media without detectable aggregation (Figure 2d). Meanwhile, free carboxylic groups existing on DA-PEG-COOH made it possible to construct tumor targeting probes via conjugating specific ligands. In our work, a kind of small molecular weight peptide, c(RGDyK), was chosen to covalently conjugate on T1-Fe3O4 surface through the formation of amide bonds (Figure 1b). This cyclized RGD peptide has been proven to specifically target to integrin αvβ3, which is overexpressed on tumor angiogenesis.[23-25]
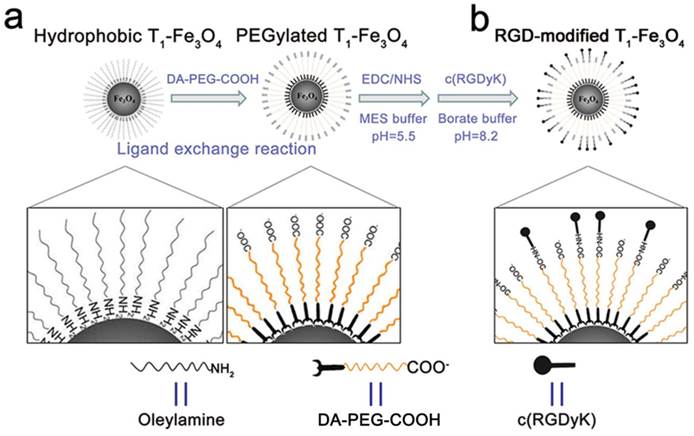
Schematic illustration of PEGylated T1-Fe3O4 and RGD-modified T1-Fe3O4 syntheses. (a) Hydrophobic T1-Fe3O4 were transformed to water-dispersible through a ligand exchange reaction with DA-PEG-COOH. (b) c(RGDyK) peptide was conjugated through the formation of amide bonds via free carboxyl groups on the surface of PEGylated T1-Fe3O4.
(a) TEM image of monodispersed PEGylated T1-Fe3O4. (b) The higher magnification TEM image of PEGylated T1-Fe3O4. (inset: HRTEM image. Scale bar, 5 nm) (c) TEM image of monodispersed RGD-modified T1-Fe3O4. (inset: HRTEM image. Scale bar, 5 nm). (d) DH of PEGylated T1-Fe3O4 and RGD-modified T1-Fe3O4. The sizes were measured from water solution by DLS. (e) Field-dependent magnetic hysteresis loops of PEGylated T1-Fe3O4 and RGD-modified T1-Fe3O4 recorded at room temperature.
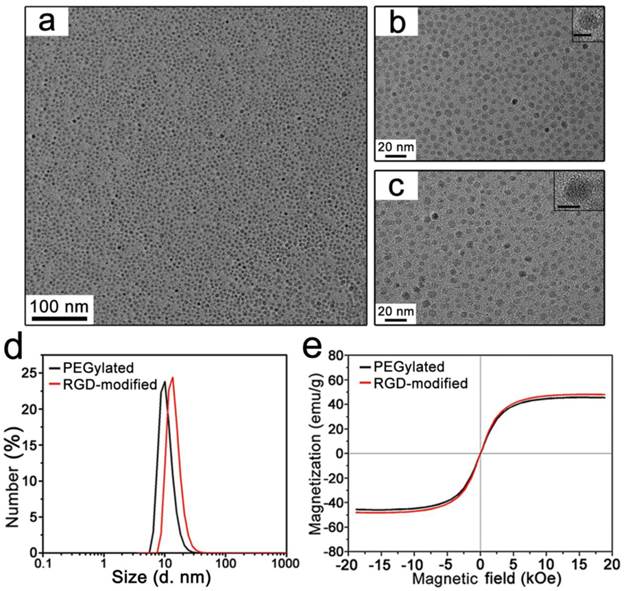
The formation of RGD-modified T1-Fe3O4 was investigated by a series of characterizations in careful comparisons with the corresponding PEGylated T1-Fe3O4. Figure 2c is representative TEM image of RGD-modified T1-Fe3O4. Nearly no differences were detected in their core morphology and RGD-modified T1-Fe3O4 was distributed with an increasing distance between the particles. These tendencies could be supported by the following DLS measurements (Figure 2d). DH of RGD-modified T1-Fe3O4 slightly increased from 10 nm to 15 nm right after the conjugation reaction. It also could be demonstrated that the conjugation reaction was well-controlled from the magnetic properties of PEGylated and RGD-modified T1-Fe3O4, which are strongly dependent on the sizes and status (dispersed or aggragated) of Fe3O4 NPs. Field-dependent magnetic hysteresis loops showed that both PEGylated and RGD-modified T1-Fe3O4 exhibited favorable superparamagnetic behavior with roughly equal saturated magnetic moment (~50 emu/g Fe element mass in Fe3O4 NPs, Figure 2e), indicating that no unexpected aggregation or clusters further formed during the conjugation reaction. IR analyses were investigated to qualitatively confirm the presence of c(RGDyK) peptide on T1-Fe3O4. From IR spectra (Figure S2), the peak at 1100 cm-1 was attributed to the stretching vibration of C-O-C group from PEG ligand. A strong peak occurred at 1600 cm-1 for RGD-modified T1-Fe3O4, which was caused by the vibration of -NH2 group and in consistent with the free RGD peptide, demonstrating the successful conjugation. Zeta potentials of PEGylated and RGD-modified T1-Fe3O4 in neutral aqueous were -41±3 mV and -11±2 mV, respectively (Figure S3a and b). The reduction in negative charge after conjugation was due to the partial absence of free carboxylic groups on RGD-modified T1-Fe3O4. TGA were performed to estimate the efficiency of the conjugation. It presented that the relative loss weight of PEGylated T1-Fe3O4 was about 54.26%, while this value increased to 57.92% for RGD-modified T1-Fe3O4 (Figure S4). According to the method reported previously,[12] the number of PEG and c(RGDyK) conjugated on the surface of one Fe3O4 NP could be roughly estimated as high as 117.8 and 78.5 respectively, and the conjugation efficiency was about 66.7% (the molar ratio between free carboxylic groups existed on PEGylated T1-Fe3O4 and the c(RGDyK) peptide successfully conjugated). In addition, RGD-modified T1-Fe3O4 exhibited excellent stability in aqueous dispersion during a long period time of 15 months without obvious aggregation (Figure S5), which was benefit from the denser PEG coating on T1-Fe3O4 in comparison with other PEGylated NPs reported previously.[12, 26, 27]
Relaxivity measurements
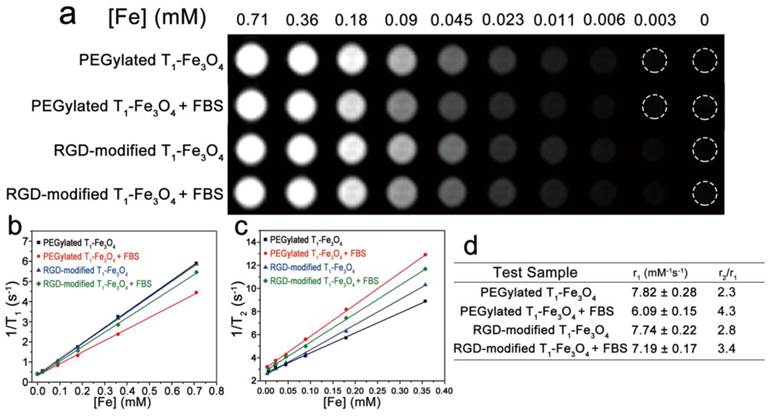
Relaxivity measurements were performed at the clinically relevant magnetic field strength of 3.0 T, which could better reflect the relaxation properties of NPs for clinic application. Figure 3a showed the T1-weighted images of PEGylated and RGD-modified T1-Fe3O4 at different Fe concentrations in distilled water or 20% fetal bovine serum (FBS) solution. Herein, NPs were incubated with FBS before MR studies in order to investigate their relaxation properties sustaining from the adsorption of serum proteins under physiological conditions. Evidently, no matter in water or FBS solution, all test samples induced favorable bright contrast enhancement even at an extremely low Fe concentration of 0.09 mM. To further evaluate their T1 contrast efficacy, r1 and r2 of PEGylated and RGD-modified T1-Fe3O4 in different medium were extracted (Figure 3b and c). PEGylated and RGD-modified T1-Fe3O4 exhibited relatively high r1 of 7.82±0.28 mM-1s-1 and 7.74±0.22 mM-1s-1 in distilled water, respectively. After incubation with FBS for 4 h, r1 of both samples slightly decreased whereas r2 had some increase and hence the r2/r1 ratio raised (Figure 3d). This phenomenon was probably due to the occurrence of the irreversible, partial aggregation of NPs during the incubation which was known to be responsible for the shortening of transversal relaxation time. In this case, RGD-modified T1-Fe3O4 with larger DH would have stronger steric hindrance effect against the aggregation processes that would affect its relaxation properties. Nevertheless, PEGylated and RGD-modified T1-Fe3O4 still held relatively low r2/r1 ratio comparing with other Fe3O4 based T1 contrast agents reported previously.[9, 11-14, 28] Generally, low r2/r1 ratio is a key parameter widely used to confirm the strong T1 contrast efficiency of one MRI contrast agent.[28] The relaxivities of these two NPs in distilled water were also measured on a 7.0 T Micro-MR scanner and the results were shown in Figure S6, respectively. At 7.0 T, the r2 value was much higher since the transversal relaxation time of Fe3O4 NPs was highly field strength dependent. However, these values were still comparable with some previous reports measured under the same condition.[9] More importantly, optimal T1 relaxation property of RGD-modified T1-Fe3O4 could kept as long as 15 months (Figure S7), which was in good agreement with its colloidal stability investigated before, and was highly essential for RGD-modified T1-Fe3O4 when used as long-term stable T1 contrast agent for further applications.
(a) T1-weighted MR images of PEGylated T1-Fe3O4 and RGD-modified T1-Fe3O4 at different Fe concentrations in water or 20% FBS solutions. Plots of (b) 1/T1 and (c) 1/T2 against Fe concentration of PEGylated T1-Fe3O4 and RGD-modified T1-Fe3O4 in water or 20% FBS solutions at 3.0 T. r1 and r2 were calculated from the slopes of the corresponding linear fits of the experimental data. (d) Relaxation properties of different test samples measured in (b) and (c).
In vitro studies
Another key consideration for the in vivo use of NPs for tumor targeting is assessing their ability to resist against the non-specific uptake by MPS in living subjects. NPs without suitable surface modification would significantly increase the non-specific recognition by macrophages and facilitate the elimination of NPs.[29-31] Herein, we conducted cellular phagocytosis experiments of PEGylated and RGD-modified T1-Fe3O4 by using RAW 264.7 cells belonging to murine macrophages as preludes to the in vivo studies. Meanwhile, another common ligand modified T1-Fe3O4, known as meso-2, 3-dimercaptosuccinic acid (DMSA)-modified T1-Fe3O4 were tested under the same condition as a comparison.[32] Representative microscopy images of RAW 264.7 cells after incubation with T1-Fe3O4 and Prussian blue staining were shown in Figure 4a-c. It could be observed that PEGylated and RGD-modified T1-Fe3O4 incubation induced non-detectable cellular uptake whereas nearly all RAW 264.7 cells incubated with DMSA-modified T1-Fe3O4 were stained with blue even at an extremely low concentration of 12.5 μg Fe/mL (Figure S8). These results confirmed the significant effect of PEGylation to drastically minimize the non-specific phagocytosis by macrophages even though there were large numbers of c(RGDyK) peptide on PEGylated NPs surface, which would endow RGD-modified T1-Fe3O4 with prolonged blood half-life to reach tumors in vivo.
Prussian blue staining of RAW 264.7 macrophages incubated with (a) PEGylated T1-Fe3O4, (b) RGD-modified T1-Fe3O4 and (c) control group for 24 h at Fe concentration of 100 μg/mL. Prussian blue staining of HUVECs incubated with (d) PEGylated T1-Fe3O4, (e) RGD-modified T1-Fe3O4, (f) RGD-modified T1-Fe3O4 plus free c(RGDyK) peptide (competition group) and (g) control group for 12 h at Fe concentration of 100 μg/mL. (h) Cytotoxic effects of RGD-modified T1-Fe3O4 on RAW 264.7, 4T1 and H1975 cells measured with the MTT assay kit by incubation with various concentrations of Fe for 24 h. The error bars represented ±s.d. of six independent experiments. Scale bar: 20 μm for all images.
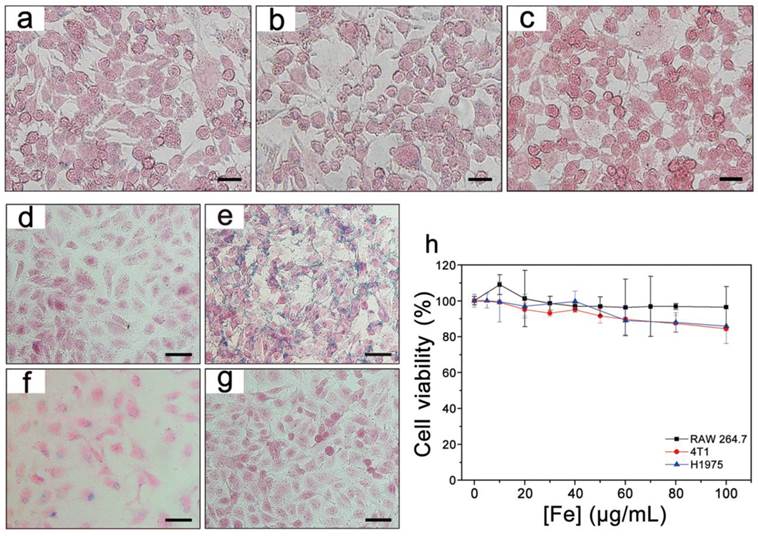
In order to evaluate the targeting specificity to integrin αvβ3, RGD-modified T1-Fe3O4 were incubated with HUVECs (a kind of cells overexpress integrin αvβ3) and PEGylated T1-Fe3O4 were incubated under the same condition as the negative control.[33] Besides, the competition experiments were conducted by adding free c(RGDyK) to RGD-modified T1-Fe3O4 group in a ratio of 100:1 to block the integrin region. Figure 4d-g presented the Prussian blue staining of HUVECs after incubation. Strong targeting specificity was observed for RGD-modified T1-Fe3O4 group whereas there was no detectable NPs for PEGylated T1-Fe3O4 group. Blocking the integrin αvβ3 with free c(RGDyK) led to significant decrease of blue deposition, providing a direct evidence that RGD-modified T1-Fe3O4 specifically targeted to HUVECs via the interaction between RGD on T1-Fe3O4 and the integrin αvβ3 expressing on HUVECs. Moreover, the prominent target specificity still maintained after 15 months (Figure S9). This was benefit from the higher stability of c(RGDyK) comparing with other tumor-specific ligands (e.g., antibodies) which are easier to loss bioactivities. Notably, this was quite long time reported so far for imaging probes to keep their targeting specificity.
MTT assays were carried out to evaluate the cytotoxicity of RGD-modified T1-Fe3O4 in vitro. Figure 4h presented that cell viability exceeded 80% even at a high incubating concentration of 100 μg Fe/mL, which was much higher than the clinical dose for Feridex (0.56 mg Fe/kg body weight), demonstrating the low cytotoxicity and favorable biocompatibility of RGD-modified T1-Fe3O4 as MRI contrast agent for in vivo applications.
Active-target T1 imaging of hepatic tumors in vivo
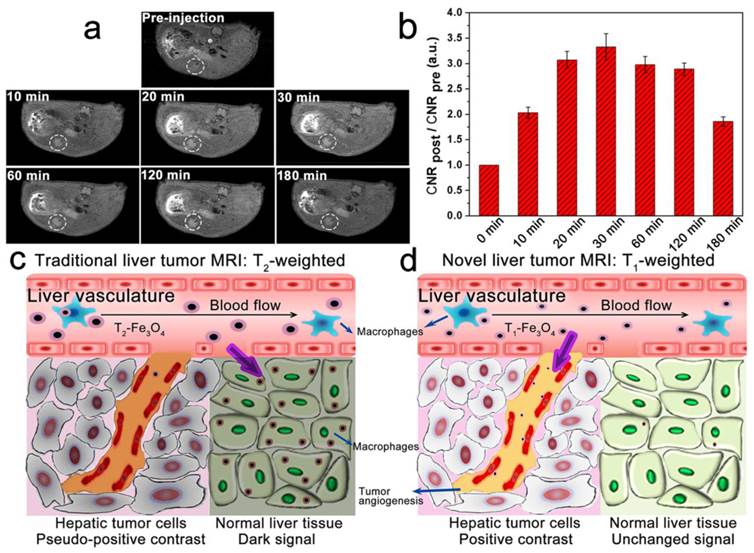
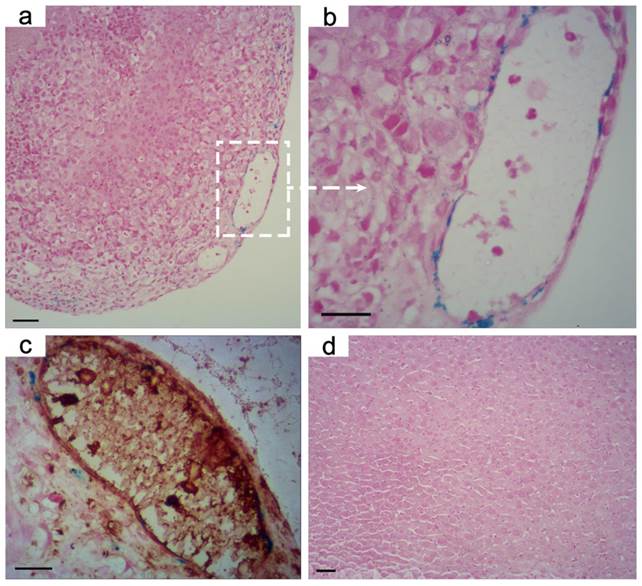
With the properties of low non-specific macrophages uptake and high targeting specificity to integrin αvβ3, we further verified the active-target T1 imaging ability of RGD-modified T1-Fe3O4 on orthotopic hepatic tumors in vivo. Notably, orthotopic tumor models conducted here not only reflected the natural environment of hepatic tumors, but also could be taken as metastases in liver.[34] After intravenous injection of RGD-modified T1-Fe3O4 at a dose of 5 mg Fe/kg (calculated in terms of Fe element mass per kilogram of mice body weight), we indeed observed the bright contrast enhancement in tumor over time, leading to easy identification of the tumor as small as 2.2 mm from surrounding normal liver tissue (Figure 5a). According to the formula in Materials and methods section, tumor-to-liver CNR was calculated to quantify signal changes and it increased rapidly to reach the maximum at 30 min which was about 3.33 times stronger than pre-injection (Figure 5b). To further demonstrate the active targeting of RGD-modified T1-Fe3O4, the corresponding PEGylated T1-Fe3O4 were injected at a same dose for comparison to conduct the non-active-target imaging (Figure S10). Obviously, the latter induced weaker bright contrast and it took longer to reach the maximum tumor-to-liver CNR, which can be interpreted by the limited tumor uptake of PEGylated T1-Fe3O4 through the enhanced permeation and retention (EPR) effect.[4] At the same time, we also examined the liver uptake of NPs by calculating liver SNR changes from T1 and T2 images because the signal in liver would obviously drop in T2 images if Fe3O4 NPs largely accumulated in liver parenchyma (Figure S11). While, SNR value in normal liver tissue, no matter in T1-weighted images or T2-weighted images, was almost unchanged after the injection of both NPs, indicating their little liver uptake (Figure S12). After imaging test, hepatic tumors and normal liver tissue from the experimental mice (injected with RGD-modified or PEGylated T1-Fe3O4) were harvested and stained with Prussian blue. Obvious blue spots appeared in tumor while they were hardly found in normal liver tissue for the active targeting group (Figure 6a, b and d), suggesting that RGD-modified T1-Fe3O4 specifically targeted to hepatic tumor with little undesired phagocytosis in liver parenchyma. Although PEGylated T1-Fe3O4 exhibited the similar liver phagocytosis result (Figure S13a), it was difficult for non-active targeting NPs to permeate into hepatic tumors due to the limited EPR effect (Figure S13b). The present RGD-modified and PEGylated T1-Fe3O4 were more resistant to non-specific uptake than the NPs reported in other studies, which still mainly accumulated in liver parenchyma,[27] can be ascribed to its ultra-small hydrodynamic size, denser PEG coating and higher stability in aqueous dispersion as revealed in our former experiments. The immunohistochemical staining of CD31 definitely disclosed that the RGD-modified T1-Fe3O4 accumulated in tumor were mostly localized on the integrin αvβ3 expressing tumor angiogenesis (brown color stained), which were the preferential and specific targets of RGD-modified T1-Fe3O4 for hepatic tumors active targeting in vivo (Figure 6c).
(a) T1-weighted MR images of mice bearing orthotopic hepatic tumor (white dashed circles) before and at the time points of 10, 20, 30, 60, 120 and 180 min after the administration of RGD-modified T1-Fe3O4. (b) Quantification of T1 signal changes of tumor-to-liver CNR at the corresponding time points. The error bars represented ±s.d. of three independent experiments. Hepatic tumors were differentiated from normal liver tissue through (c) pseudo-positive contrast effects by using traditional T2-Fe3O4 or (d) positive contrast enhancement by using novel tumor-target RGD-modified T1-Fe3O4.
Prussian blue stained images of orthotopic hepatic tumor at the magnifications of (a) 200, (b) 400 after the administration of RGD-modified T1-Fe3O4. (c) Overlay of Prussian blue and CD31 on the corresponding tumor slice in (b). Tumor angiogenesis were stained with brown. (d) Prussian blue stained image of normal liver tissue after the administration of RGD-modified T1-Fe3O4. It could be observed that RGD-modified T1-Fe3O4 mainly accumulated in tumor angiogenesis with little phagocytosis by normal liver tissue which contains a great quantity of activated macrophages. Scale bar: 20 μm for all images.
Accordingly, differ from the traditional liver tumor imaging through pseudo-positive contrast effects by using T2-Fe3O4 that induce dark signal in normal liver tissue (Figure 5c), RGD-modified T1-Fe3O4 having ultra-small DH and appropriate surface decoration could specifically target to tumor angiogenesis to produce bright contrast enhancement in hepatic tumors with little liver phagocytosis (Figure 5d). This novel active-target T1-weighted imaging could significantly improve the sensitivity of early liver cancer detection and effectively minimize the potential toxicity of heavy metals in body at the same time, which is of great importance in clinical diagnosis.
In vivo biodistribution
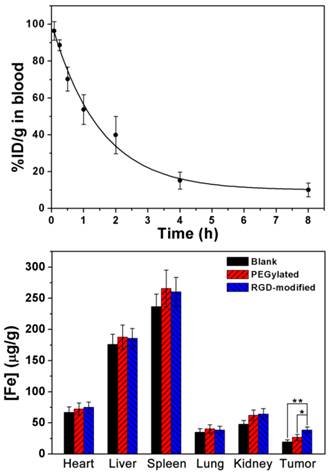
Blood half-life and biodistribution of RGD-modified T1-Fe3O4 were quantitatively analyzed to further disclose its pharmacokinetic behavior in vivo (Figure 7). By fitting the blood concentrations of RGD-modified T1-Fe3O4 acquired at different time points postinjection, the blood half-life was estimated to be 92 min, which was much longer than that of clinical Gd complex small molecules (several minutes in small animals) and other iron oxide based NPs.[9, 11] ICP-AES data revealed that some spleen accumulation of Fe occurred after the injection of RGD-modified T1-Fe3O4 while only a tiny amount of Fe was absorbed by other organs (e.g., heart and lung). Comparing with the similar NPs in the literature,[27] significantly less Fe accumulated in liver parenchyma, which was in good agreement with the MRI and Prussian blue staining results. More importantly, Fe concentration in the tumors of mice injected with the RGD-modified NPs was much higher than that injected with the non-active-target NPs (p<0.05). These results fully demonstrated the important roles of dense PEGylation and RGD targeting, which endowed RGD-modified T1-Fe3O4 with prolonged blood circulation, strong resistance to liver uptake, more effective delivery to hepatic tumors, and consequently allowed for the efficient detection of tiny hepatic tumors from the surrounding normal liver tissue through T1-weighted MR imaging.
(a) Blood clearance profile of RGD-modified T1-Fe3O4 in mice (n=6). The solid line was theoretical fitting curve depicting the clearance behavior. (b) In vivo biodistribution of Fe in some major organs (e.g., heart, liver, spleen, lung and kidney) and in hepatic tumor at 5 h after the administration of PEGylated T1-Fe3O4 or RGD-modified T1-Fe3O4 (5.0 mg Fe/kg). The error bars represented ±s.d. of three independent experiments (*, p<0.05; **, p<0.01).
Conclusions
In summary, we have successfully developed a novel strategy for the efficient active-target imaging of hepatic tumors by using T1-Fe3O4 conjugated with c(RGDyK). The bioconjugation reaction was well-controlled and RGD-modified T1-Fe3O4 possessed relatively high r1 of 7.74 mM-1s-1 and ultralow r2/r1 ratio of 2.8. Systematic stability studies revealed that RGD-modified T1-Fe3O4 could be stored as monodispersed colloid in water for 15 months with quite little changes in DH or relaxation properties. Moreover, the targeting specificity to integrin αvβ3 could be kept as long as 15 months. Dense PEG coating endowed RGD-modified T1-Fe3O4 with favorable biocompatibility and strong ability to resist against the non-specific phagocytosis. Quite outstandingly, the intravenous administrated RGD-modified T1-Fe3O4 could significantly improve the diagnostic sensitivity and made it possible to clearly identify orthotopic hepatic tumor as small as 2.2 mm through active-target T1-weighted MR imaging, which has been recognized as one enormous challenge for hepatic tumor detection so far. Therefore, we believe that RGD-modified T1-Fe3O4 have much promising applications as highly sensitive positive contrast agent for MR imaging of various diseases with ongoing angiogenesis, especially for the accurate and early detection of liver cancer in patients.
Supplementary Material
Figures S1-S13.
Acknowledgements
This work was supported by the National Key Basic Research Program of China (2013CB733800 and 2011CB933503), National Natural Science Foundation of China (31170959 and 81571806), the Basic Research Program of Jiangsu Province (BK2011036), the Jiangsu Provincial Technical Innovation Fund for Scientific and Technological Enterprises (SBC201310643) and the Jiangsu Provincial Special Program of Medical Science (BL2013029). The authors thank Dr. Jun-Jie Yin for his help and good comments concerning improvements to this work.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schroeder A, Heller DA, Winslow MM, Dahlman JE, Pratt GW, Langer R. et al. Treating metastatic cancer with nanotechnology. Nat Rev Cancer. 2012;12:39-50
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. Ca-cancer J Clin. 2015;65:87-108
3. Kumano S, Murakami T, Kim T, Hori M, Okada A, Sugiura T. et al. Using superparamagnetic iron oxide-enhanced MRI to differentiate metastatic hepatic tumors and nonsolid benign lesions. AJR Am J Roentgenol. 2003;181:1335-9
4. Liu C, Gao Z, Zeng J, Hou Y, Fang F, Li Y. et al. Magnetic/upconversion fluorescent NaGdF4:Yb,Er nanoparticle-based dual-modal molecular probes for imaging tiny tumors in vivo. Acs Nano. 2013;7:7227-40
5. Zhou Z, Huang D, Bao J, Chen Q, Liu G, Chen Z. et al. A synergistically enhanced T1-T2 dual-modal contrast agent. Adv Mater. 2012;24:6223-8
6. Bellin MF, Zaim S, Auberton E, Sarfati G, Duron JJ, Khayat D. et al. Liver metastases: safety and efficacy of detection with superparamagnetic iron oxide in MR imaging. Radiology. 1994;193:657-63
7. Zhao Z, Zhou Z, Bao J, Wang Z, Hu J, Chi X. et al. Octapod iron oxide nanoparticles as high-performance T2 contrast agents for magnetic resonance imaging. Nat Commun. 2013;4:1161-71
8. Huang J, Bu L, Xie J, Chen K, Cheng Z, Li X. et al. Effects of nanoparticle size on cellular uptake and liver MRI with polyvinylpyrrolidone-coated iron oxide nanoparticles. ACS nano. 2010;4:7151-60
9. Zhou Z, Wang L, Chi X, Bao J, Yang L, Zhao W. et al. Engineered iron-oxide-based nanoparticles as enhanced T1 contrast agents for efficient tumor imaging. ACS nano. 2013;7:3287-96
10. Sadauskas E, Wallin H, Stoltenberg M, Vogel U, Doering P, Larsen A. et al. Kupffer cells are central in the removal of nanoparticles from the organism. Part Fibre Toxicol. 2007;4:10
11. Kim BH, Lee N, Kim H, An K, Park YI, Choi Y. et al. Large-scale synthesis of uniform and extremely small-sized iron oxide nanoparticles for high-resolution T1 magnetic resonance imaging contrast agents. J Am Chem Soc. 2011;133:12624-31
12. Sandiford L, Phinikaridou A, Protti A, Meszaros LK, Cui X, Yan Y. et al. Bisphosphonate-anchored pegylation and radiolabeling of superparamagnetic iron oxide: long-circulating nanoparticles for in vivo multimodal (T1 MRI-SPECT) imaging. Acs Nano. 2012;7:500-12
13. Taboada E, Rodríguez E, Roig A, Oró J, Roch A, Muller RN. Relaxometric and magnetic characterization of ultrasmall iron oxide nanoparticles with high magnetization. Evaluation as potential T1 magnetic resonance imaging contrast agents for molecular imaging. Langmuir. 2007;23:4583-8
14. Li Z, Yi PW, Sun Q, Lei H, Li Zhao H, Zhu ZH. et al. Ultrasmall water-soluble and biocompatible magnetic iron oxide nanoparticles as positive and negative dual contrast agents. Adv Funct Mater. 2012;22:2387-93
15. Huang G, Li H, Chen J, Zhao Z, Yang L, Chi X. et al. Tunable T1 and T2 contrast abilities of manganese-engineered iron oxide nanoparticles through size control. Nanoscale. 2014;6:10404-12
16. Longmire M, Choyke PL, Kobayashi H. Clearance properties of nano-sized particles and molecules as imaging agents: considerations and caveats. Nanomedicine. 2008;3:703-17
17. Perez JM, Josephson L, Weissleder R. Use of magnetic nanoparticles as nanosensors to probe for molecular interactions. Chembiochem. 2004;5:261-4
18. Roch A, Gossuin Y, Muller RN, Gillis P. Superparamagnetic colloid suspensions: water magnetic relaxation and clustering. J Magn Magn Mater. 2005;293:532-9
19. Lee N, Choi Y, Lee Y, Park M, Moon WK, Choi SH. et al. Water-Dispersible Ferrimagnetic Iron Oxide Nanocubes with Extremely High r2 Relaxivity for Highly Sensitive in Vivo MRI of Tumors. Nano Lett. 2012;12:3127-31
20. Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148:135-46
21. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
22. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249-57
23. Brooks PC, Clark R, Cheresh DA. Requirement of vascular integrin αvβ3 for angiogenesis. Science. 1994;264:569-71
24. Friedlander M, Theesfeld CL, Sugita M, Fruttiger M, Thomas MA, Chang S. et al. Involvement of integrins αvβ3 and αvβ5 in ocular neovascular diseases. Proc Natl Acad Sci USA. 1996;93:9764-9
25. Dechantsreiter MA, Planker E, Mathä B, Lohof E, Hölzemann G, Jonczyk A. et al. N-Methylated cyclic RGD peptides as highly active and selective αvβ3 integrin antagonists. J Med Chem. 1999;42:3033-40
26. Liu D, Wu W, Ling J, Wen S, Gu N, Zhang X. Effective PEGylation of iron oxide nanoparticles for high performance in vivo cancer imaging. Adv Funct Mater. 2011;21:1498-504
27. Luo Y, Yang J, Yan Y, Li J, Shen M, Zhang G. et al. RGD-functionalized ultrasmall iron oxide nanoparticles for targeted T₁-weighted MR imaging of gliomas. Nanoscale. 2015;7:14538-46
28. Tromsdorf UI, Bruns OT, Salmen SC, Beisiegel U, Weller H. A highly effective, nontoxic T1 MR contrast agent based on ultrasmall PEGylated iron oxide nanoparticles. Nano Lett. 2009;9:4434-40
29. Li S-D, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5:496-504
30. Walkey CD, Olsen JB, Guo H, Emili A, Chan WC. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc. 2012;134:2139-47
31. Liu C, Hou Y, Gao M. Are rare-earth nanoparticles suitable for in vivo applications? Adv Mater. 2014;26:6922-32
32. Xie J, Zhang Y, Yan C, Song L, Wen S, Zang F. et al. High-performance PEGylated Mn-Zn ferrite nanocrystals as a passive-targeted agent for magnetically induced cancer theranostics. Biomaterials. 2014;35:9126-36
33. Zhang C, Jugold M, Woenne EC, Lammers T, Morgenstern B, Mueller MM. et al. Specific targeting of tumor angiogenesis by RGD-conjugated ultrasmall superparamagnetic iron oxide particles using a clinical 1.5-T magnetic resonance scanner. Cancer Res. 2007;67:1555-62
34. Teicher BA. Tumor models for efficacy determination. Mol Cancer Ther. 2006;5:571-85
Author contact
![]() Corresponding author: Yu Zhang, E-mail: zhangyuedu.cn. Tel: +86 25 8327 2496. Fax: +86 25 8327 2496
Corresponding author: Yu Zhang, E-mail: zhangyuedu.cn. Tel: +86 25 8327 2496. Fax: +86 25 8327 2496